 Wei Cai
Wei Cai Lin-Zhen Shu2
Lin-Zhen Shu2 Ding-Jie Liu
Ding-Jie Liu Huan Deng
Huan Deng- 1Department of Pathology, The Fourth Affiliated Hospital of Nanchang University, Nanchang, China
- 2Medical College, Nanchang University, Nanchang, China
- 3Zhuhai Interventional Medical Center, Zhuhai Precision Medical Center, Zhuhai People’s Hospital, Zhuhai Hospital Affiliated with Jinan University, Zhuhai, China
Cyclin D1 functions as a mitogenic sensor that specifically binds to CDK4/6, thereby integrating external mitogenic inputs and cell cycle progression. Cyclin D1 interacts with transcription factors and regulates various important cellular processes, including differentiation, proliferation, apoptosis, and DNA repair. Therefore, its dysregulation contributes to carcinogenesis. Cyclin D1 is highly expressed in papillary thyroid carcinoma (PTC). However, the particular cellular mechanisms through which abnormal cyclin D1 expression causes PTC are poorly understood. Unveiling the regulatory mechanisms of cyclin D1 and its function in PTC may help determine clinically effective strategies, and open up better opportunities for further research, leading to the development of novel PTC regimens that are clinically effective. This review explores the mechanisms underlying cyclin D1 overexpression in PTC. Furthermore, we discuss the role of cyclin D1 in PTC tumorigenesis via its interactions with other regulatory elements. Finally, recent progress in the development of therapeutic options targeting cyclin D1 in PTC is examined and summarized.
Introduction
The global incidence of thyroid cancer has increased considerably over the past few decades (1). Papillary thyroid carcinoma (PTC), derived from thyroid follicular cells, is the most common endocrine malignancy, accounting for up to 80% of all thyroid carcinomas (2, 3). Early-stage of PTC can be detected using ultrasonography and fine-needle biopsy. However, the typically indolent nature of PTC often leads to a delay in diagnosis that may substantially worsen the course of the disease (4). Consequently, the incidence of advanced large-sized PTCs has increased (2).
Despite the overall survival of patients with PTC being high, lymph node metastasis is observed during diagnosis in almost 36% of cases (5, 6). The biology of PTC is extremely diverse, ranging from non-progressive lesions to aggressive metastatic carcinomas. Although traditional thyroidectomy combined with radioactive iodine therapy is recommended as the first-line treatment for PTC, it is often incurable, leading to a high recurrence rate (4). Recurrence in the neck is a serious complication that may be considered as a sign indicating a potentially lethal outcome (7, 8). Patients at low risk for PTC death may still experience significant or even disastrous morbidities, including invasion of the paratracheal regions, large cervical vessels, and recurrent laryngeal nerves (3, 9). Thus, a deeper understanding of the various signaling pathways involved in PTC progression may help facilitate the development of effective molecular drugs.
Cyclins and their catalytic partners, cyclin-dependent kinases (CDKs), which regulate the eukaryotic cell cycle, are considered as promising candidates for primary involvement in oncogenesis (10). The cyclin D1 gene (CCND1), located on human chromosome 11q13, is an established oncogene (11). The dysregulation of cyclin D1 expression or CDK4/6 activation can directly lead to some of the hallmarks of cancer upregulation via causing proliferation or overriding of checkpoints (12, 13). In human cancer cell lines, cyclin D1 dysregulation contributes to cancer development via its interactions with more than 100 proteins (14). Cyclin D1 overexpression leads to dysregulated cell proliferation, as well as malignant tumor transformation and development, including PTC (15–17). In this review, we address the role of cyclin D1 in PTC and discuss the potential therapeutics of cyclin D1-based treatments.
Cyclin D1 in normal cells
Unique cyclins accumulate at various stages of the cell cycle, contributing to transcription and protein degradation inhibition (18). The synthesis of a single cyclin and the subsequent activation of CDK form active heterodimeric complexes at specific cell cycle phases (13, 19) that coordinate DNA replication and cell division (20). In normal cells, the processes of expression, activation, distribution, stabilization, and degradation of cyclin D1 are strictly regulated by the on/off signal response of mitosis (21, 22). In contrast to cancer cells, normal cells require extracellular signaling to proliferate via the binding of extracellular matrix components to adhesion receptors (integrins) and growth factor receptors (receptor tyrosine kinases). Thus, integrin and growth factor signaling pathways ensure that cell proliferation is restricted to cells exposed to appropriate chemical and physical cues (19, 23, 24).
The G1 phase represents the stage at which cells respond to extracellular signals (25). Cell cycle regulation requires sustained activation of signaling pathways, such as the ERK pathway. Continuous stimulation of mitogen-activated protein kinase (MAPK) is a common requirement for cyclin D1 expression in the G1 phase and cell cycle re-entry (Figure 1) (22, 26, 27). Syntheses of CCND1 mRNA and cyclin D1 protein begins when mitogenic stimulation induces quiescent cells to enter the G1 phase (28). Activation of the RAS-mediated signaling cascade and phosphoinositide-3-kinase (PI3K)/AKT induces CCND1 translation and reduces cyclin D1 degradation (23, 28–30). Growth factor signaling accelerates the formation of cyclin D1-CDK4/6 dipolymer via a Ras-dependent pathway (30). Cyclin D1-CDK4/6 phosphorylates and inactivates retinoblastoma proteins (pRB), resulting in the expression of a subset of proliferation-associated E2F target genes in response to G1 progression (13, 31). This process initiates DNA replication and regulates the transcription of specific cell proliferation genes (13). Cyclin D1 levels increase from early to late G1 phase and then decrease during the S phase (32–34). From G1 to S phase, cyclin D1 is exported to the cytoplasm and degraded by the ubiquitin-proteasome system (UPS) via the phosphorylation of a threonine residue (Thr286) at the carboxyl terminus (28, 35, 36). Studies have shown that Thr286 is mutated in a variety of cancers and that, in animal models, the production of mutant alleles generates spontaneous tumors, demonstrating the tumorigenic potential of cyclin D1 (35, 37, 38). The SCF complex, consisting of an S-phase kinase-associated protein (SKP1) adaptor, CUL1 scaffold and F-box proteins, facilitates the ubiquitination of phosphorylated cyclin D1 in the cytoplasm (33, 39–41). There are at least four E3 ubiquitin ligases in the F-box family, three of which (SKP2, FBXW8, and FBXO4) participate in the normal cell cycle, whereas the fourth (FBXO31) is involved in genotoxic stress (33, 39–41). Cyclin D1 degradation occurs via its direct interaction with the FBXO31 F-box motif and its phosphorylation at Thr286 (40). Cyclin D1-specific E3 ubiquitin ligase mutations result in the accumulation of cyclin D1 in cancer cells (41, 42).
