
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 30 June 2023
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1160764
This article is part of the Research TopicAdvances and Controversies in Skull Base Tumors: Implication for Diagnosis, Treatment and ManagementView all 19 articles
 Alberto Righi1
Alberto Righi1 Stefania Cocchi1
Stefania Cocchi1 Margherita Maioli1
Margherita Maioli1 Matteo Zoli2,3
Matteo Zoli2,3 Federica Guaraldi2
Federica Guaraldi2 Elisa Carretta1Giovanna Magagnoli1Ernesto Pasquini2Sofia Melotti3
Elisa Carretta1Giovanna Magagnoli1Ernesto Pasquini2Sofia Melotti3 Gianfranco Vornetti2
Gianfranco Vornetti2 Caterina Tonon2,3Diego Mazzatenta2,3
Caterina Tonon2,3Diego Mazzatenta2,3 Sofia Asioli2,3*
Sofia Asioli2,3*Introduction: The loss of SMARCB1/INI1 protein has been recently described in poorly differentiated chordoma, an aggressive and rare disease variant typically arising from the skull base.
Methods: Retrospective study aimed at 1) examining the differential immunohistochemical expression of SMARCB1/INI1 in conventional skull base chordomas, including the chondroid subtype; 2) evaluating SMARCB1 gene deletions/copy number gain; and 3) analyzing the association of SMARCB1/INI1 expression with clinicopathological parameters and patient survival.
Results: 65 patients (35 men and 30 women) affected by conventional skull base chordoma, 15 with chondroid subtype, followed for >48 months after surgery were collected. Median age at surgery was 50 years old (range 9-79). Mean tumor size was 3.6 cm (range 2-9.5). At immunohistochemical evaluation, a partial loss of SMARCB1/INI1 (>10% of neoplastic examined cells) was observed in 21 (32.3%) cases; the remaining 43 showed a strong nuclear expression. Fluorescence in situ hybridization (FISH) analysis was performed in 15/21 (71.4%) cases of the chordomas with partial SMARCB1/INI1 loss of expression. Heterozygous deletion of SMARCB1 was identified in 9/15 (60%) cases and was associated to copy number gain in one case; no deletion was found in the other 6 (40%) cases, 3 of which presenting with a copy number gain. No correlations were found between partial loss of SMARCB1/INI1 and the clinicopathological parameters evaluated (i.e., age, tumor size, gender, tumor size and histotype). Overall 5-year survival and 5-year disease-free rates were 82% and 59%, respectively. According to log-rank test analysis the various clinico-pathological parameters and SMARCB1/INI1 expression did not impact on overall and disease free-survival.
Discussion: Partial loss of SMARCB1/INI1, secondary to heterozygous deletion and/or copy number gain of SMARCB1, is not peculiar of aggressive forms, but can be identified by immunohistochemistry in a significant portion of conventional skull base chordomas, including the chondroid subtype. The variable protein expression does not appear to correlate with clinicopathological parameters, nor survival outcomes, but still, it could have therapeutic implications.
Skull base chordomas represent a heterogeneous group of tumors, including different histotypes (i.e., conventional, chondroid, poorly differentiated and dedifferentiated types) with different clinical behavior (1–5).
SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 (SMARCB1), also known as integrase interactor 1 (INI1), is a critical component of a chromatin-remodeling protein complex (4, 6). Recent studies have described the immunohistochemical loss of SMARCB1/INI1 protein in poorly differentiated chordoma associated with SMARCB1 gene deletions at fluorescence in situ hybridization (FISH) examination, mainly deriving from large, homozygous deletions at 22q11 locus (4, 6–8). The loss of SMARCB1/INI1 protein could potentially serve as theoretical basis for evaluating the efficacy of new targeted therapies, i.e., Enhancer of Zeste homologue 2 (EZH2) inhibitors (Tazemetostat), histone deacetylase inhibitors, and CDK4 inhibitors (4, 7, 9–11).
Some studies have recently suggested the partial loss of SMARCB1/INI1 expression at immunohistochemistry as a poor prognostic marker of outcome, being associated with higher recurrence rates and shorter survival in patients with other tumor types, including colorectal, pancreatic, uterine and sinonasal carcinomas (12–16).
Genetic studies have demonstrated that conventional chordomas are characterized by very low to modest mutation burden, and are mainly characterized by large copy number loss, typically involving chromosomes 1p, 3, 9q, 10, 13, and 14, and a small number of copy number gains on chromosome 7 and 1q (4, 9). Loss of chromosome 22 and/or heterozygous deletion of SMARCB1 seems to be a rare event in conventional chordomas, although data are referred to small series (4, 17–19).
This study aimed at evaluating the differential immunohistochemical expression of SMARCB1/INI1 in conventional skull base chordoma, including chondroid subtype, and the presence of SMARCB1 gene deletion/copy number gain by FISH. Potential associations of SMARCB1/INI1 expression with different clinicopathological parameters and survival outcomes were then analyzed.
Patients with conventional - including chondroid variant - and with poorly differentiated chordoma (1, 2), naïve for surgery and radiation therapy, operated via endoscopic endonasal approach from 1998 to 2017 in a tertiary care center (Programma Neurochirurgia Ipofisi - Pituitary Unit, IRCCS Istituto delle Scienze Neurologiche di Bologna, Italy), followed by Radiation-therapy, and with a clinico-radiological follow-up ≥48 months. Formalin-fixed paraffin-embedded (FFPE) tumor tissue of adequate size and quality was required to perform morphologic, immunohistochemical and molecular evaluations. The pathologist selected the most representative tumor fragments for size and quality (i.e., maximum representation of neoplastic cells and lowest portions of extra-chordoma tissues and necrosis). All the original tumor slides were reviewed, and the diagnosis was confirmed independently by two pathologists (SA and AR) with a confirmation of immunohistochemical expression of brachyury and pan-cytokeratin AE1/AE3. Three cases of poorly differentiated chordomas diagnosed at the Programma Neurochirurgia Ipofisi-Pituitary Unit, IRCCS Istituto delle Scienze Neurologiche di Bologna, Italy, for which FFPE tissue was available, were also included. Ethical committee approval was obtained from the Comitato Etico di Area Vasta Emilia Centro on 01/04/2019 (protocol # CE-AVEC: 184/2019/OSS/AUSLBO).
The tissue was fixed in 4% buffered formalin, processed and embedded in paraffin; 4μm-thick tissue sections were then cut and heated at 58°C for 2 h. Immunohistochemical staining was performed using an automated immunostainer following the manufacturer’s guidelines (Ventana BenchMark -Ventana Medical Systems, Tucson AZ, USA) using an antibody anti-INI-1 (MRQ-27; Cell Marque), a mouse monoclonal antibody ready to use at the concentration of 0,4 µg/ml (MRQ-27; Cell Marque). Antibody detection was performed using UltraView DAB Detection Kit (Ventana Medical Systems, Tucson AZ, USA). Immunohistochemical evaluation was performed as previously described (20). The percentage of cells stained was determined evaluating all neoplastic areas in the whole of the obtained slides in each case, independently assessed by two pathologists (AR, SA). This evaluation was done visually and a comparison between immunohistochemical expression of SMARCB1/INI1 and the signals of FISH analysis was done. Immunohistochemical staining grades were defined as intact (strong nuclear staining in malignant cells), deficient (completely unstained nuclei in malignant cells), and reduced (very weak but still noticeable nuclear staining in malignant cells), using the strong staining of normal background cells as reference (20, 21). Strong homogeneous nuclear staining in the background (including inflammatory cells, stromal fibroblasts, vascular endothelial cells, and/or normal epithelial cells) served as an internal control and was considered a prerequisite for immunohistochemical interpretation. Only unequivocal staining of the nuclei in viable tumor tissue (necrotic areas were excluded) was analyzed. The evaluations were performed on 200X of magnification, evaluating the mean of SMARCB1/INI1 loss, when present, for each mm2.
