 Shinsuke Araki
Shinsuke Araki Momoko Ohori
Momoko Ohori
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 05 June 2023
Sec. Molecular and Cellular Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1152087
Accumulating evidence has indicated that pre-mRNA splicing plays critical roles in a variety of physiological processes, including development of multiple diseases. In particular, alternative splicing is profoundly involved in cancer progression through abnormal expression or mutation of splicing factors. Small-molecule splicing modulators have recently attracted considerable attention as a novel class of cancer therapeutics, and several splicing modulators are currently being developed for the treatment of patients with various cancers and are in the clinical trial stage. Novel molecular mechanisms modulating alternative splicing have proven to be effective for treating cancer cells resistant to conventional anticancer drugs. Furthermore, molecular mechanism-based combination strategies and patient stratification strategies for cancer treatment targeting pre-mRNA splicing must be considered for cancer therapy in the future. This review summarizes recent progress in the relationship between druggable splicing-related molecules and cancer, highlights small-molecule splicing modulators, and discusses future perspectives of splicing modulation for personalized and combination therapies in cancer treatment.
Alternative pre-mRNA splicing is a critical molecular mechanism underlying the generation of diverse proteins and plays a pivotal role in various biological processes, such as development, differentiation, growth, and apoptosis (1–3). Alternative splicing of RNA-binding protein FOX1 homologue 1 plays key roles in brain development and neuronal differentiation, as the nucleic and cytoplasmic isoforms regulate approximately 500 alternative spliced cassette exons that maintain neuron function or control mRNA expression of synaptic and autism-related genes (4). In heart development, alternative spliced isoforms of Titin control the elasticity of titin, thereby determining sarcomere length (4). Alternative splicing is evolutionarily conserved, but yet diverged, as evidenced by comparisons of organ transcriptomes from vertebrate species spanning approximately 350 million years of evolution (5–7). Recent studies have linked dysregulation of the components of alternative splicing machinery with various diseases, especially cancers. Analyses of >8,000 tumors across 32 types showed that tumors have up to 30% more alternative splicing events than normal samples (8). In contrast, thousands of splicing variants were not present in nonmalignant tissues (8), suggesting potential links between splicing abnormalities and cancer progression and metastasis. Alternatively spliced isoforms associated with cancer have been implicated in tumor progression, migration, apoptosis, and angiogenesis. Several spliced isoforms, including Bcl-xL and MCL1L, are up-regulated in tumor cells as anti-apoptotic factors (3). In addition, spliced isoforms are responsible for resistance against anticancer therapies, such as androgen receptor (AR)-targeted therapy for prostate cancer (9). The aberrant isoform of AR-V7 is constitutively active, thereby contributing to the development and progression of castration-resistant prostate cancer and resistance to AR-targeted therapy (10).
Multiple strategies have been implemented to target tumor cells, including small-molecule compounds and antisense oligonucleotides. To target alternative splicing machinery is promising candidates for cancer treatment, and multiple molecules including kinases are involved in pre-mRNA splicing events in cancer (2, 11). In this review, we highlight the druggable splicing-related molecules and the newly developed compounds to target alternative splicing for cancer therapy. First, we introduce the pre-mRNA splicing machinery and mutations of splicing-related genes in cancer. Then, we discuss why targeting splicing molecules is effective for cancer treatment. Next, we describe the dysregulation of splicing-related proteins and small-molecule compounds in cancer. Finally, we discuss the therapeutic potential of splicing modulators for cancer treatment from the perspective of patient stratification and drug combination strategy.
Pre-mRNA splicing is performed by the spliceosome, which comprises multicomponent five ribonucleoprotein complexes (snRNPs; U1, U2, U4, U5, and U6), each composed of specific small nuclear RNAs (snRNAs), a number of associated proteins, and >200 additional polypeptides not directly bound to snRNPs (12–14). The spliceosome is assembled and activated through a series of ATP/GTP-dependent reactions from complex E to complexes A, B, and C via RNA–RNA and RNA–protein interactions. Spliceosome assembly starts with the recognition of the 5′ splice site (5′ss) by U1 snRNP via base paring interactions with U1 snRNA and the 5′-end of the intron. Subsequently, U2AF1, U2AF2, and SF1 interact with the 3′ss region, including the polypyrimidine tract and the AG dinucleotide, and lead to the formation of complex E (Figure 1A). The next step in spliceosome assembly involves ATP-dependent stable binding of U2 snRNP at the branch site through base pairing interactions with U2 snRNA, thereby leading to the formation of complex A (Figure 1B). SF3B1 is one of the key components of U2 snRNP that recognizes the branch site and the U2 snRNA–pre-mRNA helix and facilitates the approximation of the branch-site adenosine to the 5′ss. Other components of complex A, such as RBM10, RBM39, and RBM15, modulate 3′ss recognition, and RBM15 is regulated by protein arginine methyltransferase 1 (PRMT1). Recognition of splice sites by U1 and U2 snRNPs is assisted and modulated by several other RNA-binding proteins, such as arginine–serine-rich (SR) proteins, heterogeneous nuclear ribonucleoproteins (hnRNPs), and RNA-binding motif proteins. SR proteins, such as SRSF1 and SRSF2, bind to an exonic splicing enhancer (ESE) or intronic splicing enhancer (ISE) to promote exon selection by recruiting spliceosomal components, U1 or U2 snRNPs. After initial recognition of the splice sites by U1 and U2 snRNPs, subsequent recruitment of the U4/U6-U5 tri-snRNP complex to form complex B leads to the formation of the catalytically active conformations of the spliceosome. Then, pre-mRNA intron removal proceeds with two sequential transesterification reactions that are initiated by the nucleophilic attack of the 5′ss by the branch-site nucleotide, resulting in the formation of an intro lariat. The lariat is subsequently removed by 5′ss-mediated attack on 3′ss, producing a mature mRNA followed by spliceosome disassembly. Alternative splicing can arise from machineries, including exon skipping, intron retention, alternative 5′ splice site, alternative 3′ splice site, mutually exclusive exon, alternative promoter, alternative polyadenylation, and trans-splicing (Figure 2) (15–17).
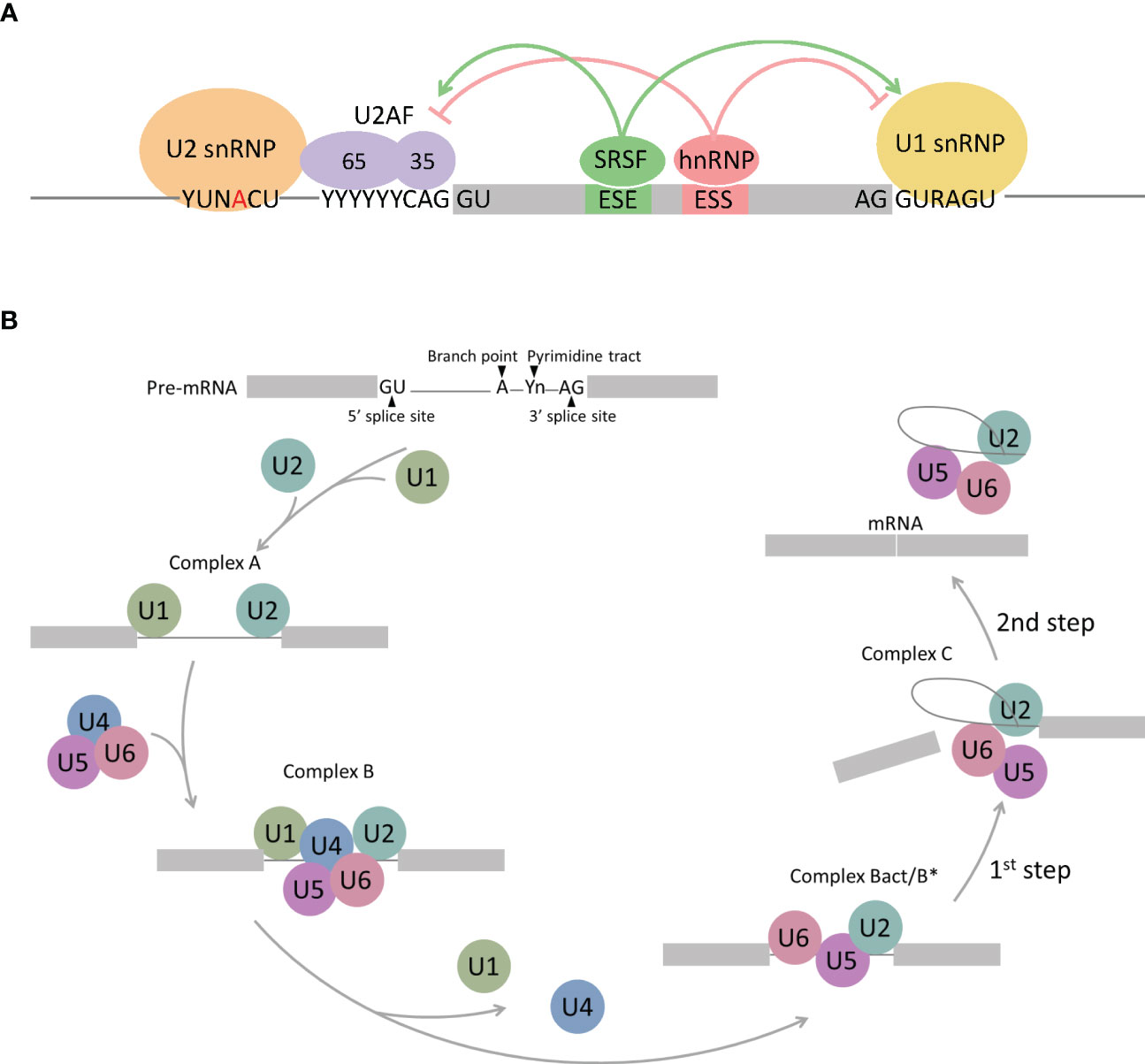
Figure 1 Schematic representation of spliceosome assembly. (A) Complex E regulated by RNA-binding proteins. SR proteins (SRSFs) bind to Exonic splicing enhancers (ESE), thereby stimulating splicing at up- and downstream splice sites by facilitating the interaction of U2AF or the U1 snRNP with the RNA. The hnRNP proteins bind to Exonic splicing silencers (ESS), subsequently suppress splicing at up- and downstream splice sites by antagonizing SR proteins. (B) Spliceosome assembly pathway. 5′ splice site is recognized by U1 snRNP, followed by U2 snRNP binding to the branch point (complex A). U4/U6/U5 tri-snRNP complex binds to complex A to form complex B and conformational rearrangements releases U1 and U4 snRNP, which results in activation of the spliceosome (complex B act/B*). Further conformational rearrangements and changes in protein composition catalyzes the first step of the splicing reaction, leading to the formation of a lariat intermediate (complex C). An additional conformational switch leads to the second catalytic step, rendering the spliced product (mRNA) and the intron lariat.
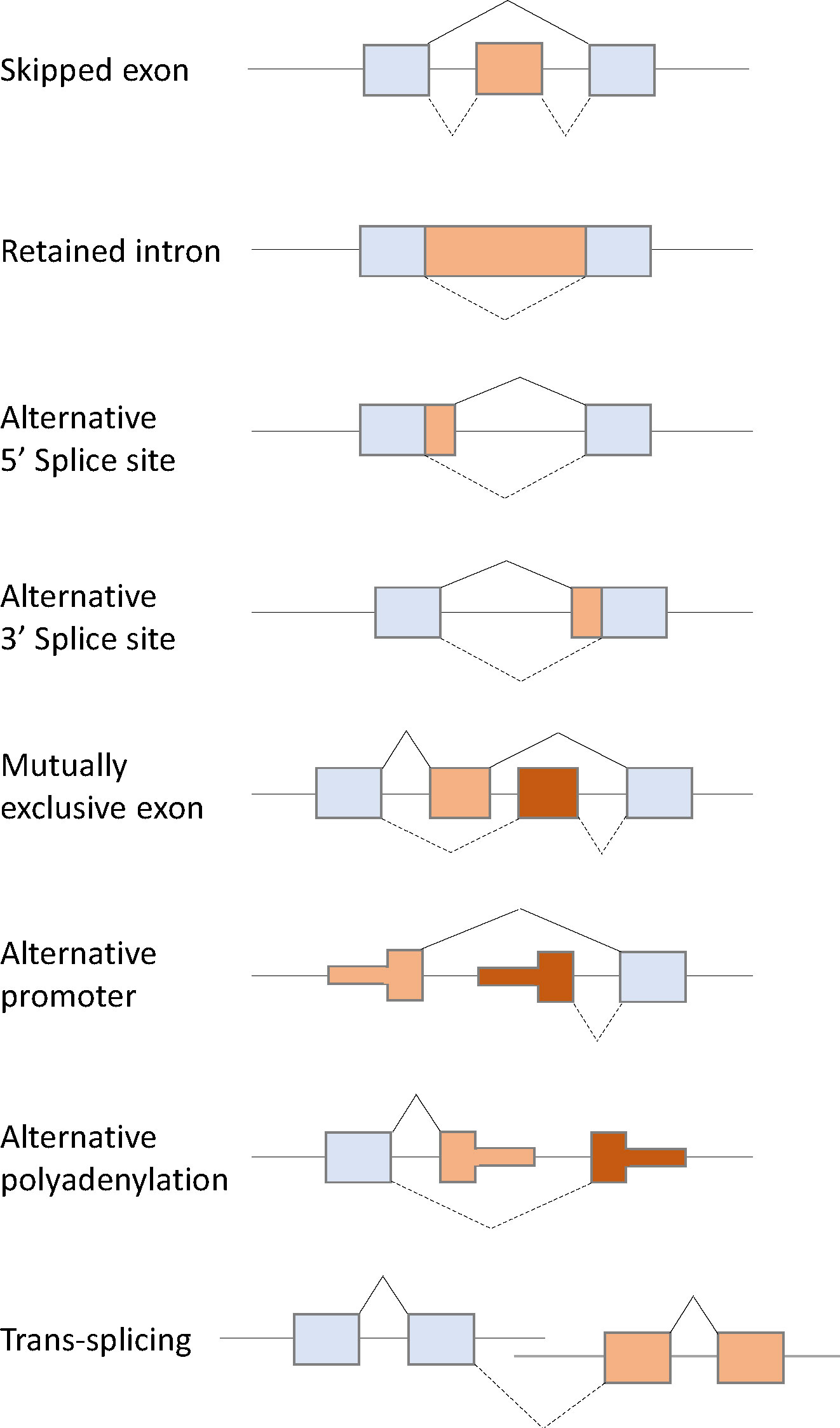
Figure 2 Schematic diagram of eight types of alternative splicing: Exon skipping, intron retention, alternative 5′ splice site, alternative 3′ splice site, mutually exclusive exon, alternative promoter, alternative polyadenylation, and trans-splicing.
