 María Matamala Montoya
María Matamala Montoya Gijs J. J. van Slobbe
Gijs J. J. van Slobbe Jung-Chin Chang1
Jung-Chin Chang1 Esther A. Zaal
Esther A. Zaal Celia R. Berkers
Celia R. Berkers- 1Division Cell Biology, Metabolism & Cancer, Department Biomolecular Health Sciences, Faculty of Veterinary Medicine, Utrecht University, Utrecht, Netherlands
- 2Biomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research and Utrecht Institute of Pharmaceutical Sciences, Utrecht University, Utrecht, Netherlands
Multiple myeloma (MM) is characterized by the clonal expansion of malignant plasma cells in the bone marrow (BM). MM remains an incurable disease, with the majority of patients experiencing multiple relapses from different drugs. The MM tumor microenvironment (TME) and in particular bone-marrow stromal cells (BMSCs) play a crucial role in the development of drug resistance. Metabolic reprogramming is emerging as a hallmark of cancer that can potentially be exploited for cancer treatment. Recent studies show that metabolism is further adjusted in MM cells during the development of drug resistance. However, little is known about the role of BMSCs in inducing metabolic changes that are associated with drug resistance. In this Perspective, we summarize current knowledge concerning the metabolic reprogramming of MM, with a focus on those changes associated with drug resistance to the proteasome inhibitor Bortezomib (BTZ). In addition, we present proof-of-concept fluxomics (glucose isotope-tracing) and Seahorse data to show that co-culture of MM cells with BMSCs skews the metabolic phenotype of MM cells towards a drug-resistant phenotype, with increased oxidative phosphorylation (OXPHOS), serine synthesis pathway (SSP), TCA cycle and glutathione (GSH) synthesis. Given the crucial role of BMSCs in conveying drug resistance, insights into the metabolic interaction between MM and BMSCs may ultimately aid in the identification of novel metabolic targets that can be exploited for therapy.
1 Introduction
Multiple myeloma (MM) is an incurable B-cell neoplasm characterized by the clonal expansion of malignant plasma cells in the bone marrow (BM) (1). MM is the 2nd most common hematological malignancy (2) and accounted for 2.1% of all cancer deaths in the USA in 2022 (3). Over the last decade therapeutic advances (4), including the proteasome inhibitor Bortezomib (BTZ, VELCADE®) (1, 5), led to improvement in overall MM survival (1). BTZ targets mainly the β5(i)/β1(i) subunits of the 26S proteosome (6), resulting in a cascade of events that include the unfolded protein response (UPR) and amino acid deprivation, ultimately leading to cell death (7–9). However, MM remains an incurable disease, with only 57.9% of MM patients reaching 5 years survival (2012–2018) (3) and ultimately most MM patients relapse after BTZ treatment (10, 11). MM cells can develop drug resistance via multifactorial mechanisms (4, 8). In case of BTZ resistance, adaptation mechanisms include alterations at the level of the proteosome (mutations in the proteosome binding pocket, reduction of the 19S proteosome subunit, up-regulation of proteasomal machinery), upregulation of heat-shock proteins, genetic changes, activation of the aggresome-autophagy pathway, interactions within the MM tumor microenvironment (TME) and metabolic alterations (6, 8, 12, 13).
Metabolic reprogramming is regarded as an emerging hallmark of cancer (14–16) and is a potential target for cancer treatment (17). Metabolic rewiring fulfils the higher requirements of cancer cells for energy, building blocks for biosynthetic pathways and helps to maintain redox balance. These metabolic changes can be driven by genetic alterations, but can also be induced by the TME (18) and support both metastasis (16) and drug resistance (19). Metabolic reprogramming is a key feature of MM (7, 20) and metabolism further changes during the development of BTZ resistance (20–27). However, the influence of the MM TME on metabolic reprogramming of MM cells and its effect on drug resistance is still poorly understood.
In this perspective, we will highlight the most prominent metabolic alterations in MM and their contribution to drug resistance. Next, we will describe the current knowledge on the metabolic interactions between the BM TME and MM cells. As a proof-of-concept, we present novel data linking MM metabolic alterations induced by bone-marrow stromal cells (BMSCs) to drug resistance. Finally, we highlight how future studies on MM TME metabolism in the context of drug resistance can open novel therapeutic avenues for (relapsed) MM.
2 Multiple myeloma metabolism and its involvement in drug resistance
2.1 Glycolysis, serine synthesis pathway and pentose phosphate pathway
Glycolysis encompasses the breakdown of glucose into pyruvate, generating ATP and NADH. Most cancers are highly dependent on glycolysis (14, 28) and characterized by an increased production of lactate from pyruvate in the presence of oxygen. This so-called Warburg effect (29, 30) entails a faster, but less productive ATP generation compared to oxidative phosphorylation (OXPHOS) (31, 32). In addition to energy production, glycolytic intermediates can branch off into the pentose phosphate pathway (PPP) or serine synthesis pathway (SSP) to support the biosynthetic needs of cancer cells (33–35).
Like most cancers, MM cells show an increased glycolic flux (Figure 1A) (19), which is sustained by increased expression of glucose transporters (GLUTs) (36) and glycolytic enzymes such as hexokinase 2 (HK2) (37, 38), phosphofructokinase (PFK) (39), pyruvate kinase M2 (PKM2) (40–42), pyruvate dehydrogenase kinase 1 (PDK1) (43, 44) and lactate dehydrogenase A (LDHA) (43, 45). The generated lactate is exported by monocarboxylate transporters (MCTs) (46–48) and promotes a pro-tumorigenic extracellular environment (49). In addition to glycolysis, both the PPP and SSP are upregulated in MM (Figure 1A). Glucose-6-phosphate dehydrogenase (G6PD), the rate limiting enzyme of the PPP, and its regulator protein disulfide isomerase family A member 3 pseudogene 1 (PDIA3P) are increased in MM patients (50). Notably, expression of glycolytic and PPP enzymes, such as LDHA, PDK1, PKM2, G6PD and PDIA3P, are also associated with poor prognosis (41, 51, 52) and low survival (50, 53) in MM patients. Furthermore, levels of 3-phosphoglycerate dehydrogenase (PHGDH), the rate-limiting enzyme of the SSP, were found to be increased in MM cell lines (54) and in MM patient cells compared to normal B-cells (35). In both plasma and BM of MM patients, serine levels decrease inversely proportional to the stage of the disease (55), which could be indicative of increased serine consumption as disease progresses.
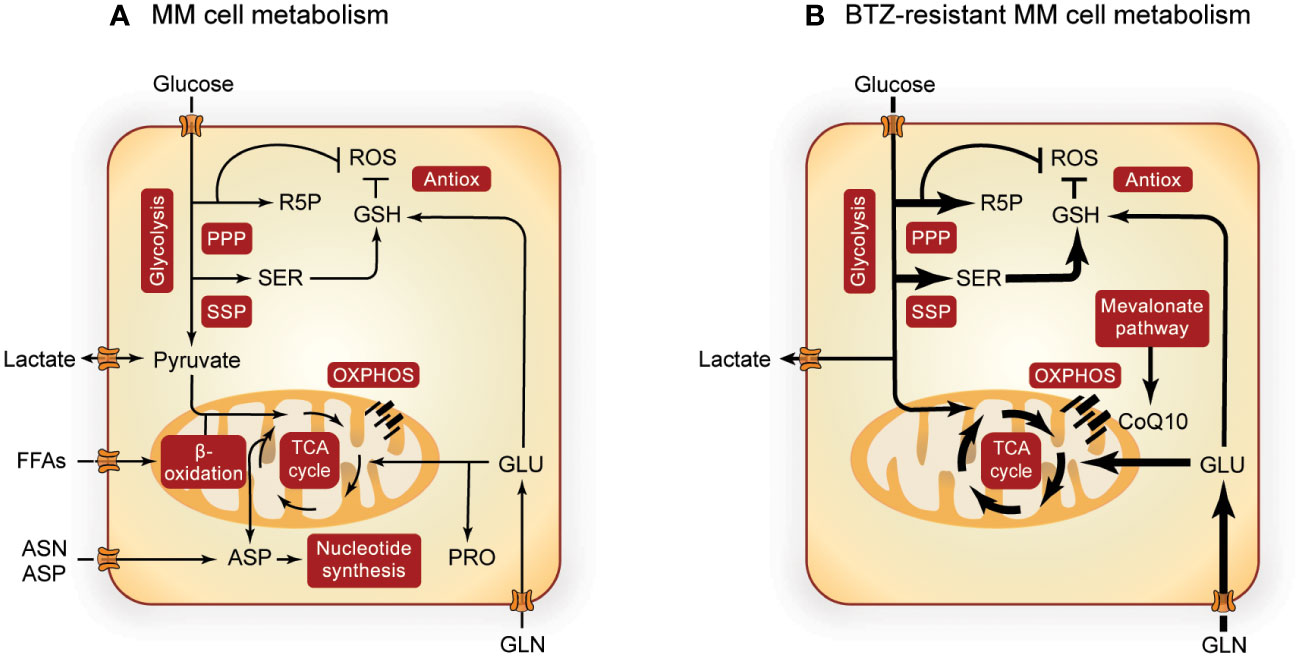
Figure 1 Overview of MM metabolism. Altered metabolic pathways in MM cells (A) and the most prominent metabolic alterations in BTZ-resistant MM cells (B). Pathways involved in central carbon metabolism are presented in red boxes and width of the arrows indicate increased flux in BTZ-resistant cells. Metabolism of MM cells is further upregulated during BTZ resistance, with special importance in the SSP, PPP, TCA cycle, OXPHOS and GSH synthesis. PPP, pentose phosphate pathway; R5P, ribose 5-phosphate; ROS, reactive oxygen species; GSH, glutathione; Antiox, antioxidative response; SER, serine; FFAs, free fatty acids; ASN, asparagine; ASP, aspartate; GLU, glutamate; PRO, proline; OXPHOS, oxidative phosphorylation; CoQ10, coenzyme Q10; MM, multiple myeloma; BTZ, bortezomib.