FISH was performed to assess SMARCB1 gene deletion using a commercial SPEC SMARCB1/22q12 Dual colour Probe (ZytoVision, Bremerhaven, Germany), according to manufacturer’s instructions. The probe included a 545 kb sequence mapping in 22q11.23 region (ZyGreen fluorochrome labeled) harboring SMARCB1 gene, and a 335 kb sequence mapping in 22q12.1-q12.2 region (ZyOrange fluorochrome labelled) harboring KREMEN1 gene, used as internal control probe, to help in detecting chromosome 22q large deletions. As previously described (22), FISH was performed on interphase nuclei using the Histology FISH accessory kit (Dako, Glostrup, Denmark), according to the manufacturers’ protocol. Briefly, 3 μm-thick FFPE tissue sections were mounted on positively charged slides. Slides were heated overnight at 60°C, deparaffinized with xylene, and dehydrated with ethanol. Samples and probes were co-denaturated in a Dako Hybridizer (Dako, Glostrup, Denmark) at 75°C for 10 minutes and incubated overnight at 37°C. Slides were then washed in stringent solution for 10 minutes at 63°C and stained with DAPI (Vector Laboratories, Inc. Burlingame CA, USA). Signal analysis was performed in combination with SMARCB1/INI1 nuclear expression correlation. For each slide, a minimum of 100 nuclei within the marked tumor area with intact morphology were scored using an Olympus BX41 fluorescent microscope (Tokyo, Japan) at 100X of magnification. Nuclei with no signal and signals in overlapped nuclei were considered non-informative and were not analyzed to avoid truncation or overlapping artifact. The presence of two copies of the SMARCB1 gene with a 1:1 ratio with the control probe was considered as the normal copy number pattern. A heterozygous co-deletion pattern (or large deletion) was defined if one allele copy of both SMARCB1 gene and control probe were lost, with a ratio of 1:1. Copy number gain was defined as the presence of extra copies of both SMARCB1 and control probe. A Color View III CCD camera soft imaging system (Olympus) was used to capture images, then analyzed with a CytoVision imaging software version 7.5 (Leica Biosystem Richmond Inc, USA).
Disease-free Survival (DFS) was defined as the time between treatment completion and first disease relapse. Patients free from disease were censored at last follow up. Overall Survival (OS) was defined as the time between treatment completion and death or last follow-up. Descriptive statistic was used to report patient and clinical characteristics. T-test or Wilcoxon Mann-Whiney test were used to analyze continuous variables; chi-squared test or Fisher’s exact test to analyze categorical variables. Kolmogorov-Smirnov and Shapiro-Wilk test were used to verify normal distribution of continuous variables. Time-to event measures were estimated using Kaplan–Meier method, and log-rank test was used to compare different parameters. All p-values were two-sided and a p<0.05 was considered as statistically significant. All statistical analyses were performed using SAS software 9.4 (SAS Institute Inc., Cary, NC).
Sixty-five patients with conventional skull base chordoma, including 35 (53.8%) men and 30 (46.2%) women, with a median age at first surgery of 50 years old (range 9-79), were enrolled. Mean tumor size at presentation was 3.6 cm (range 2-9.5 cm) (see Table 1). Histologically, 50 (76.9%) were conventional chordomas, while 15 (23.1%) were chordomas of chondroid subtype, characterized by extracellular matrix mimicking hyaline cartilage inside physalifourous neoplastic cell proliferation in the majority of the neoplastic evaluated areas (1, 2). The mean of Ki-67 labeling index was 4% (range 1-25).
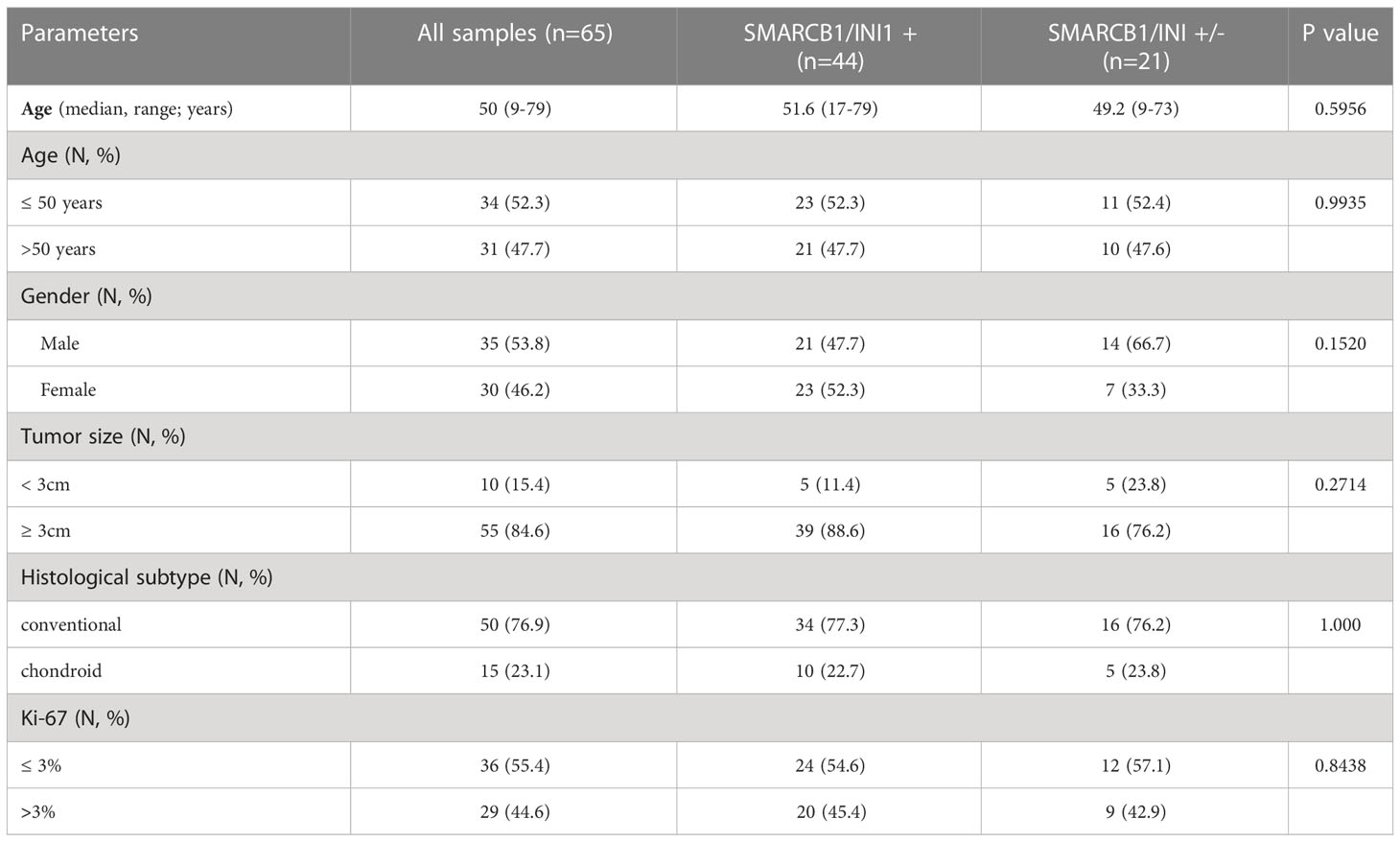
Table 1 Main clinicopathological characteristics and distribution according to SMARCB1/INI1 immunohistochemical expression.
