 Feng Lin1†
Feng Lin1† Alex Tuffour
Alex Tuffour Frank Addai Peprah
Frank Addai Peprah Aixia Huang
Aixia Huang Yang Zhou
Yang Zhou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 21 February 2023
Sec. Molecular and Cellular Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1141603
Hepcidin, a short peptide synthesized primarily by hepatocytes in response to increased body iron and inflammation, is a crucial iron-regulating factor. Hepcidin regulates intestinal iron absorption and releases iron from macrophages into plasma through a negative iron feedback mechanism. The discovery of hepcidin inspired a torrent of research into iron metabolism and related problems, which have radically altered our understanding of human diseases caused by an excess of iron, an iron deficiency, or an iron disparity. It is critical to decipher how tumor cells manage hepcidin expression for their metabolic requirements because iron is necessary for cell survival, particularly for highly active cells like tumor cells. Studies show that tumor and non-tumor cells express and control hepcidin differently. These variations should be explored to produce potential novel cancer treatments. The ability to regulate hepcidin expression to deprive cancer cells of iron may be a new weapon against cancer cells.
Iron is an element that is necessary for the survival of all living things. Iron is essential for producing enzymes and proteins that catalyze reactions, including cellular respiration, oxygen and electron transport, and DNA synthesis (1). Humans have developed unique transport systems and membrane transporters to preserve iron in a free condition conducive to blood circulation and iron transfer across cell membranes. On the other hand, an excessive amount of soluble redox-active iron might be harmful to cells. Consequently, cells have to carefully adjust the concentration of iron inside their cells to satisfy their metabolic requirements while staying below the level that would cause toxicity. The pro-oxidative effects of iron make excessive iron more harmful, mostly linked to several disorders such as diabetes, cardiovascular disease, liver cirrhosis, cancer and the like (2, 3). Because cancer cells have high iron requirements, iron depletion has been examined for its therapeutic potential. In addition, evidence suggests that modifying one’s iron metabolism may be a viable technique for preventing and treating cancer (4). Iron’s unique importance makes its metabolic and regulatory mechanisms crucial, mainly its physiological and pathological functions. To explain the interplay between anatomically different locations of iron intake, reuse, and use, the idea of controlling iron hormones was put out. In this article, we expound on the differential regulation of hepcidin as a primary regulator of iron balance and elucidate its mechanism in cancer development and its potential therapeutic modalities.
Within the human body, the hormone hepcidin acts as the primary regulator of iron homeostasis. Hepcidin is a long-chain peptide comprised of 25 amino acids (5). Hepcidin induces ferroportin internalization and degradation to control iron absorption and recycling (6). The generation of hepcidin by hepatocytes is significantly boosted by inflammation and iron excess (7). Until 2001, when animal studies showed that iron loading induced hepatic hepcidin mRNA synthesis, there was no evidence that it played a functional role in iron metabolism (8). Hepcidin was discovered in human urine and given the name hepcidin because of its synthesis site (hep-) and antibacterial characteristics in vitro (-cidin). In human urine, the most common type is 25 amino acids. However, there are also shorter 22 and 20 amino acid peptides. The core peptide has an easy hairpin structure with a ladder-like orientation, with eight cysteine residues connected by four disulfide bridges. This structure resembles other antimicrobial peptides and is typical of peptides that can damage bacterial membranes (9). This structure is comparable to other antimicrobial peptides and is typical of peptides capable of damaging bacterial membranes (10).