When MM cells become resistant to BTZ, glycolysis (45, 56, 57), PPP (26, 50, 53) and SSP (26, 35, 54) are even further upregulated. This metabolic remodeling of BTZ-resistant MM cells (Figure 1B) is characterized by higher glucose uptake and lactate secretion (26, 45, 58), with upregulation of MCTs being related to low treatment response (59). Also hypoxic environment, a characteristic of MM tumors in vivo, increases the expression of several glycolytic enzymes (60) and has been related to drug resistance in MM (45, 56, 57). Furthermore, the SSP enzyme PHGDH is upregulated in MM cells from BTZ refractory patients and in BTZ-resistant cell lines (26, 35). In line with these findings, overexpression of PHGDH induces BTZ resistance and cell growth (35), whereas PHGDH inhibition or serine starvation enhance BTZ toxicity (26, 54). BTZ exposure is linked to the overproduction of reactive oxygen species (ROS), which triggers cell death. Since both PPP and SSP play important roles in the antioxidant response, these pathways are likely to counteract BTZ-induced oxidative stress (26, 35, 54, 58).
2.2 Mitochondrial energy metabolism and associated pathways
Pyruvate can be converted to acetyl-CoA and further oxidized in the mitochondrial tricarboxylic acid (TCA) cycle to generate reducing equivalents, which in turn are used for OXPHOS to produce ATP (61). Intermediates of the TCA cycle can also serve as building blocks for the biosynthesis of lipids and nucleotides. In addition to pyruvate, other sources can replenish the TCA cycle, such as fatty acids and amino acids, such as glutamine.
OXPHOS has been described as an important energy source for MM (21, 62, 63) (Figure 1A). Several studies show that high expression of mitochondrial enzymes of TCA cycle and OXPHOS are correlated with poor survival (21, 26, 58). MM cells mostly depend on glutamine to feed the TCA cycle (64, 65), supported by overexpression of Glutaminase-1 (GLS1) and glutamine transporters ASCT2, LAT1 and SNAT1 (65). Interfering with glutamine availability through inhibition of glutamine transporters or GLS or via glutamine starvation hampered cell viability in MM cells (64–66). In addition, lipid metabolism is emerging as an important pathway for MM proliferation. Acetyl-CoA synthetase 2 (ACSS2; involved in β-oxidation) expression is increased in MM patients (67) and inhibition of β-oxidation with Etomoxir (a CPT1 inhibitor) and/or Orlistat (a Fatty acid synthase (FASN) inhibitor), decreased MM proliferation (68). Furthermore, the membrane transporter fatty acid binding protein (FABP) (69, 70) as well as fatty acid import (55, 71) are also increased in MM. Support of biosynthetic pathways appears to be another important function of the TCA cycle. For example, inhibition of de novo pyrimidine synthesis from aspartate resulted in MM cell death (72). Pyrroline-5-carboxylate reductase 1 (PYCR1), involved in proline synthesis, showed increased expression in MM patients, which correlated with poor survival (73). In line, combinational treatment of BTZ and paragyline (PYCR1 inhibitor) showed a synergistic effect on MM (73).
OXPHOS and ATP production are especially high in BTZ-resistant cells (21, 62, 74) (Figure 1B). In addition, the mevalonate pathway is upregulated in BTZ-resistant MM cells, which generates the electron carrier coenzyme Q10 (CoQ10) and is thereby important for electron transport chain (ETC) function (21). This dependence of resistant MM cells on OXPHOS makes them susceptible to its inhibition. The mevalonate pathway inhibitor simvastatin lowered CoQ10 levels in BTZ-resistant cells, which is accompanied by decreased levels of TCA cycle metabolites and an enhanced BTZ-induced cell death both in vivo and in vitro (21). In line with these findings, the use of statins in MM patients is associated with reduced mortality (75) and lower levels of serum M protein, an indicator of MM remission (76). Glutamine addiction is a second signature that is further enhanced in BTZ resistance (21, 77) as is GOT1 expression (58). In line, GLS1 inhibition in MM cells resulted in a decrease of PI resistance (72), further underscoring the importance of glutaminolysis and associated biosynthetic pathways in MM drug resistance.
3 Metabolic interactions in the MM tumor microenvironment could enhance drug resistance
MM resides in a complex permissive niche of heterogeneous cells, forming the TME (78, 79), which is composed of cellular and non-cellular components (1, 20, 80) and plays a pivotal role in promoting tumorigenesis and drug resistance (81–84). Within the MM TME, bone-marrow stromal cells (BMSCs) are thought to be crucial in promoting MM drug resistance, which has been described to be induced through direct cell adhesion (84–90) and soluble factors (79, 91–94), as well as via different signaling pathways. BMSCs, especially fibroblasts, can be activated by soluble factors and turn into cancer-associated fibroblasts (CAFs). For example, it was observed that mesenchymal stromal cells (MSCs) express tumorigenic markers such as alpha smooth muscle actin (αSMA) when co-cultured with MM cells or by addition of MM-derived factors (95). In addition, αSMA expression was increased in BM and MSCs of resistant MM patients (96). The expression of fibroblast activation protein (FAP) increases in stromal cells after co-culture with MM cells (88) or with MM exosomes (97). Together, this suggests that the presence of MM cells induces a CAF-like phenotype in BMSCs.
It is known from other tumor types that cells in the TME can engage in metabolic crosstalk and cross-feeding with tumor cells in an adaptable manner and according to the tumor’s needs. For example, autophagy in CAFs has been described to feed cancer cells via the excretion of several amino acids, including proline, alanine or glutamine (98–102), and other fuel molecules such as fatty acids, ketone bodies, pyruvate and lactate (103, 104). In MM, autophagy in BM fibroblasts has been linked to drug resistance. Proteomics data indicate that upon BTZ exposure, BM fibroblasts from BTZ-resistant patients upregulate proteins and markers that are associated with cellular stress and autophagy (105). Autophagy in these fibroblasts is induced by TGFβ, a factor secreted by both BM fibroblasts and MM cells (106, 107) and inhibition of TGFβ could overcome BMSCs derived-BTZ resistance (105). Furthermore, bidirectional mitochondrial transfer can take place in direct contact between BMSCs and MM cells, enhancing mitochondrial activity and drug resistance in MM cells (82, 91, 108, 109). Recent reports also suggest that BMSCs can engage in metabolic crosstalk with MM cells. For example, glutamine demand in MM cells induced glutamine synthesis in the neighboring MSCs (110). MM-BMSCs exosomal crosstalk is positively regulated by an increased glutamate secretion and fine-tuned according to the metabolic demands (111). Such cross-talk will likely result in metabolic changes in MM cells. Moreover, many of the signaling pathways that are regulated by MM-BMSCs interactions are known to regulate downstream metabolic pathways (45, 112–119). However, little is known about the specific metabolic changes that occur in MM upon interaction with BMSCs and, importantly, how these changes contribute to the observed drug-resistance phenotype.
