
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Oncol., 05 April 2023
Sec. Pediatric Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1167082
This article is part of the Research TopicBench to Bedside: Translating Pre-clinical Research Into Clinical Trials for Childhood Brain TumorsView all 11 articles
 Chris Jones1*
Chris Jones1* Karin Straathof2,3*
Karin Straathof2,3* Maryam Fouladi4,5Darren Hargrave3,6
Maryam Fouladi4,5Darren Hargrave3,6 Michael Prados7Adam Resnick8
Michael Prados7Adam Resnick8 Francois Doz9David T.W. Jones10,11Sabine Mueller12,13*
Francois Doz9David T.W. Jones10,11Sabine Mueller12,13*Clinical outcomes for many childhood brain tumours remain poor, despite our increasing understanding of the underlying disease biology. Advances in molecular diagnostics have refined our ability to classify tumour types and subtypes, and efforts are underway across multiple international paediatric neuro-oncology consortia to take novel biological insights in the worst prognosis entities into innovative clinical trials. Whilst for the first time we are designing such studies on the basis of disease-specific biological data, the levels of preclincial evidence in appropriate model systems on which these trials are initiated is still widely variable. We have considered these issues between CONNECT, PNOC and ITCC-Brain, and developed a framework in which we can assess novel concepts being brought forward for possible clinical translation. Whilst not intended to be proscriptive for every possible circumstance, these criteria provide a basis for self-assessment of evidence by laboratory scientists, and a platform for discussion and rational decision-making prior to moving forward clinically.
Despite remarkable advances being made in the treatment of numerous paediatric cancers over the past 40 years (1), tumours of the central nervous system remain the biggest cause of cancer-related death in children and young adults (2). For many entities, survival rates have remained unchanged for decades, and represent a major unmet clinical need (3, 4). Previous generations of clinical trials were necessarily based on an incomplete appreciation of the unique biology of childhood brain tumour entities, and a lack of preclinical evidence in appropriate model systems to show that they were likely to be effective. The failure of these studies, therefore, can now been seen as not unexpected (5). In recognition of this devastating societal impact, over the past 10-15 years partnerships between patients/families, clinical, translational and laboratory-based scientists worldwide have dramatically improved access to tissue and funding for childhood brain tumour research, which coupled with the rapid advances in next-generation sequencing and other molecular profiling techniques, has revolutionized our understanding of the underlying biology of a plethora of childhood brain tumours (6–9).
In the recent 2021 5th Edition of the WHO CNS Classification of CNS tumours, a large proportion of entities were recognized as being of paediatric ‘type’, or occurring largely in the children and young adult populations (10). Underscoring this delineation is the integrated diagnostic approach which includes key distinguishing biological data, and the appreciation of distinct drivers of the childhood disease types and the subtypes within (11). For many of these tumours, we now have both novel targets for therapeutic development, and a framework by which these children may be stratified for clinical trial enrollment, which will lead to better response assessment in a molecularly defined context. There is still substantial uncertainty, however, around the amount and type of preclinical data that is needed to develop trials that are more likely to succeed compared to their predecessors. The key tenet for moving a concept into the clinic is a strong biological rationale, with support from robust preclinical data in appropriate model systems (12). Until recently, these have been difficult to achieve, which coupled with unselected patient populations has likely contributed to the lack of success of clinical trials for children with brain tumours. As several international paediatric neuro-oncology clinical trials consortia have emerged to address the clinical issues, we now need international consensus in developing more robust preclinical platforms to provide data packages that can be reviewed objectively and systematically prior to clinical implementation.
The present article is a result of discussions between three international paediatric neuro-oncology clinical trials consortia. Each has a slightly different focus, approach, tumour-type or discipline expertise, and geographical footprint. The groups work non-competitively to ensure access to the most promising trials in the most appropriate environments, and have a degree of overlap in key personnel and centres worldwide.
CONNECT (the COllaborative Network for Neuro-oncology Clinical Trials) is a collaborative of 18 international sites across North America, Europe, UK, and Australia, with expertise in paediatric brain tumour research and clinical trials. Its purpose is to conduct scientifically rational pilot studies to assess feasibility and early efficacy of incorporating promising novel agents to established frontline therapeutic regimens in children with newly-diagnosed, high-risk brain tumours. CONNECT serves as a clinical research organisation providing concept and protocol development, data and study management, drug shipping, and all operational support. It has a diverse portfolio of trials in different childhood brain tumour entities, partnering with multiple drug companies and foundational supporters. The clinical network is supported by an active Preclinical Group, whose goal is to provide scientific assessment of novel concepts brought to the consortium for clinical translation, and to assemble collaborative research teams to provide additional experimental data as warranted.