A recent whole genome sequence analysis showed that spliceosome-related genes are mutated in multiple tumors. The most commonly mutated splicing factor was SF3B1, which is a core component of U2 snRNP (18, 19). SF3B1 mutation was observed in 90% of patients with myelodysplastic syndrome (MDS) with ring sideroblasts, which is characterized by anemia, iron-rich mitochondria surrounding the nuclei of erythroid precursors, and a favorable prognosis (18–21). SF3B1 is mutated in the early stage of myeloid malignancies, whereas SF3B1 mutations in chronic lymphocytic leukemia (CLL) are most commonly subclonal and enriched in more advanced and aggressive disease (22, 23). SF3B1 mutations induce cryptic intron-proximal 3′ss selection through usage of a different branch point, since U2 snRNPs recognize the branch point sequence within the intron (24, 25). The most common mutation in SF3B1 is the K700E mutation, which is observed in myeloid malignancies, CLL, and multiple solid tumors. The SF3B1-K700E mutation has been implicated in cancer progression (26, 27). A conditional knock-in mouse with heterozygous expression of SF3B1-K700E causes progressive macrocytic anemia (27), and a knock-in mice with hematopoietic-specific expression of SF3B1-K700E impaired erythropoiesis and progressive anemia without ringed sideroblasts, accompanied by a reduction in hematopoietic stem cells (26). Additional hot spot mutations with SF3B1, such as R625 and E902, are enriched in melanomas and bladder carcinomas; however, the lineage specificity of these hotspot mutations remains to be elucidated (28). In multiple cancers, the SF3B1 mutation has been linked to dysregulation of several cellular pathways and functions, DNA damage response, R-loop formation, telomere maintenance, and Notch signaling (29–31). In addition, SRSF2, U2AF1, and ZRSR2 mutations are observed. SRSF2, one of the SR proteins, is frequently mutated in acute myeloid leukemia (AML) (32). Canonical SRSF2 equally binds CCNG and GGNG sequences and induces alternative splicing (33). However, SRSF2 mutations preferably alter splicing of exons with C-rich sequences than with G-rich sequences (32, 34, 35). The SRSF2 mutation leads to splicing alterations of EZH2, which results in reduced expression by nonsense-mediated decay, thereby impairing hematopoietic differentiation in vivo (34). Additionally, frequent overlap of SRSF2 and IDH2 mutations alters splicing of INTS3, leading to leukaemogenesis due to reduced expression with nonsense-mediated decay (32). Hotspot mutations in U2AF1 are mainly located in the S34 or Q157 residue. S34 mutations promote inclusion of exons whose 3′ss is C-rich, whereas Q157 mutations enhance inclusion of exons whose 3′ss is G-rich. The U2AF1 mutation alters hematopoiesis and splicing in hematopoietic progenitor cells, thereby contributing to abnormal hematopoiesis in vivo (36). Mutations in ZRSR2 are identified in approximately 10% of patients with MDS (37, 38). These mutations are located sporadically across the coding regions, suggesting that these mutations are loss-of-function mutations and distinct from the change-of-function mutations in SF3B1, SRSF2, and U2AF1. ZRSR2 mutations, which predominantly occur in males, induce abnormal splicing via U12-dependent introns, leading to hematopoietic stem cell self-renewal and disorders (39, 40).
Members of kinase families, including serine–arginine protein kinases (SRPKs) and CDC-like kinases (CLKs), phosphorylate the RS domains of SR proteins, thereby regulating their subcellular localizations and interactions with ESEs or ISEs of pre-mRNAs. SRPKs (SRPK1, SRPK2, and SRPK3) are serine–threonine kinases that phosphorylate serine residues within serine/arginine (S/R) dipeptides enriched in SR proteins (41, 42). SRPKs contain a nonhomologous spacer insert domain that bifurcates the kinase domain and anchors the kinase in the cytoplasm through interactions with chaperones (43–45) (Figure 3). SRPKs phosphorylate SR proteins in the cytoplasm and induce their nuclear import by increasing their affinity to the transportin complex (46). Activated Akt induces SRPK autophosphorylation that switches the chaperones complex of SRPK, thereby enhancing SRPK nuclear translocation (45, 47). SRPKs and CLKs simultaneously regulate the shuttling of SR proteins between nuclear speckles and the nucleus via phosphorylation of different regions of SR dipeptides (48, 49). SRPK1 and SRPK2 are ubiquitously expressed in human tissues, whereas SRPK3 is predominantly expressed in muscle tissues (50, 51). Consistent with the expression pattern in humans, Srpk1-deficient mice show embryonic lethal and Srpk3-deficient mice show a type 2-specific myopathy (51, 52).
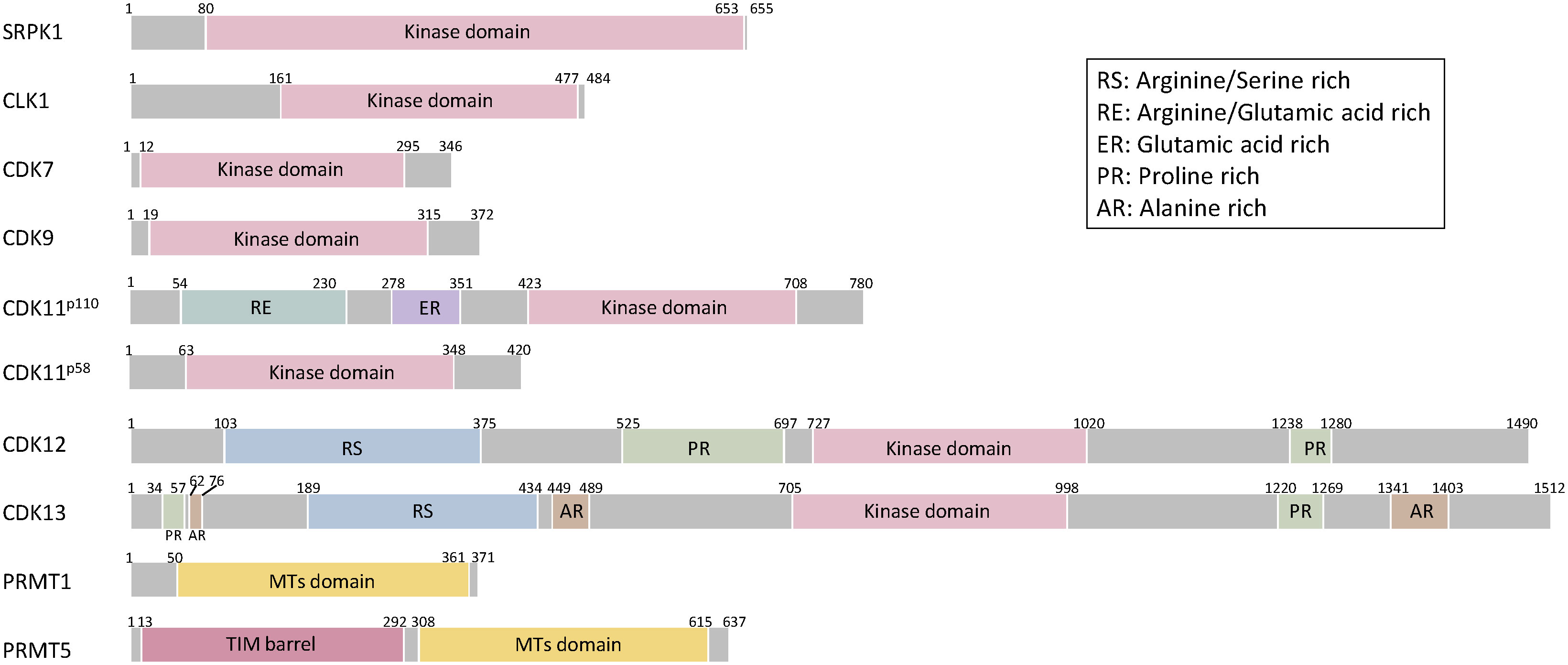
Figure 3 Schematic structures of splicing-related proteins. SRPK1, CLK1, CDK7, CDK9, CDK11, CDK12, CDK13, PRMT1, PRMT5. The Kinase domain of SRPK1 includes internal spacer sequence that separates the bipartite kinase catalytic core. CLK1 has a kinase domain at the C-terminus. CDK family proteins have consensus serine/threonine kinase domain. In addition to consensus kinase domain, CDK11 has two separate domains and arginine/glutamic acid rich (RE) domains are linked to association with RNA processing factors and glutamic acid rich (ER) domain support RE domain function and aide in keeping these proteins subnuclear. CDK11p58 is a short isoform of CDK11p110 missing 1-360 amino acids. CDK12 and CDK13 have additional arginine/serine rich (RS) domain, serve as docking sites for assembly of splicing factors and regulation of splicing and proline-rich (PR) domains serve as binding sites for Src-homoogy3, WW or profilin-domain-containing proteins. CDK13 has three alanine-rich (AR) domains. The methyl transferases domain of PRMT1 and 5 is consists of the catalytic Rossman fold and Β-barrel. PRMT5 adopts a TIM barrel structure at the N-terminal, which interacts with the C-terminal catalytic domains of adjacent monomers.
CLKs comprise four members, i.e., CLK1–4, and a phylogenetic kinome analysis revealed that CLK1 and CLK4, CLK2 and CLK3 have higher similarity (53). In addition to the phosphorylation of arginine/serine dipeptides, CLKs phosphorylate the SR proteins of serine/proline; this step is critical for binding of the SR protein by CLK1 but not for SRPK1-dependent reaction (54). CLKs have a C-terminal kinase domain and an N-terminal noncatalytic domain with a serine/arginine-rich region (55) (Figure 3). CLKs phosphorylate SR proteins in the nucleus. Subsequently, the kinase domain of SRPK1 interacts with the N-terminal domain of CLK1 and leads to the release of SR proteins from nuclear speckles and induces the interaction of SR proteins with U1 snRNP to proceed splicing machinery (56, 57). CLKs are ubiquitously expressed in human tissues and exhibit alternative splicing-independent functions. CLK2 phosphorylates the SR domain on peroxisome proliferator-activated receptor γ coactivator (PGC-1α) and functions as an insulin-regulated suppressor of hepatic gluconeogenesis through disruption of PGC-1α (58, 59). CLK4 phosphorylates nexilin and regulates cardiac function (60). Cardiac-specific Clk4-knockout mice show pathological myocardial hypertrophy with progressive left ventricular systolic dysfunction and heart dilation (60).
Cyclin-dependent kinases (CDKs) are classified into cell cycle-related and transcriptional-related subfamilies based on their substrate specificity (42, 61). Among the transcriptional-related CDKs, CDK7, CDK9, CDK11, CDK12, and CDK13 are directly or indirectly involved in the regulation of alternative splicing through reversible phosphorylation of the C-terminal domain (CTD) of RNA polymerase II (pol II). CDK7 preferentially phosphorylates Ser5 and Ser7 of RNA Pol II CTD and regulates transcriptional initiation and alternative splicing (62, 63). In addition to the phosphorylation of pol II, CDK7 phosphorylates multiple splicing factors, such as SF3B1, U2AF2, CDK9, CDK12 and CDK13 (63). Thus, a CDK7-selective inhibitor alters multiple splicing events, such as exon skipping and retained intron type of splicing (63). CDK9 preferentially phosphorylates Ser2 of RNA Pol II CTD and controls pol II pause release, elongation, alternative splicing, and polyadenylation (64–66). CDK9, together with cyclin T, forms positive transcription elongation factor b (P-TEFb), which controls the elongation phase of transcription by RNA pol II. CDK9 inhibition abrogates polyadenylation and causes an elongation defect starting at the last exon of a protein-coding gene. SILAC phospho-proteomics analysis identified that multiple splicing factors, such as SF3B1, are phosphorylated by CDK7, and CDK9 inhibition promotes the loss of interaction of SF3B1 (66). CDK9 and PP2A regulate mRNA cleavage and polyadenylation as well as alternative poly(A) site usage, since inhibition of PP2A abrogates the effect of CDK9 inhibition on transcription at the 3′end of the gene (66). CDK11 is encoded by CDK11A and CDK11B, whose mRNA is translated into two major CDK11 proteins, CDK11p110 and CDK11p58 (61, 67). CDK11–cyclin L complex involves transcription and RNA processing, in particular, alternative splicing (68). In addition to preferential phosphorylation of Ser2 of RNA Pol II CTD, CDK11 phosphorylates threonine residues of SF3B1 at its N terminus during spliceosome activation, thereby promoting binding between SF3B1 and U5/U6 snRNAs (69).
CDK12 and CDK13 exhibit 92% similarity in their kinase homology domain; thus, these are regarded as structurally and functionally redundant kinases (70). CDK12/CDK13 preferentially phosphorylate Ser2 of RNA Pol II CTD and regulate pol II elongation, termination, and alternative polyadenylation (71–75). CDK12/CDK13 bind to cyclin K and active complex while regulating transcriptional elongation and preferentially express DNA damage response and repair (DDR) genes to protect cells from genomic instability. Inhibition of CDK12 using siRNAs or small-molecule compounds leads to a predominant decrease in the expression of long genes with high numbers of exons, including breast and ovarian cancer type 1 susceptibility protein 1 (BRCA1), ataxia telangiectasia and Rad3-related (ATR), FA complementation group I (FANCI), and FA complementation group D2 (FANCD2), thereby inducing spontaneous DNA damage (76). CDK12 loss leads to elongation defects and increases intronic polyadenylation of DDR genes depending on gene length and the U1 snRNP/polyadenylation signal ratio, since the U1 snRNP complex prevents premature termination via recognition and inhibition of cryptic poly(A) sites (72, 74, 77–79). Interestingly, SILAC analysis also identified that CDK12 phosphorylates SF3B1, similar to CDK7 and CDK9. CDK12 loss of function induces minimal effect on alternative splicing (74). The phosphorylation site of SF3B1 could be critical for the regulation of alternative splicing and polyadenylation. More detailed analysis of the phosphorylation of mRNA processing-related proteins is expected to be performed in the future.
Protein arginine methyltransferase 5 (PRMT5) and 1 (PRMT1) are directly and indirectly involved in the splicing machinery, respectively. PRMT5 is an essential type II arginine methyltransferase and catalyzes the formation of symmetric demethylated arginine on a variety of proteins and histones, thereby regulating chromatin structure, gene transcription, cellular differentiation, and pre-mRNA splicing (80). Methylation by PRMT5 is required for recruiting substrate adaptors, such as spliceosome subunits, SmB/B′, SmD1, and SmD2, through a TIM barrel domain at the N terminus, thereby regulating mRNA processing, including splicing activity (81). PRMT1 is a type I arginine methyltransferase and catalyzes the formation of asymmetrically demethylated arginine (82). PRMT1 methylates the RNA-binding protein RBM15 that preferentially binds to the specific intron region of SF3b, thereby affecting alternative splicing (80). Regarding the structure, PRMT5 and PRMT1 have a conserved core including a Rossman fold for cofactor binding and a Β-barrel for substrate binding (83) (Figure 3). PRMT5 also has a triose-phosphate isomerase (TIM) barrel at the N terminus, which is required for enhanced methyltransferase activity by forming a complex with methylosome protein 50 (83, 84).
