 Marta Gómez de Cedrón
Marta Gómez de Cedrón Rocío Moreno Palomares1,3†
Rocío Moreno Palomares1,3† Ana Ramírez de Molina
Ana Ramírez de Molina
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 19 June 2023
Sec. Molecular and Cellular Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1169168
This article is part of the Research Topic Cancer Plasticity and The Microenvironment: Implications for Immunity and Therapy Response: Volume II View all 13 articles
Epigenetic modifications are chemical modifications that affect gene expression without altering DNA sequences. In particular, epigenetic chemical modifications can occur on histone proteins -mainly acetylation, methylation-, and on DNA and RNA molecules -mainly methylation-. Additional mechanisms, such as RNA-mediated regulation of gene expression and determinants of the genomic architecture can also affect gene expression. Importantly, depending on the cellular context and environment, epigenetic processes can drive developmental programs as well as functional plasticity. However, misbalanced epigenetic regulation can result in disease, particularly in the context of metabolic diseases, cancer, and ageing. Non-communicable chronic diseases (NCCD) and ageing share common features including altered metabolism, systemic meta-inflammation, dysfunctional immune system responses, and oxidative stress, among others. In this scenario, unbalanced diets, such as high sugar and high saturated fatty acids consumption, together with sedentary habits, are risk factors implicated in the development of NCCD and premature ageing. The nutritional and metabolic status of individuals interact with epigenetics at different levels. Thus, it is crucial to understand how we can modulate epigenetic marks through both lifestyle habits and targeted clinical interventions -including fasting mimicking diets, nutraceuticals, and bioactive compounds- which will contribute to restore the metabolic homeostasis in NCCD. Here, we first describe key metabolites from cellular metabolic pathways used as substrates to “write” the epigenetic marks; and cofactors that modulate the activity of the epigenetic enzymes; then, we briefly show how metabolic and epigenetic imbalances may result in disease; and, finally, we show several examples of nutritional interventions - diet based interventions, bioactive compounds, and nutraceuticals- and exercise to counteract epigenetic alterations.
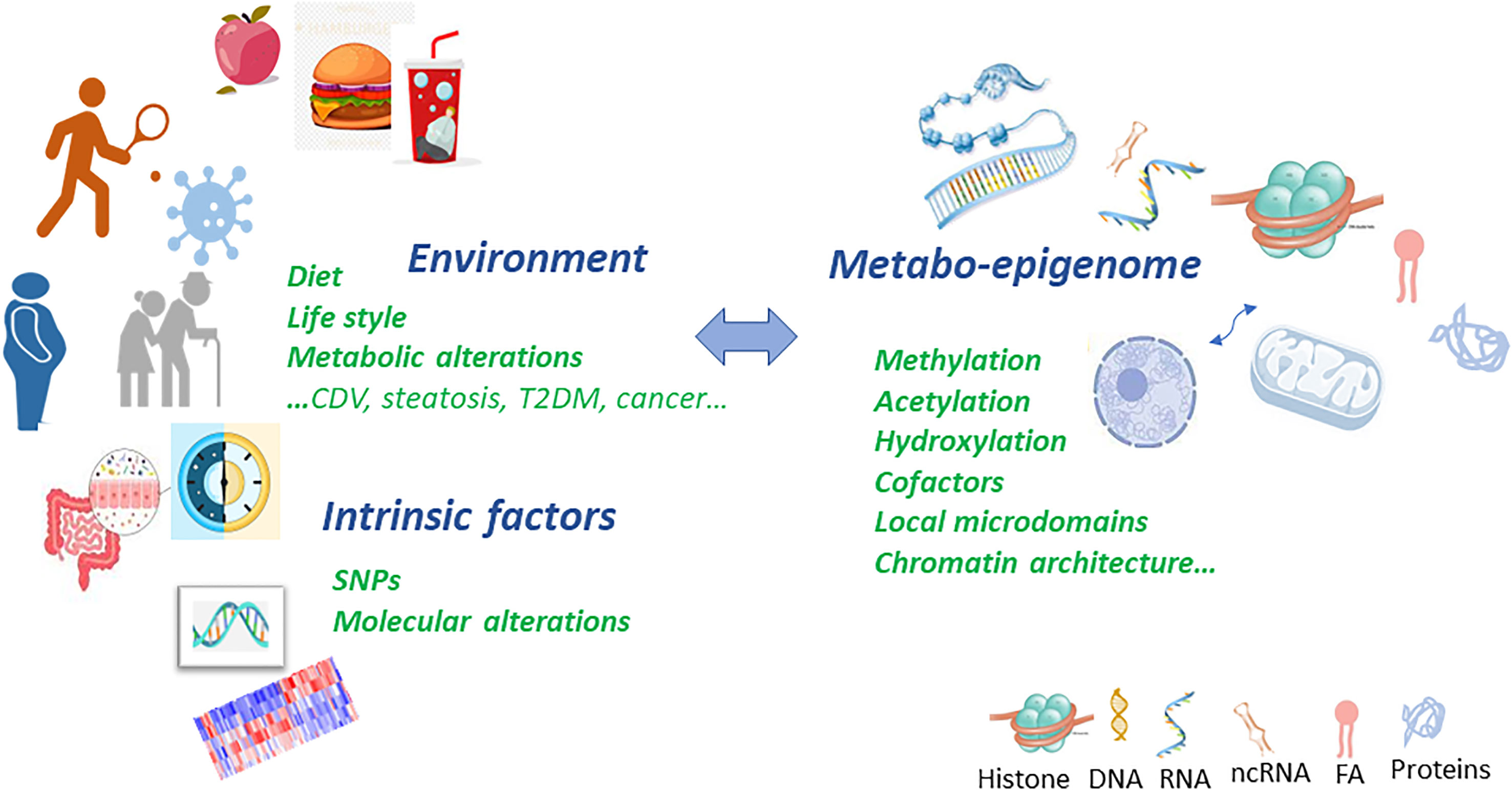
Graphical Abstract Metabolic and epigenetic imbalances result in disease. There are several factors that can cause an epigenetic imbalance and consequently different diseases. Some of these factors are intrinsic including genetic variations such as Single Nucleotide Polymorphism (SNPs) or DNA mutations caused by carcinogens and Reactive Oxygen Species (ROS); environmental factors including metabolic alterations such as obesity or ageing and lifestyle (unbalanced diet or sedentary lifestyle). This epigenetic imbalance leads to cardiovascular, neurological and cancer diseases.
Epigenetics refers to heritable changes in gene expression that are not caused by DNA sequence alterations. Although epigenetics is implicated in cell fate stability in multicellular organisms, it is highly affected by short-term environmental inputs such as stress conditions or nutrient supply (1). Alterations in the epigenome have been associated to a wide variety of disease related processes including non-communicable diseases (NCD), cancer (considered as chronic disease when it can be controlled with treatment, becoming stable and/or reaching remission) and ageing (2, 3). According to the World Health Organization (WHO), 80% of heart diseases, strokes, type 2 diabetes and over a third of cancers can be prevented by modifying life-style factors, such as cutting out tobacco, eating a healthy diet, being physically active and stopping harmful use of alcohol (4). As diet and exercise can modify the metabolic disease risk, it is crucial to understand how life-style factors contribute to shape the epigenome and, subsequently, how alterations in the epigenetic landscape contribute to disease. Indeed, the epigenome is rapidly affected by the availability of central metabolites that can be used as either substrates or allosteric cofactors, fine-tuning the activities of the epigenetic enzymes (5, 6). For this reason, diet based interventions, including nutraceuticals and bioactive compounds, and exercise may provide therapeutic tools to restore a “healthy” epigenome against metabolic diseases (7).
Life-style factors, including diet and physical activity, are the main source of metabolites, which provide the chemical moieties for DNA, RNA and histone modifications, and cofactors, which modulate the activity of epigenetic enzymes-. Indeed, central cellular metabolites, such as S-adenosyl methionine (aCETY), acetyl-coenzyme A (acetyl-CoA), adenosine triphosphate (ATP), nicotinamide adenine dinucleotide (NAD+) and flavin adenine dinucleotide (FAD) are key sensors of nutrient availability and they contribute to the regulation of gene expression by epigenetic mechanisms (8). On the other hand, epigenetics contributes to regulate metabolism by affecting gene expression. Thereby, a crosstalk between epigenetics and metabolism exists to determine molecular programs.
Over the past 20 years, there has been a great interest on the identification and characterization of enzymes responsible for adding or removing epigenetic marks. The activity of many of these chromatin-modifying enzymes -DNA methyltransferases (DNMTs), DNA hydroxylases (DNHDs), histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs), and histone demethylases (HDMs)- is regulated partially by the concentrations of their required metabolic substrates or cofactors (9). Chromatin plays important roles in DNA biology including gene expression regulation. The level of chromatin compaction has important consequences for gene transcription as it influences the accessibility of DNA sequences to transcription factors and other regulatory proteins. Thus, modifications on DNA and histones regulate the level of chromatin compaction either directly or by facilitating the binding of remodeling proteins that recognize modified sites. Modifications of RNA (epitranscriptomics) add an additional level of gene expression regulation and might influence RNA transport, splicing, stability, and translation. Furthermore, non-coding RNA sequences (mainly microRNAs and long non-coding RNAs) have been extensively shown to play a key role in the regulation of gene expression.
Nowadays, it is well established that NCD and ageing are affected by the interaction of life-style factors -diet and exercise-, metabolism and the epigenome. As such, the availability of metabolites is central for the epigenetic changes, partially determining differentiation programs, cell identity, stemness, functional plasticity and environmental responses (10).
Plasticity is crucial for all biological functions, and epigenetics is implicated in the adaptation to multiple signals and conditions by mean of several mechanisms (11, 12):
(1) Changes in the levels of cellular metabolites modulate the epigenome translating the metabolic state of a cell into changes of the chromatin pattern. For example, high levels of acetyl-CoA facilitate the acetylation of histone and non-histone proteins of transcriptional complexes (TC); high levels of NAD+ activate sirtuin-HDACs to promote transcriptional silencing; high glucose levels may increase the synthesis of UDP-GlcNac by the hexosamine pathway, stimulating the activity of the MLL5 methyltransferase to increase GlcNAcylation (13).
(2) DNA-binding factors recruit histone-modifying enzymes to specific chromosomal domains to stimulate their enzymatic activity locally. This is the case of Mat IIIa, implicated in the synthesis of SAM, for H3K9-specific histone methyltransferases to repress transcription (14).
(3) Enzymes that use the same metabolite, such as DNA or histone methyltransferases, may compete with each other resulting into distinct methylation products (15).
(4) Global and local fluctuations in the concentration of cofactors also modulate the activity of epigenetic enzymes. The relative activity of the three sirtuins present in the nucleus (SIRT1, SIRT6, and SIRT7) may modulate the local concentration of NAD+, thereby resulting in the reciprocal regulation of the other sirtuins localized nearby chromatin microdomains.
(5) Epitranscriptomic modifications, including the N6-methyladenosine (m6A), have been shown to regulate the processing, translation, and stability of mRNAs; thereby influencing cell biology (16). Posttranscriptional epigenetic modification of RNA (the so-called ‘epitranscriptome’) has emerged as a fascinating field of research (for a detailed discussion on this topic, readers can refer to several excellent reviews (17–19).
As indicated, metabolism mainly modulates the activity, the localization of chromatin remodelling enzymes, and the levels of substrates and cofactors used by chromatin modifying enzymes. For example, chromatin regulators can directly recognize specific patterns of DNA sequence, such as the percentage of GC (20), or can be affected by the local levels of specific metabolites. Although a vast network of central metabolites are substrates for chromatin regulators, such as histone and DNMTs (21), some others can bind to chromatin proteins affecting the cell metabolism and function. A recent study showed a metabolic link between the macrodomain of H2A1.1 histone regulating the mitochondrial metabolism and cell function by sequestering the NAD+ metabolite for ADP-ribose and ADP-ribosylated proteins (22, 23).
Here, we first describe key metabolites derived from the cellular metabolic pathways that are used as substrates -to “write” the epigenetic marks-; and cofactors that modulate the activity of epigenetic enzymes; then we briefly show how metabolic and epigenetic imbalances result in disease; and finally, we show several examples of nutritional interventions - diet based interventions, bioactive compounds, and nutraceuticals- and exercise to counteract epigenetic imbalances.
DNA methylation is one of the most vastly studied epigenetic marks that determines gene expression profiles, as well as germline imprinting. However, balanced DNA methylation is essential to coordinate these processes. To achieve DNA methylation balance, both, DNA methyltransferase enzymes and the TET (ten eleven translocation) family of dioxygenases mediate the deposition and removal of methyl groups on DNA, respectively.
DNA methyltransferase enzymes -DNMT1, DNMT3a, and DNMT3b- generate 5-methylcytosine (5mC), which is a major transcriptional repressive mark in many eukaryotes (24, 25). DNMT1 is mainly implicated in the germline imprinting (26) meanwhile “de novo’’ methyltransferases (27) are more prone to be activated in response to intracellular metabolic changes. On the contrary, demethylation of 5mC is driven by the TET family of dioxygenases that oxidize 5mC to form 5-hydroxymethylcytosine (5hmC) which can be further oxidized to form carbonylmethylcytosine (camC) and formylmethylcytosine (fmC), or glycosylated (28). The activity and expression levels of cytosine methyltransferases are also affected by specific epigenetic marks and post-translational modifications such as the histone methylation H3K4, H3K36 (29), highlighting the complexity of the epigenetic network.
The methylation and the demethylation of cytosines use central metabolites as substrates such as the one carbon donor SAM for methylation, and alpha ketoglutarate (α-KG) and Fe2+ for TET mediated demethylation, which is enhanced by ascorbate (vitamin C) (30). Thus, the levels of these metabolites shape the cytosine methylation profile with downstream effects on gene expression (Figure 1).
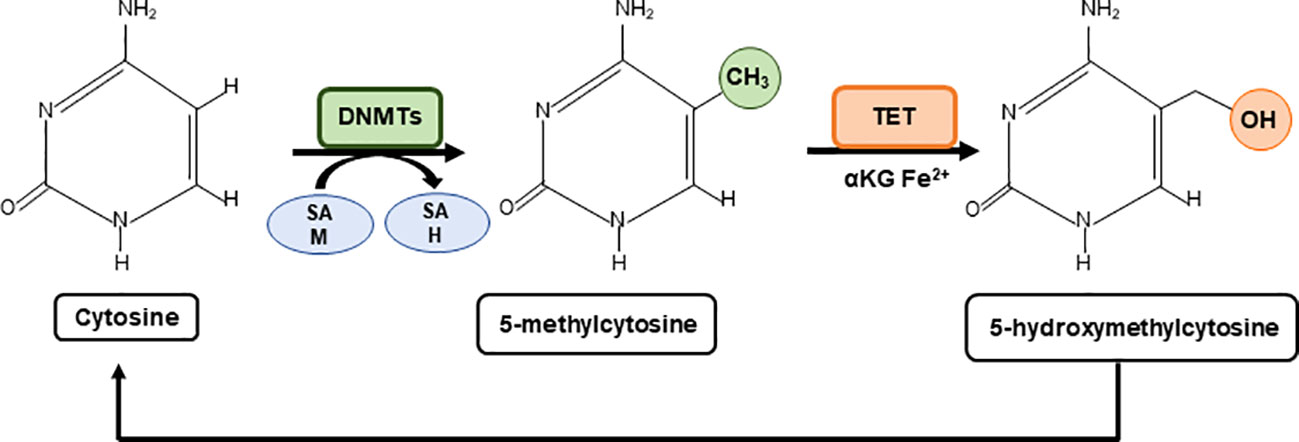
Figure 1 Metabolic regulation of cytosine methylation. A family of DNA methyltransferases (DNMTs) catalyse the transfer of a methyl group from S-adenylmethionine (SAM) to the fifth carbon of cytosine residue to form 5-methylcytosine (5mC) producing S-adenosyl-L-homocysteine (SAH). The opposite process is carried out by ten-eleven-translocation (TET) proteins which use α-Ketoglutarate (α-KG) and Fe2+ as cofactors to generate 5-hydroxymethylcytosine (5hmC). 5hmC can be further oxidised to form carbonylmethylcytosine (camC) and formylmethylcytosine (fmC), or glycosylated (not shown).
Intracellular cell signalling pathways drive rapid epigenetic mechanisms through metabolic manipulation to restore cellular homeostasis. In this line, activated AMPK favours the increase levels of α-ketoglutarate (αKG) to activate the TET-dependent demethylation of the PRDM16 promoter during brown adipogenesis (31). Interestingly, dietary α-KG promotes beige adipogenesis to prevent obesity in middle-aged mice (32), indicating the therapeutic potential of diet to shape the altered epigenome in metabolic diseases.
Increased levels of α-KG -after the administration of glucose, glutamate or glutamine- correlate with a rapid increase in 5hmC levels in the liver of mice (28). Similarly, supplementation of vitamin C in the extracellular medium of embryonic stem cells (ESC) induces a global increase in cytosine hydroxymethylation, at the expense of the 5mC (33), leading to a rapid reprogramming of fibroblasts into induced pluripotent stem cells (iPSCs). In cancer, a global hypomethylation epigenetic profile, together with inter-dispersed hypermethylated CpG islands (CIMP), constitutes a prognostic biomarker in multiple tumours (34). A specific point mutation in the isocitrate dehydrogenase (IDH1) gene in gliomas results in a mutant enzyme that generates high levels of the oncometabolite 2-hydroxyketoglurate, inhibiting α-KG-dependent TET enzymes (35).