PNOC (Pacific Pediatric Neuro-Oncology Consortium) is an international clinical trial consortium with 22 sites in the US as well as sites in Switzerland, Israel, Netherlands, Canada and Australia with recent expansion into Germany, Egypt and India. The mission of PNOC is to develop biology driven trials and expand access to innovative therapies globally for children and young adults with brain tumors, Development of clinical trials is supported by disease specific working groups composed of clinical, translational, imaging and basic science experts spanning key entities such as high grade glioma/diffuse midline glioma; ependymoma, germinoma, medulloblastoma and craniopharyngioma amongst others. Data collected as part of PNOC trials – such as imaging and genomic data – is shared in real-time with the research community through collaboration with the Children’s Brain Tumor Network (CBTN).
ITCC Brain is the CNS tumor-specific working group of ITCC (Innovative Therapies for Children with Cancer), a consortium of over 60 expert pediatric oncology centers and 25 leading research laboratories from across Europe. ITCC Brain aims to provide a framework for bringing together biologists and clinician scientists generating cutting-edge basic and translational research findings, with clinicians in large early-phase clinical trial centers, in order to accelerate the translation of novel science into effective new treatments for children with brain tumours. ITCC Brain has a portfolio of investigator-initiated trials as well as providing support for industry-led studies, and is working to expand this portfolio through well-planned studies based on strong preclinical data. The group also works closely within the larger ITCC organization to participate in entity-agnostic, biomarker-driven studies; and the group also benefits from other ITCC-led initaitives such as the ITCC-P4 pre-clinical platform [ref] and an upcoming platform for integrating data across international pediatric precision oncology progams.
Prior to the initiation of a clinical trial, we believe a robust process should be in place to critically review extant preclinical data which support the concept, as well as the strategy for clinical implementation. The intention here is not to produce binary ‘go/no-go’ decisions, but rather to assess whether a threshold of evidence has already been passed for which the clinical need mandates the concept moving forward. This may necessarily be different for distinct target patient populations, and if certain data are felt to be lacking, constructive and realistic suggestions should be made as to how to build confidence in the approach. Independent reviewers will be asked to judge any new concept proposal based on clinical significance, trial design including embedded correlative studies (e.g. CNS penetration; molecular profiling; subtype responses etc.) as well as feasibility in the context of competing trials and available patient population to conduct the proposed study. A strong biological rationale is required, with preclinical evidence benchmarked against specific idealized guidelines, with justification for any criteria not explicitly met. In this perspective, we will focus mostly on the preclinical aspects, but stress that these data dovetail with a careful, and early inclusion of the following clinical considerations:
The concept should address a clear unmet need, and represent a novel therapeutic development. This could be in the upfront setting, when an effective standard of care (SOC) has not been developed, for tumours with an extremely short overall survival, such as paediatric-type diffuse high-grade glioma (PDHGG), in particular diffuse midline glioma (DMG); subtypes of medulloblastoma (especially Group 3/4 or SHH, TP53-altered), atypical teratoid/rhabdoid tumours (ATRT), embryonal tumours with multilayered rosettes (ETMR) and others. This could also include tumours for whom the current SOC is associated with long-term burden in terms of quality of life (QOL), such as craniopharyngioma, ependymoma etc.
The concept should clearly outline the primary, secondary and exploratory aims as well as endpoints. There ought to be a valid statistical design, with innovative models to be encouraged to maximise the information gained from a minimal number of patients. It should be clearly stated how correlative studies will be used to interpret successes and failures, with inclusion of plans for access to tumour tissue (including type of material, and regulation of storage and availability for follow-up studies), genomic profiling (either as part of the study or per SOC), and digitized histology and radiology (with access and governance details). Plans for CSF and plasma/serum collection (if feasible), and details of functional (cognitive outcomes; vision, endocrine, QOL dependent on disease subtypes) and imaging endpoints ought to be provided. There should be a strategy for obtaining post-treatment tumour tissue including autopsy collection protocols, and plans for appropriate analysis of such tissue. In addition, over the last few years efforts have been made to harmonize clinical trial endpoints across consortia which will allow for more direct comparison between different study therapies. Harmonizing correlative study endpoints and biological correlates will further inform cross trial comparison within a specific disease context.