To better understand the metabolic interactions between MM and BMSCs, we developed a non-direct co-culture system using BMSCs (HS5 and HS27a) and MM (RPMI8226) cell lines (Figure 2A). Flow cytometry-based cell viability assays confirmed that MM cells in co-culture become resistant to BTZ treatment, consistent with previous reports (120, 121) (Figure 2B). We next questioned whether metabolic changes induced in MM cells by BMSCs co-culture match those changes known to be involved in the development of BTZ resistance. Previous studies have shown that direct contact between MM and BMSCs increases mitochondrial metabolism as measured with oxygen consumption rate (OCR) (108, 109), which is also known to be important in BTZ resistance (21). Seahorse experiments in our system proved that indirect co-culture also significantly induces ATP-coupled OCR respiration (ATP-linked OCR), confirming the occurrence of a metabolic switch in MM cells upon co-culture with BMSCs (Figure 2C). To study this BMSCs-induced metabolic rewiring in more detail, MM cells were cultured alone or together with BMSCs in media containing [U-13C]–glucose to track glucose metabolism under these conditions (Figure 2A). Pathway enrichment analysis of total metabolite levels (i.e. the sum of all isotopologues) showed that BMSCs co-culture induces significant changes in MM cells (Figure 2D). Of note, amino acid metabolic pathways ranked amongst the most impacted pathways in co-cultured MM cells. Alanine, aspartate and glutamate metabolic pathways, which are linked to TCA cycle metabolism, were enriched in co-cultures with both HS27a and HS5 cells, whilst glycine and serine metabolism were significantly impacted in MM-HS5 co-cultures (Figure 2D). Additionally, PPP, TCA cycle and glutathione (GSH) metabolism were significantly upregulated in both co-cultures (Figure 2D). These data show that many pathways previously described to be of importance for BTZ-resistance are also significantly altered when MM cells are co-cultured with BMSCs.
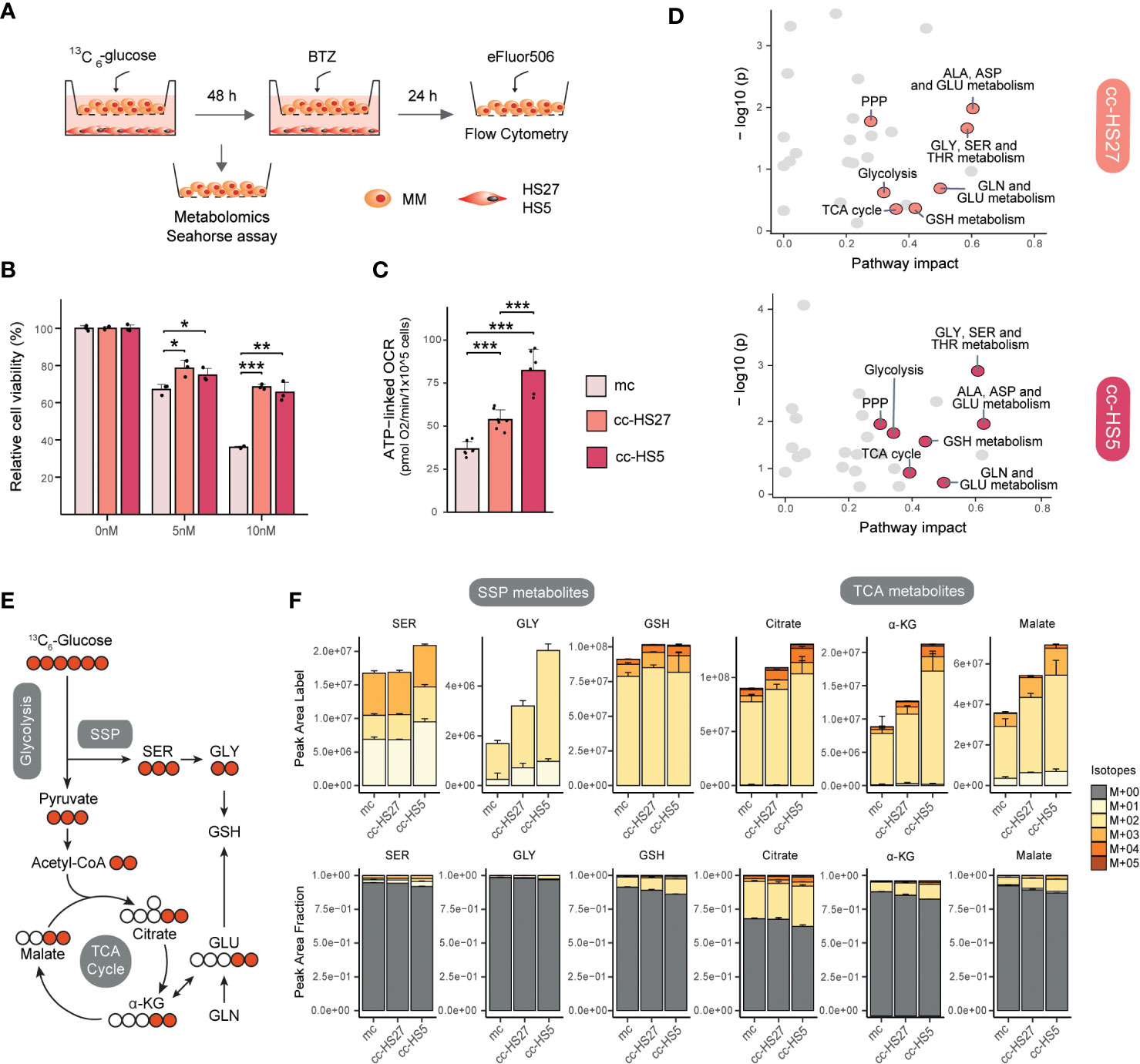
Figure 2 MM-BMSCs co-culture induces metabolic reprogramming and drug resistance-like metabolism. (A) Experimental layout for indirect co-culture of MM and BMSCs and overview of performed experiments. HS27a and HS5 human BMSCs were seeded in 12-well plates and allowed to attach overnight. Medium was replaced and RPMI8226 human MM cells were introduced in the upper chamber of Transwells® (TWs) (Corning, 0.4um, 12 well, polystyrene plates) with a seeding density of 1e5 cells/well, with a total volume of 3 mL/well. After 48h co-culture, MM cells were harvested for metabolomics or Seahorse assays or treated with Bortezomib (BTZ) for viability assays. For metabolomics experiments, media consisted of DMEM media containing 25 mM [U-13C]–glucose, 2mM glutamine and Penicillin Streptomycin. Metabolite extraction and LC-MS (pHILIC-QExactive) analysis were performed as described (26). For Seahorse experiment, MM cells were resuspended in Base DMEM Sigma D-5030 pH 7.4 supplemented with the same concentration of glucose, glutamine and Penicillin Streptomycin as metabolomics media and adding 5mM HEPES-NaOH and 21mM NaCl. After co-culture, MM cells were harvested and seeded and experiment was performed as previously described (21). For viability assays, different concentrations of BTZ (0, 5, 10 nM) were added for an additional 24h, after which MM cells were harvested, stained with the cell death die eFluor506 (BioScience; according to the manufacturer’s instructions) and then analyzed by flow cytometry (CytoFLEX). (B) Viability in MM cells after BTZ exposure in co-culture conditions: mc, mono-culture MM (light pink); cc-HS27a, co-culture MM with HS27a (orange); cc-HS5, co-culture MM with HS5 (dark pink). Data was analyzed with FlowJo™ and normalized to untreated cells (0 nM BTZ). Error bars depict SD of 3 independent wells from a representative experiment. (C) ATP-linked OCR of MM cells after co-culture conditions; mc: mono-culture MM (light pink); cc-HS27a: co-culture MM with HS27a (orange); cc-HS5: co-culture MM with HS5 (dark pink). The OCR was measured over time using the XFe24 Analyzer and ATP-linked OCR was calculated as the difference in OCR at basal conditions and after the addition of Oligomycin A. Error bars depict SD of 6-7 wells from a representative experiment. (D) Pathway enrichment analysis comparing mono- with BMSCs (HS5 or HS27a) co-cultured MM cells. Analysis was performed using Metaboanalyst package in R studio. A bigger pathway impact with smaller combined p-value (y-axis) is indicated as orange points (cc-HS27a) and dark pink points (cc-HS5) and it represents more reliably perturbed pathways in co-cultured vs. mono-cultured MM cells. (E) Schematic diagram of fluxomics, depicting the fate of 13C carbon into glycolysis, SSP, TCA cycle and GSH synthesis following [U-13C]–glucose uptake. Labeled and unlabeled C are represented with colored versus uncolored circles, respectively. (F) Metabolomic flux of [U-13C]–glucose into selected SSP and TCA cycle metabolites under mono- and co-culture conditions. Quantification of 13C-labeled peak areas (upper) and 13C-labeled fraction of total levels (lower) of serine (SER), glycine (GLY), glutathione (GSH), citrate, α-ketoglutarate (α-KG) and malate. Peaks were analyzed using TraceFinder software and isotopologue distribution was corrected for natural abundance of 13C. Data are presented as mean ± SD. Different colors represent the different isotopologues, whereby unlabeled metabolites (M+00) are grey, and 13C-labeled isotopologues are represented with yellow to red colors. Statistical significance was determined with TWO-way ANOVA. * P < 0.05, ** P < 0.01, *** P < 0.001. ALA, alanine; ASP, aspartate; GLU, glutamate; GLY, glycine; SER, serine; PPP, pentose phosphate pathway; TCA cycle, tricarboxylic acid cycle; α-KG, α-ketoglutarate; GLN, glutamine; GSH, glutathione; THR, threonine; BTZ, bortezomib; MM, multiple myeloma; mc, mono-culture MM; cc-HS27a, co-culture MM with HS27a; cc-HS5, co-culture MM with HS5; OCR, oxygen consumption rate.