RNA modifications can also affect mRNA splicing, stability, nuclear transport, and translation (36). The main RNA modification is the methylation of adenosine at position 6 to produce m6A, which is catalyzed by the methyltransferase-like 3 (METTL3)–METTL14 complex. This epigenetic mark can be removed by non-heme-Fe(II)/2-oxoglutarate(2OG)-dependent oxygenases, fat mass and obesity-associated protein (FTO) and the ALKN homolog 5 (ALKBH5) (37). Increasing evidence has shown that enzymes implicated in the m6A remodeling are deregulated in chronic diseases, ageing and cancer (38, 39). For example, METTL3 is upregulated in some tumors inducing proliferation and dissemination, and it has been proposed as a therapeutic target (40).
Thus, there is a great interest in modulating the activity of enzymes implicated in m6A remodeling. In this regard, inhibitors of FTO have been shown promising results in preclinical models of myeloid leukaemias and glioblastomas (41). Moreover, FTO inhibitors have been demonstrated to improve insulin sensitivity in high fat diets (HFDs) induced obesity (42).
Histone methylation/demethylation, similar as DNA methylation, is regulated by cellular levels of SAM, Fe2+/Fe3+ and α-KG. Histone methyltransferases (EZH2, G9A) utilize SAM as a methyl donor and histone demethylases, JmjC-family of histone demethylases and LSD-family of histone demethylases (LSDs), employ α-KG as cofactor for their enzymatic reactions.
While 5mC on DNA is a major transcriptional repressive mark in many eukaryotes, the effect of histone methylation in gene expression depends on the type of residue and the number of methyl groups added. Generally, methylation of H3K9, H3K27, and H4K20 are associated to silenced chromatin; on the contrary, H3K4, H33K36, H3K48, and H3K79 marks are associated to active gene expression (43). α-KG cofactor is required for the demethylation reactions of JmjCs, while LSDs require the FAD cofactor to demethylate the histone residues (6).
Glucose and glutamine catabolism are used to maintain a high α-KG/succinate ratio, enhancing the global demethylation of multiple histone residues (44). For this reason, manipulation of the α-KG/succinate ratio can balance the renewal versus differentiation fate in ESC cultures. Similarly, in adipocytes, various promoters of genes involved in energy expenditure, such as PGC-1A and pyruvate dehydrogenase kinase 4 (PKD4), are demethylated by the histone demethylase LSD1 (KDM1A), and, for this reason, their expression is dependent on the availability of intracellular FAD (45).
A rapid response to nutrient availability may also be mediated by the acetylation or deacetylation of histones (46). HATs use the substrate acetyl-CoA, which is produced by several catabolic pathways - glycolysis, fatty acid β-oxidation, and amino acid catabolism-. It has been described that high levels of acetyl-CoA augments the overall levels of histone acetylation to promote the expression of genes related to adipocyte differentiation (47).
While histone acetylation naturally relies on acetyl-CoA, a subset of HDACs use NAD+ as a cofactor. This feature is unique to the sirtuin family of HDACs, which cleave NAD+ to generate nicotinamide and 1-O-acetyl ADP-ribose (OAR) during histone deacetylation (48). The increase ratio of NAD+/NADH is another energy sensor for the class III histone deacetylases sirtuins, repressing the expression of genes involved in lipogenesis, glycolysis and gluconeogenesis (49, 50). On the contrary, when glucose is abundant the ratio of NAD+/NADH is diminished reducing the activity of sirtuins to favour lipogenesis and gluconeogenesis. In addition, pyruvate derived from glycolysis is used to fuel the tricarboxylic acid (TCA) augmenting the levels of acetyl-CoA and, consequently, modulating the activity of HAT (51). All in all, the metabolic control of sirtuins and HAT may provide a promising field of research affecting not only gene expression but also the mitochondrial metabolism.
In addition, other epigenetic modifications can be regulated by metabolism. There is a wide range of post-translational modifications (PTMs) on histone and non-histone proteins, integrating environmental changes into cellular responses by regulating gene expression. This is the case of acylation, lactylation, 2-hydroxy-isobutyrylation, succinylation, and malonylation of targeted proteins, which may affect cell function, proliferation, and differentiation. Histone modifications is an important type of epigenetic modification that has been widely detected and exerts different effects at different sites (52–54).
In 2019, Zhang et al. discovered a brand new epigenetic modification called lactylation on lysine residues affecting gene transcription from chromatin (55). The glycolytic-dependent metabolism found in tumours and fast-growing cells has made lactate a pivotal player in energy metabolism reprogramming, which enables cells to obtain abundant energy in a short time. Moreover, lactate shapes the acidic tumor microenvironment, recruiting immunosuppressive cells in cancer. Recently lactate-induced lactylation has been shown to modify histone proteins to alter the spatial configuration of chromatin, affect DNA accessibility and regulate the expression of corresponding genes (55). Thus, in non-small cell lung cancer cells, lactate attenuates glucose uptake, which seems to be partially affected by the increased histone lactylation in the promoters of the glycolytic enzymes (HK-1 and PKM) (56). In macrophages, lactate was reported to induce histone lactylation in the promoters of the profibrotic genes, which is concordant with the upregulation of this very epigenetic modification in the macrophages in fibrotic lungs. Moreover, the role of histone lactylation in tumorigenesis is gradually found, just as the oncogenic role found in other histone modifications (55).
Non-coding RNAs (ncRNAs) are RNA molecules which are not translated into proteins but post-transcriptionally regulate gene expression. The most functionally relevant types of non-coding RNAs studied in the context of chronic diseases are microRNAs (miRNAs) and long non-coding RNAs (lncRNAs).
ncRNAs modulate key metabolic pathways including hypoxia-inducible pathways, glycolysis, oxidative phosphorylation, lipid metabolism, amino acid metabolism and signal transduction pathways, among others. Thus, they may be biomarkers for prognosis, and personalized therapeutic interventions.
MicroRNAs -small molecules of RNA (≈22 nucleotides)- are well known regulators of adipogenesis, fatty acid metabolism, insulin signalling, inflammation, and cell development and differentiation.
Several miRNAs, such as miR-30, miR-26b, miR-199a, and miR-148a- are differentially expressed in obese people, being proposed as biomarkers of metabolic health. miR-17-5p and miR-132- are overexpressed in visceral inflammatory adipose tissue correlating with body mass index, glycosylated hemoglobin, diabetes, and dyslipidaemia.
Metabolic alterations, such as obesity, insulin resistance, ageing and cancer, affect microRNA expression. For example, miR21 is induced during obesity correlating with impaired vascular function (57). On the contrary, silencing this microRNA has been demonstrated to reduce adipogenesis and triglyceride accumulation, improving insulin sensitivity and reducing systemic inflammation.
Different studies reported that the expression of miRNAs is directly correlated with diet and lifestyle, being miR-17/20/93 family, miR-21/590-5p family, miR-200b/c family, miR-221/222 family, let-7/miR-98 family and miR-203 associated with specific diets.
The expression of miR-21 was found diminished in the white adipose tissue (WAT) of obese humans correlating with BMI (58). Interestingly, locked nucleic acid (LNA)-miR-21 treatment reduced body weight (59) in a preclinical model of diet-induced obesity (DIO). In another study, let-7 knockout mice did not develop insulin resistance after a high fat diet induced obesity (60).
In a clinical trial with obese women, reduction of body weight showed that circulating levels of several miRNAs were similar to lean controls, suggesting that some of them could be used as biomarkers (61).
More research has recently begun to unravel the biological functions of lncRNAs, which are tissue-specific long RNA transcripts (>200 bp). GYG2P1, lncRNAp21015, and lncRNA-p5549 are examples of lncRNAs that are differentially expressed in obesity (62). Their expression has been found inhibited in obese individuals. RP11-20G13.3 is among the lncRNAs required to maintain PPAR, C/EBP, and ADIPOQ levels during adipogenesis and is differentially expressed in obesity (63). Other lncRNAs, such as lnc-dPrm16 and MIST, have been shown to influence brown adipogenesis, inflammation, and lipid metabolism (64).
A central methylation pathway in eukaryotic cells is the one-carbon metabolism, implicated in the transfer of methyl groups to various substrates and cofactors within the folate and methionine cycles. The transfer of methyl groups from 5-methyltetrahydrofolate (5-mTHF) to homocysteine (Hcy) to form methionine connects the two cycles. Methionine is then converted to SAM, the universal methyl donor for DNA, proteins, and secondary metabolites. Methyltransferases use SAM as donor of methyl groups, and the resulting demethylated S-adenosyl-l-homocysteine (SAH) is hydrolyzed to form Hcy, which is then recycled to methionine using 5-MTHF as a methyl group donor, thus completing the cycle. Nicotinamide N-methyltransferase (NNMT) consumes methyl donors and makes SAM unavailable to other methyltransferases, thereby repressing H3K27me3 marks. NNMT is involved in the maintenance of the naïve state of ESC which are characterized by low levels of histone methylation (65, 66).
Both HMTs and DNMTs use SAM as a donor for the methylation reactions. Importantly, the ratio of SAM/SAH is an indicator of the status of the one-carbon metabolism to regenerate methionine (67).
Nutrients such as methionine, folate, choline, betaine, and vitamins B2, B6, and B12 are precursors of SAM affecting the SAM/SAH ratio and the global levels of DNA methylation. However, it is difficult to extrapolate the direct role of metabolism on DNA methylation, because the mechanisms regulating from DNA methylation are very complex and the relationship between methylation pattern and gene expression is tissue and context-dependent (68).
The balance of methionine, folate, and B12 from our diet regulates the activity of the folate and methionine cycles, which are mechanistically co-dependent. The folate cycle converts tetrahydrofolate (THF), into 5,10-methylene-THF by serine hydroxymethyltransferase (SHMT), an enzyme that requires B6 as a cofactor. Then, 5,10-methyleneTHF acts as a methyl-donor for thymidylate synthase (TS) to synthesize deoxythymidine monophosphate (dTMP) from deoxyuracil monophosphate (dUMP) or for methylenetetrahydrofolate reductase (MTHFR) to generate 5-mTHF (69). In the methionine cycle, methionine is converted to SAM by the activity of methyl-adenosyl transferase 2A (MAT2A). SAM is then used as a methyl donor resulting in S-adenosylhomocysteine (SAH), which can be further hydrolyzed to Hcy by S-adenosylhomocysteine hydrolase (AHCY). HCy is then converted into methionine by methionine synthase (MS) using 5-mTHF as a methyl donor and vitamin B12 as an essential cofactor (70). In addition, methionine can also be recycled from Hcy using betaine, derived from dietary choline, as a methyl donor (71), or directly from SAM through the polyamine synthesis and the methionine salvage pathway. Using vitamin B6 as a cofactor, Hcy can also be converted to cysteine for the synthesis of glutathione (72).
Vitamin B12, in addition to its role as a cofactor in the recycling of Hcy to methionine, is an important cofactor for the metabolic enzyme methyl-malonyl CoA mutase (MUT), which is located in mitochondria. MUT uses vitamin B12 in the form of adenosylcobalamin to convert L-methylmalonyl-CoA to succinyl-CoA, which can further enter the TCA cycle. Genetic loss of MUT activity can disrupt TCA cycle function and mitochondrial redox function. Thus, the levels of vitamin B12 connect the one-carbon metabolism, energy metabolism, and redox metabolism (73).
Glycolysis, glutaminolysis and fatty acid oxidation provide different metabolites that are key for epigenetic modifications. These pathways provide metabolic cofactors for epigenetic reactions and interact with each other in a complex crosstalk that links metabolic needs to gene expression responses.
Glycolysis is a key metabolic pathway implicated in the production of metabolites such as pyruvate, intermediates of the pentose phosphate pathway (PPP) and the one carbon metabolism, among others. Pyruvate may be subsequently metabolized to lactate by the enzymatic activity of lactate dehydrogenase (LDHA) or converted to acetyl-CoA through the pyruvate dehydrogenase enzyme (PDH), which enters the mitochondria to feed the TCA cycle. Interestingly, both lactate and acetyl-CoA are crucial donors of the epigenetic modifications including histone lactylation (74) and histone acetylation (75).
The PPP contributes to the synthesis of nucleotides for cell division and contribute to generate the methyl donor SAM (76).
The glycolytic flux determines the ratio of NAD+/NADH ratio which is important for the activities of sirtuin HDACs.
Cellular glucose availability is linked to acetyl-CoA abundance and histone acetylation. The uptake of glucose is mainly driven by the glucose transporters type 1 and 4 (GLUT1/4), which are frequently upregulated in activated T cells and cancer cells to support cell proliferation (77). Pyruvate decarboxylase complex (PDC) produces acetyl-CoA, connecting aerobic glycolysis to TCA cycle. LDHA produces lactate from pyruvate in highly proliferative cells where an aerobic glycolysis switch occurs over mitochondrial oxidative phosphorylation (78). The enzyme PDC is found in both mitochondria and the nucleus. Nuclear PDC regulates the expression of sterol regulatory element-binding transcription factor (SREBP) target genes by affecting histone acetylation, and mitochondrial PDC contributes to the production of cytosolic citrate for lipogenesis (79). The availability of acetyl-CoA for HATs is also modulated by the levels of GLUT1/4 -implicated in the uptake of glucose (77)- and the levels of ATP- citrate lyase (ACLY) (80), which are frequently upregulated in activated T cells and cancer cells to support cell proliferation (81).
Glucose is the main carbon source for acetyl-CoA in oxygenated cells. An exquisite mechanism is observed in cancer cells during hypoxia to promote the expression of lipogenic genes acetyl-CoA carboxylase alpha (ACACA) and fatty acid synthase (FASN), by mean of the acetate mediated histone H3 acetylation. The acetyl-CoA synthetases ACSS1/2, which catalyze the production of acetyl-CoA from acetate, contribute to the above acetate-mediated epigenetic regulation (82).
Acetyl-CoA levels change with nutrient abundance and for this reason it can be considered as a nutrient sensor to respond to metabolic changes (83). For example, acetyl-CoA is produced from distinct metabolites such as pyruvate, citrate and acetate which can be produced by the activity of the PDC, ACLY and ACSS2, respectively. In addition, acetyl-CoA levels depend on the levels of ketone bodies and fatty acid β-oxidation (84).
Glutaminolysis is a metabolic pathway that generates α-KG which, as seen above, is a cofactor for dioxygenases and histone, DNA and RNA demethylases. Glutaminolysis is required for the glutamine- dependent anaplerosis of TCA providing metabolic substrates such as α-KG. Similarly, other TCA metabolites including succinate, fumarate, malate and oxalacetate are relevant epigenetic modulators as they can inhibit cellular demethylases due to their similar chemical structure to α-KG. Furthermore, the α-KG derived metabolite 2-hydroxyglutarate (2-HG) has been shown to interfere with the activity of demethylases and thus the global epigenetic marks (85). Several TCA cycle intermediates can be exported out of mitochondria including citrate and α−KG. Cytosolic citrate is converted to acetyl-CoA, which is used as a donor for HAT-mediated histone acetylation. α−KG is used as cofactor for histone and DNA demethylation reactions by JHDM and TET, respectively.
The substrate for HMT and DNMT is SAM, which is synthesized from the essential amino acid methionine The propionate catabolic pathway breaks down branched-chain amino acids (BCAAs), odd-chain fatty acids, and cholesterol to be used in the TCA cycle in the mitochondria. MUT converts methylmalonyl-CoA to succinyl-CoA using vitamin B12, in the form of adenosylcobalamin, as a cofactor. Succinyl-CoA then enters the TCA cycle.
Fatty acid oxidation (FAO) is a key metabolic pathway affecting the levels of NAD+/NADH and FAD/FADH2 metabolites, which are key cofactors of epigenetic enzymes. In addition, augmenting FAO increases the levels of acetyl-CoA which will affect the acetylation levels of histones (86). Acetyl-CoA can also enter in the TCA to produce citrate. In the cytosol, citrate is converted to acetyl-CoA and malonyl-CoA by the ACLY enzyme. ACCS1/ACCS2 catalyze the conversion of acetyl-CoA to malonyl-CoA, which is the rate-limiting step for the synthesis of FAs. ACCs are activated by citrate and inhibited by palmitoyl-CoA and malonyl-CoA. When there are low levels of energy, AMPK phosphorylates ACCs to increase FAO in the short term. In addition, diet affects the expression of genes implicated in the synthesis of FAs or in FAO (87).
Although changes in global availability of metabolites impact on epigenetic processes globally (e.g. SAM levels regulate methylation), the enzymatic Km of the enzymes for those metabolites, and the availability of other micronutrients, such as vitamins and minerals, will fine-tune the epigenetic changes upon metabolic alterations (88). Thus, in each metabolic context, specific epigenetic marks may be favored. For this reason, balanced metabolic traits are necessary to achieve coordinated epigenetic responses that sustain health.
Figure 2 shows main intracellular metabolic pathways to provide metabolites for main epigenetic marks (methylation, acetylation, lactylation) and cofactors to modulate the activity of epigenetic enzymes.