At immunohistochemical evaluation, a partial loss of SMARCB1/INI1 (between 10% and 40% of neoplastic cells evaluated) was observed in 21 (32.3%) cases; the remaining 44 (67.7%) cases showed a strong nuclear expression in all neoplastic cells (see Supplementary File Table 1). None of conventional/chondroid chordoma cases displayed complete loss of SMARCB1/INI1 loss. Poorly differentiated chordomas presented loss of SMARCB1/INI1 in all evaluated neoplastic cells.
Conventional chordomas with focal loss of SMARCB1/INI1 displayed two different staining patterns in neoplastic areas: 13 cases showed a mosaic pattern of protein loss, with isolated single/small foci of negative cells closed to other foci of cells that retained SMARCB1/INI1 (Figure 1A); 8 cases showed protein loss in large areas, looking like ‘subclonal’ foci within the tumor (Figure 1B). No differences in clinicopathological factors between the two different staining patterns were observed. Regardless to the pattern of SMARCB1/INI1 expression, no association could be established between SMARCB1/INI1 expression and gender, age, tumor size, Ki67 and histological subtype (see Table 1). FISH analysis could be performed with a readable signal in 15/21 (71.4%) cases of conventional chordomas with a partial immunohistochemical loss of SMARCB1/INI1 expression. Six cases did not show hybridized signal due to poor tissue quality, and were thus considered inadequate for FISH scoring. FISH analysis demonstrated the presence of heterozygous deletion of SMARCB1 in 9/15 (60%) cases in over 10% of tumors cells (range 10% to 80%, Figure 2), and was associated with a copy number gain in one case. No deletion was observed in the other 6 (40%) cases, 3 of which presenting with a copy number gain of SMARCB1 (Figure 2). FISH identified homozygous SMARCB1 deletions in all 3 cases of poorly differentiated chordoma.
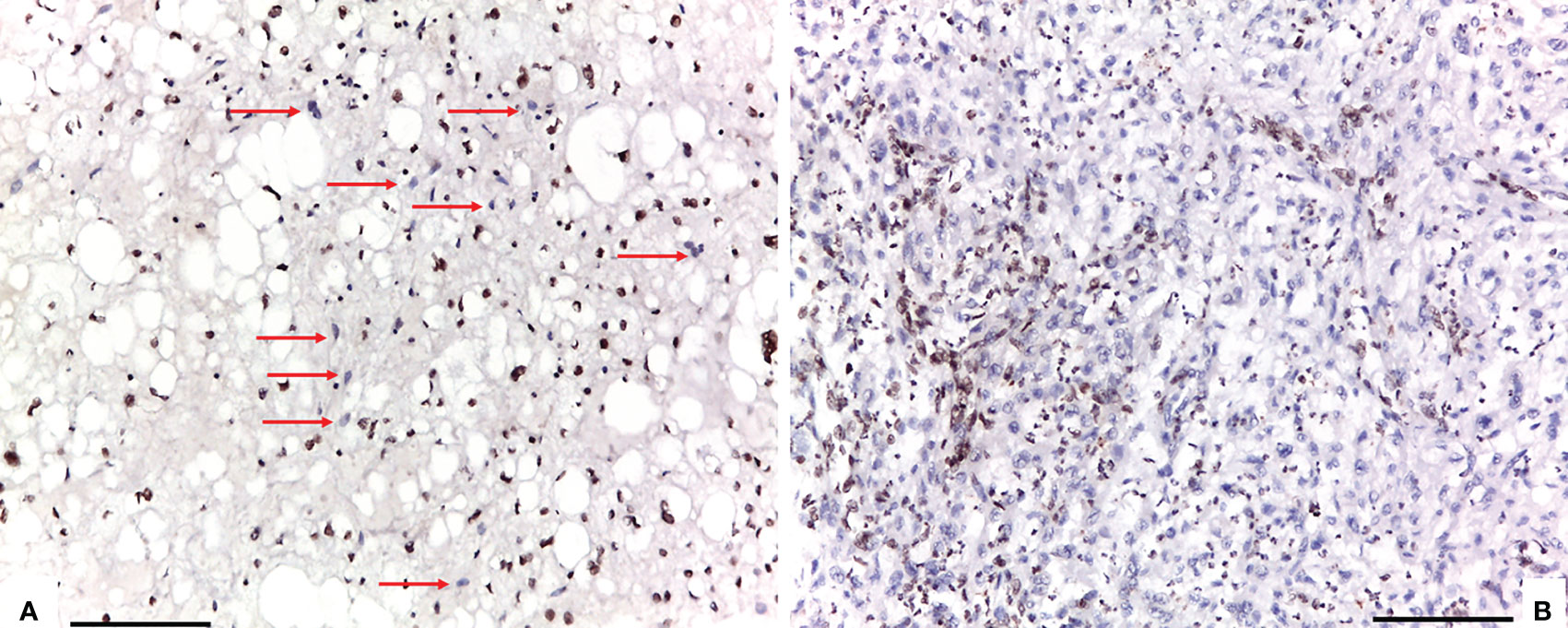
Figure 1 Two different staining patterns in neoplastic areas of focal loss of SMARCB1/INI1: (A) an example of a case that showed a mosaic pattern of protein loss, with isolated single/small foci of negative cells (red arrows) closed to other foci of cells that retained SMARCB1/INI1; (B) an example of a case that showed protein loss in large areas, looking like ‘subclonal’ foci within the tumor (A, B: 100X of magnification, Scale bar=75 μm).