Hepcidin production increases when iron levels are high, but iron released from enterocytes and macrophages decreases. This is in accordance with the hepcidin model, which states that the plasma level of hepcidin mostly governs the rate of iron efflux into the plasma. Likewise, hepcidin synthesis is suppressed as iron levels fall, and these cells produce more iron (11). Hepcidin’s discovery sparked a flood of research into iron metabolism and related problems, fundamentally altering our understanding of human diseases (non-cancerous) caused by excess iron, iron deficiency, or iron disparity (12) (Table 1). Cancer and iron homeostasis have been shown to interact occasionally for a substantial period. However, it has only been in recent years that interesting factors into the mechanisms of normal iron regulation have facilitated focused probing of fundamental mechanisms, biological rationale, and pathophysiologic repercussions of alteration in iron metabolism in cancer. There is compelling evidence for the prudent control and distribution of iron in cells and tissues. The finding of the plasma membrane protein ferroportin and the secreted liver protein hepcidin marked the beginning of an essential new era in investigating the relationship between normal iron biology and diseases. As part of the natural process of maintaining iron homeostasis, hepcidin reacts to changes in iron levels and inflammation.
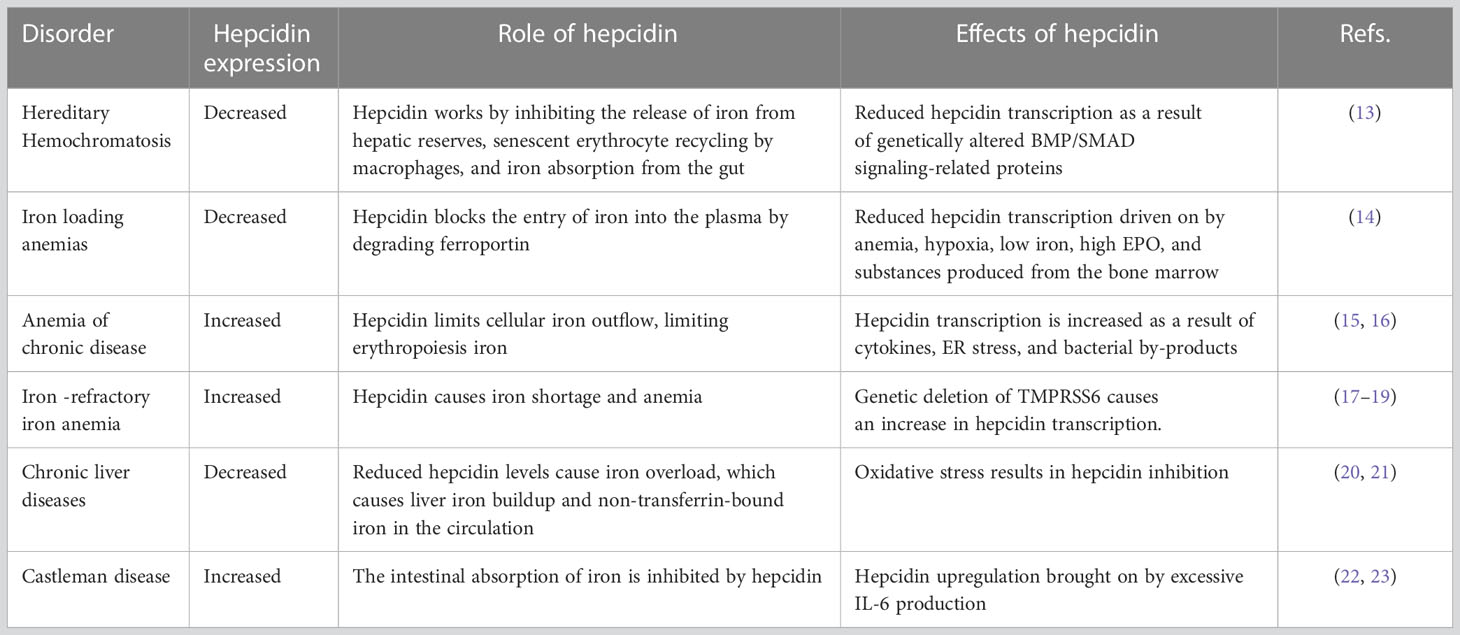
Table 1 Effect of hepcidin on iron metabolism and related disorders.
