 Rui Ni
Rui Ni Ziwei Li1†
Ziwei Li1† Dan Peng
Dan Peng Yue Ming
Yue Ming Yao Liu
Yao Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 07 March 2023
Sec. Cancer Metabolism
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1143798
This article is part of the Research TopicCancer Metabolism: Molecular Insights, Metabolic Crosstalk in the Tumor Microenvironment, and Implications for TherapyView all 24 articles
Glutamine, the most abundant non-essential amino acid in human blood, is crucial for cancer cell growth and cancer progression. Glutamine mainly functions as a carbon and nitrogen source for biosynthesis, energy metabolism, and redox homeostasis maintenance in cancer cells. Dysregulated glutamine metabolism is a notable metabolic characteristic of cancer cells. Some carcinogen-driven cancers exhibit a marked dependence on glutamine, also known as glutamine addiction, which has rendered the glutamine metabolic pathway a breakpoint in cancer therapeutics. However, some cancer cells can adapt to the glutamine unavailability by reprogramming metabolism, thus limiting the success of this therapeutic approach. Given the complexity of metabolic networks and the limited impact of inhibiting glutamine metabolism alone, the combination of glutamine metabolism inhibition and other therapeutic methods may outperform corresponding monotherapies in the treatment of cancers. This review summarizes the uptake, transport, and metabolic characteristics of glutamine, as well as the regulation of glutamine dependence by some important oncogenes in various cancers to emphasize the therapeutic potential of targeting glutamine metabolism. Furthermore, we discuss a glutamine metabolic pathway, the glutaminase II pathway, that has been substantially overlooked. Finally, we discuss the applicability of polytherapeutic strategies targeting glutamine metabolism to provide a new perspective on cancer therapeutics.
Metabolic reprogramming is a hallmark of cancer that manifests in several ways. Cancer cells exhibit substantially enhanced glucose uptake and full utilization of the glycolysis/tricarboxylic acid (TCA) cycle to generate a large amount of ATP (Warburg effect) (1). Additionally, cancer cells may exhibit accelerated uptake, transport and metabolism of glutamine, which is the most abundant non-essential amino acid in the human body. Adaptation to rapid metabolism is achieved by regulating genes that encode metabolic drivers to increase the expression of favorable transporters and metabolic enzymes. Furthermore, metabolism-regulating interactions occur between cancer cells and the tumor microenvironment (TME) (2). Since Hans Krebs first studied glutamine metabolism in animals, the biological roles of glutamine in cellular growth and cancer cell biology have gradually been recognized (3). In 1955, Eagle discovered a high dependence of cancer cells on glutamine, also known as glutamine addiction (4). Specifically, cancer cells consume 10–100-fold more glutamine than they do any other amino acids. Glutamine is the most abundant non-essential amino acid in the bloodstream. High levels of glutamine can promote cancer cell proliferation by serving as nitrogen and carbon sources for the biosynthesis of nucleotides, fatty acids, and non-essential amino acids. In addition to its primary roles in macromolecular biosynthesis, glutamine is also involved in the cellular uptake of essential amino acids, the maintenance of the mitochondrial membrane potential, and the production of glutathione and nicotinamide adenine dinucleotide phosphate (NADPH), which are required to maintain redox homeostasis (5–7). Altered glutamine metabolism is a significant outcome of changes in energy metabolism in cancer cells. Abnormal expression of regulatory genes associated with glutamine metabolism is more frequently observed in cancer cells than in healthy cells. Most abnormally expressed regulatory genes, including B-Raf proto-oncogene (BRAF), epidermal growth factor receptor (EGFR), isocitrate dehydrogenase 1 (IDH1), Kirsten rat sarcoma viral oncogene homolog (KRAS), and phosphatidylinositol 3-kinase p110 alpha (PIK3CA), are either oncogenes or tumor suppressor genes involved in the onset and development of cancer (8). Mutations in some of these oncogenes render cancer cells highly dependent on glutamine. Given the critical biologica roles of glutamine in cancer cells, an in-depth understanding of glutamine metabolism is essential to develop new cancer treatments.
Therefore, this review summarizes the uptake, transport, and metabolic characteristics of glutamine, as well as the regulatory effects of some important oncogenes on glutamine addiction in cancer to emphasize the therapeutic potential of targeting glutamine metabolism. In particular, we discuss the significance of the glutaminase II pathway, which is a historically understudied glutamine metabolic pathway in the context of cancer. Furthermore, we also discuss the potential applications of polytherapy that targets glutamine metabolism to provide novel strategies for treating cancer.
Metabolism, a fundamental process for all cellular functions, is also related to cancer cell proliferation. Unlike normal differentiated cells, cancer cells can modify many metabolic pathways—including glycolysis, glutaminolysis, the TCA cycle, the electron transport chain, and the pentose phosphate pathway—to fulfill their energy requirements (2). Following the discovery of the Warburg effect, numerous studies have confirmed the crucial role of cancer cell metabolism in tumor survival and growth. However, recent studies have demonstrated that glutamine plays a more significant role in cancer metabolism than was previously thought. The glutamine demand is usually higher in cancer cells than in normal cells owing to accelerated glutamine metabolism. Despite being a non-essential amino acid and the most abundant amino acid in humans, glutamine is considered to be conditionally essential in some instances due to its involvement in multiple cellular processes (9). Below, we summarized the uptake, transport, and functions of glutamine, and its relevant metabolic enzymes (Figure 1).
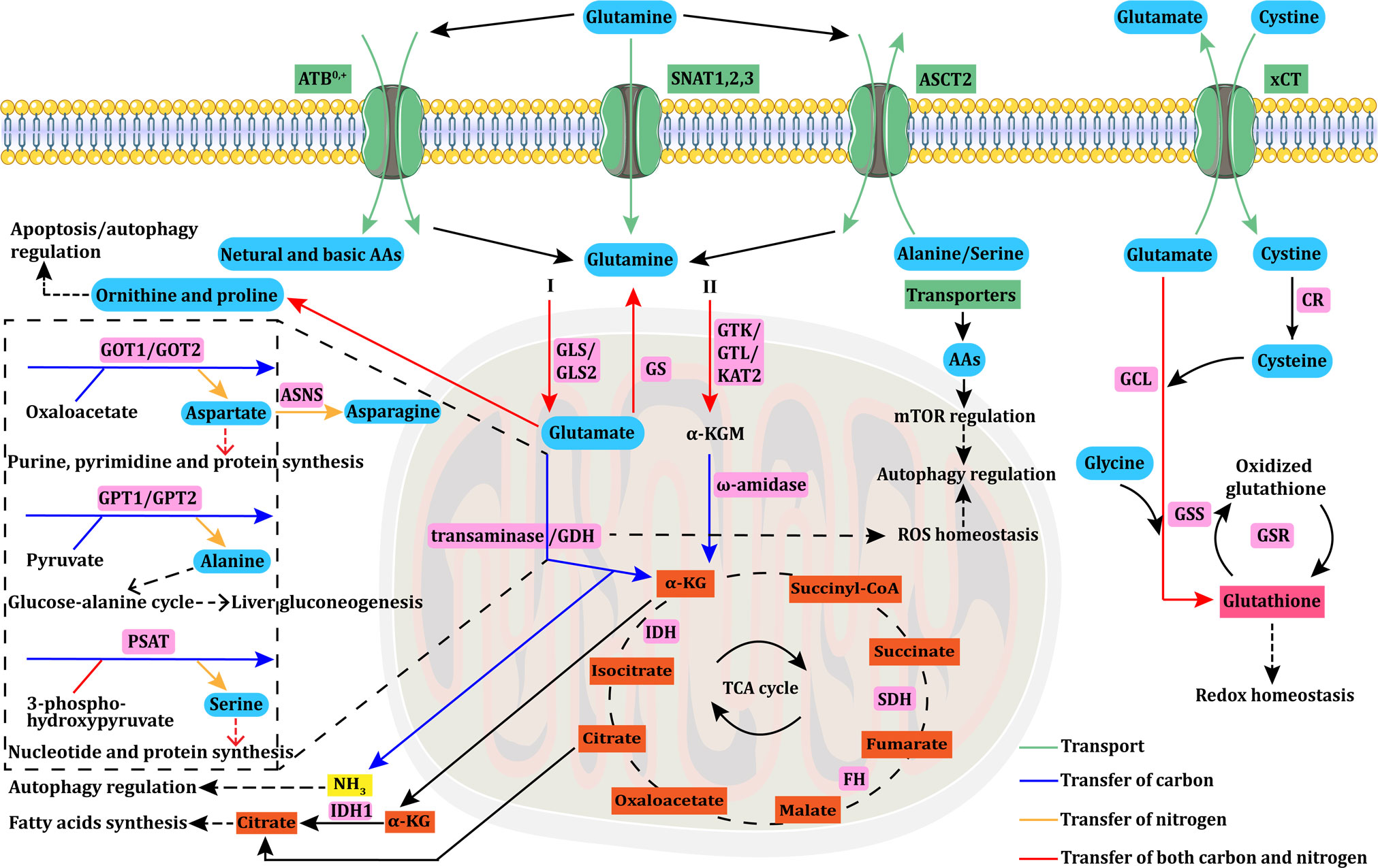
Figure 1 Glutamine uptake, transport, and metabolism. Glutamine uptake and transport in cancer cells is mediated by transporters (ASCT2/ATB0,+/SNAT1,2,3). Intracellular glutamine can be converted into α-KG through the glutaminase I (GLS+GDH/transaminase) or II (GTK/GTL/KAT2+ω-amidase) pathway. Aspartate, alanine, and serine are mainly produced via transaminases, such as GOT, GPT, and PSAT. GOT transforms glutamate into aspartate, which is essential for purine, pyrimidine, and protein synthesis. Aspartate is further converted to asparagine via ASNS. GPT catalyzes the reversible conversion of glutamate to pyruvate for α-KG and alanine generation. This GPT-catalyzed reaction plays a vital role in the glucose–alanine cycle, which is essential to support liver gluconeogenesis. PSAT transforms glutamate into serine, which is involved in nucleotide and protein synthesis. α-KG is an important anaplerotic substrate of the TCA cycle, in which FH, IDH, and SDH mutations are the main causes of cycle dysfunction and mitochondrial metabolic defects in various types of cancers. Glutamine can be synthesized from glutamate via GS. Glutamine can also be converted to ornithine and proline, which regulate apoptosis/autophagy. The byproduct of glutaminolysis, ammonia, can modulate autophagy under specific circumstances. The reductive carboxylation of α-KG into citrate can support adipogenesis, which is crucial for cancer cell survival. The GDH pathway is also linked to cellular ROS homeostasis, and further modulates autophagy, which can also be regulated by AAs through the mTOR pathway. Glutamate is exchanged for cystine via xCT, which is rapidly reduced to cysteine by CR. Then, glutamate, cysteine, and glycine react together for de novo synthesis of glutathione via GCL and GSS. The interconversion of oxidized glutathione and glutathione is catalyzed by GSR, and glutathione can directly control redox homeostasis. AAs, amino acids; ASNS, asparagine synthetase; α-KG, alpha-ketoglutarate; CR, cystine reductase; GCL, glutamylcysteine ligase; GSS, glutathione synthetase; GSR, glutathione reductase; GLS, glutaminase; GDH, glutamate dehydrogenase; GS, glutamine synthase; GTK/GTL, glutamine transaminases K/L; KAT2, kynurenine aminotransferase 2; GOT, glutamate-oxaloacetate transaminase; GPT, glutamate-pyruvate transaminase; PSAT, phosphoserine aminotransferase; FH, fumarate hydratase; IDH, isocitrate dehydrogenase; SDH, succinate dehydrogenase; ROS, reactive oxygen species.
Glutamine uptake by cancer cells is mediated by transporters (10). Alanine-Serine-Cysteine Transporter 2 (ASCT2), a sodium (Na+)-dependent transmembrane transporter encoded by the solute carrier family 1 member 5 (SLC1A5) gene, mediates the cellular uptake of glutamine and other neutral amino acids and is considered to be a primary glutamine transporter in cancer cells (11, 12). L-type amino acid transporter 1 (LAT1), which is encoded by the SLC7A5 gene, preferentially transports large branched and aromatic neutral amino acids, and its expression is upregulated in various types of cancer cells (13, 14). Intracellular glutamine can serve as an exchange substrate to import extracellular amino acids, including leucine, isoleucine, valine, phenylalanine and tyrosine via LAT1 with high affinity (15). In addition, intracellular glutamine can be metabolized into glutamate, which serves as an exchange substrate for cystine uptake mediated by the cystine/glutamate transporter xCT (SLC7A11). The imported cystine can then be converted to cysteine for the biosynthesis of glutathione and the maintenance of cellular redox homeostasis (16). ASCT2, LAT1, and xCT are antiporters that balance cytosolic amino acid composition by proportionally exchanging intracellular amino acids with extracellular amino acids. Hence, these antiporters do not mediate affect intracellular glutamine levels unless other amino acids are available for exchange.
Since the net movement of amino acids into cells can only be mediated by uniporters and symporters (17–19), their expression may also be crucial for cellular growth. Neutral amino acid transporters belong to the SLC38 gene family of sodium-coupled neutral amino acid transporters (SNATs), among which SNAT1 (SLC38A1) and SNAT2 (SLC38A2) are uniporters (19). Both SNAT1 and SNAT2 play important roles in the net uptake of glutamine in epithelial cervical cancer cells, osteosarcoma cells, human melanoma cells, and six breast cancer cell lines (19–21). The first member of the SLC38 family to be cloned was SNAT3 (SLC38A3) (22). Unlike SNAT1 and SNAT2, SNAT3 preferentially imports glutamine, asparagine, and histidine (23). SNAT3 is overexpressed in malignant glioma and non-small cell lung cancer (NSCLC) (24, 25). The amino acid transporter B0,+ (ATB0,+), a symporter encoded by the SLC6A14 gene, can transport all neutral (including glutamine) and basic amino acids. ATB0,+ is overexpressed in various types of cancer, including colorectal (CRC), breast, and pancreatic cancers (26). Additional information on glutamine transporters is provided in Table 1.

Table 1 Main membrane-anchored amino acid transporters of glutamine, their properties, and the cancers in which they are overexpressed.
Here, we address the dysregulated TCA cycle in cancer cells before summarizing glutamine metabolism in cancer cells. The TCA cycle is an intracellular central metabolic hub and a common catabolic pathway for various types of sugar, amino acids, and fatty acids (78). TCA cycle dysregulation can directly alter the metabolism of cancer cells. Alpha-ketoglutarate (α-KG), which is derived from glutamine catabolism, is an important anaplerotic substrate of the TCA cycle.