Data should be made accessible in real time to the research community without compromising clinical trial endpoints. This is of critical importance across our consortia (and others) to ensure rapid dissemination of both positive and negative data, application of important lessons learned, and provide a means for cross-validation of results, improving ongoing trial design, and identifying appropriate patient populations for trial inclusion beyond traditional research silos.
Documentation should be provided where other compounds in the same mechanism of action class have already been evaluated clinically (or preclinically) for the given or related indications. There ought to be a plan for access of the relevant patient population within the footprint of the clinical trials consortium to which the application is submitted; there should also be an evaluation of other competing trials within the consortium’s portfolio, and that of other consortia.
We strongly recognize that there is no ‘one size fits all’ approach to the process of assessment, and that each concept should be judged on its own merits on the basis of the specific clinical need of the patient populations proposed, and the feasibility of generating the idealized preclinical data package in such a context. We therefore indicate signposts for what a strong concept proposal should ideally include, and have identified five principles that could guide assessment of a data package presented for consideration (Table 1).

Table 1 Principles of preclinical assessment.
Firstly, there should be a clearly defined target population identified, based upon the mechanism of action of the investigational agent(s). In some instances, this will be relatively wide, and may span tumour entities and genotypes, whilst in others a highly restricted set of patients may be targeted. In terms of preclinical assessment, distinction is not made on this basis, so long as convincing rationale is provided. Secondly, evidence of efficacy of the agent in multiple relevant preclinical models, both in vitro and in vivo, is sought. The term ‘relevant’ here is to be contexualised by the investigators and reviewers, and may depend on disease biology and/or the agent being tested. There is complete recognition that multiple models may not be available for all tumour types or subgroups; here explanation and justification need simply be provided for both the number and identity of models chosen. Thirdly, there should be an assessment for the therapeutic window and potential safety issues by reporting target expression in the non-tumour compartments, through analysis of either novel or published data. This is of particular relevance for agents targeting wild-type targets, and for immunotherapies. Fourthly, data should be provided showing penetration of the agent into tumour tissue at clinically relevant doses; again this could be newly-generated data by the investigators or from the literature/drug company internal data, with recognition that extrapolation of doses from in vitro assays is imperfect (13). Finally, there should be demonstration of an on-target effect of the agent at clinically achievable doses in a relevant model system. Assays developed to assess this should also be evaluated for their ability to serve as predictive biomarkers for trial inclusion and/or post-hoc response assessment.
With such overarching principles driving the initial consideration of the suitability of a new concept being ‘ready’ for clinical translation, we provide specific assessment criteria in respect of in vitro and in vivo evidence that would aid prioritization of ideas. As previously stated, it is recognized not all circumstances will allow for all criteria to be met; where they cannot, the guidelines are meant to serve as discussion points rather than reasons for exclusion (Figure 1).