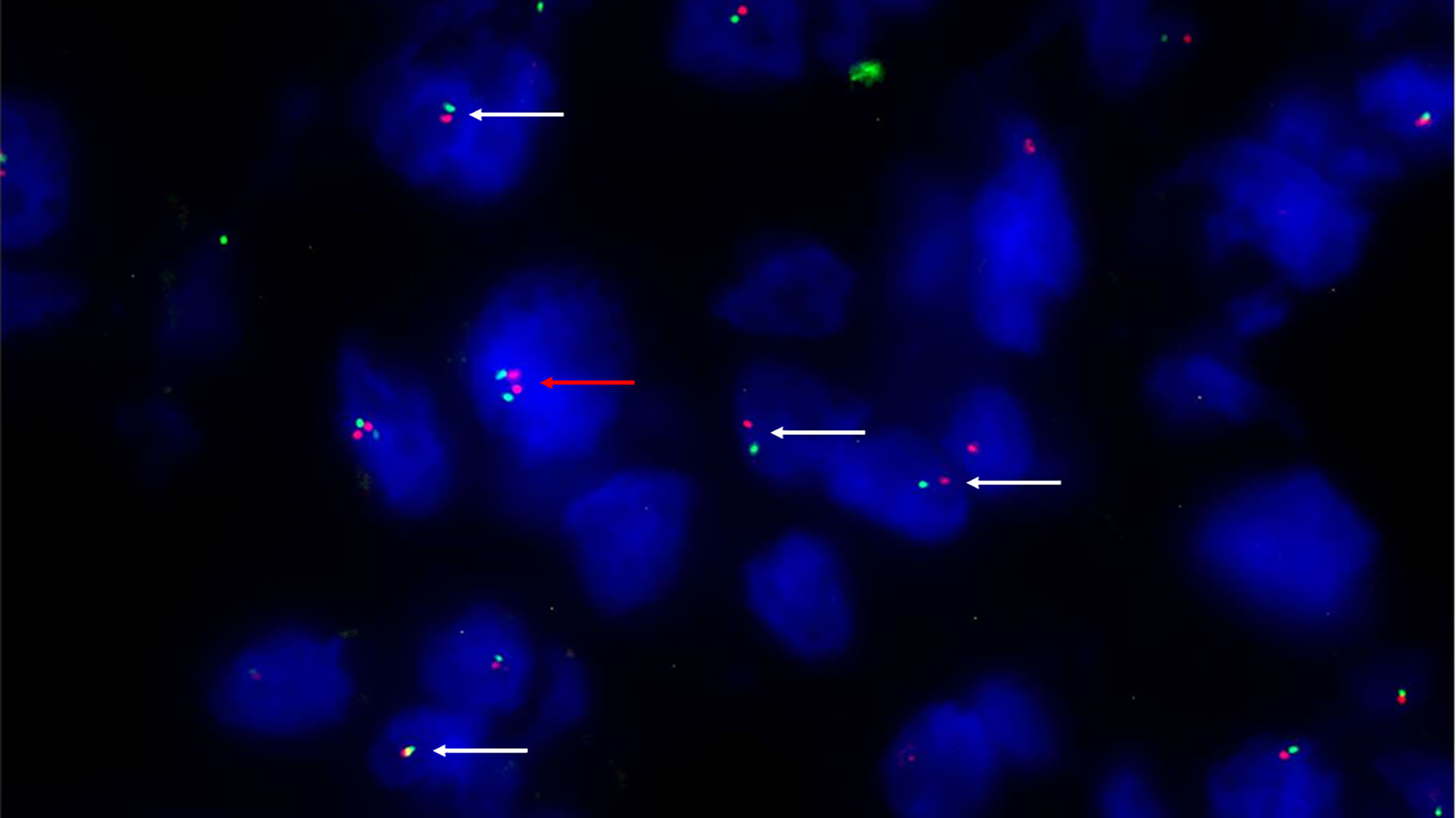
Figure 2 Fluorescence in situ hybridization (FISH) with SPEC SMARCB1/22q12 Dual Color Probe detected, in a representative tumoral area. Monoallelic co-deletion pattern: only one copy of SMARCB1/INI1 (green signal) and one copy of 22q12 (red signal) were observed in most tumor cells (white arrows). A cell without deletion is shown in the field as internal control (red arrow). (200X of magnification).
Follow-up duration after treatment completion was 80 months (range, 51-127). Overall 5-year survival and 5-year disease-free rates were 83% (95%CI: 69.9-90.5) and 59% (95%CI:44.5-71.3), respectively. Univariate analysis showed that the risk of recurrence/metastases was higher for conventional than chondroid chordoma (p=0.0281). Among all considered parameters, only the histological subtype impacted on DFS, while no predictor of OS was identified (see Tables 2, 3; Figures 3, 4).
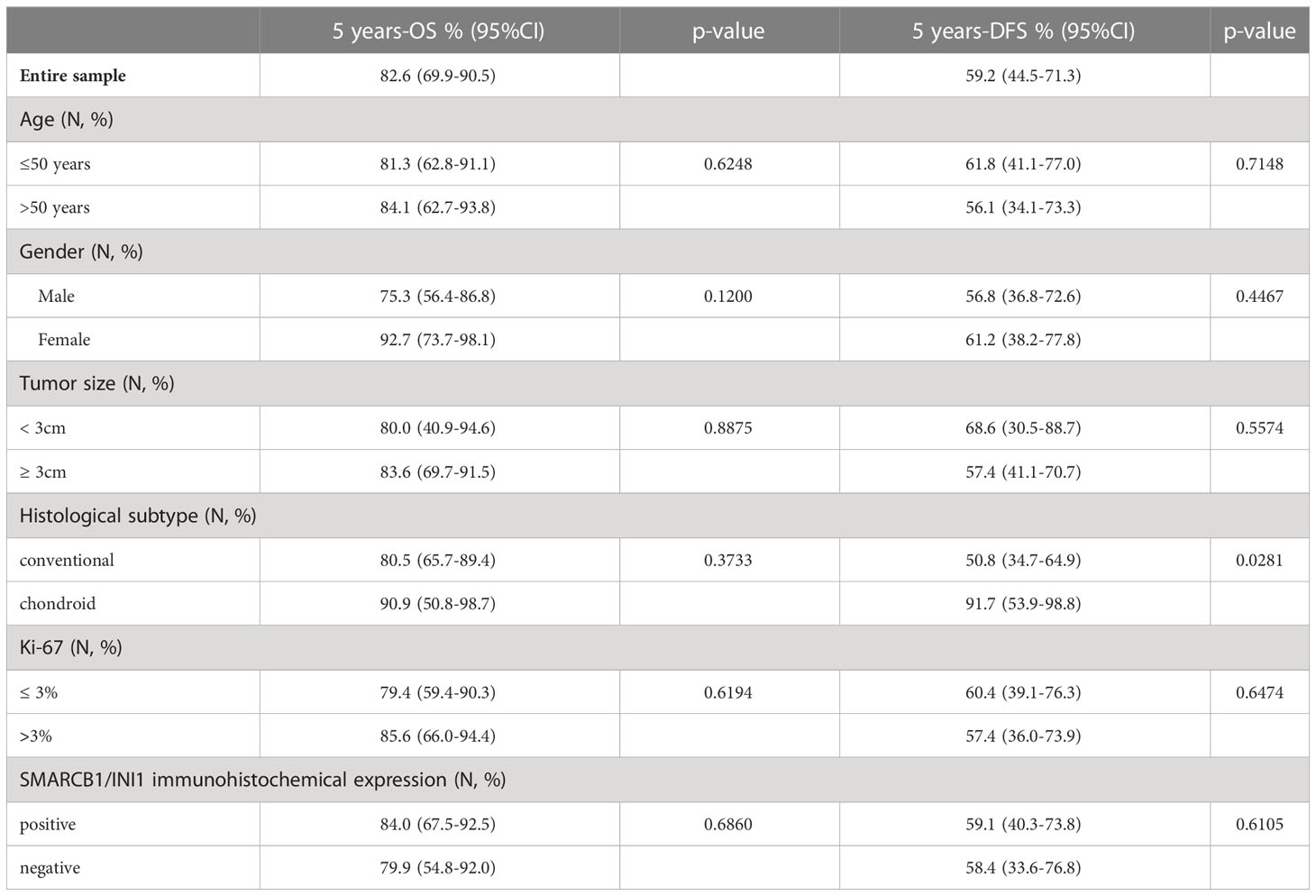
Table 2 Results from univariate Kaplan-Meier models for OS and DFS.