To understand the contribution of glucose to these pathways, we analyzed the isotopologue distribution in SSP and TCA cycle metabolites (Figures 2E, F). When cells are cultured in [U-13C]–glucose, the SSP produces 13C3-serine (M+03). Serine (M+03) can be converted into glycine and subsequently into the antioxidative tripeptide GSH, both predominantly presenting the M+02 isotope (Figure 2E). Co-culture of MM cells with BMSCs, and particularly with HS5 cells, resulted in an increased synthesis of serine from glucose compared to MM mono-culture, as evidenced by higher absolute and fractional levels of 13C-labeled serine (Figure 2F). Downstream from serine, co-culture also increased the synthesis of glycine and GSH from [U-13C]–glucose (Figure 2F), as we also observed before in BTZ-resistant MM cells (26). In the TCA cycle, labelling from [U-13C]–glucose results in the formation of (predominantly) 13C2-citrate, -α-ketoglutarate and -malate (Figure 2E). Indeed, co-culture of MM with BMSCs increased both the levels of 13C-labelled TCA cycle metabolites, as well as the fractional contribution of glucose to this pathway, indicative of a higher glucose flux towards the TCA cycle (Figure 2F). Together, these results indicate that (metabolic) interactions between MM cells and BMSCs enhance SSP, TCA cycle and OXPHOS, which are key metabolic pathways in the adaptive response to BTZ.
4 Future prospects
Despite the interest in unveiling the crosstalk between MM and BMSCs, little is currently known about the metabolic interaction between these two cell types, especially in the context of drug resistance. So far, metabolic alterations related to BTZ resistance have mainly been studied in BTZ-resistant MM cell lines in mono-culture, in which drug resistance is induced by continuous drug exposure (122). Drug resistance in MM can be multiparametric, but the importance of the MM TME and especially BMSCs in driving drug resistance is widely accepted. In fact, recent studies showed the importance of targeting CAFs (123) and stroma interactions (124) to overcome drug resistance in MM. Here, we provide proof-of-concept data that demonstrate a previously unappreciated metabolic MM-BMSCs network in which indirect co-culture induces metabolic reprogramming and drug resistance-like metabolism in MM cells. This clear correlation between metabolic rewiring and BTZ resistance in the MM TME encourages further studies to determine the causal mechanism and further metabolic effects, which will benefit novel therapeutic paradigms, ultimately improving the treatment of relapsed MM.
First, to understand which metabolic vulnerabilities can be targeted in the MM TME, there is a need to unveil both commonly and differentially altered pathways induced by direct and indirect co-culture of MM with BMSCs. Most studies in terms of MM-BMSCs interaction have been performed in direct co-culture systems (86, 121), in which also metabolic alterations associated with mitochondria transfer have been reported (108, 109). Since cells in our co-culture system have no direct contact, our data point to a role for soluble factors in mediating MM metabolic rewiring. Indeed, several studies suggest that direct and indirect metabolic communication between MM and BMSCs could show common features and induce similar changes at the transcriptome and regulome level (115), as well as similar pathway activation (116, 117) and secretion of soluble factors (89).
A second open question is the precise mechanism underlying soluble factors-triggered metabolic changes in MM. Cytokines and growth factors released by MM cells or BMSCs can induce activation of metabolic enzymes, including the ones involved in antioxidant response and mitochondrial metabolism (60, 112, 125, 126), promoting drug resistance (127). Metabolites released by BMSCs may also directly feed into the metabolism of MM cells, as has been described in MM adipocytes (71, 128, 129). MSCs from MM patients showed increased glycolytic rate and lactate export compared to healthy donors (130). As (TME-derived) lactate can be used as a fuel for OXPHOS in MM cells (131), such metabolic cross-feeding could explain the higher ATP-linked OCR observed in MM cells under co-culture.
Finally, we here focused on energy metabolism and associated pathways, but many more metabolites are linked to MM drug resistance, including amino acids such as glutamate, proline (73, 113) or aspartate (72, 132). Lipid metabolism and β-oxidation have also been reported to be further enhanced in MM drug resistance (133–137), making interfering with lipid metabolism an interesting strategy to target resistance (68, 134). In addition, metabolites with immunosuppressant properties and that can also affect MM development are increased in MM in the context of the BM, including adenosine (138) and 2-deoxy-D-ribose (139, 140). Further elucidation of the BMSCs-induced metabolic rewiring in MM, including using different 13C/15N-labelled tracers and metabolomic and lipidomic approaches, will therefore likely unveil additional metabolic vulnerabilities in the MM TME.
In conclusion, this Perspective highlights the metabolic interactions within the TME that play a substantial role in drug response. Understanding this metabolic crosstalk will ultimately open new avenues to improve MM therapy.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
MMM, EZ and CB designed the project and experiments. MMM, JCC and GS performed experiments. EZ, JCC and MMM performed data analysis and visualization. MMM, CB and EZ wrote the manuscript. All authors contributed to the article and approved the final version.
Acknowledgments
We kindly thank Jeroen Jansen at the Utrecht Metabolism Expertise Centre (Faculty of Veterinary Medicine, Utrecht University) for technical assistance with mass spectrometry experiments, Estefania Lozano Andres of the Flow Cytometry and Cell sorting facility of the Faculty of Veterinary Medicine (Utrecht University) and Miranda Triest from Universitair Medisch Centrum Utrecht for assistance with the Seahorse assay.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kumar SK, Rajkumar V, Kyle RA, van Duin M, Sonneveld P, Mateos M-V, et al. Multiple myeloma. Nat Rev Dis Primers (2017) 3:1–20. doi: 10.1038/nrdp.2017.46
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA: A Cancer J Clin (2022) 72:7–33. doi: 10.3322/caac.21708
3. Myeloma - cancer stat facts. SEER. Available at: https://seer.cancer.gov/statfacts/html/mulmy.html (Accessed December 4, 2022).