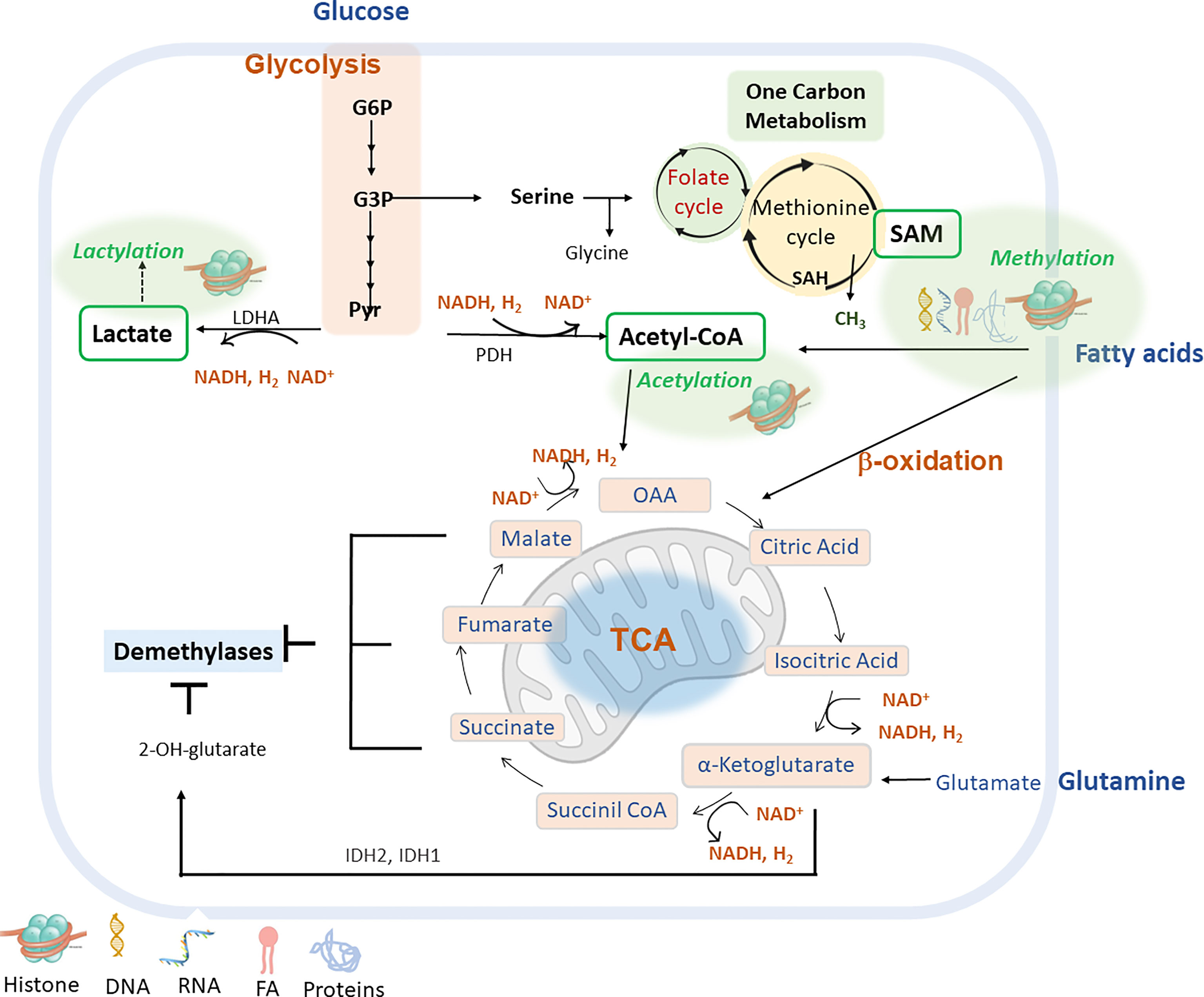
Figure 2 Main metabolic pathways can be implicated in the epigenetic remodelling include glycolysis, fatty acid β-oxidation (FAO), glutaminolysis and lactylation via derived metabolites and cofactors (lactate, acetyl-CoA, α-ketoglutarate or NAD+). G6P (glucose-6-phosphate); G3P (glycerol-3-P); Pyr (pyruvate); LDHA (lactate dehydrogenase A); PDH (pyruvate dehydrogenase); IDH1/IDH2 (isocitrate dehydrogenase 1, and 2) S-Adenosyl methionine (SAM); S-adenosyl-L-homocysteine (SAH); Nicotinamide adenine dinucleotide (NAD+); Nicotinamide adenine nucleotide reduced (NADH); Oxaloacetate acid (OAA).
Metabolic and epigenetic imbalances result in disease. There are several factors that can cause an epigenetic imbalance and consequently different diseases. Some of these factors are intrinsic including genetic variations such as Single Nucleotide Polymorphism (SNPs) or DNA mutations caused by carcinogens and Reactive Oxygen Species (ROS); environmental factors including metabolic alterations such as obesity or ageing and lifestyle (unbalanced diet or sedentary lifestyle). This epigenetic imbalance leads to cardiovascular, neurological and cancer diseases (Figure 3).
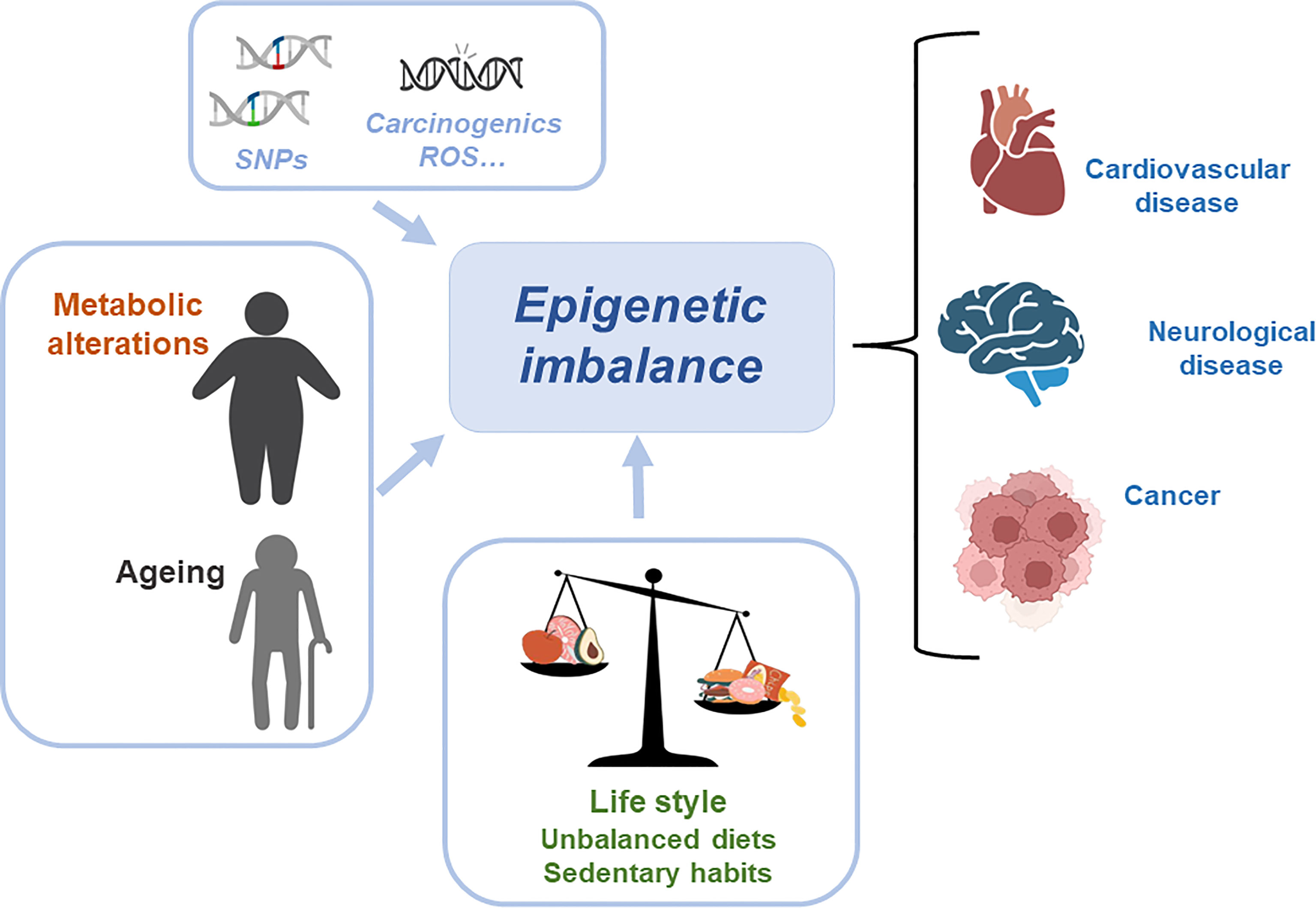
Figure 3 Metabolic and epigenetic imbalances result in disease. There are several factors that can cause an epigenetic imbalance and consequently different diseases. Some of these factors are intrinsic including genetic variations such as Single Nucleotide Polymorphism (SNPs) or DNA mutations caused by carcinogens and Reactive Oxygen Species (ROS); environmental factors including metabolic alterations such as obesity or ageing and lifestyle (unbalanced diet or sedentary lifestyle). This epigenetic imbalance leads to cardiovascular, neurological and cancer diseases.
Certain genetic variations drive metabolic alterations and thus affect the epigenetic landscape (89). For example, the C677T variant in the MTHFR gene, which synthesizes 5-mTHF, can impact DNA methylation, depending on the folate status (90). Moreover, this genetic polymorphism has been associated to several alterations including cardiovascular disease (CVD), cancer and neurological diseases (91). Similarly, mutations in the metabolic genes isocitrate-dehydrogenase 1 and 2 (IDH1 and IDH2) are linked to cancer (92), as they increase the production of the 2-hydroxyglutarate metabolite which antagonizes alpha-ketoglutarate (αKG), reduces the activity of DNA and histone demethylases and consequently the DNA and histone methylation status (93, 94). Genetic variants of the fat mass and obesity-associated protein, also known as alpha-ketoglutarate-dependent dioxygenase (FTO) gene, which was the first mRNA demethylase identified (95), have been correlated to obesity and metabolic syndrome (96).
In addition to genetic variations, ageing and obesity have detrimental effects on health by different mechanisms that include epigenetic alterations (97, 98).
Obesity is a condition caused by genetic and environmental factors, which is characterized by an increase in body weight, hyperlipidemia, hyperglycemia, hyperinsulinemia, and elevated inflammatory substances. Importantly, obesity increases the risk for other conditions including cardiovascular disease, metabolic syndrome, type II diabetes or cancer (99). At the molecular level, obesity correlates with particular DNA methylation profiles on several genes in different tissues that contribute to its pathophysiology (3, 100). Similarly, RNA methylation misbalances are associated with obesity (101). The expression of m6A regulatory factors in human adipose tissue and Peripheral Blood Mononuclear Cells (PBMCs) varies between lean and obese individuals (102). Thus, the m6A RNA methylomes of genes implicated in lipid metabolism are altered in mice upon a high-fat diet (103). Mechanistically, perturbations in the m6A methylome by FTO (104) or other m6A regulators (105) result in alterations in adipogenesis homeostasis, which likely promote obesity. In addition to alterations in epigenetic factors, the metabolome of obese and metabolically impaired individuals is also unbalanced contributing to epigenetic alterations. For example, a reduction in NAD+ levels is linked to metabolic diseases and it is therefore a potential target for intervention in humans (106, 107). In addition, the plasma alpha-ketoglutarate (AKG) levels are reduced upon increasing BMI or HbA1c (a diabetic biomarker), suggesting a possible regulatory role of AKG in pathologies such as obesity. Indeed, AKG regulates JMJD3 expression and promotes the demethylation of SERPINA1E promoter, thereby increasing gluconeogenesis (108).
Ageing and age-related diseases (cancer, cardiovascular and neurological diseases) are also a global concern, given their health and economic implications. For this reason, efforts to understand the ageing process have led to the identification of the hallmarks of ageing (97) and the postulation of different theories of ageing (109–112). One of the most deeply characterized epigenetic mark in ageing is DNA methylation, thanks to the development of DNA methylation clocks. DNA methylation profiles correlate to chronological age and clinical variables (113), although their true biological meaning remains obscure (114). Further research is needed to understand how epigenetic age and DNA methylation clocks may help us anticipate, prevent, and overcome diseases related to ageing. Clearly, epigenetic clocks have a big potential as clinical biomarkers (115, 116).
Alterations in RNA methylation are also observed in age-related diseases and, interestingly, m6A modifications have been implicated in the regulation of different biological processes associated with ageing, including inflammation (117). Often, these alterations derive from a misbalanced regulation of m6A regulatory factors. For this reason, RNA methylation and its biological regulators offer a therapeutic target for age-related diseases.
Further, during ageing, the metabolome profile gets altered, which may contribute to epigenetic alterations. In particular, NAD+ levels and AKG levels decline with age (118).
HFDs have detrimental effects on systemic metabolic homeostasis. The liver is one of the first organs affected by unbalanced diets. Related to epigenetics, it has been observed that HFDs alter the activity of HDACs and HATs in the liver of rats, diminishing hepatic regeneration and the liver gluconeogenesis which leads to insulin resistance (119). On the contrary, during fasting, the expression of HDACs implicated in the expression of lipogenic enzymes is silenced to favour hepatic gluconeogenesis (120). Furthermore, high-fat diets during pregnancy facilitate the development of metabolic syndrome in the offspring, due to higher levels of histone H3 acetylation (121). This shows the epigenetic implication in the development of metabolic diseases (a type of chronic disease) and also the detrimental and long-term effects of high fat diets.
Lastly, evidence also indicates that High-sugar diets augment the risk of metabolic diseases in offspring (122). High glucose levels induce the regulation of insulin transcription by pancreatic β-cells (123). On the contrary, low plasma glucose levels lead to PDX1 (pancreatic duodenal home box 1) binding to HDAC promoting the insulin gene acetylation to repress its transcription (124). The transcription factor HIF1A, which mediates the response of pancreatic β-cells to glucose, is destabilized by high glucose levels interfering its interaction with the coactivator p300 and thus inhibiting the transcription of HIF targets (125) (Figure 4).
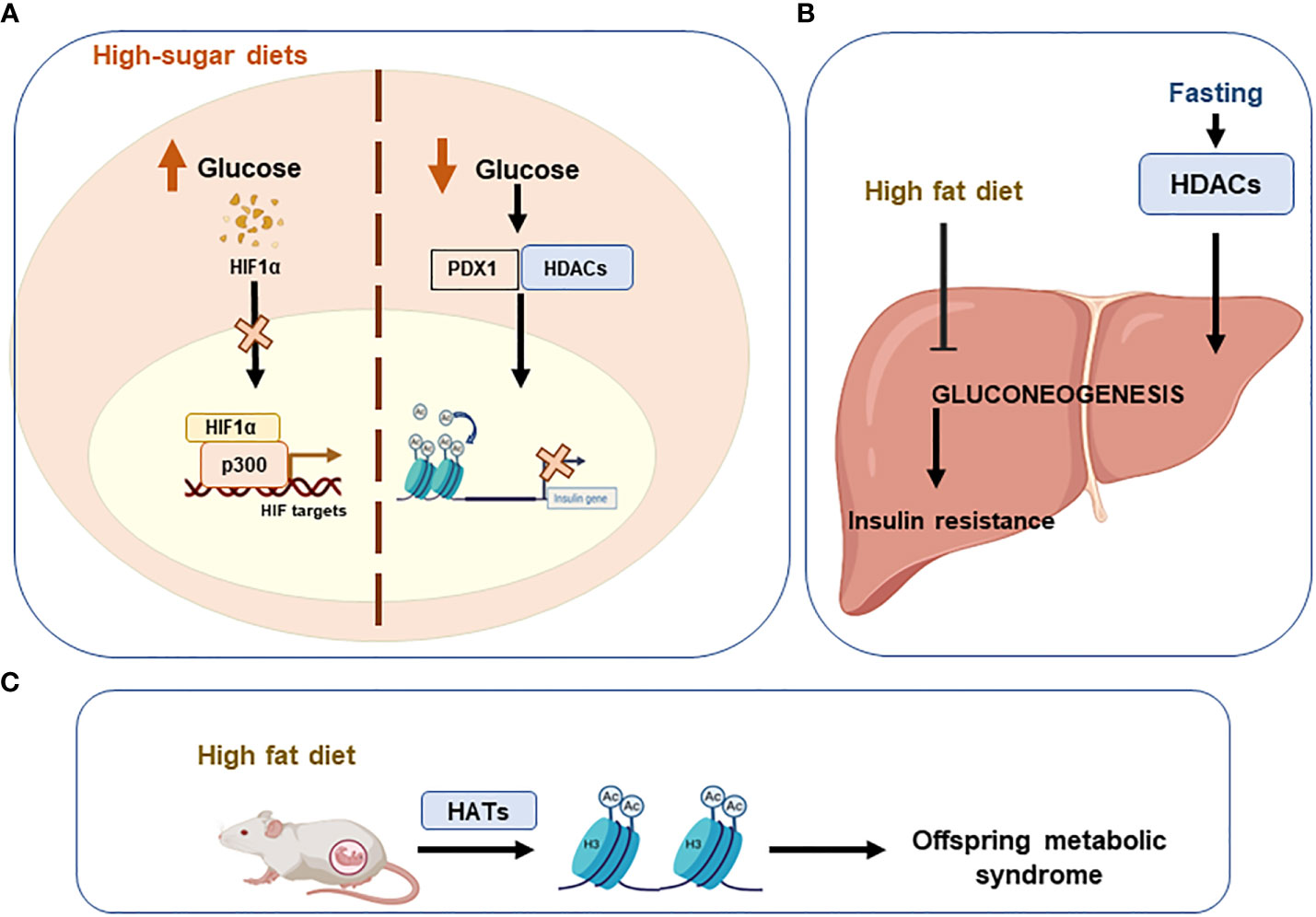
Figure 4 Unhealthy diets leading to epigenetic imbalances. Imbalanced or unhealthy diets have detrimental effects on systemic metabolic homeostasis. (A) High sugar diets increase glucose levels, which destabilise hypoxia-inducible factor 1alpha (HIF1A) and consequently inhibit the transcription of HIF. Conversely, when glucose levels are low, acetylation of the insulin gene is promoted to repress its transcription via the PDX1/HDAC interaction. (B) A high-fat diet inhibits gluconeogenesis in the liver and promotes insulin resistance. (C) Offspring can develop metabolic syndrome if high-fat diets are consumed during pregnancy. Pancreatic And Duodenal Homeobox 1 (PDX1); Histone deacetylases (HDACs); Histone acetyltransferases (HATs).