SRPK1 is upregulated in pancreatic, breast, colonic, prostate, and non-small cell lung carcinomas and leukemia (85–90). Upregulation of SRPK is correlated with poor prognosis of breast cancer metastasis-free survival and overall survival time of non-small cell lung carcinoma (91, 92). In contrast, upregulation of SRPK1 is observed in patients with germ cell tumors responding to standard chemotherapy with statistical significance (93). In addition, another study showed that reduced SRPK1 protein expression is associated with decreased response to chemotherapy in retinoblastoma patients (94). Interestingly, aberrant SRPK1 expression in either direction, up or down, is tumorigenic (52). Downregulated SRPK1 leads to constitutive Akt activation through impairing the recruitment of an Akt phosphatase, a pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP1). Overexpression of SRPK1 also activates Akt by isolating PHLPP1. These results suggest that upregulated or downregulated expression of SRPK1 is observed in human tumors and that SRPK1 is involved in tumor progression, migration, and angiogenesis via splicing-dependent and -independent machineries. In splicing-dependent machinery, SRPK1 regulates multiple splicing and leads to tumor progression. In particular, the splicing variant of the vascular endothelial growth factor (VEGF) is dominantly regulated by SRPK1 (95). A spliced isoform, VEGF165, exhibits proangiogenic activity, whereas VEGF165b exhibits antiangiogenic activity (96). Aberrant upregulation of SRPK1 induces phosphorylation and nuclear translocation of SRSF1 and leads to splicing of VEGF165 in Wilms Tumor and melanoma (95, 97). Knockdown or chemical inhibition of SRPK1 alters splicing from VEGF165 to VEGF165b, thereby inhibiting angiogenesis and tumor progression (Figure 4). CRISPR-Cas9 screen shows that SRPK1 involves genetic vulnerability in AML cells (98). Comprehensive RNA-seq alternative splicing analysis using an SRPK inhibitor revealed that multiple events of splicing, especially exon skipping events, were altered in leukemic cells (90). SPHINX31, an SRPK inhibitor, suppressed growth and induced differentiation of human AML cells without causing any severe side effects, including hematopoiesis (90). Among altered splicing events, the bromodomain 4 (BRD4) isoform was altered from the short to the long isoform, eliminating BRD4 recruitment to chromatin involved in leukemogenesis including BCL2 and MYC (Figure 4). These results suggest that SRPK plays a key role in tumor progression in terms of splicing regulation.
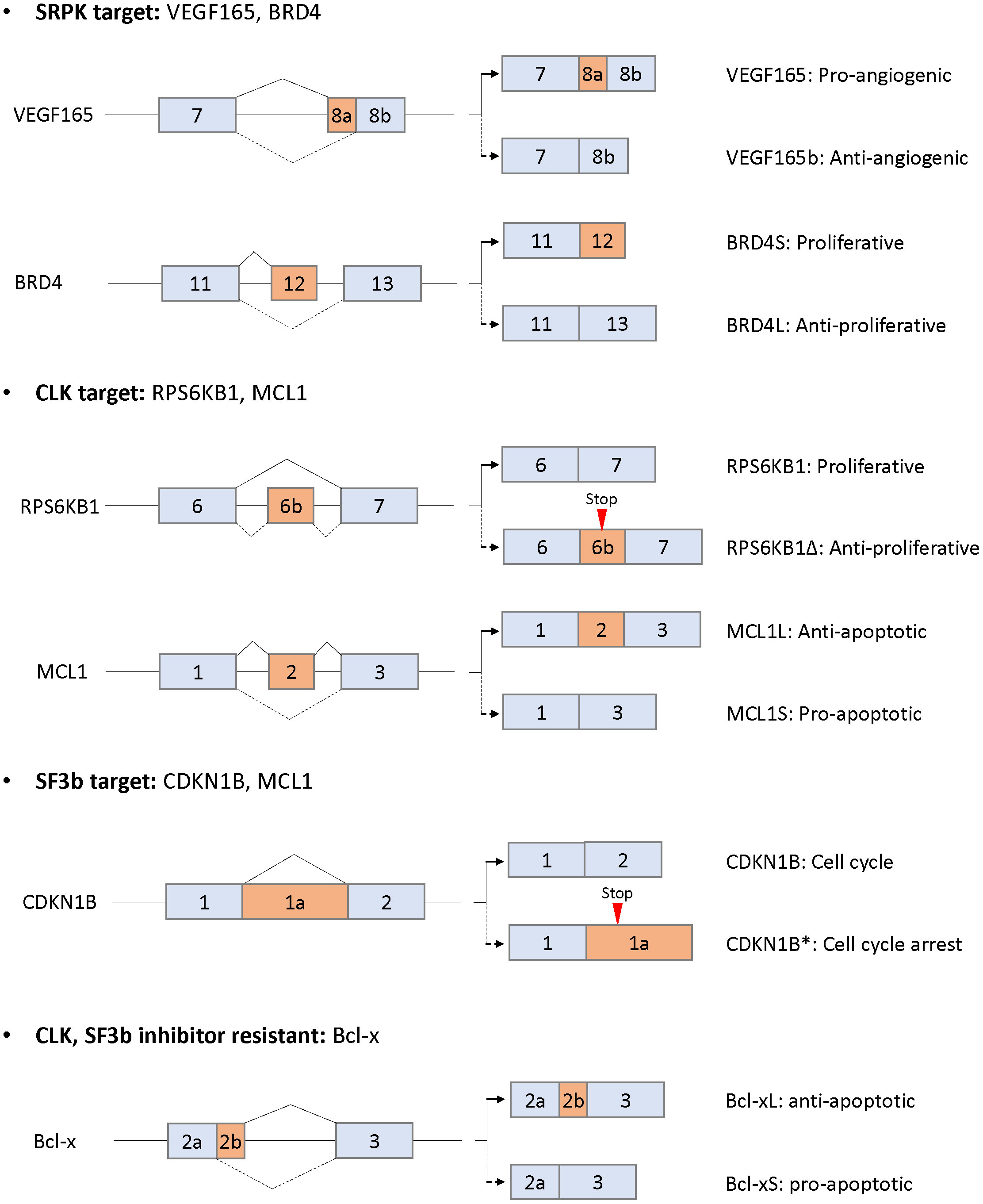
Figure 4 Alternative splicing as downstream targets for SRPK, CLK, and Sf3b and resistance to CLK or SF3b inhibitors. Only the alternatively spliced isoforms and the flanking exons are shown (not at scale). The corresponding isoforms are indicated by solid or dotted lines with the respective functions.
CLK1 and CLK2 are upregulated in breast and colorectal carcinomas and glioblastoma (99, 100). Upregulated expression of CLK1 and CLK2 is negatively correlated with prognosis in patients with kidney tumors and glioblastoma/colorectal carcinoma, respectively (100–102). The expression of CLK1 is regulated via the ubiquitin degradation mechanism during cell cycle (101). In the G2/M phase, the protein level of CLK1 is increased, whereas the protein levels of CLK2, SRPK1, and SR remain constant. Interestingly, thousands of cell cycle-dependent alternative splicing changes, including changes in the levels of RNA-binding proteins, are regulated by CLK1, suggesting that RNA metabolism is regulated by cell cycle and is aberrant in patients with tumors of higher mitotic index. Downregulation of CLKs inhibits cell growth and induces apoptosis in breast cancer and glioblastoma in vitro and in vivo (99, 102). CLKs also exhibit cancer cell growth inhibitory activity by modulating alternative splicing in multiple solid cancers (17, 103–107). A benzothiazole compound TG003 was first identified as a selective CLK inhibitor (108). TG003 inhibited cell proliferation and induced apoptosis in prostate cancer cells in vitro and in vivo through splicing changes of cancer-associated genes, including CENPE, ESCO2, CKAP2, MELK, ASPH, and CD164 (107). TG003 also switched splicing to the short isoform of estrogen-related receptor Β, which is involved in G2/M cell cycle arrest and apoptosis induction (104). In addition, several compounds (Cpd-1, Cpd-2, and Cpd-3) with CLK inhibitory activity altered multiple splicing including cancer growth and survival-related genes, such as RPS6KB1, EGFR, EIF3D, and PARP (Figure 4) (105). The splicing alteration levels were correlated with cell growth inhibitory activity. Other CLK selective inhibitors, T3 and T-025, preferentially induced exon skipping type of splicing in colorectal cancer cell lines and other multiple cancer cell lines, since phosphorylated SR proteins play a central role in exon recognition (17, 103). Interestingly, T3 induces multiple conjoined genes, which arise from the upstream transcript to the downstream transcript of partner genes with poly(A) sites (17, 109, 110), whereas there is no evidence that conjoined genes are involved in cancer progression. Oral administration of T-025 at a concentration of 50 mg/kg twice daily/2 days per week for 3 weeks showed significant antitumor efficacy in MDA-MB-468 xenograft model without body weight loss (103). These results suggest that CLKs play central roles in cancer progression through predominant exon skipping of multiple genes in splicing alteration.
Multiple studies indicating aberrant expression of CDK7 in cancer have been reported (111). CDK7 expression is correlated with poor prognosis and overall survival in triple-negative breast cancer, gastric cancer, ovarian cancer, oral squamous cell carcinoma, hepatocellular carcinoma, and glioblastoma (112–118). Similar to CDK7, the expression of CDK9 has gained attention as a prognostic biomarker in bladder cancer, papillary thyroid carcinoma, pancreatic cancer, osteosarcoma, and breast cancer (119–124). CDK11 has emerged as a target of interest for cancer therapy (67, 125). Downregulation of CDK11 expression effectively inhibits cell proliferation and induces cell death in breast cancer, osteosarcoma, ovarian cancer, and liposarcoma cells as well as xenograft tumors (126–130). Upregulation of CDK11 protein expression levels are associated with poorer patient survival in osteosarcoma and ovarian cancer (127, 130). CDK12 is involved in cancer dysregulation via both gain of function with gene amplification and loss of function with mutations encoding the CDK12 locus. CDK12 is coamplified with the neighboring Her2 gene at the RNA, protein, and phosphosite levels in breast cancer (131). CDK12 is the most common coamplification gene, along with Her2, in breast, stomach, biliary tract, and colorectal cancers (132). Amplified CDK12 is associated with disease recurrence and poor prognosis and exhibits anti-HER2 therapy resistance in patients with breast cancer (133). Since CDK12 regulates the expression of genes involved in the activation of ErbB–PI3K–AKT or Wnt signaling cascades, CDK12 inhibition is more sensitive to antitumor activity in CDK12-Her2 coamplified cells than in CDK12 no-amplified Her2 positive cells in vitro and in vivo (133). Furthermore, since CDK12 predominantly regulates the expression of DDR-related genes, CDK12 inhibitors in combination with PARP inhibitors induce synthetic lethality in cancer cells (72, 134). In contrast to genomic amplification, CDK12 mutation or deletion has been reported in <5% of prostate and ovarian adenocarcinoma cases (135, 136). CDK12 mutations/deletion are loss-of-function type by nonsense mutations or loss-of-function type by frameshift mutation due to indels and are consistently associated with a particular genomic instability pattern induced by hundreds of tandem duplications in ovarian cancer and metastatic castration-resistant prostate cancer (136, 137). Regardless of the genomic instability pattern, the expression of BRCA1, BRCA2, or other genes encoding long transcripts was not affected under the CDK12-mutation condition (137), suggesting that different genomic instability pathways exist between CDK12 mutation and amplification status.
Upregulation of PRMT5 is observed in lymphomas, breast cancer, lung cancer, colorectal cancer, and glioblastoma (138–143). The expression level of PRMT5 is associated with poor overall survival (144). Involvement of PRMT5 in glioblastoma cell growth was identified by shRNA screen (145). Disruption of PRMT5 induces apoptosis and inclusion of detained introns, which are spliced and polyadenylated transcripts. In contrast to retained introns, detained introns remain in the nucleus and are finally post-transcriptionally spliced or degraded, thereby affecting the level of mature coding mRNA (145). A PRMT5 inhibitor (GSK591 or LLY-283) inhibited the growth of patient-derived glioblastoma stem cells in accordance with splicing alteration levels (146). PRMT inhibition altered multiple splicing events, particularly encoding cell cycle-related genes. In addition, the PRMT5 inhibitor induced antitumor effects in glioblastoma patient-derived xenografts (146). Orally available PRMT5 inhibitor (EPZ015666 [GSK3235025]) inhibits SmD3 methylation and RNA splicing as well as induces antitumor efficacy in mantle cell lymphoma (MCL) cells in vitro and in vivo (147). In addition, PRMT5 silencing or inhibition (EPZ015666) induced antitumor effect in glioblastoma cells in vitro and in an in vivo PDX model by removing detained introns, thereby regulating rapid expression of transcripts associated with proliferation (145).
Multiple small-molecule compounds targeting splicing have been developed, and clinical trials have been conducted in patients with a variety of solid and hematopoietic tumors using multiple compounds. Currently, no approved compound modulating pre-mRNA splicing is available for cancer treatment. This section highlights promising small-molecule splicing modulators that can serve as anticancer therapeutics, especially those under clinical trials.
Compounds derived from natural products modulating pre-mRNA splicing have been identified as FR901464, spliceostatin A (SSA; a methyl ketal derivative of FR901464), sudemycin, pladienolide B, and E7107 (a 7-urethane derivative of plaedienolide D). These compounds bind to the SF3b subcomplex of U2 snRNP and modulate alternative splicing, thereby inhibiting cancer cell growth (148–150). Among SF3b complex binders, the detailed mechanism of action of spliceostatin A has been most clarified (149, 151–156). SSA produces the C-terminal spliced truncated form of p27 encoding CDKN1B mRNA via splicing modulation and stabilization of CDKN1B mRNA, which are the causes of G1 phase cell cycle arrest by treatment with spliceostatin A (Figure 4) (149, 151, 154). In addition, SSA downregulates the mRNA of CCNE1, CCNE2, and E2F1, which are additional genes involved in G1 arrest (152). One of the mechanisms downregulating gene expression is that SSA induces early dissociation of RNA polymerase II and decreases phosphorylation of the Ser2 of chromatin-bound RNA polymerase II (153). Other analysis showed that SSA predominantly induces intron retention type of splicing, the part of which leaks into the cytoplasm depending on the strength of the 5′ss and the length of the transcripts (156). SSA also affects the transcription of a nuclear long noncoding RNAs, including MALAT1. MALAT1 is prematurely cleaved and polyadenylated, and the truncated transcripts are exported into the cytoplasm and translated as aberrant proteins (155). Through this mechanism, the SF3b inhibitor, E7107, exerts antitumor activity in a human xenograft model without severe toxicity (157). Phase I clinical trials using E7107 in patients with solid tumors have been conducted. Intravenous treatment for 3 consecutive weeks showed that one patient had bilateral optic neuritis (158). Another administration on days 1 and 8 every 21 days revealed that two patients had visual loss due to optic nerve dysfunction at cycles 2 and 7 (159). The incidence of two cases of vision loss probably related to E7107 led to study discontinuation, although tumors in some patients responded partially to E7107. An orally available SF3b inhibitor, H3B-8800, preferentially inhibited tumor growth in spliceosome-mutant cancer cells, such as SF3B1 K700E, in vitro and in vivo by predominantly binding with short GC-rich regions (160). Currently, phase I clinical trial for H3B-8800 is ongoing in patients with MDS, AML, and chronic myelomonocytic leukemia (NCT02841540). Since SF3B1 mutation is observed in 90% of patients with MDS, good efficacy without side toxicity by administration with H3B-8800 will be expected.