4. Pinto V, Bergantim R, Caires HR, Seca H, Guimarães JE, Vasconcelos MH. Multiple myeloma: Available therapies and causes of drug resistance. Cancers (2020) 12:407. doi: 10.3390/cancers12020407
5. Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. JCO (2005) 23:630–9. doi: 10.1200/JCO.2005.11.030
6. Leonardo-Sousa C, Carvalho AN, Guedes RA, Fernandes PMP, Aniceto N, Salvador JAR, et al. Revisiting proteasome inhibitors: molecular underpinnings of their development, mechanisms of resistance and strategies to overcome anti-cancer drug resistance. Molecules (2022) 27:2201. doi: 10.3390/molecules27072201
7. Nair R, Gupta P, Shanmugam M. Mitochondrial metabolic determinants of multiple myeloma growth, survival, and therapy efficacy. Front Oncol (2022) 12:1000106. doi: 10.3389/fonc.2022.1000106
8. Robak P, Drozdz I, Szemraj J, Robak T. Drug resistance in multiple myeloma. Cancer Treat Rev (2018) 70:199–208. doi: 10.1016/j.ctrv.2018.09.001
9. Saavedra-García P, Martini F, Auner HW. Proteasome inhibition in multiple myeloma: Lessons for other cancers. Am J Physiology-Cell Physiol (2020) 318:C451–62. doi: 10.1152/ajpcell.00286.2019
10. Cantadori LO, Gaiolla RD, Nunes-Nogueira V dos S. Effect of bortezomib on the treatment of multiple myeloma: a systematic review protocol. BMJ Open (2022) 12:e061808. doi: 10.1136/bmjopen-2022-061808
11. Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: Lessons from the first decade. Clin Cancer Res (2008) 14:1649–57. doi: 10.1158/1078-0432.CCR-07-2218
12. Gonzalez-Santamarta M, Quinet G, Reyes-Garau D, Sola B, Roué G, Rodriguez MS. Resistance to the proteasome inhibitors: lessons from multiple myeloma and mantle cell lymphoma. In: Barrio R, Sutherland JD, Rodriguez MS, editors. Proteostasis and Disease: From basic mechanisms to clinics. advances in experimental medicine and biology. Cham: Springer International Publishing (2020). p. 153–74. doi: 10.1007/978-3-030-38266-7_6
13. Wallington-Beddoe CT, Sobieraj-Teague M, Kuss BJ, Pitson SM. Resistance to proteasome inhibitors and other targeted therapies in myeloma. Br J Haemato (2018) 182:11–28. doi: 10.1111/bjh.15210
14. Martínez-Reyes I, Chandel NS. Cancer metabolism: Looking forward. Nat Rev Cancer (2021) 21:669–80. doi: 10.1038/s41568-021-00378-6
15. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science (2020) 368:eaaw5473. doi: 10.1126/science.aaw5473
16. Bergers G, Fendt S-M. The metabolism of cancer cells during metastasis. Nat Rev Cancer (2021) 21:162–80. doi: 10.1038/s41568-020-00320-2
17. Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov (2022) 21:141–62. doi: 10.1038/s41573-021-00339-6
18. Elia I, Haigis MC. Metabolites and the tumour microenvironment: From cellular mechanisms to systemic metabolism. Nat Metab (2021) 3:21–32. doi: 10.1038/s42255-020-00317-z
19. Zaal EA, Berkers CR. The influence of metabolism on drug response in cancer. Front Oncol (2018) 8:500. doi: 10.3389/fonc.2018.00500
20. El Arfani C, De Veirman K, Maes K, De Bruyne E, Menu E. Metabolic features of multiple myeloma. Int J Mol Sci (2018) 19:1200. doi: 10.3390/ijms19041200
21. Zaal EA, de Grooth H-J, Oudaert I, Langerhorst P, Levantovsky S, van Slobbe GJJ, et al. Targeting coenzyme Q10 synthesis overcomes bortezomib resistance in multiple myeloma. Mol Omics (2022) 18:19–30. doi: 10.1039/D1MO00106J
22. Besse L, Besse A, Mendez-Lopez M, Vasickova K, Sedlackova M, Vanhara P, et al. A metabolic switch in proteasome inhibitor-resistant multiple myeloma ensures higher mitochondrial metabolism, protein folding and sphingomyelin synthesis. Haematologica (2019) 104:e415–9. doi: 10.3324/haematol.2018.207704
23. Song I-S, Jeong YJ, Jeong SH, Heo HJ, Kim HK, Lee SR, et al. Combination treatment with 2-methoxyestradiol overcomes bortezomib resistance of multiple myeloma cells. Exp Mol Med (2013) 45:e50. doi: 10.1038/emm.2013.104
24. Song IS, Kim HK, Lee SR, Jeong SH, Kim N, Ko KS, et al. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int J Cancer (2013) 133:1357–67. doi: 10.1002/ijc.28149
25. Tibullo D, Giallongo C, Romano A, Vicario N, Barbato A, Puglisi F, et al. Mitochondrial functions, energy metabolism and protein glycosylation are interconnected processes mediating resistance to bortezomib in multiple myeloma cells. Biomolecules (2020) 10:696. doi: 10.3390/biom10050696
26. Zaal EA, Wu W, Jansen G, Zweegman S, Cloos J, Berkers CR. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab (2017) 5:7. doi: 10.1186/s40170-017-0169-9
27. Lim JSL, Chong PSY, Chng W-J. Metabolic vulnerabilities in multiple myeloma. Cancers (2022) 14:1905. doi: 10.3390/cancers14081905
28. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer (2004) 4:891–9. doi: 10.1038/nrc1478
29. Warburg O. On the origin of cancer cells. Science (1956) 123:309–14. doi: 10.1126/science.123.3191.309
30. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
31. Xie J, Wu H, Dai C, Pan Q, Ding Z, Hu D, et al. Beyond warburg effect – dual metabolic nature of cancer cells. Sci Rep (2014) 4:4927. doi: 10.1038/srep04927
32. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi: 10.1126/science.1160809
33. Locasale JW. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat Rev Cancer (2013) 13:572–83. doi: 10.1038/nrc3557
34. Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab (2017) 25:27–42. doi: 10.1016/j.cmet.2016.08.009
35. Wu X, Xia J, Zhang J, Zhu Y, Wu Y, Guo J, et al. Phosphoglycerate dehydrogenase promotes proliferation and bortezomib resistance through increasing reduced glutathione synthesis in multiple myeloma. Br J Haemato (2020) 190:52–66. doi: 10.1111/bjh.16503
36. McBrayer SK, Cheng JC, Singhal S, Krett NL, Rosen ST, Shanmugam M. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: implications for glucose transporter-directed therapy. Blood (2012) 119:4686–97. doi: 10.1182/blood-2011-09-377846
37. Caillot M, Bourgeais J, Dakik H, Costé É, Mazure NM, Lelièvre É, et al. Cyclin D1 targets hexokinase 2 to control aerobic glycolysis in myeloma cells. Oncogenesis (2020) 9:1–13. doi: 10.1038/s41389-020-00253-3
38. Nakano A, Miki H, Nakamura S, Harada T, Oda A, Amou H, et al. Up-regulation of hexokinaseII in myeloma cells: targeting myeloma cells with 3-bromopyruvate. J Bioenerg Biomembr (2012) 44:31–8. doi: 10.1007/s10863-012-9412-9
39. Okabe S, Tanaka Y, Gotoh A. Therapeutic targeting of PFKFB3 and PFKFB4 in multiple myeloma cells under hypoxic conditions. Biomark Res (2022) 10:31. doi: 10.1186/s40364-022-00376-2
40. Gu Z, Xia J, Xu H, Frech I, Tricot G, Zhan F. NEK2 promotes aerobic glycolysis in multiple myeloma through regulating splicing of pyruvate kinase. J Hematol Oncol (2017) 10:17. doi: 10.1186/s13045-017-0392-4
41. Panchabhai S, Schlam I, Sebastian S, Fonseca R. PKM2 and other key regulators of warburg effect positively correlate with CD147 (EMMPRIN) gene expression and predict survival in multiple myeloma. Leukemia (2017) 31:991–4. doi: 10.1038/leu.2016.389
42. He Y, Wang Y, Liu H, Xu X, He S, Tang J, et al. Pyruvate kinase isoform M2 (PKM2) participates in multiple myeloma cell proliferation, adhesion and chemoresistance. Leukemia Res (2015) 39:1428–36. doi: 10.1016/j.leukres.2015.09.019
43. Fujiwara S, Kawano Y, Yuki H, Okuno Y, Nosaka K, Mitsuya H, et al. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br J Cancer (2013) 108:170–8. doi: 10.1038/bjc.2012.527