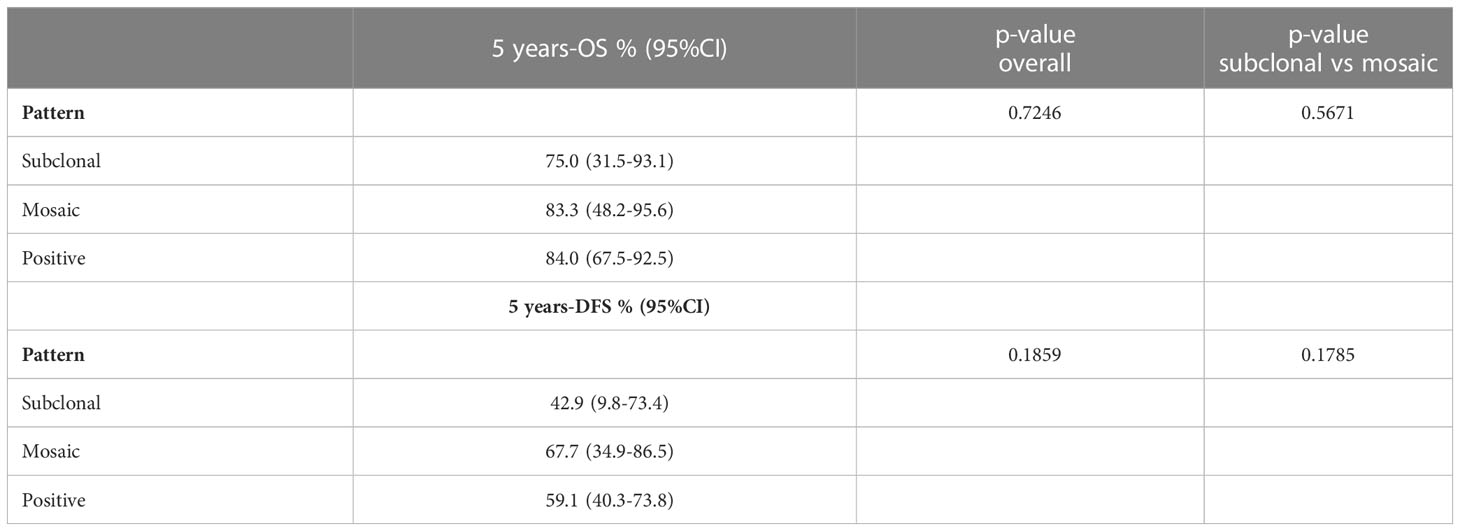
Table 3 Results from univariate Kaplan-Meier models for OS and DFS according the different pattern of partial loss of SMARCB1/INI1 by immunohistochemistry.
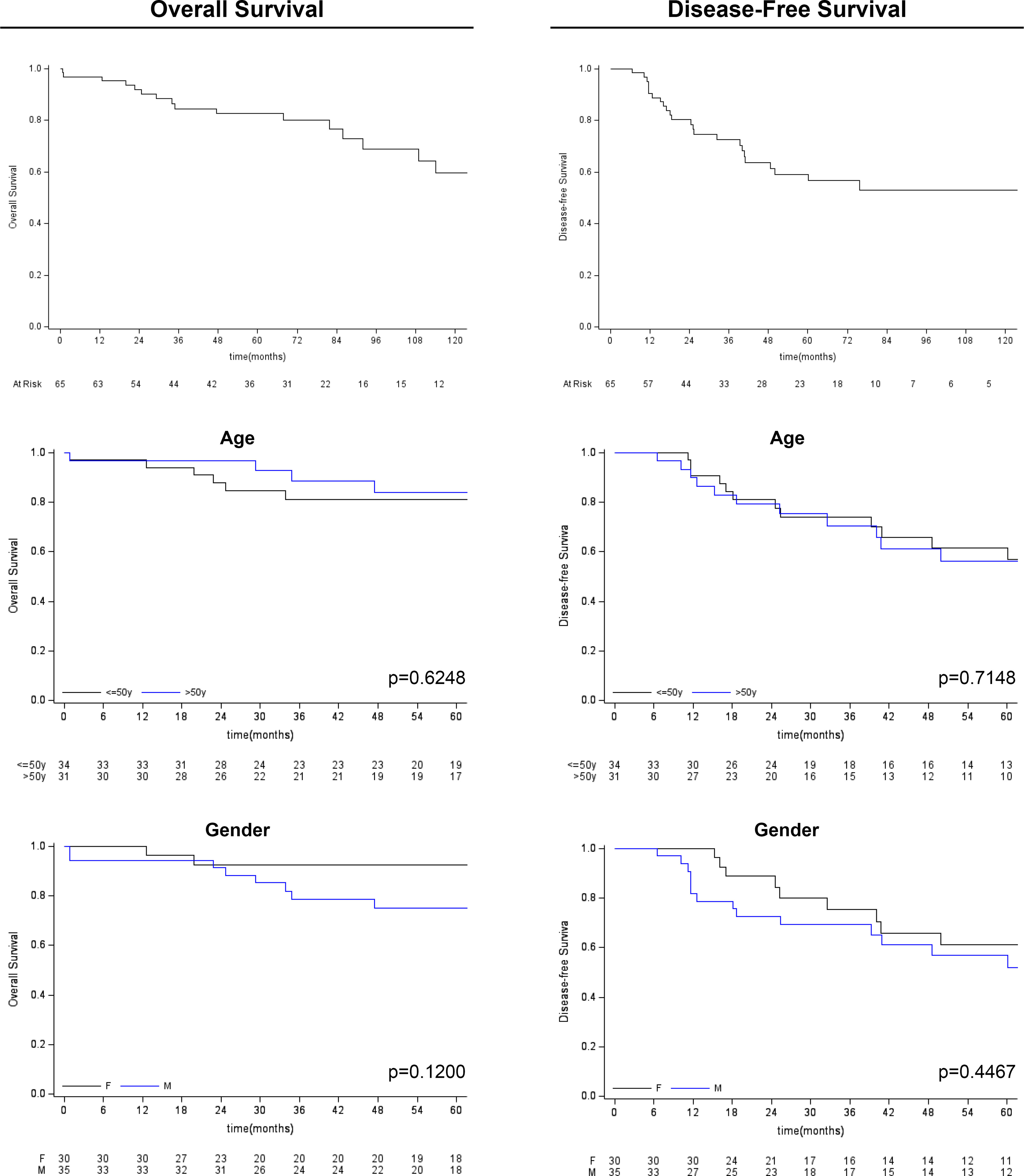
Figure 3 Kaplan-Meier survival analysis (overall survival and disease-free survival) for age and gender variables.