Among protein kinase inhibitors, the therapeutic potential of splicing-related kinases SRPKs and CLKs has gained attention. Two clinical trials (NCT04247256 and NCT04652206) are currently being conducted via oral administration of an SRPK inhibitor, SCO-101 (Table 1). SCO-101 inhibits both ATP-Binding Cassette (ABC) G2 efflux pumps and SRPK1, thereby showing antitumor potential in combination with docetaxel in triple-negative breast cancer cells (161). A phase II clinical study has been conducted in patients with 5-flurouracil, leucovorin, and irinotecan (FOLFIRI)-resistant metastatic colorectal cancer (NCT04247256). The objectives of this study are 1) to determine the maximum tolerated dose (MTD) in patients receiving FOLFIRI plus escalating doses of SCO-101 and 2) to assess the safety, toxicity, and efficacy of the combination of SCO-101 and FOLFIRI in patients receiving FOLFIRI and the MTD of SCO-101 (162). Another clinical trial is a phase Ib study in combination with gemcitabine and nab-paclitaxel for patients with pancreatic ductal adenocarcinoma to establish the safety profile and MTD of SCO-101 (NCT04652206). Current clinical trials are focusing on tumors resistant to chemotherapy, because SCO-101 has the potential to suppress drug efflux via ABCG2. Additional remarkable profile for SCO-101 is SRPK1 inhibition. Cancer cell growth inhibition is proven in pancreatic cell lines by knockdown of SRPK1 through modulating alternative splicing, and upregulation of SRSF1 in pancreatic cancer is observed in patients resistant to gemcitabine (87, 163). Since preclinical studies have shown that SRPK1 regulates multiple splicing, a pharmacodynamic (PD) biomarker to monitor splicing alteration in patients should be developed.
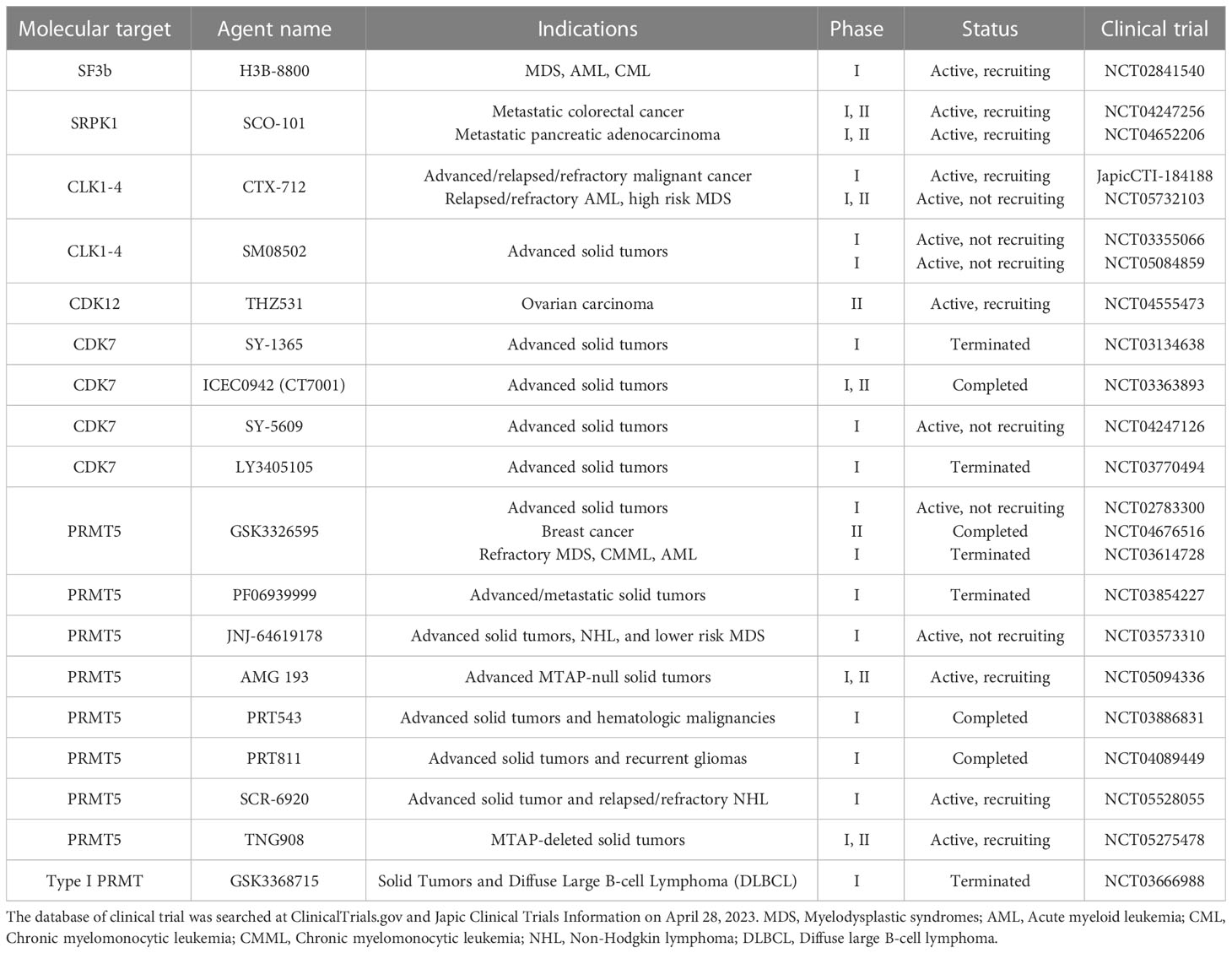
Table 1 Small-molecule splicing modulators in clinical trial.
Two orally available CLK inhibitors (SM08502 and CTX-712) have entered clinical trials (Table 1). SM08502 is selective to CLK2 and CLK3 inhibition, thereby inhibiting phosphorylation of SR proteins (SRSF6) and enlarged nuclear speckle (164). In addition, SM08502 exerts antitumor activity in colorectal cancer cells in vitro and in vivo by decreasing gene expression and altering genes of the Wnt pathway, such as splicing of LEF1, TCF7L2, and DVL1 (164). Currently, a phase I clinical trial is being conducted to evaluate the safety, tolerability, pharmacokinetics, PD, and preliminary antitumor efficacy of SM08502 in patients with advanced solid tumors resistant to standard therapy (NCT03355066), and another phase I study is being conducted in combination with hormonal therapy or chemotherapy in patients with castration-resistant prostate cancer, non-small cell lung cancer, and colorectal cancer (NCT05084859). CTX-712 is being orally administrated twice a week in a phase I study to determine the recommended dose (RD) by evaluating MTD, dose limiting toxicity, safety, pharmacokinetics, and PD profiles (JapicCTI-184188). Interim results from the phase I clinical trial revealed that two partial responses (PRs) and two complete responses (CRs) were observed in patients with ovarian cancer and AML, respectively, with a dose-dependent increase in splicing alteration (165). An additional interim phase I trial showed that four CRs and one CR with incomplete hematologic recovery were observed in eight patients with AML and MDS (166). Further phase I studies to determine the phase II dose will be started in patients with relapsed/refractory AML and higher risk MDS (NCT05732103), and detailed reports are expected to provide insightful information for cancer therapy.
A phase II/III clinical study (NCT01580228) has been conducted using a pan-CDK (CDK1/2/5/9) inhibitor, dinaciclib, in patients with relapsed/refractory CLL (167). Administration of dinaciclib at escalating doses of 7 to 10 to 14 mg/m2 (on days 1, 8, and 15, respectively) in cycle 1 and 14 mg/m2 in cycle 2 and thereafter (1 cycle, 5, 28 days) for 12 cycles provides an acceptable safety and tolerability profile with typical CDK inhibitor-oriented adverse events represented by tumor lysis syndrome; however, its efficacy was limited and clinical trials were terminated (167). Encouraging preclinical data show that dinaciclib is also a potent inhibitor of CDK12 and sensitizes to triple-negative breast cancer cells in combination with PARP inhibitors by disrupting residual homologous recombination activity (168). THZ1, a covalent CDK7/12/13 inhibitor, exerts antitumor activity through transcriptional regulation by inhibiting CDK7 in acute T cell leukemia, MYCN-amplified neuroblastoma, small cell lung cancer, and triple-negative breast cancer (169–172). In addition to CDK7 inhibition, THZ1 and THZ531, CDK12/13 inhibitors, led to the reduction of the expression of DDR genes and induction of synthetic lethality, along with PARP inhibitors, in Ewing sarcoma cells in vitro and PDX model in vivo (173). THZ531 is currently being used in a longitudinal observational phase II study to assess its sensitivity via CDK12 inhibition using organoids derived from patients with high-grade serous ovarian cancer receiving PARP inhibitors (NCT04555473) (11). Most CDK12 inhibitors are still being studied in the preclinical stage, and several CDK12 selective inhibitors have shown promising results in the preclinical stage (174, 175). Since THZ1 is a substrate of multidrug transporters ABCB1 and ABCG2, tumor cells show resistance to THZ1. To overcome the resistance of THZ1, E9 was synthesized and overcame ABC-mediated resistance (176). Additionally, new types of CDK12 inhibitors as bifunctional protein degraders have emerged (177). Molecular glue compounds can mediate protein–protein interactions between a target protein, such as CDK12 and ubiquitin ligase to induce selective protein degradation (178). Proteolysis-targeting chimeras (PROTACs) consist of three elements: a ligand for the target protein, such as CDK12, a ligand for E3 ligase, and a linker to induce proteasome-mediated degradation of the target protein (178). Molecular glues and PROTACs increase selectivity for the target protein, such as CDK12, without inhibition of CDK13 function, although the compounds are still in the pre-clinical stage. In addition to CDK12, CDK7 inhibitor sensitizes DDR-proficient cancer cells to PARP inhibitor (179). SY-1365, a selective CDK7 inhibitor, inhibited cell growth in many different cancer types at nanomolar concentrations (180). In addition, SY-1365 decreased the level of oncogenic transcripts and DDR genes, such as RAD51 and CHEK1. SY-1365 is currently under clinical trials for patients with ovarian and breast cancer (NCT03134638). Other three CDK7 selective inhibitors, namely, ICEC0942 (CT7001), SY-5609, and LY3405105, have progressed to phase I/II clinical trials (111), NCT03363893; NCT04247126; and NCT03770494). Multiple CDK9 inhibitors have also proceeded to clinical trials for patients with multiple solid and bladder tumors (181–183).
Multiple PRMT5 inhibitors, which have potential antitumor activity and regulate splicing, are under clinical trials. An orally available PRMT5 inhibitor (GSK3326595) inhibited the methylation of Sm proteins and induced cell death in MCL in vitro and in vivo (147). Three clinical trials have been conducted in patients with solid tumors (NCT5094336), relapsed/refractory myelodysplastic syndrome (MDS), chronic myelomonocytic leukemia (CMML), hypoproliferative AML (NCT03614728), solid tumors, and non-Hodgkin’s lymphoma (NCT02783300). The adverse effects observed among the abovementioned trials were common, such as fatigue and anemia, but manageable, and PRs were observed in patients with several tumor types, such as human papillomavirus+ cervical cancer (1/1 subject) and ACC (3/14 subjects) in the NCT02783300 study (184). Clinical trials for other PRMT5 inhibitors are in progress: AMG 193, JNJ-64619178, PF-06939999, PRT543, PRT811, SCR-6920, and TNG908 (185), NCT05094336, NCT03573310, NCT03854227, NCT03886831, NCT04089449, NCT05528055, and NCT05275478), although it has not been reported that these inhibitors have splicing modulation activity.
To date, a limited number of PRMT1 inhibitors have proceeded to clinical trial. GSK3368715, a PRMT1 inhibitor, exerts antitumor activity in vitro and in vivo in multiple cancer types with splicing alteration activity of exon usage (186). A phase I clinical trial of GSK3368715 has been conducted in patients with relapsed/refractory diffuse large B-cell lymphoma and solid tumors. However, according to NCT03666988 of ClinicalTrials.gov, the current status of patient recruitment is terminated because overall benefit-risk profile did not support continuation of the study. Regarding patient stratification, co-deletion with methylthioadenosine phosphorylase (MTAP) and the tumor suppressor gene CDKN2A (p16) are observed in 40% of glioblastomas, 25% of melanomas and pancreatic adenocarcinomas, and 15% of non-small cell lung carcinomas (82). Subsequently, MTAP deficiency leads to the accumulation of 2-methylthioadenosine (MTA), which has the potential to inhibit PRMT5 activity (187–189). Genetic depletion of PRMT5 leads to vulnerability in MTAP deleted cells; however, GSK3235025, a PRMT5 inhibitor, surprisingly did not recapitulate vulnerability to PRMT5 depletion in MTAP deleted cells, implying that different inhibitory modes exist between MTA and small-molecule inhibitors, S-adenosylmethionine competitive and uncompetitive, respectively (188). Since the combination of PRMT1 and PRMT5 inhibitors exerts a synergistic antitumor effect (186), MTAP deficiency is associated with decreased induction of MMA and SDMA upon inhibition of type I PRMT activity. Given that the substantial population with MTAP deficiency includes many tumor types with limited therapeutic options, inhibition of type I PRMT activity by GSK3368715 may represent a promising approach for tumors of high unmet medical need with a defined patient selection strategy.