Mutations in genes involved in the TCA cycle are associated with familial cancers (79). Mutations in the genes encoding fumarate hydratase (FH), isocitrate dehydrogenase (IDH), and succinate dehydrogenase (SDH) cause TCA cycle dysfunction and mitochondrial metabolic defects in various types of cancers (80). Mutations occur throughout the FH gene, and heterozygous mutations in this gene are related to dominantly inherited uterine fibroids, hereditary leiomyomatosis, and renal cell carcinoma (81, 82). FH acts as a tumor suppressor, and downregulation of its expression results in the accumulation of hypoxia-inducible factor-1α (HIF-1α) and fumarate (83). Fumarate is an oncometabolite and a potent inhibitor of prolyl 4-hydroxylase (P4H), which is a negative regulator of HIF-1α. Inhibition of P4H leads to HIF-1α activation under normoxic conditions, resulting in pseudohypoxia that promotes the proliferation of cancer cells (79). IDH has three isoforms: IDH1, IDH2, and IDH3. Genomic analyses have revealed IDH1 or IDH2 mutations in samples from most patients with glioblastoma multiforme and grade II-III gliomas (84). Furthermore, mutations in IDH1 at arginine 132 (R132) and in IDH2 at arginine 172 (R172) have also been identified in patients with acute myeloid leukemia (85). Virtually almost all IDH1 and IDH2 mutations are missense mutations located at R132 and R172, respectively; both isotypes are therefore promising biomarkers for cancer diagnosis and gene therapy. Mutations in IDH cause the loss of its enzymatic activity of converting isocitrate to α-KG while conferring a neo-enzymatic activity of reducing α-KG to the oncometabolite D-2-hydroxyglutarate, an excess of which favors the formation of malignant tumors (86). Similar to FH mutations, heterozygous mutations in the SDH gene are associated with hereditary paraganglioma and pheochromocytoma (87). In addition, mutations in genes encoding SDH subunits are found in other types of tumors, such as gastrointestinal stromal tumor (88), testicular seminoma (89), neuroblastoma (90), renal cell carcinoma (91), and thyroid cancer (92).
Glutamine is first converted by the glutaminase I (GLS/GLS2) system to glutamate, which is then converted to α-KG through two pathways. One pathway is mediated by the enzymatic activity of glutamate dehydrogenases (GDH). The other pathway is mediated by glutamate-oxaloacetate transaminase (GOT), glutamate-pyruvate transaminase (GPT), and phosphoserine aminotransferase (PSAT), which catalyze the conversion of glutamine to α-KG without producing ammonia. Additionally, glutamine can also be converted into α-KG through the glutaminase II pathway, which is described in detail below. Transaminases catalyze the biosynthesis of amino acids such as aspartate, alanine, and serine using the amino group of glutamine. GOT, also known as aspartate transaminase, has cytoplasmic (GOT1) and mitochondrial (GOT) isoforms. GOT catalyzes the conversion of glutamate to aspartate, which is required for biosynthesizing purines, pyrimidines, and proteins, as well as maintaining redox homeostasis by the malate–aspartate shuttle (93). Prominent studies demonstrate that aspartate biosynthesis is an essential function of mitochondrial respiration in proliferating cells (94, 95). In view of the multiple critical roles of aspartate, aspartate uptake and biosynthesis pathways have been are considered therapeutic targets in cancer (93). GPT, also called alanine transaminase, includes the cytoplasmic GPT1 and mitochondrial GPT2 isoforms and catalyzes the reversible conversion of glutamate to pyruvate for α-KG and alanine generation. This GPT-catalyzed reaction plays an important role in the glucose–alanine cycle, which is essential for supporting hepatic gluconeogenesis (96). PSAT is required to produce serine, which plays a key role in the biosynthesis of nucleotides and proteins and is an allosteric activator of several enzymes. Approximately half of all α-KG that enters the TCA cycle in breast cancer cell lines is produced via the activity of PSAT (97); thus, serine biosynthesis may play an important role in cancer cell metabolism. Glutamine can also be converted to ornithine and proline. Proline dehydrogenase/proline oxidase-dependent apoptosis/autophagy may be modulated by the interconversion of glutamate, ornithine, and proline, among which proline is the key amino acid (98). Understanding the regulatory roles of the proline in this process could facilitate the development of targeted cancer therapies. Glutamine also serves as a nitrogen donor for the biosynthesis of asparagine catalyzed by asparagine synthase (ASNS). In addition to amino acid biosynthesis, glutamine also provides nitrogen for the de novo synthesis of purines, pyrimidines, and nucleobases of DNA and RNA (99).
One of the most important metabolic pathways involving glutamine is the biosynthesis of α-KG to provide an anaplerotic supply for the TCA cycle. α-KG can be metabolized via oxidative decarboxylation and reductive carboxylation. Hypoxia (100), impaired mitochondrial respiration (101), and anchorage-independent formation of tumor spheroids (102) can promote the reductive carboxylation of glutamine-derived α-KG into citrate to support adipogenesis, which is crucial to cancer cell survival. Under such circumstances, glutamine serves as a direct carbon source for citrate and fatty acid biosynthesis (103, 104).
Glutamine produces reactive oxygen species (ROS) in addition to providing nitrogen and carbon for biosynthesis and energy metabolism. However, some glutamine metabolites can directly control ROS levels. For instance, glutathione can neutralize peroxide free radicals, and NADPH generated via GDH-catalyzed glutamine metabolism can regulate ROS homeostasis by directly scavenging excess ROS (105). Glutamine also plays an important regulatory role in autophagy suppression through various processes affected by its metabolism. ROS promote autophagy in response to stress. Meanwhile, glutamine modulates autophagy through the production of glutathione and NADPH, which affect ROS levels (106). Additionally, glutamine is involved in the activation of mammalian target of rapamycin (mTOR), which inhibits autophagy. The latest research demonstrates that glutaminolysis is one of two mechanisms through which glutamine metabolism modulates the mTORC1-autophagy pathway (107). A byproduct of glutaminolysis, ammonia, induces and inhibits autophagy at low and high concentrations, respectively (108). Thus, ammonia regulates autophagy in a concentration-dependent manner without relying on inhibited mTOR or ULK1/2 activity (108, 109). The GDH-catalyzed conversion of glutamate to α-KG generates a second molecule of ammonia in some types of cancer cells. However, others preferentially use transaminases for this reaction, thereby bypassing the generation of the second ammonia molecule. Changes in this metabolic pathway could thus indirectly affect autophagy regulation.
Greenstein discovered the two glutaminase enzyme systems in rats (110). The glutaminase I system, which is now known as the glutaminase or phosphate-activated glutaminase system, comprises kidney (GLS or KGA) and liver (GLS2 or LGA) isozymes activated by phosphate (111), while the glutaminase II system is activated by α-keto acids (112).
The mitochondrial glutaminase I system converts imported glutamine to glutamate and is considered the first key enzymatic reaction in the catalysis of glutamine in cancer cells. The ubiquitous expression of GLS in normal tissues is upregulated in various cancers, and it might play a key role in cancer onset and progression (113). GLS has thus been extensively explored as a target for drug development. In contrast, GLS2 is primarily expressed in the liver, brain, pituitary gland, and pancreas. Whether GLS2 promotes or suppresses tumorigenesis remains uncertain, and it has rarely been targeted in drug discovery (111, 114). However, GLS significantly promotes cancer cell growth. The expression of GLS is abnormally elevated in multiple cancer types, including breast (115), colorectal (116), and prostate (117) cancers, especially when compared with adjacent tissues. Knocking down or inhibiting GLS can restrict the growth of various types of cancer cells and suppress cancer progression (113, 115, 118). The GLS inhibitor CB-839 is currently entering phase I and II clinical trials as monotherapy and as polytherapy when combined with chemotherapy and/or immunotherapy.
Glutaminase II is an enzymatic complex comprising glutamine transaminase and omega (ω)-amidase (119, 120). The main glutamine transaminases in humans and rodents are glutamine transaminases K (GTK) and L (GTL) (121, 122). Kynurenine aminotransferase 2 (KAT2), also exerts some enzymatic action on glutamine (123). In the presence of various α-keto acids, glutamine transaminases catalyze the conversion of glutamine into alpha-ketoglutaramate (α-KGM), which is then deamidated by ω-amidase into α-KG. To date, cancer cell glutamine addiction is still widely and naturally thought to involve the glutaminase I pathway, by which glutamine is first converted to glutamate via a GLS-catalyzed reaction and before being converted into α-KG through GDH or transaminases (GOT/GPT/PSAT). However, the glutaminase II pathway can also realize the conversion of glutamine into α-KG. This pathway’s functions in cancer cells have been substantially overlooked, with only a few relevant studies having been reported. For instance, it has been newly confirmed that the glutaminase II pathway exists in human pancreatic cancers, and genetic suppression of GTK completely inhibits pancreatic tumorigenesis in vivo (124). Almost concurrently, the glutaminase II pathway was identified in prostate cancer cells, and expression of the GTK and ω-amidase genes in this pathway has been shown to be more upregulated with increased cancer cell invasiveness (125). The glutaminase II pathway also plays important roles in providing anaplerotic carbon to the TCA cycle, supplying citrate carbon in prostate cancers, and closing the methionine salvage pathway (125). These findings suggest that glutamine transaminase (GTK) and ω-amidase could be novel metabolic targets for cancer treatment. However, the broad substrate specificity of glutamine transaminase towards α-keto acids and amino acids should be considered. Inhibiting glutamine transaminase might interfere with other biological properties. Additionally, glutamine metabolism through the glutaminase I pathway is notably catalyzed by GLS+GDH requires an aerobic environment due to the involvement of nicotinamide adenine dinucleotide (NAD+). In contrast, α-KG production through the glutaminase II pathway does not involve net oxidation, indicating that it can function in hypoxic regions of tumors. In summary, the glutaminase II pathway in cancers has provided a new perspective for studying cancer cell glutamine addiction and for developing corresponding therapeutic strategies.
Glutamate dehydrogenase (GDH) catalyzes the second step in glutamine metabolism, which is the oxidative deamination of glutamate into α-KG. The GDH1 isozyme is expressed ubiquitously across tissues and cells, whereas GDH2 is specifically expressed in brain, testis, and embryonic tissues (126). Aside from producing anaplerotic α-KG for the TCA cycle, the GDH pathway is also associated with various cellular processes, including acid–base balance, ammonia metabolism, lactate production, redox homeostasis, and lipid biosynthesis (127). Specifically, recent research indicates that GDH is crucial for metabolic ammonia recycling in breast cancer cells, especially estrogen-receptor-positive cells, to support their growth and proliferation (128). Additionally, GDH1 plays a vital role in maintaining redox homeostasis in breast and lung cancer (129). GDH1 expression is significantly upregulated in tumor samples from patients with advanced breast or lung cancer, resulting in the accumulation of fumarate. Fumarate can bind to and activate the ROS-scavenging enzyme glutathione peroxidase 1 (GPx1), conferring metabolic advantages for cancer cell growth and proliferation. The small-molecule inhibitor R162, which targets GDH1, can reduce the proliferative capacity of cancer cells by causing redox homeostasis imbalance (129). The survival of glioblastoma cells with glucose metabolism disorders due to glucose deprivation, glycolytic inhibition, or Ak strain transforming (Akt) signaling inhibition requires GDH (130). Overexpressed GDH promotes the proliferation, migration, and invasion of CRC cells, and might serve as a novel independent prognostic biomarker for CRC progression and metastasis (131). Glutamine enhances the proliferative capacity of ovarian cancer cells in a dose-dependent manner and increases the activities of GLS and GDH by modulating the mTOR/ribosomal S6 kinase (S6) and mitogen-activated protein kinase 1 (MAPK) pathways (132). GDH expression is upregulated in extrahepatic cholangiocarcinoma tissues, whereas silencing it significantly reduces the proliferative, migratory, and invasive capacity of cancer cells. Hence, GDH is considered an important prognostic marker and therapeutic target in extrahepatic cholangiocarcinoma (133).
The roles of GDH2 in cancer growth and metabolism have not been fully investigated. However, GDH2 plays an important role in eliminating the growth-inhibiting effect of IDH1 (R132H) mutant gliomas (134). This suggests that targeting GDH2 could be a beneficial strategy for treating patients with IDH1 mutant gliomas.
GOT1/GOT2, GPT1/GPT2, and PSAT are important enzymes in glutamine metabolism and amino acid biosynthesis, and have crucial functions in various types of cancers (135). GOT1-mediated pathways play vital roles in maintaining redox homeostasis in pancreatic cancer, and increased enzymatic activity of GOT1 favors the growth of cancer cells (136). A recent study demonstrates that inhibiting GOT1 activity hinders the growth of several pancreatic ductal adenocarcinoma cell lines, primary tumor models, and tumor xenografts (137). GOT2 acetylation is essential for regulating the mitochondrial NADH/NAD+ ratio and stimulating the production of NADPH to maintain the redox state of pancreatic cancer cells (138). Acetylation of GOT2 at the K404 lysine residue promotes the proliferation of pancreatic cancer cells and tumor growth in vivo, and GOT2 acetylation at the lysine residue K159 is increased in human pancreatic tumors. Triple-negative breast cancer (TNBC) cell lines also overexpress GOT1, which controls intracellular ROS levels by producing NADPH, in turn promoting tumor growth. The shRNA-mediated inhibition of GOT1 expression enhances cancer cell sensitivity to doxorubicin by causing doxorubicin-induced ROS generation (139). The transaminase inhibitor aminooxyacetate (AOA) can inhibit the proliferation of breast cancer cells (140).
The enzymatic activity of GPT2 is pivotal to the anchorage-independent growth of KRAS-transformed colon cancer cells (141). PIK3CA mutations can trigger glutamine metabolism reprogramming in CRC cells by upregulating GPT2 expression (142). GPT2 overexpression enhances the tumorigenicity and stemness of breast cancer cells by activating the sonic hedgehog signaling pathway, suggesting its potential as a therapeutic target (143).
Recent studies show that elevated ratios of aspartate to alanine transaminases (AST/ALT or GOT/GPT) correlate with a poor prognosis in bladder (144), colorectal (145), hepatic (146), head and neck (147), oral and oropharyngeal (148), prostate (149), and pancreatic (150) cancers. The metabolic roles of PSAT in cancer cells have not yet been comprehensively explored. It has been reported that significantly more PSAT is expressed in colon tumors than in normal tissues (151), but its specific mechanism and prognostic value remain to be elucidated.
Glutamine can be synthesized de novo from glutamate via glutamine synthase (GS). The selective, irreversible GS inhibitor L-methionine sulfoximine (MSO) has anticancer potential due to its ability to inhibit cancer cell proliferation in vitro (152, 153).
Cancer-associated genes are broadly categorized as oncogenes and tumorsuppressor genes that promote and suppress carcinogenesis, respectively. Interactions between these two types of genes result in oncogenesis (154). Hence, cancers are ultimately outcomes of dysregulated gene expression. Genes can be activated or inactivated by mutations. Some of the prominent mutated oncogenes in cancers include BRAF, ErbB2, JAK2, KRAS, MYC, and PIK3CA. Mutations in these oncogenes can result in a high reliance of cancer cells on glutamine for survival and proliferation. Therefore, depleting glutamine and inhibiting glutamine metabolism eventually lead to the growth arrest or even death of these glutamine-addicted cancer cells. Below we summarize the regulatory effects of some important oncogenes on glutamine addiction in cancers.