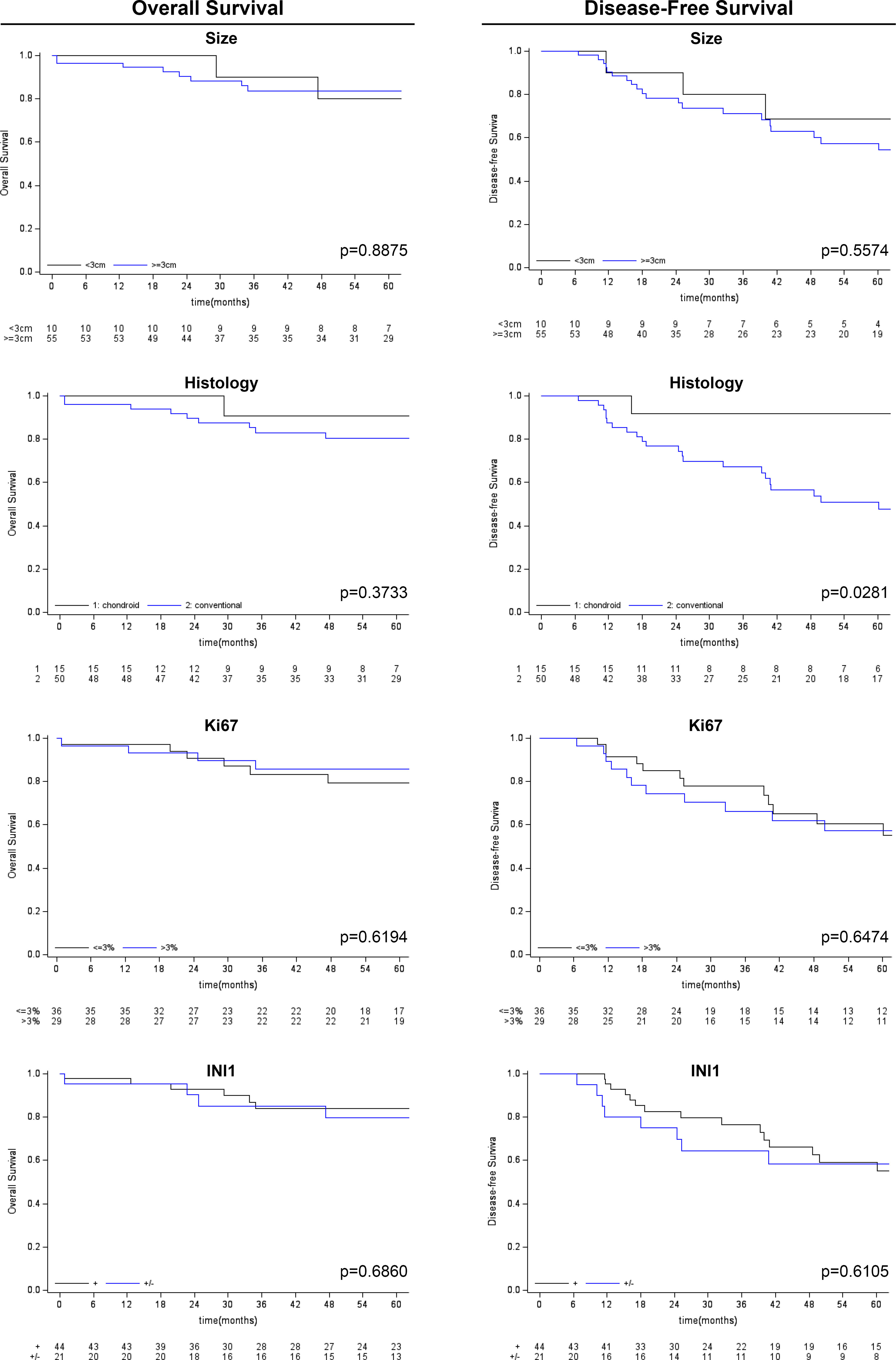
Figure 4 Kaplan-Meier survival analysis (overall survival and disease-free survival) for other clinico-pathological variables considered (tumor size, histological subtype, Ki-67 and SMARCB1/INI1 immunohistochemical expression).
Skull base chordomas represent a heterogeneous group of neoplasia, extremely difficult to be eradicated by surgical and adjuvant means, although typically slow-growing. New therapeutic targeted therapies are currently under investigation, including EZH2 inhibitors (Tazemetostat) (4, 10, 11, 23). EZH2 is a catalytic subunit of the histone methyltransferase PCR2 polycomb repressive complex whose overexpression promotes oncogenesis (24). Agents targeting EZH2 have shown to induce tumor regression and promote radiation sensitivity in models of SMARCB1/INI1-deficient tumors, including poorly differentiated chordomas, malignant rhabdoid tumors and epithelioid sarcomas (4, 10, 11). Differently from most of atypical teratoid/rhabdoid tumors, in chordomas, loss of SMARCB1/INI1 expression at immunohistochemistry results from a homozygous deletion of the SMARCB1 gene (4, 9, 25), and has been reported not only in poorly differentiated variants (in which it represents a diagnostic hallmark) (4, 6, 9, 26, 27), but also in a case of conventional chordoma with transformation to poorly differentiated chordoma (17), and in another case of conventional chordoma with dedifferentiated sarcomatous components (28).
Based on literature data, total loss of SMARCB1/INI1 immunohistochemical expression associated with the presence of a homozygous deletion of the SMARCB1 gene is correlated with aggressive clinical behavior of chordomas (8, 17, 27–29). Only few series have evaluated SMARCB1/INI1 immunohistochemical expression in association with FISH analysis in conventional chordoma. Overall, the study by Mobley et al. (30) and by Hassellbatt et al. (4) found the retention of SMARCB1/INI1 without a recurrent deletion of SMARCB1 region in 14 out of 24 cases. Conversely, Yadav et al. (27) described 2 cases of conventional chordomas with loss of immunohistochemical expression of SMARCB1/INI1 associated with loss of SMARCB1 locus, and Wen reported a single case of extra-axial conventional chordoma with a partial loss of SMARCB1/INI1 at immunohistochemistry despite no deletion of SMARCB1 detected by FISH analysis (9). Therefore, to best of our knowledge, this is the largest study aimed at investigation the incidence of SMARCB1/INI1 loss in of conventional skull base chordomas, accounting for >95% of chordomas (1, 2, 8). Immunohistochemical analysis demonstrated a partial loss of SMARCB1/INI1 in 10 to 40% of neoplastic cells in 21/65 (32.3%) cases, and no correlations between partial SMARCB1/INI1 loss and clinicopathological parameters. Furthermore, unlike poorly differentiated chordoma and other types of carcinomas (8, 12, 13, 21, 31), log-rank test analysis showed no impact of SMARCB1/INI1 expression on overall and disease free-survival.