Recent progress in multiple clinical trials targeting pre-mRNA splicing indicates that various splicing modulators have potential value as a novel class of antitumor agents. In this review, specific splicing-related kinases and related inhibitors entering clinical trials are described. It is one of the critical factors to consider patient stratification strategy for increasing the success rate in clinical trials. As described in this review, splicing factor-mutated tumors have recently been observed and attracted considerable attention, although the detailed mechanisms underlying splicing dysregulation in cancer remain unclear. Some of the SF3b complex inhibitors, such as H3B-8800, have the potential to be preferentially effective to splicing factor-mutated tumors compared to nonmutated tumors (Table 2). In addition, recent evidence has indicated that PRMT inhibitors also have potential for possibility of patient stratification for tumors with mutation of splicing factors since PRMT inhibitors of class I (PRMT1) or class II (PRMT5) preferentially inhibits cell viability and delayed disease progression in splicing factor, SRSF2P95H, SF3B1Y765C, or SF3B1K700E mutated cells in vitro leukemia cells and in vivo AML PDX model compared to wild type cells (190) (Table 2). The results of ongoing clinical trial will be expected, and detailed molecular mechanism should be clarified to consider biomarker strategy. Other potential possibility for patient stratification is SRPK inhibitor, which specifically showed antitumor efficacy in AML cells since alternative splicing of BRD4 was altered by SRPK inhibitor (90). The inhibitors targeting bromo- and extra-terminal domain (BET) including BRD4 have remarkable antitumor activity in preclinical studies, and more than 20 clinical trials have been completed and ongoing (191). To date, limited clinical efficacies for BET inhibitor have been observed implying that predictive biomarker, pharmacodynamics marker, and combination strategy is lacking in clinical trials. Since SRPK inhibitors modulate multiple splicing events involving aberrantly expressed oncogenes, it is conceivable that SRPK inhibitor is more effective compared to BET inhibitor through combination of antitumor effect by multiple splicing changes. In addition, BRD4 forms fusion proteins with nuclear protein in testis (NUT). The BRD4-NUT fusion proteins have aggressive oncogenic property and involve in NUT-midline carcinoma (NMC) with a poor prognosis (192). In addition to AML, SRPK inhibitor could be effective to NMC, though toxicity should be carefully validated and monitored.
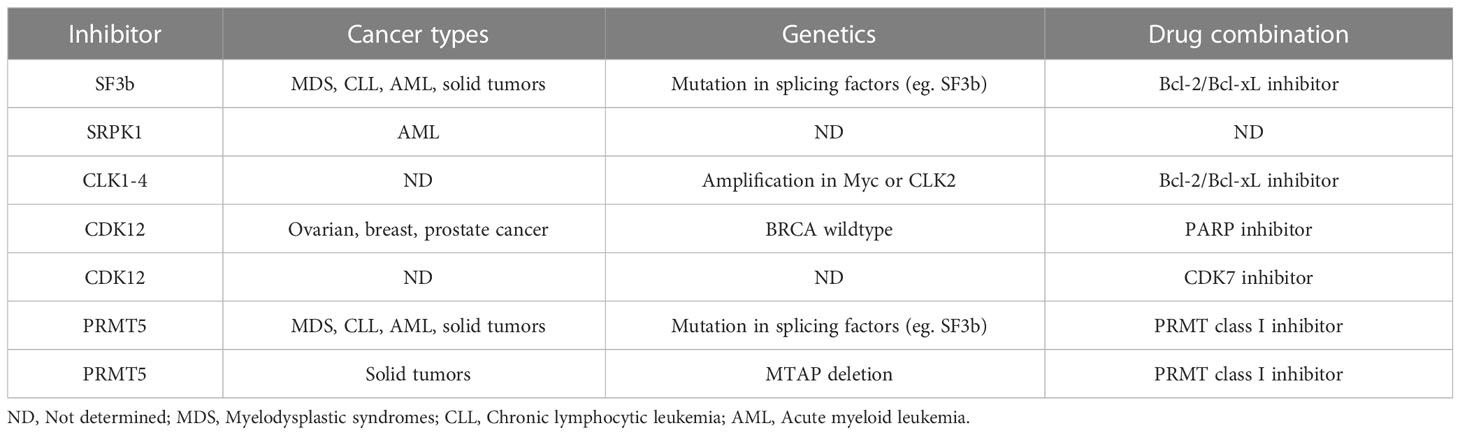
Table 2 Potential patient stratification and drug combination strategy in clinical.
Additional potential possibility of patient stratification suggests that, in MYC-driven cancer cells, increase of total RNA synthesis and protein translation lead to increased burden on the core spliceosome (193). This evidence supports that CLK inhibitor (T-025) sensitives to MYC-amplified tumors as well as CLK2 upregulated tumors (103). The AACR Project GENIE, an international data-sharing consortium, showing clinical-grade cancer genomic data with clinical outcome data for tens of thousands of cancer patients revealed that the amplification of MYC is predominantly found in breast invasive ductal carcinoma, lung adenocarcinoma, colon adenocarcinoma, prostate adenocarcinoma, and invasive breast carcinoma (194). Independent analysis based on Cancer Genome Atlas using 489 high-grade serous ovarian adenocarcinomas (HGSOC) showed that chromosome 8q including MYC has the most significant gains and occurred in 65% of HGSOC (195), and MYC amplification was the highest frequency in ovarian cancer compared to other tumor types (196). Additionally, MYC transcription is reduced by THZ531, a CDK12/13 inhibitor in vitro and in vivo, patient-derived xenografts from ovarian cancer patients, not CDK7 inhibitor (196). These evidences suggest that MYC dependent tumors, such as HGSOC, could be effective for CLK inhibitor and CDK12/13 inhibitor (Table 2).
In addition to patient stratification strategy, it is of particular importance to consider molecular mechanism-based combination therapy strategy. Independent group shows that SF3b inhibitor, E7107, or CLK inhibitor (T3) alters splicing of anti-apoptotic MCL1, but not that of anti-apoptotic Bcl-2 family, such as Bcl-2 and Bcl-xL (Figure 4) (197, 198). Combination of E7107 or T3 with Bcl-xL/Bcl-2 inhibitor synergistically induced apoptosis in cancer cells. Therefore, drug combination strategies with splicing modulator and Bcl-xL/Bcl-2 inhibitors could be effective. However, more detailed analysis should be needed to clarify the reason why splicing inhibitors are insensitive to splicing of Bcl-xL and Bcl-2, and identify the predictive biomarkers in clinics.
CDK12 inhibitor could be ideal to induce synthetic lethal in combination with PARP inhibitor in BRCA wildtype of tumor patients. Although CDK7 inhibitor also sensitizes DDR-proficient gene to PARP inhibitor, it has been suggested that the molecular mechanism between CDK12 and CDK7 would be different. Whereas CDK7 involves in oncogene-related transcription and alternative splicing, CDK12 regulates alternative premature intronic polyadenylation. This evidence implies that combination with CDK12 inhibitor and CDK7 inhibitor has synergistic antitumor effect, in particular, in ovarian and breast DDR-proficient tumors. PRMT inhibitors also have potential for possibility of drug combination. Combination of PRMT1 inhibitor and PRMT5 inhibitor shows stronger synergistic cell growth effect and antitumor efficacy in vitro and in vivo splicing factor-mutated cells compared to wild type cells (190). To date, the number of CDK12 selective inhibitor or PRMT1 inhibitor is limited in clinical trial. In addition to ATP-competitive inhibitors, molecular glue and PROTACs technology have recently gained attention to obtain selective CDK12 inhibitors due to the high homology between kinase domains of CDK12 and other CDKs, especially CDK13 (134, 199–202). It is highly expected that more selective CDK12 inhibitors will proceed to clinical trials to show antitumor effect through precise molecular mechanism.
Further validation of the accuracy of patient stratification in a clinical trial is awaited and will pave the way to cure cancers through modulating pre-mRNA splicing. To achieve these, molecular machinery modulating alternative splicing should be clarified more precisely via development of new type of selective compounds, such as molecular glues and PROTACs, and modalities targeting pre-mRNA splicing-related molecules. The evidence will lead to define patient stratification and combination therapy strategy for patients with cancer in near future.
SA conceived and designed the overall manuscript. SA, MO, and MY wrote the manuscript. All authors contributed to the article and approved the submitted version.
Authors SA, MO, and MY were employed by the company Takeda Pharmaceutical Co. Ltd. Takeda Pharmaceutical Co. Ltd. provided support in the form of salaries for the authors but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stanley RF, Abdel-Wahab O. Dysregulation and therapeutic targeting of rna splicing in cancer. Nat Cancer (2022) 3(5):536–46. doi: 10.1038/s43018-022-00384-z
2. Desterro J, Bak-Gordon P, Carmo-Fonseca M. Targeting mrna processing as an anticancer strategy. Nat Rev Drug Discov (2020) 19(2):112–29. doi: 10.1038/s41573-019-0042-3
3. Bonnal SC, Lopez-Oreja I, Valcarcel J. Roles and mechanisms of alternative splicing in cancer - implications for care. Nat Rev Clin Oncol (2020) 17(8):457–74. doi: 10.1038/s41571-020-0350-x
4. Baralle FE, Giudice J. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol (2017) 18(7):437–51. doi: 10.1038/nrm.2017.27
5. Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science (2012) 338(6114):1587–93. doi: 10.1126/science.1230612
6. Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science (2012) 338(6114):1593–9. doi: 10.1126/science.1228186
7. Ule J, Blencowe BJ. Alternative splicing regulatory networks: functions, mechanisms, and evolution. Mol Cell (2019) 76(2):329–45. doi: 10.1016/j.molcel.2019.09.017
8. Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell (2018) 34(2):211–24.e6. doi: 10.1016/j.ccell.2018.07.001
9. Konda P, Viswanathan SR. How splicing confers treatment resistance in prostate cancer. eLife (2022) 11:e82070. doi: 10.7554/eLife.82070
10. Qu Y, Dai B, Ye D, Kong Y, Chang K, Jia Z, et al. Constitutively active ar-V7 plays an essential role in the development and progression of castration-resistant prostate cancer. Sci Rep (2015) 5:7654. doi: 10.1038/srep07654
11. Nero C, Vizzielli G, Lorusso D, Cesari E, Daniele G, Loverro M, et al. Patient-derived organoids and high grade serous ovarian cancer: from disease modeling to personalized medicine. J Exp Clin Cancer Res (2021) 40(1):116. doi: 10.1186/s13046-021-01917-7
12. Gehring NH, Roignant JY. Anything but ordinary - emerging splicing mechanisms in eukaryotic gene regulation. Trends Genet (2021) 37(4):355–72. doi: 10.1016/j.tig.2020.10.008
13. Daguenet E, Dujardin G, Valcárcel J. The pathogenicity of splicing defects: mechanistic insights into pre-mrna processing inform novel therapeutic approaches. EMBO Rep (2015) 16(12):1640–55. doi: 10.15252/embr.201541116
14. Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic rnp machine. Cell (2009) 136(4):701–18. doi: 10.1016/j.cell.2009.02.009
15. Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature (2008) 456(7221):470–6. doi: 10.1038/nature07509
16. Araki S, Nakayama Y, Sano O, Nakao S, Shimizu-Ogasawara M, Toyoshiba H, et al. Decoding transcriptome dynamics of genome-encoded polyadenylation and autoregulation with small-molecule modulators of alternative polyadenylation. Cell Chem Biol (2018) 25(12):1470–84.e5. doi: 10.1016/j.chembiol.2018.09.006
17. Funnell T, Tasaki S, Oloumi A, Araki S, Kong E, Yap D, et al. Clk-dependent exon recognition and conjoined gene formation revealed with a novel small molecule inhibitor. Nat Commun (2017) 8(1):7. doi: 10.1038/s41467-016-0008-7
18. Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic Sf3b1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med (2011) 365(15):1384–95. doi: 10.1056/NEJMoa1103283
19. Yoshimi A, Abdel-Wahab O. Splicing factor mutations in mds rars and Mds/Mpn-Rs-T. Int J Hematol (2017) 105(6):720–31. doi: 10.1007/s12185-017-2242-0
20. Yamauchi H, Nishimura K, Yoshimi A. Aberrant rna splicing and therapeutic opportunities in cancers. Cancer Sci (2022) 113(2):373–81. doi: 10.1111/cas.15213
21. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature (2011) 478(7367):64–9. doi: 10.1038/nature10496
22. Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor Sf3b1 gene in chronic lymphocytic leukemia. Nat Genet (2011) 44(1):47–52. doi: 10.1038/ng.1032
23. Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. Sf3b1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med (2011) 365(26):2497–506. doi: 10.1056/NEJMoa1109016
24. Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, et al. Cancer-associated Sf3b1 hotspot mutations induce cryptic 3' splice site selection through use of a different branch point. Cell Rep (2015) 13(5):1033–45. doi: 10.1016/j.celrep.2015.09.053
25. DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, et al. Transcriptome sequencing reveals potential mechanism of cryptic 3' splice site selection in Sf3b1-mutated cancers. PLoS Comput Biol (2015) 11(3):e1004105. doi: 10.1371/journal.pcbi.1004105
26. Mupo A, Seiler M, Sathiaseelan V, Pance A, Yang Y, Agrawal AA, et al. Hemopoietic-specific Sf3b1-K700e knock-in mice display the splicing defect seen in human mds but develop anemia without ring sideroblasts. Leukemia (2017) 31(3):720–7. doi: 10.1038/leu.2016.251
27. Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, et al. Physiologic expression of Sf3b1(K700e) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell (2016) 30(3):404–17. doi: 10.1016/j.ccell.2016.08.006
28. Liu Z, Yoshimi A, Wang J, Cho H, Chun-Wei Lee S, Ki M, et al. Mutations in the rna splicing factor Sf3b1 promote tumorigenesis through myc stabilization. Cancer Discovery (2020) 10(6):806–21. doi: 10.1158/2159-8290.Cd-19-1330
29. Pellagatti A, Armstrong RN, Steeples V, Sharma E, Repapi E, Singh S, et al. Impact of spliceosome mutations on rna splicing in myelodysplasia: dysregulated Genes/Pathways and clinical associations. Blood (2018) 132(12):1225–40. doi: 10.1182/blood-2018-04-843771
30. Singh S, Ahmed D, Dolatshad H, Tatwavedi D, Schulze U, Sanchi A, et al. Sf3b1 mutations induce r-loop accumulation and DNA damage in mds and leukemia cells with therapeutic implications. Leukemia (2020) 34(9):2525–30. doi: 10.1038/s41375-020-0753-9
31. Wang L, Brooks AN, Fan J, Wan Y, Gambe R, Li S, et al. Transcriptomic characterization of Sf3b1 mutation reveals its pleiotropic effects in chronic lymphocytic leukemia. Cancer Cell (2016) 30(5):750–63. doi: 10.1016/j.ccell.2016.10.005
32. Yoshimi A, Lin KT, Wiseman DH, Rahman MA, Pastore A, Wang B, et al. Coordinated alterations in rna splicing and epigenetic regulation drive leukaemogenesis. Nature (2019) 574(7777):273–7. doi: 10.1038/s41586-019-1618-0
33. Daubner GM, Cléry A, Jayne S, Stevenin J, Allain FH. A syn-anti conformational difference allows Srsf2 to recognize guanines and cytosines equally well. EMBO J (2012) 31(1):162–74. doi: 10.1038/emboj.2011.367
34. Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, et al. Srsf2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell (2015) 27(5):617–30. doi: 10.1016/j.ccell.2015.04.006
35. Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, et al. Disease-associated mutation in Srsf2 misregulates splicing by altering rna-binding affinities. Proc Natl Acad Sci USA (2015) 112(34):E4726–34. doi: 10.1073/pnas.1514105112
36. Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, et al. Mutant U2af1 expression alters hematopoiesis and pre-mrna splicing in vivo. Cancer Cell (2015) 27(5):631–43. doi: 10.1016/j.ccell.2015.04.008
37. Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2af1 reveals commonly altered splicing events. PLoS One (2014) 9(1):e87361. doi: 10.1371/journal.pone.0087361
38. Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, et al. U2af1 mutations alter splice site recognition in hematological malignancies. Genome Res (2015) 25(1):14–26. doi: 10.1101/gr.181016.114
39. Inoue D, Polaski JT, Taylor J, Castel P, Chen S, Kobayashi S, et al. Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat Genet (2021) 53(5):707–18. doi: 10.1038/s41588-021-00828-9
40. Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, et al. Aberrant splicing of U12-type introns is the hallmark of Zrsr2 mutant myelodysplastic syndrome. Nat Commun (2015) 6:6042. doi: 10.1038/ncomms7042
41. Giannakouros T, Nikolakaki E, Mylonis I, Georgatsou E. Serine-arginine protein kinases: a small protein kinase family with a Large cellular presence. FEBS J (2011) 278(4):570–86. doi: 10.1111/j.1742-4658.2010.07987.x
42. Naro C, Bielli P, Sette C. Oncogenic dysregulation of pre-mrna processing by protein kinases: challenges and therapeutic opportunities. FEBS J (2021) 288(21):6250–72. doi: 10.1111/febs.16057
43. Ding JH, Zhong XY, Hagopian JC, Cruz MM, Ghosh G, Feramisco J, et al. Regulated cellular partitioning of Sr protein-specific kinases in mammalian cells. Mol Biol Cell (2006) 17(2):876–85. doi: 10.1091/mbc.e05-10-0963
44. Plocinik RM, Li S, Liu T, Hailey KL, Whitesides J, Ma CT, et al. Regulating Sr protein phosphorylation through regions outside the kinase domain of Srpk1. J Mol Biol (2011) 410(1):131–45. doi: 10.1016/j.jmb.2011.04.077
45. Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. Regulation of Sr protein phosphorylation and alternative splicing by modulating kinetic interactions of Srpk1 with molecular chaperones. Genes Dev (2009) 23(4):482–95. doi: 10.1101/gad.1752109
46. Lai MC, Lin RI, Tarn WY. Transportin-Sr2 mediates nuclear import of phosphorylated Sr proteins. Proc Natl Acad Sci USA (2001) 98(18):10154–9. doi: 10.1073/pnas.181354098
47. Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, et al. The akt-Srpk-Sr axis constitutes a major pathway in transducing egf signaling to regulate alternative splicing in the nucleus. Mol Cell (2012) 47(3):422–33. doi: 10.1016/j.molcel.2012.05.014
48. Aubol BE, Fattet L, Adams JA. A conserved sequence motif bridges two protein kinases for enhanced phosphorylation and nuclear function of a splicing factor. FEBS J (2021) 288(2):566–81. doi: 10.1111/febs.15351
49. Ngo JC, Chakrabarti S, Ding JH, Velazquez-Dones A, Nolen B, Aubol BE, et al. Interplay between srpk and Clk/Sty kinases in phosphorylation of the splicing factor Asf/Sf2 is regulated by a docking motif in Asf/Sf2. Mol Cell (2005) 20(1):77–89. doi: 10.1016/j.molcel.2005.08.025
50. Kuroyanagi N, Onogi H, Wakabayashi T, Hagiwara M. Novel Sr-Protein-Specific kinase, Srpk2, disassembles nuclear speckles. Biochem Biophys Res Commun (1998) 242(2):357–64. doi: 10.1006/bbrc.1997.7913
51. Nakagawa O, Arnold M, Nakagawa M, Hamada H, Shelton JM, Kusano H, et al. Centronuclear myopathy in mice lacking a novel muscle-specific protein kinase transcriptionally regulated by Mef2. Genes Dev (2005) 19(17):2066–77. doi: 10.1101/gad.1338705
52. Wang P, Zhou Z, Hu A, Ponte de Albuquerque C, Zhou Y, Hong L, et al. Both decreased and increased Srpk1 levels promote cancer by interfering with phlpp-mediated dephosphorylation of akt. Mol Cell (2014) 54(3):378–91. doi: 10.1016/j.molcel.2014.03.007
53. Agnew C, Liu L, Liu S, Xu W, You L, Yeung W, et al. The crystal structure of the protein kinase Hipk2 reveals a unique architecture of its cmgc-insert region. J Biol Chem (2019) 294(37):13545–59. doi: 10.1074/jbc.RA119.009725
54. Aubol BE, Plocinik RM, Hagopian JC, Ma CT, McGlone ML, Bandyopadhyay R, et al. Partitioning rs domain phosphorylation in an Sr protein through the clk and srpk protein kinases. J Mol Biol (2013) 425(16):2894–909. doi: 10.1016/j.jmb.2013.05.013
55. Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, et al. The Clk/Sty protein kinase phosphorylates Sr splicing factors and regulates their intranuclear distribution. EMBO J (1996) 15(2):265–75. doi: 10.1002/j.1460-2075.1996.tb00357.x
56. Aubol BE, Wu G, Keshwani MM, Movassat M, Fattet L, Hertel KJ, et al. Release of Sr proteins from Clk1 by Srpk1: a symbiotic kinase system for phosphorylation control of pre-mrna splicing. Mol Cell (2016) 63(2):218–28. doi: 10.1016/j.molcel.2016.05.034
57. Cho S, Hoang A, Sinha R, Zhong XY, Fu XD, Krainer AR, et al. Interaction between the rna binding domains of ser-arg splicing factor 1 and U1-70k snrnp protein determines early spliceosome assembly. Proc Natl Acad Sci USA (2011) 108(20):8233–8. doi: 10.1073/pnas.1017700108
58. Rodgers JT, Haas W, Gygi SP, Puigserver P. Cdc2-like kinase 2 is an insulin-regulated suppressor of hepatic gluconeogenesis. Cell Metab (2010) 11(1):23–34. doi: 10.1016/j.cmet.2009.11.006
59. Tabata M, Rodgers JT, Hall JA, Lee Y, Jedrychowski MP, Gygi SP, et al. Cdc2-like kinase 2 suppresses hepatic fatty acid oxidation and ketogenesis through disruption of the pgc-1α and Med1 complex. Diabetes (2014) 63(5):1519–32. doi: 10.2337/db13-1304
60. Huang J, Wang L, Shen Y, Zhang S, Zhou Y, Du J, et al. Cdc-like kinase 4 deficiency contributes to pathological cardiac hypertrophy by modulating nexn phosphorylation. Nat Commun (2022) 13(1):4433. doi: 10.1038/s41467-022-31996-9
62. Clopper KC, Taatjes DJ. Chemical inhibitors of transcription-associated kinases. Curr Opin Chem Biol (2022) 70:102186. doi: 10.1016/j.cbpa.2022.102186
63. Rimel JK, Poss ZC, Erickson B, Maas ZL, Ebmeier CC, Johnson JL, et al. Selective inhibition of Cdk7 reveals high-confidence targets and new models for tfiih function in transcription. Genes Dev (2020) 34(21-22):1452–73. doi: 10.1101/gad.341545.120
64. Bacon CW, D'Orso I. Cdk9: a signaling hub for transcriptional control. Transcription (2019) 10(2):57–75. doi: 10.1080/21541264.2018.1523668
65. Hu Q, Poulose N, Girmay S, Helevä A, Doultsinos D, Gondane A, et al. Inhibition of Cdk9 activity compromises global splicing in prostate cancer cells. RNA Biol (2021) 18(sup2):722–9. doi: 10.1080/15476286.2021.1983287
66. Tellier M, Zaborowska J, Neve J, Nojima T, Hester S, Fournier M, et al. Cdk9 and Pp2a regulate rna polymerase ii transcription termination and coupled rna maturation. EMBO Rep (2022) 23(10):e54520. doi: 10.15252/embr.202154520
67. Ahmed RL, Shaughnessy DP, Knutson TP, Vogel RI, Ahmed K, Kren BT, et al. Cdk11 loss induces cell cycle dysfunction and death of braf and nras melanoma cells. Pharm (Basel) (2019) 12(2):50. doi: 10.3390/ph12020050
68. Gajdušková P, Ruiz de Los Mozos I, Rájecký M, Hluchý M, Ule J, Blazek D. Cdk11 is required for transcription of replication-dependent histone genes. Nat Struct Mol Biol (2020) 27(5):500–10. doi: 10.1038/s41594-020-0406-8
69. Hluchý M, Gajdušková P, Ruiz de Los Mozos I, Rájecký M, Kluge M, Berger BT, et al. Cdk11 regulates pre-mrna splicing by phosphorylation of Sf3b1. Nature (2022) 609(7928):829–34. doi: 10.1038/s41586-022-05204-z
70. Greenleaf AL. Human Cdk12 and Cdk13, multi-tasking ctd kinases for the new millenium. Transcription (2019) 10(2):91–110. doi: 10.1080/21541264.2018.1535211
71. Davidson L, Muniz L, West S. 3' end formation of pre-mrna and phosphorylation of Ser2 on the rna polymerase ii ctd are reciprocally coupled in human cells. Genes Dev (2014) 28(4):342–56. doi: 10.1101/gad.231274.113
72. Dubbury SJ, Boutz PL, Sharp PA. Cdk12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature (2018) 564(7734):141–5. doi: 10.1038/s41586-018-0758-y
73. Fan Z, Devlin JR, Hogg SJ, Doyle MA, Harrison PF, Todorovski I, et al. Cdk13 cooperates with Cdk12 to control global rna polymerase ii processivity. Sci Adv (2020) 6(18):eaaz5041. doi: 10.1126/sciadv.aaz5041
74. Krajewska M, Dries R, Grassetti AV, Dust S, Gao Y, Huang H, et al. Cdk12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat Commun (2019) 10(1):1757. doi: 10.1038/s41467-019-09703-y
75. Tien JF, Mazloomian A, Cheng SG, Hughes CS, Chow CCT, Canapi LT, et al. Cdk12 regulates alternative last exon mrna splicing and promotes breast cancer cell invasion. Nucleic Acids Res (2017) 45(11):6698–716. doi: 10.1093/nar/gkx187
76. Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, et al. The cyclin K/Cdk12 complex maintains genomic stability Via regulation of expression of DNA damage response genes. Genes Dev (2011) 25(20):2158–72. doi: 10.1101/gad.16962311
77. Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, et al. U1 snrnp determines mrna length and regulates isoform expression. Cell (2012) 150(1):53–64. doi: 10.1016/j.cell.2012.05.029
78. Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, et al. U1 snrnp protects pre-mrnas from premature cleavage and polyadenylation. Nature (2010) 468(7324):664–8. doi: 10.1038/nature09479
79. Oh JM, Di C, Venters CC, Guo J, Arai C, So BR, et al. U1 snrnp telescripting regulates a size-Function-Stratified human genome. Nat Struct Mol Biol (2017) 24(11):993–9. doi: 10.1038/nsmb.3473
80. Zhang L, Tran NT, Su H, Wang R, Lu Y, Tang H, et al. Cross-talk between Prmt1-mediated methylation and ubiquitylation on Rbm15 controls rna splicing. Elife (2015) 4:e07938. doi: 10.7554/eLife.07938
81. Meister G, Eggert C, Bühler D, Brahms H, Kambach C, Fischer U. Methylation of Sm proteins by a complex containing Prmt5 and the putative U snrnp assembly factor picln. Curr Biol (2001) 11(24):1990–4. doi: 10.1016/s0960-9822(01)00592-9
82. Wu Q, Schapira M, Arrowsmith CH, Barsyte-Lovejoy D. Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nat Rev Drug Discov (2021) 20(7):509–30. doi: 10.1038/s41573-021-00159-8
83. Schapira M, Ferreira de Freitas R. Structural biology and chemistry of protein arginine methyltransferases. Medchemcomm (2014) 5(12):1779–88. doi: 10.1039/c4md00269e
84. Mulvaney KM, Blomquist C, Acharya N, Li R, Ranaghan MJ, O'Keefe M, et al. Molecular basis for substrate recruitment to the Prmt5 methylosome. Mol Cell (2021) 81(17):3481–95.e7. doi: 10.1016/j.molcel.2021.07.019
85. Bullock N, Potts J, Simpkin AJ, Koupparis A, Harper SJ, Oxley J, et al. Serine-arginine protein kinase 1 (Srpk1), a determinant of angiogenesis, is upregulated in prostate cancer and correlates with disease stage and invasion. J Clin Pathol (2016) 69(2):171–5. doi: 10.1136/jclinpath-2015-203125
86. Gout S, Brambilla E, Boudria A, Drissi R, Lantuejoul S, Gazzeri S, et al. Abnormal expression of the pre-mrna splicing regulators Srsf1, Srsf2, Srpk1 and Srpk2 in non small cell lung carcinoma. PLoS One (2012) 7(10):e46539. doi: 10.1371/journal.pone.0046539
87. Hayes GM, Carrigan PE, Beck AM, Miller LJ. Targeting the rna splicing machinery as a novel treatment strategy for pancreatic carcinoma. Cancer Res (2006) 66(7):3819–27. doi: 10.1158/0008-5472.Can-05-4065
88. Hayes GM, Carrigan PE, Miller LJ. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res (2007) 67(5):2072–80. doi: 10.1158/0008-5472.Can-06-2969
89. Siqueira RP, Barbosa Éde A, Polêto MD, Righetto GL, Seraphim TV, Salgado RL, et al. Potential antileukemia effect and structural analyses of srpk inhibition by n-(2-(Piperidin-1-Yl)-5-(Trifluoromethyl)Phenyl)Isonicotinamide (Srpin340). PLoS One (2015) 10(8):e0134882. doi: 10.1371/journal.pone.0134882
90. Tzelepis K, De Braekeleer E, Aspris D, Barbieri I, Vijayabaskar MS, Liu WH, et al. Srpk1 maintains acute myeloid leukemia through effects on isoform usage of epigenetic regulators including Brd4. Nat Commun (2018) 9(1):5378. doi: 10.1038/s41467-018-07620-0
91. Gong L, Song J, Lin X, Wei F, Zhang C, Wang Z, et al. Serine-arginine protein kinase 1 promotes a cancer stem cell-like phenotype through activation of Wnt/Β-catenin signalling in nsclc. J Pathol (2016) 240(2):184–96. doi: 10.1002/path.4767
92. van Roosmalen W, Le Dévédec SE, Golani O, Smid M, Pulyakhina I, Timmermans AM, et al. Tumor cell migration screen identifies Srpk1 as breast cancer metastasis determinant. J Clin Invest (2015) 125(4):1648–64. doi: 10.1172/jci74440
93. Schenk PW, Stoop H, Bokemeyer C, Mayer F, Stoter G, Oosterhuis JW, et al. Resistance to platinum-containing chemotherapy in testicular germ cell tumors is associated with downregulation of the protein kinase Srpk1. Neoplasia (2004) 6(4):297–301. doi: 10.1593/neo.03406
94. Krishnakumar S, Mohan A, Kandalam M, Ramkumar HL, Venkatesan N, Das RR. Srpk1: a cisplatin sensitive protein expressed in retinoblastoma. Pediatr Blood Cancer (2008) 50(2):402–6. doi: 10.1002/pbc.21088
95. Gammons MV, Lucas R, Dean R, Coupland SE, Oltean S, Bates DO. Targeting Srpk1 to control vegf-mediated tumour angiogenesis in metastatic melanoma. Br J Cancer (2014) 111(3):477–85. doi: 10.1038/bjc.2014.342
96. Bates DO, Cui TG, Doughty JM, Winkler M, Sugiono M, Shields JD, et al. Vegf165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res (2002) 62(14):4123–31.