Inflammatory cytokines like IL-6, bacterial infections, and lipopolysaccharide (LPS) cause transcriptional induction in the hepcidin gene. This connection between inflammatory pathways and hepcidin has a molecular basis for the pathophysiology of chronic illness anemia, a prevalent and, up until now inadequately comprehended side effect of many common medical disorders, including cancer (Figure 1). Hepcidin expression in a tumor may be primarily regulated by external and intracellular iron. The results go against the previously established theory for iron homeostasis in cancer cells since cancer cells need to retain iron to proliferate and ingest more iron for development, even though the mechanism causing the decrease of hepcidin mRNA expression in a tumor is still unknown.
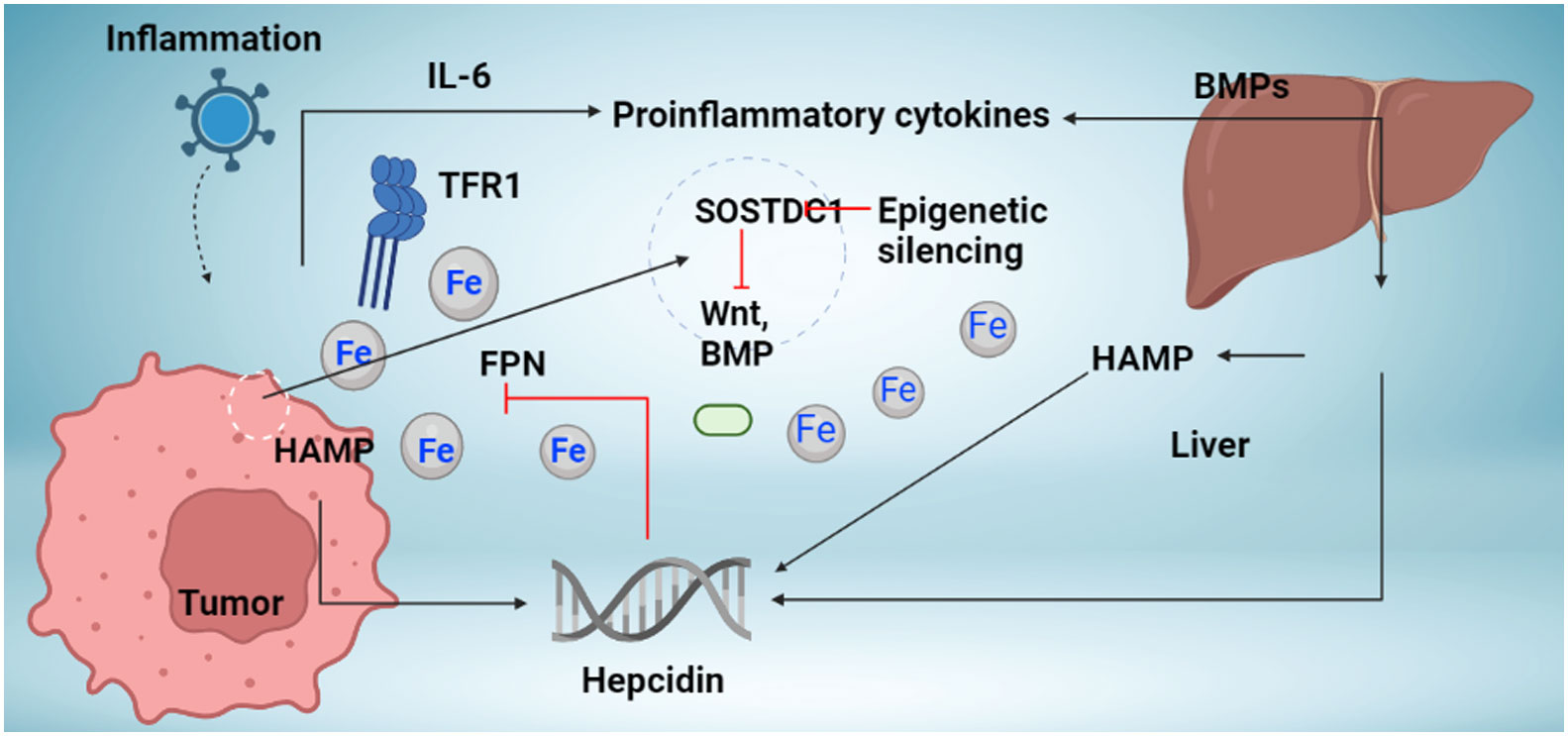
Figure 1 Mechanism regulation hepcidin synthesis in cancer. The liver and local tumor cells in cancer tissue produce hepcidin. Different bone morphogenetic protein (BMP) molecules, including BMP7, and inflammatory triggers, like interleukin-6, are associated with hepcidin overexpression in tumor tissue (IL-6). Studies indicate the presence of novel hepcidin regulators in cancer tissue, including the Wnt pathway and the sclerostin domain-containing protein 1 (SOSTDC1), which is downregulated in cancer due to epigenetic silencing (which is upregulated in cancer). Increased hepcidin in the tumor microenvironment causes ferroportin to behave in a way that causes iron sequestration in tumor cells (FPN). TFR1 overexpression, which increases iron availability to tumor cells, occurs concurrently with an increase in hepcidin. Increased iron depots aid tumor cells in surviving and proliferating.