From a molecular point of view, SMARCB1/INI1 loss of function may be caused by gene deletions, inactivating mutations, or epigenetic modifications. SMARCB1 heterozygosity is considered the main underlying mechanism and can be revealed by FISH analysis with very high sensitivity (7, 9, 32). FISH analysis on FFPE tissue in SMARCB1-deficient tumors has been proven to be a reliable test to investigate large homozygous or heterozygous deletions at 22q11.12 (33). Due to cross-hybridizations of chromosome 22 alpha satellites to other centromeric regions, probes specific for 22q12.1-q12.2 region are frequently used as control for chromosome 22 copy number detection. However, since the SMARCB1 gene and the control probe used are only 5.5 Mb away (chromosome bands 22q11.23 and 22q12.1-q12.2, respectively), secondary regional deletions may occur in SMARCB1-deleted tumors. Many studies have stated that in SMARCB1-deficient tumors large deletions covering also the EWSR1 gene locus (chromosome band 22q12.2) can occur (7, 30, 33–35), so demonstrating the deletion of a large portion of the long arm of chromosome 22. More advanced genomics and epigenetics sequencing approaches should be used in future in depth studies of chromatin modifier SMARCB1/INI1. In our series, 21 cases of conventional chordoma displayed partial loss of SMARCB1/INI1 expression at immunohistochemistry; 6 (28.6%) showed no readable signal at FISH analysis, due to the poor tissue quality. Of the remaining 15, 9 showed heterozygous large 22q deletion encompassing the entire SMARCB1 gene locus, while 6 cases had no deletion, confirming a previous observation (9). Consistent with previous studies (4, 9), homozygous SMARCB1 loss was observed in the 3 cases of poorly differentiated, but not in conventional chordoma. Interestingly, one case with SMARCB1 heterozygous deletion showed copy number gain of SMARCB1; control probe suggested a gain in the 22q region. This pattern was observed also in 3 cases without SMARCB1 deletion. Anyway, since the 12 patients with heterozygous 22q deletion and/or a copy number gain of SMARCB1 showed reduced expression of SMARCB1/INI1 at immunohistochemistry, it is possible that this partial loss of SMARCB1/INI1 is a marker of the accumulation of additional mutations as suggested by Bai et al., who reported complex copy number alterations, including also the deletion of 22q, without apparent recurrent oncogene mutations in conventional chordoma (29). Therefore, EZH2 inhibitors (Tazemetostat) may prove to be beneficial in treating conventional chordoma, as recently demonstrated in vitro and in vivo studies and in patient-derived xenograft model (23, 36). Although promising, these data are preliminary and collected retrospectively, thus need to be confirmed by larger prospective studies, possibly multicentric because of the rarity of the disease.
In conclusion, the partial loss of SMARCB1/INI1, secondary to heterozygous deletion and/or copy number gain of SMARCB1, can be identified by immunohistochemistry in a significant portion of conventional chordomas, and is not peculiar of aggressive cases. The different protein expression does not appear to correlate with clinicopathological parameters, nor survival outcomes. However, it could still have therapeutic implications.
The publication of this article was supported by the “Ricerca Corrente” funding from the Italian Ministry of Health. This work was supported by funds for selected research topics from the Fondazione CARISBO Project (#19344).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1160764/full#supplementary-material
1. WHO Classification of Tumours Editorial Board. WHO classification of tumours. In: Soft tissue and bone tumours, 5th Edition. Lyon, France: WHO Press (2020).
2. WHO Classification of Tumours Editorial Board. Central nervous system tumours. Lyon (France): International Agency for Research on Cancer (2021). (WHO classification of tumours series, 5th ed.; vol. 6). Available at: https://publications.iarc.fr/601.
3. Asioli S, Zoli M, Guaraldi F, Sollini G, Bacci A, Gibertoni D, et al. Peculiar pathological, radiological and clinical features of skull-base de-differentiated chordomas. results from a referral centre case-series and literature review. Histopathology (2020) 76(5):731–9. doi: 10.1111/his.14024
4. Hasselblatt M, Thomas C, Hovestadt V, Schrimpf D, Johann P, Bens S, et al. Poorly differentiated chordoma with SMARCB1/INI1 loss: a distinct molecular entity with dismal prognosis. Acta Neuropathol (2016) 132(1):149–51. doi: 10.1007/s00401-016-1574-9
5. Yoneoka Y, Tsumanuma I, Fukuda M, Tamura T, Morii K, Tanaka R, et al. Cranial base chordoma–long term outcome and review of the literature. Acta Neurochir (Wien) (2008) 150(8):773–8. doi: 10.1007/s00701-008-1600-3
6. Antonelli M, Raso A, Mascelli S, Gessi M, Nozza P, Coli A, et al. SMARCB1/INI1 involvement in pediatric chordoma: a mutational and immunohistochemical analysis. Am J Surg Pathol (2017) 41(1):56–61. doi: 10.1097/PAS.0000000000000741
7. Owosho AA, Zhang L, Rosenblum MK, Antonescu CR. High sensitivity of FISH analysis in detecting homozygous SMARCB1 deletions in poorly differentiated chordoma: a clinicopathologic and molecular study of nine cases. Genes Chromosomes Cancer (2018) 57(2):89–95. doi: 10.1002/gcc.22511
8. Shih AR, Cote GM, Chebib I, Choy E, DeLaney T, Deshpande V, et al. Clinicopathologic characteristics of poorly differentiated chordoma. Mod Pathol (2018) 31(8):1237–1245. doi: 10.1038/s41379-018-0002-1
9. Wen X, Cimera R, Aryeequaye R, Abhinta M, Athanasian E, Healey J, et al. Recurrent loss of chromosome 22 and SMARCB1 deletion in extra-axial chordoma: a clinicopathological and molecular analysis. Genes Chromosomes Cancer (2021) 60(12):796–807. doi: 10.1002/gcc.22992
10. Kalimuthu SN, Chetty R. Gene of the month: SMARCB1. J Clin Pathol (2016) 69(06):484–9. doi: 10.1136/jclinpath-2016-203650
11. Tarpey PS, Behjati S, Young MD, Martincorena I, Alexandrov LB, Farndon SJ, et al. The driver landscape of sporadic chordoma. Nat Commun (2017) 8(1):890. doi: 10.1038/s41467-017-01026-0
12. Agaimy A, Rau TT, Hartmann A, Stoehr R. SMARCB1 (INI1)-negative rhabdoid carcinomas of the gastrointestinal tract: clinicopathologic and molecular study of a highly aggressive variant with literature review. Am J Surg Pathol (2014) 38(07):910–20. doi: 10.1097/PAS0000000000000173
13. Agaimy A, Haller F, Frohnauer J, Schaefer I-M, Ströbel P, Hartmann A, et al. Pancreatic undifferentiated rhabdoid carcinoma: KRAS alterations and SMARCB1 expression status define two subtypes. Mod Pathol (2015) 28:248–60. doi: 10.1038/modpathol.2014.100