The BRAF gene is one of three rapidly accelerated fibrosarcoma (RAF) isoforms that encodes the serine/threonine kinase of the RAS family. The BRAF mutation rate in all cancers is about 7%, but the rate varies, being about 66% in melanoma and 10%–25% in CRC (155). Over 90% of mutations in the BRAF gene occur in codon 600; the substitution of valine (V) with glutamic acid (E) (BRAF V600E) is the most common mutation, followed by the substitution of V with lysine (K) (BRAF V600K) (156). Therefore, the National Comprehensive Cancer Network (NCCN) recommends that patients with advanced CRC should be tested for BRAF mutations before starting first-line treatment. Targeted polytherapies for patients with mutated BRAF include the triplet regimens of Dabrafenib (BRAF inhibitor) + Trametinib (MEK inhibitor) + Panitumumab or Cetuximab and Encorafenib (BRAF inhibitor) + Binimetinib (MEK inhibitor) + Panitumumab or Cetuximab.
Melanoma cells, which contain mutated BRAF and are resistant to the BRAF inhibitor PLX4720, exhibit increased oxidative metabolism and mitochondrial dependence for survival (157). The increased oxidative metabolism is related to a shift from glucose to glutamine metabolism and a greater dependence on glutamine over glucose. Such cells are more sensitive to glutamine metabolism inhibitors and mitochondrial poisons. Hence, combining inhibitors of BRAF and glutamine metabolism has a more prominent therapeutic effect. Melanoma cell lines with single resistance to Vemurafenib (BRAF inhibitor) and double resistance to Vemurafenib/Selumetinib (MEK inhibitor)exhibit increased glutamine uptake and NH4+ production without changes in glucose uptake (158). Furthermore, glutamine deprivation induces the apoptosis of drug-resistant cell lines. Taking advantage of glutamine addiction, the glutaminase inhibitor BPTES and the glutamine-mimetic antimetabolite L-DON have yielded superior antitumor effects in vivo.
KRAS, HRAS and NRAS, which are three isoforms of the mammalian rat sarcoma (RAS) GTPase, encode KRAS4A, KRAS4B, HRAS, and NRAS. These four small G proteins bind to GTP/GDP and exhibit GTP hydrolase activity. As a molecular switch, RAS activates downstream signaling pathways, such as the MAPK and PI3K-Akt pathways, by binding to GTP to regulate the proliferation, differentiation, and apoptosis of cells. RAS mutations account for one-third of all human cancers (159, 160). The analysis of data retrieved from four cancer databases (COSMIC, cBioPortal, ICGC, and TCGA) reveals significantly higher mutation rates for KRAS than for HRAS and NRAS (160). A KRAS mutation is an important driver leading to metabolic reprogramming in cancer cells, and it is closely related to glutamine and its metabolic changes.
KRAS mutations increase glutamine demand in cancer cells that use glutamine to accelerate energy metabolism and maintain redox homeostasis (161). The selectively upregulated expression of ASCT2, LAT1 and SNAT2 in CRC cells containing mutated KRAS enhances the uptake of glutamine, leucine, and other amino acids. Knocking out KRAS downregulates the expression of the above amino acid transporters, thereby reducing the uptake of amino acids by CRC cells (70). Metabolomic analyses have revealed higher concentrations of amino acids, especially glutamine, in CRC cells containing mutated KRAS than in wild-type CRC cells (162). A subsequent study of amino acid transporters in CRC cells with mutated KRAS found that KRAS signaling mainly regulated the expression of ASCT2 through the PI3-Akt-mTOR pathway. Additionally, the prognosis of patients with mutated KRAS and high ASCT2 expression is worse than patients with mutated KRAS and low ASCT2 expression (28). A monoclonal antibody antagonist, Ab3-8, has been developed that recognizes the extracellular domain of ASCT2, reduces cellular glutamine import and Akt/ERK phosphorylation in SW1116 and HCT116 human CRC cell lines containing mutated KRAS, and inhibits the growth of tumor xenografts in mice (163). However, Ab3-8 does not inhibit the in vivo growth of tumor xenografts of the HT29 human CRC cells with wild-type KRAS. These findings collectively suggest that ASCT2 could be a useful target for treating cancers with mutated KRAS.
The effects of oncogenic KRAS mutations on glutamine metabolism have been confirmed. In human breast cancer cells with mutated KRAS, anabolic glutamine utilization is increased, and the expression of genes associated with glutamine metabolism is significantly upregulated, and there exists a high dependence on glutamine for cellular growth (164). Oncogenic KRAS promotes glutamine metabolism reprogramming, and glutamine deprivation can increase ROS levels and decrease reductive glutathione levels in pancreatic cancer cells (136). The anaplerotic feeding of the glutaminolysis metabolite α-KG into the TCA cycle is essential for the growth of various anchorage-independent, KRAS-induced cancer cells (141). In addition, LKB1 and KEAP1/NRF2 pathways synergistically promote metabolic reprogramming toward enhanced glutamine dependence in lung adenocarcinoma with mutated KRAS, and they also enhance the sensitivity of cancer cells to CB-839 (GLS inhibitor) in vitro and in vivo (165). Combining CB-839 with Selumetinib (MEK inhibitor) takes advantage of the increased glutamine utilization in NSCLC cells with mutated KRAS to improve therapeutic efficacy (166).
The myelocytomatosis (MYC) proto-oncogenes include c-MYC, n-MYC, l-MYC, and r-MYC, among which c-MYC is the most commonly activated proto-oncogene. As a transcription factor, MYC protein strictly responds to and integrates mitogenic and developmental signals into broad changes in gene expression to support cell growth and proliferation (167). In fact, most cancers harbor altered MYC genes. For instance, MYC is amplified in up to 78% of osteosarcomas, 65% of ovarian serous cystadenocarcinomas, 48% of breast cancers, 45% of esophageal cancers and 37% of lung squamous cell carcinomas (167). TCGA data show that MYC amplification accounts for 21% of all tumor samples (167). Furthermore, MYC signaling in cancer cells enables abnormal TME regulation and evasion of the host immune response. The inactivation of MYC in preclinical models might lead to sustained tumor regression due to oncogene addiction (168). Hence, MYC activation elicits numerous hallmarks required for autonomous tumor growth. Yuneva et al. first discovered that MYC-driven proliferating cells exhibit glutamine addiction (169). The expression of genes associated with glutamine metabolism can be positively stimulated by MYC. Sensitivity to glutamine deprivation is c-MYC-dependent in glioma cells and can be suppressed by targeting MYC expression (170). Furthermore, various cell lines derived from cancers such as P493-6 B-cell lymphoma, PC3 prostate cancer (171), osteosarcoma (172), Ramos and Raji B-cell lymphoma (173), renal cell carcinoma (174), HCT116 CRC (175), and U-1906 small cell lung carcinoma (176) cells rely on glutamine for cellular survival and growth under MYC activation. Interestingly, glutamine itself can also regulate c-MYC protein expression in HCT116 CRC (175), U266 and INA-6 multiple myeloma (177), and SK-N-AS and SH-SY5Y neuroblastoma (178) cells. Glutamine addiction extends from the context of MYC, and mutations in KRAS —a key gene related to the stability and activity of c-MYC protein— in cancer cells similarly cause dependence on exogenous glutamine for cellular growth and proliferation, as does MYC amplification (162, 179, 180). Therefore, combining therapy targeting the MYC pathway with intervention in glutamine metabolism should be key to reversing MYC-driven tumor growth and restoring the antitumor immune response.
The mammalian target of rapamycin complex 1 (mTORC1) is a highly conserved protein kinase complex that regulates cellular growth, metabolism, and autophagy in response to exogenous signals from nutrients and growth factors (181). As a key downstream effector of many oncogenic pathways, mTORC1 is associated with cancer progression (182). Mutations in mTORC1 are often hyperactivating in cancer (183, 184), and mTOR inhibitors Rapalogs (rapamycin and its analogs) have been clinically approved by FDA for treating some cancers (185). Amino acids are likely required to activate mTORC1, which in turn can regulate amino acid metabolism (186, 187). With further research, activation of the mTORC1 pathway has been shown to be related to glutamine addiction in cancer cells (188). mTORC1 promotes glutamine anaplerosis by activating GDH in human epithelial cancer cell lines such as DLD1 colon cancer, as well as the prostate cancer cell lines LNCaP and DU145 (189). Mechanistically, mTORC1 promotes the proteasome-mediated destabilization of CREB2 to suppress SIRT4 (a mitochondria-localized member of the sirtuin family), which inhibits GDH, thereby enhancing GDH enzymatic activity. Furthermore, mTORC1 not only regulates GDH, but also promotes glutamine uptake by cancer cells by positively regulating GLS through S6K1-dependent c-MYC regulation (190). At the molecular level, S6K1 enhances the efficiency of MYC translation by regulating phosphorylation of the eukaryotic initiation factor eIF4B. The inhibitors of mTOR and GLS can significantly attenuate the growth of BxPC3 pancreatic cancer cells.
Phosphatidylinositol 3-kinase p110 alpha (PIK3CA) is an important proto-oncogene in the PI3K/Akt signaling pathway because it participates in the modulation of numerous cellular functions, including proliferation, differentiation, apoptosis, and glucose transport. It was initially detected by in situ hybridization, and is a 34-kb gene located in 3q26.3, which contains 20 exons (191). Among mutations in the PIK3CA gene, about80% occur in the helical and kinase domains; H1047R on exon 20 and E542K and E545K on exon 9 are the three most prevalent mutations (192). Mutations in PIK3CA play important roles in the onset and development of cancer and are found to have a rate of 2–7% in NSCLC, especially squamous cell lung carcinoma. PIK3CA mutations occur in CRC at a rate of 20–30% and often occur concomitantly with KRAS and BRAF mutations. Indeed, PIK3CA is one of the most commonly mutated genes, with a rate of about 30% in breast cancer (193). NCCN guidelines (2019) recommend PIK3CA mutation tests for patients with ER+/HER2- breast cancer. Polytherapy with Alpelisib (PIK3CA inhibitor) and Fulvestrant (estrogen receptor antagonist) can improve the survival rates of patients with breast cancer contaning PIK3CA mutations.
Mutations in PIK3CA can reprogram glutamine metabolism by upregulating GPT2 expression, thereby increasing glutamine dependence of CRC cells. Specifically, the findings of metabolic flux analyses identified a higher rate of glutamine conversion to α-KG in CRC cells with mutated PIK3CA than in wild-type CRC cells. Mutations in the catalytic subunit p110α can upregulate GPT2 expression through the PDK1-RSK2-ATF4 signaling axis, thus increasing CRC cell dependence on glutamine. Blocking this signaling axis can inhibit the growth of CRC cells with mutated PIK3CA. The GPT2 inhibitor AOA can also inhibit the growth of CRC with mutated PIK3CA, but not in wild-type CRC xenografts (142). The results of [13C5]-glutamine tracer studies using mice with subcutaneous, orthotopic, and spontaneous CRC xenografts reveal that glutamine primarily enters the TCA cycle in tumors. Utilization rates of excess glutamine in tissue culture and subcutaneous xenografts, are higher for CRC with mutated PIK3CA than wild-type CRC. Levels of TCA cycle intermediates were shown to be more enriched in an orthotopic model with mutated PIK3CA, than in wild-type tumors (194). PIK3CA mutations induced in the MCF10A human mammary epithelial cell line result in a 50% increase in glutamine uptake and a significant increase in glutamate production (195). Therefore, PIK3CA mutations lead to glutamine addiction in tumors. Intervention in glutamine metabolism could facilitate treatment for cancers with mutated PIK3CA.
Given that CRC cells with mutated PIK3CA are more dependent on glutamine, CB-839 (GLS inhibitor) combined with 5-fluorouracil (5-FU) can significantly induce CRC cell apoptosis when compared with the corresponding monotherapies (196). The most recent results of an ongoing phase I clinical trial (NCT02861300) on CB-839 with Capecitabine (a 5-FU prodrug) for advanced CRC and other solid tumors yielded a better therapeutic outcome for patients with mutated PIK3CA than those with non-mutated cancers.
The activated proto-oncogene ErbB2 (also termed neu or HER2) is a leading cause of breast cancer. More GLS mRNA and protein are expressed in ErbB2-transformed MCF10A than in MCF-10A cells, whereas ErbB2 knockdown downregulates GLS expression. Further research has shown that activated ErbB2 stimulates GLS expression in breast cancer cells through the PI3K/Akt-independent NF-κB pathway (197). If these findings are validated in models in vivo, this could facilitate the identification of novel targets for cancer prevention and treatment.
A V617F mutation in the JAK2 gene mutation is found in most patients with myeloproliferative neoplasms (MPNs). Among the peripheral blood cluster of differentiation CD34(+) cells from patients with MPN, more GLS is expressed in progenitor cells with mutated JAK2 than in progenitor cells with wild-type JAK2 [198]. More vigorous glutamine metabolism and significantly higher GLS expression are also confirmed in murine pro-lymphoid (BaF3) cells with mutated JAK2 when compared with that in cells with wild-type JAK2. Therefore, GLS inhibitors can improve the therapeutic effects of the JAK2 inhibitor Ruxolitinib by enhancing its inhibitory effect against CD34(+) and growth of cells with mutated JAK2 in patients with MPN (198).
Similar to cancer cells, activated T cells enhance glutamine uptake and metabolism to support mitochondrial anaplerosis, nucleotide synthesis, amino acid production, and redox homeostasis (199–201). Competitive depletion of glutamine by cancer cells in the TME triggers the starvation of activated T lymphocytes and suppresses their proliferation and cytokine production (202). Alternatively, glutamine deprivation impairs the T-lymphocyte-mediated anticancer immune response by promoting Treg cell activation and proliferation (203). Deleting GLS in cancer cells increases interstitial glutamine concentrations to near physiological plasma levels and enhances the overall activation and effector capacity of T lymphocytes (199, 204), suggesting that reducing glutamine confers immunosuppressive effects in the TME. Therefore, the specific inhibition of glutamine metabolism in tumor cells not only inhibits tumor growth but also improves the T cell-mediated antitumor immune response by reversing the “glutamine steal” scenario of tumors (204, 205). The glutamine-mimetic antimetabolite JHU083 disrupts metabolism in various types of tumors and reverses hypoxic, acidic, and nutrient-deprived conditions in the TME. Furthermore, it restores antitumor immunity by inducing T cell activation, extending their lifespan, and promoting memory T cell differentiation (206). These findings have confirmed glutamine metabolism as a metabolic checkpoint in cancer immunotherapy. Transient GLS inhibition can also improve the function of CAR-T cells in a mouse model administered with cellular immunotherapy (207).
The main inhibitors of glutamine metabolism are glutamine uptake, GLS, GDH, transaminase, and GS inhibitors; glutamine-mimetic antimetabolites; and systemic glutamine-depleting drugs (Table 2). Most metabolic inhibitors targeting cancer remain in preclinical phases, but glutamine-depleting L-asparaginases have already been approved as a standard component of a therapeutic regimen for acute lymphoblastic leukemia (223). Additionally, the GLS inhibitor CB-839 has been used to develop monotherapy and polytherapy with chemotherapy and/or immunotherapy that have entered phase I and II of clinical trials, respectively (Table 3).