Recently, the interaction between the host microbiome and diet has been shown to influence the epigenetic landscape. Diet modulates the gut microbiome affecting the microbial-produced metabolites and the development of metabolic diseases. The production of short-chain fatty acids, mainly acetate and butyrate, by the microbiome affects the levels of acetyl-CoA and histone acetylation. It has been demonstrated the relevant role of β-hydroxybutyrate, an HDAC inhibitor, in the inhibition of CRC progression in mice by mean of the upregulation of H3 acetylation and the activation of apoptosis. Butyrate is produced by the colonic fermentation of dietary fibers. C57BL/6 mice fed a high-fat diet supplemented with butyrate (5% w/w) consumed more energy than those that did not receive the supplement (126). Butyrate stimulated mitochondrial function and biogenesis in skeletal muscle cells and brown adipocytes, protecting against diet-induced obesity and insulin resistance (Lin et al., 2012). In part, this effect has been demonstrated to be related to the inhibitory effects on HDAC augmenting the expression of FGF21 and fatty acid oxidation (127) (Figure 5).
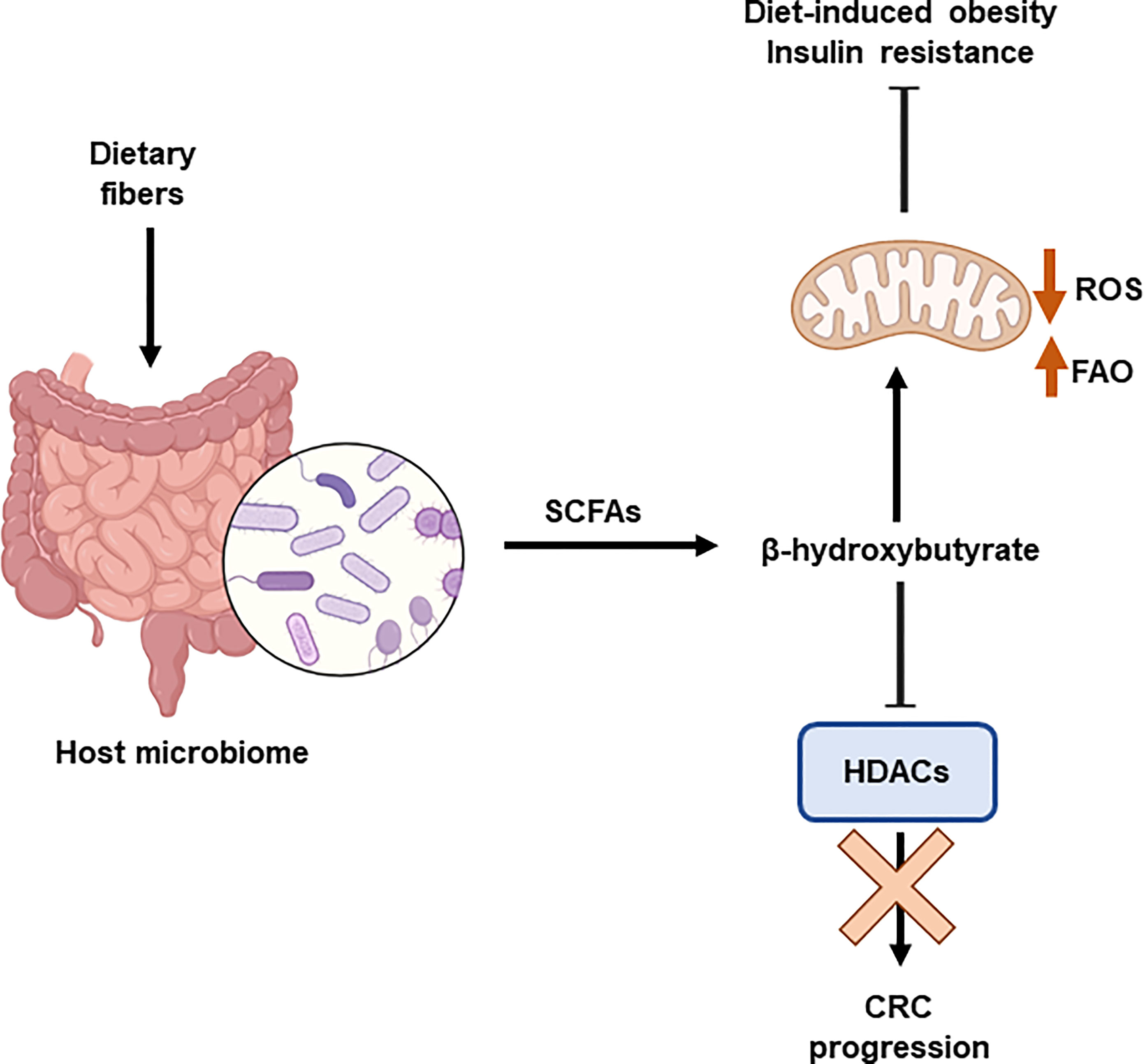
Figure 5 Interplay between epigenetics and microbiome. The host microbiome is influenced by the diet. Dietary fibre induces the production of shirt chain fatty acids (SCFAs), including β-hydroxybutyrate. This metabolite is an HDAC inhibitor that prevents the progression of colorectal cancer (CRC). In addition, β-hydroxybutyrate stimulates mitochondrial function and biogenesis, which protects against obesity and insulin resistance. Histone deacetylases (HDACs); Reactive Oxygen Species (ROS); fatty acid β-oxidation (FAO).
As indicated previously, a large number of miRNAs have been found differently expressed in obese people, suggesting that restoring the systemic metabolic health (mainly glucose and lipid homeostasis) may be partially mediated by miRNAs, as many of them are key modulators of adipogenesis, glycolysis and inflammation (57). Although the list of miRNAs associated with a diet is important, several studies suggest that the miR-17/20/93 family, miR-21/590-5p family, miR-200b/c family, miR-221/222 family, let-7/miR-98 family and miR-203 are the most dysregulated in this context (128, 129).
The expression of miR-21 was found diminished in the WAT of obese humans, correlating with BMI (58). Interestingly, treatment with locked nucleic acid (LNA)-miR-21 treatment reduced weight in a preclinical model of DIO (59). In another study, let-7 knockout mice did not develop insulin resistance after a high-fat diet induced obesity (60).
In a clinical trial with obese women, reduction of body weight showed that circulating levels of several miRNAs were similar to lean controls, suggesting that some of them could be used as biomarkers (61).
Diet is the main source of metabolites -substrates and cofactors- used for the epigenetic programs. Thus, unbalanced diets may affect the epigenome contributing to the development and progression of chronic diseases, as well as longevity. On the other hand, controlling metabolism through nutrient sensing pathways or particular metabolites´ availability may contribute to counteract chronic and age-related diseases. Figure 6 provides examples of protective diets to modulate epigenetics.
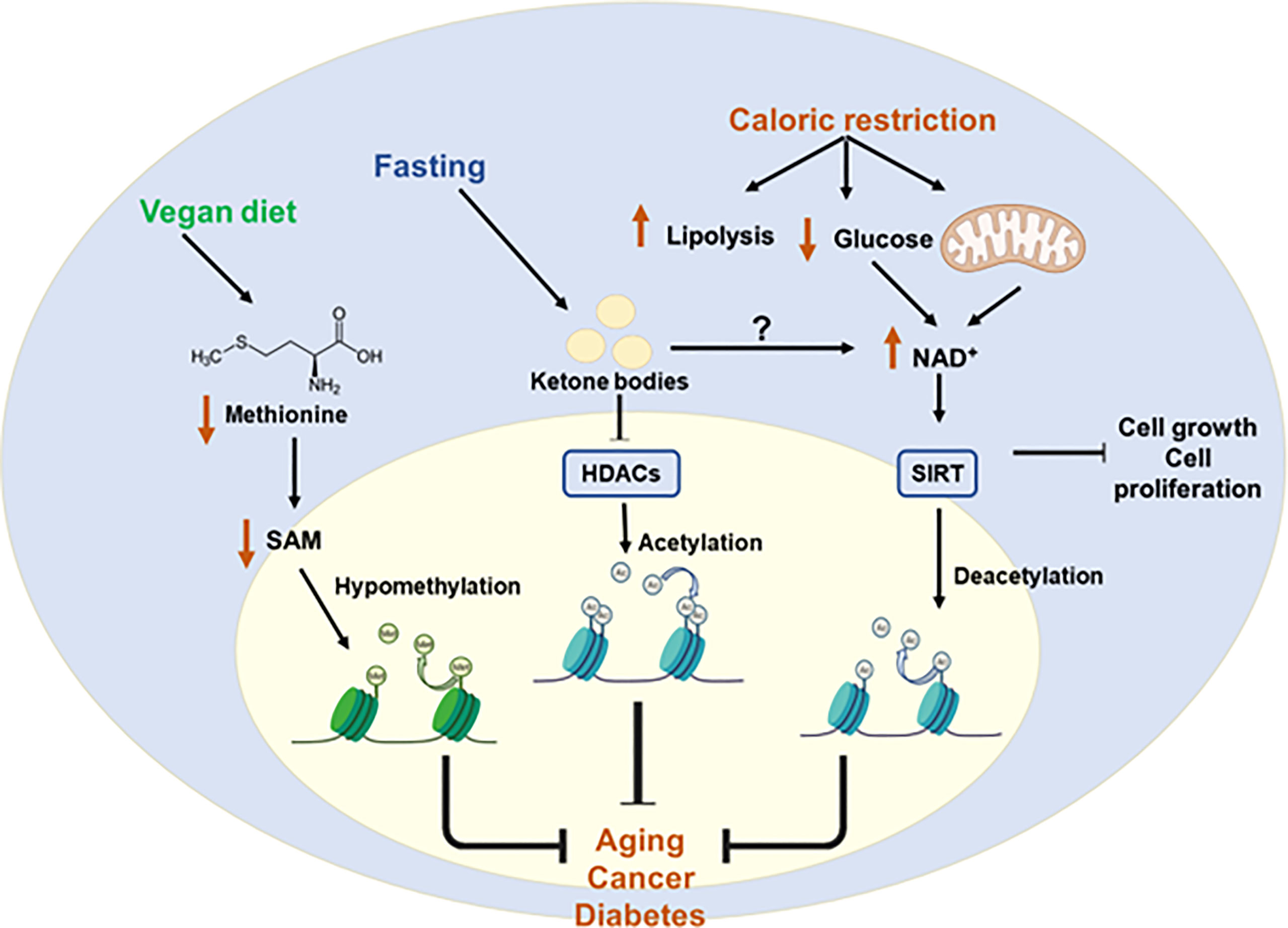
Figure 6 Protective diets to restore epigenetic imbalances. Metabolic diseases caused by epigenetic imbalances can be controlled by protective diets. Caloric restriction increases NAD+ levels by increasing lipolysis and decreasing glucose availability. This increase in the NAD+ leads to the activation of HATs and subsequent deacetylation. Fasting is associated with increased production of ketone bodies which is related to the inhibition of class I HDACs. Finally, a vegan diet is associated with reduced levels of methionine and SAM which leads to hypomethylation of some genes. All these diets may help to counteract chronic and age-related diseases. S-Adenosylmethionine (SAM); Nicotinamide adenine dinucleotide (NAD+); Histone deacetylases (HDACs); sirtuin (SIRT); Histone acetyltransferases (HATs).
Calorie restriction (CR) - reduction of approximately a 20% of the total calorie intake - has been shown to expand lifespan. In mice, the beneficial effects of CR have been associated to the metabolic regulation of epigenetics, although the precise underlying mechanisms are not fully understood. Nevertheless, DNA methylation in specific genomic regions have been described implicated on the beneficial effects of CR on age-related diseases. For example, in the kidney of aged rats, CR was shown to attenuate inflammation, cancer, or diabetes, by restoring the methylation patterns altered during ageing (130). In a similar manner, CR restored liver triglyceride content by mean of epigenetic effects on lipid metabolism related pathways (131). Some of the metabolic adaptations to fasting are associated to the increased production of ketone bodies and/or the inhibition of the class I HDACs, which are implicated in the activation of the expression of metabolic starvation responsive genes (132). A similar effect related to ketogenesis and histone acetylation has been observed during exercise (133). Altogether, diets that limit caloric intake may be efficient to prolong health-span through epigenetic control, thus reducing the impact of age-related diseases. A low-carb ketogenic diet that rescues the hippocampal memory defects in a mouse model of Kabuki syndrome through microbial production of β-hydroxybutyrate, an HDAC inhibitor, leads to changes in H3ac and H3K4me3 in the hippocampus and rescues the neurogenesis and memory phenotypes of these mice models (134). On the contrary, a “Western-type” diet diminishes the microbial SCFAs production, affecting hepatic gene expression by epigenetic mechanisms.
In addition, the regulation of particular metabolites´ availability may modulate epigenetic marks. Therefore, dietary interventions that limit or enhance certain nutrients or metabolites may be effective tools to affect the epigenetic landscape of individuals. For example, regulating methionine availability through a whole food vegan diet increases longevity in rodents (135, 136). Cancer cells, in addition to the Warburg effect, present a high avidity for methionine which is used to write the epigenetic mark H3K4me3 in specific oncogenic genes (137), and targeting the production of methyl donors from the methionine cycle may provide a therapeutic opportunity in cancer (138), as shown by the inhibition of tumor growth in mice after methionine reduction from diet (139). Other studies have shown the therapeutic benefit of a methionine restricted (MR) diet on the improved systemic glucose and fatty acid profiles and reduction of inflammation and oxidative stress (140). Dietary choline is oxidized to betaine by the betaine homocysteine methyltransferase (BHMT), affecting the methionine homeostasis. Disruption of choline/betaine metabolism, by the reduction of BHMT gene expression and/or the dietary intake of choline, has been associated with hepatocellular carcinogenesis in animal models (141).
The epigenetic machinery can also be regulated by vitamins from diet, including A, B, C and D vitamins (142). Vitamin A is not synthesized by humans and must be obtained from the diet. This vitamin or its derivatives, including retinoic acid (RA), are involved in DNA methylation, histone modification and miRNA regulation. RA inhibits transcription by blocking DNMTs and induces some miRNAs to regulate DNA methylation. Vitamin A can both convert 5hmc to 5mc by altering TET activity and block HDAC while activating HATs (142, 143). B group vitamins are involved in DNMTs activity by being converted to SAM, such as B9, or by acting as cofactors of one carbon metabolism pathway (B6, B3 and B12). Biotin (Vitamin B7) causes transcriptionally repressed heterochromatin formation through the biotinylation process (144). Vitamin C or L-ascorbate is synthesized from ascorbate in mammals and can also be obtained from the diet (145). In relation to epigenetics, this vitamin plays an important role in the demethylation process of DNA and histones in a TET dependent or independent manner, respectively. Like other vitamins, vitamin C can promote the expression of certain miRNAs. For example, vitamin C represses DNMT3A via miR-143 (142, 146). Finally, vitamin D, produced endogenously or ingested with food, represses p21 by methylating its promoter, while enhancing E-cadherin transcription by demethylating its promoter. It is also involved in histone modifications by activating both HAT and HDACs (142, 147).
Exercise can reverse chronic metabolic diseases by triggering epigenetic modifications (Figure 7). For example, exercise augments the methylation of DNA at the regulatory region of PGC1A gene promoting fatty acid oxidation and mitochondrial biogenesis in skeletal muscle (148).