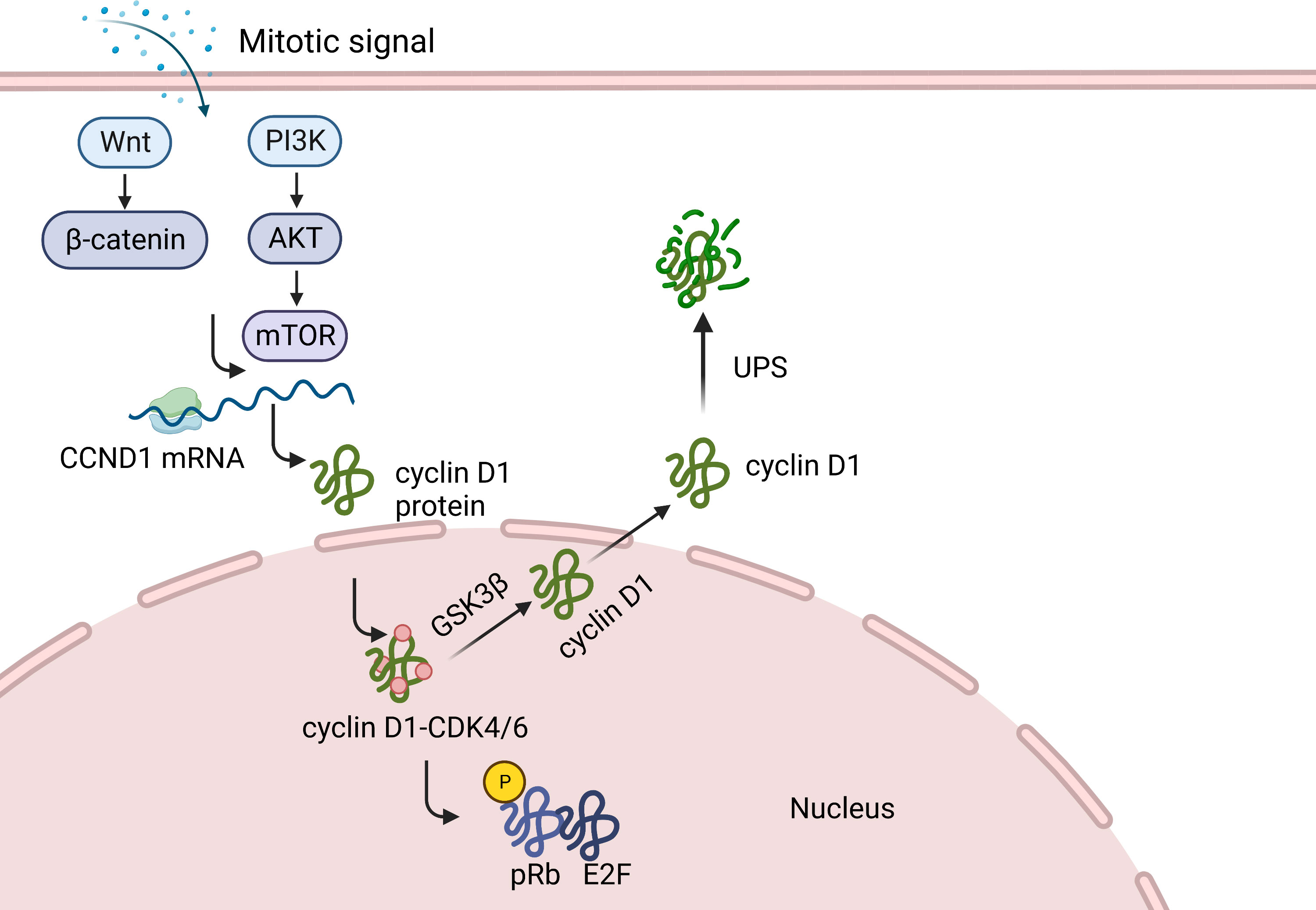
Figure 1 Function of cyclin D1 in normal cells. When mitogenic stimulation induces quiescent cells to enter G1, the synthesis of CCND1 mRNA and cyclin D1 protein begins. Cyclin D1, by forming different heterodimeric complexes with CDK4/6, phosphorylates and inactivates retinoblastoma protein (pRB), causing the expression of a subset of proliferation associated E2F target genes. This, in turn activate some genes in response to the G1 phase progression, thereby initiating DNA replication and regulating the transcription of specific cell proliferation genes. Cyclin D1 is then transported to the cytoplasm and degraded by UPS.
Cyclin D1 as a human oncogene
In line with high CCND1 expression in solid cancers, cyclin D1 is more frequently dysregulated than cyclin D2 or D3 (43–46). There are several explanations for upregulation of cyclin D1, including gene amplification, chromosomal rearrangement, increased gene transcription and protein translation, decreased miRNA expression, and ubiquitination-mediated protein degradation inefficiency or loss (11, 32). Chromosomal translocation in the mantle cell lymphoma (MCL) places the CCND1 under the control of the immunoglobulin heavy chain enhancer, resulting in the abnormal accumulation of cyclin D1 in tumor cells (46–48). In breast cancer, the cyclin D1 overexpression may be attributed to an increase in CCND1 copy numbers (49, 50). In addition, the positive association between cyclin D1 expression and tumor progression has been validated in different cancers, including lung adenocarcinoma, head and neck squamous cell carcinoma, and esophageal squamous cell carcinoma (ESCC) (51–53).
Cyclin D1 and PTC
Dysregulation of cyclin D1 in PTC
Tumor markers constitute the principle of rationalizing the complexity of occurrence and development of tumor diseases that includes, maintaining proliferation signals, evading growth inhibitors, resisting cell death, achieving replication immortality, inducing angiogenesis, and activating invasion and metastasis (12).
A high prevalence of cyclin D1 overexpression in PTC has been demonstrated by several studies (54–60). It is possible that various forms of primary clonal damage may secondarily lead to the dysregulation of cyclin D1, thus providing alternative pathways for cells to develop similar tumor characteristics. Although cyclin D1 is aberrantly overexpressed in PTC, neither translocation nor gene amplification has been reported (61, 62), indicating that pathogenic activation of cyclin D1 may occur via additional mechanisms, including transcriptional and post-transcriptional dysregulation (63, 64). Jeon S et al., showed that CCND1 mRNA levels in PTC are higher than those in benign diseases (65). It is hypothesized that overexpression of cyclin D1 in PTC with high cyclin D1 levels, may not be due to CCND1 amplification, but rather to deregulation of a trans-acting inducer of expression or mRNA degradation machinery.
The expression of cyclin D1 can be regulated by miRNAs (57, 66, 67) that negatively modulate gene expression by binding to the 3′-UTRs of targeted mRNA (68). MiR-211 has been found to bind directly to cyclin D1 mRNA and inhibit its expression in cancers (69, 70). Molecular evidence obtained from clinical PTC samples has revealed a significant inverse correlation between MiR-195 and CCND1 (71). A dual-luciferase reporter assay revealed that co-transfection of MiR-195 inhibits the activity of the luciferase reporter with wild-type 3′-UTR of CCND1, resulting in a consistent negative correlation with the above clinical samples (71). In addition, MiR-195 suppresses the Wnt/β-catenin pathway in PTC, thus significantly reducing the protein levels (cyclin D1) involved in this pathway (71). Targeting MiR-195 may reverse cyclin D1-mediated cellular effects, including cell proliferation, apoptosis, migration, and invasion in PTC (71). Paired box gene 8 (PAX8) plays a critical role in thyroid development (72). MiR-144-3p binds to PAX8, that indirectly regulates the expression of cyclin D1 in PTC, thereby promoting cell cycle progression (72). MiR-1256 inhibits PTC cell growth and induces G0/G1 phase arrest. MiR-1256 is downregulated in PTC and its inhibitory effect on 5-hydroxy tryptamine receptor 3A (HTR3A) is dysregulated. High levels of HTR3A in PTC cells can partially eliminate the inhibitory effect of MiR-1256 on cyclin D1 expression (73). HTR3A knockdown significantly induces cell cycle arrest at the G0/G1 phase, hampering PTC cell proliferation (73). Lou et al., established the hsa_circ_0088494-miR-876-3p-CTNNB1/CCND1 axis using database analysis and found that it was associated with PTC carcinogenesis and progression (67).
Cyclin D1 is a target molecule in the aberrantly activated Wnt/β-catenin signaling pathway (74, 75). Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (PIN1) contributes to the upregulation of cyclin D1, either directly or via the abnormal accumulation of β-catenin in tumor cells (76). PIN1 increases cyclin D1 expression, both transcriptionally and post-transcriptionally (77, 78). PIN1 interacts with c-Jun that is phosphorylated on Ser63/67-Pro motifs by activated oncogenic Ras/JNK, and subsequently boosts transcriptional activity of c-Jun to the cyclin D1 promoter (77). Moreover, PIN1 improves the protein stability and increases nuclear accumulation of cyclin D1 via heterodimerization (78). PIN1 inhibits nuclear export and protein degradation of β-catenin (79). Increased nuclear accumulation of β-catenin during Wnt/β-catenin cell signaling leads to enhanced transcriptional activity of β-catenin on downstream target genes, including CCND1 (74). APC gene mutation is a common event in familial adenomatous polyposis coil (FAP)-related PTC, accompanied by nuclear β-catenin translocation (76, 80). In PTC cell lines, the formation of a destruction complex with APC is inhibited by PIN1 (76). Therefore, overexpression of PIN1 may be a factor of cyclin D1 upregulation and abnormal β-catenin expression during malignant thyroid gland transformation.
The constitutive activation of the MAPK pathway plays an important role in PTC development (81, 82). MAPK induces several mitotic and survival processes, including proliferation and protection from apoptosis via cell membrane substrate interactions and subsequent phosphorylation of transcription factors (83). The mutant genes involved encode the cell membrane-receptor tyrosine kinases, RET and NTRK1, as well as the intracellular signal transduction genes, BRAF and RAS (82). These mutations, which occur in approximately 70% of patients with PTC, are associated with specific clinical and biological tumor characteristics (16, 84–86). Important oncogenic mechanisms of the MAPK pathway in PTC include modulation of type 3 deiodinase (DIO3) to increase cyclin D1 expression (81). Nelfinavir (NFV), a MAPK/ERK pathway inhibitor, accelerates PTC cell arrest at the G0/G1 phase and downregulates cyclin D1 and CDK4 (87).