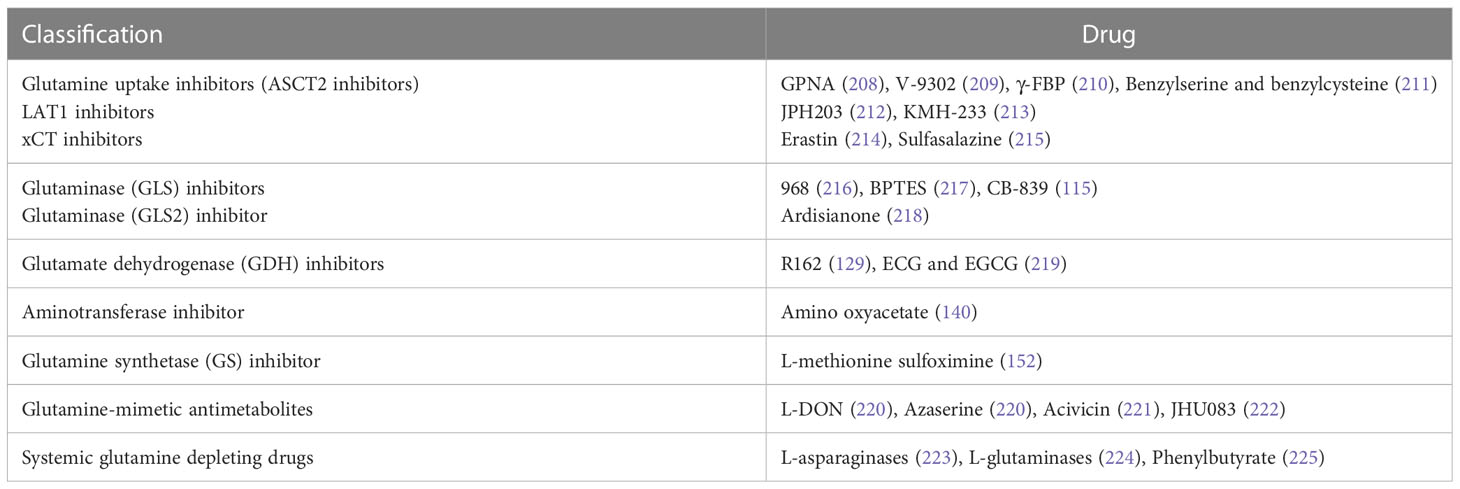
Table 2 Strategies for inhibiting glutamine metabolism.
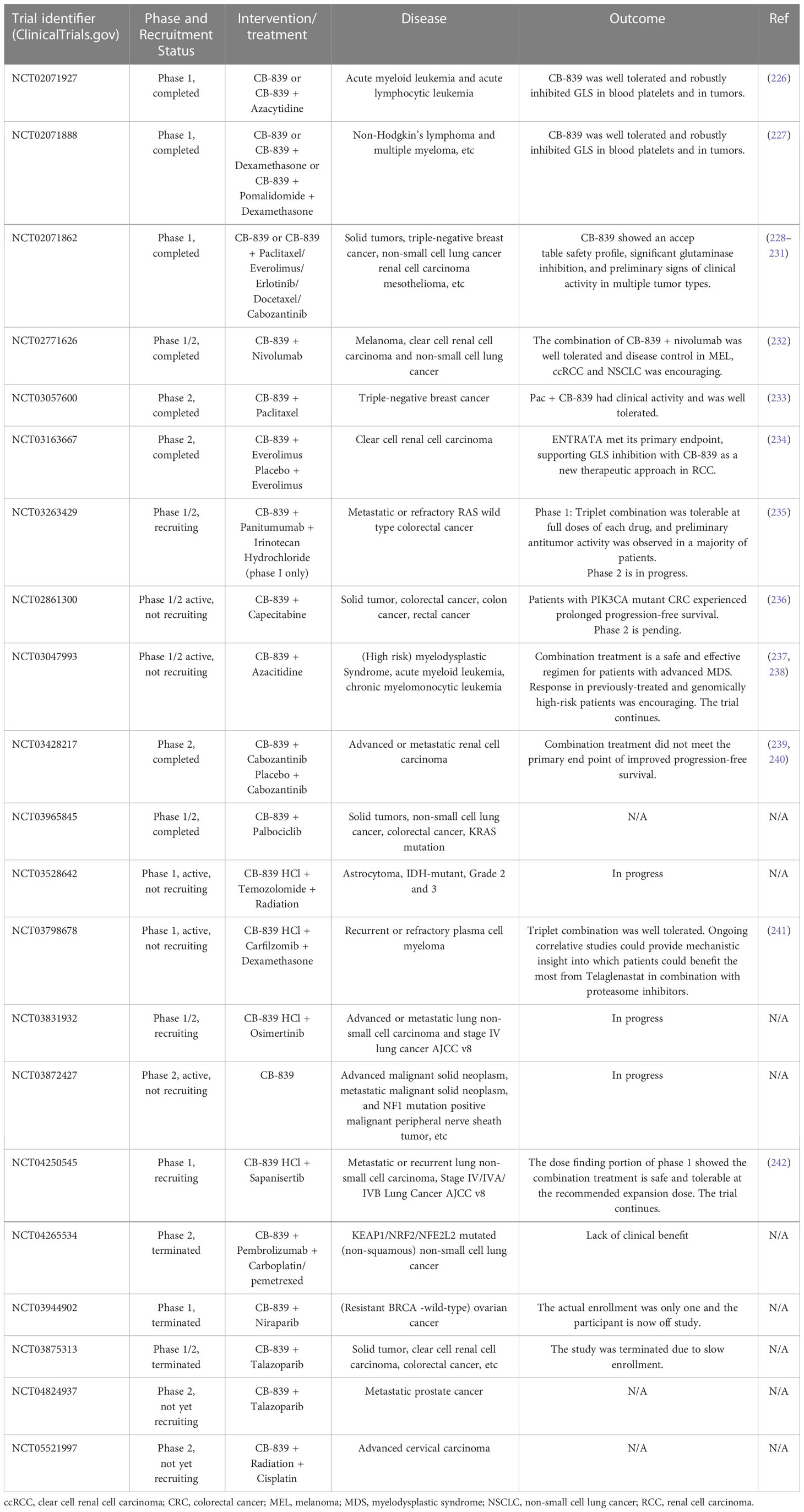
Table 3 Clinical trials of glutaminase inhibitor CB-839 (Telaglenastat) against various types of cancers.
L-asparaginases also exhibit glutaminase activity required for durable therapeutic effects against acute lymphoblastic leukemia (243). However, glutamine depletion could cause some adverse effects such as acute pancreatitis, thrombotic complication, and immunosuppression (244). The clinical hypersensitivity led to the development of L-asparaginases with different bacterial origins (245). Human-derived L-asparaginase may be a solution in the future considering the problems of immunosuppression and hypersensitivity. Reversible and asymptomatic elevations in transaminases were the primary adverse effects when CB-839 monotherapy was used to treat hematologic malignancies and solid tumors in clinical trials (226–228). On balance, CB-839 was well tolerated and produced robustly inhibition of GLS in blood platelets and in tumors. Targeted nano-delivery systems have been developed to further improve antitumor efficacy and reduce systemic effect. CB-839 loaded nanoparticles could preferentially accumulate in tumor tissue through enhanced penetration and retention (EPR) effect, known as passive targeting (246). Furthermore, ligand/antibody-modified nanoparticles could recognize overexpressed tumor cell receptor/antigen, which actively targets tumor glutamine metabolism and achieves tumor-specific accumulation of CB-839 (247).
Many dogmas have been overturned and refined since the discovery of oncogenic metabolic alterations and the rediscovery of the roles played by glutamine and glucose in cancer cell proliferation. Here, we provide an overview of the uptake, transport and metabolism of glutamine, as well as the regulation of glutamine addiction by oncogenes in cancer. The oncogene-driven types of cancer summarized herein are highly dependent on glutamine. Hence, targeting glutamine metabolism could facilitate the pharmacological improvement of cancer therapeutics. In contrast, some oncogenic drivers could allow cancer cells to bypass the need for glutamine by upregulating other metabolic pathways for their cellular growth and proliferation. However, targeted inhibition of some oncogenic drivers can restore cellular reliance on glutamine. Therefore, inhibiting glutamine metabolism and these oncogenic drivers could collectively induce synthetic lethality in cancer cells.
The potent ability of glutamine metabolic inhibitors to enhance the anticancer immune response may be a feasible mechanism through which their therapeutic effectiveness can be improved. Thus, the most favorable glutamine-blocking strategy should be considered. The latest studies show that immune checkpoint blockades exhibit synergistic effects with glutamine uptake inhibitors (248, 249), GLS inhibitor (250), and glutamine-mimetic antimetabolites (206, 251). Additionally, nanowire sensors can be used to monitor changes in the level of cancer-associated proteins and mRNAs (252). Therefore, real-time dynamic monitoring of intratumoral metabolic processes may enable dynamic optimization or adjustment of therapeutic strategies, which is an important step towards developing precision medicine against cancers.
To date, the outcomes of clinical trials using glutamine metabolism inhibitors to treat cancer remain unsatisfactory because of the metabolic plasticity exhibited by cancer cells. Nevertheless, a scientific foundation has been lain for the further assessment of potential targeted molecules and the rational design of polytherapies to maximize clinical efficacy.
Idea and design: RN. Evidence collection, analysis, and arrangement: RN, ZL and LiL. Manuscript drafting: RN and ZL. Critical revision of the manuscript: LinL and YL. Obtained funding: YL. Technical support: DP and YM. All authors contributed to the article and approved the submitted version.
This work was financially supported by The Chongqing Special Project for Technological Innovation and Application Development (NO. CSTC2021jscx-gksb-N0013) and The Chongqing Traditional Chinese Medicine Research Project (NO. 2023ZDXM032).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Warburg O. On the origin of cancer cells. Sci (New York NY) (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
2. Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: A therapeutic perspective. Nat Rev Clin Oncol (2017) 14(1):11–31. doi: 10.1038/nrclinonc.2016.60
3. Krebs HA, Eggleston LV, Hems R. Distribution of glutamine and glutamic acid in animal tissues. Biochem J (1949) 44(2):159–63. doi: 10.1042/bj0440159
4. Eagle H. Nutrition needs of mammalian cells in tissue culture. Sci (New York NY) (1955) 122(3168):501–14. doi: 10.1126/science.122.3168.501
5. Shanware NP, Mullen AR, DeBerardinis RJ, Abraham RT. Glutamine: Pleiotropic roles in tumor growth and stress resistance. J Mol Med (Berlin Germany) (2011) 89(3):229–36. doi: 10.1007/s00109-011-0731-9
6. Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-Flight mass spectrometry. Cancer Res (2009) 69(11):4918–25. doi: 10.1158/0008-5472.Can-08-4806
7. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J Clin Invest (2013) 123(9):3678–84. doi: 10.1172/jci69600
8. Iranzo J, Martincorena I, Koonin EV. Cancer-mutation network and the number and specificity of driver mutations. Proc Natl Acad Sci USA (2018) 115(26):E6010–e9. doi: 10.1073/pnas.1803155115
9. Lacey JM, Wilmore DW. Is glutamine a conditionally essential amino acid? Nutr Rev (1990) 48(8):297–309. doi: 10.1111/j.1753-4887.1990.tb02967.x
10. Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, et al. A variant of Slc1a5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab (2020) 31(2):267–83.e12. doi: 10.1016/j.cmet.2019.11.020
11. Freidman N, Chen I, Wu Q, Briot C, Holst J, Font J, et al. Amino acid transporters and exchangers from the Slc1a family: Structure, mechanism and roles in physiology and cancer. Neurochemical Res (2020) 45(6):1268–86. doi: 10.1007/s11064-019-02934-x
12. Teixeira E, Silva C, Martel F. The role of the glutamine transporter Asct2 in antineoplastic therapy. Cancer chemotherapy Pharmacol (2021) 87(4):447–64. doi: 10.1007/s00280-020-04218-6
13. Nishikubo K, Ohgaki R, Okanishi H, Okuda S, Xu M, Endou H, et al. Pharmacologic inhibition of Lat1 predominantly suppresses transport of Large neutral amino acids and downregulates global translation in cancer cells. J Cell Mol Med (2022) 26(20):5246–56. doi: 10.1111/jcmm.17553
14. Kanai Y. Amino acid transporter Lat1 (Slc7a5) as a molecular target for cancer diagnosis and therapeutics. Pharmacol Ther (2022) 230:107964. doi: 10.1016/j.pharmthera.2021.107964
15. Yanagida O, Kanai Y, Chairoungdua A, Kim DK, Segawa H, Nii T, et al. Human l-type amino acid transporter 1 (Lat1): Characterization of function and expression in tumor cell lines. Biochim Biophys Acta (2001) 1514(2):291–302. doi: 10.1016/s0005-2736(01)00384-4
16. Bröer S. Amino acid transporters as targets for cancer therapy: Why, where, when, and how. Int J Mol Sci (2020) 21(17):6156–75. doi: 10.3390/ijms21176156
17. Bröer A, Wagner C, Lang F, Bröer S. Neutral amino acid transporter Asct2 displays substrate-induced na+ exchange and a substrate-gated anion conductance. Biochem J (2000) 346 Pt 3(Pt 3):705–10.