14. Chitguppi C, Rabinowitz MR, Johnson J, Bar-Ad V, Fastenberg JH, Molligan J, et al. Loss of SMARCB1 expression confers poor prognosis to sinonasal undifferentiated carcinoma. J Neurol Surg B Skull Base (2020) 81(6):610–9. doi: 10.1055/s-0039-1693659
15. Donner LR, Wainwright LM, Zhang F, Biegel JA. Mutation of the INI1 gene in composite rhabdoid tumor of the endometrium. Hum Pathol (2007) 38:935–9. doi: 10.1016/j.humpath.2016.12.003
16. Wang J, Andrici J, Sioson L, Clarkson A, Sheen A, Farzin M, et al. Loss of INI1 expression in colorectal carcinoma is associated with high tumor grade, poor survival, BRAFV600E mutation, and mismatch repair deficiency. Hum Pathol (2016) 55:83–90. doi: 10.1016/j.humpath.2016.04.018
17. Curcio C, Cimera R, Aryeequaye R, Rao M, Fabbri N, Zhang Y, et al. Poorly differentiated chordoma with whole-genome doubling evolving from a SMARCB1- deficient conventional chordoma: a case report. Genes Chromosomes Cancer (2021) 60(1):43–8. doi: 10.1002/gcc.22895
18. Choy E, MacConaill LE, Cote GM, Le LP, Shen JK, Nielsen GP, et al. Genotyping cancer-associated genes in chordoma identifies mutations in oncogenes and areas of chromosomal loss involving CDKN2A, PTEN, and SMARCB1. PloS One (2014) 9:e101283. doi: 10.1371/journal.pone.0101283
19. Wang L, Zehir A, Nafa K, Zhou N, Berger MF, Casanova J, et al. Genomic aberrations frequently alter chromatin regulatory genes in chordoma. Genes Chromosomes Cancer (2016) 55(7):591–600. doi: 10.1002/gcc.22362
20. Strehl JD, Wachter DL, Fiedler J, Heimerl E, Beckmann MW, Hartmann A, et al. Pattern of SMARCB1 (INI1) and SMARCA4 (BRG1) in poorly differentiated endometrioid adenocarcinoma of the uterus: analysis of a series with emphasis on a novel SMARCA4-deficient dedifferentiated rhabdoid variant. Ann Diagn Pathol (2015) 19(04):198–202. doi: 10.1016/j.anndiagpath.2015.04.001
21. Agaimy A, Hartmann A, Antonescu CR, Chiosea SI, El-Mofty SK, Geddert H, et al. SMARCB1 (INI-1)-deficient sinonasal carcinoma: a series of 39 cases expanding the morphologic and clinicopathologic spectrum of a recently described entity. Am J Surg Pathol (2017) 41(04):458–71. doi: 10.1097/PAS0000000000000797
22. Cocchi S, Gamberi G, Magagnoli G, Maioli M, Righi A, Frisoni T, et al. CIC rearranged sarcomas: a single institution experience of the potential pitfalls in interpreting CIC FISH results. Pathol Res Pract (2022) 231:153773. doi: 10.1016/j.prp.2022.153773
23. Passeri T, Dahmani A, Masliah-Planchon J, Naguez A, Michou M, El Botty R, et al. Dramatic In vivo efficacy of the EZH2-inhibitor tazemetostat in PBRM1-mutated human chordoma xenograft. Cancers (Basel) (2022) 14(6):1486. doi: 10.3390/cancers14061486
24. Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho Y-J, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell (2010) 18(4):316–28. doi: 10.1016/j.ccr.2010.09.006
25. Walhart TA, Vacca B, Hepperla JA, Hamad SH, Petrongelli J, Wang Y, et al. SMARCB1 loss in poorly differentiated chordomas drives tumor progression. Am J Pathol (2023) 16:S0002–9440(23)00028-7. doi: 10.1016/j.ajpath.2022.12.012
26. Rekhi B, Michal M, Ergen FB, Roy P, Puls F, Haugland HK, et al. Poorly differentiated chordoma showing loss of SMARCB1/INI1: clinicopathological and radiological spectrum of nine cases, including uncommon features of a relatively under-recognized entity. Ann Diagn Pathol (2021) 55:151809. doi: 10.1016/j.anndiagpath.2021.151809
27. Yadav R, Sharma MC, Malgulwar PB, Pathak P, Sigamani E, Suri V, et al. Prognostic value of MIB-1, p53, epidermal growth factor receptor, and INI1 in childhood chordomas. Neuro Oncol (2014) 16(3):372–81. doi: 10.1093/neuonc/not228
28. Hung YP, Diaz-Perez JA, Cote GM, Wejde J, Schwab JH, Nardi V, et al. Dedifferentiated chordoma: clinicopathologic and molecular characteristics with integrative analysis. Am J Surg Pathol (2020) 44(9):1213–23. doi: 10.1097/PAS.0000000000001501
29. Bai J, Shi J, Li C, Wang S, Zhang T, Hua X, et al. Whole genome sequencing of skull-base chordoma reveals genomic alterations associated with recurrence and chordoma-specific survival. Nat Commun (2021) 12(1):757. doi: 10.1038/s41467-021-21026-5
30. Mobley BC, McKenney JK, Bangs CD, Callahan K, Yeom KW, Schneppenheim R, et al. Loss of SMARCB1/INI1 expression in poorly differentiated chordomas. Acta Neuropathol (2010) 120(6):745–53. doi: 10.1007/s00401-010-0767
31. Yeter HG, Kosemehmetoglu K, Soylemezoglu F. Poorly differentiated chordoma: review of 53 cases. APMIS (2019) 127(9):607–15. doi: 10.1111/apm.12978
32. Dermawan JK, Singer S, Tap WD, Nacev BA, Chi P, Wexler LH, et al. The genetic landscape of SMARCB1 alterations in SMARCB1-deficient spectrum of mesenchymal neoplasms. Mod Pathol (2022) 35(12):1900–9. doi: 10.1038/s41379-022-01148-x
33. Huang SC, Zhang L, Sung YS, Chen CL, Kao YC, Agaram NP, et al. Secondary EWSR1 gene abnormalities in SMARCB1-deficient tumors with 22q11-12 regional deletions: potential pitfalls in interpreting EWSR1 FISH results. Genes Chromosomes Cancer (2016) 55(10):767–76. doi: 10.1002/gcc.22376
34. Le Loarer F, Zhang L, Fletcher CD, Ribeiro A, Singer S, Italiano A, et al. Consistent SMARCB1 homozygous deletions in epithelioid sarcoma and in a subset of myoepithelial carcinomas can be reliably detected by FISH in archival material. Genes Chromosomes Cancer (2014) 53(6):475–86. doi: 10.1002/gcc.22159
35. Cha YJ, Hong CK, Kim DS, Lee SK, Park HJ, Kim SH. Poorly differentiated chordoma with loss of SMARCB1/INI1 expression in pediatric patients: a report of two cases and review of the literature. Neuropathology (2018) 38(1):47–53. doi: 10.1111/neup.12407
Keywords: skull base, chordoma, prognosis, SMARCB1/INI1, FISH analysis
Citation: Righi A, Cocchi S, Maioli M, Zoli M, Guaraldi F, Carretta E, Magagnoli G, Pasquini E, Melotti S, Vornetti G, Tonon C, Mazzatenta D and Asioli S (2023) SMARCB1/INI1 loss in skull base conventional chordomas: a clinicopathological and molecular analysis. Front. Oncol. 13:1160764. doi: 10.3389/fonc.2023.1160764
Received: 07 February 2023; Accepted: 20 June 2023;
Published: 30 June 2023.
Edited by:
Haotian Zhao, New York Institute of Technology, United StatesReviewed by:
Kamil Krystkiewicz, Copernicus Memorial Hospital, PolandCopyright © 2023 Righi, Cocchi, Maioli, Zoli, Guaraldi, Carretta, Magagnoli, Pasquini, Melotti, Vornetti, Tonon, Mazzatenta and Asioli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sofia Asioli, c29maWEuYXNpb2xpM0B1bmliby5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.