Cyclin D1 expression is also transcriptionally regulated by signal transducers and activators of transcription 3 (STAT3), known to be important for angiogenesis, tumorigenesis, and metastasis (88–92). STAT3 regulates the expression of cyclin D1 by binding to the CCND1 promoter (93). Cyclin D1 mRNA levels are increased in tumor cells that express the STAT3 oncogenic variant (STAT3-C) or vSrc that constitutively phosphorylate STAT3 (93). Moreover, it has been proved that cyclin D1 is necessary for STAT3-C and vSrc-mediated anchorage-independent growth (93). In addition, both STAT3 and cyclin D1 protein levels in human PTC tissues are higher than those in adjacent non-tumorous thyroid tissues (88, 94).
The association between cyclin D1 and clinical parameters of PTC have been studied. In PTC, high cyclin D1 levels are associated with larger tumor size, intraglandular metastasis, extrathyroidal extension, lymph node metastasis, and aggressive behavior (17, 95–98). High cyclin D1 levels are linked to poor prognoses and high recurrence rates (56). These observations indicate that deregulated cyclin D1 expression may play an important role in determining the clinical course of PTC.
Oncogenic consequences of cyclin D1 dysregulation in PTC
An essential characteristic of tumor cells is their ability to progress through proliferation limits set by tumor suppressors under conditions of cellular stress (18). In PTC, the dysregulation of cyclin D1 represents the initiation of sustained proliferative signaling acquisition and the malignant transformation of thyroid follicular cells. Activation of cyclin D1-CDK4/6 phosphorylates various substrates that regulate PTC cell proliferation, growth, migration, DNA repair, and centrosome duplication (11, 87). The cyclin D1-CDK4/6-Rb-E2F pathway regulates the G1 to S phase transition. During the S phase, cyclin D1 is exported to the cytoplasm and degraded (99). Dysregulation of these processes leads to the abnormal accumulation of cyclin D1 (100). Cell proliferation becomes independent of extracellular signals and bypasses cell cycle checkpoints responsible for ensuring genomic integrity (101, 102). Increased cyclin D1-CDK4/6 induces E2F-dependent transcription and promotes continuous proliferation of PTC cells by preventing cell cycle exit (18, 102–105).
Transcriptional regulation is one of the major non-catalytic functions of cyclin D1. Cyclin D1 affects the promoters of many genes by interacting with various transcription factors (106). Thrombospondin 1 (TSP-1), a multifunctional matricellular ECM and secreted protein (107), effectively inhibits tumorigenesis, cellular metastasis, and in vivo neovascularization (107, 108). Its promoter activity is repressed by cyclin D1 in a dose-dependent manner (108). Cyclin D1 inhibits TSP-1 transcription and mRNA expression and promotes cell migration (108, 109). Compared with those in normal thyroid tissues, TSP-1 mRNA levels are significantly lower in PTC tissues (110). The extrathyroidal infiltration of PTC is inversely correlated with TSP-1 expression (111). It has been shown that TSP-1 suppresses angiogenesis in PTC (17). These results further confirm that cyclin D1 promotes PTC carcinogenesis by downregulating TSP-1 expression.
Many studies have suggested the presence of a tight link between estrogen receptor-α (Erα) and cyclin D1 in PTC (112, 113). Cyclin D1 interacts with several members of the steroid hormone receptor superfamily and their co-regulators (63, 114). Cyclin D1 enhances the activity of Erα by interacting with its co-regulators, SRC1 (NCOA1) and AIB1 (NCOA3), in tumor cells (11). It also interacts with SRC1 and SRC3 to recruit additional transcriptional cofactors that increase the transcriptional activity of ER (115). Consequently, abnormally upregulated ER signaling leads to increased proliferation of PTC cell lines (112). These findings suggest that cyclin D1 overexpression promotes PTC development via the enhancement of Erα function.
Cyclin D1-targeted treatments
Oncoproteins are appealing therapeutic targets due to their involvement in malignant cell behavior (116). Given its importance in PTC, cyclin D1 has come to be considered as an attractive drug target for clinical treatments. Fibroblasts, epithelial cells, and macrophages exhibit increased cellular adhesion and decreased motility in the absence of cyclin D1 (108, 117, 118). Previous studies have shown that suppression of cyclin D1 in PTC cells reduces cell proliferation, migration, and invasion (108, 119, 120).
If an important component of the oncogenic action of cyclin D1 is accomplished via CDK-independent mechanisms, anti-cyclin D1 may be expected to be especially efficacious in cyclin D1-driven PTCs. The effective use of such new approaches is centered on the tailored selection of PTC subgroups that are likely to respond to treatment. In PTC, high nuclear cyclin D1 levels are associated with aggressive clinicopathological features, including lymph node metastasis, tumor recurrence, extrathyroidal invasion, and more advanced initial tumor stages. As described, aberration of activated Wnt/β-catenin signaling pathway enhances the transcriptional activity of CCND1 and increases nuclear accumulation of cyclin D1. Activation of MAPK and PI3K/AKT pathways, which is usually induced by BRAF and RAS family mutations, are the two most fundamental causative factors of PTC (121, 122). The RAS–MAPK pathway regulates cell cycle entry by upregulating cyclin D1 expression in PTC cells (56). Therefore, β-catenin, BRAF, and RAS may serve as useful biomarkers in patients requiring cyclin D1-based therapy for PTC. Patients receiving this therapy may have longer progression-free and a better overall survival than those receiving standard therapies that are accepted in cases of unsuccessful surgery and radioiodine therapy. However, targeting cyclin D1 is often considered difficult because of its lack of intrinsic enzymatic activity (11). Drug combination therapies that affect CCND1 transcription or cyclin D1 degradation, target several end points of cyclin D1 function. Alternative methods may be employed to target cyclin D1 and block PTC progression (Figure 2).
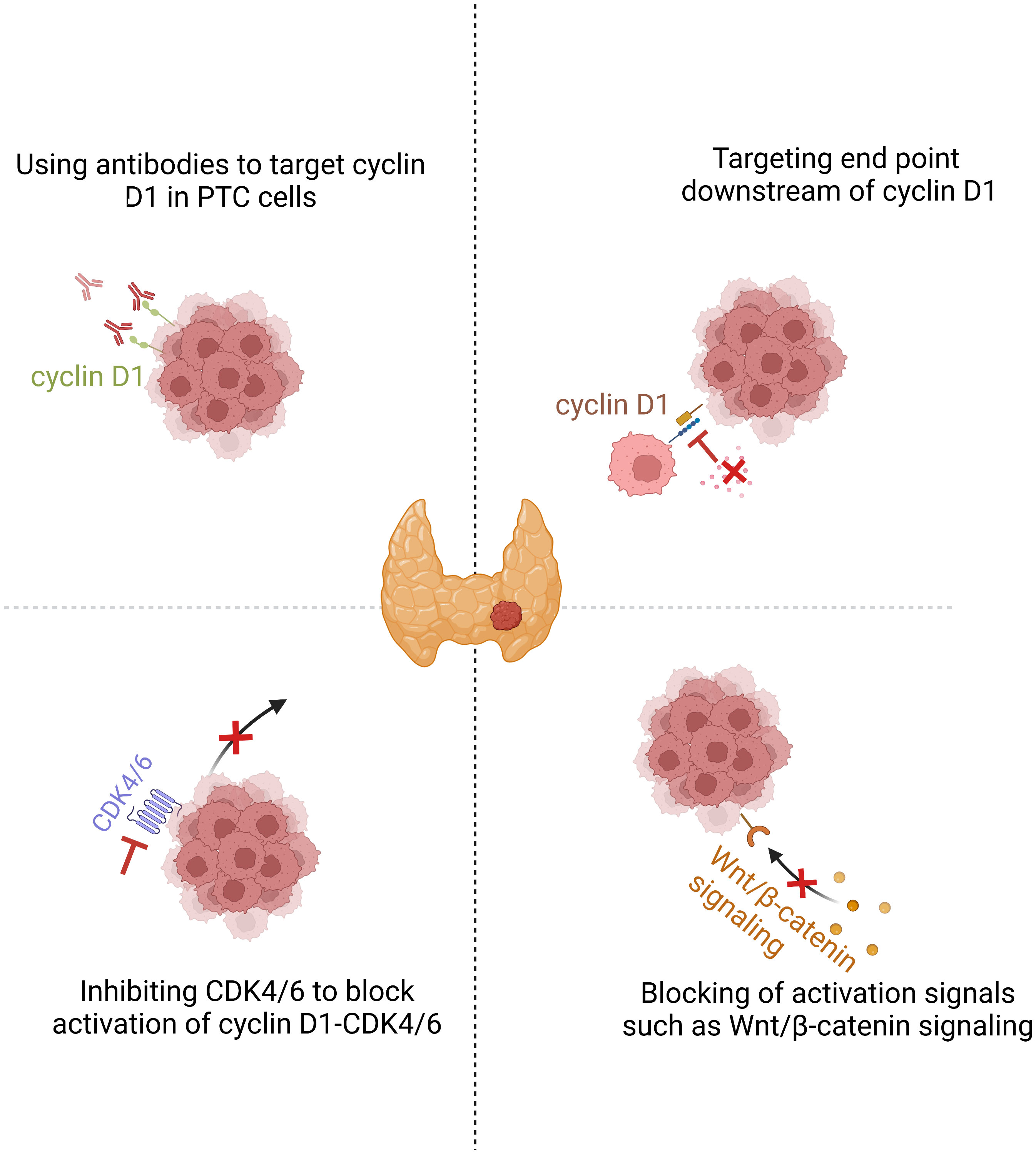
Figure 2 Cyclin D1 is a promising target in PTC. Downregulating cyclin D1 or inhibiting the end points of cyclin D1 action are two possible therapeutic approaches for cyclin D1-dependent cancers. Inhibition of PTC cell proliferation by CDK4/6 inhibition or broader targeting of cyclin D1 action by drugs that cause cyclin D1 downregulation or protein degradation.
