 Lingyan Wang
Lingyan Wang Vipin Shankar Chelakkot1
Vipin Shankar Chelakkot1 Kensuke Hirasawa
Kensuke Hirasawa
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 02 October 2023
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1156763
This article is part of the Research TopicRoles of Pan-cell Death of Tumor Cells in Regulating Tumor Immune MicroenvironmentView all 14 articles
Background: Inflammatory cell death is a form of programmed cell death (PCD) that induces inflammatory mediators during the process. The production of inflammatory mediators during cell death is beneficial in standard cancer therapies as it can break the immune silence in cancers and induce anticancer immunity. Photodynamic therapy (PDT) is a cancer therapy with photosensitizer molecules and light sources to destroy cancer cells, which is currently used for treating different types of cancers in clinical settings. In this study, we investigated if PDT using 5-aminolevulinic (5-ALA-PDT) causes inflammatory cell death and, subsequently, increases the immunogenicity of cancer cells.
Methods: Mouse breast cancer (4T1) and human colon cancer (DLD-1) cells were treated with 5-ALA for 4 hours and then irradiated with a light source. PCD induction was measured by western blot analysis and FACS. Morphological changes were determined by transmission electron microscopy (TEM). BALB/c mice were injected with cell-free media, supernatant of freeze/thaw cells or supernatant of PDT cells intramuscular every week for 4 weeks and then challenged with 4T1 cells at the right hind flank of BALB/c. Tumor growth was monitored for 12 days.
Results: We found that 5-ALA-PDT induces inflammatory cell death, but not apoptosis, in 4T1 cells and DLD-1 cells in vitro. Moreover, when mice were pretreated with 5-ALA-PDT culture supernatant, the growth of 4T1 tumors was significantly suppressed compared to those pretreated with freeze and thaw (F/T) 4T1 culture supernatant.
Conclusion: These results indicate that 5-ALA-PDT induces inflammatory cell death which promotes anticancer immunity in vivo.
Photodynamic therapy (PDT) is a minimally invasive treatment to destroy cancer cells by combining treatment with photosensitizers (PSs) and light exposure at an appropriate wavelength (1). PSs accumulated in cancer cells absorb light energy and are excited from a ground singlets state to an excited singlet state, followed by a transition to a relatively long-lived excited triplet state (2). At the excited triplet state, the PSs initiate type I and type II photosensitized oxidation reactions. In the type I reaction, PSs transfer electrons to neighbouring biomolecules to produce reactive oxygen species (ROS). Moreover, type II reactions generate singlet oxygen by direct interaction of PSs with molecular oxygen which produces a variety of ROS. The production of ROS plays a primary role in causing cancer cell death induced by PDT (3).
5-aminolevulinic acid (5-ALA) is a natural amino acid which can lead to the generation of protoporphyrin IX (PpIX) via the heme biosynthesis pathway (4). PpIX is further converted into heme in the mitochondria by ferrochelatase (FECH) (5). As the heme biosynthesis pathway is highly activated in transformed cells, PpIX, which is a photosensitizer, can be accumulated in most cancer cells more efficiently than in normal cells when treated with exogenous 5-ALA (6). Accumulated PpIX in cancer cells can be utilized during PDT to induce ROS when excited with light at the specific wavelength, leading to cancer cell death. PDT using 5-ALA (5-ALA-PDT) has been established as a promising alternative to conventional cancer therapies for treating different types of cancers such as bladder, brain, colon, head and neck, lung, oesophagus, oral and skin (7–11). Additionally, PpIX is an endogenous fluorophore that exhibits red fluorescence, which can be used for intraoperative visualization of tumors known as photodynamic diagnosis (PDD) (12). PDD using 5-ALA (5-ALA-PDD) is recently approved for an optical imaging method for Grade III and IV gliomas during surgery by the FDA (13).
Most cancer cells die by activation of programmed cell death (PCD) mechanisms during cancer treatments (Figure 1) (14, 15). Apoptosis, the first identified PCD, is characterized by chromatin condensation and DNA fragmentation, which is considered as non-immunogenic cell death (16, 17). Apoptosis can be triggered by the stimulation of death receptors (extrinsic pathway) or by various internal or external stimulation such as cell damage, DNA damage, cellular stresses, and irradiation (intrinsic pathway). Upon activation of the extrinsic pathway by binding of death ligands with death receptors, Fas-associate death domain (FADD) and caspase-8 form a death-inducing signaling complex to cleave caspase-8 (18). Activation of the intrinsic pathway leads to the release of cytochrome C in the mitochondria, which induces the apoptosome to cleave caspase-9 (19). Both activated caspases-8 and 9 cleave caspase-3, 6 and 7, and subsequently causing the formation of apoptotic bodies that lead to cell death (20). Calreticulin (CRT), which is known as another apoptosis marker, is translocated from the endoplasmic reticulum (ER) to the cellular membrane when cells undergo apoptosis, which stimulates phagocytosis (21). On the other hand, pyroptosis and necroptosis are classified as inflammatory cell death that produce cytokines and inflammatory mediators during their PCD process (22, 23). For induction of pyroptosis, danger signals derived from host cells or intracellular pathogens induce the formation of the inflammasome, which cleaves and activates caspase-1 (24). Activated caspase-1 further cleaves gasdermine D, which translocates to the cell membrane and forms pores to release intracellular contents and induce membrane rupture (25). Activated caspase-1 also cleaves the precursors of interleukin-1β (IL-1β) and IL-18 into their active forms, which are released during pyroptosis (26). Necroptosis is defined as a programmed form of necrosis. Necroptosis can be induced by a variety of stimulation such as activation of Toll-like receptor (TLR), tumor necrosis factor (TNF), cellular stresses, and microbial or viral infection (27). As a classic necroptosis inducer, TNF binds with the TNF receptor, which recruits the receptor interacting protein kinases (RIPK1) and TNFRSF1A associated via death domain (TRADD), inducing the phosphorylation of RIPK1 and RIPK3 (28). The phosphorylated complex of RIPK1 and RIPK3 phosphorylates MLKL (29). Alternatively, microbial or viral infection and cellular stresses that phosphorylates RIPK3 through Z-DNA binding protein 1(ZBP1) (30) or TIR-domain-containing adapter-inducing interferon-β (TRIF) (31). Phosphorylated MLKL translocates to the membrane and causes membrane rupture. Phosphorylated RIPK3 is also capable of inducing MLKL phosphorylation (32). Cells undergoing necroptosis release inflammatory cytokines, damage-associate molecular patterns (DAMPs), high mobility box 1 protein (HMGB1) and ATPs to promote inflammatory responses (33–35).
