 Jiasheng Wang
Jiasheng Wang Leland Metheny
Leland Metheny
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 18 May 2023
Sec. Hematologic Malignancies
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1167266
This article is part of the Research Topic Immune Function and Immune Reconstitution After Cord Blood Transplantation View all 4 articles
While cord blood (CB) is primarily utilized in allogeneic hematopoietic cell transplantation (HCT), the development of novel cell therapy products from CB is a growing and developing field. Compared to adult blood, CB is characterized by a higher percentage of hematopoietic stem cells (HSCs) and progenitor cells, less mature immune cells that retain a high capacity of proliferation, and stronger immune tolerance that requires less stringent HLA-matching when used in the allogenic setting. Given that CB is an FDA regulated product and along with its unique cellular composition, CB lends itself as a readily available and safe starting material for the development of off-the-shelf cell therapies. Moreover, non-hematologic cells such as mesenchymal stem cell (MSCs) residing in CB or CB tissue also have potential in regenerative medicine and inflammatory and autoimmune conditions. In this review, we will focus on recent clinical development on CB-derived cellular therapies in the field of oncology, including T-cell therapies such as chimeric antigen receptor (CAR) T-cells, regulatory T-cells, and virus-specific T-cells; NK-cell therapies, such as NK cell engagers and CAR NK-cells; CB-HCT and various modifications; as well as applications of MSCs in HCT.
The umbilical cord (UC) connects the fetus to the placenta for nutrient uptake, waste elimination, and gas exchange. The UC contains one vein, two arteries, and is protected by a surrounding gelatinous substance called Wharton’s jelly. At term, the UC has a mean length of 50cm, a mean diameter of 14mm, an approximate weight of 40g, and together with the placenta contains 50 to 200mL of cord blood (CB) (1). Compared to adult blood, CB contains a higher percentage of hematopoietic stem cells (HSCs) (2); moreover, the immune cells, such as T-cells and NK-cells, are phenotypically less mature (3, 4) These CB-specific features allow for more robust ex vivo and in vivo hematopoietic expansions and greater immune tolerance when used as a source for allogeneic cellular therapies. Apart from blood cells, nonhematopoietic progenitor cells such as mesenchymal stem cells (MSCs) can also be readily obtained from CB or CB tissue compartments such as Wharton’s jelly, which have great potential in regenerative medicine and immune modulation.
Following the first successful CB hematopoietic cell transplantation (CB-HCT) in 1988 and the establishment of the first public CB bank in New York in 1991 (5), more than 100 public CB banks were established worldwide with millions of CB units donated altruistically, greatly expanding the availability of CB (6). Moreover, it was estimated that more than 4 million units are being cryopreserved in private CB banks (7). Additionally, CB is an FDA regulated product that adheres to regulatory requirements. Therefore, the CB banks can provide readily available, high quality, and immunologically compatible hematopoietic cells to the majority of the population (8). CB is a particularly important cell source for patients who are racial or ethnic minorities, where matched adult donors are lacking in the registry (9). Although CB-HCT remains the main application of public CB banks, recent progress in cellular therapy has shown great potential in other CB-derived cellular products. Therefore, CB banks are an important resource to quickly upscale CB-derived cellular therapy. In addition to public cord blood banks, private cord blood banks offer the option of storing and using autologous cells for future use. However, preserving cord blood cells at birth for personal use poses several challenges. Firstly, the likelihood of needing CB-HCT, which is currently the only FDA-approved use for CB cells, is very low for people without a family history of blood disorders, estimated to be around 1 in 20,000 in their lifetime (10). Moreover, autologous CB-HCT is less effective than allogeneic CB-HCT in treating malignancies due to the absence of GvL effect. Thirdly, the quantities of stored single unit autologous cells may not be sufficient for cellular therapy. Finally, the impact of prolonged cryopreservation on the functionality of CB cells remains unclear. While the transplantation outcomes appear unaffected when CB cells were cryopreserved for up to 10 years, longer preservation may affect the viability of CB cells (11).
CB-derived cellular therapy harbors certain limitations. First, biologically, CB might not be the ideal source for all cellular therapies. For example, virus-specific T-cells (VSTs) have been used in severely immunosuppressed patients with resistant viral infections. However, given the naïve nature of CB, it lacks virus-experienced T-cells to manufacture VSTs (12). Second, practically, CB is limited by the small volume and total amount of hematopoietic cells. Therefore, ex vivo expansion may be required prior to adoptive cellular transfer. Moreover, good manufacturing practice (GMP)-compliant expansion protocols need to be established for each type of cellular therapy (13). Third, acquisition, processing, and storage of CB are expensive, which may result in low utilization of CB units from CB banks (7). Therefore, the high cost of each CB unit may translate into expensive CB-derived cellular products. Indeed, the current cost of a CB unit for HCT in developed countries ranges from thirty- to sixty-thousand US dollars (14).
In this review, we will discuss cellular therapies utilizing different hematologic components of CB, including T-cells, NK-cells, HSCs, and hematopoietic progenitor cells, as well as non-hematologic components such as MSCs (Figure 1). This review will focus on their applications in oncology based on clinical evidence from published clinical trials, and discuss advantages, challenges, and limitations of each CB-derived cellular product (Table 1)
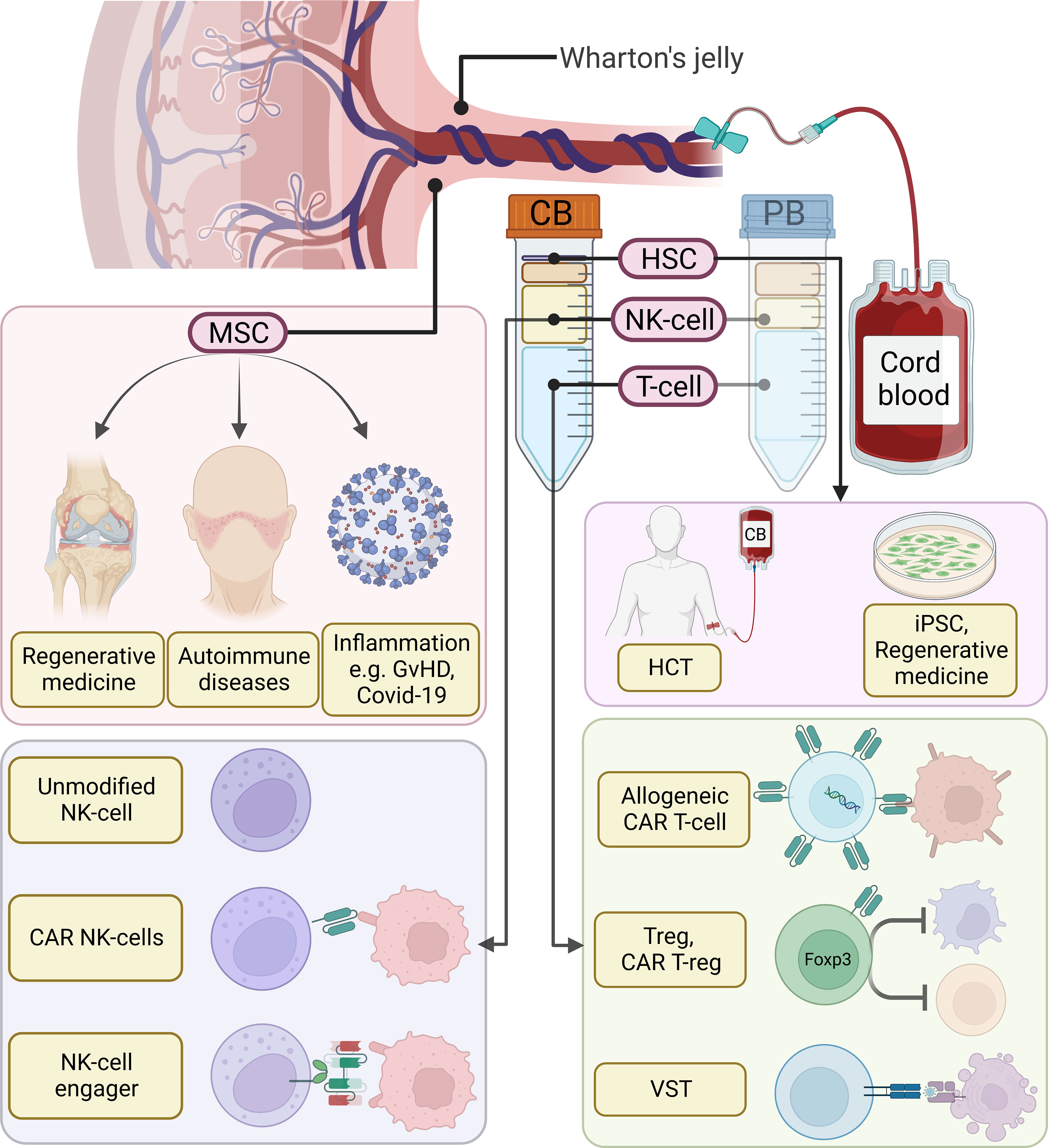
Figure 1 Overview of cord blood (CB) derived cellular therapies. Compared to adult peripheral blood (PB), CB is characterized by a higher percentage of NK-cells and the presence of hematopoietic stem cells (HSCs). Moreover, blood cells in CB are more naïve and harbor greater proliferation potential. Among them, HSCs can be used in hematopoietic stem cell transplantation (HCT) or as a source for induced pluripotent stem cells (iPSCs) and regenerative medicine. T-cells can be utilized to generate allogeneic chimeric antigen receptor (CAR) T-cells, regulatory T-cells (Tregs), CAR T-regs, or virus-specific T-cells (VSTs). CB-derived NK-cells have been shown to be a great source for allogeneic NK-cell infusion, CAR NK-cells, as well as pre-activated NK-cells pre-complexed with NK-cell engagers. In addition to CB-derived blood cells, nonhematologic cells in the Wharton’s jelly, such as mesenchymal stem cells (MSCs), process regenerative and immunomodulatory effects. MSCs can be used in tissue engineering, autoimmune diseases, as well as inflammatory diseases.
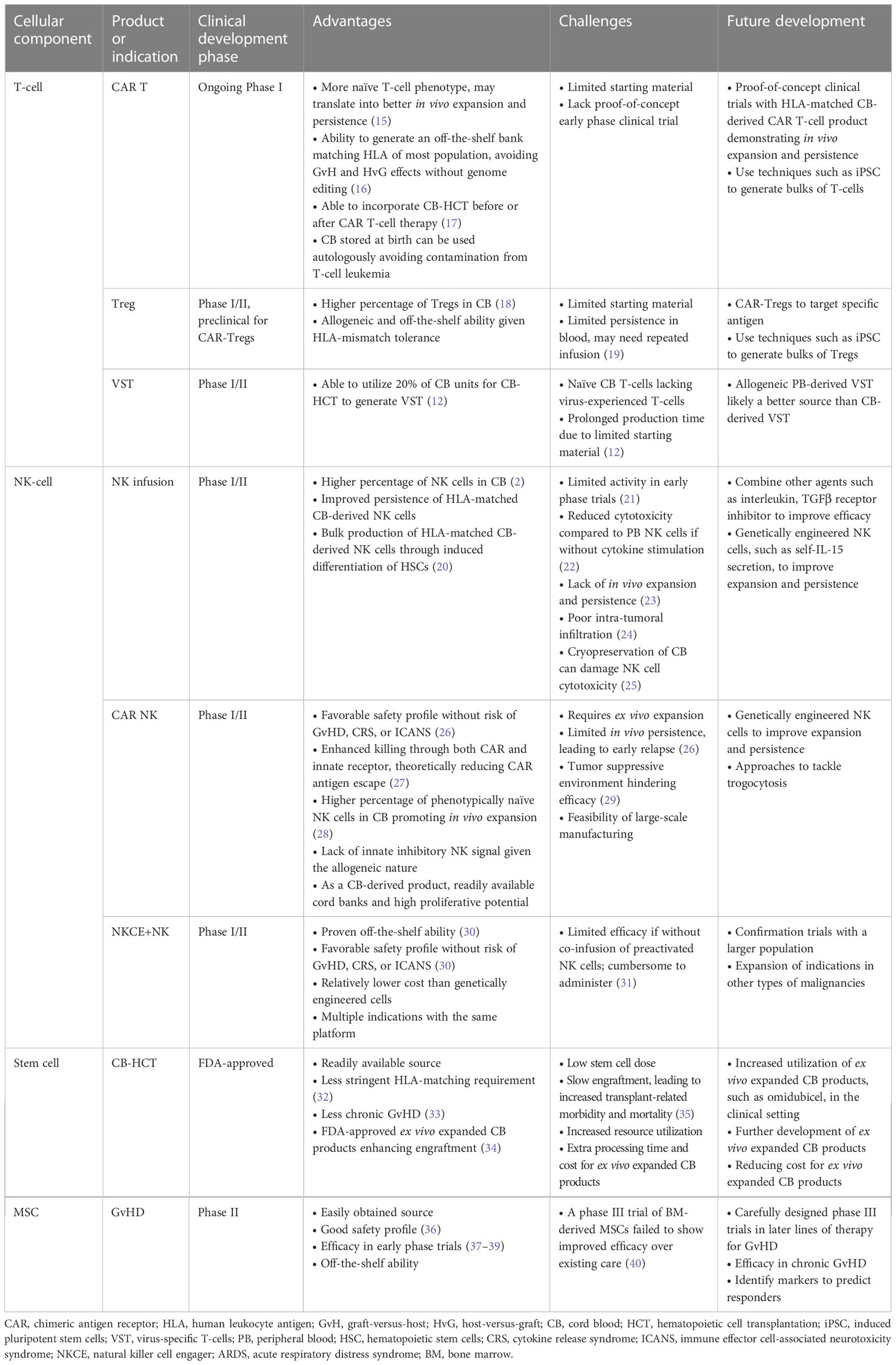
Table 1 Overview of advantages and challenges of cord blood-derived cellular therapies .