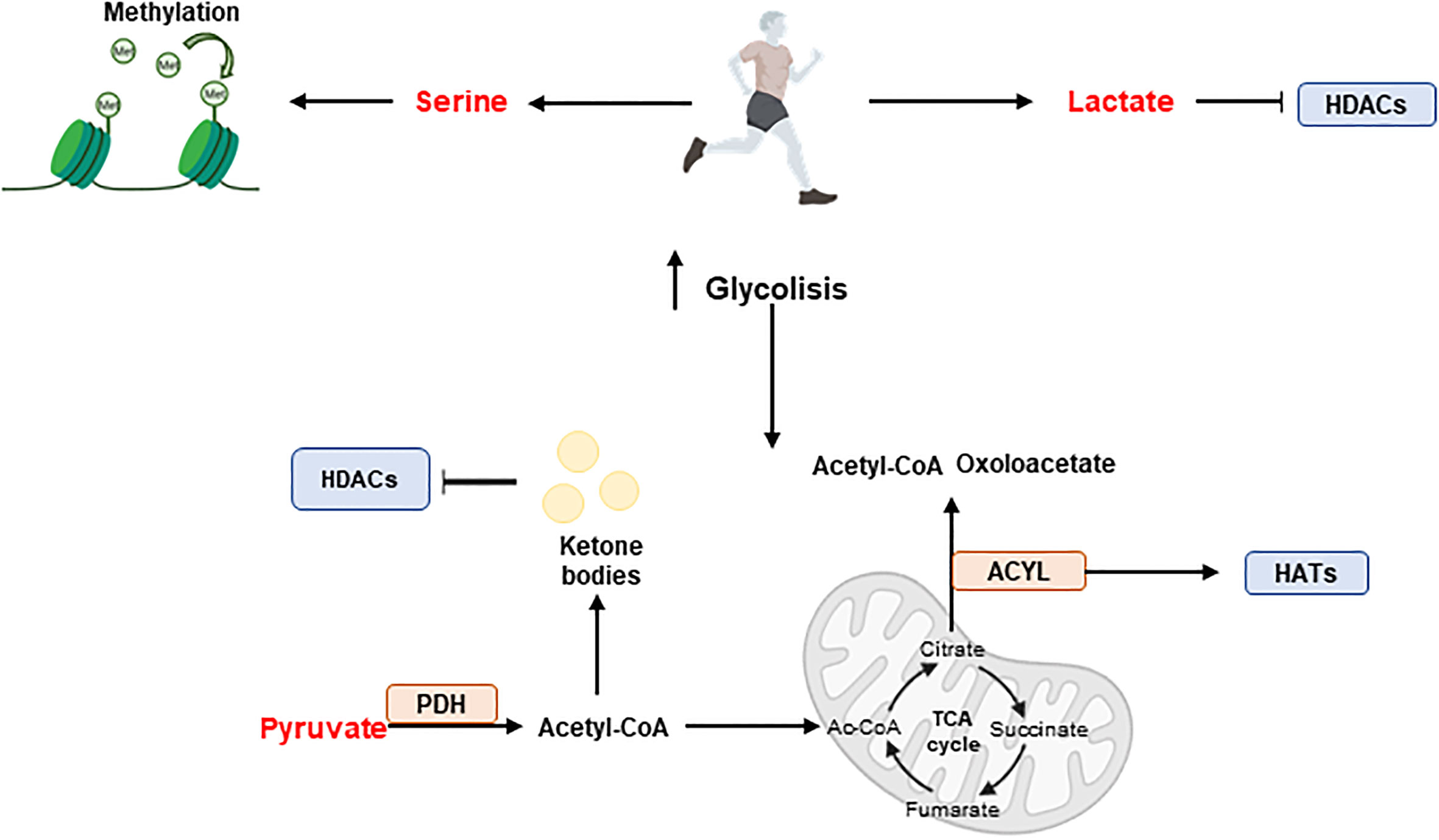
Figure 7 Exercise and epigenetic regulation. During exercise, skeletal muscle produces metabolites such as serine, pyruvate and lactate, which have effects on epigenetics. Serine promotes methylation while pyruvate and lactate have an influence on acetylation. Acetyl-CoA produced from pyruvate enters the tricarboxylic acid cycle (TCA) cycle where key intermediates such as citrate, succinate or fumarate are synthesised. In addition, the ACYL enzyme can produce Ac-CoA and oxaloacetate from citrate and promote hyperacetylation by interacting with HATs. Furthermore, ketone bodies can be formed from acetyl-CoA and participate in HDACs inhibition. Histone acetyl transferases (HATs); Histone deacetylases (HDACs); Pyruvate dehydrogenase (PDH).
In a clinical study, a six-month exercise intervention influenced the genome-wide DNA methylation pattern in human adipose tissue in more than 60 genes related to obesity and insulin resistance (149). In another study, the DNA methylation and miRNA profiles from skeletal muscles in obese and diabetic volunteers were compared at the before and after 4 months of exercise. Interestingly, depending on the type of exercise -aerobic or resistance training- changes in the patterns of DNA methylated genes and miRNAs were observed. Thus, aerobic exercise affected the DNA methylation levels of metabolic genes such as FASN and NRF1, which correlated with a reduction on the levels of circulating free fatty acids. In addition, miR-29a and miR-132 expression levels were found altered. On the other hand, resistance training augmented the DNA methylation of SLC2A4, improving respiratory capacity with a reduction of intramuscular lipid accumulation (150). In another study, acute endurance exercise augmented the expression of miR-1 and miR-133a promoting mitochondrial biogenesis and respiratory capacity (151).
In general, several studies have demonstrated the role of exercise in reducing meta-inflammation in chronic metabolic diseases (152).
During exercise and depending on the intensity, skeletal muscles rapidly metabolize available nutrients -including fats, carbohydrates, and amino acids-, resulting in the production of metabolites such as Ac-CoA, serine, ATP, lactate, butyrate, and α-KG, with effects on the epigenetic programs.
Carbohydrates are the main source of energy during exercise, where glycogen stores are used over muscular triglyceride reserves (153). Upregulation of glycolysis during exercise -high intensity aerobic exercise and resistance exercise- augments the production of metabolites such as serine, pyruvate, and lactate with effects on epigenetics. The upregulation of serine increases the methylation potential in skeletal muscle, meanwhile pyruvate upregulation can further increase the levels of acetyl to produce Ac-CoA. PDH catalyzes the decarboxylation of pyruvate to Ac-CoA during periods of high glucose metabolism. The active form of PDH has been shown to significantly increase almost instantly following exercise - acute aerobic exercise at ranging intensities, and during long term aerobic training- in skeletal muscle (154), suggesting a significant increase in glucose-derived Ac-CoA. Ac-CoA enters the TCA cycle and undergoes a chain of reactions to augment the pool of key intermediates such as citrate, succinate, and fumarate, that have distinct roles in regulating the activity of epigenetic enzymes.
Mitochondrial derived citrate provides nuclear Ac-CoA via a reaction with ACLY, which releases Ac-CoA and oxaloacetate from citrate. In a skeletal muscles ACLY is implicated in myoblast cell differentiation. Recently, overexpression of ACLY has been shown to promote the hyperacetylation of K9, 14, and 27 on histone protein 3, allowing the local concentration of the key myogenic regulatory factor MyoD for the transcription of myoblast cell differentiation genes (155).
Although PDH complex is the main pathway to produce Ac-CoA from glucose, during exercise, the increased demands of energy, specifically in acute and long-term aerobic exercise, induces lipolysis to release FAs for Ac-CoA production through β-oxidation.
Ac-CoA can enter the TCA cycle for ATP production or, during periods of high energy demand, for the synthesis of ketone bodies in the liver, such as acetoacetate and β-OHB, which will be available for energy demanding tissues such as skeletal muscle. During aerobic endurance exercise, fat becomes the dominant energy substrate, and the remaining carbohydrate resources remain intact (156). The cellular mechanisms responsible for this metabolic shift are, however, not yet fully understood. Ketone bodies have important signalling benefits in the skeletal muscle health acting inhibitors of HDAC activity, augmenting the hyperacetylation of histones for the expression genes, such as the forkhead box O3 (Foxo3) (157).
Lactate is an endogenous HDAC inhibitor, sharing common regulatory pathways with SCFA metabolites, such as butyrate and acetoacetate (39). In addition, the epigenetic mark lysine lactylation in the promoter regions of coding genes augments their expression, although the consequences of lactylation epigenetic remodelling during intense exercise requires further research.
Bioactive compounds from the diet have been extensively described to modulate the epigenome and contribute significantly to nutritional programs to prevent and/or treat metabolic diseases.
Table 1 summarizes well known bioactive compounds from diet with health promoting benefits partially mediated by their epigenetic effects.
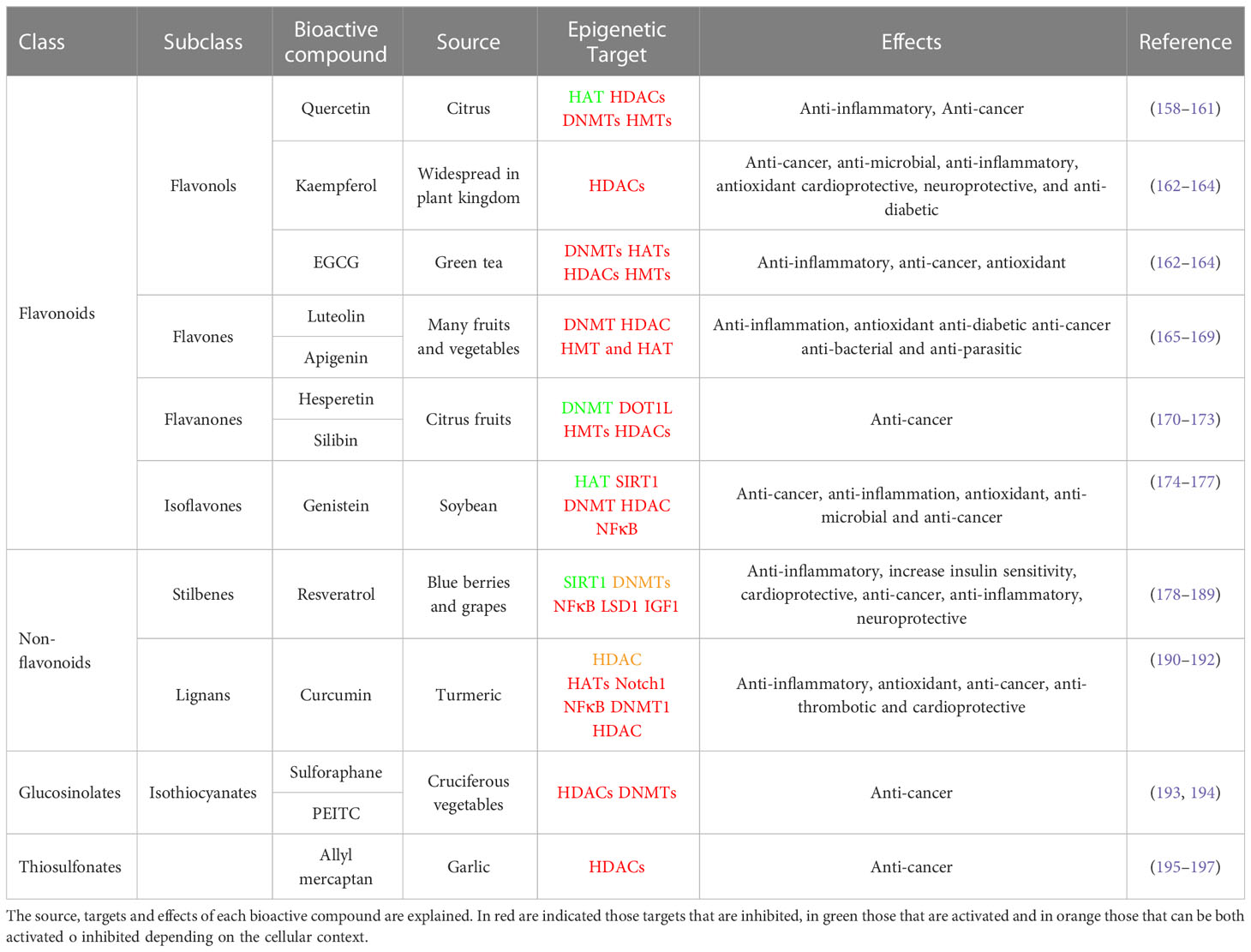
Table 1 Summary of main bioactive compounds implicated in the modulation of epigenetic enzymes.
Quercetin, a dietary polyphenol present in citrus, activates the NAD-dependent deacetylase SIRT1. In addition, it inhibits the expression of pro-inflammatory genes by mean of the inhibition of the interaction of p300/CBP and post-translational modifications, such as acetylation of H3 histones associated with the promoters of pro-inflammatory genes (158, 159). Quercetin has also been demonstrated to reduce the activity of HDACs, DNMTs and HMTs in a dose-dependent manner in human cervical cancer (160). Through its role in the inhibition of HDAC and DNMT1, quercetin has also been shown to inhibit the cell cycle and induce apoptosis, thus suppressing tumor growth and angiogenesis in preclinical mouse models (161).
Kaempferol (3,4′,5,7-tetrahydroxyflavone) is a potential HDAC inhibitor and an anti-cancer agent against many types of cancer (162). Kaempferol stimulates hyperacetylation of histone H3 in HepG2 and Hep3B (hepatoma cancer cell lines) as well as on HCT-116 (colon cancer cell line) (163). Kaempferol induced autophagic cell death via the inhibition of the HDAC/G9a axis and IRE1-JNK-CHOP in gastric cancer cell lines (164).
EGCG, present in green tea, has been extensively shown to affect the activity of distinct histone modifiers and DNA methyltransferases -DNMT1, DNMT3A, DNMT3B, AURKA, AURKB, AURKC, PRMT6, PRMT7, KDM4A, KDM5C, HDAC5, HDAC6, HDAC7, HDAC11 and UBE2B- activating the expression of various tumor suppressor genes (198). In prostate cancer cells, EGCG has been shown to reduce the activity of class I HDACs promoting the expression of the tumor suppressor genes p21 and Bax (199). EGCG inhibits inflammation by modulating the activity of HATs inhibiting the activity of NF-kB and downstream inflammatory targets (200, 201). EGCG improves insulin sensitivity and reduces obesity by epigenetic mechanisms modulating the muscle function (201). More specifically, EGCG directly inhibits DNMT by interacting with the catalytic site of the DNMT molecule (202).
Apigenin has been used in studies of many diseases because of its low toxicity and strong biological effects, including anti-inflammation, anti-oxidation, anti-diabetic, anti-tumor, anti-bacterial, and anti-parasitic effects (165). In MB-231 breast cancer cell line, apigenin promoted the acetylation of histone H3 and thus the transcription of P21WAF1. Importantly, apigenin inhibited breast cancer tumor growth in a xenograft nude mouse model (166). In human prostate cancer cells, apigenin inhibited HDAC1 and HDAC3, inducing growth arrest and apoptosis (167). Luteolin inhibited the proliferation and metastasis of androgen receptor-positive triple-negative breast cancer cell by mean of the epigenetic regulation -increased H3K56 and H3K27 acetylation- diminishing the AKT/mTOR dependent MMP9 expression (168).
The epigenetic modulatory effects of luteolin have been analyzed on HeLa cells, where it modulated the enzymatic activity of DNMT, HDAC, HMT, and HAT to reduce the global DNA methylation resulting in the reactivation of silenced tumor suppressor genes including FHIT, DAPK1, PTEN, CDH1, SOCS1, TIMPS, VHL, TP53, TP73 (169).
Citrus fruits are rich in aromatic flavonones such as hesperetin and silibinin.
Hesperetin has been shown to inhibit gastric cancer through epigenetic mechanisms including degradation of DOT1L, which is a Lysine N-Methyltransferase, and thereby reducing histone H3K79 methylation (170). Moreover hesperetin is an activator of SIRT1 contributing to the inhibition of hepatic inflammation via AMPK/CREB pathway (171).
Silibinin inhibits the growth of cancer cells, synergizing with DNA methyltransferase and histone deacetylase inhibitors to augment the expression of epithelial markers such as N-cadherin in non-small cell lung cancer cells (172). In human prostate cancer cells (DU145 and PC3), silibinin reduced gene expression levels of EZH2 by increasing H3K27me3 levels, decreased histone deacetylases 1–2 (HDACs1-2) expression levels, and increased total DNMTs activity (173).
Genistein (4’,5,7-trihydroxyisoflavone) is the most abundant isoflavone found in the soybean. Genistein has been shown to mediate post-translational changes in histones. Genistein inhibits SIRT1 leading to increase acetylation of histone H3K9 in PTEN, CYCD, and FOXO3A promoters in prostate cancer cells. Thus, in human prostate cancer cells genistein increases the expression of tumor suppressor genes such as p21WAF1/CIP1 and p16INK4a by regulating chromatin condensation via HAT expression (174). In LNCaP human prostate adenocarcinoma cells, genistein mediates post-translational changes such as the ubiquitination of androgen receptor by modulating the HDAC6-Hsp90 function (175). In prostate cancer cell lines, soy phytoestrogens have been shown to reduce DNA methylation at EPHB2, BRCA1 and GSTP1 promoters to inhibit cell proliferation and to induce cell death (176). Other genes, such as hTERT, MAD1L1, KDM4B and TRAF7 have shown reduced methylation profiles after genistein treatment (177).
Resveratrol is a polyphenol mainly found in blue berries and grapes. Some studies indicate that the anti-inflammatory effects of resveratrol are mediated by its epigenetic effects on the activity of HDAC11, SIRT1, and p300. Induction of SIRT1 by resveratrol has been demonstrated in several studies, where resveratrol augmented glucose-stimulated insulin secretion in both INS-1E cells (insulin-secreting beta cells) and human islets (178). In another work, resveratrol improved insulin sensitivity in mice fed with a high-fat diet, which seemed to be associated to the increased activity of SIRT1, that repressed the negative regulator protein tyrosine phosphatase 1B (PTP1B) of insulin secretion (179). Similarly, in rhesus monkeys, resveratrol led to an increase in SIRT1 expression and to an improvement in insulin sensitivity in visceral white adipose tissue (180). Many studies suggest that the activation of SIRT1 by resveratrol also contributes to the reduction of the lipid metabolism targets ACC (acetyl-CoA carboxylase), FAS (fatty acid synthase) in human HepG2 hepatocytes exposed to high glucose (181). SIRT1 has been shown to negatively regulate the expression of survivin, a member of the inhibitor of apoptosis (IAP) family, and to play an important role in ageing. Resveratrol has been shown to increase the expression of breast cancer 1, early onset (BRCA1), a human tumor suppressor gene, via histone H3 acetylation (182). In prostate cancer cells, resveratrol reduces growth and stimulates apoptosis through activation of FOXO transcription factors. The cardioprotective effects of resveratrol derive from its anti-apoptotic and anti-oxidant effects and its positive modulation of SIRT1 expression (183). In a mouse model of senescent heart, resveratrol improved the cardiac function by mean of its effects on the reduction of Foxo1 acetylation (184). The anti-tumorigenic activity of resveratrol by mean of its epigenetic effects on the HDAC pathway has been demonstrated in tumor models, both in vitro and in vivo (185, 186). In prostate cancer, resveratrol reactivated the expression of PTEN by abrogating the activity of the MTA1/HDAC complex. In addition, the MTA1 downregulation by resveratrol was shown to be partially mediated through its role in increasing p53 acetylation in PCa cells (187). The role of resveratrol on SIRT1 activation to extend lifespan has been extensively studied. Similar to caloric restriction, resveratrol increases autophagy through SIRT1 activation increasing lifespan both in human cells and C. elegans (188). Although several studies suggest that resveratrol is not able to extend lifespan in healthy mice its beneficial effects on metabolic-compromised mammals have demonstrated to be successful (189).