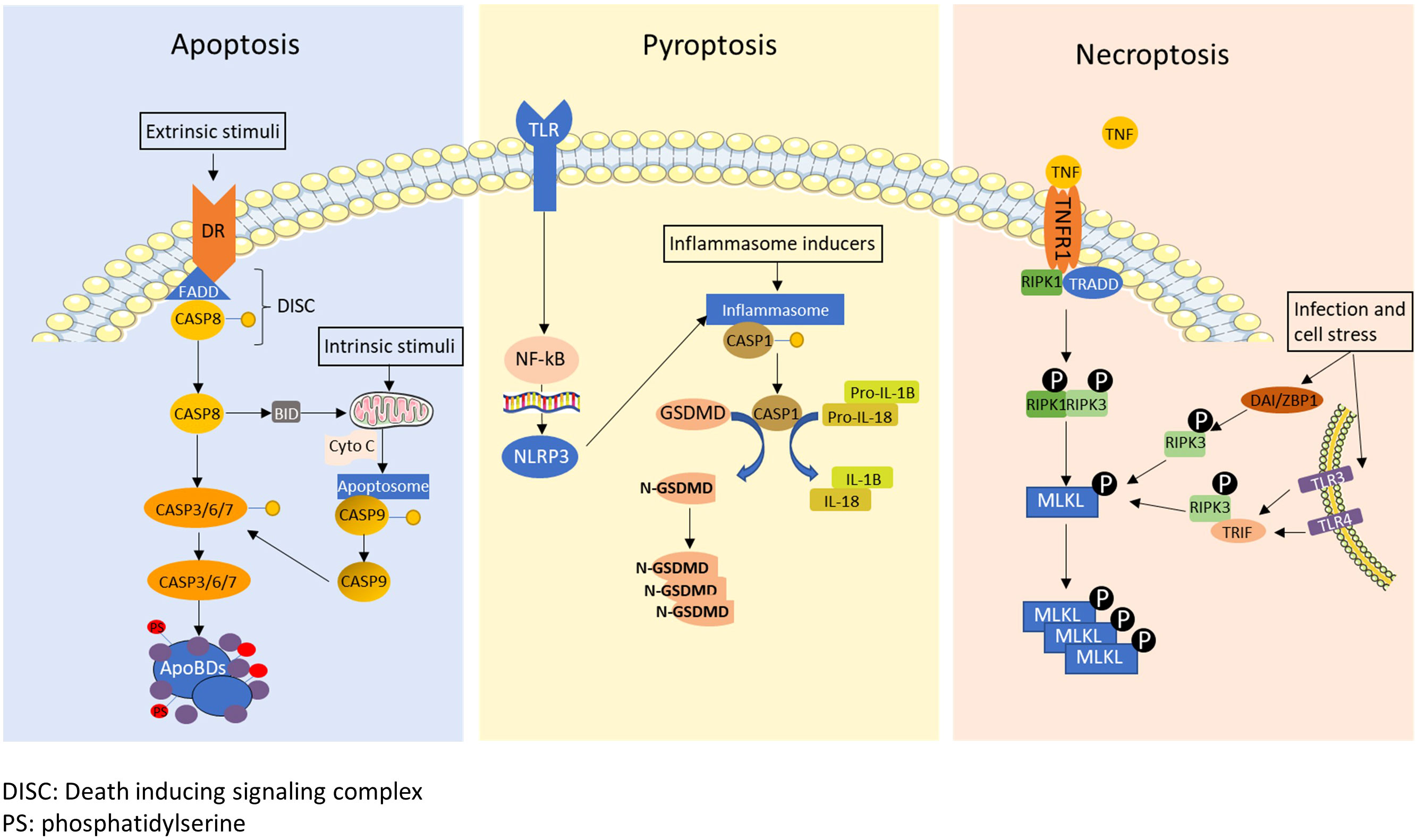
Figure 1 A schematic diagram of apoptosis, pyroptosis and necroptosis. Apoptosis: The death receptors recruit Fas-associate death domain (FADD) and caspase-8 to form death-inducing signaling complex (DISC) under extrinsic stimuli. The DISC cleaves caspase-8, and then cleaved caspase-8 initiates the formation of apoptotic bodies by activating caspase-3, 6 and 7. Caspase-8 also cleaves BH3 interacting-domain death agonist (BID), which translocate to mitochondria to induce cytochrome C (Cyto C) release. Internal stimuli such as cell damage, DNA damage, cellular stresses, and irradiation also trigger release of cytochrome C as the intrinsic pathway. Released cytochrome c forms apoptosome which cleaves caspase-9. Cleaved caspase-9 further cleaves and activates caspase-3, 6 and 7. Pyroptosis: Inflammatory signals or infections trigger pyroptosis by activating inflammasome formation. Activated inflammasome cleaves and activates caspase-1 and subsequently gasdermin D (GSDMD). Cleaved GSDMD translocates to the membrane and causes membrane rupture. Activated caspase-1 also cleave pro-IL1β and pro-IL-18 into mature IL-1β and IL-18. Necroptosis: Necroptosis is typically induced by tumor necrosis factor alpha (TNF-α), infection or cellular stress. TNF-α signal induces phosphorylation of receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and 3 (RIPK3), resulting in the subsequent phosphorylation of mixed lineage kinase domain like pseudokinase (MLKL). Moreover, MLKL can be phosphorylated through Z-DNA-binding protein 1 (ZBP1)/RIPK3, Toll-like receptors 3 (TLR3)/TIR-domain-containing adapter-inducing interferon-β (TRIF)/RIPK3 or TLR4/TRIF/RIPK3 pathway upon cellular stress and microbial or viral infection.
As cancer cells undergo multiple genetic changes, they generate mutated proteins that can be presented on their MHC class I molecules to immune cells (36). On the other hand, to avoid immune elimination, tumors evade the immune system through various mechanisms such as suppressing immune system (37), causing immune exhaustion (38), and limiting antigen presentation (39). As standard cancer therapies have been developed to directly kill cancer cells and shrink tumors, it would be beneficial for cancer survival and cure if immune evasion is broken, and anticancer immunity is induced. Cancer therapies such as chemotherapy (40), radiotherapy (41, 42) and oncolytic virus therapy (43) have been reported to activate cancer immune surveillance and anticancer immunity. To evoke anticancer immunity within tumors during cancer therapies, induction of inflammatory cell death is the crucial first step. Until now, it remains to be determined whether 5-ALA-PDT could induce inflammatory cell death and break immune silence caused by tumors though previous studies reported that 5-ALA-PDT induces apoptosis (44, 45). Here, we report that 5-ALA-PDT induces pyroptosis and necroptosis in mouse and human cancer cells, leading to the induction of anticancer immunity in an animal model of cancer.
Mouse 4T1 mammary gland tumor cells were obtained from Dr. Jean Marshall (Dalhousie University, Halifax, Canada). Human colon cancer DLD-1 cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). 5-aminolevulinic acid (5-ALA), NLRP inhibitor (MCC950), RIPK 1 inhibitor (Nec-1s), RIPK 3 inhibitor (Dabrafenib) were purchased from Sigma (Oakville, ON). Caspase-1 inhibitor (Z-WEHD-FMK) and caspase-3 inhibitor (Z-DEVD-FMK) were purchased from R&D systems (Minneapolis, MN). Cell counting Kit (WST-8/CCK8) (ab228554) (Abcam, Boston, MA) was used to determine the cell viability. LDH assay kit (Thermo Fisher Scientific, Mississauga, ON) was used to measure amounts of the lactate dehydrogenase released into the cell culture medium to determine the plasma membrane damages.
4T1 and DLD-1 cells were maintained in high glucose Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (FBS) and antibiotic-antimycotic mixture (Thermo Fisher Scientific) (100 units/mL penicillin G sodium). Cells plated in 24 or 48 well-plates were treated with 5-ALA (50 μM (4T1) or 250 μM (DLD-1)) for 4 hours and then irradiated using a Theralase TLC 3000A modular light source for 1.5 mins (Theralase Technologies Inc., Toronto, Canada; λ = 618–630 nm, fluence rate = 150 mW/cm2, energy density (ED) = 27 J/cm2). Cell viability and LDH release were measured using a CCK-8 assay kit and an LDH cytotoxicity assay kit, respectively, following the manufacturer’s instructions.
Cells were lysed using RIPA buffer supplemented with aprotinin (Sigma), and Halt Protease Inhibitor Cocktail (100X) (Thermo Scientific). Protein samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (Bio-Rad, Mississauga, ON). Primary antibodies used in this study include caspase-1 (Sant Cruz Biotechnology, Dallas, TX), cleaved caspase-1 (AdipoGen Life Science, San Diego, CA), caspase-3 (Sant Cruz Biotechnology, Dallas, TX), cleaved caspase-3 (Cell signaling technology, Danvers, MA), HMGB1 (Cell signaling technology, Danvers, MA), MLKL (Abcam, Boston, MA), phosphorylated MLKL (Abcam, Boston, MA), CRT (Abcam, Boston, MA), and GAPDH (Sant Cruz Biotechnology, Dallas, TX). The expression levels Relative expression of proteins were visualized using the corresponding secondary HRP-conjugated antibodies and Amersham ECL select western blotting detection reagent, as described previously (46).
TUNEL staining was conducted on 4T1 cells at 0.5, 1, 2 and 4 hours after 5-ALA-PDT according to the manufacturer’s protocol (Abcam; Cambridge, MA). Briefly, the cells were collected, resuspend in PBS and then fixed with 4% paraformaldehyde. The cells were labelled with Br-dUTP for 60 mins at 37°C, and then incubated in anti-BrdU-Red antibody for 30 mins. TUNEL positive cells were analyzed by flow cytometry using a BD FACSCalibur (BD Biosciences, San Jose, CA, USA).
Cells were fixed in Karnovsky fixative (2% paraformaldehyde and 2.5% Glutaraldehyde in 0.1 M sodium cacodylate buffer (Sigma-Aldrich) for 20 mins, and post-fixed in 1% osmium tetroxide (Sigma-Aldrich) dissolved in 0.1 M sodium cacodylate (Sigma-Aldrich) buffer pH 7.4. Fixed cells were dehydrated in increasing concentrations (70–100%) of ethanol and acetone, and then embedded in BEEM resin capsules and polymerized overnight at 70°C. Ultrathin sections were cut at 80-100 nm and mounted on 300 mesh copper grids, stained with lead citrate (Electron Microscopy Sciences, Hatfield, PA), and observed using an FEI Tecnai G2 Spirit transmission electron microscope (FEI, Hillsboro, OR) operating at 80 KV.