CB T-cells differ in composition and functional properties when compared to peripheral blood (PB) T-cells. Close to 90% of T-cells in the CB are naïve T-cells since they have not encountered any foreign antigens (3). Moreover, CB harbors a higher percentage of CD8+CCR7+ T-central memory (TCM) cells, which mediates enhanced antitumor responses through increased tumor-homing and rapid gain of cytotoxic function, making it an excellent source for adoptive T-cell therapies (41, 42). However, CB T-cells are not simply immature versions of adult T-cells, but a distinct population with different gene expression profile and functional properties (43). They are more tolerant-prone and poised for rapid effector differentiation (44), which are ideally suited for allogeneic T-cell therapies.
T-cell therapies in CB can be broadly divided into two categories, one involves isolation and expansion of existing T-cells (Tregs, or virus-specific or tumor antigen-specific cytotoxic T-cells), and the other involves genetic engineering of T-cells. The latter, which in the early years utilized T-cell receptor (TCR) engineering, has been associated with suboptimal clinical efficacy and considerable toxicities (45). The recent years have witnessed a rapid development of chimeric antigen receptor (CAR) T-cell therapy, which has demonstrated superior clinical efficacy especially in B-cell malignancies (46), making it an ideal platform for CB-based T-cell therapies.
CAR T-cell therapy has revolutionized the treatment of refractory/relapsed B-cell malignancies. However, current FDA-approved CAR T-cells are all autologous products derived from patients, limited by long production time, chance of production failure, product variability, possible contamination by tumor cells, and high production cost. Therefore, “off-the-shelf” allogeneic CAR T-cells have attracted great attention in recent years, albeit encountering many challenges, such as graft (CAR T)-versus-host (GvH) effects due to Human Leukocyte Antigens (HLA) mismatch, and lack of in vivo expansion and persistence due to host-versus-graft (HvG) effects (47). Using CB as the source for allogeneic CAR T-cells may overcome some of the challenges and offer unique advantages over PB from healthy donors. First, CB CAR T-cells retained a less differentiated phenotype (such as TSCM and TCM) than PB CAR T-cells (15), which could translate into a longer persistence of CAR T-cells. Second, CB T-cells have been known to have greater immune tolerance to HLA mismatch (16). In CB HCT, matching for alleles HLA-A, HLA-B and HLA-DR (i.e. ≥ 4 of 6 HLA match) is sufficient to reduce the incidence of graft-versus-host disease (GvHD) to a low level (48). Therefore, it has been demonstrated that an CB bank of only 150 selected samples could provide full matches (6 of 6 HLA loci) for 93% of the UK population (49); if partial mismatch is allowed (4 or 5 of 6 HLA loci), the required number can be further reduced. Therefore, it is possible to generate an “off-the-shelf” allogeneic CB-derived CAR T-cell bank that can be readily used for most of the population with minimal GvH effects. On the other hand, to reduce GvH effects in PB-derived CAR T-cells, T-cell receptor (TCR) knockout using genome editing is required, for which the long-term safety is still unknown (50). Another advantage of CB-derived CAR T-cell therapy is the ease to incorporate consolidative CB HCT after CB CAR T-cell therapy, which might provide improved progression-free survival (17).
Several preclinical studies have demonstrated the feasibility of manufacturing CAR T-cells from CB (15, 41, 51). In an earlier study by Pegram et al., banked CB T-cells were expanded with IL-12/IL-15 stimulation, and transduced with anti-CD19 and IL-12 secreting modules (51). The CAR T-cells retained a central memory-effector phenotype, and demonstrated activity in a B-cell acute lymphoblastic leukemia mouse model. In a study by Cael et al., fresh or thawed CB T-cells were transduced by lentivirus encoding CD123-CAR to target blastic plasmacytoid dendritic cell neoplasm (BPDCN) mouse models (15). They found that CB CAR T-cells exhibited potent anti-tumor activities while retaining a less differentiated profile than PB CAR T-cells; moreover, fresh or cryopreserved CB CAR T-cells demonstrated similar functionalities. In another study by Liu et al., cryopreserved, CB-derived, CD19-targeting CAR T-cells had more TN and TCM than PB CAR T-cells derived from the patients, and harbored fewer T-cell exhaustion markers, leading to a better antitumor efficacy and longer tumor-suppressive effects (41).
However, allogeneic use of CB as an “off-the-shelf” product in the clinical setting is still lacking. A search of ClinicalTrials.gov in December 2022 returned only one ongoing clinical trial (NCT03881774). Indeed, several limitations of CB-derived CAR T-cells need be addressed before broader clinical applications. First, like other allogeneic CAR T-cell therapies, solutions need be developed to tackle GvH and HvG effects. Although TCR knockout has been effective to reduce GvH in PB-derived (52) and CB-derived (53) CAR T-cells, it is possible to avoid genome editing, as mentioned above, by obtaining an HLA-matched CB unit from a limited and highly selected CB bank as the cell source for CAR T-cells (49). However, proof-of-concept clinical studies are lacking at this point. Second, due to limited availability of selected CB units, techniques such as induced pluripotent stem cells (iPSCs) (54) or directed differentiation of hematopoietic stem cells (55) are required to produce the bulk population of T-cells. However, further developments of these methods are still needed to generate a self-renewing and clinically applicable source of T-cells.
Tregs are a subset of natural occurring CD4+ T-cells that suppresses the activation and expansion of overactive lymphocytes, thus preventing excessive immune activation and autoimmunity. While Tregs comprise less than 3% of mononuclear cells in the PB, CB contains a high proportion of circulating natural Tregs, obviating multiple selection processes to reach the required purity of Tregs (18). Moreover, compared to PB-derived Tregs, CB-derived Tregs have greater post-expansion purity, FOXP3 stability, and proliferative capacity (56). Another advantage is the potential of CB units as an allogeneic source of Tregs due to great tolerability of HLA-mismatch. Adoptive cellular therapy using Tregs can be divided into two categories: selection and expansion of natural occurring polyclonal Tregs, and manufacturing antigen specific Tregs using TCR or CAR gene transfer.
Polyclonal Tregs have been reported in human trials in treating autoimmune diseases such as multiple sclerosis (57) and type 1 diabetes mellitus (58, 59), preventing rejection and reducing needs for immunosuppressant agents after solid-organ transplantation (20, 60, 61), and preventing and treating GvHD after HCT (19, 62–64). While most of the aforementioned studies used autologous PB mononuclear cells (PBMC) as the source of Treg, in a study by Brunstein et al., 11 patients undergoing CB-HCT received CB-derived, HLA-matched Tregs from a cord bank; they observed very low incidence of Grade II-IV acute GvHD of only 9%, and none developed chronic GvHD (19). However, although Tregs have shown a favorable safety profile and early signs of clinical efficacy, several challenges must be overcome before broader applications. The first challenge is to determine the timing and frequency of administration. Although in some studies Tregs can still be detected in the PB one year after administration (59), other studies have shown the persistence of only 14 days (19). Therefore, for conditions like autoimmune disease or solid-organ transplantation where persistent immunosuppression is required, it is unknown whether and when Treg products should be re-dosed after the initial infusion. Indeed, in the landmark ONE trial among patients with kidney transplantation, after a single dose of autologous Treg infusion, immunosuppressants were able to be tapered but not completely weaned off (60). Second, due to limited volume of CB, it is not uncommon to fail to achieve the targeted cell dose, which occurres in a frequency of more than 10% in previous studies (18, 19). Therefore, a better understanding of Treg biology and improvements of manufacturing process are needed for future larger trials. While identifying new Treg markers can simplify Treg selection and purification, techniques such as 3D culture, automated bioreactors, or robotic platforms may aid in the large-scale production of lineage-stable CB-derived Treg cells.
While polyclonal Tregs may be suitable for the treatment of autoimmune conditions where a variety of tissues are targeted, antigen-specific Tregs may be particularly useful in situations where the disease-relevant antigens are known or limited tissues are involved (65). Through engineered antigen-specific receptors (CARs or TCRs), Tregs have enhanced trafficking and targeting to specific microenvironment. With the success of CD19-targeting CAR T-cell therapy, CAR-Tregs have attracted great attention in recent years, albeit current studies have been limited to the preclinical phase. Major conditions being studied include HCT/solid organ transplantation (66), type 1 diabetes (67), inflammatory bowel disease (68), and hemophilia (69), targeting HLA-A2, insulin/HiP2, carcinoembryonic antigen (CEA), and factor VIII, respectively. A search of ClinicalTrials.gov in December 2022 showed two ongoing clinical trials (NCT04817774 and NCT05234190), both using autologous, HLA-A2 targeting CAR Tregs to prevent rejection in solid-organ transplantation recipients who are HLA-A2 negative receiving an A2 positive graft. Hurdles for manufacturing autologous CAR Tregs for clinical use include prolonged production time, contamination of conventional T-cells, and higher chance of production failure due to limited amount in the PB (70). Moreover, in a population often receiving high dose immunosuppressants, the function of Tregs might be impaired. Therefore, “Off-the-shelf” allogeneic CAR Tregs products, such as those manufactured from CB, are promising alternatives. However, further studies are needed to demonstrate their safety and efficacy.
Over the past three decades, adoptive transfer of VSTs has been proven to be safe and effective for the treatment of refractory viral infections in patients with severely compromised immune system, such as those underwent HCT/solid-organ transplantation (71) or with primary immunodeficiency disorders (72). Moreover, the indications of VSTs have been extended to viral-associated malignancies such as EBV-positive post-transplantation lymphoma (73–75). These VSTs are usually organ or stem-cell donor derived, either through ex vivo stimulation and expansion, or direct sorting and selection from apheresis products of seropositive donors (76). True allogeneic VSTs obtained from healthy third-party donors have been increasingly reported in recent years. Even with low level HLA match (1 or 2 out of 6 or 8), studies have shown excellent clinical efficacy without causing significant GvHD (77–79). The extremely low rates of acute GvHD (<10%) are probably due to the fact that VSTs are largely central or effector memory T cells that are less likely to be alloreactive (80). However, studies have shown that persistence of third-party allogenic VSTs was only 12 weeks (78), which might not be enough to cover the whole immune reconstitution phase. Therefore, future studies are needed to examine the necessity and timing of redosing or investigate methods to promote allogeneic VST’s persistence.
However, CB has several major limitations as a source for VSTs. First, CB contains predominantly naïve T-cells lacking virus-experienced T-cells, which makes direct sorting and selection difficult. Second, a single CB unit has a small volume; with the inability to procure further donations, this leads to prolonged expansion and production time with high chances of production failure. However, for patients undergoing CB HCT, CB becomes the only available source for autologous VSTs. Abraham et al. reported the use of 20% fraction of CB units that were reserved prior to the stem cell infusion, and they used them to manufacture CB-derived multivirus-specific T-cells in pediatric patients (12). Using ex vivo antigen stimulation, production was successful in 18 of the 21 pediatric patients. However, the median production time was 52.5 days, which remains the major obstacle for widespread application given the urgency of antiviral treatment. Further development has resulted in reduced production time, but it could still take around 30 days (81). Moreover, the expanded cell dose is likely insufficient for adult patients. Therefore, although it is feasible to produce VSTs from CB, challenges remain; given the efficacy and safety of allogeneic PBMC-derived VSTs, they will likely come out as a better option for patients undergoing CB HCT complicated by refractory viral infections.
Autologous HCT using stored self CB have been reported (82). Therefore, it is also possible to manufacture autologous CAR T-cells from preserved CB at birth. This may be particularly useful for patients with T-cell leukemia where it is vital to avoid CAR T-cell product contamination from circulating leukemic T-cells.
γδ T-cells is a unique subpopulation of T-cells that function in between innate and adaptive immunity – they mediate cytotoxicity through antigen-pattern recognition independent of MHC presentation (83). Over the past two decades, adoptive transfer of PB-derived autologous γδ T-cells have been tested in a wide range of malignancies without good clinical response (84). Interestingly, given the HLA-independent killing, allogenic γδ T-cells have been proven to be safe with a low risk of GvHD. Therefore, repeated infusion of healthy donor derived allogeneic γδ T-cells has become possible. Indeed, two recent studies showed improved clinical efficacy with repeated γδ T-cells infusion in patients with late-stage malignancies (85, 86). Furthermore, targeted therapy using CAR-engineered γδ T-cells have attracted great attention, with a recent phase I study showed promising safety and efficacy (87). On the other hand, CB has several disadvantages as the source for γδ T-cell therapies, including low frequency of γδ T-cells (<1%) and lack of the functional subset of γδ T-cells (88). Although successful expansion of CB-derived γδ T-cells has been reported (89, 90), its clinical usage remains to be defined, given that PB-derived γδ T-cells can be more easily manufactured.