The BMP-mothers against the decapentaplegic homolog (SMAD) pathway is essential for controlling iron homeostasis because it controls hepcidin production by modulating transcription (24, 25). Hepcidin signaling, which happens in reaction to increased iron accumulation in the tissue, depends on the proteins BMP2 and BMP6, produced by sinusoidal endothelial cells in the liver. (Figure 2) (26, 27). In mice, BMP6 and SMAD 1/5/8 phosphorylation activity were associated with hepcidin mRNA expression, which was regulated by iron status (24). Moreover, it was discovered that BMP6 is a positive regulator for the expression of hepcidin in reaction to iron status and that the lack of BMP6 led to an excess of iron in the body (28). It was finally discovered that BMP6 could actively bind to hemojuvelin (HJV). This protein is found on the membrane of hepatocyte cells and is a component of the iron sensing complex together with the hemochromatosis gene (HFE) and transferrin receptor 2 (TFR2) (25, 28). Activin receptor-like kinase 3 (ALK3) and ALK2 and BMPR2, perhaps in heterodimers with HJV, appear to bind to a receptor complex composed of BMP 2 and 6, which are produced in response to iron. This binding results in SMAD 1/5/8 phosphorylation, which then causes SMAD4 to form a complex with SMAD1. The complex enters the nucleus and attaches to promoters, increasing the production of hepcidin mRNA. (Figure 3) (25, 29). However, HJV is rendered inactive by being converted to its soluble form by a serine protease encoded by matriptase-2 (TMPRSS6), which also lowers the transcriptional level of hepcidin and prevents this positive pathway for hepcidin (25, 30). The idea is that BMP2 and 6 are secreted by liver sinusoidal endothelial cells in response to tissue iron overload, whereas TFR1 and TFR2, along with HFE, are the detectors for circulating iron stores estimated through transferrin-iron saturation (27); Others have asserted that BMP2 helps monitor circulating iron while BMP6 serves as a sensor for storing iron.