Female BALB/c mice obtained from Charles River Laboratories (Montreal, QC) were housed in a barrier unit within the central animal care facility in the Health Sciences Center at the Memorial University of Newfoundland. All animal experiments were conducted following the guidelines set by the Canadian Council on Animal Care and as approved by the Institutional Animal Care Committee of Memorial University Animal care committee. 4T1 cells were grown in DMEM with 10% FBS and antibiotics in a humidified atmosphere of 5% CO2 at 37°C. 5-ALA-PDT supernatant was collected from 4T1 cells treated with 5-ALA at the concentration of 50 μM for 4 hours prior to 1.5 mins of irradiation using a Theralase TLC 3000A modular light source. The supernatant of treated cells was collected at 24 hours after PDT treatment and centrifuged at 1300 rpm to remove the cell debris. For the freeze and thaw group, 4T1 cells were trypsinized and centrifuged to obtain cell pellets. Cells were resuspended in serum-free media and then subjected to three freeze and thaw cycles, followed by centrifugation at 1300 rpm to remove cell debris. At 8 weeks of age, female BALB/c mice randomly separated into 3 groups were injected with 30μl of cell-free media, supernatant of freeze/thaw cells, or supernatant of PDT cells intramuscular (i.m.) every week for 4 weeks. Resting for a week, at week 6, mouse mammary carcinoma 4T1 cells prepared in sterile normal saline (2x106 cells/100μl) were injected into the right hind flank of BALB/c mice. We monitored the tumor growth for 12 days before the mice were sacrificed.
Two-way ANOVA with post-hoc Tukey, Student’s t-test or Kruskal-Wallis test were performed using Prism (9.5.0) or IBM SPSS Statistics 27 as indicated in figure legends.
4T1 and DLD-1 cells were incubated with 5-ALA at concentrations of 50 μM and 250 μM for 4 hours followed by 1.5 mins light exposure, respectively. Cell viability was detected at 0.5, 1, 2, 4 and 24 hours after light treatment and LDH release was analyzed at 0.5, 1, 2 and 4 hours after light treatment for 4T1 cells and DLD-1 cells. Distinct 5-ALA-PDT conditions were used for 4T1 and DLD-1 cells as their sensitivities to 5-ALA-PDT were different. 5-ALA-PDT significantly reduced cell viability and increased LDH levels in the supernatant at 0.5, 1, 2 and 4 hours after light exposure in 4T1 cells (Figures 2A, B). Cell death was similarly induced from 0.5 hour after light exposure in DLD-1 cells (Figures 2C, D).
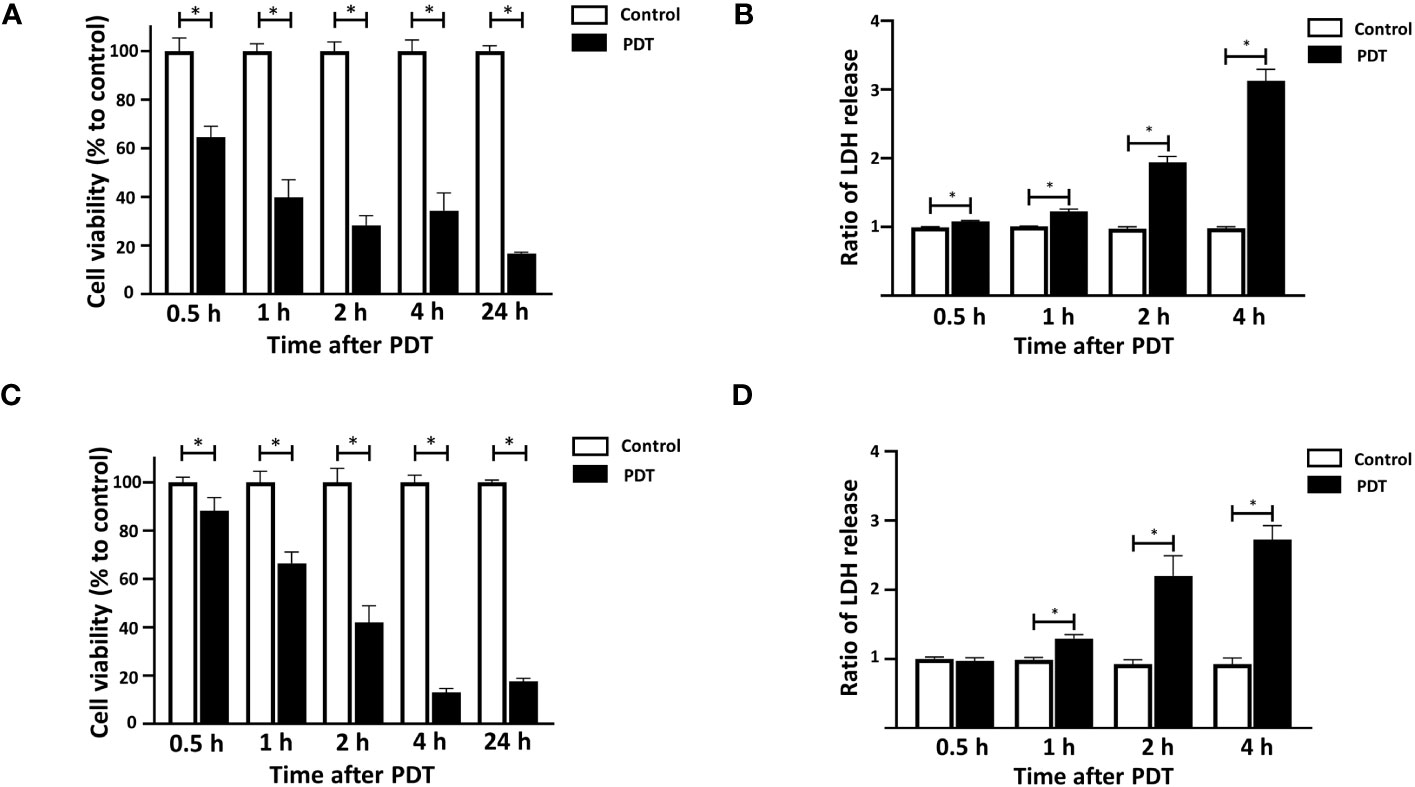
Figure 2 Time courses of 5-ALA-PDT induced cell death in 4T1 and DLD-1 cells. Mouse mammary carcinoma 4T1 cells (A, B) or human colon cancer DLD-1 cells (C, D) were treated with 5-ALA (50 μM for 4T1 and 250 μM for DLD-1) (PDT) or left untreated (Control) and for 4 hours, and then exposed to the light (618–630 nm/wavelength) for 1.5 mins. At the indicated time points after the light exposure, cell viability was measured using the CCK-8 assay kit (A, C) and LDH in culture was quantified using LDH cytotoxicity assay kit (B, D). Mean ± SD relative cell viability and LDH release to control from 4 independent experiments. *p<0.01 by student’s t-test.
To clarify if 5-ALA-PDT activates programmed cell death (PCD) pathways, we determined the expression of PCD markers such as phosphorylated MLKL (necroptosis), cleaved caspase-1 (pyroptosis) and cleaved caspase-3 (apoptosis) in 4T1 cells treated with or without 5-ALA-PDT (50 μM 5-ALA for 4 hours, 1.5 mins of light exposure). MLKL was phosphorylated at 0.5, 1, 2 and 4 hours after light exposure. 5-ALA-PDT decreased caspase-1 but increased cleaved caspase-1, indicating that caspase-1 is activated. In contrast, the apoptosis pathway was not activated as the amount of caspase-3 and cleaved caspase-3 did not show any difference between control cells and those treated with 5-ALA-PDT (Figure 3A and Supplementary Figure 1).