A broader application of CAR T-cell therapy in solid tumors is limited due to the lack of tumor-specific surface antigens. However, frequently mutated intracellular protein can be presented to the cell surface through coupling with MHC I molecules, forming neoantigens that can be recognized by TCRs; this provides an opportunity for genetically engineered TCR-T cell therapy. A recent study using autologous T cells transduced with TCRs recognizing the mutant KRAS G12D-MHC I complex showed a partial response in a patient with metastatic pancreatic cancer (91). CB-derived TCR T-cell therapy has remained in the pre-clinical phase (92).
Lastly, tumor antigen-specific cytotoxic lymphocytes (CTLs) can be selectively expanded (93) or primed (94) using techniques similar to manufacturing VSTs. Compared to CTLs obtained from PB, CB-derived CTLs showed higher proliferative and cytotoxic activities in samples obtained from patients (95). Antigen-specific CTLs might be useful in patients who have received CB HCT to prevent or treat cancer relapse, in a role similar to donor lymphocyte infusion (DLI). However, research in this field is extremely limited.
NK cells are part of the innate immune system that target malignant or virus-infected cells. Unlike T cells, NK cells lack specificity to a certain antigen. Rather, their activation is based on a “signal threshold” from the integration of inhibitory and activating receptors – bindings between NK inhibitory receptors and target cell HLA molecules provide the inhibitory signals, while the engagements of NK activating receptors with stress-induced self-ligands provide the activating signals (96). When HLA expression is decreased due to infection or malignant transformation, or when NK cells are used in the allogeneic setting, the inhibitory signals are weakened; along with increased activating signals, NK cells are activated.
While NK cells are the main effector for the early graft-versus-leukemia reactions, they do not directly cause GvHD even with HLA-mismatch (97), making them an ideal source for allogeneic adoptive cell therapy (98). NK cells are enriched in CB comprising up to 30%, whereas in the PB they only constitute 10% of lymphocytes (2). Furthermore, it has been observed that NK cells reconstitute more rapidly after CB HCT than PB HCT (99), which is likely due to better homing capability of HCB NK cells to the bone marrow (4) and the presence of a unique progenitor-like subpopulation (28). In addition, much like CB T cells, CB NK cells are less mature than PB NK cells, as characterized by the reduced expression of activation receptors (4). While this leads to reduced cytotoxicity compared to PB NK cells, their activities can be enhanced with cytokine stimulation (22). Therefore, allogeneic NK cell therapies especially using CB as the source have attracted great attention in recent years. However, limited persistence and modest antitumor activities remain the two biggest barriers in the field of NK cell therapy.
Earlier clinical trials using unmodified autologous NK cell infusion demonstrated only modest success (21). Advancement in allogeneic HCT has helped us better understand the physiology of NK cells – the mismatch between NK receptors and ligands are the key for the graft-versus-leukemia effect. In the past two decades, haploidentical NK cell therapies tested in patients with hematologic malignancies with or without prior HCT, have demonstrated feasibility, safety, and signals of efficacy (100–108). However, most of the studies used PB from healthy donors as the source for NK cells; clinical studies on infusion of unmodified NK cells from a CB source are scarce. Shah et al. reported the infusion of ex vivo expanded, 4/6 HLA-matched, cryopreserved CB-derived NK cells to patients with multiple myeloma, in between high-dose melphalan conditioning and autologous HCT (109). They observed no dose-limiting toxicities. Recently, our group reported the use of HLA-mismatched NK cells in patients with refractory malignancies; while we observed a good safety profile without GVHD, NK cells only persisted less than 4 weeks (97). Therefore, HLA-match or partial match is likely not required from the safety standpoint, but it may play a role in preventing allorejection and ensuring long-term persistence.
In addition to the “isolation-expansion” approach described above, NK cells can also be obtained through differentiation of hematopoietic stem and progenitor cells (HSPCs). This approach has been shown to result in a higher purity of NK cells with an improved bone marrow homing capacity (110). Dolstra et al. reported the first-in-human use of isolating CD34+ HSPCs from partially HLA-matched CB units, and inducing ex vivo NK cell differentiation by applying IL-2 and IL-15 (111). The final product was infused in 10 elderly patients with acute myeloid leukemia in remission, and they observed a good safety profile with preliminary efficacy as 2 of the 4 patients with positive measurable residual disease (MRD) turned negative. Nevertheless, larger and comparative studies are needed to determine whether induced NK cells have better clinical outcomes than isolated NK cells.
Due to the modest antitumor activities of unmodified NK cell infusion, various strategies have been developed in the past few years to improve NK cell recognition and activation, including overexpressing chemokines or chemokine receptors to enhance NK cell tumor infiltration (112, 113), concurrent use of immune checkpoint inhibitors to reverse NK cell exhaustion (114), blocking the immunosuppressive TGF-β signaling from the TME (115), and promoting NK cell tumor-specific engagement through NK cell engagers (NKCEs) (31, 116–119). Of note, almost all have remained in the preclinical/early clinical phase.
Among them, NKCEs have received most attention in recent years due to excellent preclinical results, with multiple ongoing early phase clinical trials in various types of malignancies (Table 2). NK cells exert their cytotoxic activities through three main mechanisms: direct killing by perforin/granzyme release, cytokine secretion, and antibody-dependent cell-mediated cytotoxicity (ADCC) though Fc-region binding receptor CD16 (FcγRIIA). The third mechanism provides a unique opportunity to pair monoclonal antibodies and NK cells to enhance their anti-tumor activities, prompting the development of NKCEs. Depending on the number of modules, NKCEs can be bispecific killer cell engagers (BiKEs) targeting CD16 expressed on NK cells and antigens of interest on tumor cells, or trispecific killer cell engagers (TriKEs), where an additional IL-15 module was added to promote NK cell expansion and persistence (31). Although NKCEs can be given as a monotherapy, the response rate was suboptimal. For example, when an anti-CD16/anti-CD30 bispecific NKCE, AFM13, was administered in patients with relapsed/refractory Hodgkin lymphoma, only 16.7% response rate was observed (120). However, a preclinical study by Kerbauy et al. combined AFM 13 with cytokine-induced memory-like NK cells and infused them in a pre-complexed manner (117). The authors found the combination of NKCE and pre-activated NK cells significantly improved the efficacy against CD30+ cancers compared to using either NKCE or NK cells alone. Moreover, when CB-derived NK cells were pre-activated by a set of cytokines and complexed with NKCE, they mounted an augmented response against CD30+ cancers (117). Currently, a phase I/II clinical trial (NCT04074746) is actively investigating this approach using CB-derived NK cells in patients with CD30+ T-cell lymphoma and Hodgkin lymphoma. Preliminary results of 30 patients with relapsed/refractory CD30+ lymphoma showed that this approach was safe – without infusion reaction, CRS, ICANS, or GvHD of any grade – and demonstrated an impressive efficacy with an 97% overall response rate and 63% complete response rate (30).
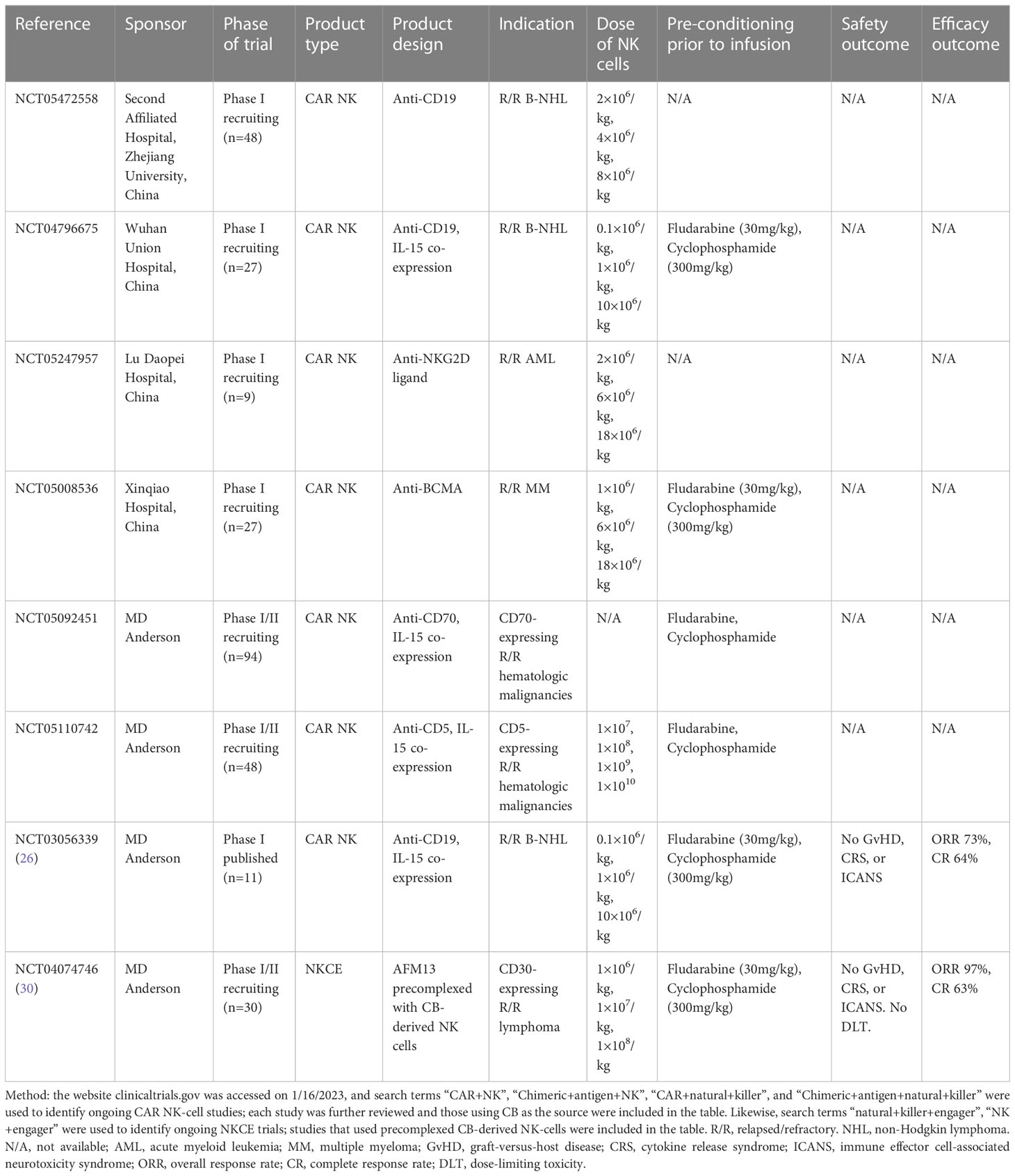
Table 2 Ongoing trials of cord blood-derived CAR NK-cells or NK-cells pre-complexed with NK-cell engagers (NKCEs).
CAR NK-cells have several advantages over CAR T-cells. First, they have a favorable safety profile due to the lack of TCR, with low risks of GvHD, cytokine release syndrome and neurotoxicity (26). Second, CAR NK-cells possess dual mechanisms of killing, through both CAR and innate receptors (27), which can potentially reduce the chance of CAR-targeted antigen escape. Compared with autologous CAR NK-cells, allogeneic CAR NK-cells also have advantages. First, “off-the-shelf” allogeneic products can significantly reduce the cost and increase availability. Second, as the activation of autologous CAR NK-cells is negatively affected by their innate inhibitory receptors (27); the use of allogeneic NK-cells can avoid the innate inhibition signal and enhance CAR NK activities.
Liu et al. reported the first-in-human trial of CAR-NK cells derived from unmatched or partially matched HLA cord blood in 2020 (26). The CAR-NK cells were transduced with a vector encoding anti-CD19 CAR, CD28 co-stimulatory domain, IL-15, and a safety switch of inducible caspase 9, and infused in 11 patients with B-cell lymphoma. No GvHD, CRS, or neurotoxicity was observed even at the highest dose; a response rate of 73% was reported, although the duration of response was unable to be assessed due to subsequent treatments. Of note, although CAR copies were still detectable in the peripheral blood after one year, on flow cytometry, CAR NK-cell rapidly declined after two weeks. Li et al. from the same group found that trogocytosis, a process involving transfer of tumor surface proteins (CD19) to CAR NK-cells upon anti-CD19 CAR/CD19 ligation, resulted in fratricide among CAR NK-cells and led to limited persistence (29). Co-infusion of KIR-based inhibitory CARs were able to counteract this process (29). Other ongoing CB-derived CAR NK-cell are summarized in Table 2.