Targeting of cyclin D1 using a combination of the retinoid X receptor (RXR) activator (Bexarotene) and a EGFR inhibitor (Erlotinib) has shown satisfactory levels of efficacy in clinical studies (123). Rosiglitazone diminishes the expression of cyclin D1, increases the expression of cell cycle inhibitors p21 and p27, and suppresses the proliferation and migration of tumor cells in a dose-dependent manner (124, 125). Both rosiglitazone and bexarotene exhibit anti-cancer activities against human PTC cells (126). The sodium/iodine symporter (NIS) is positively expressed in thyroid cancers with a good prognosis and plays a key role in response to radioiodine therapy. Moreover, NIS downregulation usually predicts tumor recurrence or treatment failure (126). In the setting of normal or hypoxic conditions, bexarotene synergizes with rosiglitazone to inhibit malignant cell growth and increase NIS levels in PTC, thereby demonstrating its potential as a chemotherapeutic candidate for PTC. However, despite their promising anti-proliferative effects, the efficacy and safety of rosiglitazone and bexarotene for PTC in humans require further verification.
Translation of CCND1 mRNA is dependent on mTOR, raising the possibility that mTOR inhibitors may hamper the cell cycle and tumor progression via cyclin D1 (101, 127). A recent clinical trial found, rapamycin, an mTOR inhibitor, to be therapeutically beneficial for nearly 40% of patients with advanced MCLs (128). An elaborate study demonstrated that the mTOR kinase inhibitor, CZ415, decreases cyclin D1 expression and induces G1-S arrest in human PTC cells. Moreover, it proved that CZ415 plays a role in the inhibition of cell proliferation and xenograft tumor growth in PTC (129). Thus, future studies on mTOR kinase inhibitor-based therapies may help improve the survival of patients with PTC.
Abnormal dysregulation of cyclin-CDK perturbs cell cycle control and allows continuous cell division (31, 130, 131). The earliest identified function of cyclin D1 was its role as a regulatory partner of CDK4/6 (11). Owing to increasing clinical applications of specific kinase inhibitors, a more direct and feasible method is to target cyclin D1 by inhibiting CDK4/6 (25, 132). Many first-generation CDK inhibitors have been excluded from research studies, partly because nonselective pan-CDK inhibition causes cytotoxicity in noncancerous cells (133, 134). However, these issues have been largely addressed by the newly developed CDK 4/6 pharmaceuticals with high specificity (132). Manageable cytotoxicity makes CDK 4/6 inhibitors (palbociclib, abemaciclib, and ribociclib) attractive candidates for anti-cancer drugs (135, 136). A key phase III trial of palbociclib for advanced ER-positive breast cancer showed improved progression-free as well as overall survival, with well tolerated toxicity. Testing for CDK4/6 inhibitors in low-risk patients with PTC might be difficult for ethical reasons. Extending the use of CDK4/6 inhibitors beyond ER-positive breast cancer is challenging and requires biomarkers that are predictive of the applicable standards. Antonello et al., demonstrated that targeting CDK4/6 in aggressive PTC helps overcome CDK4/6-dependent cell cycle checkpoint dysfunction and triggers apoptosis (137). Combination therapy with CDK4/6 inhibitors downregulates pAKT levels, resulting in changes in its downstream effectors. In addition, combination treatments with CDK4/6 inhibitors showed impressive improvements in the ability to overcome both primary and secondary resistance offered to treatments with single agents in invasive human PTC cell lines (137). Radioactive iodine (RAI) is administered for differentiated thyroid cancer (DTC) with intermediate to high-risk features (138). However, some patients with DTC and distant metastases exhibit dedifferentiation and decreased iodine uptake, rendering adjuvant treatment with RAI ineffective (138). Anaplastic thyroid carcinoma (ATC) is the most aggressive type of thyroid cancer. It accounts for a disproportionate number of thyroid cancer-related deaths, because of its resistance to current therapeutic approaches (138, 139). The selective CDK4/6 inhibitor, ribociclib, induces cell cycle arrest at the G0-G1 phase, facilitates cell apoptosis, and inhibits cell proliferation in ATC (140). Lack of expression or dysfunction of NIS is considered to be an important causative factor of iodine-resistant tumors (138). Studies have indicated that mutations in MAPK and/or PI3K (BRAF V600E and RAS) may decrease NIS expression by directly affecting its transcription (141, 142). BRAF V600E and MEK inhibitors can re-sensitize dedifferentiated thyroid cancer to iodine, thus allowing RAI treatment, leading to improved disease control (143, 144). The selective BRAF inhibitor (dabrafenib) can stimulate radioiodine uptake in patients with metastatic BRAF V600E mutant iodine-refractory PTC (143). Taken together, these studies suggest that BRAF V600E may serve as a promising biomarker for predicting high-risk radioiodine-resistant PTC, in patients enrolled for CDK4/6 inhibitor clinical trials. Specific CDK4/6 inhibitors have fewer therapeutic targets and are, therefore, more specific than multi-kinase inhibitors. Although, a clinical study of combined therapy with CDK4/6 inhibitors against PTC, is yet to be conducted, this approach may provide new insights to the possibility of improving the overall survival of patients with PTC and minimizing the risk of drug resistance and side effects.
Conclusion
Since the discovery of CCND1 in 1991, considerable progress has been made towards a better understanding of its physiology and role in various diseases. Cyclin D1 is frequently deregulated in PTC and serves as a biomarker of cancer phenotype and disease progression. In this review, we discuss recent studies that uncovered the detailed role of cyclin D1 in cell cycle control and specific changes in PTC cells. With due consideration given to the oncogenic role of cyclin D1 in PTC carcinogenesis, cyclin D1 shows promise as a potential therapeutic target against PTC. Through high-throughput screening techniques, it is possible to identify novel and effective cyclin D1 inhibitors from a diverse library of chemical compounds. Meanwhile, the effective use of potential therapies towards cyclin D1 or CDK4/6 will rely on the development of biomarkers for the therapeutic response. Considering that the efficacy of most targeted therapies is limited due to drug resistance, it is important to understand the mechanisms underlying resistance to cyclin D1 or CDK4/6 inhibition. Thus, further studies leading to the development of optimization methods that enhance the anti-cancer effects of cyclin D1 inhibitors in PTC are warranted.
Author contributions
HD and WC conceived the study. WC wrote the manuscript. WC and D-JL participated in the figure production. WC, L-ZS, D-JL, LZ, M-MW, and HD improved the manuscript quality. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Science Foundation of China, No.82160546; the Science Foundation of Jiangxi Province, No. 20202BBG73027; the Foundation of Jiangxi Province for Distinguished Scholars No. JXSQ2023201020.