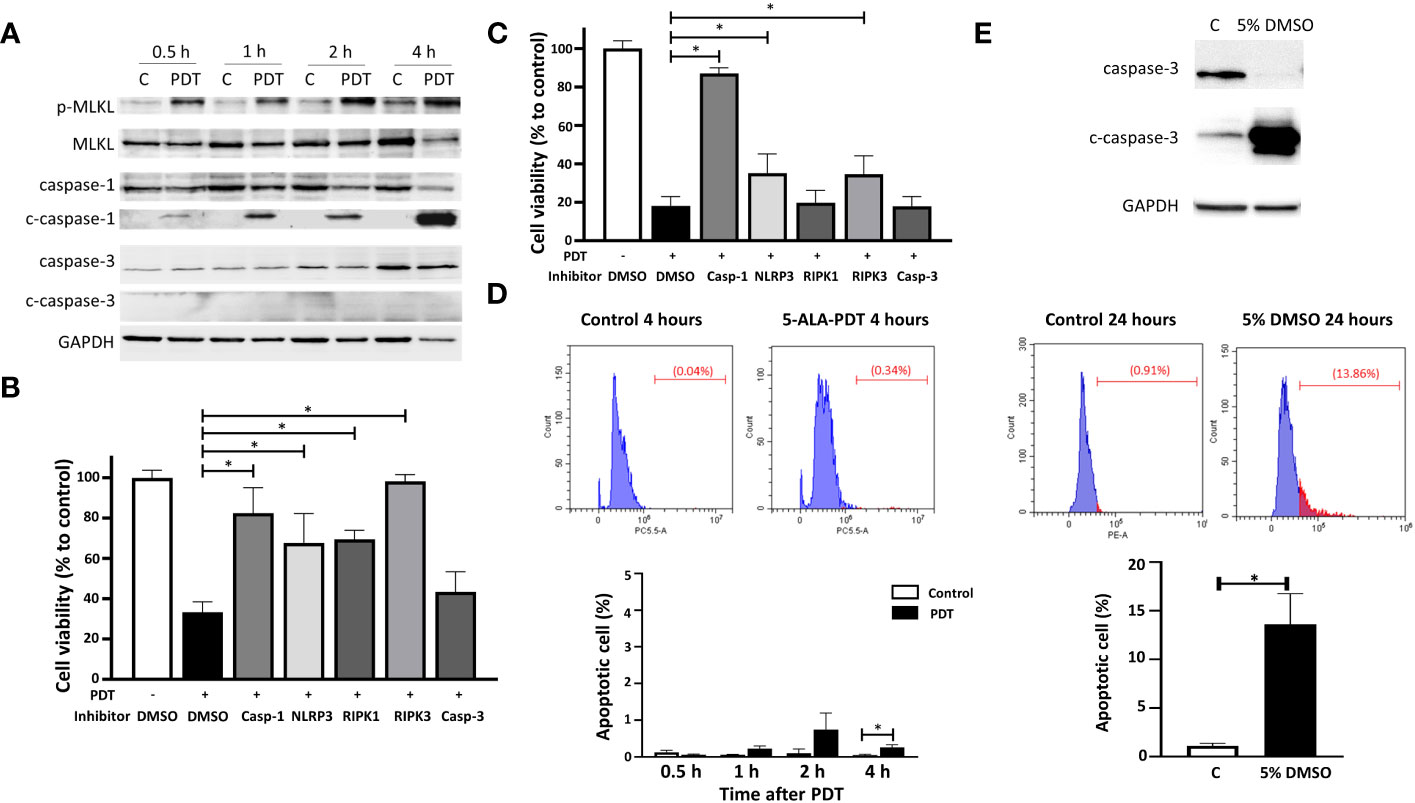
Figure 3 5-ALA-PDT causes cancer cell death via necroptosis and pyroptosis, but not apoptosis. (A) 4T1 cells were treated with 5-ALA (50 μM) (PDT) or left untreated (C) for 4 hours, and then exposed to light (618–630 nm/wavelength) for 1.5 mins. At 0.5, 1, 2, and 4 hours after light exposure, cell lysates were collected for western blot analysis using antibodies against phosphorylated MLKL (p-MLKL), MLKL, caspase-1, cleaved caspase-1 (c-caspase-1), caspase-3, cleaved caspase-3 (c-caspase-3) and GAPDH. (B) 4T1 or (C) DLD-1 cells were treated with vehicle control (DMSO), caspase-1 inhibitor (Z-WEHD-FMK, 20 μM), NLRP inhibitor (MCC950, 20 μM), RIPK1 inhibitor (Nec-1s, 20 μM), RIPK3 inhibitor (Dabrafenib, 10 μM) or caspase-3 inhibitor (Z-DEVD-FMK, 20 μM) in combination with 5-ALA treatment (50 μM (4T1) and 250 μM (DLD-1)) (PDT +) or left untreated (PDT -) for 4 hours. Cells were then exposed to light (618–630 nm/wave) for 1.5 mins. At 4 hours post light exposure, the cell viability was measured using the CCK-8 assay kit. Mean ± SD relative cell viability to control from 4 independent experiments. *p<0.01 by Two-way ANOVA. (D) 4T1 cells treated with or without 5-ALA-PDT were subjected to TUNEL staining followed by flow cytometry analysis. Mean ± SD from 3 independent experiments. *p<0.01 by the student’s t test. (E) Apoptotic 4T1 cells treated with or without 5% DMSO for 24 hours were determined by western blot analysis using antibodies against caspase-3, cleaved caspase-3 and GAPDH, and by TUNEL staining followed by flow cytometry analysis. Mean ± SD from 3 independent experiments. *p<0.01 by the student’s t test.
To determine if necroptosis and/or pyroptosis are responsible for cell death induced by 5-ALA-PDT, 5-ALA-PDT was conducted on 4T1 or DLD-1 cells in the presence or absence of the PCD inhibitors. 34% of 4T1 cells treated with vehicle control (DMSO) survived at 4 hours after 5-ALA-PDT. Treatment with caspase-1 (pyroptosis), NLRP3 (pyroptosis), RIPK1 (necroptosis) or RIPK3 (necroptosis) inhibitor significantly reduced cell death induced by 5-ALA-PDT while caspase-3 inhibitor (apoptosis) did not increase cell viability of 5-ALA-PDT treated 4T1 cells (Figure 3B). These results demonstrate that necroptosis (RIPK1 and RIPK3 dependent) and pyroptosis, but not apoptosis, are responsible for 5-ALA-PDT induced cell death in 4T1 cells. On the other hand, cell death induced by 5-ALA-PDT was reduced in DLD-1 cells treated with caspase-1, NLRP3 or RIPK3 inhibitors, while caspase-3 or PIPK1 inhibitor did not influence the cell death (Figure 3C), suggesting that necroptosis (RIPK3 dependent) and pyroptosis are responsible for 5-ALA-PDT induced cell death in DLD-1 cells. These results agree with the previous study demonstrating RIPK-dependent cell death in glioblastoma (47). To further determine apoptosis induction in 4T1 cells treated with 5-ALA-PDT, TUNEL staining followed by flow cytometry analysis was conducted (Figure 3D). We found that TUNEL positive cells induced by 5-ALA-PDT were less than 2% throughout the experiment while apoptotic cells were significantly more in 4T1 cells at 4 hours after 5-ALA-PDT than in control 4T1 cells. Moreover, we confirmed that apoptosis induction in 4T1 cells treated with 5% DMSO was detected by western blot analysis using cleaved caspase-3 antibody and TUNEL staining (Figure 3E), demonstrating that our experimental system can detect apoptotic cells efficiently.
Accumulation of CRT in the cellular membrane is observed in apoptotic or pre-apoptotic cells (48). HMGB1 is one of the inflammatory mediators released during inflammatory cell death (49). To determine whether these changes of CRT and HMGB1 were induced in 4T1 cells treated with 5-ALA-PDT, we conducted western blot analysis of CRT in the cellular cytoplasmic and membrane fractions, and of HMGB1 in the cells and the supernatants (Figure 4 and Supplementary Figure 2). As results, we did not observe the translocation of CRT from the cytoplasm to the membrane caused by 5-ALA-PDT. In contrast, HMGB1 expression was decreased in the cells and increased in the supernatant from 1 hour after 5-ALA-PDT, indicating that inflammatory cell death is induced.