18. Bröer S, Bröer A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem J (2017) 474(12):1935–63. doi: 10.1042/bcj20160822
19. Bröer A, Rahimi F, Bröer S. Deletion of amino acid transporter Asct2 (Slc1a5) reveals an essential role for transporters Snat1 (Slc38a1) and Snat2 (Slc38a2) to sustain glutaminolysis in cancer cells. J Biol Chem (2016) 291(25):13194–205. doi: 10.1074/jbc.M115.700534
20. Böhme-Schäfer I, Lörentz S, Bosserhoff AK. Role of amino acid transporter Snat1/Slc38a1 in human melanoma. Cancers (2022) 14(9):2151–66. doi: 10.3390/cancers14092151
21. Morotti M, Zois CE, El-Ansari R, Craze ML, Rakha EA, Fan SJ, et al. Increased expression of glutamine transporter Snat2/Slc38a2 promotes glutamine dependence and oxidative stress resistance, and is associated with worse prognosis in triple-negative breast cancer. Br J Cancer (2021) 124(2):494–505. doi: 10.1038/s41416-020-01113-y
22. Chaudhry FA, Reimer RJ, Krizaj D, Barber D, Storm-Mathisen J, Copenhagen DR, et al. Molecular analysis of system n suggests novel physiological roles in nitrogen metabolism and synaptic transmission. Cell (1999) 99(7):769–80. doi: 10.1016/s0092-8674(00)81674-8
23. Bröer S. The Slc38 family of sodium-amino acid Co-transporters. Pflugers Archiv Eur J Physiol (2014) 466(1):155–72. doi: 10.1007/s00424-013-1393-y
24. Sidoryk M, Matyja E, Dybel A, Zielinska M, Bogucki J, Jaskólski DJ, et al. Increased expression of a glutamine transporter Snat3 is a marker of malignant gliomas. Neuroreport (2004) 15(4):575–8. doi: 10.1097/00001756-200403220-00001
25. Wang Y, Fu L, Cui M, Wang Y, Xu Y, Li M, et al. Amino acid transporter Slc38a3 promotes metastasis of non-small cell lung cancer cells by activating Pdk1. Cancer Lett (2017) 393:8–15. doi: 10.1016/j.canlet.2017.01.036
26. Nałęcz KA. Amino acid transporter Slc6a14 (Atb(0,+)) - a target in combined anti-cancer therapy. Front Cell Dev Biol (2020) 8:594464. doi: 10.3389/fcell.2020.594464
27. van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, et al. Asct2/Slc1a5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene (2016) 35(24):3201–8. doi: 10.1038/onc.2015.381
28. Toda K, Nishikawa G, Iwamoto M, Itatani Y, Takahashi R, Sakai Y, et al. Clinical role of Asct2 (Slc1a5) in kras-mutated colorectal cancer. Int J Mol Sci (2017) 18(8):1632–44. doi: 10.3390/ijms18081632
29. Lin J, Yang T, Peng Z, Xiao H, Jiang N, Zhang L, et al. Slc1a5 silencing inhibits esophageal cancer growth Via cell cycle arrest and apoptosis. Cell Physiol Biochem Int J Exp Cell physiology biochemistry Pharmacol (2018) 48(1):397. doi: 10.1159/000491769
30. Lu J, Chen M, Tao Z, Gao S, Li Y, Cao Y, et al. Effects of targeting Slc1a5 on inhibiting gastric cancer growth and tumor development in vitro and in vivo. Oncotarget (2017) 8(44):76458–67. doi: 10.18632/oncotarget.19479
31. Wu J, Li Z, Yang Z, Guo L, Zhang Y, Deng H, et al. A glutamine-rich carrier efficiently delivers anti-Cd47 sirna driven by a "Glutamine trap" to inhibit lung cancer cell growth. Mol pharmaceutics (2018) 15(8):3032–45. doi: 10.1021/acs.molpharmaceut.8b00076
32. Guo H, Xu Y, Wang F, Shen Z, Tuo X, Qian H, et al. Clinical associations between Asct2 and P−Mtor in the pathogenesis and prognosis of epithelial ovarian cancer. Oncol Rep (2018) 40(6):3725–33. doi: 10.3892/or.2018.6729
33. Wang Q, Hardie RA, Hoy AJ, van Geldermalsen M, Gao D, Fazli L, et al. Targeting Asct2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol (2015) 236(3):278–89. doi: 10.1002/path.4518
34. Ren P, Yue M, Xiao D, Xiu R, Gan L, Liu H, et al. Atf4 and n-myc coordinate glutamine metabolism in mycn-amplified neuroblastoma cells through Asct2 activation. J Pathol (2015) 235(1):90–100. doi: 10.1002/path.4429
35. Marshall AD, van Geldermalsen M, Otte NJ, Lum T, Vellozzi M, Thoeng A, et al. Asct2 regulates glutamine uptake and cell growth in endometrial carcinoma. Oncogenesis (2017) 6(7):e367. doi: 10.1038/oncsis.2017.70
36. Liu Y, Yang L, An H, Chang Y, Zhang W, Zhu Y, et al. High expression of solute carrier family 1, member 5 (Slc1a5) is associated with poor prognosis in clear-cell renal cell carcinoma. Sci Rep (2015) 5:16954. doi: 10.1038/srep16954
37. Sun HW, Yu XJ, Wu WC, Chen J, Shi M, Zheng L, et al. Glut1 and Asct2 as predictors for prognosis of hepatocellular carcinoma. PloS One (2016) 11(12):e0168907. doi: 10.1371/journal.pone.0168907
38. Zhang Z, Liu R, Shuai Y, Huang Y, Jin R, Wang X, et al. Asct2 (Slc1a5)-dependent glutamine uptake is involved in the progression of head and neck squamous cell carcinoma. Br J Cancer (2020) 122(1):82–93. doi: 10.1038/s41416-019-0637-9
39. Luo Y, Li W, Ling Z, Hu Q, Fan Z, Cheng B, et al. Asct2 overexpression is associated with poor survival of oscc patients and Asct2 knockdown inhibited growth of glutamine-addicted oscc cells. Cancer Med (2020) 9(10):3489–99. doi: 10.1002/cam4.2965
40. Chen Z, Gao Y, Huang X, Yao Y, Chen K, Zeng S, et al. Tissue-based metabolomics reveals metabolic biomarkers and potential therapeutic targets for esophageal squamous cell carcinoma. J Pharm Biomed Anal (2021) 197:113937. doi: 10.1016/j.jpba.2021.113937
41. Kurozumi S, Kaira K, Matsumoto H, Kurosumi M, Yokobori T, Kanai Y, et al. Association of l-type amino acid transporter 1 (Lat1) with the immune system and prognosis in invasive breast cancer. Sci Rep (2022) 12(1):2742. doi: 10.1038/s41598-022-06615-8
42. Kaira K, Sunose Y, Ohshima Y, Ishioka NS, Arakawa K, Ogawa T, et al. Clinical significance of l-type amino acid transporter 1 expression as a prognostic marker and potential of new targeting therapy in biliary tract cancer. BMC Cancer (2013) 13:482. doi: 10.1186/1471-2407-13-482
43. Najumudeen AK, Ceteci F, Fey SK, Hamm G, Steven RT, Hall H, et al. The amino acid transporter Slc7a5 is required for efficient growth of kras-mutant colorectal cancer. Nat Genet (2021) 53(1):16–26. doi: 10.1038/s41588-020-00753-3
44. Wang J, Chen X, Su L, Li P, Liu B, Zhu Z. Lat-1 functions as a promotor in gastric cancer associated with clinicopathologic features. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie (2013) 67(8):693–9. doi: 10.1016/j.biopha.2013.05.003
45. Liu YH, Li YL, Shen HT, Chien PJ, Sheu GT, Wang BY, et al. L-type amino acid transporter 1 regulates cancer stemness and the expression of programmed cell death 1 ligand 1 in lung cancer cells. Int J Mol Sci (2021) 22(20):10955–69. doi: 10.3390/ijms222010955
46. Altan B, Kaira K, Watanabe A, Kubo N, Bao P, Dolgormaa G, et al. Relationship between Lat1 expression and resistance to chemotherapy in pancreatic ductal adenocarcinoma. Cancer chemotherapy Pharmacol (2018) 81(1):141–53. doi: 10.1007/s00280-017-3477-4
47. Xu M, Sakamoto S, Matsushima J, Kimura T, Ueda T, Mizokami A, et al. Up-regulation of Lat1 during antiandrogen therapy contributes to progression in prostate cancer cells. J Urol (2016) 195(5):1588–97. doi: 10.1016/j.juro.2015.11.071
48. Rosilio C, Nebout M, Imbert V, Griessinger E, Neffati Z, Benadiba J, et al. L-type amino-acid transporter 1 (Lat1): A therapeutic target supporting growth and survival of T-cell lymphoblastic Lymphoma/T-cell acute lymphoblastic leukemia. Leukemia (2015) 29(6):1253–66. doi: 10.1038/leu.2014.338
49. Cappoli N, Jenkinson MD, Dello Russo C, Dickens D. Lat1, a novel pharmacological target for the treatment of glioblastoma. Biochem Pharmacol (2022) 201:115103. doi: 10.1016/j.bcp.2022.115103
50. Srisongkram T, Bahrami K, Järvinen J, Timonen J, Rautio J, Weerapreeyakul N. Development of sesamol carbamate-L-Phenylalanine prodrug targeting l-type amino acid Transporter1 (Lat1) as a potential antiproliferative agent against melanoma. Int J Mol Sci (2022) 23(15):8446–58. doi: 10.3390/ijms23158446
51. Sato K, Miyamoto M, Takano M, Furuya K, Tsuda H. Significant relationship between the Lat1 expression pattern and chemoresistance in ovarian clear cell carcinoma. Virchows Archiv an Int J Pathol (2019) 474(6):701–10. doi: 10.1007/s00428-019-02520-0
52. Namikawa M, Kakizaki S, Kaira K, Tojima H, Yamazaki Y, Horiguchi N, et al. Expression of amino acid transporters (Lat1, Asct2 and xct) as clinical significance in hepatocellular carcinoma. Hepatol Res Off J Japan Soc Hepatol (2015) 45(9):1014–22. doi: 10.1111/hepr.12431
53. Nobusawa A, Kim M, Kaira K, Miyashita G, Negishi A, Oriuchi N, et al. Diagnostic usefulness of ¹⁸F-famt pet and l-type amino acid transporter 1 (Lat1) expression in oral squamous cell carcinoma. Eur J Nucl Med Mol Imaging (2013) 40(11):1692–700. doi: 10.1007/s00259-013-2477-9
54. Nikkuni O, Kaira K, Toyoda M, Shino M, Sakakura K, Takahashi K, et al. Expression of amino acid transporters (Lat1 and Asct2) in patients with stage Iii/Iv laryngeal squamous cell carcinoma. Pathol Oncol Res POR (2015) 21(4):1175–81. doi: 10.1007/s12253-015-9954-3
55. Yadav P, Sharma P, Sundaram S, Venkatraman G, Bera AK, Karunagaran D. Slc7a11/ xct is a target of mir-5096 and its restoration partially rescues mir-5096-Mediated ferroptosis and anti-tumor effects in human breast cancer cells. Cancer Lett (2021) 522:211–24. doi: 10.1016/j.canlet.2021.09.033
56. Shi ZZ, Tao H, Fan ZW, Song SJ, Bai J. Prognostic and immunological role of key genes of ferroptosis in pan-cancer. Front Cell Dev Biol (2021) 9:748925. doi: 10.3389/fcell.2021.748925
57. Hong T, Lei G, Chen X, Li H, Zhang X, Wu N, et al. Parp inhibition promotes ferroptosis Via repressing Slc7a11 and synergizes with ferroptosis inducers in brca-proficient ovarian cancer. Redox Biol (2021) 42:101928. doi: 10.1016/j.redox.2021.101928
58. He J, Ding H, Li H, Pan Z, Chen Q. Intra-tumoral expression of Slc7a11 is associated with immune microenvironment, drug resistance, and prognosis in cancers: A pan-cancer analysis. Front Genet (2021) 12:770857. doi: 10.3389/fgene.2021.770857
59. He Q, Liu M, Huang W, Chen X, Zhang B, Zhang T, et al. Il-1β-Induced elevation of solute carrier family 7 member 11 promotes hepatocellular carcinoma metastasis through up-regulating programmed death ligand 1 and colony-stimulating factor 1. Hepatol (Baltimore Md) (2021) 74(6):3174–93. doi: 10.1002/hep.32062
60. Hu K, Li K, Lv J, Feng J, Chen J, Wu H, et al. Suppression of the Slc7a11/Glutathione axis causes synthetic lethality in kras-mutant lung adenocarcinoma. J Clin Invest (2020) 130(4):1752–66. doi: 10.1172/jci124049
61. Umans RA, Martin J, Harrigan ME, Patel DC, Chaunsali L, Roshandel A, et al. Transcriptional regulation of amino acid transport in glioblastoma multiforme. Cancers (2021) 13(24):6169–83. doi: 10.3390/cancers13246169
62. Hémon A, Louandre C, Lailler C, Godin C, Bottelin M, Morel V, et al. Slc7a11 as a biomarker and therapeutic target in hpv-positive head and neck squamous cell carcinoma. Biochem Biophys Res Commun (2020) 533(4):1083–7. doi: 10.1016/j.bbrc.2020.09.134
63. Wang K, Cao F, Fang W, Hu Y, Chen Y, Ding H, et al. Activation of Snat1/Slc38a1 in human breast cancer: Correlation with p-akt overexpression. BMC Cancer (2013) 13:343. doi: 10.1186/1471-2407-13-343
64. Yu J, Chen X, Li J, Wang F. Circrunx1 functions as an oncogene in colorectal cancer by regulating Circrunx1/Mir-485-5p/Slc38a1 axis. Eur J Clin Invest (2021) 51(7):e13540. doi: 10.1111/eci.13540
65. Xie J, Li P, Gao HF, Qian JX, Yuan LY, Wang JJ. Overexpression of Slc38a1 is associated with poorer prognosis in Chinese patients with gastric cancer. BMC Gastroenterol (2014) 14:70. doi: 10.1186/1471-230x-14-70
66. Sudo H, Tsuji AB, Sugyo A, Okada M, Kato K, Zhang MR, et al. Direct comparison of 2−Amino[3−11c]Isobutyric acid and 2−Amino[11c]Methyl−Isobutyric acid uptake in eight lung cancer xenograft models. Int J Oncol (2018) 53(6):2737–44. doi: 10.3892/ijo.2018.4596
67. Baczewska M, Supruniuk E, Bojczuk K, Guzik P, Milewska P, Konończuk K, et al. Energy substrate transporters in high-grade ovarian cancer: Gene expression and clinical implications. Int J Mol Sci (2022) 23(16):8968–88. doi: 10.3390/ijms23168968
68. Li Y, Shao H, Da Z, Pan J, Fu B. High expression of Slc38a1 predicts poor prognosis in patients with De novo acute myeloid leukemia. J Cell Physiol (2019) 234(11):20322–8. doi: 10.1002/jcp.28632
69. Liu Y, Yang Y, Jiang L, Xu H, Wei J. High expression levels of Slc38a1 are correlated with poor prognosis and defective immune infiltration in hepatocellular carcinoma. J Oncol (2021) 2021:5680968. doi: 10.1155/2021/5680968
70. Kandasamy P, Zlobec I, Nydegger DT, Pujol-Giménez J, Bhardwaj R, Shirasawa S, et al. Oncogenic kras mutations enhance amino acid uptake by colorectal cancer cells Via the hippo signaling effector Yap1. Mol Oncol (2021) 15(10):2782–800. doi: 10.1002/1878-0261.12999
71. Nie K, Cai M. Snat2/Slc38a2 confers the stemness of gastric cancer cells Via regulating glutamine level. Digestive Dis Sci (2022) 67(7):2948–56. doi: 10.1007/s10620-021-07110-2
72. Nielsen CU, Krog NF, Sjekirica I, Nielsen SS, Pedersen ML. Snat2 is responsible for hyperosmotic induced sarcosine and glycine uptake in human prostate pc-3 cells. Pflugers Archiv Eur J Physiol (2022) 474(12):1249–62. doi: 10.1007/s00424-022-02752-1
73. Gupta N, Miyauchi S, Martindale RG, Herdman AV, Podolsky R, Miyake K, et al. Upregulation of the amino acid transporter Atb0,+ (Slc6a14) in colorectal cancer and metastasis in humans. Biochim Biophys Acta (2005) 1741(1-2):215–23. doi: 10.1016/j.bbadis.2005.04.002
74. Guo Q, Xu W, Li X, Sun JL, Gu XC, Jing FB. Slc6a14 depletion contributes to amino acid starvation to suppress emt-induced metastasis in gastric cancer by perturbing the Pi3k/Akt/Mtorc1 pathway. BioMed Res Int (2022) 2022:7850658. doi: 10.1155/2022/7850658
75. Coothankandaswamy V, Cao S, Xu Y, Prasad PD, Singh PK, Reynolds CP, et al. Amino acid transporter Slc6a14 is a novel and effective drug target for pancreatic cancer. Br J Pharmacol (2016) 173(23):3292–306. doi: 10.1111/bph.13616
76. Karunakaran S, Ramachandran S, Coothankandaswamy V, Elangovan S, Babu E, Periyasamy-Thandavan S, et al. Slc6a14 (Atb0,+) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor-positive breast cancer. J Biol Chem (2011) 286(36):31830–8. doi: 10.1074/jbc.M111.229518
77. Gupta N, Prasad PD, Ghamande S, Moore-Martin P, Herdman AV, Martindale RG, et al. Up-regulation of the amino acid transporter Atb(0,+) (Slc6a14) in carcinoma of the cervix. Gynecologic Oncol (2006) 100(1):8–13. doi: 10.1016/j.ygyno.2005.08.016
78. Martínez-Reyes I, Chandel NS. Mitochondrial tca cycle metabolites control physiology and disease. Nat Commun (2020) 11(1):102. doi: 10.1038/s41467-019-13668-3
79. Cardaci S, Ciriolo MR. Tca cycle defects and cancer: When metabolism tunes redox state. Int J Cell Biol (2012) 2012:161837. doi: 10.1155/2012/161837
80. Laurenti G, Tennant DA. Isocitrate dehydrogenase (Idh), succinate dehydrogenase (Sdh), fumarate hydratase (Fh): Three players for one phenotype in cancer? Biochem Soc Trans (2016) 44(4):1111–6. doi: 10.1042/bst20160099
81. Yu Y, Zheng M, Zhu W, Zhao F, Guan B, Shen Q, et al. Hereditary leiomyomatosis and renal cell cancer (Hlrcc): Case series and review of the literature. Urologic Oncol (2021) 39(11):791.e9–.e16. doi: 10.1016/j.urolonc.2021.07.026
82. Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in fh predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet (2002) 30(4):406–10. doi: 10.1038/ng849
83. Shanmugasundaram K, Nayak B, Shim EH, Livi CB, Block K, Sudarshan S. The oncometabolite fumarate promotes pseudohypoxia through noncanonical activation of nf-Kb signaling. J Biol Chem (2014) 289(35):24691–9. doi: 10.1074/jbc.M114.568162
84. Quinones A, Le A. The multifaceted glioblastoma: From genomic alterations to metabolic adaptations. Adv Exp Med Biol (2021) 1311:59–76. doi: 10.1007/978-3-030-65768-0_4
85. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated Idh1 and Idh2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell (2010) 17(3):225–34. doi: 10.1016/j.ccr.2010.01.020
86. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated Idh1 mutations produce 2-hydroxyglutarate. Nature (2010) 465(7300):966. doi: 10.1038/nature09132
87. Takács-Vellai K, Farkas Z, Ősz F, Stewart GW. Model systems in sdhx-related Pheochromocytoma/Paraganglioma. Cancer Metastasis Rev (2021) 40(4):1177–201. doi: 10.1007/s10555-021-10009-z
88. Yebra M, Bhargava S, Kumar A, Burgoyne AM, Tang CM, Yoon H, et al. Establishment of patient-derived succinate dehydrogenase-deficient gastrointestinal stromal tumor models for predicting therapeutic response. Clin Cancer Res an Off J Am Assoc Cancer Res (2022) 28(1):187–200. doi: 10.1158/1078-0432.Ccr-21-2092
89. Galera-Ruiz H, Gonzalez-Campora R, Rey-Barrera M, Rollón-Mayordomo A, Garcia-Escudero A, Fernández-Santos JM, et al. W43x sdhd mutation in sporadic head and neck paraganglioma. Analytical quantitative cytology Histol (2008) 30(2):119–23.