97. Amin EM, Oltean S, Hua J, Gammons MV, Hamdollah-Zadeh M, Welsh GI, et al. Wt1 mutants reveal Srpk1 to be a downstream angiogenesis target by altering vegf splicing. Cancer Cell (2011) 20(6):768–80. doi: 10.1016/j.ccr.2011.10.016
98. Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, et al. A crispr dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep (2016) 17(4):1193–205. doi: 10.1016/j.celrep.2016.09.079
99. Yoshida T, Kim JH, Carver K, Su Y, Weremowicz S, Mulvey L, et al. Clk2 is an oncogenic kinase and splicing regulator in breast cancer. Cancer Res (2015) 75(7):1516–26. doi: 10.1158/0008-5472.Can-14-2443
100. Zhou HB, Yang L, Liu SF, Xu XH, Chen Z, Li YX, et al. Cdc like kinase 2 plays an oncogenic role in colorectal cancer Via modulating the Wnt/Β-catenin signaling. Neoplasma (2022) 69(3):657–69. doi: 10.4149/neo_2022_220206N138
101. Dominguez D, Tsai YH, Weatheritt R, Wang Y, Blencowe BJ, Wang Z. An extensive program of periodic alternative splicing linked to cell cycle progression. elife (2016) 5:e10288. doi: 10.7554/eLife.10288
102. Park SY, Piao Y, Thomas C, Fuller GN, de Groot JF. Cdc2-like kinase 2 is a key regulator of the cell cycle Via Foxo3a/P27 in glioblastoma. Oncotarget (2016) 7(18):26793–805. doi: 10.18632/oncotarget.8471
103. Iwai K, Yaguchi M, Nishimura K, Yamamoto Y, Tamura T, Nakata D, et al. Anti-tumor efficacy of a novel clk inhibitor Via targeting rna splicing and myc-dependent vulnerability. EMBO Mol Med (2018) 10(6):e8289. doi: 10.15252/emmm.201708289
104. Tiek DM, Khatib SA, Trepicchio CJ, Heckler MM, Divekar SD, Sarkaria JN, et al. Estrogen-related receptor Β activation and isoform shifting by Cdc2-like kinase inhibition restricts migration and intracranial tumor growth in glioblastoma. FASEB J (2019) 33(12):13476–91. doi: 10.1096/fj.201901075R
105. Araki S, Dairiki R, Nakayama Y, Murai A, Miyashita R, Iwatani M, et al. Inhibitors of clk protein kinases suppress cell growth and induce apoptosis by modulating pre-mrna splicing. PLoS One (2015) 10(1):e0116929. doi: 10.1371/journal.pone.0116929
106. Babu N, Pinto SM, Biswas M, Subbannayya T, Rajappa M, Mohan SV, et al. Phosphoproteomic analysis identifies Clk1 as a novel therapeutic target in gastric cancer. Gastric Cancer (2020) 23(5):796–810. doi: 10.1007/s10120-020-01062-8
107. Uzor S, Porazinski SR, Li L, Clark B, Ajiro M, Iida K, et al. Cdc2-like (Clk) protein kinase inhibition as a novel targeted therapeutic strategy in prostate cancer. Sci Rep (2021) 11(1):7963. doi: 10.1038/s41598-021-86908-6
108. Muraki M, Ohkawara B, Hosoya T, Onogi H, Koizumi J, Koizumi T, et al. Manipulation of alternative splicing by a newly developed inhibitor of clks. J Biol Chem (2004) 279(23):24246–54. doi: 10.1074/jbc.M314298200
109. Akiva P, Toporik A, Edelheit S, Peretz Y, Diber A, Shemesh R, et al. Transcription-mediated gene fusion in the human genome. Genome Res (2006) 16(1):30–6. doi: 10.1101/gr.4137606
110. Prakash T, Sharma VK, Adati N, Ozawa R, Kumar N, Nishida Y, et al. Expression of conjoined genes: another mechanism for gene regulation in eukaryotes. PLoS One (2010) 5(10):e13284. doi: 10.1371/journal.pone.0013284
111. Sava GP, Fan H, Coombes RC, Buluwela L, Ali S. Cdk7 inhibitors as anticancer drugs. Cancer Metastasis Rev (2020) 39(3):805–23. doi: 10.1007/s10555-020-09885-8
112. Jiang L, Huang R, Wu Y, Diao P, Zhang W, Li J, et al. Overexpression of Cdk7 is associated with unfavourable prognosis in oral squamous cell carcinoma. Pathology (2019) 51(1):74–80. doi: 10.1016/j.pathol.2018.10.004
113. Li B, Ni Chonghaile T, Fan Y, Madden SF, Klinger R, O'Connor AE, et al. Therapeutic rationale to target highly expressed Cdk7 conferring poor outcomes in triple-negative breast cancer. Cancer Res (2017) 77(14):3834–45. doi: 10.1158/0008-5472.Can-16-2546
114. Meng W, Wang J, Wang B, Liu F, Li M, Zhao Y, et al. Cdk7 inhibition is a novel therapeutic strategy against gbm both in vitro and in vivo. Cancer Manag Res (2018) 10:5747–58. doi: 10.2147/cmar.S183696
115. Naseh G, Mohammadifard M, Mohammadifard M. Upregulation of cyclin-dependent kinase 7 and matrix metalloproteinase-14 expression contribute to metastatic properties of gastric cancer. IUBMB Life (2016) 68(10):799–805. doi: 10.1002/iub.1543
116. Tsang FH, Law CT, Tang TC, Cheng CL, Chin DW, Tam WV, et al. Aberrant super-enhancer landscape in human hepatocellular carcinoma. Hepatology (2019) 69(6):2502–17. doi: 10.1002/hep.30544
117. Wang Q, Li M, Zhang X, Huang H, Huang J, Ke J, et al. Upregulation of Cdk7 in gastric cancer cell promotes tumor cell proliferation and predicts poor prognosis. Exp Mol Pathol (2016) 100(3):514–21. doi: 10.1016/j.yexmp.2016.05.001
118. Zhang Z, Peng H, Wang X, Yin X, Ma P, Jing Y, et al. Preclinical efficacy and molecular mechanism of targeting Cdk7-dependent transcriptional addiction in ovarian cancer. Mol Cancer Ther (2017) 16(9):1739–50. doi: 10.1158/1535-7163.Mct-17-0078
119. Borowczak J, Szczerbowski K, Maniewski M, Zdrenka M, Słupski P, Antosik P, et al. The prognostic role of Cdk9 in bladder cancer. Cancers (Basel) (2022) 14(6):1492. doi: 10.3390/cancers14061492
120. Guo T, Liu DF, Peng SH. Cdk9 is up-regulated and associated with prognosis in patients with papillary thyroid carcinoma. Med (Baltimore) (2022) 101(5):e28309. doi: 10.1097/md.0000000000028309
121. Kretz AL, Schaum M, Richter J, Kitzig EF, Engler CC, Leithäuser F, et al. Cdk9 is a prognostic marker and therapeutic target in pancreatic cancer. Tumour Biol (2017) 39(2):1010428317694304. doi: 10.1177/1010428317694304
122. Ma H, Seebacher NA, Hornicek FJ, Duan Z. Cyclin-dependent kinase 9 (Cdk9) is a novel prognostic marker and therapeutic target in osteosarcoma. EBioMedicine (2019) 39:182–93. doi: 10.1016/j.ebiom.2018.12.022
123. Schlafstein AJ, Withers AE, Rudra S, Danelia D, Switchenko JM, Mister D, et al. Cdk9 expression shows role as a potential prognostic biomarker in breast cancer patients who fail to achieve pathologic complete response after neoadjuvant chemotherapy. Int J Breast Cancer (2018) 2018:6945129. doi: 10.1155/2018/6945129
124. Wang J, Dean DC, Hornicek FJ, Shi H, Duan Z. Cyclin-dependent kinase 9 (Cdk9) is a novel prognostic marker and therapeutic target in ovarian cancer. FASEB J (2019) 33(5):5990–6000. doi: 10.1096/fj.201801789RR
125. Zhou Y, Shen JK, Hornicek FJ, Kan Q, Duan Z. The emerging roles and therapeutic potential of cyclin-dependent kinase 11 (Cdk11) in human cancer. Oncotarget (2016) 7(26):40846–59. doi: 10.18632/oncotarget.8519
126. Kren BT, Unger GM, Abedin MJ, Vogel RI, Henzler CM, Ahmed K, et al. Preclinical evaluation of cyclin dependent kinase 11 and casein kinase 2 survival kinases as rna interference targets for triple negative breast cancer therapy. Breast Cancer Res (2015) 17:19. doi: 10.1186/s13058-015-0524-0
127. Duan Z, Zhang J, Choy E, Harmon D, Liu X, Nielsen P, et al. Systematic kinome shrna screening identifies Cdk11 (Pitslre) kinase expression is critical for osteosarcoma cell growth and proliferation. Clin Cancer Res (2012) 18(17):4580–8. doi: 10.1158/1078-0432.Ccr-12-1157
128. Jia B, Choy E, Cote G, Harmon D, Ye S, Kan Q, et al. Cyclin-dependent kinase 11 (Cdk11) is crucial in the growth of liposarcoma cells. Cancer Lett (2014) 342(1):104–12. doi: 10.1016/j.canlet.2013.08.040
129. Zhou Y, Han C, Li D, Yu Z, Li F, Li F, et al. Cyclin-dependent kinase 11(P110) (Cdk11(P110)) is crucial for human breast cancer cell proliferation and growth. Sci Rep (2015) 5:10433. doi: 10.1038/srep10433
130. Liu X, Gao Y, Shen J, Yang W, Choy E, Mankin H, et al. Cyclin-dependent kinase 11 (Cdk11) is required for ovarian cancer cell growth in vitro and in vivo, and its inhibition causes apoptosis and sensitizes cells to paclitaxel. Mol Cancer Ther (2016) 15(7):1691–701. doi: 10.1158/1535-7163.Mct-16-0032
131. Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature (2016) 534(7605):55–62. doi: 10.1038/nature18003
132. Wang H, Miao J, Wen Y, Xia X, Chen Y, Huang M, et al. Molecular landscape of Erbb2 alterations in 14,956 solid tumors. Pathol Oncol Res (2022) 28:1610360. doi: 10.3389/pore.2022.1610360
133. Choi HJ, Jin S, Cho H, Won HY, An HW, Jeong GY, et al. Cdk12 drives breast tumor initiation and trastuzumab resistance Via wnt and Irs1-Erbb-Pi3k signaling. EMBO Rep (2019) 20(10):e48058. doi: 10.15252/embr.201948058
134. Quereda V, Bayle S, Vena F, Frydman SM, Monastyrskyi A, Roush WR, et al. Therapeutic targeting of Cdk12/Cdk13 in triple-negative breast cancer. Cancer Cell (2019) 36(5):545–58.e7. doi: 10.1016/j.ccell.2019.09.004
135. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature (2012) 487(7406):239–43. doi: 10.1038/nature11125
136. Popova T, Manié E, Boeva V, Battistella A, Goundiam O, Smith NK, et al. Ovarian cancers harboring inactivating mutations in Cdk12 display a distinct genomic instability pattern characterized by Large tandem duplications. Cancer Res (2016) 76(7):1882–91. doi: 10.1158/0008-5472.Can-15-2128
137. Wu YM, Cieślik M, Lonigro RJ, Vats P, Reimers MA, Cao X, et al. Inactivation of Cdk12 delineates a distinct immunogenic class of advanced prostate cancer. Cell (2018) 173(7):1770–82.e14. doi: 10.1016/j.cell.2018.04.034
138. Chen Y, Shao X, Zhao X, Ji Y, Liu X, Li P, et al. Targeting protein arginine methyltransferase 5 in cancers: roles, inhibitors and mechanisms. BioMed Pharmacother (2021) 144:112252. doi: 10.1016/j.biopha.2021.112252
139. Litzler LC, Zahn A, Meli AP, Hébert S, Patenaude AM, Methot SP, et al. Prmt5 is essential for b cell development and germinal center dynamics. Nat Commun (2019) 10(1):22. doi: 10.1038/s41467-018-07884-6
140. Cao L, Wu G, Zhu J, Tan Z, Shi D, Wu X, et al. Genotoxic stress-triggered Β-Catenin/Jdp2/Prmt5 complex facilitates reestablishing glutathione homeostasis. Nat Commun (2019) 10(1):3761. doi: 10.1038/s41467-019-11696-7
141. Wang Y, Li Y, He H, Wang F. Circular rna circ-Prmt5 facilitates non-small cell lung cancer proliferation through upregulating Ezh2 Via sponging mir-377/382/498. Gene (2019) 720:144099. doi: 10.1016/j.gene.2019.144099
142. Li WJ, He YH, Yang JJ, Hu GS, Lin YA, Ran T, et al. Profiling prmt methylome reveals roles of Hnrnpa1 arginine methylation in rna splicing and cell growth. Nat Commun (2021) 12(1):1946. doi: 10.1038/s41467-021-21963-1
143. Banasavadi-Siddegowda YK, Welker AM, An M, Yang X, Zhou W, Shi G, et al. Prmt5 as a druggable target for glioblastoma therapy. Neuro Oncol (2018) 20(6):753–63. doi: 10.1093/neuonc/nox206
144. Tan L, Xiao K, Ye Y, Liang H, Chen M, Luo J, et al. High Prmt5 expression is associated with poor overall survival and tumor progression in bladder cancer. Aging (Albany NY) (2020) 12(9):8728–41. doi: 10.18632/aging.103198
145. Braun CJ, Stanciu M, Boutz PL, Patterson JC, Calligaris D, Higuchi F, et al. Coordinated splicing of regulatory detained introns within oncogenic transcripts creates an exploitable vulnerability in malignant glioma. Cancer Cell (2017) 32(4):411–26.e11. doi: 10.1016/j.ccell.2017.08.018
146. Sachamitr P, Ho JC, Ciamponi FE, Ba-Alawi W, Coutinho FJ, Guilhamon P, et al. Prmt5 inhibition disrupts splicing and stemness in glioblastoma. Nat Commun (2021) 12(1):979. doi: 10.1038/s41467-021-21204-5
147. Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, et al. A selective inhibitor of Prmt5 with in vivo and in vitro potency in mcl models. Nat Chem Biol (2015) 11(6):432–7. doi: 10.1038/nchembio.1810
148. Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM. Sudemycins, novel small molecule analogues of Fr901464, induce alternative gene splicing. ACS Chem Biol (2011) 6(6):582–9. doi: 10.1021/cb100356k
149. Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, et al. Spliceostatin a targets Sf3b and inhibits both splicing and nuclear retention of pre-mrna. Nat Chem Biol (2007) 3(9):576–83. doi: 10.1038/nchembio.2007.18
150. Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, et al. Splicing factor Sf3b as a target of the antitumor natural product pladienolide. Nat Chem Biol (2007) 3(9):570–5. doi: 10.1038/nchembio.2007.16