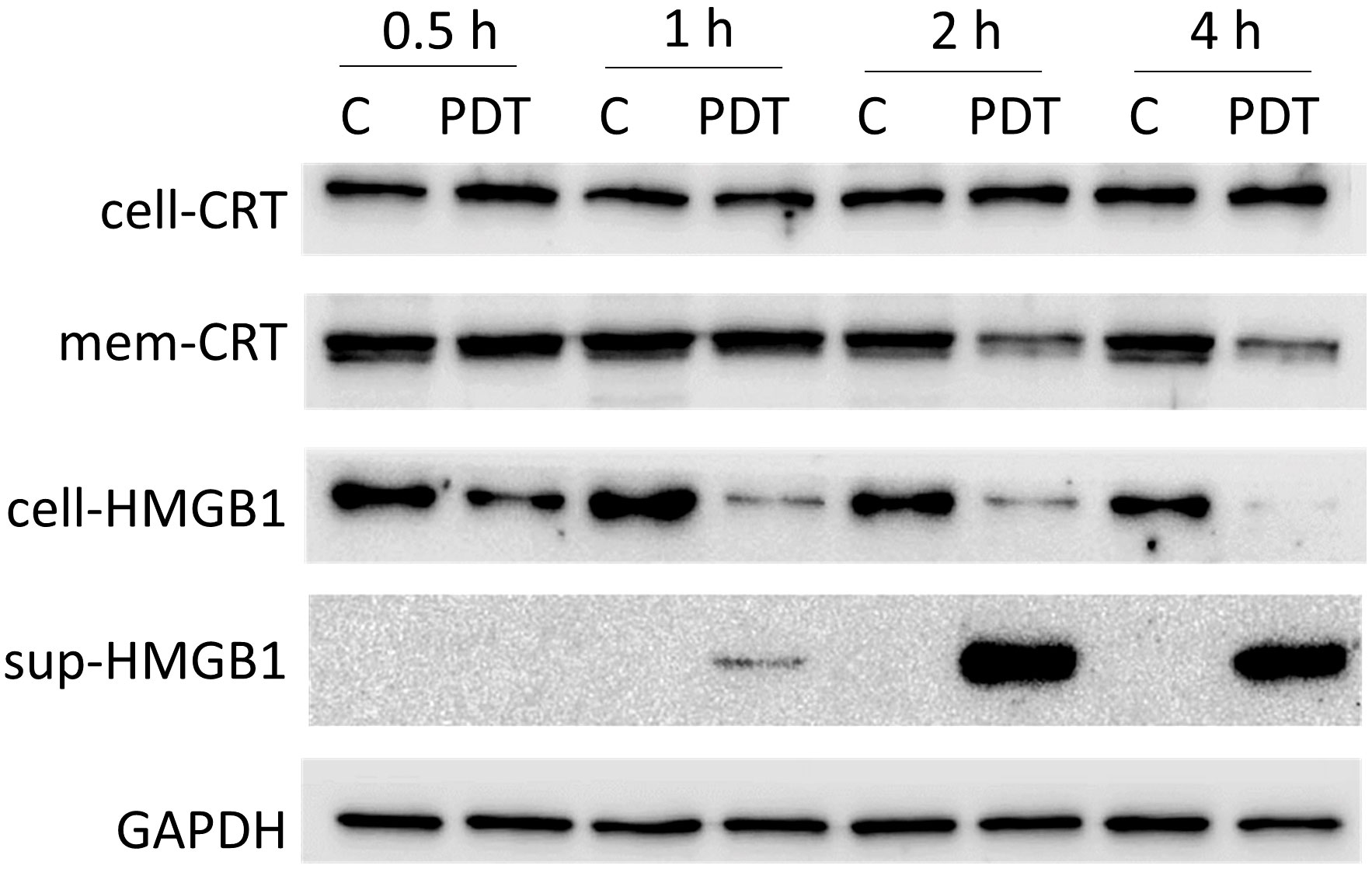
Figure 4 Expression of CRT and HMGB1 in 4T1 cells treated with 5-ALA-PDT. 4T1 cells were treated with 5-ALA (50 μM) (PDT) or left untreated (C) for 4 hours, and then exposed to light (618–630 nm/wavelength) for 1.5 mins. At 0.5, 1, 2, and 4 hours after light exposure, proteins from the cytoplasm, from the cellular membrane and in the supernatants were collected for western blot analysis using antibodies against CRT (whole cell CRT, cell-CRT; membrane CRT, mem-CRT), HMGB1 (whole cell HMGB1, cell-HMGB1; supernatant HMGB1, sup-HMGB1) and GAPDH.
Morphological changes induced by 5-ALA-PDT were examined in 4T1 and DLD-1 cells by transmission electron microscopy (TEM) (Figure 5). In both cell lines, we observed typical morphological features of pyroptosis and necroptosis such as plasma membrane permeabilization (red arrow), organelle swelling and vacuolization (green arrow), mitochondrial swelling (blue arrow) and deficiency of nuclear chromatin (yellow arrow) (50, 51). In contrast, morphological features of apoptosis such as chromatin condensation and relocation were not found in the TEM analysis. The results in Figures 3, 5 clearly suggest that 5-ALA-PDT induces pyroptosis and necroptosis but not apoptosis.
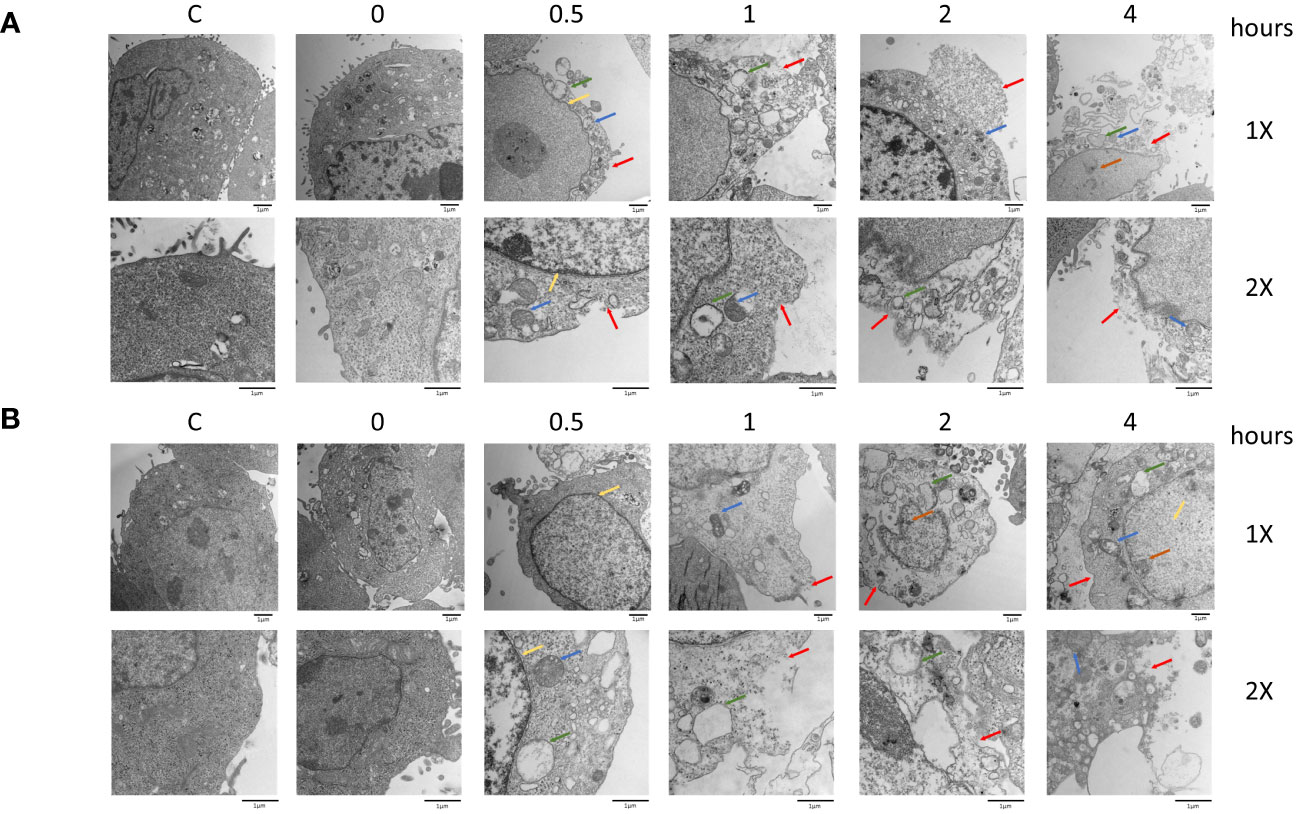
Figure 5 Morphological changes after PDT in 4T1 and DLD-1 cells. Mouse mammary carcinoma 4T1 cells (A) or human colon cancer DLD-1 cells (B) were treated with 5-ALA (50 μM for 4T1 and 250 μM for DLD-1) (PDT) or left untreated (Control) for 4 hours, and then exposed to light (618–630 nm/wavelength) for 1.5 mins. At the indicated time points after 5-ALA-PDT, morphological changes were investigated by TEM analysis. Plasma membrane permeabilization (red arrow), organelle swelling and vacuolization (green arrow), mitochondrial swelling (blue arrow), swollen nucleus (yellow arrow), and deficiency of nuclear chromatin (orange arrow) are indicated.