Conflict of interest
The authors declare that this study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer YX declared a shared parent affiliation with the authors WC, HD, L-ZS, LZ, M-MW to the handling editor at the time of review.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin (2011) 61(2):69–90. doi: 10.3322/caac.20107
2. Enewold L, Zhu K, Ron E, Marrogi AJ, Stojadinovic A, Peoples GE, et al. Rising thyroid cancer incidence in the united states by demographic and tumor characteristics, 1980-2005. Cancer Epidemiol Biomarkers Prev (2009) 18(3):784–91. doi: 10.1158/1055-9965.EPI-08-0960
3. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer (2013) 13(3):184–99. doi: 10.1038/nrc3431
4. Tuttle RM, Ball DW, Byrd D, Dilawari RA, Doherty GM, Duh QY, et al. Thyroid carcinoma. J Natl Compr Canc Netw (2010) 8(11):1228–74. doi: 10.6004/jnccn.2010.0093
5. Esposito T, Lucariello A, Hay E, Contieri M, Tammaro P, Varriale B, et al. Effects of curcumin and its adjuvant on TPC1 thyroid cell line. Chem Biol Interact (2019) 305:112–8. doi: 10.1016/j.cbi.2019.03.031
6. Mazzaferri EL. Management of a solitary thyroid nodule. N Engl J Med (1993) 328(8):553–9. doi: 10.1056/NEJM199302253280807
7. Nixon IJ, Whitcher MM, Palmer FL, Tuttle RM, Shaha AR, Shah JP, et al. The impact of distant metastases at presentation on prognosis in patients with differentiated carcinoma of the thyroid gland. Thyroid (2012) 22(9):884–9. doi: 10.1089/thy.2011.0535
8. Robie DK, Dinauer CW, Tuttle RM, Ward DT, Parry R, McClellan D, et al. The impact of initial surgical management on outcome in young patients with differentiated thyroid cancer. J Pediatr Surg (1998) 33(7):1134–8. doi: 10.1016/S0022-3468(98)90546-2
9. Xing M, Westra WH, Tufano RP, Cohen Y, Rosenbaum E, Rhoden KJ, et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J Clin Endocrinol Metab (2005) 90(12):6373–9. doi: 10.1210/jc.2005-0987
10. Hunter T, Pines J. Cyclins and cancer. Cell (1991) 66(6):1071–4. doi: 10.1016/0092-8674(91)90028-W
11. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin d as a therapeutic target in cancer. Nat Rev Cancer (2011) 11(8):558–72. doi: 10.1038/nrc3090
12. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
13. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer (2009) 9(3):153–66. doi: 10.1038/nrc2602
14. Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature (2011) 474(7350):230–4. doi: 10.1038/nature10155
15. Sherr CJ. Cancer cell cycles. Science (1996) 274(5293):1672–7. doi: 10.1126/science.274.5293.1672
16. Frattini M, Ferrario C, Bressan P, Balestra D, De Cecco L, Mondellini P, et al. Alternative mutations of BRAF, RET and NTRK1 are associated with similar but distinct gene expression patterns in papillary thyroid cancer. Oncogene (2004) 23(44):7436–40. doi: 10.1038/sj.onc.1207980
17. Khoo ML, Beasley NJ, Ezzat S, Freeman JL, Asa SL. Overexpression of cyclin D1 and underexpression of p27 predict lymph node metastases in papillary thyroid carcinoma. J Clin Endocrinol Metab (2002) 87(4):1814–8. doi: 10.1210/jcem.87.4.8353
18. Matthews HK, Bertoli C, de Bruin RAM. Cell cycle control in cancer. Nat Rev Mol Cell Biol (2021), 23(1):74–88. doi: 10.1038/s41580-021-00404-3
19. Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer (2001) 1(3):222–31. doi: 10.1038/35106065
20. Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell (2008) 14(2):159–69. doi: 10.1016/j.devcel.2008.01.013
21. Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev (2004) 18(22):2699–711. doi: 10.1101/gad.1256504
22. Balmanno K, Cook SJ. Sustained MAP kinase activation is required for the expression of cyclin D1, p21Cip1 and a subset of AP-1 proteins in CCL39 cells. Oncogene (1999) 18(20):3085–97. doi: 10.1038/sj.onc.1202647
23. Takuwa N, Fukui Y, Takuwa Y. Cyclin D1 expression mediated by phosphatidylinositol 3-kinase through mTOR-p70(S6K)-independent signaling in growth factor-stimulated NIH 3T3 fibroblasts. Mol Cell Biol (1999) 19(2):1346–58. doi: 10.1128/MCB.19.2.1346
24. Perry JE, Grossmann ME, Tindall DJ. Epidermal growth factor induces cyclin D1 in a human prostate cancer cell line. Prostate (1998) 35(2):117–24. doi: 10.1002/(SICI)1097-0045(19980501)35:2<117::AID-PROS5>3.0.CO;2-G
25. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discovery (2016) 6(4):353–67. doi: 10.1158/2159-8290.CD-15-0894
26. Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem J (1997) 326(Pt 1):61–8. doi: 10.1042/bj3260061
27. Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell (1995) 80(2):179–85. doi: 10.1016/0092-8674(95)90401-8
28. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev (1998) 12(22):3499–511. doi: 10.1101/gad.12.22.3499
29. Daniel P, Filiz G, Brown DV, Hollande F, Gonzales M, D’Abaco G, et al. Selective CREB-dependent cyclin expression mediated by the PI3K and MAPK pathways supports glioma cell proliferation. Oncogenesis (2014) 3:e108. doi: 10.1038/oncsis.2014.21
30. Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin d to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin d-dependent kinase CDK4. Genes Dev (1993) 7(3):331–42. doi: 10.1101/gad.7.3.331
31. Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene (2005) 24(17):2909–15. doi: 10.1038/sj.onc.1208618
32. Qie S, Diehl JA. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (Berl) (2016) 94(12):1313–26. doi: 10.1007/s00109-016-1475-3
33. Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell (2006) 24(3):355–66. doi: 10.1016/j.molcel.2006.09.007
34. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev (1999) 13(12):1501–12. doi: 10.1101/gad.13.12.1501
35. Benzeno S, Lu F, Guo M, Barbash O, Zhang F, Herman JG, et al. Identification of mutations that disrupt phosphorylation-dependent nuclear export of cyclin D1. Oncogene (2006) 25(47):6291–303. doi: 10.1038/sj.onc.1209644
36. Yoshida A, Choi J, Jin HR, Li Y, Bajpai S, Qie S, et al. Fbxl8 suppresses lymphoma growth and hematopoietic transformation through degradation of cyclin D3. Oncogene (2021) 40(2):292–306. doi: 10.1038/s41388-020-01532-4
37. Moreno-Bueno G, Rodriguez-Perales S, Sanchez-Estevez C, Hardisson D, Sarrio D, Prat J, et al. Cyclin D1 gene (CCND1) mutations in endometrial cancer. Oncogene (2003) 22(38):6115–8. doi: 10.1038/sj.onc.1206868
38. Augello MA, Berman-Booty LD, Carr R 3rd, Yoshida A, Dean JL, Schiewer MJ, et al. Consequence of the tumor-associated conversion to cyclin D1b. EMBO Mol Med (2015) 7(5):628–47. doi: 10.15252/emmm.201404242
39. Okabe H, Lee SH, Phuchareon J, Albertson DG, McCormick F, Tetsu O. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PloS One (2006) 1:e128. doi: 10.1371/journal.pone.0000128
40. Santra MK, Wajapeyee N, Green MR. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature (2009) 459(7247):722–5. doi: 10.1038/nature08011
41. Barbash O, Zamfirova P, Lin DI, Chen X, Yang K, Nakagawa H, et al. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer. Cancer Cell (2008) 14(1):68–78. doi: 10.1016/j.ccr.2008.05.017
42. Vaites LP, Lee EK, Lian Z, Barbash O, Roy D, Wasik M, et al. The Fbx4 tumor suppressor regulates cyclin D1 accumulation and prevents neoplastic transformation. Mol Cell Biol (2011) 31(22):4513–23. doi: 10.1128/MCB.05733-11
43. Qie S, Diehl JA. Cyclin d degradation by E3 ligases in cancer progression and treatment. Semin Cancer Biol (2020) 67(Pt 2):159–70. doi: 10.1016/j.semcancer.2020.01.012
44. Qie S, Yoshida A, Parnham S, Oleinik N, Beeson GC, Beeson CC, et al. Targeting glutamine-addiction and overcoming CDK4/6 inhibitor resistance in human esophageal squamous cell carcinoma. Nat Commun (2019) 10(1):1296. doi: 10.1038/s41467-019-09179-w
45. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463(7283):899–905. doi: 10.1038/nature08822
46. Rosenberg CL, Wong E, Petty EM, Bale AE, Tsujimoto Y, Harris NL, et al. PRAD1, a candidate BCL1 oncogene: mapping and expression in centrocytic lymphoma. Proc Natl Acad Sci U S A (1991) 88(21):9638–42. doi: 10.1073/pnas.88.21.9638
47. Komatsu H, Iida S, Yamamoto K, Mikuni C, Nitta M, Takahashi T, et al. A variant chromosome translocation at 11q13 identifying PRAD1/cyclin D1 as the BCL-1 gene. Blood (1994) 84(4):1226–31. doi: 10.1182/blood.V84.4.1226.1226
48. Bergsagel PL, Kuehl WM. Critical roles for immunoglobulin translocations and cyclin d dysregulation in multiple myeloma. Immunol Rev (2003) 194:96–104. doi: 10.1034/j.1600-065X.2003.00052.x
49. Courjal F, Cuny M, Simony-Lafontaine J, Louason G, Speiser P, Zeillinger R, et al. Mapping of DNA amplifications at 15 chromosomal localizations in 1875 breast tumors: definition of phenotypic groups. Cancer Res (1997) 57(19):4360–7.