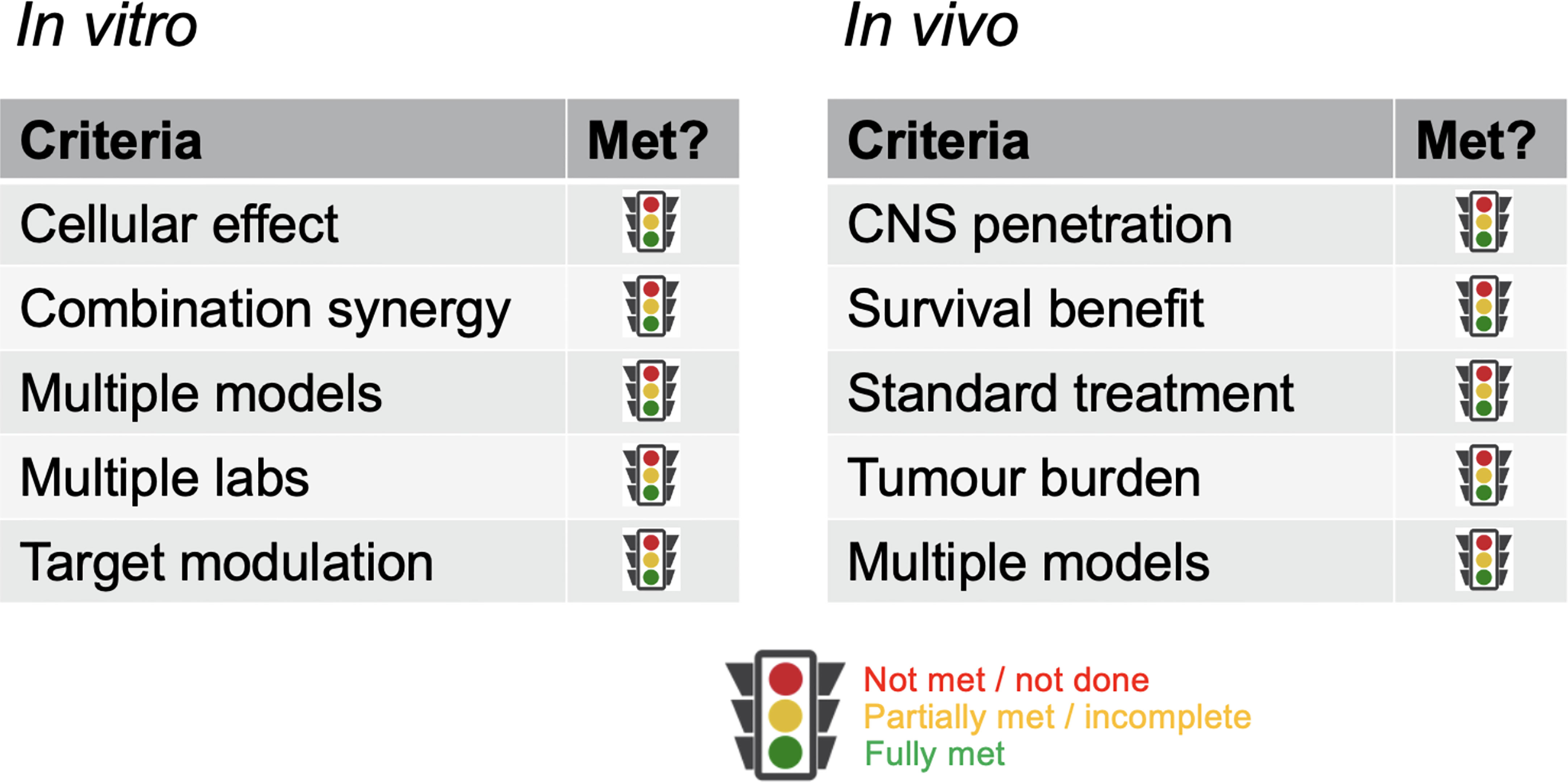
Figure 1 Criteria for critical review. Key parameters against which data packages should be assessed. Rather than a metrics-based scoring system, a fully justified benchmarking against each category should be provided.
◼ The agent to be tested should be potent in the models tested, with evidence of a clear cellular effect in terms of cell viability, cell death, cell differentiation or other appropriate end-points. This may be demonstrated in terms of effects observed (IC50/GI50 etc.) at sub-micromolar concentrations and/or showing a greater than two-fold statistically significant differential sensitivity in models representing the target population compared to (i) an appropriate ‘normal’ cell type and/or (ii) other disease subtypes and/or (iii) other disease entities.
◼ Assays should be carried out in multiple appropriately-accredited models representing the heterogeneity of the target population(s). Where available for a given entity or subtype etc., this should be carried out in at least n=4-6 distinct models, with phenotypic/genotypic data for each provided. Where available, both patient-derived and genetically-engineered models are desirable for a given target.
◼ There should be evidence of target modulation at doses producing a cellular effect by an appropriate assay, western blot, ELISA, mass spectrometry, etc.) in at least n=2 models. Although not a prerequisite for preclinical assessment, indications should be given as to the applicability of such an assay that is translatable to the clinical setting a predictive biomarker.
◼ For immunotherapies, evidence should be provided of target antigen-specific tumour cell lysis, including where possible of (primary) tumour cells with endogenous target antigen expression. Here, level of target expression reflective of that of primary tumours should be taken into account given that immunotherapeutics commonly have a target density threshold for efficacy (14).