151. Kaida D, Shida K. Spliceostatin a stabilizes Cdkn1b mrna through the 3' utr. Biochem Biophys Res Commun (2022) 608:39–44. doi: 10.1016/j.bbrc.2022.03.085
152. Kikuchi K, Kaida D. Ccne1 and E2f1 partially suppress G1 phase arrest caused by spliceostatin a treatment. Int J Mol Sci (2021) 22(21):11623. doi: 10.3390/ijms222111623
153. Koga M, Hayashi M, Kaida D. Splicing inhibition decreases phosphorylation level of Ser2 in pol ii ctd. Nucleic Acids Res (2015) 43(17):8258–67. doi: 10.1093/nar/gkv740
154. Satoh T, Kaida D. Upregulation of P27 cyclin-dependent kinase inhibitor and a c-terminus truncated form of P27 contributes to G1 phase arrest. Sci Rep (2016) 6:27829. doi: 10.1038/srep27829
155. Yoshimoto R, Chhipi-Shrestha JK, Schneider-Poetsch T, Furuno M, Burroughs AM, Noma S, et al. Spliceostatin a interaction with Sf3b limits U1 snrnp availability and causes premature cleavage and polyadenylation. Cell Chem Biol (2021) 28(9):1356–65.e4. doi: 10.1016/j.chembiol.2021.03.002
156. Yoshimoto R, Kaida D, Furuno M, Burroughs AM, Noma S, Suzuki H, et al. Global analysis of pre-mrna subcellular localization following splicing inhibition by spliceostatin a. Rna (2017) 23(1):47–57. doi: 10.1261/rna.058065.116
157. Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med (2016) 22(6):672–8. doi: 10.1038/nm.4097
158. Eskens FA, Ramos FJ, Burger H, O'Brien JP, Piera A, de Jonge MJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-Class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res (2013) 19(22):6296–304. doi: 10.1158/1078-0432.Ccr-13-0485
159. Hong DS, Kurzrock R, Naing A, Wheler JJ, Falchook GS, Schiffman JS, et al. Open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (Pre-mrna) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs (2014) 32(3):436–44. doi: 10.1007/s10637-013-0046-5
160. Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, et al. H3b-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med (2018) 24(4):497–504. doi: 10.1038/nm.4493
161. Nøhr-Nielsen A, Bagger SO, Brünner N, Stenvang J, Lund TM. Pharmacodynamic modelling reveals synergistic interaction between docetaxel and sco-101 in a docetaxel-resistant triple negative breast cancer cell line. Eur J Pharm Sci (2020) 148:105315. doi: 10.1016/j.ejps.2020.105315
162. Bergmann TK, Stage TB, Stenvang J, Christophersen P, Jacobsen TA, Roest NL, et al. Four phase 1 trials to evaluate the safety and pharmacokinetic profile of single and repeated dosing of sco-101 in adult Male and female volunteers. Basic Clin Pharmacol Toxicol (2020) 127(4):329–37. doi: 10.1111/bcpt.13466
163. Adesso L, Calabretta S, Barbagallo F, Capurso G, Pilozzi E, Geremia R, et al. Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the Mnk2/Eif4e pathway. Oncogene (2013) 32(23):2848–57. doi: 10.1038/onc.2012.306
164. Tam BY, Chiu K, Chung H, Bossard C, Nguyen JD, Creger E, et al. The clk inhibitor Sm08502 induces anti-tumor activity and reduces wnt pathway gene expression in gastrointestinal cancer models. Cancer Lett (2020) 473:186–97. doi: 10.1016/j.canlet.2019.09.009
165. Shimizu T, Yonemori K, Koyama T, Katsuya Y, Sato J, Fukuhara N, et al. First-in-Human phase I study of ctx-712 in patients with advanced, relapsed or refractory malignant tumors. J Clin Oncol (2022) 40(16_suppl):3080. doi: 10.1200/JCO.2022.40.16_suppl.3080
166. Yokoyama H, Ando K, Fukuhara N, Iida H, Fukuhara S, Miyake H, et al. A first-in-Human phase I study of ctx-712 in patients with advanced, relapsed or refractory malignant tumors - hematologic malignancies dose escalation cohort. Blood (2022) 140(Supplement 1):6211–2. doi: 10.1182/blood-2022-157241
167. Ghia P, Scarfò L, Perez S, Pathiraja K, Derosier M, Small K, et al. Efficacy and safety of dinaciclib vs ofatumumab in patients with Relapsed/Refractory chronic lymphocytic leukemia. Blood (2017) 129(13):1876–8. doi: 10.1182/blood-2016-10-748210
168. Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, et al. Cdk12 inhibition reverses De novo and acquired parp inhibitor resistance in brca wild-type and mutated models of triple-negative breast cancer. Cell Rep (2016) 17(9):2367–81. doi: 10.1016/j.celrep.2016.10.077
169. Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent Cdk7 inhibitor. Nature (2014) 511(7511):616–20. doi: 10.1038/nature13393
170. Chipumuro E, Marco E, Christensen Camilla L, Kwiatkowski N, Zhang T, Hatheway Clark M, et al. Cdk7 inhibition suppresses super-Enhancer-Linked oncogenic transcription in mycn-driven cancer. Cell (2014) 159(5):1126–39. doi: 10.1016/j.cell.2014.10.024
171. Christensen Camilla L, Kwiatkowski N, Abraham Brian J, Carretero J, Al-Shahrour F, Zhang T, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent Cdk7 inhibitor. Cancer Cell (2014) 26(6):909–22. doi: 10.1016/j.ccell.2014.10.019
172. Wang Y, Zhang T, Kwiatkowski N, Abraham Brian J, Lee Tong I, Xie S, et al. Cdk7-dependent transcriptional addiction in triple-negative breast cancer. Cell (2015) 163(1):174–86. doi: 10.1016/j.cell.2015.08.063
173. Iniguez AB, Stolte B, Wang EJ, Conway AS, Alexe G, Dharia NV, et al. Ews/Fli confers tumor cell synthetic lethality to Cdk12 inhibition in Ewing sarcoma. Cancer Cell (2018) 33(2):202–16.e6. doi: 10.1016/j.ccell.2017.12.009
174. Ito M, Tanaka T, Toita A, Uchiyama N, Kokubo H, Morishita N, et al. Discovery of 3-Benzyl-1-( trans-4-((5-Cyanopyridin-2-Yl)Amino)Cyclohexyl)-1-Arylurea derivatives as novel and selective cyclin-dependent kinase 12 (Cdk12) inhibitors. J Med Chem (2018) 61(17):7710–28. doi: 10.1021/acs.jmedchem.8b00683
175. Johannes JW, Denz CR, Su N, Wu A, Impastato AC, Mlynarski S, et al. Structure-based design of selective noncovalent Cdk12 inhibitors. ChemMedChem (2018) 13(3):231–5. doi: 10.1002/cmdc.201700695
176. Gao Y, Zhang T, Terai H, Ficarro SB, Kwiatkowski N, Hao MF, et al. Overcoming resistance to the thz series of covalent transcriptional cdk inhibitors. Cell Chem Biol (2018) 25(2):135–42.e5. doi: 10.1016/j.chembiol.2017.11.007
177. Lei P, Zhang J, Liao P, Ren C, Wang J, Wang Y. Current progress and novel strategies that target Cdk12 for drug discovery. Eur J Med Chem (2022) 240:114603. doi: 10.1016/j.ejmech.2022.114603
178. Dong G, Ding Y, He S, Sheng C. Molecular glues for targeted protein degradation: from serendipity to rational discovery. J Medicinal Chem (2021) 64(15):10606–20. doi: 10.1021/acs.jmedchem.1c00895
179. Shan W, Yuan J, Hu Z, Jiang J, Wang Y, Loo N, et al. Systematic characterization of recurrent genomic alterations in cyclin-dependent kinases reveals potential therapeutic strategies for cancer treatment. Cell Rep (2020) 32(2):107884. doi: 10.1016/j.celrep.2020.107884
180. Hu S, Marineau JJ, Rajagopal N, Hamman KB, Choi YJ, Schmidt DR, et al. Discovery and characterization of sy-1365, a selective, covalent inhibitor of Cdk7. Cancer Res (2019) 79(13):3479–91. doi: 10.1158/0008-5472.Can-19-0119
181. Anshabo AT, Milne R, Wang S, Albrecht H. Cdk9: a comprehensive review of its biology, and its role as a potential target for anti-cancer agents. Front Oncol (2021) 11:678559. doi: 10.3389/fonc.2021.678559
182. Mandal R, Becker S, Strebhardt K. Targeting Cdk9 for anti-cancer therapeutics. Cancers (Basel) (2021) 13(9):2181. doi: 10.3390/cancers13092181
183. Zhang M, Zhang L, Hei R, Li X, Cai H, Wu X, et al. Cdk inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res (2021) 11(5):1913–35.
184. Siu LL, Rasco DW, Vinay SP, Romano PM, Menis J, Opdam FL, et al. 438o - meteor-1: a phase I study of Gsk3326595, a first-in-Class protein arginine methyltransferase 5 (Prmt5) inhibitor, in advanced solid tumours. Ann Oncol (2019) 30:v159. doi: 10.1093/annonc/mdz244
185. Feustel K, Falchook GS. Protein arginine methyltransferase 5 (Prmt5) inhibitors in oncology clinical trials: a review. J Immunother Precis Oncol (2022) 5(3):58–67. doi: 10.36401/jipo-22-1
186. Fedoriw A, Rajapurkar SR, O'Brien S, Gerhart SV, Mitchell LH, Adams ND, et al. Anti-tumor activity of the type I prmt inhibitor, Gsk3368715, synergizes with Prmt5 inhibition through mtap loss. Cancer Cell (2019) 36(1):100–14.e25. doi: 10.1016/j.ccell.2019.05.014
187. Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, et al. Mtap deletion confers enhanced dependency on the Prmt5 arginine methyltransferase in cancer cells. Science (2016) 351(6278):1214–8. doi: 10.1126/science.aad5214
188. Marjon K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, et al. Mtap deletions in cancer create vulnerability to targeting of the Mat2a/Prmt5/Riok1 axis. Cell Rep (2016) 15(3):574–87. doi: 10.1016/j.celrep.2016.03.043
189. Mavrakis KJ, McDonald ER 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A, et al. Disordered methionine metabolism in Mtap/Cdkn2a-deleted cancers leads to dependence on Prmt5. Science (2016) 351(6278):1208–13. doi: 10.1126/science.aad5944
190. Fong JY, Pignata L, Goy PA, Kawabata KC, Lee SC, Koh CM, et al. Therapeutic targeting of rna splicing catalysis through inhibition of protein arginine methylation. Cancer Cell (2019) 36(2):194–209.e9. doi: 10.1016/j.ccell.2019.07.003
191. Shorstova T, Foulkes WD, Witcher M. Achieving clinical success with bet inhibitors as anti-cancer agents. Br J Cancer (2021) 124(9):1478–90. doi: 10.1038/s41416-021-01321-0
192. French CA. Nut carcinoma: clinicopathologic features, pathogenesis, and treatment. Pathol Int (2018) 68(11):583–95. doi: 10.1111/pin.12727
193. Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, et al. The spliceosome is a therapeutic vulnerability in myc-driven cancer. Nature (2015) 525(7569):384–8. doi: 10.1038/nature14985
194. AACR Project GENIE Consortium. Aacr project genie: powering precision medicine through an international consortium. Cancer Discov (2017) 7(8):818–31. doi: 10.1158/2159-8290.Cd-17-0151
195. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature (2011) 474(7353):609–15. doi: 10.1038/nature10166
196. Zeng M, Kwiatkowski NP, Zhang T, Nabet B, Xu M, Liang Y, et al. Targeting myc dependency in ovarian cancer through inhibition of Cdk7 and Cdk12/13. Elife (2018) 7:e39030. doi: 10.7554/eLife.39030
197. Aird D, Teng T, Huang CL, Pazolli E, Banka D, Cheung-Ong K, et al. Sensitivity to splicing modulation of Bcl2 family genes defines cancer therapeutic strategies for splicing modulators. Nat Commun (2019) 10(1):137. doi: 10.1038/s41467-018-08150-5
198. Murai A, Ebara S, Sasaki S, Ohashi T, Miyazaki T, Nomura T, et al. Synergistic apoptotic effects in cancer cells by the combination of clk and bcl-2 family inhibitors. PLoS One (2020) 15(10):e0240718. doi: 10.1371/journal.pone.0240718
199. Słabicki M, Kozicka Z, Petzold G, Li Y-D, Manojkumar M, Bunker RD, et al. The cdk inhibitor Cr8 acts as a molecular glue degrader that depletes cyclin K. Nature (2020) 585(7824):293–7. doi: 10.1038/s41586-020-2374-x
200. Dieter SM, Siegl C, Codo PL, Huerta M, Ostermann-Parucha AL, Schulz E, et al. Degradation of Ccnk/Cdk12 is a druggable vulnerability of colorectal cancer. Cell Rep (2021) 36(3):109394. doi: 10.1016/j.celrep.2021.109394
201. Jiang B, Gao Y, Che J, Lu W, Kaltheuner IH, Dries R, et al. Discovery and resistance mechanism of a selective Cdk12 degrader. Nat Chem Biol (2021) 17(6):675–83. doi: 10.1038/s41589-021-00765-y
Keywords: alternative splicing, splicing modulator, small molecule, SRPK, CLK, CDK12, patient stratification
Citation: Araki S, Ohori M and Yugami M (2023) Targeting pre-mRNA splicing in cancers: roles, inhibitors, and therapeutic opportunities. Front. Oncol. 13:1152087. doi: 10.3389/fonc.2023.1152087
Received: 27 January 2023; Accepted: 09 May 2023;
Published: 05 June 2023.
Edited by:
Avaniyapuram Kannan Murugan, King Faisal Specialist Hospital & Research Centre, Saudi ArabiaReviewed by:
Michael R. Ladomery, University of the West of England, United KingdomCopyright © 2023 Araki, Ohori and Yugami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shinsuke Araki, c2hpbnN1a2UuYXJha2lAdGFrZWRhLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.