50. Schuuring E. The involvement of the chromosome 11q13 region in human malignancies: cyclin D1 and EMS1 are two new candidate oncogenes–a review. Gene (1995) 159(1):83–96. doi: 10.1016/0378-1119(94)00562-7
51. Jiang W, Kahn SM, Tomita N, Zhang YJ, Lu SH, Weinstein IB. Amplification and expression of the human cyclin d gene in esophageal cancer. Cancer Res (1992) 52(10):2980–3.
52. Michalides R, van Veelen N, Hart A, Loftus B, Wientjens E, Balm A. Overexpression of cyclin D1 correlates with recurrence in a group of forty-seven operable squamous cell carcinomas of the head and neck. Cancer Res (1995) 55(5):975–8.
53. Yao G, Tang J, Yang X, Zhao Y, Zhou R, Meng R, et al. Cyclin K interacts with beta-catenin to induce cyclin D1 expression and facilitates tumorigenesis and radioresistance in lung cancer. Theranostics (2020) 10(24):11144–58. doi: 10.7150/thno.42578
54. Muro-Cacho CA HT, Klotch D, Mora L, Livingston S, Futran N. Cyclin D1 expression as a prognostic parameter in papillary carcinoma of the thyroid. Otolaryngol Head Neck Surg (1999), 200–7. doi: 10.1016/S0194-5998(99)70407-9
55. Khoo ML, Ezzat S, Freeman JL, Asa SL. Cyclin D1 protein expression predicts metastatic behavior in thyroid papillary microcarcinomas but is not associated with gene amplification. J Clin Endocrinol Metab (2002), 87(4):1810–3. doi: 10.1210/jcem.87.4.8352
56. Sanjari M, Kordestani Z, Safavi M, Mashrouteh M, FekriSoofiAbadi M, Ghaseminejad Tafreshi A. Enhanced expression of cyclin D1 and c-myc, a prognostic factor and possible mechanism for recurrence of papillary thyroid carcinoma. Sci Rep (2020) 10(1):5100. doi: 10.1038/s41598-020-61985-1
57. Nakajima K, Inagawa M, Uchida C, Okada K, Tane S, Kojima M, et al. Coordinated regulation of differentiation and proliferation of embryonic cardiomyocytes by a jumonji (Jarid2)-cyclin D1 pathway. Development (2011) 138(9):1771–82. doi: 10.1242/dev.059295
58. Lazzereschi D, Sambuco L, Carnovale Scalzo C, Ranieri A, Mincione G, Nardi F, et al. Cyclin D1 and cyclin e expression in malignant thyroid cells and in human thyroid carcinomas. Int J Cancer (1998) 76(6):806–11. doi: 10.1002/(SICI)1097-0215(19980610)76:6<806::AID-IJC7>3.0.CO;2-1
59. Basolo F, Caligo MA, Pinchera A, Fedeli F, Baldanzi A, Miccoli P, et al. Cyclin D1 overexpression in thyroid carcinomas: relation with clinico-pathological parameters, retinoblastoma gene product, and Ki67 labeling index. Thyroid (2000) 10(9):741–6. doi: 10.1089/thy.2000.10.741
60. Bieche I, Franc B, Vidaud D, Vidaud M, Lidereau R. Analyses of MYC, ERBB2, and CCND1 genes in benign and malignant thyroid follicular cell tumors by real-time polymerase chain reaction. Thyroid (2001) 11(2):147–52. doi: 10.1089/105072501300042802
61. Cancer Genome Atlas Research N. Integrated genomic characterization of papillary thyroid carcinoma. Cell (2014) 159(3):676–90. doi: 10.1016/j.cell.2014.09.050
62. Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest (2016) 126(3):1052–66. doi: 10.1172/JCI85271
63. Coqueret O. Linking cyclins to transcriptional control. Gene (2002) 299(1-2):35–55. doi: 10.1016/S0378-1119(02)01055-7
64. Albanese C, Wu K, D’Amico M, Jarrett C, Joyce D, Hughes J, et al. IKKalpha regulates mitogenic signaling through transcriptional induction of cyclin D1 via tcf. Mol Biol Cell (2003) 14(2):585–99. doi: 10.1091/mbc.02-06-0101
65. Jeon S, Kim Y, Jeong YM, Bae JS, Jung CK. CCND1 splice variant as a novel diagnostic and predictive biomarker for thyroid cancer. Cancers (Basel) (2018) 10(11):437. doi: 10.3390/cancers10110437
66. Lazaro JB, Bailey PJ, Lassar AB. Cyclin d-cdk4 activity modulates the subnuclear localization and interaction of MEF2 with SRC-family coactivators during skeletal muscle differentiation. Genes Dev (2002) 16(14):1792–805. doi: 10.1101/gad.U-9988R
67. Lou W, Ding B, Wang J, Xu Y. The involvement of the hsa_circ_0088494-miR-876-3p-CTNNB1/CCND1 axis in carcinogenesis and progression of papillary thyroid carcinoma. Front Cell Dev Biol (2020) 8:605940. doi: 10.3389/fcell.2020.605940
68. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol (2018) 141(4):1202–7. doi: 10.1016/j.jaci.2017.08.034
69. Xia B, Yang S, Liu T, Lou G. miR-211 suppresses epithelial ovarian cancer proliferation and cell-cycle progression by targeting cyclin D1 and CDK6. Mol Cancer (2015) 14:57. doi: 10.1186/s12943-015-0322-4
70. Guo Y, Chen Y, Liu H, Yan W. Alpinetin inhibits oral squamous cell carcinoma proliferation via miR-211-5p upregulation and notch pathway deactivation. Nutr Cancer (2020) 72(5):757–67. doi: 10.1080/01635581.2019.1651878
71. Yin Y, Hong S, Yu S, Huang Y, Chen S, Liu Y, et al. MiR-195 inhibits tumor growth and metastasis in papillary thyroid carcinoma cell lines by targeting CCND1 and FGF2. Int J Endocrinol (2017) 2017:6180425. doi: 10.1155/2017/6180425
72. Liu C, Su C, Chen Y, Li G. MiR-144-3p promotes the tumor growth and metastasis of papillary thyroid carcinoma by targeting paired box gene 8. Cancer Cell Int (2018) 18:54. doi: 10.1186/s12935-018-0550-y
73. Wu C, Ma L, Wei H, Nie F, Ning J, Jiang T. MiR-1256 inhibits cell proliferation and cell cycle progression in papillary thyroid cancer by targeting 5-hydroxy tryptamine receptor 3A. Hum Cell (2020) 33(3):630–40. doi: 10.1007/s13577-020-00325-x
74. Kafri P, Hasenson SE, Kanter I, Sheinberger J, Kinor N, Yunger S, et al. Quantifying beta-catenin subcellular dynamics and cyclin D1 mRNA transcription during wnt signaling in single living cells. Elife (2016) 5:e16748. doi: 10.7554/eLife.16748
75. Yu J, Liu D, Sun X, Yang K, Yao J, Cheng C, et al. CDX2 inhibits the proliferation and tumor formation of colon cancer cells by suppressing wnt/beta-catenin signaling via transactivation of GSK-3beta and Axin2 expression. Cell Death Dis (2019) 10(1):26. doi: 10.1038/s41419-018-1263-9
76. Nakashima M, Meirmanov S, Naruke Y, Kondo H, Saenko V, Rogounovitch T, et al. Cyclin D1 overexpression in thyroid tumours from a radio-contaminated area and its correlation with Pin1 and aberrant beta-catenin expression. J Pathol (2004) 202(4):446–55. doi: 10.1002/path.1534
77. Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, et al. Pin1 is overexpressed in breast cancer and cooperates with ras signaling in increasing the transcriptional activity of c-jun towards cyclin D1. EMBO J (2001) 20(13):3459–72. doi: 10.1093/emboj/20.13.3459
78. Liou YC, Ryo A, Huang HK, Lu PJ, Bronson R, Fujimori F, et al. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc Natl Acad Sci U S A (2002) 99(3):1335–40. doi: 10.1073/pnas.032404099
79. Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat Cell Biol (2001) 3(9):793–801. doi: 10.1038/ncb0901-793
80. Cole AM, Myant K, Reed KR, Ridgway RA, Athineos D, Van den Brink GR, et al. Cyclin D2-cyclin-dependent kinase 4/6 is required for efficient proliferation and tumorigenesis following apc loss. Cancer Res (2010) 70(20):8149–58. doi: 10.1158/0008-5472.CAN-10-0315
81. Romitti M, Wajner SM, Ceolin L, Ferreira CV, Ribeiro RV, Rohenkohl HC, et al. MAPK and SHH pathways modulate type 3 deiodinase expression in papillary thyroid carcinoma. Endocr Relat Cancer (2016) 23(3):135–46. doi: 10.1530/ERC-15-0162
82. Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol (2011) 7(10):569–80. doi: 10.1038/nrendo.2011.142
83. Sekulic A, Haluska P Jr., Miller AJ, Genebriera De Lamo J, Ejadi S, Pulido JS, et al. Malignant melanoma in the 21st century: the emerging molecular landscape. Mayo Clin Proc (2008) 83(7):825–46. doi: 10.4065/83.7.825
84. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res (2003) 63(7):1454–7.