◼ For combination studies, there should be evidence of at least additivity, or better formal synergy, of the agents to be combined by one or more appropriately designed assays, including but not restricted to the Chou-Talalay median effects model (15), BLISS independence score (16), isobologram (17) etc.). This should be carried out in at least n=2 models, where possible.
◼ Collaborative studies across laboratories are encouraged in order to demonstrate reproducibility, as well as maximise resources and expand the number of models available. Where data is pooled in such a way, at least n=1 of the models/assays should be consistently assessed across all partner laboratories in order to assess comparability.
◼ If clinical data from human studies is unavailable, demonstration of drug penetration into the relevant normal brain and/or tumour tissue of appropriate model organisms. These experiments should be carried out at tolerable doses resulting in concentrations at least greater than the in vitro IC50/GI50 values, assessed by direct measurement (using assays such as LC-MS, MALDI-TOF, etc.) and/or appropriate biomarker modulation (e.g. western blot, immunohistochemistry, etc.).
◼ For immunotherapies, if clinical data from human studies is unavailable, demonstration of homing to and penetration into tumour tissue of appropriate model organisms at tolerable doses should be provided. Presence of the immunotherapeutic agent at tumour sites can be demonstrated by immunostaining using e.g. anti-idiotype antibodies or detection of linked marker genes).
◼ There should be demonstration of a statistically significant survival benefit of treated animals, typically >20% prolongation of the median survival over vehicle (or control biologic)-treated controls. For combination studies, in addition, a statistically significant survival benefit for the combination of >10% of the median survival over the most effective single agent should be seen.
◼ As for in vitro, assays should be carried out in multiple appropriately-accredited where available. This should be undertaken at least n=3 distinct models representing the target population(s), and grown in the relevant orthotopic location where feasible. Where available, both patient-derived and genetically-engineered models, with at least n=1 in an immunocompetent background, are desirable. These could be carried out in models in the same or different species, with the latter encouraged.
◼ There should be evidence of a statistically significant reduction in tumour burden on treatment provided, assessed by an appropriate assay (e.g. MRI, bioluminescent imaging, ddPCR etc.). As with in vitro, indications should be given as to clinical translation of any predictive biomarkers.
◼ Priority will be given to treatments which can be shown to provide a survival advantage greater than SOC treatments for a given patient population, in a preclinical trial mimicking the appropriate clinical protocols (18). This could include addition and/or comparison to a standard radio/chemotherapy regimen, including surgical resection where practicable, as well as ‘mouse hospital’ designs of multiple individual patient-derived models at, e.g. n=1 mouse each (19).
Within our consortia, elements of these principles have been generically applied since initiation, but not in a systematic way. By formalizing standards, we aim to achieve two things. The first is to provide an unbiased methodology for assessing concepts brought forward from multiple sources and across disparate entities and therapeutic targets, such that cross-review between the various collaborative groups can be undertaken to the same criteria. Such a harmonized but flexible approach also seeks to provide investigators with a clear set of guidelines against which they may judge their own extant data, and help to plan additional experimental work. Inherent in this is a desire to encourage and facilitate data sharing to avoid unnecessary duplication of effort. The second critical goal is to provide a framework for discussion of novel concepts, rather than a strict metrics-based exercise. A key point is that that concepts may come from many different sources, and certainly external to any of our consortia (or others). It is the hope that having such guidelines would encourage researchers not otherwise connected to such groupings to self-evalaute their own data prior to engagement with clinical trials groups, but not in any exclusionary way; the hope is to stimulate discussion and not to restrict good ideas being brought forward at any stage.
We recognize the present limitations inherent in certain fields which make adherence to certain points impossible, and aim to highlight these caveats for frank conversation as to their importance relative to the other evidence presented, and clinical need of the target population. In this way, we also hope to flag areas that are in need of further development by the field. It should also, however, hold to account other areas in which the criteria could be, but are often not, routinely met. An example is in the desire for demonstration of efficacy and on-target effects etc. in multiple disease models. We appreciate that for certain high-risk childhood brain tumour entities such as ETMR and ATRT they may be limited (20, 21), or for others like Group 3/4 medulloblastoma they may be imperfect representations of the human disease (22). For others such as DMG however, large panels of patient-derived cells, PDXs and GEMMs are widely available, and single cell line studies are hard to justify (23–25). In all cases, we anticipate an iterative process whereby the criteria provide a checklist for early discussion, template for initial benchmarking, and a guidebook for eventual translational decision-making.