44. Kawano Y, Sasano T, Arima Y, Kushima S, Tsujita K, Matsuoka M, et al. A novel PDK1 inhibitor, JX06, inhibits glycolysis and induces apoptosis in multiple myeloma cells. Biochem Biophys Res Commun (2022) 587:153–9. doi: 10.1016/j.bbrc.2021.11.102
45. Maiso P, Huynh D, Moschetta M, Sacco A, Aljawai Y, Mishima Y, et al. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res (2015) 75:2071–82. doi: 10.1158/0008-5472.CAN-14-3400
46. Hanson DJ, Nakamura S, Amachi R, Hiasa M, Oda A, Tsuji D, et al. Effective impairment of myeloma cells and their progenitors by blockade of monocarboxylate transportation. Oncotarget (2015) 6:33568–86. doi: 10.18632/oncotarget.5598
47. Fujiwara S, Wada N, Kawano Y, Okuno Y, Kikukawa Y, Endo S, et al. Lactate, a putative survival factor for myeloma cells, is incorporated by myeloma cells through monocarboxylate transporters 1. Exp Hematol Oncol (2015) 4:12. doi: 10.1186/s40164-015-0008-z
48. Walters DK, Arendt BK, Jelinek DF. CD147 regulates the expression of MCT1 and lactate export in multiple myeloma cells. Cell Cycle (2013) 12:3364–72. doi: 10.4161/cc.26193
49. Ustun C, Fall P, Szerlip HM, Jillella A, Hendricks L, Burgess R, et al. Multiple myeloma associated with lactic acidosis. Leukemia Lymphoma (2002) 43:2395–7. doi: 10.1080/1042819021000040116
50. Yang X, Ye H, He M, Zhou X, Sun N, Guo W, et al. LncRNA PDIA3P interacts with c-myc to regulate cell proliferation via induction of pentose phosphate pathway in multiple myeloma. Biochem Biophys Res Commun (2018) 498:207–13. doi: 10.1016/j.bbrc.2018.02.211
51. Gkotzamanidou M, Kastritis E, Gavriatopoulou MRMMM, Nikitas N, Gika D, Mparmparousi D, et al. Increased serum lactate dehydrongenase should be included among the variables that define very-High-Risk multiple myeloma. Clin Lymphoma Myeloma Leukemia (2011) 11:409–13. doi: 10.1016/j.clml.2011.07.001
52. Gu Y, Yuan Y-H, Xu J, Shi Q-L, Qu X-Y, Guo R, et al. High serum lactate dehydrogenase predicts an unfavorable outcome in Chinese elderly patients with multiple myeloma. Oncotarget (2017) 8:48350–61. doi: 10.18632/oncotarget.16237
53. Li R, Ke M, Qi M, Han Z, Cao Y, Deng Z, et al. G6PD promotes cell proliferation and dexamethasone resistance in multiple myeloma via increasing anti-oxidant production and activating wnt/β-catenin pathway. Exp Hematol Oncol (2022) 11:77. doi: 10.1186/s40164-022-00326-6
54. Elsaadi S, Steiro I, Abdollahi P, Vandsemb EN, Yang R, Slørdahl TS, et al. Targeting phosphoglycerate dehydrogenase in multiple myeloma. Exp Hematol Oncol (2021) 10:3. doi: 10.1186/s40164-020-00196-w
55. Fei F, Ma T, Zhou X, Zheng M, Cao B, Li J. Metabolic markers for diagnosis and risk-prediction of multiple myeloma. Life Sci (2021) 265:118852. doi: 10.1016/j.lfs.2020.118852
56. Ikeda S, Kitadate A, Abe F, Takahashi N, Tagawa H. Hypoxia-inducible KDM3A addiction in multiple myeloma. Blood Adv (2018) 2:323–34. doi: 10.1182/bloodadvances.2017008847
57. Ikeda S, Abe F, Matsuda Y, Kitadate A, Takahashi N, Tagawa H. Hypoxia-inducible hexokinase-2 enhances anti-apoptotic function via activating autophagy in multiple myeloma. Cancer Sci (2020) 111:4088–101. doi: 10.1111/cas.14614
58. Soriano GP, Besse L, Li N, Kraus M, Besse A, Meeuwenoord N, et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia (2016) 30:2198–207. doi: 10.1038/leu.2016.102
59. Nathwani N, Palmer J, Synold TW, Salehian B, Rosenzweig M, Sanchez JF, et al. Toxicities associated with Metformin/Ritonavir combination treatment in Relapsed/Refractory multiple myeloma. Clin Lymphoma Myeloma Leukemia (2020) 20:e667–72. doi: 10.1016/j.clml.2020.05.017
60. Clees A-S, Stolp V, Häupl B, Fuhrmann DC, Wempe F, Seibert M, et al. Identification of the cysteine protease legumain as a potential chronic hypoxia-specific multiple myeloma target gene. Cells (2022) 11:292. doi: 10.3390/cells11020292
61. Pavlova NN, Zhu J, Thompson CB. The hallmarks of cancer metabolism: Still emerging. Cell Metab (2022) 34:355–77. doi: 10.1016/j.cmet.2022.01.007
62. Xiang Y, Fang B, Liu Y, Yan S, Cao D, Mei H, et al. SR18292 exerts potent antitumor effects in multiple myeloma via inhibition of oxidative phosphorylation. Life Sci (2020) 256:117971. doi: 10.1016/j.lfs.2020.117971
63. Dalva-Aydemir S, Bajpai R, Martinez M, Adekola KUA, Kandela I, Wei C, et al. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin Cancer Res (2015) 21:1161–71. doi: 10.1158/1078-0432.CCR-14-1088
64. Gonsalves WI, Jang JS, Jessen E, Hitosugi T, Evans LA, Jevremovic D, et al. In vivo assessment of glutamine anaplerosis into the TCA cycle in human pre-malignant and malignant clonal plasma cells. Cancer Metab (2020) 8:29. doi: 10.1186/s40170-020-00235-4
65. Bolzoni M, Chiu M, Accardi F, Vescovini R, Airoldi I, Storti P, et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: a new attractive target. Blood (2016) 128:667–79. doi: 10.1182/blood-2016-01-690743
66. Effenberger M, Bommert KS, Kunz V, Kruk J, Leich E, Rudelius M, et al. Glutaminase inhibition in multiple myeloma induces apoptosis via MYC degradation. Oncotarget (2017) 8:85858–67. doi: 10.18632/oncotarget.20691
67. Li Z, Liu H, He J, Wang Z, Yin Z, You G, et al. Acetyl-CoA synthetase 2: A critical linkage in obesity-induced tumorigenesis in myeloma. Cell Metab (2021) 33:78–93.e7. doi: 10.1016/j.cmet.2020.12.011
68. Tirado-Vélez JM, Joumady I, Sáez-Benito A, Cózar-Castellano I, Perdomo G. Inhibition of fatty acid metabolism reduces human myeloma cells proliferation. PloS One (2012) 7:e46484. doi: 10.1371/journal.pone.0046484
69. Shimizu M, Tachikawa S, Saitoh N, Nakazono K, Yu-Jung L, Suga M, et al. Thalidomide affects limb formation and multiple myeloma related genes in human induced pluripotent stem cells and their mesoderm differentiation. Biochem Biophysics Rep (2021) 26:100978. doi: 10.1016/j.bbrep.2021.100978
70. Farrell M, Fairfield H, Murphy CS, D’Amico A. Targeting bone marrow adipose tissue and the FABP family increases efficacy of dexamethasone in multiple myeloma. Blood (2020) 136:13–4. doi: 10.1182/blood-2020-142512
71. Panaroni C, Fulzele K, Mori T, Siu KT, Onyewadume C, Maebius A, et al. Multiple myeloma cells induce lipolysis in adipocytes and uptake fatty acids through fatty acid transporter proteins. Blood (2022) 139:876–88. doi: 10.1182/blood.2021013832
72. Bardeleben C, Sharma S, Reeve JR, Bassilian S, Frost P, Hoang B, et al. Metabolomics identifies pyrimidine starvation as the mechanism of 5-Aminoimidazole-4-Carboxamide-1-β-Riboside-Induced apoptosis in multiple myeloma cells. Mol Cancer Ther (2013) 12:1310–21. doi: 10.1158/1535-7163.MCT-12-1042
73. Oudaert I, Satilmis H, Vlummens P, De Brouwer W, Maes A, Hose D, et al. Pyrroline-5-Carboxylate reductase 1: a novel target for sensitizing multiple myeloma cells to bortezomib by inhibition of PRAS40-mediated protein synthesis. J Exp Clin Cancer Res (2022) 41:45. doi: 10.1186/s13046-022-02250-3
74. Thompson RM, Dytfeld D, Reyes L, Robinson RM, Smith B, Manevich Y, et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget (2017) 8:35863–76. doi: 10.18632/oncotarget.16262
75. Sanfilippo KM, Keller J, Gage BF, Luo S, Wang T-F, Moskowitz G, et al. Statins are associated with reduced mortality in multiple myeloma. JCO (2016) 34:4008–14. doi: 10.1200/JCO.2016.68.3482
76. Schmidmaier R, Baumann P, Bumeder I, Meinhardt G, Straka C, Emmerich B. First clinical experience with simvastatin to overcome drug resistance in refractory multiple myeloma. Eur J Haemato (2007) 79:240–3. doi: 10.1111/j.1600-0609.2007.00902.x