The limited persistence of CAR NK-cells can also be an advantage from a safety standpoint. Therefore, CAR NK-cells may be an ideal tool for bridging to HCT. If a patient was ineligible for HCT, repeated infusions of CAR NK-cells might be needed to achieve long-term immune surveillance. However, previous studies using allogeneic unmodified NK cells have demonstrated that repeated NK-cell infusion could result in even shorter persistence (106). Therefore, clinical studies are needed to test whether repeated CAR NK-cell infusions are safe and effective.
Autologous NK cells obtained from patients with cancer have suppressed effector function and poor clinical activities, likely due to the presence of “self” MHC molecules and NK cell exhaustion from cancer TME (121). Allogeneic NK cells provide an intriguing alternative despite still facing many challenges – some are intrinsic to the NK cell therapy; others are specific to the allogeneic nature.
First, in the absence of cytokine support, NK cells lack initial expansion after adoptive transfer (23). Although this can be improved by co-administration of IL-2, it is associated with increased toxicity (100, 122). In recent years, applying high intensity lymphodepleting chemotherapy prior to NK cell infusion has become a standard protocol, which creates a cytokine sink and leads to a surge of IL-15, facilitating NK cell expansion (104). However, the overall expansion is still poor especially in certain solid tumors (104).
Second, long-term persistence of functional allogeneic NK cells has not been established. On the other hand, it has been shown that CAR T-cells are able to form a stable clone persisting over 10 years, which is likely the key to long-term remissions in certain indolent cancers (123). Without long-term tumor immunosurveillance, a high relapse rate from NK cell therapy has been observed (124). Therefore, most clinical studies have only us`ed allogeneic NK cells as a bridging treatment to more definitive therapies such as HCT (26, 125). In recent years, a combination of cytokines were found to be able to induce NK-cell differentiation into memory-like NK cells, which have improved persistence to months in the matched-donor setting after HCT (126, 127). However, its use in the true allogeneic setting needs to be further explored (107). In another attempt, NK cells transduced with an IL-15 gene with the ability of endogenous IL-15 secretion have shown improved persistence to over a year (26, 128). Moreover, knockout of a negative regular of IL-15 signaling (cytokine checkpoint) can further improve NK cell persistence (129). However, more clinical studies are needed to confirm the functional status of persistent NK cells. Another factor leading to the short persistence is T-cell and host NK cell allorejection. To overcome this, knockout of HLA class I molecule and overexpression of inhibitory NK receptors have been proposed (130).
Third, as shown in tumor biopsy samples, NK cells have limited intra-tumoral infiltration or homing ability (24); moreover, NK cells are usually dysfunctional within the tumor due to its immunosuppressive TME (131). These factors likely lead to limited clinical activities of NK cell therapy in solid tumors. Strategies to enhance NK cell trafficking to tumor tissue and restore their function within TME are being developed in animal models, awaiting to be tested in the clinical setting (132).
Fourth, NK cells are subject to cryoinjury – cryopreserved and thawed post-expansion NK cells have reduced cytotoxicity which might become a hurdle for large-scale allogeneic NK cell production – although the significance of such impaired activity remains to be studied in the clinical setting (25).
The most common application of banked CB is to use as a source for allogeneic HCT — around 500 CB-HCT are performed in the US each year (133). Although limited by the quantity as a single unit, CB hematopoietic stem cells possess a higher capability for self-renewal and proliferation than bone marrow stem cells (134). Moreover, the immune tolerant state and low frequency of alloreactive T cells in the graft have likely led to a lower incidence of GVHD and less restrictive HLA matching for successful engraftment (33, 135). Ever since CB-HCT was first performed in 1988 (5), several landmark studies have significantly improved the safety and efficacy of CB-HCT. For example, studies have shown that 1 or 2 allele mismatches did not impair engraftment, albeit a slightly increased risk of aGVHD and non-relapse mortality (32, 136); the development of double partially HLA-matched CB units (dCB) has expanded the use of CB-HCT to adults, where the cell dose in a single unit is frequently insufficient (137). Nevertheless, limited number of initial stem cells and slow engraftment remain the two biggest challenges in CB-HCT. Indeed, the current guideline from American Society for Transplantation and Cellular Therapy (ASTCT) recommends filtering out CB units with low total nucleated cell and CD34+ cell count as the initial steps for selection, irrespective of good HLA matching (138). Moreover, a study found that only 4% of CB units in the US CB bank inventory were suitable for a single unit CB-HCT (139). Therefore, techniques to improve CB ex vivo and in vivo expansion are needed to expand the use of CB-HCT.
With recent advancements in haploidentical HCT, which greatly expanded the donor source, the need for CB-HCT has been questioned due to its high cost and limitations. Indeed, the recently published prospective BMT CTN 1101 trial showed that compared to haploidentical HCT with post-transplantation cyclophosphamide, dCB-HCT had similar rates of GVHD, but a higher non-relapse mortality rate and lower overall survival, in even experienced CB transplant centers (35). However, CB-HCT still has its role in certain situations. First, about 10% patients in the US do not have a sibling, matched unrelated, or haploidentical donor, making CB the only feasible choice (140). Second, the BMT CTN 1101 trial allowed partially mismatched CB units and the median infused CD34+ cell dose was notably lower than the current ASTCT guideline (138); it is unknown if patients had fully matched CB units and received higher CB doses, their outcomes would be improved. Third, different strategies have been developed or are being developed to improve CB expansion and early hematopoietic recovery, which can further improve the safety of CB-HCT.
In addition to HCT, CB stem cells have also been studied in regenerative medicine, including organs that typically lack self-regenerative abilities, such as nerve, heart, or cirrhotic liver. However, a detailed discussion on this topic is beyond the scope of this review; we will refer reader to other updated review on this topic (141).
To improve the in vivo expansion of CB after transplantation, several strategies have been tested in clinical trials. One attempt was to directly inject CB stem cells into the bone marrow (intrabone injection). It was first developed by Frassoni et al. with the hypothesis of reducing trapping in organs such as liver after intravenous injection, improving homing to the bone marrow, and lowering required cell dose for CB HCT (142). Multiple phase I/II studies have confirmed feasibility of this approach, with neutrophil engraftment time ranging from 17 to 23 days with lower than standard total nucleated cell (TNC) and CD34+ cell doses (142–148). In a retrospective study comparing intrabone injection with traditional dCB-HCT, the intrabone approach was associated with an improved neutrophil engraftment (23 vs 28 days) and a lower relapse rate (25 vs 29% at 2 years), despite receiving significantly lower cell doses (median TNC count 2.5 vs 3.9 × 107/kg) (149). Therefore, intrabone injection is particularly useful when CB cell dose is insufficient and no alternative HCT approach is available. Phase III studies are needed to further investigate the efficacy of this approach.
Another method is to combine a small dose of CB with CD34-selected PB HSCs from a haploidentical donor. This haplo-cord approach resulted in a rapid early engraftment from the haploidentical donor, which was later replaced by the more durable engraftment of CB cells (150, 151). A retrospective study compared haplo-cord approach with haploidentical HCT with BM graft and post-transplantation cyclophosphamide, and found that the two approaches were associated with similar progression-free survival and overall survival, but the haplo-cord approach was associated with faster neutrophil engraftment and lower incidences of GvHD (152). However, the haplo-cord approach requires extra resources to obtain both CB and haploidentical donor cells; further, its efficacy needs to be confirmed in a prospective trial. Another approach to augment CB-HCT that has been developed recently was to infuse third-party granulocytes, with the goal to provide allogeneic MHC class I signaling, which would stimulate CD8+ T-cell proliferation to increase graft-versus-leukemia effects (153). In a proof-of-concept trial of 4 pediatric patients undergoing CB-HCT, the infusion of granulocytes led to more than 20-folds of CD8+ T-cell expansion (153). However, the safety of this approach, such as the occurrence of pre-engraftment syndrome, needs to be further studied.
Progress has been made in the past few years to improve the ex vivo expansion of CB through manipulation of culture conditions. These techniques have resulted in a higher stem cell dose at the time of infusion, with accumulating clinical studies demonstrating improved outcomes.
One strategy to optimize CB expansion is to co-culture with mesenchymal stem cells (MSCs), which can provide necessary microenvironment and cytokine support (154, 155). de Lima et al. showed that this approach led to a 12-fold increase in nucleated cells and a 30-fold increase in CD34+ stem cells, with the final product containing a median of 1.8×106 CD34+ cells per kilogram (154). The improvement of initial cell dose translated into a significantly faster neutrophils engraftment than those undergoing single unit CB HCT (15 days vs 24 days) (154).
Another strategy to improve CB expansion is to add certain agents to the culture media (156). A variety of molecules, including Notch ligand (157, 158), copper chelation agents such as TEPA (159), vitamin B derivatives such as nicotinamide (160), aryl hydrocarbon receptor antagonists (AHRs) (161), and pyrimidoindole derivative UM171 (162), have shown improved expansion in clinical studies (Table 3). For example, nicotinamide can inhibit differentiation of hematopoietic stem cells; when added to CB culture media, an outgrowth of phenotypically primitive stem cells has been observed (163). A phase III clinical trial using nicotinamide expanded CB stem cells, known as the omidubicel, has shown significantly improved engraftment when compared with single or double-unit standard CB HCT (median neutrophil engraftment time 10 vs 20 days) (34). Omidubicel has also resulted in fewer bacterial and viral infections and less time in the hospital. Based on the trial results, omidubicel is currently seeking FDA approval for patients with hematologic malignancies in need of allo-HCT. However, it is important to note that these techniques require ex vivo manipulation of CB, which can lead to increased complexity and cost.
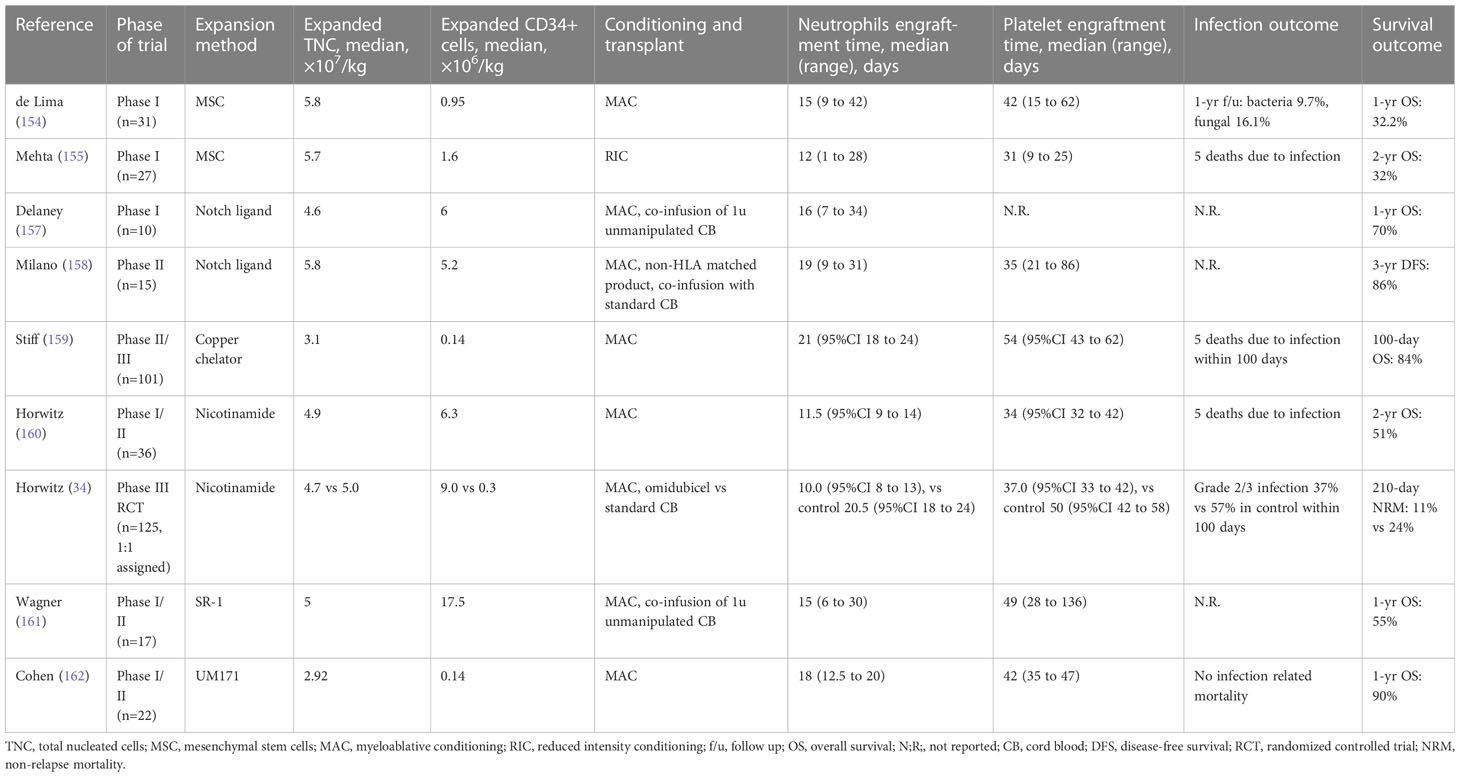
Table 3 Completed trials of ex vivo expansion of cord blood for hematopoietic stem cell transplantation.