The proposed criteria are intended as positive and achievable, with the goal of leading to therapies that are more likely to be successful in the clinic (26, 27). They are meant to encompass the most common treatment concepts brought forward to our consortia, and will likely need refinement to include more innovative modalities. We do not expressly provide specific proposals for how to assess novel drug delivery methodologies, for example, including such disparate approaches as nanoparticles (28), convection-enhanced delivery (29) and focused ultrasound-mediated opening of the blood-brain barrier (30), etc. Another emerging area which may require distinct end-points clinically, and therefore unique criteria preclinically, is that of cancer neuroscience (31). We do not explicitly lay out a framework for assessing the modulation of cancer cell – neuron interactions, nor what our expectations should be in the preclinical context for such agents to have a beneficial effect in patients. The same could be said to be true of other microenvironment modulation strategies, such as targeting tumour-associated immune cells or angiogenesis (32). Here, novel model systems such as ex vivo tumour explant or organotypic models which recapitulate the complex tumour milieu are being developed to provide information complementary to that of current models (33, 34). Further refinements to our in vivo strategy will likely come to include a more thorough consideration of the age of animals used for such studies, to better replicated the developmental context in which these tumours arise (35), as well as evaluating therapies in both male and female models, given the sex-related biological differences which are beginning to emerge (36). Moreover, as we aim to develop therapies which can spare children and young adults from the toxic effects of chemotherapy and radiation, we would need to include additional means to assess how we improve QoL measures and control for the late effects of therapies (27).
As our biological understanding of paediatric CNS tumours increases, and we subclassify them into ever-more subtypes with distinct drivers warranting unique therapies (37), we face a challenge both in terms of generating preclinical data and moving these concepts into clinical trial. Idealised criteria in which we hope to see evidence of efficacy in multiple models, in vitro and in vivo, makes little sense for ultra-rare, newly-defined subtypes, and patient numbers for such entities mean traditional clinical trial designs are unlikely to recruit sufficient numbers, even with international co-operation. It will be a challenge for the community to determine to what extent we can relax or refine our standards to assess novel concepts in these entities, and how they may be robustly and safely tested in patients. We need also to be cogniscent that positive results in model systems do not necessarily predict for success in clinical trial (38–40). Although we assume that the patient-centric and biologically-driven models we have recently developed will be a substantial advance on what went before, this is as yet unproven. Careful credentialling of the models (and assays) required as part of the criteria are inherent in generating preclinical data which will eventually prove effective in the clinic, and need constant assessment and benchmarking in the manner of the criteria we apply here to the data generated with them.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
CJ, KS and SM jointly conceived, wrote and edited the article; all other authors wrote and edited the article. All authors contributed to the article and approved the submitted version.
The authors wish to thank the preclinical groups and steering committees of CONNECT, PNOC and ITCC-Brain for numerous helpful discussions relating to the development of the guidelines described in this perspective. We also like to thank the families and patients for their participation in our clinical trials and providing input in the design of these studies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
2. Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin (2014) 64(2):83–103. doi: 10.3322/caac.21219
3. Girardi F, Allemani C, Coleman MP. Worldwide trends in survival from common childhood brain tumors: A systematic review. J Glob Oncol (2019) 5:1–25. doi: 10.1200/JGO.19.00140
4. Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, et al. Cbtrus statistical report: Primary brain and other central nervous system tumors diagnosed in the united states in 2015-2019. Neuro Oncol (2022) 24(Suppl 5):v1–v95. doi: 10.1093/neuonc/noac202
5. Jones C, Karajannis MA, Jones DT, Kieran MW, Monje M, Baker SJ, et al. Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro Oncol (2016) 19:153–61. doi: 10.1093/neuonc/now101
6. Hovestadt V, Ayrault O, Swartling FJ, Robinson GW, Pfister SM, Northcott PA. Medulloblastomics revisited: Biological and clinical insights from thousands of patients. Nat Rev Cancer (2020) 20(1):42–56. doi: 10.1038/s41568-019-0223-8
7. Saleh AH, Samuel N, Juraschka K, Saleh MH, Taylor MD, Fehlings MG. The biology of ependymomas and emerging novel therapies. Nat Rev Cancer (2022) 22(4):208–22. doi: 10.1038/s41568-021-00433-2
8. Sturm D, Pfister SM, Jones DTW. Pediatric gliomas: Current concepts on diagnosis, biology, and clinical management. J Clin Oncol (2017) 35(21):2370–7. doi: 10.1200/JCO.2017.73.0242
9. Pfister SM, Korshunov A, Kool M, Hasselblatt M, Eberhart C, Taylor MD. Molecular diagnostics of cns embryonal tumors. Acta Neuropathol (2010) 120(5):553–66. doi: 10.1007/s00401-010-0751-5