Curcumin has been extensively demonstrated to have anti-inflammatory, antioxidant, and anti-cancer effects. During diabetes, high glucose levels break the equilibrium between HAT and HDAC, augmenting the transcriptional activity of NF-kB and it´s downstream pro-inflammatory genes IL-6 and TNF-α. This effect has been shown to be partially reverted by curcumin by mean of its role on the inhibition of HDAC as demonstrated in preclinical mouse models (190). In PC3-M prostate cancer cells, curcumin promoted the proteasome-dependent degradation of p300/CBP without affecting the levels of other HATs such as PCAF or GCN5, being proposed as a therapeutic compound to specifically inhibit p300/CBP HAT (191). In hematopoietic Raji cells, curcumin suppressed the activity of NF-kB and Notch1 proteins, by inhibiting the protein levels of p300/CBP, HDAC13, HDAC1, HDAC3, and HDAC8, contributing to the reduction of inflammatory markers and cell proliferation (192).
Cruciferous vegetables are one of the main sources of the isothiocyanate sulforaphane, which is a well-known bioactive compound implicated in the induction of phase-II detoxification enzymes. Recently, sulforaphane has been described as an epigenetic modulator in several human diseases, including cancer. Sulforaphane inhibits histone deacetylases HDAC2 and 3, leading to the increase acetylation of histones H3 and H4 on p21 promoter inhibiting breast cancer cell proliferation (193). In LnCaP prostate cancer cells and in breast cancer cells, sulforaphane reduces the expression of the DNA methyltransferases DNMT1 and DNMT3a, contributing to the increased expression of several tumor suppressor genes (194).
Phenethyl isothiocyanate (PEITC) has been shown to be an HDAC inhibitor in prostate cancer, leukemia, and myeloma cells (203, 204). PEITC was also shown to inhibit leukemia development in mice. Treatment of LNCaP cells with PEITC inhibited the activity and levels of HDACs and induced selective histone acetylation, whereas in mouse erythroleukemia DS19 cells, allyl isothiocyanate increased histone acetylation without affecting HDAC activity (205).
Several garlic-derived small organosulfur compounds such as allyl mercaptan (AM) have been described to inhibit the HDAC activity in vitro. In AM-treated human colon cancer cells, HDAC inhibition led to a rapid and sustained accumulation of acetylated histones in total cellular chromatin (195). Chromatin immunoprecipitation assays demonstrated the role of AM on histone H3 acetylation at the P21WAF1 promoter, favoring the increased binding of the transcription factor Sp3. Interestingly, AM also augmented the binding of p53 in the distal enhancer region of the P21WAF1 gene promoter, inducing a G1 cell cycle arrest (195). The anti-cancer effects of diallyl disulfide (DADS) and its active metabolite S-allyl mercaptocysteine (SAMC) have been also associated to H3K14 acetylation at the P21WAF1 gene in colon and breast cancer (196, 197).
Nutrition, diet, and metabolism regulate epigenetic responses and integrate environmental cues with cellular responses. Healthy diets sustaining balanced epigenetic landscapes have been demonstrated to promote healthspan and to counteract ageing and metabolic diseases. On the contrary, unbalanced diets such as Western diets can alter metabolic and epigenetic traits leading to the development and progression of metabolic diseases and ageing. Diet, exercise, and bioactive compounds from natural sources may provide metabolites and nutrients -vitamins and minerals- to restore epigenetic homeostasis.
Unbalanced diets affect the epigenome contributing to the development and progression of chronic diseases, cancer, and ageing. Therefore, the relationship between lifestyle habits (mainly diet and exercise) and epigenetic regulation is a promising landscape to develop personalized nutrition strategies (nutritional epigenetics). Recently, epitranscriptomics -epigenetic marks in mRNA- is a novel field of research. m6A RNA modification has been shown to play a critical role in regulating gene expression and signalling pathways for physiological homeostasis. The molecular interactions between gut microbiota and the metabolo-epigenetic regulation need to be further investigated as part of diagnosis, prognosis, and treatment of human metabolic diseases. (5) In addition, some metabolites have been shown to be affected by circadian clock, establishing a direct link between cyclic rhythms and metabolism in the cell. The circadian clock in cells and the systemic level in a variety of metabolic and physiological processes is in harmony with the external 24-hour cycle of light and dark. Thus, alteration of the circadian rhythm may be associated with the development of NCDs, and ageing through its effects on the cyclic availability of coenzymes and metabolites (206). For example, although the expression levels of SIRT1 are non-cyclic, NAD+ synthesis is directly regulated by the circadian clock machinery, which controls the transcription of the Nampt gene. These findings suggest that several SIRT1 targets are likely to display circadian oscillations in their acetylation. This is the case for K9/K14 histone H3 sites at circadian gene promoters, as well as BMAL1, a non-histone target of SIRT1, that operates as a transcriptional co-activator of the circadian regulator CLOCK (207).
MGdC and ARdM contributed to conception and design of the study. MGdC wrote the first draft of the manuscript. RMP wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by grants to AdM from the Ministerio de Ciencia e Innovacion, Spain (PID2019-110183RB-C21), Ramon Areces Foundation (CIVP19A5937) and Regional Government of Community of Madrid (P2018/BAA-4343-ALIBIRD2020-CM and NutriSION-CM Y2020/BIO-6350), REACT EU Program (FACINGLCOVID-CM project, Comunidad de Madrid and The European Regional Development Fund (ERDF) European Union), and EU Structural Funds.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Radman-Livaja M, Liu CL, Friedman N, Schreiber SL, Rando OJ. Replication and active demethylation represent partially overlapping mechanisms for erasure of H3K4me3 in budding yeast. PloS Genet (2010) 6:e1000837. doi: 10.1371/journal.pgen.1000837
2. Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature (2019) 571:489–99. doi: 10.1038/s41586-019-1411-0
3. Ling C, Ronn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab (2019) 29:1028–44. doi: 10.1016/j.cmet.2019.03.009
4. Global Burden of Disease Collaborative Network, G. B. o. D. S. G. R, Institute for Health Metrics and Evaluation - IHME. .
5. Janke R, Dodson AE, Rine J. Metabolism and epigenetics. Annu Rev Cell Dev Biol (2015) 31:473–96. doi: 10.1146/annurev-cellbio-100814-125544
6. Etchegaray JP, Mostoslavsky R. Interplay between metabolism and epigenetics: A nuclear adaptation to environmental changes. Mol Cell (2016) 62:695–711. doi: 10.1016/j.molcel.2016.05.029
7. Divella R, Daniele A, Savino E, Paradiso A. Anticancer effects of nutraceuticals in the mediterranean diet: An epigenetic diet model. Cancer Genomics Proteomics (2020) 17:335–50. doi: 10.21873/cgp.20193
8. Zhang L, Lu Q, Chang C. Epigenetics in health and disease. Adv Exp Med Biol (2020) 1253:3–55. doi: 10.1007/978-981-15-3449-2_1
9. Dai Z, Ramesh V, Locasale JW. The evolving metabolic landscape of chromatin biology and epigenetics. Nat Rev Genet (2020) 21:737–53. doi: 10.1038/s41576-020-0270-8
10. Boon R. Metabolic fuel for epigenetic: Nuclear production meets local consumption. Front Genet (2021) 12:768996. doi: 10.3389/fgene.2021.768996
11. Gomez-Pinilla F, Tyagi E. Diet and cognition: interplay between cell metabolism and neuronal plasticity. Curr Opin Clin Nutr Metab Care (2013) 16:726–33. doi: 10.1097/MCO.0b013e328365aae3
12. Katada S, Imhof A, Sassone-Corsi P. Connecting threads: epigenetics and metabolism. Cell (2012) 148:24–8. doi: 10.1016/j.cell.2012.01.001
13. Harvey A, Caretti G, Moresi V, Renzini A, Adamo S. Interplay between metabolites and the epigenome in regulating embryonic and adult stem cell potency and maintenance. Stem Cell Rep (2019) 13:573–89. doi: 10.1016/j.stemcr.2019.09.003
14. Smith E, Shilatifard A. The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol Cell (2010) 40:689–701. doi: 10.1016/j.molcel.2010.11.031
15. Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature (2013) 502:472–9. doi: 10.1038/nature12750
16. Kumar S, Mohapatra T. Deciphering epitranscriptome: Modification of mRNA bases provides a new perspective for post-transcriptional regulation of gene expression. Front Cell Dev Biol (2021) 9:628415. doi: 10.3389/fcell.2021.628415
17. Geng X, Li Z, Yang Y. Emerging role of epitranscriptomics in diabetes mellitus and its complications. Front Endocrinol (2022) 13:907060. doi: 10.3389/fendo.2022.907060
18. Barrero MJ, Cejas P, Long HW, de Molina AR. Nutritional epigenetics in cancer. Adv Nutr (2022). doi: 10.1093/advances/nmac039
19. Li Y, et al. m(6)A regulates liver metabolic disorders and hepatogenous diabetes. Genomics Proteomics Bioinf (2020) 18:371–83. doi: 10.1016/j.gpb.2020.06.003
20. Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. Genome regulation by polycomb and trithorax: 70 years and counting. Cell (2017) 171:34–57. doi: 10.1016/j.cell.2017.08.002
21. Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep (2015) 16:1467–81. doi: 10.15252/embr.201540945
22. Posavec Marjanovic M, Hurtado-Bagès S, Lassi M, Valero V, Malinverni R, Delage H, et al. MacroH2A1.1 regulates mitochondrial respiration by limiting nuclear NAD(+) consumption. Nat Struct Mol Biol (2017) 24:902–10. doi: 10.1038/nsmb.3481
23. Guberovic I, Hurtado-Bagès S, Rivera-Casas C, Knobloch G, Malinverni R, Valero V, et al. Evolution of a histone variant involved in compartmental regulation of NAD metabolism. Nat Struct Mol Biol (2021) 28:1009–19. doi: 10.1038/s41594-021-00692-5
24. Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science (2001) 294:2536–9. doi: 10.1126/science.1065848
25. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet (2012) 13:484–92. doi: 10.1038/nrg3230
26. Svedruzic ZM. Dnmt1 structure and function. Prog Mol Biol Transl Sci (2011) 101:221–54. doi: 10.1016/B978-0-12-387685-0.00006-8
27. Gao L, Emperle M, Guo Y, Grimm SA, Ren W, Adam S, et al. Comprehensive structure-function characterization of DNMT3B and DNMT3A reveals distinctive de novo DNA methylation mechanisms. Nat Commun (2020) 11:3355. doi: 10.1038/s41467-020-17109-4
28. Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet (2017) 18:517–34. doi: 10.1038/nrg.2017.33
29. Jeltsch A, Broche J, Bashtrykov P. Molecular processes connecting DNA methylation patterns with DNA methyltransferases and histone modifications in mammalian genomes. Genes (Basel) (2018) 9. doi: 10.3390/genes9110566
30. Gillberg L, Orskov AD, Liu M, Harslof LBS, Jones PA, Gronbaek K. Vitamin c - a new player in regulation of the cancer epigenome. Semin Cancer Biol (2018) 51:59–67. doi: 10.1016/j.semcancer.2017.11.001
31. Yang Q, Liang X, Sun X, Zhang L, Fu X, Rogers CJ, et al. AMPK/alpha-ketoglutarate axis dynamically mediates DNA demethylation in the Prdm16 promoter and brown adipogenesis. Cell Metab (2016) 24:542–54. doi: 10.1016/j.cmet.2016.08.010
32. Tian Q, Zhao J, Yang Q, Wang B, Deavila JM, Zhu MJ, et al. Dietary alpha-ketoglutarate promotes beige adipogenesis and prevents obesity in middle-aged mice. Aging Cell (2020) 19:e13059. doi: 10.1111/acel.13059
33. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martinez JA, Goyal P, Mahapatra S, et al. Vitamin c induces tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature (2013) 500:222–6. doi: 10.1038/nature12362
34. Loaeza-Loaeza J, Illades-Aguiar B, Del Moral-Hernandez O, Castro-Coronel Y, Leyva-Vazquez MA, Dircio-Maldonado R, et al. The CpG island methylator phenotype increases the risk of high-grade squamous intraepithelial lesions and cervical cancer. Clin Epigenet (2022) 14:4. doi: 10.1186/s13148-021-01224-0
35. Madala HR, Punganuru SR, Arutla V, Misra S, Thomas TJ, Srivenugopal KS. Beyond brooding on oncometabolic havoc in IDH-mutant gliomas and AML: Current and future therapeutic strategies. Cancers (2018) 10. doi: 10.3390/cancers10020049
36. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol (2014) 10:93–5. doi: 10.1038/nchembio.1432
37. Dai XY, Shi L, Li Z, Yang HY, Wei JF, Ding Q. Main N6-methyladenosine readers: YTH family proteins in cancers. Front Oncol (2021) 11:635329. doi: 10.3389/fonc.2021.635329
38. Han X, Wang L, Han Q. Advances in the role of m(6)A RNA modification in cancer metabolic reprogramming. Cell bioscience (2020) 10:117. doi: 10.1186/s13578-020-00479-z
39. Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell (2020) 38:79–96.e11. doi: 10.1016/j.ccell.2020.04.017
40. Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature (2021) 593:597–601. doi: 10.1038/s41586-021-03536-w
41. Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell (2019) 35:677–691.e610. doi: 10.1016/j.ccell.2019.03.006
42. Peng S, Xiao W, Ju D, Sun B, Hou N, Liu Q, et al. Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Sci Trans Med (2019) 11. doi: 10.1126/scitranslmed.aau7116
43. Roberti A, Valdes AF, Torrecillas R, Fraga MF, Fernandez AF. Epigenetics in cancer therapy and nanomedicine. Clin Epigenet (2019) 11:81. doi: 10.1186/s13148-019-0675-4
44. Cascante A, Klum S, Biswas M, Antolin-Fontes B, Barnabé-Heider F, Hermanson O. Gene-specific methylation control of H3K9 and H3K36 on neurotrophic BDNF versus astroglial GFAP genes by KDM4A/C regulates neural stem cell differentiation. J Mol Biol (2014) 426:3467–77. doi: 10.1016/j.jmb.2014.04.008
45. Niwa H, Umehara T. Structural insight into inhibitors of flavin adenine dinucleotide-dependent lysine demethylases. Epigenetics (2017) 12:340–52. doi: 10.1080/15592294.2017.1290032
46. Molina-Serrano D, Kyriakou D, Kirmizis A. Histone modifications as an intersection between diet and longevity. Front Genet (2019) 10:192. doi: 10.3389/fgene.2019.00192
47. Sivanand S, Viney I, Wellen KE. Spatiotemporal control of acetyl-CoA metabolism in chromatin regulation. Trends Biochem Sci (2018) 43:61–74. doi: 10.1016/j.tibs.2017.11.004
48. Liou GG, Tanny JC, Kruger RG, Walz T, Moazed D. Assembly of the SIR complex and its regulation by o-acetyl-ADP-ribose, a product of NAD-dependent histone deacetylation. Cell (2005) 121:515–27. doi: 10.1016/j.cell.2005.03.035
49. Walker AK, Yang F, Jiang K, Ji JY, Watts JL, Purushotham A, et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev (2010) 24:1403–17. doi: 10.1101/gad.1901210
50. Kugel S, Mostoslavsky R. Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem Sci (2014) 39:72–81. doi: 10.1016/j.tibs.2013.12.002
51. Donohoe DR, Bultman SJ. Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol (2012) 227:3169–77. doi: 10.1002/jcp.24054
52. Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell (2014) 26:577–90. doi: 10.1016/j.ccr.2014.07.028
53. Bowman GD, Poirier MG. Post-translational modifications of histones that influence nucleosome dynamics. Chem Rev (2015) 115:2274–95. doi: 10.1021/cr500350x
54. Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol (2014) 15:703–8. doi: 10.1038/nrm3890
55. Wang T, Ye Z, Li Z, Jing DS, Fan GX, Liu MQ, et al. Lactate-induced protein lactylation: A bridge between epigenetics and metabolic reprogramming in cancer. Cell proliferation (2023) e13478. doi: 10.1111/cpr.13478