Mesenchymal stem cells (MSCs) are multipotent perivascular progenitors that can differentiate into osteogenic, adipogenic, myogenic, or chondrogenic cells, making them an ideal resource for regenerative medicine. In addition, as part of the bone marrow niche microenvironment, MSCs possess immune modulatory properties by secreting cytokines, chemokines, and extracellular vesicles, rendering them great potential in treating inflammatory or autoimmune conditions (164). Although MSCs can be obtained from adult tissues such as the bone marrow or adipose tissue, CB and its compartments derived MSCs have advantages of ease in harvesting, increased stemness potential, and improved expansion (165). MSC therapies have consistently been proven safe, but clinical outcomes have been mixed and later phase studies are often disappointing despite efficacy signals in early phase studies. The discrepancy can be partially explained by MSC heterogeneity between donors, different manufacturing protocols for MSCs, and handling protocols after manufacture (166, 167). Moreover, due to short persistence of MSCs after infusion, repeated administrations are needed to achieve long-term effects. Despite these limitations, MSCs have been increasingly studied in regenerative medicine, inflammatory and autoimmune diseases including Covid-19. A detailed discussion on these topics is beyond the scope of this review, and we refer readers to other recently published review articles in this journal (168–170).MSCs have demonstrated two clinical applications in the field HCT. One is to promote ex vivo expansion and sustain in vivo hematopoiesis of HSCs, and the other is to prevent and treat GvHD given their immunomodulation ability. Several studies utilized co-infusion of CB-derived MSCs with allogeneic HSCs in patients with aplastic anemia – a condition known to be associated with poor graft function and a high risk of graft failure – in order to improve transplant outcomes (171–173). For example, Wang et al. showed that in 22 patients undergoing alloHCT for aplastic anemia who also received co-infusion of CB-derived MSCs, all achieved successful neutrophil engraftment in a median of 14 days; after 15 months’ follow-up, 21 patients were still alive and all maintained full donor chimerism (173).
MSCs have also been applied in GvHD. In the seminal study by Le Blanc et al. in 2004, bone marrow derived MSCs were first used in a patient with grade IV treatment-resistant acute GvHD and achieved great outcomes (36). Since the initial case report, numerous studies have been conducted to investigate various sources of MSCs in both acute and chronic GvHD. Although most studies were performed with BM-derived MSCs (174), a few studies have shown promising results from CB or its compartments derived MSCs. For example, in a phase I trial of 10 patients with de novo high-risk or steroid-refractory acute GvHD, Soder et al. showed that Wharton’s jelly-derived MSCs achieved an overall response and complete response rate of 70% and 40%, respectively (175). However, in a large phase III trial of 260 patients with steroid-refractory acute GvHD who were randomized to receive BM-derived MSCs versus placebo added to second-line therapy, there was no significant improvement of durable complete response (40). Therefore, more research is needed to fully understand the potential of these cells in the treatment of GvHD. CB-derived MSCs have also been tested to prevent GvHD (37, 38). In a phase II trial by Gao et al., 124 patients were randomized to receive monthly CB-derived MSCs or placebo for a maximum of 4 cycles starting at least 4 months after haploidentical alloHCT (38). They reported that the MSC group had significantly lower 2-year cumulative incidence of chronic GvHD than the placebo (27.4 vs 49.0%). Of note, the optimal timing of MSC infusion to prevent GvHD especially acute GvHD needs to be further investigated, as too early infusion might increase the risk of disease relapse (39).
With recent advancement in adoptive cell transfer, CB has demonstrated great and unique potential in various types of cellular therapy. As a source for allogeneic CAR T-cell therapy, it is possible to generate an HLA-homozygous CAR T-cell bank that matches most of the population, providing off-the-shelf CAR T-cell products with minimal GvH side effect and without genome editing. As a source for NK-cell therapy, recent early-phase studies have shown promising results with CAR NK-cells, as well as co-infusion of NK-cell engager and preactivated NK-cells. As a source for HCT, recent development in various techniques have greatly shorten the engraftment time resulting in improved safety. As a source of MSC, clinical studies for various autoimmune and inflammatory conditions such as GvHD are underway. However, most of the clinical studies are early phase; further developments are needed to facilitate more CB-derived cellular therapies into the market. Moreover, many of the CB-derived cellular products are regulated as advanced therapy medicinal products (ATMPs) both in the US and in the EU due to more-than-minimal or substantial biological manipulations of the CB cells. Although the regulatory frameworks were installed to ensure the safety and quality of cellular products, the complexity of regulatory requirements imposes significant challenges particularly for academic researchers who lack the resources to meet them.
LM conceived the project. JW and LM reviewed the literature and drafted the article. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Spurway J, Logan P, Pak S. The development, structure and blood flow within the umbilical cord with particular reference to the venous system. Australas J Ultrasound Med (2012) 15(3):97–102. doi: 10.1002/j.2205-0140.2012.tb00013.x
2. Theilgaard-Monch K, Raaschou-Jensen K, Palm H, Schjodt K, Heilmann C, Vindelov L, et al. Flow cytometric assessment of lymphocyte subsets, lymphoid progenitors, and hematopoietic stem cells in allogeneic stem cell grafts. Bone Marrow Transplant (2001) 28(11):1073–82. doi: 10.1038/sj.bmt.1703270
3. Szabolcs P, Park KD, Reese M, Marti L, Broadwater G, Kurtzberg J. Coexistent naive phenotype and higher cycling rate of cord blood T cells as compared to adult peripheral blood. Exp Hematol (2003) 31(8):708–14. doi: 10.1016/S0301-472X(03)00160-7
4. Luevano M, Daryouzeh M, Alnabhan R, Querol S, Khakoo S, Madrigal A, et al. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum Immunol (2012) 73(3):248–57. doi: 10.1016/j.humimm.2011.12.015
5. Gluckman E, Broxmeyer HA, Auerbach AD, Friedman HS, Douglas GW, Devergie A, et al. Hematopoietic reconstitution in a patient with fanconi’s anemia by means of umbilical-cord blood from an HLA-identical sibling. N Engl J Med (1989) 321(17):1174–8. doi: 10.1056/NEJM198910263211707
6. Querol S, Rubinstein P, Madrigal A. The wider perspective: cord blood banks and their future prospects. Br J Haematol (2021) 195(4):507–17. doi: 10.1111/bjh.17468
7. Dessels C, Alessandrini M, Pepper MS. Factors influencing the umbilical cord blood stem cell industry: an evolving treatment landscape. Stem Cells Transl Med (2018) 7(9):643–50. doi: 10.1002/sctm.17-0244
8. Gragert L, Eapen M, Williams E, Freeman J, Spellman S, Baitty R, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U. S Registry N Engl J Med (2014) 371(4):339–48. doi: 10.1056/NEJMsa1311707
9. Barker JN, Boughan K, Dahi PB, Devlin SM, Maloy MA, Naputo K, et al. Racial disparities in access to HLA-matched unrelated donor transplants: a prospective 1312-patient analysis. Blood Adv (2019) 3(7):939–44. doi: 10.1182/bloodadvances.2018028662
10. Annas GJ. Waste and longing–the legal status of placental-blood banking. N Engl J Med (1999) 340(19):1521–4. doi: 10.1056/NEJM199905133401923
11. Mitchell R, Wagner JE, Brunstein CG, Cao Q, McKenna DH, Lund TC, et al. Impact of long-term cryopreservation on single umbilical cord blood transplantation outcomes. Biol Blood Marrow Transplant (2015) 21(1):50–4. doi: 10.1016/j.bbmt.2014.09.002
12. Abraham AA, John TD, Keller MD, Cruz CRN, Salem B, Roesch L, et al. Safety and feasibility of virus-specific T cells derived from umbilical cord blood in cord blood transplant recipients. Blood Adv (2019) 3(14):2057–68. doi: 10.1182/bloodadvances.2019000201
13. Pham PV, Vu NB, Pham VM, Truong NH, Pham TL, Dang LT, et al. Good manufacturing practice-compliant isolation and culture of human umbilical cord blood-derived mesenchymal stem cells. J Transl Med (2014) 12:56. doi: 10.1186/1479-5876-12-56
14. Bart T. Cost effectiveness of cord blood versus bone marrow and peripheral blood stem cells. Clinicoecon Outcomes Res (2010) 2:141–7. doi: 10.2147/CEOR.S11210
15. Cael B, Galaine J, Bardey I, Marton C, Fredon M, Biichle S, et al. Umbilical cord blood as a source of less differentiated T cells to produce CD123 CAR-T cells. Cancers (Basel) (2022) 14(13). doi: 10.3390/cancers14133168
16. Barker JN, Davies SM, DeFor T, Ramsay NK, Weisdorf DJ, Wagner JE. Survival after transplantation of unrelated donor umbilical cord blood is comparable to that of human leukocyte antigen-matched unrelated donor bone marrow: results of a matched-pair analysis. Blood (2001) 97(10):2957–61. doi: 10.1182/blood.V97.10.2957
17. Sun G, Tang B, Wan X, Yao W, Song K, Tu M, et al. Chimeric antigen receptor T cell therapy followed by unrelated cord blood transplantation for the treatment of Relapsed/Refractory b cell acute lymphoblastic leukemia in children and young adults: superior survival but relatively high post-transplantation relapse. Transplant Cell Ther (2022) 28(2):71 e1– e8. doi: 10.1016/j.jtct.2021.11.011
18. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117(3):1061–70. doi: 10.1182/blood-2010-07-293795
19. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood (2016) 127(8):1044–51. doi: 10.1182/blood-2015-06-653667
20. Roemhild A, Otto NM, Moll G, Abou-El-Enein M, Kaiser D, Bold G, et al. Regulatory T cells for minimising immune suppression in kidney transplantation: phase I/IIa clinical trial. BMJ (2020) 371:m3734. doi: 10.1136/bmj.m3734
21. Rosenberg SA, Lotze MT, Muul LM, Chang AE, Avis FP, Leitman S, et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N Engl J Med (1987) 316(15):889–97. doi: 10.1056/NEJM198704093161501
22. Dalle JH, Menezes J, Wagner E, Blagdon M, Champagne J, Champagne MA, et al. Characterization of cord blood natural killer cells: implications for transplantation and neonatal infections. Pediatr Res (2005) 57(5 Pt 1):649–55. doi: 10.1203/01.PDR.0000156501.55431.20
23. Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res (2009) 69(9):4010–7. doi: 10.1158/0008-5472.CAN-08-3712
24. Stankovic B, Bjorhovde HAK, Skarshaug R, Aamodt H, Frafjord A, Muller E, et al. Immune cell composition in human non-small cell lung cancer. Front Immunol (2018) 9:3101. doi: 10.3389/fimmu.2018.03101
25. Mark C, Czerwinski T, Roessner S, Mainka A, Horsch F, Heublein L, et al. Cryopreservation impairs 3-d migration and cytotoxicity of natural killer cells. Nat Commun (2020) 11(1):5224. doi: 10.1038/s41467-020-19094-0
26. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
27. Oei VYS, Siernicka M, Graczyk-Jarzynka A, Hoel HJ, Yang W, Palacios D, et al. Intrinsic functional potential of NK-cell subsets constrains retargeting driven by chimeric antigen receptors. Cancer Immunol Res (2018) 6(4):467–80. doi: 10.1158/2326-6066.CIR-17-0207
28. Rutella S, Bonanno G, Marone M, De Ritis D, Mariotti A, Voso MT, et al. Identification of a novel subpopulation of human cord blood CD34-CD133-CD7-CD45+lineage- cells capable of lymphoid/NK cell differentiation after in vitro exposure to IL-15. J Immunol (2003) 171(6):2977–88. doi: 10.4049/jimmunol.171.6.2977
29. Li Y, Basar R, Wang G, Liu E, Moyes JS, Li L, et al. KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nat Med (2022) 28(10):2133–44. doi: 10.1038/s41591-022-02003-x
30. Nieto Y, Banerjee PP, Kaur I, Griffin L, Ganesh C, Kerbauy L, et al. Innate cell engager AFM13 combined with preactivated and expanded cord blood-derived NK cells for patients with double refractory CD30+ lymphoma. Blood (2022) 140(Supplement 1):415–6. doi: 10.1182/blood-2022-156125
31. Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol Biol (2016) 1441:333–46. doi: 10.1007/978-1-4939-3684-7_28
32. Eapen M, Klein JP, Sanz GF, Spellman S, Ruggeri A, Anasetti C, et al. Effect of donor-recipient HLA matching at HLA a, b, c, and DRB1 on outcomes after umbilical-cord blood transplantation for leukaemia and myelodysplastic syndrome: a retrospective analysis. Lancet Oncol (2011) 12(13):1214–21. doi: 10.1016/S1470-2045(11)70260-1
33. Rocha V, Wagner JE Jr., Sobocinski KA, Klein JP, Zhang MJ, Horowitz MM, et al. Graft-versus-host disease in children who have received a cord-blood or bone marrow transplant from an HLA-identical sibling. eurocord and international bone marrow transplant registry working committee on alternative donor and stem cell sources. N Engl J Med (2000) 342(25):1846–54. doi: 10.1056/NEJM200006223422501
34. Horwitz ME, Stiff PJ, Cutler C, Brunstein C, Hanna R, Maziarz RT, et al. Omidubicel vs standard myeloablative umbilical cord blood transplantation: results of a phase 3 randomized study. Blood (2021) 138(16):1429–40. doi: 10.1182/blood.2021011719
35. Fuchs EJ, O’Donnell PV, Eapen M, Logan B, Antin JH, Dawson P, et al. Double unrelated umbilical cord blood vs HLA-haploidentical bone marrow transplantation: the BMT CTN 1101 trial. Blood (2021) 137(3):420–8. doi: 10.1182/blood.2020007535
36. Le Blanc K, Rasmusson I, Sundberg B, Gotherstrom C, Hassan M, Uzunel M, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet (2004) 363(9419):1439–41. doi: 10.1016/S0140-6736(04)16104-7
37. Wu Y, Wang Z, Cao Y, Xu L, Li X, Liu P, et al. Cotransplantation of haploidentical hematopoietic and umbilical cord mesenchymal stem cells with a myeloablative regimen for refractory/relapsed hematologic malignancy. Ann Hematol (2013) 92(12):1675–84. doi: 10.1007/s00277-013-1831-0
38. Gao L, Zhang Y, Hu B, Liu J, Kong P, Lou S, et al. Phase II multicenter, randomized, double-blind controlled study of efficacy and safety of umbilical cord-derived mesenchymal stromal cells in the prophylaxis of chronic graft-Versus-Host disease after HLA-haploidentical stem-cell transplantation. J Clin Oncol (2016) 34(24):2843–50. doi: 10.1200/JCO.2015.65.3642
39. Zhu L, Wang Z, Zheng X, Ding L, Han D, Yan H, et al. Haploidentical hematopoietic stem cell transplant with umbilical cord-derived multipotent mesenchymal cell infusion for the treatment of high-risk acute leukemia in children. Leuk Lymphoma (2015) 56(5):1346–52. doi: 10.3109/10428194.2014.939970
40. Kebriaei P, Hayes J, Daly A, Uberti J, Marks DI, Soiffer R, et al. A phase 3 randomized study of remestemcel-l versus placebo added to second-line therapy in patients with steroid-refractory acute graft-versus-Host disease. Biol Blood Marrow Transplant (2020) 26(5):835–44. doi: 10.1016/j.bbmt.2019.08.029
41. Liu DD, Hong WC, Qiu KY, Li XY, Liu Y, Zhu LW, et al. Umbilical cord blood: a promising source for allogeneic CAR-T cells. Front Oncol (2022) 12:944248. doi: 10.3389/fonc.2022.944248
42. Hiwarkar P, Qasim W, Ricciardelli I, Gilmour K, Quezada S, Saudemont A, et al. Cord blood T cells mediate enhanced antitumor effects compared with adult peripheral blood T cells. Blood (2015) 126(26):2882–91. doi: 10.1182/blood-2015-06-654780
43. Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina SA, et al. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science (2010) 330(6011):1695–9. doi: 10.1126/science.1196509
44. Rudd BD. Neonatal T cells: a reinterpretation. Annu Rev Immunol (2020) 38:229–47. doi: 10.1146/annurev-immunol-091319-083608
45. Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer (2013) 13(8):525–41. doi: 10.1038/nrc3565
46. Wang J, Hu Y, Huang H. Current development of chimeric antigen receptor T-cell therapy. Stem Cell Investig (2018) 5:44. doi: 10.21037/sci.2018.11.05
47. Caldwell KJ, Gottschalk S, Talleur AC. Allogeneic CAR cell therapy-more than a pipe dream. Front Immunol (2020) 11:618427. doi: 10.3389/fimmu.2020.618427
48. Rubinstein P, Carrier C, Scaradavou A, Kurtzberg J, Adamson J, Migliaccio AR, et al. Outcomes among 562 recipients of placental-blood transplants from unrelated donors. N Engl J Med (1998) 339(22):1565–77. doi: 10.1056/NEJM199811263392201
49. Taylor CJ, Peacock S, Chaudhry AN, Bradley JA, Bolton EM. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell (2012) 11(2):147–52. doi: 10.1016/j.stem.2012.07.014
50. Razeghian E, Nasution MKM, Rahman HS, Gardanova ZR, Abdelbasset WK, Aravindhan S, et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res Ther (2021) 12(1):428. doi: 10.1186/s13287-021-02510-7
51. Pegram HJ, Purdon TJ, van Leeuwen DG, Curran KJ, Giralt SA, Barker JN, et al. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of b-cell acute lymphoblastic leukemia. Leukemia (2015) 29(2):415–22. doi: 10.1038/leu.2014.215
52. Lekakis LJ, Locke FL, Tees M, Neelapu SS, Malik SA, Hamadani M, et al. ALPHA2 study: ALLO-501A allogeneic CAR T in LBCL, updated results continue to show encouraging safety and efficacy with consolidation dosing. Blood (2021) 138(Supplement 1):649–. doi: 10.1182/blood-2021-146045
53. Karasiewicz K, He S, Ng M, Tess K, Ling W, Kaufmann GF, et al. Preclinical evaluation of human placental-derived allogeneic CD19 CAR-T cells against b cell malignancies. Blood (2019) 134(Supplement_1):3222–. doi: 10.1182/blood-2019-130782
54. Iriguchi S, Yasui Y, Kawai Y, Arima S, Kunitomo M, Sato T, et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat Commun (2021) 12(1):430. doi: 10.1038/s41467-020-20658-3
55. Awong G, Herer E, La Motte-Mohs RN, Zuniga-Pflucker JC. Human CD8 T cells generated in vitro from hematopoietic stem cells are functionally mature. BMC Immunol (2011) 12:22. doi: 10.1186/1471-2172-12-22
56. Seay HR, Putnam AL, Cserny J, Posgai AL, Rosenau EH, Wingard JR, et al. Expansion of human tregs from cryopreserved umbilical cord blood for GMP-compliant autologous adoptive cell transfer therapy. Mol Ther Methods Clin Dev (2017) 4:178–91. doi: 10.1016/j.omtm.2016.12.003
57. Chwojnicki K, Iwaszkiewicz-Grzes D, Jankowska A, Zielinski M, Lowiec P, Gliwinski M, et al. Administration of CD4(+)CD25(high)CD127(-)FoxP3(+) regulatory T cells for relapsing-remitting multiple sclerosis: a phase 1 study. BioDrugs (2021) 35(1):47–60. doi: 10.1007/s40259-020-00462-7
58. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care (2012) 35(9):1817–20. doi: 10.2337/dc12-0038
59. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
60. Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, et al. Regulatory cell therapy in kidney transplantation (The ONE study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet (2020) 395(10237):1627–39. doi: 10.1016/S0140-6736(20)30167-7
61. Sanchez-Fueyo A, Whitehouse G, Grageda N, Cramp ME, Lim TY, Romano M, et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am J Transplant (2020) 20(4):1125–36. doi: 10.1111/ajt.15700
62. MacMillan ML, Hippen KL, McKenna DH, Kadidlo D, Sumstad D, DeFor TE, et al. First-in-human phase 1 trial of induced regulatory T cells for graft-versus-host disease prophylaxis in HLA-matched siblings. Blood Adv (2021) 5(5):1425–36. doi: 10.1182/bloodadvances.2020003219
63. Theil A, Tuve S, Oelschlagel U, Maiwald A, Dohler D, Ossmann D, et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy (2015) 17(4):473–86. doi: 10.1016/j.jcyt.2014.11.005
64. Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol (2009) 133(1):22–6. doi: 10.1016/j.clim.2009.06.001
65. Bluestone JA, Tang Q. Treg cells-the next frontier of cell therapy. Science (2018) 362(6411):154–5. doi: 10.1126/science.aau2688
66. Bezie S, Charreau B, Vimond N, Lasselin J, Gerard N, Nerriere-Daguin V, et al. Human CD8+ tregs expressing a MHC-specific CAR display enhanced suppression of human skin rejection and GVHD in NSG mice. Blood Adv (2019) 3(22):3522–38. doi: 10.1182/bloodadvances.2019000411
67. Radichev IA, Yoon J, Scott DW, Griffin K, Savinov AY. Towards antigen-specific tregs for type 1 diabetes: construction and functional assessment of pancreatic endocrine marker, HPi2-based chimeric antigen receptor. Cell Immunol (2020) 358:104224. doi: 10.1016/j.cellimm.2020.104224
68. Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther (2014) 22(5):1018–28. doi: 10.1038/mt.2014.41
69. Yoon J, Schmidt A, Zhang AH, Konigs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and b-cell responses to FVIII. Blood (2017) 129(2):238–45. doi: 10.1182/blood-2016-07-727834
70. Fritsche E, Volk HD, Reinke P, Abou-El-Enein M. Toward an optimized process for clinical manufacturing of CAR-treg cell therapy. Trends Biotechnol (2020) 38(10):1099–112. doi: 10.1016/j.tibtech.2019.12.009
71. Kaeuferle T, Krauss R, Blaeschke F, Willier S, Feuchtinger T. Strategies of adoptive T -cell transfer to treat refractory viral infections post allogeneic stem cell transplantation. J Hematol Oncol (2019) 12(1):13. doi: 10.1186/s13045-019-0701-1
72. Keller MD, Bollard CM. Virus-specific T-cell therapies for patients with primary immune deficiency. Blood (2020) 135(9):620–8. doi: 10.1182/blood.2019000924
73. McLaughlin LP, Rouce R, Gottschalk S, Torrano V, Carrum G, Wu MF, et al. EBV/LMP-specific T cells maintain remissions of T- and b-cell EBV lymphomas after allogeneic bone marrow transplantation. Blood (2018) 132(22):2351–61. doi: 10.1182/blood-2018-07-863654
74. Bollard CM, Gottschalk S, Torrano V, Diouf O, Ku S, Hazrat Y, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol (2014) 32(8):798–808. doi: 10.1200/JCO.2013.51.5304
75. Prockop S, Doubrovina E, Suser S, Heller G, Barker J, Dahi P, et al. Off-the-shelf EBV-specific T cell immunotherapy for rituximab-refractory EBV-associated lymphoma following transplantation. J Clin Invest (2020) 130(2):733–47. doi: 10.1172/JCI121127