10. Central nervous system tumours. 5th Edition. Lyon: International Agency for Research on Cancer (2021).
11. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 who classification of tumors of the central nervous system: A summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
12. Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol (2019) 16(8):509–20. doi: 10.1038/s41571-019-0177-5
13. Bell SM, Chang X, Wambaugh JF, Allen DG, Bartels M, Brouwer KLR, et al. In vitro to in vivo extrapolation for high throughput prioritization and decision making. Toxicol In Vitro (2018) 47:213–27. doi: 10.1016/j.tiv.2017.11.016
14. Heitzeneder S, Bosse KR, Zhu Z, Zhelev D, Majzner RG, Radosevich MT, et al. Gpc2-car T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell (2022) 40(1):53–69.e9. doi: 10.1016/j.ccell.2021.12.005
15. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul (1984) 22:27–55. doi: 10.1016/0065-2571(84)90007-4
16. Bliss CI. The calculation of microbial assays. Bacteriol Rev (1956) 20(4):243–58. doi: 10.1128/br.20.4.243-258.1956
17. Loewe S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung (1953) 3(6):285–90.
18. Nimmervoll BV, Boulos N, Bianski B, Dapper J, DeCuypere M, Shelat A, et al. Establishing a preclinical multidisciplinary board for brain tumors. Clin Cancer Res (2018) 24(7):1654–66. doi: 10.1158/1078-0432.CCR-17-2168
19. Ghilu S, Li Q, Fontaine SD, Santi DV, Kurmasheva RT, Zheng S, et al. Prospective use of the single-mouse experimental design for the evaluation of Plx038a. Cancer Chemother Pharmacol (2020) 85(2):251–63. doi: 10.1007/s00280-019-04017-8
20. Ho B, Johann PD, Grabovska Y, De Dieu Andrianteranagna MJ, Yao F, Fruhwald M, et al. Molecular subgrouping of atypical Teratoid/Rhabdoid tumors-a reinvestigation and current consensus. Neuro Oncol (2020) 22(5):613–24. doi: 10.1093/neuonc/noz235
21. Lambo S, von Hoff K, Korshunov A, Pfister SM, Kool M. Etmr: A tumor entity in its infancy. Acta Neuropathol (2020) 140(3):249–66. doi: 10.1007/s00401-020-02182-2
22. Smith KS, Bihannic L, Gudenas BL, Haldipur P, Tao R, Gao Q, et al. Unified rhombic lip origins of group 3 and group 4 medulloblastoma. Nature (2022) 609(7929):1012–20. doi: 10.1038/s41586-022-05208-9
23. Chen Z, Peng P, Zhang X, Mania-Farnell B, Xi G, Wan F. Advanced pediatric diffuse pontine glioma murine models pave the way towards precision medicine. Cancers (Basel) (2021) 13(5):1114. doi: 10.3390/cancers13051114
24. Tsoli M, Shen H, Mayoh C, Franshaw L, Ehteda A, Upton D, et al. International experience in the development of patient-derived xenograft models of diffuse intrinsic pontine glioma. J Neurooncol (2019) 141(2):253–63. doi: 10.1007/s11060-018-03038-2
25. Welby JP, Kaptzan T, Wohl A, Peterson TE, Raghunathan A, Brown DA, et al. Current murine models and new developments in H3k27m diffuse midline gliomas. Front Oncol (2019) 9:92. doi: 10.3389/fonc.2019.00092
26. Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol (2014) 32(1):40–51. doi: 10.1038/nbt.2786
27. Byer L, Kline C, Mueller S. Clinical trials in pediatric neuro-oncology: What is missing and how we can improve. CNS Oncol (2016) 5(4):233–9. doi: 10.2217/cns-2016-0016
28. Fisusi FA, Schatzlein AG, Uchegbu IF. Nanomedicines in the treatment of brain tumors. Nanomedicine (Lond) (2018) 13(6):579–83. doi: 10.2217/nnm-2017-0378