56. Jiang J, Huang D, Jiang Y, Hou J, Tian M, Li J, et al. Lactate modulates cellular metabolism through histone lactylation-mediated gene expression in non-small cell lung cancer. Front Oncol (2021) 11:647559. doi: 10.3389/fonc.2021.647559
57. Landrier JF, Derghal A, Mounien L. MicroRNAs in obesity and related metabolic disorders. Cells (2019) 8. doi: 10.3390/cells8080859
58. Kloting N, et al. MicroRNA expression in human omental and subcutaneous adipose tissue. PloS One (2009) 4:e4699. doi: 10.1371/journal.pone.0004699
59. Seeger T, Fischer A, Muhly-Reinholz M, Zeiher AM, Dimmeler S. Long-term inhibition of miR-21 leads to reduction of obesity in db/db mice. Obes (Silver Spring) (2014) 22:2352–60. doi: 10.1002/oby.20852
60. Frost RJ, Olson EN. Control of glucose homeostasis and insulin sensitivity by the let-7 family of microRNAs. Proc Natl Acad Sci United States America (2011) 108:21075–80. doi: 10.1073/pnas.1118922109
61. Manning P, Munasinghe PE, Bellae Papannarao J, Gray AR, Sutherland W, Katare R. Acute weight loss restores dysregulated circulating MicroRNAs in individuals who are obese. J Clin Endocrinol Metab (2019) 104:1239–48. doi: 10.1210/jc.2018-00684
62. Sun J, Ruan Y, Wang M, Chen R, Yu N, Sun L, et al. Differentially expressed circulating LncRNAs and mRNA identified by microarray analysis in obese patients. Sci Rep (2016) 6:35421. doi: 10.1038/srep35421
63. Stapleton K, Novel Long Noncoding RNA. Macrophage inflammation-suppressing transcript (MIST), regulates macrophage activation during obesity. Arteriosclerosis thrombosis Vasc Biol (2020) 40:914–28. doi: 10.1161/ATVBAHA.119.313359
64. Squillaro T, Peluso G, Galderisi U, Di Bernardo G. Long non-coding RNAs in regulation of adipogenesis and adipose tissue function. eLife (2020) 9. doi: 10.7554/eLife.59053
65. Chrysanthou S, Flores JC, Dawlaty MM. Tet1 suppresses p21 to ensure proper cell cycle progression in embryonic stem cells. Cells (2022) 11. doi: 10.3390/cells11081366
66. Saha D, Norvil AB, Lanman NA, Gowher H. Simplified MethylRAD sequencing to detect changes in DNA methylation at enhancer elements in differentiating embryonic stem cells. Epigenomes (2020) 4. doi: 10.3390/epigenomes4040024
67. Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab (2015) 22:861–73. doi: 10.1016/j.cmet.2015.08.024
68. Zhang N. Role of methionine on epigenetic modification of DNA methylation and gene expression in animals. Anim Nutr (2018) 4:11–6. doi: 10.1016/j.aninu.2017.08.009
69. Froese DS, Fowler B, Baumgartner MR. Vitamin B12 , folate, and the methionine remethylation cycle-biochemistry, pathways, and regulation. J Inherit Metab Dis (2019) 42:673–85. doi: 10.1002/jimd.12009
70. Gueant JL, Oussalah A, Zgheib R, Siblini Y, Hsu SB, Namour F. Genetic, epigenetic and genomic mechanisms of methionine dependency of cancer and tumor-initiating cells: What could we learn from folate and methionine cycles. Biochimie (2020) 173:123–8. doi: 10.1016/j.biochi.2020.03.015
71. Chiuve SE, Giovannucci EL, Hankinson SE, Zeisel SH, Dougherty LW, Willett WC, et al. The association between betaine and choline intakes and the plasma concentrations of homocysteine in women. Am J Clin Nutr (2007) 86:1073–81. doi: 10.1093/ajcn/86.4.1073
72. Garcia-Gimenez JL, Roma-Mateo C, Perez-Machado G, Peiro-Chova L, Pallardo FV. Role of glutathione in the regulation of epigenetic mechanisms in disease. Free Radic Biol Med (2017) 112:36–48. doi: 10.1016/j.freeradbiomed.2017.07.008
73. Costanzo M, Cevenini A, Marchese E, Imperlini E, Raia M, Del Vecchio L, et al. Label-free quantitative proteomics in a methylmalonyl-CoA mutase-silenced neuroblastoma cell line. Int J Mol Sci (2018) 19. doi: 10.3390/ijms19113580
74. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
75. Feron O. The many metabolic sources of acetyl-CoA to support histone acetylation and influence cancer progression. Ann Transl Med (2019) 7:S277. doi: 10.21037/atm.2019.11.140
76. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet (2011) 43:869–74. doi: 10.1038/ng.890
77. Echeverria C, Nualart F, Ferrada L, Smith GJ, Godoy AS. Hexose transporters in cancer: From multifunctionality to diagnosis and therapy. Trends Endocrinol Metab (2021) 32:198–211. doi: 10.1016/j.tem.2020.12.006
78. Stacpoole PW. Therapeutic targeting of the pyruvate dehydrogenase Complex/Pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J Natl Cancer Inst (2017) 109. doi: 10.1093/jnci/djx071
79. Chen J, Guccini I, Di Mitri D, Brina D, Revandkar A, Sarti M, et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat Genet (2018) 50:219–28. doi: 10.1038/s41588-017-0026-3
80. Wong CC, Qian Y, Yu J. Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches. Oncogene (2017) 36:3359–74. doi: 10.1038/onc.2016.485
81. Moffett JR, Puthillathu N, Vengilote R, Jaworski DM, Namboodiri AM. Acetate revisited: A key biomolecule at the nexus of metabolism, epigenetics, and oncogenesis - part 2: Acetate and ACSS2 in health and disease. Front Physiol (2020) 11:580171. doi: 10.3389/fphys.2020.580171
82. Gao X, Lin SH, Ren F, Li JT, Chen JJ, Yao CB, et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat Commun (2016) 7:11960. doi: 10.1038/ncomms11960
83. Campbell SL, Wellen KE. Metabolic signaling to the nucleus in cancer. Mol Cell (2018) 71:398–408. doi: 10.1016/j.molcel.2018.07.015
84. Li X, Egervari G, Wang Y, Berger SL, Lu Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol (2018) 19:563–78. doi: 10.1038/s41580-018-0029-7
85. Du X, Hu H. The roles of 2-hydroxyglutarate. Front Cell Dev Biol (2021) 9:651317. doi: 10.3389/fcell.2021.651317
86. McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA, et al. Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep (2016) 17:1463–72. doi: 10.1016/j.celrep.2016.10.012
87. Jiang X, Ye X, Guo W, Lu H, Gao Z. Inhibition of HDAC3 promotes ligand-independent PPARgamma activation by protein acetylation. J Mol Endocrinol (2014) 53:191–200. doi: 10.1530/JME-14-0066
88. Kim JA, Yeom YI. Metabolic signaling to epigenetic alterations in cancer. Biomolecules Ther (2018) 26:69–80. doi: 10.4062/biomolther.2017.185
89. Carson C, Lawson HA. Epigenetics of metabolic syndrome. Physiol Genomics (2018) 50:947–55. doi: 10.1152/physiolgenomics.00072.2018
90. Prinz-Langenohl R, Bramswig S, Tobolski O, Smulders YM, Smith DE, Finglas PM, et al. [6S]-5-methyltetrahydrofolate increases plasma folate more effectively than folic acid in women with the homozygous or wild-type 677C–>T polymorphism of methylenetetrahydrofolate reductase. Br J Pharmacol (2009) 158:2014–21. doi: 10.1111/j.1476-5381.2009.00492.x
91. Tanaka T, Scheet P, Giusti B, Bandinelli S, Piras MG, Usala G, et al. Genome-wide association study of vitamin B6, vitamin B12, folate, and homocysteine blood concentrations. Am J Hum Genet (2009) 84:477–82. doi: 10.1016/j.ajhg.2009.02.011
92. Yen KE, Bittinger MA, Su SM, Fantin VR. Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene (2010) 29:6409–17. doi: 10.1038/onc.2010.444
93. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell (2010) 18:553–67. doi: 10.1016/j.ccr.2010.11.015
94. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature (2012) 483:474–8. doi: 10.1038/nature10860
95. Loos RJ, Yeo GS. The bigger picture of FTO: the first GWAS-identified obesity gene. Nat Rev Endocrinol (2014) 10:51–61. doi: 10.1038/nrendo.2013.227
96. Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, et al. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med (2015) 373:895–907. doi: 10.1056/NEJMoa1502214
97. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
98. Tiffon C. The impact of nutrition and environmental epigenetics on human health and disease. Int J Mol Sci (2018) 19. doi: 10.3390/ijms19113425
99. Scully T, Ettela A, LeRoith D, Gallagher EJ. Obesity, type 2 diabetes, and cancer risk. Front Oncol (2020) 10:615375. doi: 10.3389/fonc.2020.615375
100. Gao W, Liu JL, Lu X, Yang Q. Epigenetic regulation of energy metabolism in obesity. J Mol Cell Biol (2021) 13:480–99. doi: 10.1093/jmcb/mjab043
101. Wu J, Frazier K, Zhang J, Gan Z, Wang T, Zhong X. Emerging role of m(6) a RNA methylation in nutritional physiology and metabolism. Obes Rev an Off J Int Assoc Study Obes (2020) 21:e12942. doi: 10.1111/obr.12942
102. Ronningen T, Dahl MB, Valderhaug TG, Cayir A, Keller M, Tonjes A, et al. m6A regulators in human adipose tissue - depot-specificity and correlation with obesity. Front Endocrinol (2021) 12:778875. doi: 10.3389/fendo.2021.778875
103. Luo Z, Zhang Z, Tai L, Zhang L, Sun Z, Zhou L. Comprehensive analysis of differences of N(6)-methyladenosine RNA methylomes between high-fat-fed and normal mouse livers. Epigenomics (2019) 11:1267–82. doi: 10.2217/epi-2019-0009
104. Azzam SK, Alsafar H, Sajini AA. FTO m6A demethylase in obesity and cancer: Implications and underlying molecular mechanisms. Int J Mol Sci (2022) 23. doi: 10.3390/ijms23073800
105. Zhang Z, Zhang C, Yang Z, Zhang G, Wu P, Luo Y, et al. m(6)A regulators as predictive biomarkers for chemotherapy benefit and potential therapeutic targets for overcoming chemotherapy resistance in small-cell lung cancer. J Hematol Oncol (2021) 14:190. doi: 10.1186/s13045-021-01173-4
106. Okabe K, Yaku K, Tobe K, Nakagawa T. Implications of altered NAD metabolism in metabolic disorders. J Biomed Sci (2019) 26:34. doi: 10.1186/s12929-019-0527-8
107. Katsyuba E, Romani M, Hofer D, Auwerx J. NAD(+) homeostasis in health and disease. Nat Metab (2020) 2:9–31. doi: 10.1038/s42255-019-0161-5
108. Yuan Y, Zhu C, Wang Y, Sun J, Feng J, Ma Z, et al. Alpha-ketoglutaric acid ameliorates hyperglycemia in diabetes by inhibiting hepatic gluconeogenesis via serpina1e signaling. Sci Adv (2022) 8:eabn2879. doi: 10.1126/sciadv.abn2879
110. Kane AE, Sinclair DA. Epigenetic changes during aging and their reprogramming potential. Crit Rev Biochem Mol Biol (2019) 54:61–83. doi: 10.1080/10409238.2019.1570075
111. Ni XL, Yuan HP, Jiao J, Wang ZP, Su HB, Lyu Y, et al. [An epigenetic clock model for assessing the human biological age of healthy aging]. Zhonghua yi xue za zhi (2022) 102:119–24. doi: 10.3760/cma.j.cn112137-20210817-01862
112. Kabacik S, Horvath S, Cohen H, Raj K. Epigenetic ageing is distinct from senescence-mediated ageing and is not prevented by telomerase expression. Aging (2018) 10:2800–15. doi: 10.18632/aging.101588
113. Seale K, Horvath S, Teschendorff A, Eynon N, Voisin S. Making sense of the ageing methylome. Nat Rev Genet (2022) 23:585–605. doi: 10.1038/s41576-022-00477-6
114. Rutledge J, Oh H, Wyss-Coray T. Measuring biological age using omics data. Nat Rev Genet (2022) 23:715–27. doi: 10.1038/s41576-022-00511-7
115. Beynon RA, et al. Epigenetic biomarkers of ageing are predictive of mortality risk in a longitudinal clinical cohort of individuals diagnosed with oropharyngeal cancer. Clin Epigenet (2022) 14:1. doi: 10.1186/s13148-021-01220-4
116. Kuzub N, Smialkovska V, Momot V, Moseiko V, Lushchak O, Koliada A. Evaluation of epigenetic age based on DNA methylation analysis of several CpG sites in ukrainian population. Front Genet (2021) 12:772298. doi: 10.3389/fgene.2021.772298
117. Ouyang Y, Tu Y, Chen S, Min H, Wen Z, Zheng G, et al. Characterization of immune microenvironment infiltration and m(6)A regulator-mediated RNA methylation modification patterns in osteoarthritis. Front Immunol (2022) 13:1018701. doi: 10.3389/fimmu.2022.1018701
118. Covarrubias AJ, Kale A, Perrone R, Lopez-Dominguez JA, Pisco AO, Kasler HG, et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat Metab (2020) 2:1265–83. doi: 10.1038/s42255-020-00305-3
119. Rodriguez-Sanabria JS, Escutia-Gutierrez R, Rosas-Campos R, Armendariz-Borunda JS, Sandoval-Rodriguez A. An update in epigenetics in metabolic-associated fatty liver disease. Front Med (Lausanne) (2021) 8:770504. doi: 10.3389/fmed.2021.770504
120. Asif S, Morrow NM, Mulvihill EE, Kim KH. Understanding dietary intervention-mediated epigenetic modifications in metabolic diseases. Front Genet (2020) 11:590369. doi: 10.3389/fgene.2020.590369
121. Urbonaite G, Knyzeliene A, Bunn FS, Smalskys A, Neniskyte U. The impact of maternal high-fat diet on offspring neurodevelopment. Front Neurosci (2022) 16:909762. doi: 10.3389/fnins.2022.909762
122. Pham TX, Lee J. Dietary regulation of histone acetylases and deacetylases for the prevention of metabolic diseases. Nutrients (2012) 4:1868–86. doi: 10.3390/nu4121868
123. Xu F, Liu J, Na L, Chen L. Roles of epigenetic modifications in the differentiation and function of pancreatic beta-cells. Front Cell Dev Biol (2020) 8:748. doi: 10.3389/fcell.2020.00748
124. Maganti AV, Maier B, Tersey SA, Sampley ML, Mosley AL, Ozcan S, et al. Transcriptional activity of the islet beta cell factor Pdx1 is augmented by lysine methylation catalyzed by the methyltransferase Set7/9. J Biol Chem (2015) 290:9812–22. doi: 10.1074/jbc.M114.616219
125. Li T, Mao C, Wang X, Shi Y, Tao Y. Epigenetic crosstalk between hypoxia and tumor driven by HIF regulation. J Exp Clin Cancer Res (2020) 39:224. doi: 10.1186/s13046-020-01733-5
126. Berni Canani R, Di Costanzo M, Leone L. The epigenetic effects of butyrate: potential therapeutic implications for clinical practice. Clin Epigenet (2012) 4:4. doi: 10.1186/1868-7083-4-4
127. Martinez-Garza U, Torres-Oteros D, Yarritu-Gallego A, Marrero PF, Haro D, Relat J. Fibroblast growth factor 21 and the adaptive response to nutritional challenges. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20194692
128. Palmer JD, Soule BP, Simone BA, Zaorsky NG, Jin L, Simone NL. MicroRNA expression altered by diet: can food be medicinal? Ageing Res Rev (2014) 17:16–24. doi: 10.1016/j.arr.2014.04.005
129. Slattery ML, Herrick JS, Mullany LE, Stevens JR, Wolff RK. Diet and lifestyle factors associated with miRNA expression in colorectal tissue. Pharmacogenomics personalized Med (2017) 10:1–16. doi: 10.2147/PGPM.S117796
130. Kim CH, Lee EK, Choi YJ, An HJ, Jeong HO, Park D, et al. Short-term calorie restriction ameliorates genomewide, age-related alterations in DNA methylation. Aging Cell (2016) 15:1074–81. doi: 10.1111/acel.12513
131. Hahn O, Gronke S, Stubbs TM, Ficz G, Hendrich O, Krueger F, et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol (2017) 18:56. doi: 10.1186/s13059-017-1187-1
132. Ruan HB, Crawford PA. Ketone bodies as epigenetic modifiers. Curr Opin Clin Nutr Metab Care (2018) 21:260–6. doi: 10.1097/MCO.0000000000000475
133. Evans M, Cogan KE, Egan B. Metabolism of ketone bodies during exercise and training: physiological basis for exogenous supplementation. J Physiol (2017) 595:2857–71. doi: 10.1113/JP273185
134. Marosi K, Kim SW, Moehl K, Scheibye-Knudsen M, Cheng A, Cutler R, et al. 3-hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J Neurochem (2016) 139:769–81. doi: 10.1111/jnc.13868
135. McCarty MF, Barroso-Aranda J, Contreras F. The low-methionine content of vegan diets may make methionine restriction feasible as a life extension strategy. Med Hypotheses (2009) 72:125–8. doi: 10.1016/j.mehy.2008.07.044
136. Ables GP, Hens JR, Nichenametla SN. Methionine restriction beyond life-span extension. Ann N Y Acad Sci (2016) 1363:68–79. doi: 10.1111/nyas.13014
137. Dai Z, Mentch SJ, Gao X, Nichenametla SN, Locasale JW. Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat Commun (2018) 9:1955. doi: 10.1038/s41467-018-04426-y