85. Soares P, Trovisco V, Rocha AS, Lima J, Castro P, Preto A, et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene (2003) 22(29):4578–80. doi: 10.1038/sj.onc.1206706
86. Adeniran AJ, Zhu Z, Gandhi M, Steward DL, Fidler JP, Giordano TJ, et al. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am J Surg Pathol (2006) 30(2):216–22. doi: 10.1097/01.pas.0000176432.73455.1b
87. Jensen K, Bikas A, Patel A, Kushchayeva Y, Costello J, McDaniel D, et al. Nelfinavir inhibits proliferation and induces DNA damage in thyroid cancer cells. Endocr Relat Cancer (2017) 24(3):147–56. doi: 10.1530/ERC-16-0568
88. Hwang JH, Kim DW, Suh JM, Kim H, Song JH, Hwang ES, et al. Activation of signal transducer and activator of transcription 3 by oncogenic RET/PTC (rearranged in transformation/papillary thyroid carcinoma) tyrosine kinase: roles in specific gene regulation and cellular transformation. Mol Endocrinol (2003) 17(6):1155–66. doi: 10.1210/me.2002-0401
89. Park S, Cho S, Kim M, Park JU, Jeong EC, Choi E, et al. Dermatofibrosarcoma protuberans: a retrospective study of clinicopathologic features and related Akt/mTOR, STAT3, ERK, cyclin D1, and PD-L1 expression. J Am Acad Dermatol (2018) 79(5):843–52. doi: 10.1016/j.jaad.2018.05.016
90. Chen Z, Chen X, Xie R, Huang M, Dong W, Han J, et al. DANCR promotes metastasis and proliferation in bladder cancer cells by enhancing IL-11-STAT3 signaling and CCND1 expression. Mol Ther (2019) 27(2):326–41. doi: 10.1016/j.ymthe.2018.12.015
91. Huo N, Cong R, Sun ZJ, Li WC, Zhu X, Xue CY, et al. STAT3/LINC00671 axis regulates papillary thyroid tumor growth and metastasis via LDHA-mediated glycolysis. Cell Death Dis (2021) 12(9):799. doi: 10.1038/s41419-021-04081-0
92. Huang J, Sun W, Wang Z, Lv C, Zhang T, Zhang D, et al. FTO suppresses glycolysis and growth of papillary thyroid cancer via decreasing stability of APOE mRNA in an N6-methyladenosine-dependent manner. J Exp Clin Cancer Res (2022) 41(1):42. doi: 10.1186/s13046-022-02254-z
93. Leslie K, Lang C, Devgan G, Azare J, Berishaj M, Gerald W, et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res (2006) 66(5):2544–52. doi: 10.1158/0008-5472.CAN-05-2203
94. Kim DW, Chung HK, Park KC, Hwang JH, Jo YS, Chung J, et al. Tumor suppressor LKB1 inhibits activation of signal transducer and activator of transcription 3 (STAT3) by thyroid oncogenic tyrosine kinase rearranged in transformation (RET)/papillary thyroid carcinoma (PTC). Mol Endocrinol (2007) 21(12):3039–49. doi: 10.1210/me.2007-0269
95. Pesutic-Pisac V, Punda A, Gluncic I, Bedekovic V, Pranic-Kragic A, Kunac N. Cyclin D1 and p27 expression as prognostic factor in papillary carcinoma of thyroid: association with clinicopathological parameters. Croat Med J (2008) 49(5):643–9. doi: 10.3325/cmj.2008.5.643
96. Balta AZ, Filiz AI, Kurt Y, Sucullu I, Yucel E, Akin ML. Prognostic value of oncoprotein expressions in thyroid papillary carcinoma. Med Oncol (2012) 29(2):734–41. doi: 10.1007/s12032-011-9969-x
97. Teshima M, Tokita K, Ryo E, Matsumoto F, Kondo M, Ikegami Y, et al. Clinical impact of a cytological screening system using cyclin D1 immunostaining and genomic analysis for the diagnosis of thyroid nodules. BMC Cancer (2019) 19(1):245. doi: 10.1186/s12885-019-5452-4
98. Cheng S, Serra S, Mercado M, Ezzat S, Asa SL. A high-throughput proteomic approach provides distinct signatures for thyroid cancer behavior. Clin Cancer Res (2011) 17(8):2385–94. doi: 10.1158/1078-0432.CCR-10-2837
99. Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev (2000) 14(24):3102–14. doi: 10.1101/gad.854900
100. Tchakarska G, Sola B. The double dealing of cyclin D1. Cell Cycle (2020) 19(2):163–78. doi: 10.1080/15384101.2019.1706903
101. Musgrove EA. Cyclins: roles in mitogenic signaling and oncogenic transformation. Growth Factors (2006) 24(1):13–9. doi: 10.1080/08977190500361812
102. Matthews HK, Bertoli C, de Bruin RAM. Cell cycle control in cancer. Nat Rev Mol Cell Biol (2022) 23(1):74–88. doi: 10.1038/s41580-021-00404-3
103. Zerjatke T, Gak IA, Kirova D, Fuhrmann M, Daniel K, Gonciarz M, et al. Quantitative cell cycle analysis based on an endogenous all-in-One reporter for cell tracking and classification. Cell Rep (2017) 19(9):1953–66. doi: 10.1016/j.celrep.2017.05.022
104. Jeong S, Lee J, Kim D, Seol MY, Lee WK, Jeong JJ, et al. Relationship of focally amplified long noncoding on chromosome 1 (FAL1) lncRNA with E2F transcription factors in thyroid cancer. Med (Baltimore) (2016) 95(4):e2592. doi: 10.1097/MD.0000000000002592
105. Sun J, Shi R, Zhao S, Li X, Lu S, Bu H, et al. E2F8, a direct target of miR-144, promotes papillary thyroid cancer progression via regulating cell cycle. J Exp Clin Cancer Res (2017) 36(1):40. doi: 10.1186/s13046-017-0504-6
106. Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, et al. Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature (2010) 463(7279):374–8. doi: 10.1038/nature08684
107. Murphy-Ullrich JE, Suto MJ. Thrombospondin-1 regulation of latent TGF-beta activation: a therapeutic target for fibrotic disease. Matrix Biol (2018) 68-69:28–43. doi: 10.1016/j.matbio.2017.12.009
108. Li Z, Wang C, Jiao X, Lu Y, Fu M, Quong AA, et al. Cyclin D1 regulates cellular migration through the inhibition of thrombospondin 1 and ROCK signaling. Mol Cell Biol (2006) 26(11):4240–56. doi: 10.1128/MCB.02124-05
109. Li Z, Wang C, Prendergast GC, Pestell RG. Cyclin D1 functions in cell migration. Cell Cycle (2006) 5(21):2440–2. doi: 10.4161/cc.5.21.3428
110. Scarpino S, Di Napoli A, Taraboletti G, Cancrini A, Ruco LP. Hepatocyte growth factor (HGF) downregulates thrombospondin 1 (TSP-1) expression in thyroid papillary carcinoma cells. J Pathol (2005) 205(1):50–6. doi: 10.1002/path.1675
111. Tanaka K, Sonoo H, Kurebayashi J, Nomura T, Ohkubo S, Yamamoto Y, et al. Inhibition of infiltration and angiogenesis by thrombospondin-1 in papillary thyroid carcinoma. Clin Cancer Res (2002) 8(5):1125–31.