90. Rapizzi E, Ercolino T, Fucci R, Zampetti B, Felici R, Guasti D, et al. Succinate dehydrogenase subunit b mutations modify human neuroblastoma cell metabolism and proliferation. Hormones Cancer (2014) 5(3):174–84. doi: 10.1007/s12672-014-0172-3
91. Yoo A, Tang C, Zucker M, Fitzgerald K, DiNatale RG, Rappold PM, et al. Genomic and metabolic hallmarks of sdh- and fh-deficient renal cell carcinomas. Eur Urol Focus (2022) 8(5):1278–88. doi: 10.1016/j.euf.2021.12.002
92. Ashtekar A, Huk D, Magner A, La Perle K, Zhang X, Piruat JI, et al. Sdhd ablation promotes thyroid tumorigenesis by inducing a stem-like phenotype. Endocrine-related Cancer (2017) 24(11):579–91. doi: 10.1530/erc-17-0229
93. Helenius IT, Madala HR, Yeh JJ. An asp to strike out cancer? therapeutic possibilities arising from aspartate's emerging roles in cell proliferation and survival. Biomolecules (2021) 11(11):1666–76. doi: 10.3390/biom11111666
94. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell (2015) 162(3):552–63. doi: 10.1016/j.cell.2015.07.017
95. Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell (2015) 162(3):540–51. doi: 10.1016/j.cell.2015.07.016
96. Guo W, Tan HY, Li S, Wang N, Feng Y. Glutamic-pyruvic transaminase 1 facilitates alternative fuels for hepatocellular carcinoma growth-a small molecule inhibitor, berberine. Cancers (2020) 12(7):1854–71. doi: 10.3390/cancers12071854
97. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature (2011) 476(7360):346–50. doi: 10.1038/nature10350
98. Huynh TYL, Zareba I, Baszanowska W, Lewoniewska S, Palka J. Understanding the role of key amino acids in regulation of proline Dehydrogenase/Proline oxidase (Prodh/Pox)-dependent Apoptosis/Autophagy as an approach to targeted cancer therapy. Mol Cell Biochem (2020) 466(1-2):35–44. doi: 10.1007/s11010-020-03685-y
99. Lane AN, Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res (2015) 43(4):2466–85. doi: 10.1093/nar/gkv047
100. Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of A-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci United States America (2011) 108(49):19611–6. doi: 10.1073/pnas.1117773108
101. Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature (2011) 481(7381):385–8. doi: 10.1038/nature10642
102. Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature (2016) 532(7598):255–8. doi: 10.1038/nature17393
103. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by Idh1 mediates lipogenesis under hypoxia. Nature (2011) 481(7381):380–4. doi: 10.1038/nature10602
104. Sun RC, Denko NC. Hypoxic regulation of glutamine metabolism through Hif1 and Siah2 supports lipid synthesis that is necessary for tumor growth. Cell Metab (2014) 19(2):285–92. doi: 10.1016/j.cmet.2013.11.022
105. Jiang B, Zhang J, Zhao G, Liu M, Hu J, Lin F, et al. Filamentous Gls1 promotes ros-induced apoptosis upon glutamine deprivation Via insufficient asparagine synthesis. Mol Cell (2022) 82(10):1821–35.e6. doi: 10.1016/j.molcel.2022.03.016
106. Mukha A, Kahya U, Linge A, Chen O, Löck S, Lukiyanchuk V, et al. Gls-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and Atg5-mediated autophagy. Theranostics (2021) 11(16):7844–68. doi: 10.7150/thno.58655
107. Bodineau C, Tomé M, Murdoch PDS, Durán RV. Glutamine, mtor and autophagy: A multiconnection relationship. Autophagy (2022) 18(11):2749–50. doi: 10.1080/15548627.2022.2062875
108. Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signaling (2010) 3(119):ra31. doi: 10.1126/scisignal.2000911
109. Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of Ulk1/Ulk2 kinases. Proc Natl Acad Sci United States America (2011) 108(27):11121–6. doi: 10.1073/pnas.1107969108
110. Errera M, Greenstein JP. Phosphate-activated glutaminase in kidney and other tissues. J Biol Chem (1949) 178(1):495–502. doi: 10.1016/S0021-9258(18)56979-9
111. Katt WP, Lukey MJ, Cerione RA. A tale of two glutaminases: Homologous enzymes with distinct roles in tumorigenesis. Future medicinal Chem (2017) 9(2):223–43. doi: 10.4155/fmc-2016-0190
112. Greenstein JP, Price VE. Alpha-keto acid-activated glutaminase and asparaginase. J Biol Chem (1949) 178(2):695–705. doi: 10.1016/S0021-9258(18)56887-3
113. Yu W, Yang X, Zhang Q, Sun L, Yuan S, Xin Y. Targeting Gls1 to cancer therapy through glutamine metabolism. Clin Trans Oncol Off Publ Fed Spanish Oncol Societies Natl Cancer Institute Mexico (2021) 23(11):2253–68. doi: 10.1007/s12094-021-02645-2
114. Matés JM, Campos-Sandoval JA, Márquez J. Glutaminase isoenzymes in the metabolic therapy of cancer. Biochim Biophys Acta Rev Cancer (2018) 1870(2):158–64. doi: 10.1016/j.bbcan.2018.07.007
115. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor cb-839 in triple-negative breast cancer. Mol Cancer Ther (2014) 13(4):890–901. doi: 10.1158/1535-7163.Mct-13-0870
116. Huang F, Zhang Q, Ma H, Lv Q, Zhang T. Expression of glutaminase is upregulated in colorectal cancer and of clinical significance. Int J Clin Exp Pathol (2014) 7(3):1093–100.
117. Pan T, Gao L, Wu G, Shen G, Xie S, Wen H, et al. Elevated expression of glutaminase confers glucose utilization Via glutaminolysis in prostate cancer. Biochem Biophys Res Commun (2015) 456(1):452–8. doi: 10.1016/j.bbrc.2014.11.105
118. Xiang L, Mou J, Shao B, Wei Y, Liang H, Takano N, et al. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis (2019) 10(2):40. doi: 10.1038/s41419-018-1291-5
119. Meister A, Tice SV. Transamination from glutamine to alpha-keto acids. J Biol Chem (1950) 187(1):173–87. doi: 10.1016/S0021-9258(19)50942-5
120. Meister A, Sober HA, Tice SV, Fraser PE. Transamination and associated deamidation of asparagine and glutamine. J Biol Chem (1952) 197(1):319–30. doi: 10.1016/S0021-9258(18)55681-7
121. Cooper JL, Meister A. Isolation and properties of highly purified glutamine transaminase. Biochemistry (1972) 11(5):661–71. doi: 10.1021/bi00755a001
122. Cooper AJ, Meister A. Isolation and properties of a new glutamine transaminase from rat kidney. J Biol Chem (1974) 249(8):2554–61. doi: 10.1016/S0021-9258(19)42765-8
123. Han Q, Cai T, Tagle DA, Robinson H, Li J. Substrate specificity and structure of human aminoadipate Aminotransferase/Kynurenine aminotransferase ii. Bioscience Rep (2008) 28(4):205–15. doi: 10.1042/bsr20080085
124. Udupa S, Nguyen S, Hoang G, Nguyen T, Quinones A, Pham K, et al. Upregulation of the glutaminase ii pathway contributes to glutamate production upon glutaminase 1 inhibition in pancreatic cancer. Proteomics (2019) 19(21-22):e1800451. doi: 10.1002/pmic.201800451
125. Dorai T, Dorai B, Pinto JT, Grasso M, Cooper AJL. High levels of glutaminase ii pathway enzymes in normal and cancerous prostate suggest a role in 'Glutamine addiction'. Biomolecules (2019) 10(1):2–17. doi: 10.3390/biom10010002
126. Spanaki C, Kotzamani D, Plaitakis A. Widening spectrum of cellular and subcellular expression of human Glud1 and Glud2 glutamate dehydrogenases suggests novel functions. Neurochemical Res (2017) 42(1):92–107. doi: 10.1007/s11064-016-1986-x
127. Plaitakis A, Kalef-Ezra E, Kotzamani D, Zaganas I, Spanaki C. The glutamate dehydrogenase pathway and its roles in cell and tissue biology in health and disease. Biology (2017) 6(1):11–36. doi: 10.3390/biology6010011
128. Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, Haigis MC. Metabolic recycling of ammonia Via glutamate dehydrogenase supports breast cancer biomass. Sci (New York NY) (2017) 358(6365):941–6. doi: 10.1126/science.aam9305
129. Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z, et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell (2015) 27(2):257–70. doi: 10.1016/j.ccell.2014.12.006
130. Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or akt signaling. Cancer Res (2009) 69(20):7986–93. doi: 10.1158/0008-5472.Can-09-2266
131. Liu G, Zhu J, Yu M, Cai C, Zhou Y, Yu M, et al. Glutamate dehydrogenase is a novel prognostic marker and predicts metastases in colorectal cancer patients. J Trans Med (2015) 13:144. doi: 10.1186/s12967-015-0500-6
132. Yuan L, Sheng X, Willson AK, Roque DR, Stine JE, Guo H, et al. Glutamine promotes ovarian cancer cell proliferation through the Mtor/S6 pathway. Endocrine-related Cancer (2015) 22(4):577–91. doi: 10.1530/erc-15-0192
133. Su Z, Liu G, Fang T, Zhang K, Yang S, Zhang H, et al. Expression and prognostic value of glutamate dehydrogenase in extrahepatic cholangiocarcinoma patients. Am J Trans Res (2017) 9(5):2106–18.