77. Gonsalves WI, Ramakrishnan V, Hitosugi T, Ghosh T, Jevremovic D, Dutta T, et al. Glutamine-derived 2-hydroxyglutarate is associated with disease progression in plasma cell malignancies. JCI Insight (2018) 3(1):e94543. doi: 10.1172/jci.insight.94543
78. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Communication Signaling (2020) 18:59. doi: 10.1186/s12964-020-0530-4
79. García-Ortiz A, Rodríguez-García Y, Encinas J, Maroto-Martín E, Castellano E, Teixidó J, et al. The role of tumor microenvironment in multiple myeloma development and progression. Cancers (2021) 13:217. doi: 10.3390/cancers13020217
80. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer (2007) 7:585–98. doi: 10.1038/nrc2189
81. Corre J, Mahtouk K, Attal M, Gadelorge M, Huynh A, Fleury-Cappellesso S, et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia (2007) 21:1079–88. doi: 10.1038/sj.leu.2404621
82. Schwestermann J, Besse A, Driessen C, Besse L. Contribution of the tumor microenvironment to metabolic changes triggering resistance of multiple myeloma to proteasome inhibitors. Front Oncol (2022) 12:899272. doi: 10.3389/fonc.2022.899272
83. Terpos E, Ntanasis-Stathopoulos I, Gavriatopoulou M, Dimopoulos MA. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J (2018) 8:1–12. doi: 10.1038/s41408-017-0037-4
84. Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, et al. Integrin β7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood (2011) 117:6202–13. doi: 10.1182/blood-2010-06-292243
85. Schmidmaier R, Baumann P, Simsek M, Dayyani F, Emmerich B, Meinhardt G. The HMG-CoA reductase inhibitor simvastatin overcomes cell adhesion–mediated drug resistance in multiple myeloma by geranylgeranylation of rho protein and activation of rho kinase. Blood (2004) 104:1825–32. doi: 10.1182/blood-2003-12-4218
86. Yanamandra N, Colaco NM, Parquet NA, Buzzeo RW, Boulware D, Wright G, et al. Tipifarnib and bortezomib are synergistic and overcome cell adhesion–mediated drug resistance in multiple myeloma and acute myeloid leukemia. Clin Cancer Res (2006) 12:591–9. doi: 10.1158/1078-0432.CCR-05-1792
87. Hathi D, Chanswangphuwana C, Cho N, Fontana F, Maji D, Ritchey J, et al. Ablation of VLA4 in multiple myeloma cells redirects tumor spread and prolongs survival. Sci Rep (2022) 12:30. doi: 10.1038/s41598-021-03748-0
88. Zi F-M, He J-S, Li Y, Wu C, Wu W-J, Yang Y, et al. Fibroblast activation protein protects bortezomib-induced apoptosis in multiple myeloma cells through β-catenin signaling pathway. Cancer Biol Ther (2014) 15:1413–22. doi: 10.4161/cbt.29924
89. Gupta D, Treon S, Shima Y, Hideshima T, Podar K, Tai Y, et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: Therapeutic applications. Leukemia (2001) 15:1950–61. doi: 10.1038/sj.leu.2402295
90. Suzuki R, Ogiya D, Ogawa Y, Kawada H, Ando K. Targeting CAM-DR and mitochondrial transfer for the treatment of multiple myeloma. Curr Oncol (2022) 29:8529–39. doi: 10.3390/curroncol29110672
91. Barbato A, Scandura G, Puglisi F, Cambria D, La Spina E, Palumbo GA, et al. Mitochondrial bioenergetics at the onset of drug resistance in hematological malignancies: An overview. Front Oncol (2020) 10:604143. doi: 10.3389/fonc.2020.604143
92. Voorhees PM, Manges RF, Sonneveld P, Jagannath S, Somlo G, Krishnan A, et al. A phase 2 multicentre study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with relapsed or refractory multiple myeloma. Br J Haemato (2013) 161:357–66. doi: 10.1111/bjh.12266
93. Oudaert I, van der Vreken A, Maes A, De Bruyne E, De Veirman K, Vanderkerken K, et al. Metabolic cross-talk within the bone marrow milieu: focus on multiple myeloma. Exp Hematol Oncol (2022) 11:49. doi: 10.1186/s40164-022-00303-z
94. Saltarella I, Lamanuzzi A, Desantis V, Di Marzo L, Melaccio A, Curci P, et al. Myeloma cells regulate miRNA transfer from fibroblast-derived exosomes by expression of lncRNAs. J Pathol (2022) 256:402–13. doi: 10.1002/path.5852
95. Duan C-W, Shi J, Chen J, Wang B, Yu Y-H, Qin X, et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell (2014) 25:778–93. doi: 10.1016/j.ccr.2014.04.015
96. Enukashvily NI, Semenova N, Chubar AV, Ostromyshenskii DI, Gushcha EA, Gritsaev S, et al. Pericentromeric non-coding DNA transcription is associated with niche impairment in patients with ineffective or partially effective multiple myeloma treatment. Int J Mol Sci (2022) 23:3359. doi: 10.3390/ijms23063359
97. Frassanito MA, Desantis V, Di Marzo L, Craparotta I, Beltrame L, Marchini S, et al. Bone marrow fibroblasts overexpress miR-27b and miR-214 in step with multiple myeloma progression, dependent on tumour cell-derived exosomes. J Pathol (2019) 247:241–53. doi: 10.1002/path.5187
98. Bai J, Liu T, Tu B, Yuan M, Shu Z, Fan M, et al. Autophagy loss impedes cancer-associated fibroblast activation via downregulating proline biosynthesis. Autophagy (2023) 19(2):632–43. doi: 10.1080/15548627.2022.2093026
99. Yang L, Achreja A, Yeung T-L, Mangala LS, Jiang D, Han C, et al. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab (2016) 24:685–700. doi: 10.1016/j.cmet.2016.10.011
100. Li Z, Sun C, Qin Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics (2021) 11:8322–36. doi: 10.7150/thno.62378
101. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016) 536:479–83. doi: 10.1038/nature19084
102. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
103. Sakamoto A, Kunou S, Shimada K, Tsunoda M, Aoki T, Iriyama C, et al. Pyruvate secreted from patient-derived cancer-associated fibroblasts supports survival of primary lymphoma cells. Cancer Sci (2019) 110:269–78. doi: 10.1111/cas.13873
104. Martinez-Outschoorn U, Sotgia F, Lisanti MP. Tumor microenvironment and metabolic synergy in breast cancers: Critical importance of mitochondrial fuels and function. Semin Oncol (2014) 41:195–216. doi: 10.1053/j.seminoncol.2014.03.002
105. Frassanito MA, De Veirman K, Desantis V, Marzo LD, Vergara D, Ruggieri S, et al. Halting pro-survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia (2016) 30:640–8. doi: 10.1038/leu.2015.289
106. Hayashi T, Hideshima T, Nguyen AN, Munoz O, Podar K, Hamasaki M, et al. Transforming growth factor β receptor I kinase inhibitor down-regulates cytokine secretion and multiple myeloma cell growth in the bone marrow microenvironment. Clin Cancer Res (2004) 10:7540–6. doi: 10.1158/1078-0432.CCR-04-0632
107. Lu A, Pallero MA, Lei W, Hong H, Yang Y, Suto MJ, et al. Inhibition of transforming growth factor-β activation diminishes tumor progression and osteolytic bone disease in mouse models of multiple myeloma. Am J Pathol (2016) 186:678–90. doi: 10.1016/j.ajpath.2015.11.003
108. Marlein CR, Piddock RE, Mistry JJ, Zaitseva L, Hellmich C, Horton RH, et al. CD38-driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res (2019) 79:2285–97. doi: 10.1158/0008-5472.CAN-18-0773
109. Matula Z, Mikala G, Lukácsi S, Matkó J, Kovács T, Monostori É, et al. Stromal cells serve drug resistance for multiple myeloma via mitochondrial transfer: a study on primary myeloma and stromal cells. Cancers (2021) 13:3461. doi: 10.3390/cancers13143461
110. Chiu M, Toscani D, Marchica V, Taurino G, Costa F, Bianchi MG, et al. Myeloma cells deplete bone marrow glutamine and inhibit osteoblast differentiation limiting asparagine availability. Cancers (2020) 12:3267. doi: 10.3390/cancers12113267