Next, we determined whether the inflammatory cell death initiated by 5-ALA-PDT leads to the induction of anticancer immunity in vivo. We injected female 8-weeks-old BALB/c mice i.m. with serum-free medium (Control), freeze and thaw 4T1 supernatant (F/T) or 5-ALA-PDT 4T1 supernatant (PDT) once a week for 4 weeks. Resting for a week, we then challenged mice with s.c. injections of 4T1 cells at the right hind flank (Figure 6A). Tumor growth in mice treated with PDT supernatant was inhibited compared to those treated with the cell-free medium at 10 and 12 days after 4T1 cell injection (Figure 6B). In contrast, tumor growth was significantly inhibited in mice treated with the F/T supernatant only 12 days after 4T1 cells injection. The result of tumor measurements was confirmed visually by images of tumors removed from the mice 12 days after 4T1 cell injection (Figure 6C). Moreover, we found that the weight of tumors from mice treated with PDT supernatant were significantly smaller than those from mice treated with F/T supernatant or cell free medium, while the F/T supernatant treatment did not reduce the tumor weight significantly (Figure 6D). These results demonstrate that cell death caused by 5-ALA-PDT initiates anticancer immunity in an animal model of cancer.
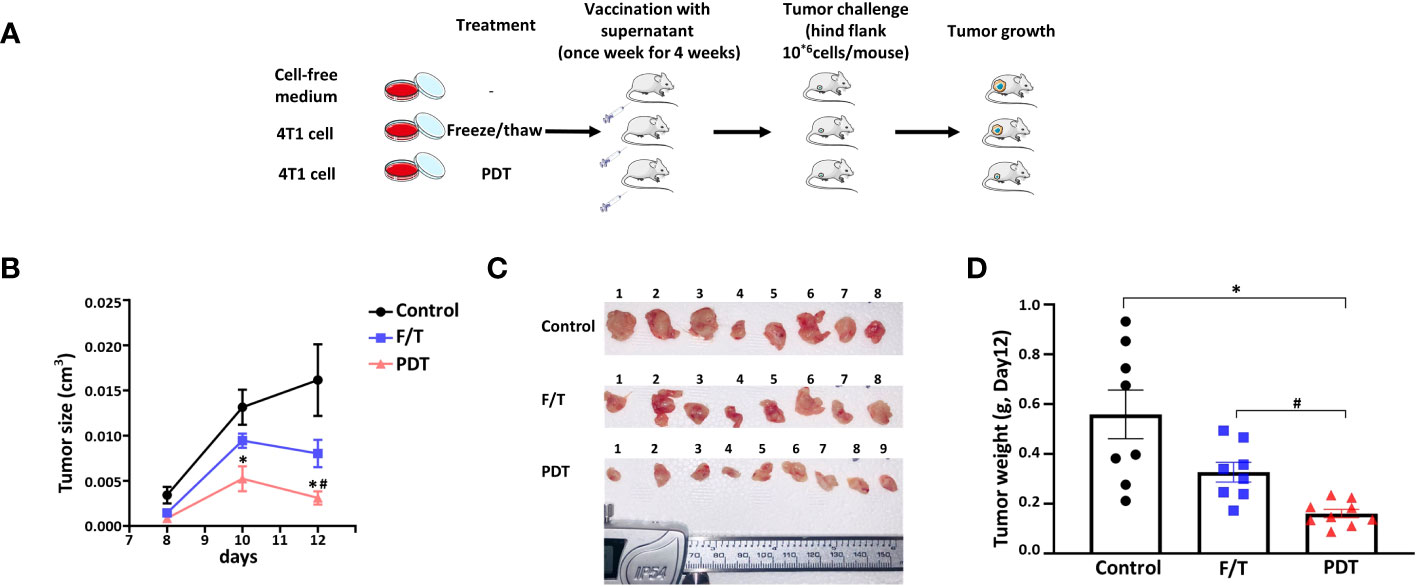
Figure 6 5-ALA-PDT stimulates antitumor immunity in mice. (A) BALB/c mice were injected intramuscular (i.m.) with 30 μl of cell-free media (Control), supernatant of freeze/thaw 4T1 cells (FT) or supernatant of 5-ALA-PDT treated 4T1 cells (PDT) every week for 4 weeks (n=8-9/group). Resting for a week, at week 6, 4T1 cells were injected (2x106 cells/100 μL) into the right hind flank of the mice. (B) Tumor measurement at 8, 10 and 12 days after 4T1 cell injection, (C) images of the tumors removed from mice at 12 days after 4T1 cell injection and (D) tumor weight at 12 days after 4T1 cell injection. *p<0.05 and # p<0.05 by Two-way ANOVA, * compared with control group, # compared with F/Z group (B) and *p<0.05 by Kruskal-Wallis test (D).
The immunosuppressive microenvironment of tumors is one of the major obstacles for cancer therapies. Standard cancer therapies, such as chemotherapy and radiotherapy, could cause inflammatory responses and break the immune silence of tumors, which may further promote therapeutic efficacies in cancer patients and lead to cancer cure (40, 52, 53). Induction of inflammatory cell death by the cancer therapies is the first essential step to establish systemic anticancer immunity. The objective of the current study was to investigate whether 5-ALA-PDT could initiate anticancer immunity by inducing inflammatory cell death of cancer cells. We found that 5-ALA-PDT causes cancer cell death by inducing pyroptosis and necroptosis, which are a form of inflammatory cell death. Moreover, the supernatant of cells treated with 5-ALA-PDT was effective to induce anticancer immunity in vivo. These results demonstrate that 5-ALA-PDT can kill cancer cells in an immunogenic manner that initiates anticancer immunity, which sheds light on a new therapeutic aspect of 5-ALA-PDT.
We used a lower amount of 5-ALA in this study than those used in our previous studies which focused on the efficacy of direct cancer cell killing by 5-ALA-PDT (5). In fact, 5-ALA-PDT does not induce inflammatory cell death when higher concentrations of 5-ALA were used (data not shown), suggesting that cancer cells undergo other types of cell death, such as apoptosis and necrosis. This finding agrees with a previous study reported by M Korbelik and his colleagues that the cancer vaccine generated by chlorin e6-based PDT was more effective to reduce mouse SCCVII squamous cell carcinoma cells when lower concentrations of the photosensitizer were used (54). It is unknown how the degree of cell damage could influence the induction of different types of cancer cell death. Nevertheless, it should be noted that 5-ALA-PDT conditions could be optimized independently for induction of cell lysis (direct cancer killing) and of anticancer immunity (breaking immune silence) in clinical settings.
Previous studies demonstrated that 5-ALA-PDT causes cancer cell death by inducing apoptosis (17, 44, 55–59). However, we did not find the expression of the apoptosis marker (cleaved caspase-3) or positive TUNEL staining in 5-ALA-PDT treated cells (Figures 3A, D) or the reduction of 5-ALA-PDT-induced cell death by the apoptosis inhibitor (Figures 3B, C). Moreover, RIPK1 was involved in 5-ALA-PDT-induced cell death of 4T1 cells but not that of DLD-1 cells (Figures 3B, C). We believe that these discrepancies may be caused by differences in potency of 5-ALA-PDT or cancer cell lines used in the studies. As mentioned above, high dosage of 5-ALA-PDT could lead to non-inflammatory cell death such as apoptosis and necrosis while low doses of 5-ALA-PDT, which kills cancer cell slowly, may be the key to induce inflammatory cell death. It is also known that the induction of the PCD is dependent on cell lines, which might lead to the differences between 4T1 and DLD-1 cells (60, 61). Furthermore, caspase-1 inhibitor increased cell viability more than NLRP3 inhibitor in 5-ALA-PDT treated DLD-1 cells (Figure 3C), suggesting that other upstream elements of caspase-1 may initiate pyroptosis in DLD-1 cells. More detailed studies are required to identify precise cellular signaling pathways of PCD activated by 5-ALA-PDT.