134. Chen R, Nishimura MC, Kharbanda S, Peale F, Deng Y, Daemen A, et al. Hominoid-specific enzyme Glud2 promotes growth of Idh1r132h glioma. Proc Natl Acad Sci United States America (2014) 111(39):14217–22. doi: 10.1073/pnas.1409653111
135. Wei Z, Liu X, Cheng C, Yu W, Yi P. Metabolism of amino acids in cancer. Front Cell Dev Biol (2020) 8:603837. doi: 10.3389/fcell.2020.603837
136. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a kras-regulated metabolic pathway. Nature (2013) 496(7443):101–5. doi: 10.1038/nature12040
137. Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, et al. Got1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun (2021) 12(1):4860. doi: 10.1038/s41467-021-24859-2
138. Yang H, Zhou L, Shi Q, Zhao Y, Lin H, Zhang M, et al. Sirt3-dependent Got2 acetylation status affects the malate-aspartate nadh shuttle activity and pancreatic tumor growth. EMBO J (2015) 34(8):1110–25. doi: 10.15252/embj.201591041
139. Yang Y. Enhancing doxorubicin efficacy through inhibition of aspartate transaminase in triple-negative breast cancer cells. Biochem Biophys Res Commun (2016) 473(4):1295–300. doi: 10.1016/j.bbrc.2016.04.061
140. Korangath P, Teo WW, Sadik H, Han L, Mori N, Huijts CM, et al. Targeting glutamine metabolism in breast cancer with aminooxyacetate. Clin Cancer Res an Off J Am Assoc Cancer Res (2015) 21(14):3263–73. doi: 10.1158/1078-0432.Ccr-14-1200
141. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ros generation are essential for kras-mediated tumorigenicity. Proc Natl Acad Sci United States America (2010) 107(19):8788–93. doi: 10.1073/pnas.1003428107
142. Hao Y, Samuels Y, Li Q, Krokowski D, Guan BJ, Wang C, et al. Oncogenic Pik3ca mutations reprogram glutamine metabolism in colorectal cancer. Nat Commun (2016) 7:11971. doi: 10.1038/ncomms11971
143. Cao Y, Lin SH, Wang Y, Chin YE, Kang L, Mi J. Glutamic pyruvate transaminase Gpt2 promotes tumorigenesis of breast cancer cells by activating sonic hedgehog signaling. Theranostics (2017) 7(12):3021–33. doi: 10.7150/thno.18992
144. Ghahari M, Salari A, Ghafoori Yazdi M, Nowroozi A, Fotovat A, Momeni SA, et al. Association between preoperative de ritis (Ast/Alt) ratio and oncological outcomes following radical cystectomy in patients with urothelial bladder cancer. Clin genitourinary Cancer (2022) 20(2):e89–93. doi: 10.1016/j.clgc.2021.10.007
145. Scheipner L, Smolle MA, Barth D, Posch F, Stotz M, Pichler M, et al. The Ast/Alt ratio is an independent prognostic marker for disease-free survival in stage ii and iii colorectal carcinoma. Anticancer Res (2021) 41(1):429–36. doi: 10.21873/anticanres.14792
146. Zhang LX, Lv Y, Xu AM, Wang HZ. The prognostic significance of serum gamma-glutamyltransferase levels and Ast/Alt in primary hepatic carcinoma. BMC Cancer (2019) 19(1):841. doi: 10.1186/s12885-019-6011-8
147. Sansa A, Venegas MDP, Valero C, Pardo L, Avilés-Jurado FX, Terra X, et al. The aspartate Aminotransaminase/Alanine aminotransaminase (De ritis) ratio predicts sensitivity to radiotherapy in head and neck carcinoma patients. Head Neck (2021) 43(7):2091–100. doi: 10.1002/hed.26673
148. Knittelfelder O, Delago D, Jakse G, Reinisch S, Partl R, Stranzl-Lawatsch H, et al. The Ast/Alt (De ritis) ratio predicts survival in patients with oral and oropharyngeal cancer. Diagnostics (Basel Switzerland) (2020) 10(11):973–86. doi: 10.3390/diagnostics10110973
149. Zhou J, He Z, Ma S, Liu R. Ast/Alt ratio as a significant predictor of the incidence risk of prostate cancer. Cancer Med (2020) 9(15):5672–7. doi: 10.1002/cam4.3086
150. Riedl JM, Posch F, Prager G, Eisterer W, Oehler L, Sliwa T, et al. The Ast/Alt (De ritis) ratio predicts clinical outcome in patients with pancreatic cancer treated with first-line nab-paclitaxel and gemcitabine: Post hoc analysis of an Austrian multicenter, noninterventional study. Ther Adv Med Oncol (2020) 12:1–12. doi: 10.1177/1758835919900872
151. Yoon S, Kim JG, Seo AN, Park SY, Kim HJ, Park JS, et al. Clinical implication of serine metabolism-associated enzymes in colon cancer. Oncology (2015) 89(6):351–9. doi: 10.1159/000439571
152. Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol (2015) 17(12):1556–68. doi: 10.1038/ncb3272
153. Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, et al. Oncogenic myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab (2015) 22(6):1068–77. doi: 10.1016/j.cmet.2015.09.025
154. Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harbor Perspect Biol (2016) 8(9):a019505. doi: 10.1101/cshperspect.a019505
155. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the braf gene in human cancer. Nature (2002) 417(6892):949–54. doi: 10.1038/nature00766
156. Zaman A, Wu W, Bivona TG. Targeting oncogenic braf: Past, present, and future. Cancers (2019) 11(8):1197–215. doi: 10.3390/cancers11081197
157. Baenke F, Chaneton B, Smith M, Van Den Broek N, Hogan K, Tang H, et al. Resistance to braf inhibitors induces glutamine dependency in melanoma cells. Mol Oncol (2016) 10(1):73–84. doi: 10.1016/j.molonc.2015.08.003
158. Hernandez-Davies JE, Tran TQ, Reid MA, Rosales KR, Lowman XH, Pan M, et al. Vemurafenib resistance reprograms melanoma cells towards glutamine dependence. J Trans Med (2015) 13:210. doi: 10.1186/s12967-015-0581-2
159. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. Ras oncogenes: Weaving a tumorigenic web. Nat Rev Cancer (2011) 11(11):761–74. doi: 10.1038/nrc3106
160. Prior IA, Hood FE, Hartley JL. The frequency of ras mutations in cancer. Cancer Res (2020) 80(14):2969–74. doi: 10.1158/0008-5472.Can-19-3682
161. Bernfeld E, Foster DA. Glutamine as an essential amino acid for kras-driven cancer cells. Trends Endocrinol metabolism: TEM (2019) 30(6):357–68. doi: 10.1016/j.tem.2019.03.003
162. Toda K, Kawada K, Iwamoto M, Inamoto S, Sasazuki T, Shirasawa S, et al. Metabolic alterations caused by kras mutations in colorectal cancer contribute to cell adaptation to glutamine depletion by upregulation of asparagine synthetase. Neoplasia (New York NY) (2016) 18(11):654–65. doi: 10.1016/j.neo.2016.09.004
163. Hara Y, Minami Y, Yoshimoto S, Hayashi N, Yamasaki A, Ueda S, et al. Anti-tumor effects of an antagonistic mab against the Asct2 amino acid transporter on kras-mutated human colorectal cancer cells. Cancer Med (2020) 9(1):302–12. doi: 10.1002/cam4.2689
164. Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, et al. Oncogenic K-ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol (2011) 7:523. doi: 10.1038/msb.2011.56
165. Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, et al. Lkb1 and Keap1/Nrf2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in kras-mutant lung adenocarcinoma. Cancer Res (2019) 79(13):3251–67. doi: 10.1158/0008-5472.Can-18-3527
166. Xia M, Li X, Diao Y, Du B, Li Y. Targeted inhibition of glutamine metabolism enhances the antitumor effect of selumetinib in kras-mutant nsclc. Trans Oncol (2021) 14(1):100920. doi: 10.1016/j.tranon.2020.100920
167. Schaub FX, Dhankani V, Berger AC, Trivedi M, Richardson AB, Shaw R, et al. Pan-cancer alterations of the myc oncogene and its proximal network across the cancer genome atlas. Cell Syst (2018) 6(3):282–300.e2. doi: 10.1016/j.cels.2018.03.003
168. Dhanasekaran R, Deutzmann A, Mahauad-Fernandez WD, Hansen AS, Gouw AM, Felsher DW. The myc oncogene - the grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol (2022) 19(1):23–36. doi: 10.1038/s41571-021-00549-2
169. Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces myc-dependent apoptosis in human cells. J Cell Biol (2007) 178(1):93–105. doi: 10.1083/jcb.200703099
170. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci United States America (2008) 105(48):18782–7. doi: 10.1073/pnas.0810199105
171. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. C-myc suppression of mir-23a/B enhances mitochondrial glutaminase expression and glutamine metabolism. Nature (2009) 458(7239):762–5. doi: 10.1038/nature07823
172. Anso E, Mullen AR, Felsher DW, Matés JM, Deberardinis RJ, Chandel NS. Metabolic changes in cancer cells upon suppression of myc. Cancer Metab (2013) 1(1):7. doi: 10.1186/2049-3002-1-7
173. Jeong SM, Lee A, Lee J, Haigis MC. Sirt4 protein suppresses tumor formation in genetic models of myc-induced b cell lymphoma. J Biol Chem (2014) 289(7):4135–44. doi: 10.1074/jbc.M113.525949
174. Shroff EH, Eberlin LS, Dang VM, Gouw AM, Gabay M, Adam SJ, et al. Myc oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci United States America (2015) 112(21):6539–44. doi: 10.1073/pnas.1507228112
175. Dejure FR, Royla N, Herold S, Kalb J, Walz S, Ade CP, et al. The myc mrna 3'-utr couples rna polymerase ii function to glutamine and ribonucleotide levels. EMBO J (2017) 36(13):1854–68. doi: 10.15252/embj.201796662
176. Munksgaard Thorén M, Vaapil M, Staaf J, Planck M, Johansson ME, Mohlin S, et al. Myc-induced glutaminolysis bypasses hif-driven glycolysis in hypoxic small cell lung carcinoma cells. Oncotarget (2017) 8(30):48983–95. doi: 10.18632/oncotarget.16904
177. Effenberger M, Bommert KS, Kunz V, Kruk J, Leich E, Rudelius M, et al. Glutaminase inhibition in multiple myeloma induces apoptosis Via myc degradation. Oncotarget (2017) 8(49):85858–67. doi: 10.18632/oncotarget.20691
178. Le Grand M, Mukha A, Püschel J, Valli E, Kamili A, Vittorio O, et al. Interplay between mycn and c-myc regulates radioresistance and cancer stem cell phenotype in neuroblastoma upon glutamine deprivation. Theranostics (2020) 10(14):6411–29. doi: 10.7150/thno.42602
179. Brunelli L, Caiola E, Marabese M, Broggini M, Pastorelli R. Capturing the metabolomic diversity of kras mutants in non-Small-Cell lung cancer cells. Oncotarget (2014) 5(13):4722–31. doi: 10.18632/oncotarget.1958
180. Mukhopadhyay S, Goswami D, Adiseshaiah PP, Burgan W, Yi M, Guerin TM, et al. Undermining glutaminolysis bolsters chemotherapy while Nrf2 promotes chemoresistance in kras-driven pancreatic cancers. Cancer Res (2020) 80(8):1630–43. doi: 10.1158/0008-5472.Can-19-1363
181. Harwood FC, Klein Geltink RI, O'Hara BP, Cardone M, Janke L, Finkelstein D, et al. Etv7 is an essential component of a rapamycin-insensitive mtor complex in cancer. Sci Adv (2018) 4(9):eaar3938. doi: 10.1126/sciadv.aar3938
182. Saxton RA, Sabatini DM. Mtor signaling in growth, metabolism, and disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
183. Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, et al. Transcriptional activation of ragd gtpase controls Mtorc1 and promotes cancer growth. Sci (New York NY) (2017) 356(6343):1188–92. doi: 10.1126/science.aag2553
184. Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, et al. Mechanisms of Mtorc1 activation by rheb and inhibition by Pras40. Nature (2017) 552(7685):368–73. doi: 10.1038/nature25023
185. Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, et al. A diverse array of cancer-associated mtor mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discovery (2014) 4(5):554–63. doi: 10.1158/2159-8290.Cd-13-0929
186. Bar-Peled L, Sabatini DM. Regulation of Mtorc1 by amino acids. Trends Cell Biol (2014) 24(7):400–6. doi: 10.1016/j.tcb.2014.03.003
187. Zhang T, Wang R, Wang Z, Wang X, Wang F, Ding J. Structural basis for ragulator functioning as a scaffold in membrane-anchoring of rag gtpases and Mtorc1. Nat Commun (2017) 8(1):1394. doi: 10.1038/s41467-017-01567-4
188. Choo AY, Kim SG, Vander Heiden MG, Mahoney SJ, Vu H, Yoon SO, et al. Glucose addiction of tsc null cells is caused by failed Mtorc1-dependent balancing of metabolic demand with supply. Mol Cell (2010) 38(4):487–99. doi: 10.1016/j.molcel.2010.05.007
189. Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, et al. The Mtorc1 pathway stimulates glutamine metabolism and cell proliferation by repressing Sirt4. Cell (2013) 153(4):840–54. doi: 10.1016/j.cell.2013.04.023
190. Csibi A, Lee G, Yoon SO, Tong H, Ilter D, Elia I, et al. The Mtorc1/S6k1 pathway regulates glutamine metabolism through the Eif4b-dependent control of c-myc translation. Curr Biol CB (2014) 24(19):2274–80. doi: 10.1016/j.cub.2014.08.007
191. Volinia S, Hiles I, Ormondroyd E, Nizetic D, Antonacci R, Rocchi M, et al. Molecular cloning, cdna sequence, and chromosomal localization of the human phosphatidylinositol 3-kinase P110 alpha (Pik3ca) gene. Genomics (1994) 24(3):472–7. doi: 10.1006/geno.1994.1655
192. Zhao L, Vogt PK. Class I Pi3k in oncogenic cellular transformation. Oncogene (2008) 27(41):5486–96. doi: 10.1038/onc.2008.244
193. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the Pik3ca gene in human cancers. Sci (New York NY) (2004) 304(5670):554. doi: 10.1126/science.1096502
194. Zhao Y, Zhao X, Chen V, Feng Y, Wang L, Croniger C, et al. Colorectal cancers utilize glutamine as an anaplerotic substrate of the tca cycle in vivo. Sci Rep (2019) 9(1):19180. doi: 10.1038/s41598-019-55718-2
195. Lau CE, Tredwell GD, Ellis JK, Lam EW, Keun HC. Metabolomic characterisation of the effects of oncogenic Pik3ca transformation in a breast epithelial cell line. Sci Rep (2017) 7:46079. doi: 10.1038/srep46079
196. Zhao Y, Feng X, Chen Y, Selfridge JE, Gorityala S, Du Z, et al. 5-fluorouracil enhances the antitumor activity of the glutaminase inhibitor cb-839 against Pik3ca-mutant colorectal cancers. Cancer Res (2020) 80(21):4815–27. doi: 10.1158/0008-5472.Can-20-0600
197. Qie S, Chu C, Li W, Wang C, Sang N. Erbb2 activation upregulates glutaminase 1 expression which promotes breast cancer cell proliferation. J Cell Biochem (2014) 115(3):498–509. doi: 10.1002/jcb.24684
198. Zhan H, Ciano K, Dong K, Zucker S. Targeting glutamine metabolism in myeloproliferative neoplasms. Blood cells molecules Dis (2015) 55(3):241–7. doi: 10.1016/j.bcmd.2015.07.007
199. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14(5):500–8. doi: 10.1038/ni.2556
200. Buck MD, O'Sullivan D, Pearce EL. T Cell metabolism drives immunity. J Exp Med (2015) 212(9):1345–60. doi: 10.1084/jem.20151159
201. Ron-Harel N, Ghergurovich JM, Notarangelo G, LaFleur MW, Tsubosaka Y, Sharpe AH, et al. T Cell activation depends on extracellular alanine. Cell Rep (2019) 28(12):3011–21.e4. doi: 10.1016/j.celrep.2019.08.034
202. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by Erk/Mapk during T lymphocyte activation. J Immunol (Baltimore Md 1950) (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
203. Fu Q, Xu L, Wang Y, Jiang Q, Liu Z, Zhang J, et al. Tumor-associated macrophage-derived interleukin-23 interlinks kidney cancer glutamine addiction with immune evasion. Eur Urol (2019) 75(5):752–63. doi: 10.1016/j.eururo.2018.09.030
204. Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest (2021) 131(4):e140100. doi: 10.1172/jci140100
205. Yang WH, Qiu Y, Stamatatos O, Janowitz T, Lukey MJ. Enhancing the efficacy of glutamine metabolism inhibitors in cancer therapy. Trends Cancer (2021) 7(8):790–804. doi: 10.1016/j.trecan.2021.04.003
206. Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Sci (New York NY) (2019) 366(6468):1013–21. doi: 10.1126/science.aav2588
207. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell (2018) 175(7):1780–95.e19. doi: 10.1016/j.cell.2018.10.001
208. Esslinger CS, Cybulski KA, Rhoderick JF. Ngamma-aryl glutamine analogues as probes of the Asct2 neutral amino acid transporter binding site. Bioorganic medicinal Chem (2005) 13(4):1111–8. doi: 10.1016/j.bmc.2004.11.028
209. Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, et al. Pharmacological blockade of Asct2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med (2018) 24(2):194–202. doi: 10.1038/nm.4464
210. Colas C, Grewer C, Otte NJ, Gameiro A, Albers T, Singh K, et al. Ligand discovery for the alanine-Serine-Cysteine transporter (Asct2, Slc1a5) from homology modeling and virtual screening. PloS Comput Biol (2015) 11(10):e1004477. doi: 10.1371/journal.pcbi.1004477
211. Grewer C, Grabsch E. New inhibitors for the neutral amino acid transporter Asct2 reveal its na+-dependent anion leak. J Physiol (2004) 557(Pt 3):747–59. doi: 10.1113/jphysiol.2004.062521
212. Choi DW, Kim DK, Kanai Y, Wempe MF, Endou H, Kim JK. Jph203, a selective l-type amino acid transporter 1 inhibitor, induces mitochondria-dependent apoptosis in Saos2 human osteosarcoma cells. Korean J Physiol Pharmacol Off J Korean Physiol Soc Korean Soc Pharmacol (2017) 21(6):599–607. doi: 10.4196/kjpp.2017.21.6.599
213. Markowicz-Piasecka M, Huttunen J, Montaser A, Huttunen KM. Hemocompatible Lat1-inhibitor can induce apoptosis in cancer cells without affecting brain amino acid homeostasis. Apoptosis an Int J programmed Cell Death (2020) 25(5-6):426–40. doi: 10.1007/s10495-020-01603-7
214. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
215. Timmerman LA, Holton T, Yuneva M, Louie RJ, Padró M, Daemen A, et al. Glutamine sensitivity analysis identifies the xct antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell (2013) 24(4):450–65. doi: 10.1016/j.ccr.2013.08.020
216. Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell (2010) 18(3):207–19. doi: 10.1016/j.ccr.2010.08.009
217. Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-Phenylacetamido-1,2,4-Thiadiazol-2-Yl)Ethyl sulfide (Bptes). Biochem J (2007) 406(3):407–14. doi: 10.1042/bj20070039
218. Chang HS, Lin YJ, Lee SJ, Yang CW, Lin WY, Tsai IL, et al. Cytotoxic alkyl benzoquinones and alkyl phenols from ardisia virens. Phytochemistry (2009) 70(17-18):2064–71. doi: 10.1016/j.phytochem.2009.09.006
219. Li C, Li M, Chen P, Narayan S, Matschinsky FM, Bennett MJ, et al. Green tea polyphenols control dysregulated glutamate dehydrogenase in transgenic mice by hijacking the adp activation site. J Biol Chem (2011) 286(39):34164–74. doi: 10.1074/jbc.M111.268599
220. Magill GB, Myers WP, Reilly HC, Putnam RC, Magill JW, Sykes MP, et al. Pharmacological and initial therapeutic observations on 6-Diazo-5-Oxo-1-Norleucine (Don) in human neoplastic disease. Cancer (1957) 10(6):1138–50. doi: 10.1002/1097-0142(195711/12)10:6<1138::aid-cncr2820100608>3.0.co;2-k
221. Neil GL, Berger AE, Bhuyan BK, Blowers CL, Kuentzel SL. Studies of the biochemical pharmacology of the fermentation-derived antitumor agent, (Alpha s, 5s)-Alpha-Amino-3-Chloro-4,5-Dihydro-5-Isoxazoleacetic acid (at-125). Adv Enzyme Regul (1978) 17:375–98. doi: 10.1016/0065-2571(79)90023-2
222. Rais R, Jančařík A, Tenora L, Nedelcovych M, Alt J, Englert J, et al. Discovery of 6-Diazo-5-Oxo-L-Norleucine (Don) prodrugs with enhanced csf delivery in monkeys: A potential treatment for glioblastoma. J medicinal Chem (2016) 59(18):8621–33. doi: 10.1021/acs.jmedchem.6b01069
223. Butler M, van Ingen Schenau DS, Yu J, Jenni S, Dobay MP, Hagelaar R, et al. Btk inhibition sensitizes acute lymphoblastic leukemia to asparaginase by suppressing the amino acid response pathway. Blood (2021) 138(23):2383–95. doi: 10.1182/blood.2021011787
224. Mueller C, Al-Batran S, Jaeger E, Schmidt B, Bausch M, Unger C, et al. A phase iia study of pegylated glutaminase (Peg-pga) plus 6-Diazo-5-Oxo-L-Norleucine (Don) in patients with advanced refractory solid tumors. J Clin Oncol (2008) 26(15_suppl):2533. doi: 10.1200/jco.2008.26.15_suppl.2533
225. Darmaun D, Welch S, Rini A, Sager BK, Altomare A, Haymond MW. Phenylbutyrate-induced glutamine depletion in humans: Effect on leucine metabolism. Am J Physiol (1998) 274(5):E801–7. doi: 10.1152/ajpendo.1998.274.5.E801
226. Wang ES, Frankfurt O, Orford KW, Bennett M, Flinn IW, Maris M, et al. Phase 1 study of cb-839, a first-in-Class, orally administered small molecule inhibitor of glutaminase in patients with Relapsed/Refractory leukemia. Blood (2015) 126(23):2566–. doi: 10.1182/blood.V126.23.2566.2566
227. Vogl DT, Younes A, Stewart K, Orford KW, Bennett M, Siegel D, et al. Phase 1 study of cb-839, a first-in-Class, glutaminase inhibitor in patients with multiple myeloma and lymphoma. Blood (2015) 126(23):3059. doi: 10.1182/blood.V126.23.3059.3059
228. Harding JJ, Telli ML, Munster PN, Le MH, Molineaux C, Bennett MK, et al. Safety and tolerability of increasing doses of cb-839, a first-in-Class, orally administered small molecule inhibitor of glutaminase, in solid tumors. J Clin Oncol (2015) 33(15_suppl):2512. doi: 10.1200/jco.2015.33.15_suppl.2512
229. DeMichele A, Harding JJ, Telli ML, Munster PN, McKay R, Iliopoulos O, et al. Phase 1 study of cb-839, a small molecule inhibitor of glutaminase (Gls) in combination with paclitaxel (Pac) in patients (Pts) with triple negative breast cancer (Tnbc). J Clin Oncol (2016) 34(15_suppl):1011. doi: 10.1200/JCO.2016.34.15_suppl.1011
230. Tannir NM, Fan AC, Lee RJ, Carthon BC, Iliopoulos O, Mier JW, et al. Phase 1 study of glutaminase (Gls) inhibitor cb-839 combined with either everolimus (E) or cabozantinib (Cabo) in patients (Pts) with clear cell (Cc) and papillary (Pap) metastatic renal cell cancer (Mrcc). J Clin Oncol (2018) 36(6_suppl):603. doi: 10.1200/JCO.2018.36.6_suppl.603
231. Meric-Bernstam F, Lee RJ, Carthon BC, Iliopoulos O, Mier JW, Patel MR, et al. Cb-839, a glutaminase inhibitor, in combination with cabozantinib in patients with clear cell and papillary metastatic renal cell cancer (Mrcc): Results of a phase I study. J Clin Oncol (2019) 37(7_suppl):549. doi: 10.1200/JCO.2019.37.7_suppl.549
232. Meric-Bernstam F, Gordon M, Tykodi S, Lam E, Vaishampayan U, Chaves J, et al. Cx-839-004 a phase 1/2 study of cb-839, a first-in-Class glutaminase inhibitor, combined with nivolumab in patients with advanced melanoma (Mel), renal cell carcinoma (Rcc), or non-small cell lung cancer (Nsclc). Soc Immunotherapy Cancer Annu Meeting (2017). doi: 10.1016/S0959-8049(16)32626-0
233. Vidal G, Kalinsky K, Stringer-Reasor E, Lynce F, Cole J, Valdes-Albini F, et al. Abstract P6-20-07: Efficacy and safety of cb-839, a small molecule inhibitor of glutaminase, in combination with paclitaxel in patients with advanced triple negative breast cancer (Tnbc): Initial findings from a multicenter, open-label phase 2 study. Cancer Res (2019) 79(4_Supplement). doi: 10.1158/1538-7445.Sabcs18-p6-20-07
234. Motzer RJ, Lee CH, Emamekhoo H, Matrana M, Percent I, Hsieh JJ, et al. Lba54 - entrata: Randomized, double-blind, phase ii study of telaglenastat (Tela; cb-839) + everolimus (E) vs placebo (Pbo) + e in patients (Pts) with Advanced/Metastatic renal cell carcinoma (Mrcc). Ann Oncol (2019) 30:v889–v90. doi: 10.1093/annonc/mdz394.048
235. Ciombor KK, Whisenant J, Cardin DB, Goff LW, Das S, Schulte M, et al. Cb-839, panitumumab, and irinotecan in ras wildtype (Wt) metastatic colorectal cancer (Mcrc): Phase I results. J Clin Oncol (2019) 37(4_suppl):574. doi: 10.1200/JCO.2019.37.4_suppl.574
236. Eads JR, Krishnamurthi SS, Saltzman JN, Bajor DL, Vinayak S, Barnholtz-Sloan J, et al. Phase I clinical trial of the glutaminase inhibitor cb-839 plus capecitabine in patients with advanced solid tumors. J Clin Oncol (2018) 36(15_suppl):2562. doi: 10.1200/JCO.2018.36.15_suppl.2562
237. Guerra VA, Burger JA, Borthakur GM, Jabbour E, Pemmaraju N, Kadia TM, et al. Interim analysis of a phase ii study of the glutaminase inhibitor telaglenastat (Cb-839) in combination with azacitidine in advanced myelodysplastic syndrome (Mds). Blood (2019) 134:567. doi: 10.1182/blood-2019-125970
238. Saxena K, Konopleva M, Bhagat TD, Guerra VA, Maduike R, Tiziani S, et al. Aza + glutaminase inhibition with telaglenastat (Cb-839) for advanced mds: An updated interim analysis. Blood (2020) 136:31–2. doi: 10.1182/blood-2020-137266
239. Tannir NM, Agarwal N, Dawson NA, Motzer RJ, Jacobs CM, Choueiri TK, et al. Cantata: Randomized, international, double-blind study of cb-839 plus cabozantinib versus cabozantinib plus placebo in patients with metastatic renal cell carcinoma. J Clin Oncol (2019) 37(7_suppl):TPS682–TPS. doi: 10.1200/JCO.2019.37.7_suppl.TPS682
240. Tannir NM, Agarwal N, Porta C, Lawrence NJ, Motzer R, McGregor B, et al. Efficacy and safety of telaglenastat plus cabozantinib vs placebo plus cabozantinib in patients with advanced renal cell carcinoma: The cantata randomized clinical trial. JAMA Oncol (2022) 8(10):1411–8. doi: 10.1001/jamaoncol.2022.3511
241. Gonsalves WI, Neparidze N, Allred JB, Kumar SK, Reid JM, Shapiro G, et al. A multicenter phase I dose-escalation trial of a novel glutaminase inhibitor telaglenastat in combination with carfilzomib and dexamethasone in relapsed and refractory multiple myeloma. Blood (2022) 140:7315–6. doi: 10.1182/blood-2022-164966
242. Riess J, Frankel P, Massarelli E, Nieva J, Lai WCV, Koczywas M, et al. Ma13.08 a phase 1 trial of sapanisertib and telaglenastat (Cb-839) in patients with advanced nsclc (Nci 10327): Results from dose escalation. J Thorac Oncol (2022) 17(9, Supplement):S91–S2. doi: 10.1016/j.jtho.2022.07.153
243. Chan WK, Horvath TD, Tan L, Link T, Harutyunyan KG, Pontikos MA, et al. Glutaminase activity of l-asparaginase contributes to durable preclinical activity against acute lymphoblastic leukemia. Mol Cancer Ther (2019) 18(9):1587–92. doi: 10.1158/1535-7163.Mct-18-1329
244. Samudio I, Konopleva M. Asparaginase unveils glutamine-addicted aml. Blood (2013) 122(20):3398–400. doi: 10.1182/blood-2013-09-526392
245. Pieters R, Hunger SP, Boos J, Rizzari C, Silverman L, Baruchel A, et al. L-asparaginase treatment in acute lymphoblastic leukemia: A focus on erwinia asparaginase. Cancer (2011) 117(2):238–49. doi: 10.1002/cncr.25489
246. Wu M, Wang Q, Chen S, Zhou Z, Li J, Sun H, et al. Metabolic intervention liposome for targeting glutamine-addiction of breast cancer. J Controlled release Off J Controlled Release Soc (2022) 350:1–10. doi: 10.1016/j.jconrel.2022.07.034
247. Poonaki E, Nickel AC, Shafiee Ardestani M, Rademacher L, Kaul M, Apartsin E, et al. Cd133-functionalized gold nanoparticles as a carrier platform for telaglenastat (Cb-839) against tumor stem cells. Int J Mol Sci (2022) 23(10):5479–93. doi: 10.3390/ijms23105479
248. Byun JK, Park M, Lee S, Yun JW, Lee J, Kim JS, et al. Inhibition of glutamine utilization synergizes with immune checkpoint inhibitor to promote antitumor immunity. Mol Cell (2020) 80(4):592–606.e8. doi: 10.1016/j.molcel.2020.10.015
249. Li Q, Zhong X, Yao W, Yu J, Wang C, Li Z, et al. Inhibitor of glutamine metabolism V9302 promotes ros-induced autophagic degradation of B7h3 to enhance antitumor immunity. J Biol Chem (2022) 298(4):101753. doi: 10.1016/j.jbc.2022.101753
250. Wang JJ, Siu MK, Jiang YX, Leung TH, Chan DW, Wang HG, et al. A combination of glutaminase inhibitor 968 and pd-L1 blockade boosts the immune response against ovarian cancer. Biomolecules (2021) 11(12):1749–66. doi: 10.3390/biom11121749
251. Oh MH, Sun IH, Zhao L, Leone RD, Sun IM, Xu W, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest (2020) 130(7):3865–84. doi: 10.1172/jci131859
Keywords: glutamine metabolism, oncogene, glutamine addiction, metabolism inhibiton, cancer therapy
Citation: Ni R, Li Z, Li L, Peng D, Ming Y, Li L and Liu Y (2023) Rethinking glutamine metabolism and the regulation of glutamine addiction by oncogenes in cancer. Front. Oncol. 13:1143798. doi: 10.3389/fonc.2023.1143798
Received: 13 January 2023; Accepted: 24 February 2023;
Published: 07 March 2023.
Edited by:
Parmanand Malvi, University of Alabama at Birmingham, United StatesReviewed by:
Sanjay Pandey, Albert Einstein College of Medicine, United StatesCopyright © 2023 Ni, Li, Li, Peng, Ming, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Li, Y3FmeWxsQDE2My5jb20=; Yao Liu, c3dobGl1eWFvQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.