112. Kumar A, Klinge CM, Goldstein RE. Estradiol-induced proliferation of papillary and follicular thyroid cancer cells is mediated by estrogen receptors alpha and beta. Int J Oncol (2010) 36(5):1067–80. doi: 10.3892/ijo_00000588
113. Rajoria S, Suriano R, Shanmugam A, Wilson YL, Schantz SP, Geliebter J, et al. Metastatic phenotype is regulated by estrogen in thyroid cells. Thyroid (2010) 20(1):33–41. doi: 10.1089/thy.2009.0296
114. Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: cyclin D1: normal and abnormal functions. Endocrinology (2004) 145(12):5439–47. doi: 10.1210/en.2004-0959
115. McMahon C, Suthiphongchai T, DiRenzo J, Ewen ME. P/CAF associates with cyclin D1 and potentiates its activation of the estrogen receptor. Proc Natl Acad Sci U S A (1999) 96(10):5382–7. doi: 10.1073/pnas.96.10.5382
116. Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction–a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol (2006) 3(8):448–57. doi: 10.1038/ncponc0558
117. Li Z, Jiao X, Wang C, Ju X, Lu Y, Yuan L, et al. Cyclin D1 induction of cellular migration requires p27(KIP1). Cancer Res (2006) 66(20):9986–94. doi: 10.1158/0008-5472.CAN-06-1596
118. Neumeister P, Pixley FJ, Xiong Y, Xie H, Wu K, Ashton A, et al. Cyclin D1 governs adhesion and motility of macrophages. Mol Biol Cell (2003) 14(5):2005–15. doi: 10.1091/mbc.02-07-0102
119. Shan J, Zhao W, Gu W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol Cell (2009) 36(3):469–76. doi: 10.1016/j.molcel.2009.10.018
120. Chen B, Lei S, Yin X, Fei M, Hu Y, Shi Y, et al. Mitochondrial respiration inhibition suppresses papillary thyroid carcinoma Via PI3K/Akt/FoxO1/Cyclin D1 pathway. Front Oncol (2022) 12:900444. doi: 10.3389/fonc.2022.900444
121. Yadav V, Burke TF, Huber L, Van Horn RD, Zhang Y, Buchanan SG, et al. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol Cancer Ther (2014) 13(10):2253–63. doi: 10.1158/1535-7163.MCT-14-0257
122. Anania MC, Di Marco T, Mazzoni M, Greco A. Targeting non-oncogene addiction: focus on thyroid cancer. Cancers (Basel) (2020) 12(1):129. doi: 10.3390/cancers12010129
123. Kim ES, Lee JJ, Wistuba II. Cotargeting cyclin D1 starts a new chapter in lung cancer prevention and therapy. Cancer Prev Res (Phila) (2011) 4(6):779–82. doi: 10.1158/1940-6207.CAPR-11-0143
124. Ferruzzi P, Ceni E, Tarocchi M, Grappone C, Milani S, Galli A, et al. Thiazolidinediones inhibit growth and invasiveness of the human adrenocortical cancer cell line H295R. J Clin Endocrinol Metab (2005) 90(3):1332–9. doi: 10.1210/jc.2004-0978
125. Barchiesi F, Lucchinetti E, Zaugg M, Ogunshola OO, Wright M, Meyer M, et al. Candidate genes and mechanisms for 2-methoxyestradiol-mediated vasoprotection. Hypertension (2010) 56(5):964–72. doi: 10.1161/HYPERTENSIONAHA.110.152298
126. Chen JY, Wang JJ, Lee HC, Chi CW, Lee CH, Hsu YC. Combination of peroxisome proliferator-activated receptor gamma and retinoid X receptor agonists induces sodium/iodide symporter expression and inhibits cell growth of human thyroid cancer cells. J Chin Med Assoc (2020) 83(10):923–30. doi: 10.1097/JCMA.0000000000000389
127. Averous J, Fonseca BD, Proud CG. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene (2008) 27(8):1106–13. doi: 10.1038/sj.onc.1210715
128. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer (2006) 6(9):729–34. doi: 10.1038/nrc1974
129. Li X, Li Z, Song Y, Liu W, Liu Z. The mTOR kinase inhibitor CZ415 inhibits human papillary thyroid carcinoma cell growth. Cell Physiol Biochem (2018) 46(2):579–90. doi: 10.1159/000488625
130. Pack LR, Daigh LH, Meyer T. Putting the brakes on the cell cycle: mechanisms of cellular growth arrest. Curr Opin Cell Biol (2019) 60:106–13. doi: 10.1016/j.ceb.2019.05.005
131. Zatulovskiy E, Zhang S, Berenson DF, Topacio BR, Skotheim JM. Cell growth dilutes the cell cycle inhibitor Rb to trigger cell division. Science (2020) 369(6502):466–71. doi: 10.1126/science.aaz6213
132. O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol (2016) 13(7):417–30. doi: 10.1038/nrclinonc.2016.26
133. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discovery (2015) 14(2):130–46. doi: 10.1038/nrd4504
134. Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discovery (2009) 8(7):547–66. doi: 10.1038/nrd2907
135. DeMichele A, Clark AS, Tan KS, Heitjan DF, Gramlich K, Gallagher M, et al. CDK 4/6 inhibitor palbociclib (PD0332991) in rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res (2015) 21(5):995–1001. doi: 10.1158/1078-0432.CCR-14-2258
136. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol (2015) 16(1):25–35. doi: 10.1016/S1470-2045(14)71159-3
137. Antonello ZA, Hsu N, Bhasin M, Roti G, Joshi M, Van Hummelen P, et al. Vemurafenib-resistance via de novo RBM genes mutations and chromosome 5 aberrations is overcome by combined therapy with palbociclib in thyroid carcinoma with BRAF(V600E). Oncotarget (2017) 8(49):84743–60. doi: 10.18632/oncotarget.21262
138. Fullmer T, Cabanillas ME, Zafereo M. Novel therapeutics in radioactive iodine-resistant thyroid cancer. Front Endocrinol (Lausanne) (2021) 12:720723. doi: 10.3389/fendo.2021.720723
139. Wong K, Di Cristofano F, Ranieri M, De Martino D, Di Cristofano A. PI3K/mTOR inhibition potentiates and extends palbociclib activity in anaplastic thyroid cancer. Endocr Relat Cancer (2019) 26(4):425–36. doi: 10.1530/ERC-19-0011
140. Lee HJ, Lee WK, Kang CW, Ku CR, Cho YH, Lee EJ. A selective cyclin-dependent kinase 4, 6 dual inhibitor, ribociclib (LEE011) inhibits cell proliferation and induces apoptosis in aggressive thyroid cancer. Cancer Lett (2018) 417:131–40. doi: 10.1016/j.canlet.2017.12.037
141. Liu D, Hu S, Hou P, Jiang D, Condouris S, Xing M. Suppression of BRAF/MEK/MAP kinase pathway restores expression of iodide-metabolizing genes in thyroid cells expressing the V600E BRAF mutant. Clin Cancer Res (2007) 13(4):1341–9. doi: 10.1158/1078-0432.CCR-06-1753
142. Cass LA, Meinkoth JL. Ras signaling through PI3K confers hormone-independent proliferation that is compatible with differentiation. Oncogene (2000) 19(7):924–32. doi: 10.1038/sj.onc.1203393
143. Rothenberg SM, Daniels GH, Wirth LJ. Redifferentiation of iodine-refractory BRAF V600E-mutant metastatic papillary thyroid cancer with dabrafenib-response. Clin Cancer Res (2015) 21(24):5640–1. doi: 10.1158/1078-0432.CCR-15-2298
Keywords: cell cycle, cyclin D1, CCND1, papillary thyroid carcinoma, pathogenesis
Citation: Cai W, Shu L-Z, Liu D-J, Zhou L, Wang M-M and Deng H (2023) Targeting cyclin D1 as a therapeutic approach for papillary thyroid carcinoma. Front. Oncol. 13:1145082. doi: 10.3389/fonc.2023.1145082
Received: 15 January 2023; Accepted: 09 June 2023;
Published: 22 June 2023.
Edited by:
Monica Fedele, National Research Council (CNR), ItalyReviewed by:
Yuanping Xiong, First Affiliated Hospital of Nangchang University, ChinaEtienne Marbaix, Université Catholique de Louvain, Belgium
Copyright © 2023 Cai, Shu, Liu, Zhou, Wang and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huan Deng, YmVhbmRlbmdAbmN1LmVkdS5jbg==