76. Peggs KS, Thomson K, Samuel E, Dyer G, Armoogum J, Chakraverty R, et al. Directly selected cytomegalovirus-reactive donor T cells confer rapid and safe systemic reconstitution of virus-specific immunity following stem cell transplantation. Clin Infect Dis (2011) 52(1):49–57. doi: 10.1093/cid/ciq042
77. Withers B, Blyth E, Clancy LE, Yong A, Fraser C, Burgess J, et al. Long-term control of recurrent or refractory viral infections after allogeneic HSCT with third-party virus-specific T cells. Blood Adv (2017) 1(24):2193–205. doi: 10.1182/bloodadvances.2017010223
78. Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, et al. Off-the-Shelf virus-specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, Epstein-Barr virus, and adenovirus infections after allogeneic hematopoietic stem-cell transplantation. J Clin Oncol (2017) 35(31):3547–57. doi: 10.1200/JCO.2017.73.0655
79. Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood (2013) 121(26):5113–23. doi: 10.1182/blood-2013-02-486324
80. Melenhorst JJ, Leen AM, Bollard CM, Quigley MF, Price DA, Rooney CM, et al. Allogeneic virus-specific T cells with HLA alloreactivity do not produce GVHD in human subjects. Blood (2010) 116(22):4700–2. doi: 10.1182/blood-2010-06-289991
81. Dave H, Luo M, Blaney JW, Patel S, Barese C, Cruz CR, et al. Toward a rapid production of multivirus-specific T cells targeting BKV, adenovirus, CMV, and EBV from umbilical cord blood. Mol Ther Methods Clin Dev (2017) 5:13–21. doi: 10.1016/j.omtm.2017.02.001
82. Fruchtman SM, Hurlet A, Dracker R, Isola L, Goldman B, Schneider BL, et al. The successful treatment of severe aplastic anemia with autologous cord blood transplantation. Biol Blood Marrow Transplant (2004) 10(11):741–2. doi: 10.1016/j.bbmt.2004.07.003
83. Barros MS, de Araujo ND, Magalhaes-Gama F, Pereira Ribeiro TL, Alves Hanna FS, Tarrago AM, et al. Gammadelta T cells for leukemia immunotherapy: new and expanding trends. Front Immunol (2021) 12:729085. doi: 10.3389/fimmu.2021.729085
84. Saura-Esteller J, de Jong M, King LA, Ensing E, Winograd B, de Gruijl TD, et al. Gamma delta T-cell based cancer immunotherapy: past-Present-Future. Front Immunol (2022) 13:915837. doi: 10.3389/fimmu.2022.915837
85. Lin M, Zhang X, Liang S, Luo H, Alnaggar M, Liu A, et al. Irreversible electroporation plus allogenic Vgamma9Vdelta2 T cells enhances antitumor effect for locally advanced pancreatic cancer patients. Signal Transduct Target Ther (2020) 5(1):215. doi: 10.1038/s41392-020-00260-1
86. Xu Y, Xiang Z, Alnaggar M, Kouakanou L, Li J, He J, et al. Allogeneic Vgamma9Vdelta2 T-cell immunotherapy exhibits promising clinical safety and prolongs the survival of patients with late-stage lung or liver cancer. Cell Mol Immunol (2021) 18(2):427–39. doi: 10.1038/s41423-020-0515-7
87. Neelapu SS, Hamadani M, Miklos DB, Holmes H, Hinkle J, Kennedy-Wilde J, et al. A phase 1 study of ADI-001: anti-CD20 CAR-engineered allogeneic gamma delta (γδ) T cells in adults with b-cell malignancies. J Clin Oncol (2022) 40(16_suppl):7509–. doi: 10.1200/JCO.2022.40.16_suppl.7509
88. Willcox CR, Davey MS, Willcox BE. Development and selection of the human Vgamma9Vdelta2(+) T-cell repertoire. Front Immunol (2018) 9:1501. doi: 10.3389/fimmu.2018.01501
89. Berglund S, Gaballa A, Sawaisorn P, Sundberg B, Uhlin M. Expansion of gammadelta T cells from cord blood: a therapeutical possibility. Stem Cells Int (2018) 2018:8529104. doi: 10.1155/2018/8529104
90. Cairo C, Sagnia B, Cappelli G, Colizzi V, Leke RG, Leke RJ, et al. Human cord blood gammadelta T cells expressing public Vgamma2 chains dominate the response to bisphosphonate plus interleukin-15. Immunology (2013) 138(4):346–60. doi: 10.1111/imm.12039
91. Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih YP, et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N Engl J Med (2022) 386(22):2112–9. doi: 10.1056/NEJMoa2119662
92. Ma Q, Garber HR, Lu S, He H, Tallis E, Ding X, et al. A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy (2016) 18(8):985–94. doi: 10.1016/j.jcyt.2016.05.001
93. St John LS, Wan L, He H, Garber HR, Clise-Dwyer K, Alatrash G, et al. PR1-specific cytotoxic T lymphocytes are relatively frequent in umbilical cord blood and can be effectively expanded to target myeloid leukemia. Cytotherapy (2016) 18(8):995–1001. doi: 10.1016/j.jcyt.2016.05.007
94. Kwoczek J, Riese SB, Tischer S, Bak S, Lahrberg J, Oelke M, et al. Cord blood-derived T cells allow the generation of a more naive tumor-reactive cytotoxic T-cell phenotype. Transfusion (2018) 58(1):88–99. doi: 10.1111/trf.14365
95. Okas M, Gertow J, Uzunel M, Karlsson H, Westgren M, Karre K, et al. Clinical expansion of cord blood-derived T cells for use as donor lymphocyte infusion after cord blood transplantation. J Immunother (2010) 33(1):96–105. doi: 10.1097/CJI.0b013e3181b291a4
96. Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol (2017) 8:1124. doi: 10.3389/fimmu.2017.01124
97. Otegbeye F, Cooper B, Caimi P, Zamborsky K, Reese-Koc J, Hillian A, et al. A phase I study to determine the maximum tolerated dose of ex vivo expanded natural killer cells derived from unrelated, HLA-disparate adult donors. Transplant Cell Ther (2022) 28(5):250 e1– e8. doi: 10.1016/j.jtct.2022.02.008
98. Simonetta F, Alvarez M, Negrin RS. Natural killer cells in graft-versus-Host-Disease after allogeneic hematopoietic cell transplantation. Front Immunol (2017) 8:465. doi: 10.3389/fimmu.2017.00465
99. Jacobson CA, Turki AT, McDonough SM, Stevenson KE, Kim HT, Kao G, et al. Immune reconstitution after double umbilical cord blood stem cell transplantation: comparison with unrelated peripheral blood stem cell transplantation. Biol Blood Marrow Transplant (2012) 18(4):565–74. doi: 10.1016/j.bbmt.2011.08.018
100. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood (2014) 123(25):3855–63. doi: 10.1182/blood-2013-10-532531
101. Bachanova V, Sarhan D, DeFor TE, Cooley S, Panoskaltsis-Mortari A, Blazar BR, et al. Haploidentical natural killer cells induce remissions in non-Hodgkin lymphoma patients with low levels of immune-suppressor cells. Cancer Immunol Immunother (2018) 67(3):483–94. doi: 10.1007/s00262-017-2100-1
102. Shaffer BC, Le Luduec JB, Forlenza C, Jakubowski AA, Perales MA, Young JW, et al. Phase II study of haploidentical natural killer cell infusion for treatment of relapsed or persistent myeloid malignancies following allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant (2016) 22(4):705–9. doi: 10.1016/j.bbmt.2015.12.028
103. Rizzieri DA, Storms R, Chen DF, Long G, Yang Y, Nikcevich DA, et al. Natural killer cell-enriched donor lymphocyte infusions from a 3-6/6 HLA matched family member following nonmyeloablative allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2010) 16(8):1107–14. doi: 10.1016/j.bbmt.2010.02.018
104. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105(8):3051–7. doi: 10.1182/blood-2004-07-2974
105. Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol (2010) 28(6):955–9. doi: 10.1200/JCO.2009.24.4590
106. Curti A, Ruggeri L, D’Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood (2011) 118(12):3273–9. doi: 10.1182/blood-2011-01-329508
107. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med (2016) 8(357):357ra123. doi: 10.1126/scitranslmed.aaf2341
108. Shi J, Tricot G, Szmania S, Rosen N, Garg TK, Malaviarachchi PA, et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol (2008) 143(5):641–53. doi: 10.1111/j.1365-2141.2008.07340.x
109. Shah N, Li L, McCarty J, Kaur I, Yvon E, Shaim H, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol (2017) 177(3):457–66. doi: 10.1111/bjh.14570
110. Cany J, van der Waart AB, Tordoir M, Franssen GM, Hangalapura BN, de Vries J, et al. Natural killer cells generated from cord blood hematopoietic progenitor cells efficiently target bone marrow-residing human leukemia cells in NOD/SCID/IL2Rg(null) mice. PloS One (2013) 8(6):e64384. doi: 10.1371/journal.pone.0064384
111. Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, Maas F, et al. Successful transfer of umbilical cord blood CD34(+) hematopoietic stem and progenitor-derived NK cells in older acute myeloid leukemia patients. Clin Cancer Res (2017) 23(15):4107–18. doi: 10.1158/1078-0432.CCR-16-2981
112. Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother Cancer (2017) 5(1):73. doi: 10.1186/s40425-017-0275-9
113. Lavergne E, Combadiere C, Iga M, Boissonnas A, Bonduelle O, Maho M, et al. Intratumoral CC chemokine ligand 5 overexpression delays tumor growth and increases tumor cell infiltration. J Immunol (2004) 173(6):3755–62. doi: 10.4049/jimmunol.173.6.3755
114. Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The mechanism of anti-PD-L1 antibody efficacy against PD-L1-Negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discovery (2019) 9(10):1422–37. doi: 10.1158/2159-8290.CD-18-1259
115. Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, Ozerov IV, et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFbeta enhance the efficacy of cancer immunotherapy. Nat Commun (2018) 9(1):741. doi: 10.1038/s41467-017-02696-6
116. Yun HD, Felices M, Vallera DA, Hinderlie P, Cooley S, Arock M, et al. Trispecific killer engager CD16xIL15xCD33 potently induces NK cell activation and cytotoxicity against neoplastic mast cells. Blood Adv (2018) 2(13):1580–4. doi: 10.1182/bloodadvances.2018018176
117. Kerbauy LN, Marin ND, Kaplan M, Banerjee PP, Berrien-Elliott MM, Becker-Hapak M, et al. Combining AFM13, a bispecific CD30/CD16 antibody, with cytokine-activated blood and cord blood-derived NK cells facilitates CAR-like responses against CD30(+) malignancies. Clin Cancer Res (2021) 27(13):3744–56. doi: 10.1158/1078-0432.CCR-21-0164
118. Felices M, Kodal B, Hinderlie P, Kaminski MF, Cooley S, Weisdorf DJ, et al. Novel CD19-targeted TriKE restores NK cell function and proliferative capacity in CLL. Blood Adv (2019) 3(6):897–907. doi: 10.1182/bloodadvances.2018029371
119. McCall AM, Adams GP, Amoroso AR, Nielsen UB, Zhang L, Horak E, et al. Isolation and characterization of an anti-CD16 single-chain fv fragment and construction of an anti-HER2/neu/anti-CD16 bispecific scFv that triggers CD16-dependent tumor cytolysis. Mol Immunol (1999) 36(7):433–45. doi: 10.1016/S0161-5890(99)00057-7
120. Sasse S, Brockelmann PJ, Momotow J, Plutschow A, Huttmann A, Basara N, et al. AFM13 in patients with relapsed or refractory classical Hodgkin lymphoma: final results of an open-label, randomized, multicenter phase II trial. Leuk Lymphoma (2022) 63(8):1871–8. doi: 10.1080/10428194.2022.2095623
121. Bi J, Tian Z. NK cell dysfunction and checkpoint immunotherapy. Front Immunol (2019) 10:1999. doi: 10.3389/fimmu.2019.01999
122. Gluck WL, Hurst D, Yuen A, Levine AM, Dayton MA, Gockerman JP, et al. Phase I studies of interleukin (IL)-2 and rituximab in b-cell non-hodgkin’s lymphoma: IL-2 mediated natural killer cell expansion correlations with clinical response. Clin Cancer Res (2004) 10(7):2253–64. doi: 10.1158/1078-0432.CCR-1087-3
123. Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature (2022) 602(7897):503–9. doi: 10.1038/s41586-021-04390-6
124. Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res (2018) 8(6):1083–9.