138. Bose S, Allen AE, Locasale JW. The molecular link from diet to cancer cell metabolism. Mol Cell (2020) 78:1034–44. doi: 10.1016/j.molcel.2020.05.018
139. Wanders D, Hobson K, Ji X. Methionine restriction and cancer biology. Nutrients (2020) 12. doi: 10.3390/nu12030684
140. Liu G, Yu L, Fang J, Hu CA, Yin J, Ni H, et al. Methionine restriction on oxidative stress and immune response in dss-induced colitis mice. Oncotarget (2017) 8:44511–20. doi: 10.18632/oncotarget.17812
141. Pellanda H, Namour F, Fofou-Caillierez M, Bressenot A, Alberto JM, Chery C, et al. A splicing variant leads to complete loss of function of betaine-homocysteine methyltransferase (BHMT) gene in hepatocellular carcinoma. Int J Biochem Cell Biol (2012) 44:385–92. doi: 10.1016/j.biocel.2011.11.014
142. Nur SM, Rath S, Ahmad V, Ahmad A, Ateeq B, Khan MI. Nutritive vitamins as epidrugs. Crit Rev Food Sci Nutr (2021) 61:1–13. doi: 10.1080/10408398.2020.1712674
143. Bar-El Dadon S, Reifen R. Vitamin a and the epigenome. Crit Rev Food Sci Nutr (2017) 57:2404–11. doi: 10.1080/10408398.2015.1060940
144. Lyon P, Strippoli V, Fang B, Cimmino ,LB. Vitamins and one-carbon metabolism: Implications in human health and disease. Nutrients (2020) 12. doi: 10.3390/nu12092867
145. Dosedel M, Jirkovsky E, Macakova K, Krcmova LK, Javorska L, Pourova J, et al. Vitamin c-sources, physiological role, kinetics, deficiency, use, toxicity, and determination. Nutrients (2021) 13. doi: 10.3390/nu13020615
146. Lee Chong T, Ahearn EL, Cimmino L. Reprogramming the epigenome with vitamin c. Front Cell Dev Biol (2019) 7:128. doi: 10.3389/fcell.2019.00128
148. Bajpeyi S, Covington JD, Taylor EM, Stewart LK, Galgani JE, Henagan TM. Skeletal muscle PGC1alpha -1 nucleosome position and -260 nt DNA methylation determine exercise response and prevent ectopic lipid accumulation in men. Endocrinology (2017) 158:2190–9. doi: 10.1210/en.2017-00051
149. Ronn T, Volkov P, Davegardh C, Dayeh T, Hall E, Olsson AH, et al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. PloS Genet (2013) 9:e1003572. doi: 10.1371/journal.pgen.1003572
150. Rowlands DS, Page RA, Sukala WR, Giri M, Ghimbovschi SD, Hayat I, et al. Multi-omic integrated networks connect DNA methylation and miRNA with skeletal muscle plasticity to chronic exercise in type 2 diabetic obesity. Physiol Genomics (2014) 46:747–65. doi: 10.1152/physiolgenomics.00024.2014
151. Nielsen S, Scheele C, Yfanti C, Akerstrom T, Nielsen AR, Pedersen BK, et al. Muscle specific microRNAs are regulated by endurance exercise in human skeletal muscle. J Physiol (2010) 588:4029–37. doi: 10.1113/jphysiol.2010.189860
152. Barron-Cabrera E, Ramos-Lopez O, Gonzalez-Becerra K, Riezu-Boj JI, Milagro FI, Martinez-Lopez E, et al. Epigenetic modifications as outcomes of exercise interventions related to specific metabolic alterations: A systematic review. Lifestyle Genomics (2019) 12:25–44. doi: 10.1159/000503289
153. Flores-Opazo M, McGee SL, Hargreaves M. Exercise and GLUT4. Exercise sport Sci Rev (2020) 48:110–8. doi: 10.1249/JES.0000000000000224
154. LeBlanc PJ, Peters SJ, Tunstall RJ, Cameron-Smith D, Heigenhauser GJ. Effects of aerobic training on pyruvate dehydrogenase and pyruvate dehydrogenase kinase in human skeletal muscle. J Physiol (2004) 557:559–70. doi: 10.1113/jphysiol.2003.058263
155. Das S, Morvan F, Morozzi G, Jourde B, Minetti GC, Kahle P, et al. ATP citrate lyase regulates myofiber differentiation and increases regeneration by altering histone acetylation. Cell Rep (2017) 21:3003–11. doi: 10.1016/j.celrep.2017.11.038
156. Phinney SD, Bistrian BR, Evans WJ, Gervino E, Blackburn GL. The human metabolic response to chronic ketosis without caloric restriction: preservation of submaximal exercise capability with reduced carbohydrate oxidation. Metabolism: Clin Exp (1983) 32:769–76. doi: 10.1016/0026-0495(83)90106-3
157. Volek JS, Freidenreich DJ, Saenz C, Kunces LJ, Creighton BC, Bartley JM, et al. Metabolic characteristics of keto-adapted ultra-endurance runners. Metabolism: Clin Exp (2016) 65:100–10. doi: 10.1016/j.metabol.2015.10.028
158. Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chem Rev (2015) 115:2419–52. doi: 10.1021/cr500452k
159. Ruiz PA, Braune A, Holzlwimmer G, Quintanilla-Fend L, Haller D. Quercetin inhibits TNF-induced NF-kappaB transcription factor recruitment to proinflammatory gene promoters in murine intestinal epithelial cells. J Nutr (2007) 137:1208–15. doi: 10.1093/jn/137.5.1208
160. Kedhari Sundaram M, Hussain A, Haque S, Raina R, Afroze N. Quercetin modifies 5'CpG promoter methylation and reactivates various tumor suppressor genes by modulating epigenetic marks in human cervical cancer cells. J Cell Biochem (2019) 120:18357–69. doi: 10.1002/jcb.29147
161. Bouyahya A, El Hachlafi N, Aanniz T, Bourais I, Mechchate H, Benali T, et al. Natural bioactive compounds targeting histone deacetylases in human cancers: Recent updates. Molecules (2022) 27. doi: 10.3390/molecules27082568
162. Imran M, Salehi B, Sharifi-Rad J, Aslam Gondal T, Saeed F, Imran A, et al. Kaempferol: A key emphasis to its anticancer potential. Molecules (2019) 24. doi: 10.3390/molecules24122277
163. Berger A, Venturelli S, Kallnischkies M, Bocker A, Busch C, Weiland T, et al. Kaempferol, a new nutrition-derived pan-inhibitor of human histone deacetylases. J Nutr Biochem (2013) 24:977–85. doi: 10.1016/j.jnutbio.2012.07.001
164. Kim TW, Lee SY, Kim M, Cheon C, Ko SG. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis (2018) 9:875. doi: 10.1038/s41419-018-0930-1
165. Kedhari Sundaram M, Haque S, Somvanshi P, Bhardwaj T, Hussain A. Epigallocatechin gallate inhibits HeLa cells by modulation of epigenetics and signaling pathways. 3 Biotech (2020) 10:484. doi: 10.1007/s13205-020-02473-1
166. Thakur VS, Deb G, Babcook MA, Gupta S. Plant phytochemicals as epigenetic modulators: role in cancer chemoprevention. AAPS J (2014) 16:151–63. doi: 10.1208/s12248-013-9548-5
167. Choi KC, Jung MG, Lee YH, Yoon JC, Kwon SH, Kang HB, et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced b lymphocyte transformation via suppression of RelA acetylation. Cancer Res (2009) 69:583–92. doi: 10.1158/0008-5472.CAN-08-2442
168. Casanova E, Salvado J, Crescenti A, Gibert-Ramos A. Epigallocatechin gallate modulates muscle homeostasis in type 2 diabetes and obesity by targeting energetic and redox pathways: A narrative review. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20030532
169. Lee WJ, Shim JY, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol (2005) 68:1018–30. doi: 10.1124/mol.104.008367
170. Kowalczyk A, Bodalska A, Miranowicz M, Karlowicz-Bodalska K. Insights into novel anticancer applications for apigenin. Adv Clin Exp Med (2017) 26:1143–6. doi: 10.17219/acem/41978
171. Tseng TH, Chien MH, Lin WL, Wen YC, Chow JM, Chen CK, et al. Inhibition of MDA-MB-231 breast cancer cell proliferation and tumor growth by apigenin through induction of G2/M arrest and histone H3 acetylation-mediated p21(WAF1/CIP1) expression. Environ Toxicol (2017) 32:434–44. doi: 10.1002/tox.22247
172. Pandey M, Kaur P, Shukla S, Abbas A, Fu P, Gupta S. Plant flavone apigenin inhibits HDAC and remodels chromatin to induce growth arrest and apoptosis in human prostate cancer cells: in vitro and in vivo study. Mol Carcinog (2012) 51:952–62. doi: 10.1002/mc.20866
173. Wu HT, Lin J, Liu YE, Chen HF, Hsu KW, Lin SH, et al. Luteolin suppresses androgen receptor-positive triple-negative breast cancer cell proliferation and metastasis by epigenetic regulation of MMP9 expression via the AKT/mTOR signaling pathway. Phytomedicine (2021) 81:153437. doi: 10.1016/j.phymed.2020.153437
174. Pramodh S, Raina R, Hussain A, Bagabir SA, Haque S, Raza ST, et al. Luteolin causes 5'CpG demethylation of the promoters of TSGs and modulates the aberrant histone modifications, restoring the expression of TSGs in human cancer cells. Int J Mol Sci (2022) 23. doi: 10.3390/ijms23074067
175. Wang SW, Sheng H, Zheng F, Zhang F. Hesperetin promotes DOT1L degradation and reduces histone H3K79 methylation to inhibit gastric cancer metastasis. Phytomedicine (2021) 84:153499. doi: 10.1016/j.phymed.2021.153499
176. Wang SW, Sheng H, Zheng F, Zhang F. Hesperetin, a SIRT1 activator, inhibits hepatic inflammation via AMPK/CREB pathway. Int Immunopharmacol (2020) 89:107036. doi: 10.1016/j.intimp.2020.107036
177. Mateen S, Raina K, Agarwal R. Chemopreventive and anti-cancer efficacy of silibinin against growth and progression of lung cancer. Nutr Cancer (2013) 65(Suppl 1):3–11. doi: 10.1080/01635581.2013.785004
178. Anestopoulos I, Sfakianos AP, Franco R, Chlichlia K, Panayiotidis MI, Kroll DJ, et al. A novel role of silibinin as a putative epigenetic modulator in human prostate carcinoma. Molecules (2016) 22. doi: 10.3390/molecules22010062
179. Ye D, Li Z, Wei C. Genistein inhibits the s-phase kinase-associated protein 2 expression in breast cancer cells. Exp Ther Med (2018) 15:1069–75. doi: 10.3892/etm.2017.5489
180. Basak S, Pookot D, Noonan EJ, Dahiya R. Genistein down-regulates androgen receptor by modulating HDAC6-Hsp90 chaperone function. Mol Cancer Ther (2008) 7:3195–202. doi: 10.1158/1535-7163.MCT-08-0617
181. Coussement L, Bolca S, Van Criekinge W, Trooskens G, Mensaert K, Poels K, et al. Exploratory analysis of the human breast DNA methylation profile upon soymilk exposure. Sci Rep (2018) 8:13617. doi: 10.1038/s41598-018-31767-x
182. Karsli-Ceppioglu S, Ngollo M, Adjakly M, Dagdemir A, Judes G, Lebert A, et al. Genome-wide DNA methylation modified by soy phytoestrogens: role for epigenetic therapeutics in prostate cancer? OMICS (2015) 19:209–19. doi: 10.1089/omi.2014.0142
183. Casimir M, Chaffard G, Maechler P. Resveratrol long-term treatment differentiates INS-1E beta-cell towards improved glucose response and insulin secretion. Pflugers Arch (2019) 471:337–45. doi: 10.1007/s00424-018-2215-z
184. Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, et al. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab (2007) 6:307–19. doi: 10.1016/j.cmet.2007.08.014
185. Jimenez-Gomez Y, Mattison JA, Pearson KJ, Martin-Montalvo A, Palacios HH, Sossong AM, et al. Resveratrol improves adipose insulin signaling and reduces the inflammatory response in adipose tissue of rhesus monkeys on high-fat, high-sugar diet. Cell Metab (2013) 18:533–45. doi: 10.1016/j.cmet.2013.09.004
186. Fernandes GFS, Silva GDB, Pavan AR, Chiba DE, Chin CM, Dos Santos JL. Epigenetic regulatory mechanisms induced by resveratrol. Nutrients (2017) 9. doi: 10.3390/nu9111201
187. Liu TF, McCall CE. Deacetylation by SIRT1 reprograms inflammation and cancer. Genes Cancer (2013) 4:135–47. doi: 10.1177/1947601913476948
188. Cattelan A, Ceolotto G, Bova S, Albiero M, Kuppusamy M, De Martin S, et al. NAD(+)-dependent SIRT1 deactivation has a key role on ischemia-reperfusion-induced apoptosis. Vascul Pharmacol (2015) 70:35–44. doi: 10.1016/j.vph.2015.02.004
189. Sin TK, Yu AP, Yung BY, Yip SP, Chan LW, Wong CS, et al. Modulating effect of SIRT1 activation induced by resveratrol on Foxo1-associated apoptotic signalling in senescent heart. J Physiol (2014) 592:2535–48. doi: 10.1113/jphysiol.2014.271387
190. Dhar S, Kumar A, Li K, Tzivion G, Levenson AS. Resveratrol regulates PTEN/Akt pathway through inhibition of MTA1/HDAC unit of the NuRD complex in prostate cancer. Biochim Biophys Acta (2015) 1853:265–75. doi: 10.1016/j.bbamcr.2014.11.004
191. Kumar A, Dhar S, Rimando AM, Lage JM, Lewin JR, Zhang X, et al. Epigenetic potential of resveratrol and analogs in preclinical models of prostate cancer. Ann N Y Acad Sci (2015) 1348:1–9. doi: 10.1111/nyas.12817
192. Kai L, Samuel SK, Levenson AS. Resveratrol enhances p53 acetylation and apoptosis in prostate cancer by inhibiting MTA1/NuRD complex. Int J Cancer (2010) 126:1538–48. doi: 10.1002/ijc.24928
193. Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell (2006) 126:257–68. doi: 10.1016/j.cell.2006.07.002
194. Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature (2006) 444:337–42. doi: 10.1038/nature05354
195. Yun JM, Jialal I, Devaraj S. Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J Nutr Biochem (2011) 22:450–8. doi: 10.1016/j.jnutbio.2010.03.014
196. Marcu MG, Jung YJ, Lee S, Chung EJ, Lee MJ, Trepel J, et al. Curcumin is an inhibitor of p300 histone acetylatransferase. Med Chem (2006) 2:169–74. doi: 10.2174/157340606776056133
197. Liu HL, Chen Y, Cui GH, Zhou JF. Curcumin, a potent anti-tumor reagent, is a novel histone deacetylase inhibitor regulating b-NHL cell line raji proliferation. Acta Pharmacol Sin (2005) 26:603–9. doi: 10.1111/j.1745-7254.2005.00081.x
198. Royston KJ, Paul B, Nozell S, Rajbhandari R, Tollefsbol TO. Withaferin a and sulforaphane regulate breast cancer cell cycle progression through epigenetic mechanisms. Exp Cell Res (2018) 368:67–74. doi: 10.1016/j.yexcr.2018.04.015
199. Wong CP, Hsu A, Buchanan A, Palomera-Sanchez Z, Beaver LM, Houseman EA, et al. Effects of sulforaphane and 3,3'-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PloS One (2014) 9:e86787. doi: 10.1371/journal.pone.0086787
200. Zou Y, Ma X, Huang Y, Hong L, Chiao JW. Effect of phenylhexyl isothiocyanate on aberrant histone H3 methylation in primary human acute leukemia. J Hematol Oncol (2012) 5:36. doi: 10.1186/1756-8722-5-36
201. Wang LG, Beklemisheva A, Liu XM, Ferrari AC, Feng J, Chiao JW. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Mol Carcinog (2007) 46:24–31. doi: 10.1002/mc.20258
202. Park JE, Sun Y, Lim SK, Tam JP, Dekker M, Chen H, et al. Dietary phytochemical PEITC restricts tumor development via modulation of epigenetic writers and erasers. Sci Rep (2017) 7:40569. doi: 10.1038/srep40569
203. Nian H, Delage B, Pinto JT, Dashwood RH. Allyl mercaptan, a garlic-derived organosulfur compound, inhibits histone deacetylase and enhances Sp3 binding on the P21WAF1 promoter. Carcinogenesis (2008) 29:1816–24. doi: 10.1093/carcin/bgn165
204. Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol (2016) 8:a019521. doi: 10.1101/cshperspect.a019521
205. Rajendran P, Ho E, Williams DE, Dashwood RH. Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clin Epigenet (2011) 3:4. doi: 10.1186/1868-7083-3-4
206. Kinouchi K, Sassone-Corsi P. Metabolic rivalry: circadian homeostasis and tumorigenesis. Nat Rev Cancer (2020) 20:645–61. doi: 10.1038/s41568-020-0291-9
Keywords: epigenetics, chronic diseases, bioactive compounds, metabolism, ageing
Citation: Gómez de Cedrón M, Moreno Palomares R and Ramírez de Molina A (2023) Metabolo-epigenetic interplay provides targeted nutritional interventions in chronic diseases and ageing. Front. Oncol. 13:1169168. doi: 10.3389/fonc.2023.1169168
Received: 18 February 2023; Accepted: 24 May 2023;
Published: 19 June 2023.
Edited by:
Monica Fedele, National Research Council (CNR), ItalyReviewed by:
Yuyan Zhu, The First Affiliated Hospital of China Medical University, ChinaCopyright © 2023 Gómez de Cedrón, Moreno Palomares and Ramírez de Molina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Ramírez de Molina, YW5hLnJhbWlyZXpAaW1kZWEub3Jn; Marta Gómez de Cedrón, bWFydGEuZ29tZXpkZWNlZHJvbkBpbWRlYS5vcmc=
†These authors have contributed equally to this work
‡ORCID: Marta Gómez de Cedrón, orcid.org/0000-0002-0894-970X
Ana Ramírez de Molina, orcid.org/0000-0003-1439-7494
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.