29. Aquilina K, Chakrapani A, Carr L, Kurian MA, Hargrave D. Convection-enhanced delivery in children: Techniques and applications. Adv Tech Stand Neurosurg (2022) 45:199–228. doi: 10.1007/978-3-030-99166-1_6
30. Burgess A, Shah K, Hough O, Hynynen K. Focused ultrasound-mediated drug delivery through the blood-brain barrier. Expert Rev Neurother (2015) 15(5):477–91. doi: 10.1586/14737175.2015.1028369
31. Monje M, Borniger JC, D'Silva NJ, Deneen B, Dirks PB, Fattahi F, et al. Roadmap for the emerging field of cancer neuroscience. Cell (2020) 181(2):219–22. doi: 10.1016/j.cell.2020.03.034
32. Bejarano L, Jordao MJC, Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Discovery (2021) 11(4):933–59. doi: 10.1158/2159-8290.CD-20-1808
33. Shekarian T, Zinner CP, Bartoszek EM, Duchemin W, Wachnowicz AT, Hogan S, et al. Immunotherapy of glioblastoma explants induces interferon-gamma responses and spatial immune cell rearrangements in tumor center, but not periphery. Sci Adv (2022) 8(26):eabn9440. doi: 10.1126/sciadv.abn9440
34. Pasqualini C, Kozaki T, Bruschi M, Nguyen THH, Minard-Colin V, Castel D, et al. Modeling the interaction between the microenvironment and tumor cells in brain tumors. Neuron (2020) 108(6):1025–44. doi: 10.1016/j.neuron.2020.09.018
35. Behjati S, Gilbertson RJ, Pfister SM. Maturation block in childhood cancer. Cancer Discovery (2021) 11(3):542–4. doi: 10.1158/2159-8290.CD-20-0926
36. Massey SC, Whitmire P, Doyle TE, Ippolito JE, Mrugala MM, Hu LS, et al. Sex differences in health and disease: A review of biological sex differences relevant to cancer with a spotlight on glioma. Cancer Lett (2021) 498:178–87. doi: 10.1016/j.canlet.2020.07.030
37. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA Methylation-based classification of central nervous system tumours. Nature (2018) 469–474. doi: 10.1038/nature26000
38. Lowenstein PR, Castro MG. Uncertainty in the translation of preclinical experiments to clinical trials. why do most phase iii clinical trials fail? Curr Gene Ther (2009) 9(5):368–74. doi: 10.2174/156652309789753392
39. Franshaw L, Tsoli M, Byrne J, Mayoh C, Sivarajasingam S, Norris M, et al. Predictors of success of phase ii pediatric oncology clinical trials. Oncologist (2019) 24(8):e765–e74. doi: 10.1634/theoncologist.2017-0666
Keywords: pediatric, CNS, preclinical, models, translational, in vitro, in vivo
Citation: Jones C, Straathof K, Fouladi M, Hargrave D, Prados M, Resnick A, Doz F, Jones DTW and Mueller S (2023) Evaluating preclinical evidence for clinical translation in childhood brain tumours: Guidelines from the CONNECT, PNOC, and ITCC brain networks. Front. Oncol. 13:1167082. doi: 10.3389/fonc.2023.1167082
Received: 15 February 2023; Accepted: 22 March 2023;
Published: 05 April 2023.
Edited by:
Brandon Wainwright, The University of Queensland, AustraliaReviewed by:
Magimairajan Issai Vanan, CancerCare Manitoba, CanadaCopyright © 2023 Jones, Straathof, Fouladi, Hargrave, Prados, Resnick, Doz, Jones and Mueller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chris Jones, Y2hyaXMuam9uZXNAaWNyLmFjLnVr; Karin Straathof, ay5zdHJhYXRob2ZAdWNsLmFjLnVr; Sabine Mueller, c2FiaW5lLm11ZWxsZXJAdWNzZi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.