111. Wang F, Oudaert I, Tu C, Maes A, van der Vreken A, Vlummens P, et al. System xc– inhibition blocks bone marrow-multiple myeloma exosomal crosstalk, thereby countering bortezomib resistance. Cancer Lett (2022) 535:215649. doi: 10.1016/j.canlet.2022.215649
112. Ortiz-Ruiz A, Ruiz-Heredia Y, Morales ML, Aguilar-Garrido P, García-Ortiz A, Valeri A, et al. Myc-related mitochondrial activity as a novel target for multiple myeloma. Cancers (2021) 13:1662. doi: 10.3390/cancers13071662
113. Schwörer S, Berisa M, Violante S, Qin W, Zhu J, Hendrickson RC, et al. Proline biosynthesis is a vent for TGFβ-induced mitochondrial redox stress. EMBO J (2020) 39:e103334. doi: 10.15252/embj.2019103334
114. Zhang D, Wang Y, Shi Z, Liu J, Sun P, Hou X, et al. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep (2015) 10:1335–48. doi: 10.1016/j.celrep.2015.02.006
115. Dziadowicz SA, Wang L, Akhter H, Aesoph D, Sharma T, Adjeroh DA, et al. Bone marrow stroma-induced transcriptome and regulome signatures of multiple myeloma. Cancers (2022) 14:927. doi: 10.3390/cancers14040927
116. Markovina S, Callander NS, O’Connor SL, Xu G, Shi Y, Leith CP, et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-κB activity in myeloma cells. Mol Cancer (2010) 9:176. doi: 10.1186/1476-4598-9-176
117. Yuan L, Chan GCF, Fung KL, Chim CS. RANKL expression in myeloma cells is regulated by a network involving RANKL promoter methylation, DNMT1, microRNA and TNFα in the microenvironment. Biochim Biophys Acta (BBA) - Mol Cell Res (2014) 1843:1834–8. doi: 10.1016/j.bbamcr.2014.05.010
118. Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with “Warburg-like” cancer metabolism and l-lactate production. Cell Cycle (2012) 11:3019–35. doi: 10.4161/cc.21384
119. Nigdelioglu R, Hamanaka RB, Meliton AY, O’Leary E, Witt LJ, Cho T, et al. Transforming growth factor (TGF)-β promotes de Novo serine synthesis for collagen production *. J Biol Chem (2016) 291:27239–51. doi: 10.1074/jbc.M116.756247
120. Perez LE, Parquet N, Meads M, Anasetti C, Dalton W. Bortezomib restores stroma-mediated APO2L/TRAIL apoptosis resistance in multiple myeloma. Eur J Haematology (2010) 84:212–22. doi: 10.1111/j.1600-0609.2009.01381.x
121. Bar-Natan M, Stroopinsky D, Luptakova K, Coll MD, Apel A, Rajabi H, et al. Bone marrow stroma protects myeloma cells from cytotoxic damage via induction of the oncoprotein MUC1. Br J Haemato (2017) 176:929–38. doi: 10.1111/bjh.14493
122. Franke NE, Niewerth D, Assaraf YG, van Meerloo J, Vojtekova K, van Zantwijk CH, et al. Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia (2012) 26:757–68. doi: 10.1038/leu.2011.256
123. Sakemura R, Hefazi M, Siegler EL, Cox MJ, Larson DP, Hansen MJ, et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood (2022) 139:3708–21. doi: 10.1182/blood.2021012811
124. Ludwig H, Weisel K, Petrucci MT, Leleu X, Cafro AM, Garderet L, et al. Olaptesed pegol, an anti-CXCL12/SDF-1 spiegelmer, alone and with bortezomib–dexamethasone in relapsed/refractory multiple myeloma: a phase IIa study. Leukemia (2017) 31:997–1000. doi: 10.1038/leu.2017.5
125. Croonquist PA, Linden MA, Zhao F, Van Ness BG. Gene profiling of a myeloma cell line reveals similarities and unique signatures among IL-6 response, n-ras-activating mutations, and coculture with bone marrow stromal cells. Blood (2003) 102:2581–92. doi: 10.1182/blood-2003-04-1227
126. Aass KR, Mjelle R, Kastnes MH, Tryggestad SS, van den Brink LM, Aass Roseth I, et al. Intracellular IL-32 regulates mitochondrial metabolism, proliferation, and differentiation of malignant plasma cells. iScience (2022) 25:103605. doi: 10.1016/j.isci.2021.103605
127. Ghobrial IM, Detappe A, Anderson KC, Steensma DP. The bone-marrow niche in MDS and MGUS: implications for AML and MM. Nat Rev Clin Oncol (2018) 15:219–33. doi: 10.1038/nrclinonc.2017.197
128. Liu Z, Xu J, He J, Liu H, Lin P, Wan X, et al. Mature adipocytes in bone marrow protect myeloma cells against chemotherapy through autophagy activation. Oncotarget (2015) 6:34329–41. doi: 10.18632/oncotarget.6020
129. Trotter TN, Gibson JT, Sherpa TL, Gowda PS, Peker D, Yang Y. Adipocyte-lineage cells support growth and dissemination of multiple myeloma in bone. Am J Pathol (2016) 186:3054–63. doi: 10.1016/j.ajpath.2016.07.012
130. Giallongo C, Dulcamare I, Tibullo D, Del Fabro V, Vicario N, Parrinello N, et al. CXCL12/CXCR4 axis supports mitochondrial trafficking in tumor myeloma microenvironment. Oncogenesis (2022) 11:1–13. doi: 10.1038/s41389-022-00380-z
131. Barbato A, Giallongo C, Giallongo S, Romano A, Scandura G, Concetta S, et al. Lactate trafficking inhibition restores sensitivity to proteasome inhibitors and orchestrates immuno-microenvironment in multiple myeloma. Cell Proliferation, e13388. doi: 10.1111/cpr.13388
132. Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell (2015) 162:540–51. doi: 10.1016/j.cell.2015.07.016
133. Faict S, Oudaert I, D’Auria L, Dehairs J, Maes K, Vlummens P, et al. The transfer of sphingomyelinase contributes to drug resistance in multiple myeloma. Cancers (2019) 11:1823. doi: 10.3390/cancers11121823
134. Besse L, Besse A, Stolze SC, Sobh A, Zaal EA, van der Ham AJ, et al. Treatment with HIV-protease inhibitor nelfinavir identifies membrane lipid composition and fluidity as a therapeutic target in advanced multiple myeloma. Cancer Res (2021) 81:4581–93. doi: 10.1158/0008-5472.CAN-20-3323
135. Bhattacharya A, Brea RJ, Niederholtmeyer H, Devaraj NK. A minimal biochemical route towards de novo formation of synthetic phospholipid membranes. Nat Commun (2019) 10:300. doi: 10.1038/s41467-018-08174-x
136. Wei Y, Wang J, Chen F, Li X, Zhang J, Shen M, et al. Serum abnormal metabolites for evaluating therapeutic response and prognosis of patients with multiple myeloma. Front Oncol (2022) 12:808290. doi: 10.3389/fonc.2022.808290
137. Xu G, Huang S, Peng J, Gao X, Li M, Yu S, et al. Targeting lipid metabolism in multiple myeloma cells: Rational development of a synergistic strategy with proteasome inhibitors. Br J Pharmacol (2021) 178:4741–57. doi: 10.1111/bph.15653
138. Yang R, Elsaadi S, Misund K, Abdollahi P, Vandsemb EN, Moen SH, et al. Conversion of ATP to adenosine by CD39 and CD73 in multiple myeloma can be successfully targeted together with adenosine receptor A2A blockade. J Immunother Cancer (2020) 8:e000610. doi: 10.1136/jitc-2020-000610
139. Liu H, He J, Bagheri-Yarmand R, Li Z, Liu R, Wang Z, et al. Osteocyte CIITA aggravates osteolytic bone lesions in myeloma. Nat Commun (2022) 13:3684. doi: 10.1038/s41467-022-31356-7
Keywords: cancer metabolism, multiple myeloma, drug resistance, bone marrow stromal cell (BMSC), fluxomics, metabolomics, tumor microenvironment, Bortezomib
Citation: Matamala Montoya M, van Slobbe GJJ, Chang J-C, Zaal EA and Berkers CR (2023) Metabolic changes underlying drug resistance in the multiple myeloma tumor microenvironment. Front. Oncol. 13:1155621. doi: 10.3389/fonc.2023.1155621
Received: 31 January 2023; Accepted: 21 March 2023;
Published: 06 April 2023.
Edited by:
Parmanand Malvi, University of Alabama at Birmingham, United StatesReviewed by:
Jasvinder Singh, University of Minnesota Twin Cities, United StatesHarish Kumar, University of Alabama at Birmingham, United States
Copyright © 2023 Matamala Montoya, van Slobbe, Chang, Zaal and Berkers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Celia R. Berkers, Yy5yLmJlcmtlcnNAdXUubmw=; Esther A. Zaal, ZS5hLnphYWxAdXUubmw=
†These authors have contributed equally to this work and share last authorship