Supernatant from PDT treated cells was more effective to inhibit 4T1 tumor growth than the F/T supernatant, suggesting that the PDT supernatant contains components that initiate anticancer immunity (Figure 6). Oxidative stress induced by 5-ALA-PDT could generate oxidation-associated molecular patterns, which may increase the immunogenicity of 4T1 tumor cells (3, 62). Furthermore, cytokines and inflammatory mediators in PDT supernatant, which are produced during pyroptosis or necroptosis, could also promote antitumor immunity in the presence of the tumor antigens. Cytokines and inflammatory mediators induced by pyroptosis, or necroptosis include IL-1β, IL-6, IL-18, TNF-α, DAMPs and PAMPs. It is of interest to identify which of these cytokines and inflammatory mediators play critical roles in initiating anticancer immunity following 5-ALA-PDT.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was approved by Institutional Animal Care Committee. The study was conducted in accordance with the local legislation and institutional requirements.
The authors confirm their contribution to the paper as follows: study conception and design: KH and LW; data collection: LW, VC, NN, and ST; analysis and interpretation of results: LW and KH; draft manuscript preparation: LW and KH. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1156763/full#supplementary-material
Supplementary Figure 1 | Original gel images of Figure 3A. (A) phosphorylated-MLKL, (B) total MLKL, (C) total caspapse-1, (D) cleaved caspase-1, (E) total caspase-3 and (F) GAPDH.
Supplementary Figure 2 | Original gel images of Figure 4. (A) whole cell CRT, (B)membrane CRT, (C)whole cell HMGB1, (D) supernatant HMGB1 and (E) GAPDH.
1. Kwiatkowski S, Knap B, Przystupski D, Saczko J, Kedzierska E, Knap-Czop K, et al. Photodynamic therapy - mechanisms, photosensitizers and combinations. BioMed Pharmacother (2018) 106:1098–107. doi: 10.1016/j.biopha.2018.07.049
2. Poiroux G, Barre A, van Damme EJM, Benoist H, Rouge P. Plant lectins targeting O-glycans at the cell surface as tools for cancer diagnosis, prognosis and therapy. Int J Mol Sci (2017) 18(6). doi: 10.3390/ijms18061232
3. Donohoe C, Senge MO, Arnaut LHG, and Gomes-da-Silva LC. Cell death in photodynamic therapy: From oxidative stress to anti-tumor immunity. Biochim Biophys Acta Rev Cancer (2019) 1872(2):188308. doi: 10.1016/j.bbcan.2019.07.003
4. Traylor JI, Pernik MN, Sternisha AC, McBrayer SK, Abdullah KG, et al. Molecular and metabolic mechanisms underlying selective 5-aminolevulinic acid-induced fluorescence in gliomas. Cancers (Basel) (2021) 13(3). doi: 10.3390/cancers13030580
5. Chelakkot VS, Liu K, Yoshioka E, Saha S, Xu D, Licursi M, et al. MEK reduces cancer-specific PpIX accumulation through the RSK-ABCB1 and HIF-1alpha-FECH axes. Sci Rep (2020) 10(1):22124. doi: 101038/s41598-020-79144-x
6. Sansaloni-Pastor S, Varesio E, Lange N. Modulation and proteomic changes on the heme pathway following treatment with 5-aminolevulinic acid. J Photochem Photobiol B (2022) 233:112484. doi: 10.1016/j.jphotobiol.2022.112484
7. Casas A. Clinical uses of 5-aminolaevulinic acid in photodynamic treatment and photodetection of cancer: A review. Cancer Lett (2020) 490:165–73. doi: 10.1016/j.canlet.2020.06.008
8. Mahmoudi K, Garvey KL, Bouras A, Cramer G, Stepp H, Jesu Raj JG, et al. 5-aminolevulinic acid photodynamic therapy for the treatment of high-grade gliomas. J Neurooncol (2019) 141(3):595–607. doi: 10.1007/s11060-019-03103-4
9. Tan WC, Fulljames C, Stone N, Dix AJ, Shepherd N, Roberts DJ, et al. Photodynamic therapy using 5-aminolaevulinic acid for oesophageal adenocarcinoma associated with Barrett's metaplasia. J Photochem Photobiol B (1999) 53(1-3):75–80. doi: 10.1016/S1011-1344(99)00129-3
10. Kawczyk-Krupka A, Latos W, Oles P, Czuba ZP, Latos M, Krupka M, et al. The influence of 5-aminolevulinic photodynamic therapy on colon cancer cell interleukin secretion in hypoxia-like condition in vitro. Photodiagnosis Photodyn Ther (2018) 23:240–3. doi: 10.1016/j.pdpdt.2018.07.007
11. Ahn PH, Finlay JC, Gallagher-Colombo SM, Quon H, O'Malley BW Jr, Weinstein GS, et al. Lesion oxygenation associates with clinical outcomes in premalignant and early stage head and neck tumors treated on a phase 1 trial of photodynamic therapy. Photodiagnosis Photodyn Ther (2018) 21:28–35. doi: 10.1016/j.pdpdt.2017.10.015
12. Owari T, Iwamoto T, Anai S, Miyake M, Nakai Y, Hori S, et al. The sustaining of fluorescence in photodynamic diagnosis after the administration of 5-aminolevulinic acid in carcinogen-induced bladder cancer orthotopic rat model and urothelial cancer cell lines. Photodiagnosis Photodyn Ther (2021) 34:102309. doi: 10.1016/j.pdpdt.2021.102309
13. Hadjipanayis CG, Stummer W. 5-ALA and FDA approval for glioma surgery. J Neurooncol (2019) 141(3):479–86. doi: 10.1007/s11060-019-03098-y
14. Kolb JP, Oguin TH 3rd, Oberst A, Martinez J. Programmed cell death and inflammation: winter is coming. Trends Immunol (2017) 38(10):705–18. doi: 10.1016/j.it.2017.06.009
15. Bedoui S, Herold MJ, Strasser A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol (2020) 21(11):678–95. doi: 10.1038/s41580-020-0270-8
16. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med (2009) 361(16):1570–83. doi: 10.1056/NEJMra0901217
17. Christgen S, Tweedell RE, Kanneganti TD. Programming inflammatory cell death for therapy. Pharmacol Ther (2022) 232:108010. doi: 10.1016/j.pharmthera.2021.108010
18. Ranjan K, Pathak C. FADD regulates NF-kappaB activation and promotes ubiquitination of cFLIPL to induce apoptosis. Sci Rep (2016) 6:22787. doi: 10.1038/srep22787
19. Yuan S, Yu X, Asara JM, Heuser JE, Ludtke SJ, Akey CW. The holo-apoptosome: activation of procaspase-9 and interactions with caspase-3. Structure (2011) 19(8):1084–96. doi: 10.1016/j.str.2011.07.001
20. Cavalcante GC, Schaan AP, Cabral GF, Santana-da-Silva MN, Pinto P, Vidal AF, et al. A cell's fate: an overview of the molecular biology and genetics of apoptosis. Int J Mol Sci (2019) 20(17). doi: 10.3390/ijms20174133
21. Jo SH, Choi JA, Lim YJ, Lee J, Cho SN, Oh SM, et al. Calreticulin modulates the intracellular survival of mycobacteria by regulating ER-stress-mediated apoptosis. Oncotarget (2017) 8(35):58686–98. doi: 10.18632/oncotarget.17419
22. Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: A new frontier in cancer. BioMed Pharmacother (2020) 121:109595. doi: 10.1016/j.biopha.2019.109595
23. Yan J, Wan P, Choksi S, Liu ZG. Necroptosis and tumor progression. Trends Cancer (2022) 8(1):21–7. doi: 10.1016/j.trecan.2021.09.003
24. Case CL, Shin S, Roy CR. Asc and Ipaf Inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect Immun (2009) 77(5):1981–91. doi: 10.1128/IAI.01382-08
25. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
26. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell (2014) 157(5):1013–22. doi: 10.1016/j.cell.2014.04.007
27. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol (2021) 18(5):1106–21. doi: 10.1038/s41423-020-00630-3
28. Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol (2015) 25(6):347–53. doi: 10.1016/j.tcb.2015.01.001
29. Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat Commun (2020) 11(1):3151. doi: 10.1038/s41467-020-16887-1
30. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature (2020) 580(7803):391–5. doi: 10.1038/s41586-020-2129-8
31. Linkermann A, Green DR. Necroptosis. N Engl J Med (2014) 370(5):455–65. doi: 10.1056/NEJMra1310050
32. Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, et al. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell (2020) 180(6):1115–1129 e13. doi: 10.1016/j.cell.2020.02.050
33. Newton K, Manning G. Necroptosis and inflammation. Annu Rev Biochem (2016) 85:743–63. doi: 10.1146/annurev-biochem-060815-014830
34. Fang Y, Tang Y, Huang B. Pyroptosis: A road to next-generation cancer immunotherapy. Semin Immunol (2023) 68:101782. doi: 10.1016/j.smim.2023.101782
35. Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O'Rourke K, Li Q, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature (2021) 591(7848):131–6. doi: 10.1038/s41586-021-03218-7
36. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
37. Grabowski MM, Sankey EW, Ryan KJ, Chongsathidkiet P, Lorrey SJ, Wilkinson DS, et al. Immune suppression in gliomas. J Neurooncol (2021) 151(1):3–12. doi: 10.1007/s11060-020-03483-y
38. Nguyen PHD, Wasser M, Tan CT, Lim CJ, Lai HLH, Seow JJW, et al. Trajectory of immune evasion and cancer progression in hepatocellular carcinoma. Nat Commun (2022) 13(1):1441. doi: 10.1038/s41467-022-29122-w
39. Jongsma MLM, de Waard AA, Raaben M, Zhang T, Cabukusta B, Platzer R, et al. The SPPL3-defined glycosphingolipid repertoire orchestrates HLA class I-mediated immune responses. Immunity (2021) 54(1):132–150 e9. doi: 10.1016/j.immuni.2020.11.003
40. Taniura T, Iida Y, Kotani H, Ishitobi K, Tajima Y, Harada M, et al. Immunogenic chemotherapy in two mouse colon cancer models. Cancer Sci (2020) 111(10):3527–39. doi: 10.1111/cas.14624
41. McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer (2020) 20(4):203–17. doi: 10.1038/s41568-020-0246-1
42. Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med (2012) 366(10):925–31. doi: 10.1056/NEJMoa1112824
43. West EJ, Scott KJ, Tidswell E, Bendjama K, Stojkowitz N, Lusky M, et al. Intravenous oncolytic vaccinia virus therapy results in a differential immune response between cancer patients. Cancers (Basel) (2022) 14(9). doi: 10.3390/cancers14092181
44. Qiao L, Mei Z, Yang Z, Li X, Cai H, Liu W, et al. ALA-PDT inhibits proliferation and promotes apoptosis of SCC cells through STAT3 signal pathway. Photodiagnosis Photodyn Ther (2016) 14:66–73. doi: 10.1016/j.pdpdt.2016.01.008
45. Wang H, Xiong L, Xia Y, Wang X. 5-aminolaevulinic acid-based photodynamic therapy induces both necrosis and apoptosis of keratinocytes in plantar warts. J Cosmet Laser Ther (2020) 22(3):165–70. doi: 10.1080/14764172.2020.1785626
46. Chelakkot VS, Som J, Yoshioka E, Rice CP, Rutihinda SG, Hirasawa K, et al. Systemic MEK inhibition enhances the efficacy of 5-aminolevulinic acid-photodynamic therapy. Br J Cancer (2019) 121(9):758–67. doi: 10.1038/s41416-019-0586-3
47. Coupienne I, Fettweis G, Rubio N, Agostinis P, Piette J. 5-ALA-PDT induces RIP3-dependent necrosis in glioblastoma. Photochem Photobiol Sci (2011) 10(12):1868–78. doi: 10.1039/c1pp05213f
48. Tanaka M, Kataoka H, Yano S, Sawada T, Akashi H, Inoue M, et al. Immunogenic cell death due to a new photodynamic therapy (PDT) with glycoconjugated chlorin (G-chlorin). Oncotarget (2016) 7(30):47242–51. doi: 10.18632/oncotarget.9725
49. Chen R, Kang R, Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med (2022) 54(2):91–102. doi: 10.1038/s12276-022-00736-w
50. Han J, Cheng C, Zhang J, Fang J, Yao W, Zhu Y, et al. Myricetin activates the Caspase-3/GSDME pathway via ER stress induction of pyroptosis in lung cancer cells. Front Pharmacol (2022) 13:959938. doi: 10.3389/fphar.2022.959938
51. Chen D, Yu J, Zhang L. Necroptosis: an alternative cell death program defending against cancer. Biochim Biophys Acta (2016) 1865(2):228–36. doi: 10.1016/j.bbcan.2016.03.003
52. Lauret Marie Joseph E, Kirilovsky A, Lecoester B, El Sissy C, Boullerot L, Rangan L, et al. Chemoradiation triggers antitumor Th1 and tissue resident memory-polarized immune responses to improve immune checkpoint inhibitors therapy. J Immunother Cancer (2021) 9(7). doi: 10.1136/jitc-2020-002256
53. Lhuillier C, Rudqvist NP, Yamazaki T, Zhang T, Charpentier M, Galluzzi L, et al. Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J Clin Invest (2021) 131(5). doi: 10.1172/JCI138740
54. Korbelik M, Stott B, Sun J. Photodynamic therapy-generated vaccines: relevance of tumour cell death expression. Br J Cancer (2007) 97(10):1381–7. doi: 10.1038/sj.bjc.6604059
55. Kuzelova K, Grebenova D, Pluskalova M, Marinov I, Hrkal Z. Early apoptotic features of K562 cell death induced by 5-aminolaevulinic acid-based photodynamic therapy. J Photochem Photobiol B (2004) 73(1-2):67–78. doi: 10.1016/j.jphotobiol.2003.07.007
56. Grebenova D, Kuzelova K, Smetana K, Pluskalova M, Cajthamlova H, Marinov I, et al. Mitochondrial and endoplasmic reticulum stress-induced apoptotic pathways are activated by 5-aminolevulinic acid-based photodynamic therapy in HL60 leukemia cells. J Photochem Photobiol B (2003) 69(2):71–85. doi: 10.1016/S1011-1344(02)00410-4
57. Jee SH, Shen SC, Chiu HC, Tsai WL, Kuo ML, et al. Overexpression of interleukin-6 in human basal cell carcinoma cell lines increases anti-apoptotic activity and tumorigenic potency. Oncogene (2001) 20(2):198–208. doi: 10.1038/sj.onc.1204076
58. Lilge L, Portnoy M, Wilson BC. Apoptosis induced in vivo by photodynamic therapy in normal brain and intracranial tumour tissue. Br J Cancer (2000) 83(8):1110–7. doi: 10.1054/bjoc.2000.1426
59. Noodt BB, Berg K, Stokke T, Peng Q, Nesland JM. Apoptosis and necrosis induced with light and 5-aminolaevulinic acid-derived protoporphyrin IX. Br J Cancer (1996) 74(1):22–9. doi: 10.1038/bjc.1996.310
60. Hashida T, Kotake Y, Ohta S. Protein disulfide isomerase knockdown-induced cell death is cell-line-dependent and involves apoptosis in MCF-7 cells. J Toxicol Sci (2011) 36(1):1–7. doi: 10.2131/jts.36.1
61. Soriano J, Mora-Espi I, Alea-Reyes ME, Perez-Garcia L, Barrios L, Ibanez E, et al. Cell death mechanisms in tumoral and non-tumoral human cell lines triggered by photodynamic treatments: apoptosis, necrosis and parthanatos. Sci Rep (2017) 7:41340. doi: 10.1038/srep41340
62. Lin SH, Tsai GY, Wang MJ, Chen SY. Effects of cancer cell-derived nanovesicle vaccines produced by the oxidative stress-induced expression of DAMP and spontaneous release/filter extrusion in the interplay of cancer cells and macrophages. Biomedicines (2022) 10(8). doi: 10.3390/biomedicines10081977
Keywords: 5-aminolevulinic acid, photodynamic therapy, pyroptosis, necroptosis, anticancer immunity
Citation: Wang L, Chelakkot VS, Newhook N, Tucker S and Hirasawa K (2023) Inflammatory cell death induced by 5-aminolevulinic acid-photodynamic therapy initiates anticancer immunity. Front. Oncol. 13:1156763. doi: 10.3389/fonc.2023.1156763
Received: 01 February 2023; Accepted: 07 September 2023;
Published: 02 October 2023.
Edited by:
Jiajie Peng, Northwestern Polytechnical University, ChinaReviewed by:
Pål Kristian Selbo, Oslo University Hospital, NorwayCopyright © 2023 Wang, Chelakkot, Newhook, Tucker and Hirasawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kensuke Hirasawa, a2Vuc3VrZUBtdW4uY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.