125. Vela M, Corral D, Carrasco P, Fernandez L, Valentin J, Gonzalez B, et al. Haploidentical IL-15/41BBL activated and expanded natural killer cell infusion therapy after salvage chemotherapy in children with relapsed and refractory leukemia. Cancer Lett (2018) 422:107–17. doi: 10.1016/j.canlet.2018.02.033
126. Shapiro RM, Birch GC, Hu G, Vergara Cadavid J, Nikiforow S, Baginska J, et al. Expansion, persistence, and efficacy of donor memory-like NK cells infused for posttransplant relapse. J Clin Invest (2022) 132(11). doi: 10.1172/JCI154334
127. Bednarski JJ, Zimmerman C, Berrien-Elliott MM, Foltz JA, Becker-Hapak M, Neal CC, et al. Donor memory-like NK cells persist and induce remissions in pediatric patients with relapsed AML after transplant. Blood (2022) 139(11):1670–83. doi: 10.1182/blood.2021013972
128. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia (2018) 32(2):520–31. doi: 10.1038/leu.2017.226
129. Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood (2021) 137(5):624–36. doi: 10.1182/blood.2020007748
130. Hoerster K, Uhrberg M, Wiek C, Horn PA, Hanenberg H, Heinrichs S. HLA class I knockout converts allogeneic primary NK cells into suitable effectors for “Off-the-Shelf” immunotherapy. Front Immunol (2020) 11:586168. doi: 10.3389/fimmu.2020.586168
131. Melero I, Rouzaut A, Motz GT, Coukos G. T-Cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discovery (2014) 4(5):522–6. doi: 10.1158/2159-8290.CD-13-0985
132. Cozar B, Greppi M, Carpentier S, Narni-Mancinelli E, Chiossone L, Vivier E. Tumor-infiltrating natural killer cells. Cancer Discovery (2021) 11(1):34–44. doi: 10.1158/2159-8290.CD-20-0655
133. D’Souza A, Fretham C, Lee SJ, Arora M, Brunner J, Chhabra S, et al. Current use of and trends in hematopoietic cell transplantation in the United States. Biol Blood Marrow Transplant (2020) 26:e177–e82. doi: 10.1016/j.bbmt.2020.04.013
134. Wang JC, Doedens M, Dick JE. Primitive human hematopoietic cells are enriched in cord blood compared with adult bone marrow or mobilized peripheral blood as measured by the quantitative in vivo SCID-repopulating cell assay. Blood (1997) 89(11):3919–24. doi: 10.1182/blood.V89.11.3919
135. Eapen M, Rubinstein P, Zhang MJ, Stevens C, Kurtzberg J, Scaradavou A, et al. Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet (2007) 369(9577):1947–54. doi: 10.1016/S0140-6736(07)60915-5
136. Eapen M, Klein JP, Ruggeri A, Spellman S, Lee SJ, Anasetti C, et al. Impact of allele-level HLA matching on outcomes after myeloablative single unit umbilical cord blood transplantation for hematologic malignancy. Blood (2014) 123(1):133–40. doi: 10.1182/blood-2013-05-506253
137. Wagner JE Jr., Eapen M, Carter S, Wang Y, Schultz KR, Wall DA, et al. One-unit versus two-unit cord-blood transplantation for hematologic cancers. N Engl J Med (2014) 371(18):1685–94. doi: 10.1056/NEJMoa1405584
138. Politikos I, Davis E, Nhaissi M, Wagner JE, Brunstein CG, Cohen S, et al. Guidelines for cord blood unit selection. Biol Blood Marrow Transplant (2020) 26(12):2190–6. doi: 10.1016/j.bbmt.2020.07.030
139. Barker JN, Kempenich J, Kurtzberg J, Brunstein CG, Delaney C, Milano F, et al. CD34(+) cell content of 126 341 cord blood units in the US inventory: implications for transplantation and banking. Blood Adv (2019) 3(8):1267–71. doi: 10.1182/bloodadvances.2018029157
140. Kosuri S, Wolff T, Devlin SM, Byam C, Mazis CM, Naputo K, et al. Prospective evaluation of unrelated donor cord blood and haploidentical donor access reveals graft availability varies by patient ancestry: practical implications for donor selection. Biol Blood Marrow Transplant (2017) 23(6):965–70. doi: 10.1016/j.bbmt.2017.03.001
141. Rizk M, Aziz J, Shorr R, Allan DS. Cell-based therapy using umbilical cord blood for novel indications in regenerative therapy and immune modulation: an updated systematic scoping review of the literature. Biol Blood Marrow Transplant (2017) 23(10):1607–13. doi: 10.1016/j.bbmt.2017.05.032
142. Frassoni F, Gualandi F, Podesta M, Raiola AM, Ibatici A, Piaggio G, et al. Direct intrabone transplant of unrelated cord-blood cells in acute leukaemia: a phase I/II study. Lancet Oncol (2008) 9(9):831–9. doi: 10.1016/S1470-2045(08)70180-3
143. Brunstein CG, Barker JN, Weisdorf DJ, Defor TE, McKenna D, Chong SY, et al. Intra-BM injection to enhance engraftment after myeloablative umbilical cord blood transplantation with two partially HLA-matched units. Bone Marrow Transplant (2009) 43(12):935–40. doi: 10.1038/bmt.2008.417
144. Nishida T, Kobayashi T, Sawa M, Masuda S, Shibasaki Y, Goto T, et al. A multicenter phase II study of intrabone single-unit cord blood transplantation without antithymocyte globulin. Ann Hematol (2021) 100(3):743–52. doi: 10.1007/s00277-020-04365-z
145. Murata M, Maeda Y, Masuko M, Onishi Y, Endo T, Terakura S, et al. Phase II study of intrabone single unit cord blood transplantation for hematological malignancies. Cancer Sci (2017) 108(8):1634–9. doi: 10.1111/cas.13291
146. Kurita N, Gosho M, Yokoyama Y, Kato T, Obara N, Sakata-Yanagimoto M, et al. A phase I/II trial of intrabone marrow cord blood transplantation and comparison of the hematological recovery with the Japanese nationwide database. Bone Marrow Transplant (2017) 52(4):574–9. doi: 10.1038/bmt.2016.319
147. Okada M, Tasaka T, Ikegame K, Aotsuka N, Kobayashi T, Najima Y, et al. A prospective multicenter phase II study of intrabone marrow transplantation of unwashed cord blood using reduced-intensity conditioning. Eur J Haematol (2018) 100(4):335–43. doi: 10.1111/ejh.12999
148. Bonifazi F, Dan E, Labopin M, Sessa M, Guadagnuolo V, Ferioli M, et al. Intrabone transplant provides full stemness of cord blood stem cells with fast hematopoietic recovery and low GVHD rate: results from a prospective study. Bone Marrow Transplant (2019) 54(5):717–25. doi: 10.1038/s41409-018-0335-x
149. Rocha V, Labopin M, Ruggeri A, Podesta M, Gallamini A, Bonifazi F, et al. Unrelated cord blood transplantation: outcomes after single-unit intrabone injection compared with double-unit intravenous injection in patients with hematological malignancies. Transplantation (2013) 95(10):1284–91. doi: 10.1097/TP.0b013e318288ca4d
150. Liu H, Rich ES, Godley L, Odenike O, Joseph L, Marino S, et al. Reduced-intensity conditioning with combined haploidentical and cord blood transplantation results in rapid engraftment, low GVHD, and durable remissions. Blood (2011) 118(24):6438–45. doi: 10.1182/blood-2011-08-372508
151. Hsu J, Artz A, Mayer SA, Guarner D, Bishop MR, Reich-Slotky R, et al. Combined haploidentical and umbilical cord blood allogeneic stem cell transplantation for high-risk lymphoma and chronic lymphoblastic leukemia. Biol Blood Marrow Transplant (2018) 24(2):359–65. doi: 10.1016/j.bbmt.2017.10.040
152. van Besien K, Artz A, Champlin RE, Guarneri D, Bishop MR, Chen J, et al. Haploidentical vs haplo-cord transplant in adults under 60 years receiving fludarabine and melphalan conditioning. Blood Adv (2019) 3(12):1858–67. doi: 10.1182/bloodadvances.2019000200
153. Hiwarkar P, Adams S, Gilmour K, Nataraj R, Bonney D, Poulton K, et al. Cord blood CD8+ T-cell expansion following granulocyte transfusions eradicates refractory leukemia. Blood Adv (2020) 4(17):4165–74. doi: 10.1182/bloodadvances.2020001737
154. de Lima M, McNiece I, Robinson SN, Munsell M, Eapen M, Horowitz M, et al. Cord-blood engraftment with ex vivo mesenchymal-cell coculture. N Engl J Med (2012) 367(24):2305–15. doi: 10.1056/NEJMoa1207285
155. Mehta RS, Saliba RM, Cao K, Kaur I, Rezvani K, Chen J, et al. Ex vivo mesenchymal precursor cell-expanded cord blood transplantation after reduced-intensity conditioning regimens improves time to neutrophil recovery. Biol Blood Marrow Transplant (2017) 23(8):1359–66. doi: 10.1016/j.bbmt.2017.05.002
156. Saiyin T, Kirkham AM, Bailey AJM, Shorr R, Pineault N, Maganti HB, et al. Clinical outcomes of umbilical cord blood transplantation using ex vivo expansion: a systematic review and meta-analysis of controlled studies. Transplant Cell Ther (2023) 29(2):129 e1– e9. doi: 10.1016/j.jtct.2022.11.007
157. Delaney C, Heimfeld S, Brashem-Stein C, Voorhies H, Manger RL, Bernstein ID. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med (2010) 16(2):232–6. doi: 10.1038/nm.2080
158. Milano F, Thur LA, Blake J, Delaney C. Infusion of non-HLA-Matched off-the-Shelf ex vivo expanded cord blood progenitors in patients undergoing cord blood transplantation: result of a phase II clinical trial. Front Cell Dev Biol (2022) 10:835793. doi: 10.3389/fcell.2022.835793
159. Stiff PJ, Montesinos P, Peled T, Landau E, Goudsmid NR, Mandel J, et al. Cohort-controlled comparison of umbilical cord blood transplantation using carlecortemcel-l, a single progenitor-enriched cord blood, to double cord blood unit transplantation. Biol Blood Marrow Transplant (2018) 24(7):1463–70. doi: 10.1016/j.bbmt.2018.02.012
160. Horwitz ME, Wease S, Blackwell B, Valcarcel D, Frassoni F, Boelens JJ, et al. Phase I/II study of stem-cell transplantation using a single cord blood unit expanded ex vivo with nicotinamide. J Clin Oncol (2019) 37(5):367–74. doi: 10.1200/JCO.18.00053
161. Wagner JE Jr., Brunstein CG, Boitano AE, DeFor TE, McKenna D, Sumstad D, et al. Phase I/II trial of StemRegenin-1 expanded umbilical cord blood hematopoietic stem cells supports testing as a stand-alone graft. Cell Stem Cell (2016) 18(1):144–55. doi: 10.1016/j.stem.2015.10.004
162. Cohen S, Roy J, Lachance S, Delisle JS, Marinier A, Busque L, et al. Hematopoietic stem cell transplantation using single UM171-expanded cord blood: a single-arm, phase 1-2 safety and feasibility study. Lancet Haematol (2020) 7(2):e134–e45. doi: 10.1016/S2352-3026(19)30202-9
163. Peled T, Shoham H, Aschengrau D, Yackoubov D, Frei G, Rosenheimer GN, et al. Nicotinamide, a SIRT1 inhibitor, inhibits differentiation and facilitates expansion of hematopoietic progenitor cells with enhanced bone marrow homing and engraftment. Exp Hematol (2012) 40(4):342–55 e1. doi: 10.1016/j.exphem.2011.12.005
164. Harrell CR, Jovicic N, Djonov V, Arsenijevic N, Volarevic V. Mesenchymal stem cell-derived exosomes and other extracellular vesicles as new remedies in the therapy of inflammatory diseases. Cells (2019) 8(12). doi: 10.3390/cells8121605
165. Erices A, Conget P, Minguell JJ. Mesenchymal progenitor cells in human umbilical cord blood. Br J Haematol (2000) 109(1):235–42. doi: 10.1046/j.1365-2141.2000.01986.x
166. de Wolf C, van de Bovenkamp M, Hoefnagel M. Regulatory perspective on in vitro potency assays for human mesenchymal stromal cells used in immunotherapy. Cytotherapy (2017) 19(7):784–97. doi: 10.1016/j.jcyt.2017.03.076
167. Wiese DM, Wood CA, Braid LR. From vial to vein: crucial gaps in mesenchymal stromal cell clinical trial reporting. Front Cell Dev Biol (2022) 10:867426. doi: 10.3389/fcell.2022.867426
168. Merimi M, El-Majzoub R, Lagneaux L, Moussa Agha D, Bouhtit F, Meuleman N, et al. The therapeutic potential of mesenchymal stromal cells for regenerative medicine: current knowledge and future understandings. Front Cell Dev Biol (2021) 9:661532. doi: 10.3389/fcell.2021.661532
169. Garcia-Bernal D, Garcia-Arranz M, Yanez RM, Hervas-Salcedo R, Cortes A, Fernandez-Garcia M, et al. The current status of mesenchymal stromal cells: controversies, unresolved issues and some promising solutions to improve their therapeutic efficacy. Front Cell Dev Biol (2021) 9:650664. doi: 10.3389/fcell.2021.650664
170. Xu Z, Huang Y, Zhou J, Deng X, He W, Liu X, et al. Current status of cell-based therapies for COVID-19: evidence from mesenchymal stromal cells in sepsis and ARDS. Front Immunol (2021) 12:738697. doi: 10.3389/fimmu.2021.738697
171. Wu Y, Cao Y, Li X, Xu L, Wang Z, Liu P, et al. Cotransplantation of haploidentical hematopoietic and umbilical cord mesenchymal stem cells for severe aplastic anemia: successful engraftment and mild GVHD. Stem Cell Res (2014) 12(1):132–8. doi: 10.1016/j.scr.2013.10.001
172. Wu KH, Tsai C, Wu HP, Sieber M, Peng CT, Chao YH. Human application of ex vivo expanded umbilical cord-derived mesenchymal stem cells: enhance hematopoiesis after cord blood transplantation. Cell Transplant (2013) 22(11):2041–51. doi: 10.3727/096368912X663533
173. Wang H, Wang Z, Zheng X, Ding L, Zhu L, Yan H, et al. Hematopoietic stem cell transplantation with umbilical cord multipotent stromal cell infusion for the treatment of aplastic anemia–a single-center experience. Cytotherapy (2013) 15(9):1118–25. doi: 10.1016/j.jcyt.2013.04.007
174. Li Y, Hao J, Hu Z, Yang YG, Zhou Q, Sun L, et al. Current status of clinical trials assessing mesenchymal stem cell therapy for graft versus host disease: a systematic review. Stem Cell Res Ther (2022) 13(1):93. doi: 10.1186/s13287-021-02683-1
175. Soder RP, Dawn B, Weiss ML, Dunavin N, Weir S, Mitchell J, et al. A phase I study to evaluate two doses of wharton’s jelly-derived mesenchymal stromal cells for the treatment of De novo high-risk or steroid-refractory acute graft versus host disease. Stem Cell Rev Rep (2020) 16(5):979–91. doi: 10.1007/s12015-020-10015-8
Keywords: cord blood, chimeric antigen receptor, hematopoietic stem cell transplantation, NK-cells, mesenchymal stem cells
Citation: Wang J and Metheny L (2023) Umbilical cord blood derived cellular therapy: advances in clinical development. Front. Oncol. 13:1167266. doi: 10.3389/fonc.2023.1167266
Received: 16 February 2023; Accepted: 04 May 2023;
Published: 18 May 2023.
Edited by:
Liren Qian, Fifth Medical Center of the PLA General Hospital, ChinaReviewed by:
Concha Herrera, Hospital Reina Sofía de Córdoba, SpainCopyright © 2023 Wang and Metheny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leland Metheny, TGVsYW5kLk1ldGhlbnlAVUhob3NwaXRhbHMub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.