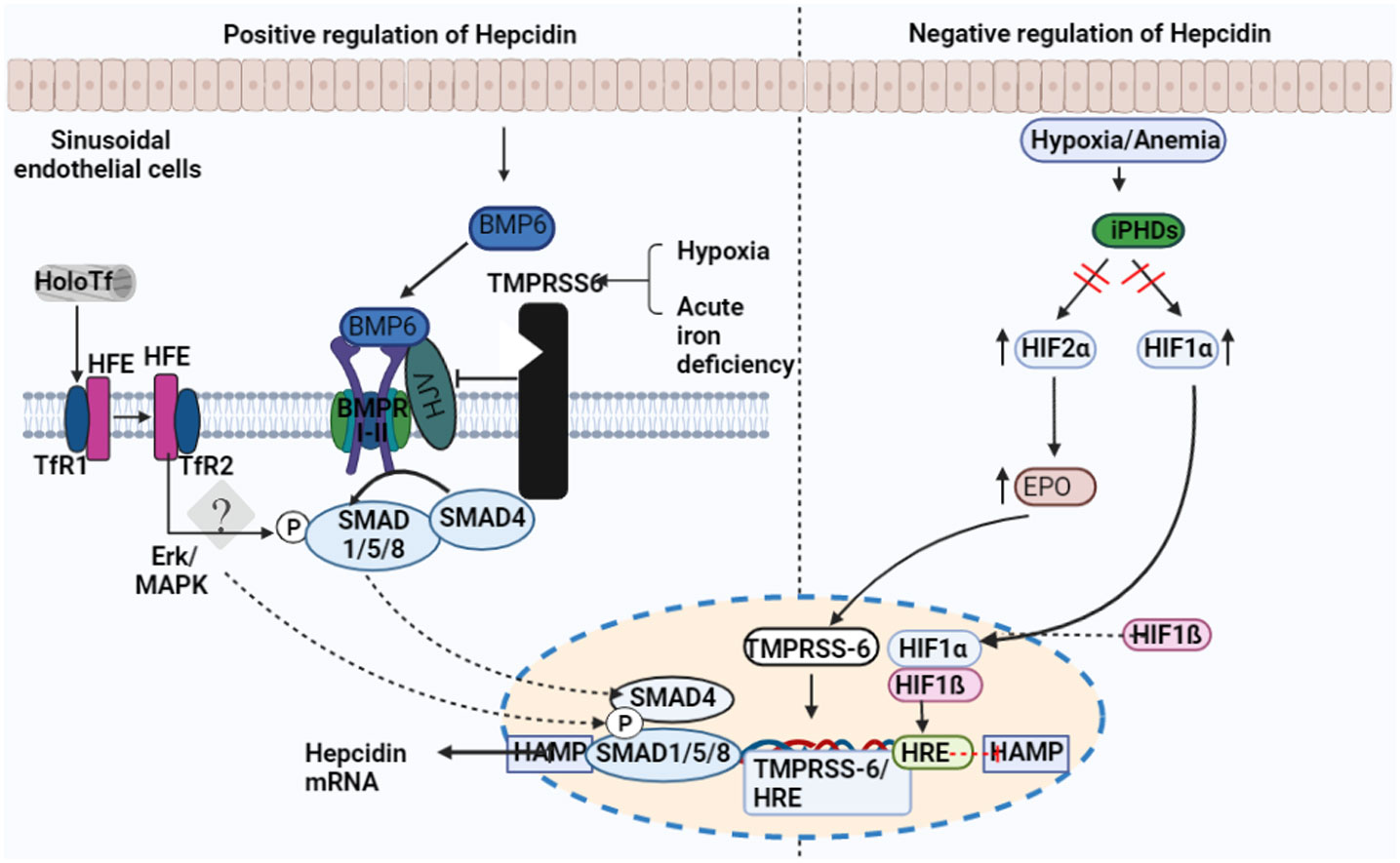
Figure 2 Factors regulating the expression of hepcidin. Transferrin receptor-1 binds Diferric (Holo) transferrin (TfR1). TfR1 induces HFE to interact with cell-surface-stabilized TfR2. Hepcidin transcription is regulated by the interaction between HFE and TfR2. Sinusoid endothelial cells (SEC) and other non-parenchymal cells produce BMP6 when intracellular iron is present. In order to activate BMP6, hemojuvelin (HJV) interacts with BMPRI-II, a receptor for BMP type I and II. The development of the hepatocyte multiprotein complex promotes the phosphorylation of SMAD1/5/8 and their interaction with SMAD4. The nucleus is where SMAD enters to activate hepcidin. The transmembrane serine protease TMPRSS6, which cleaves membrane HJV and suppresses hepcidin mRNA, is made more active by hypoxia and acute iron deficiency.

Figure 3 Schematic representation of therapeutic treatments targeting hepcidin. Hepcidin antagonists (Pink). Mediators that inhibit hepcidin expression, anti-hepcidin antibodies, or substances that interfere with the interaction between hepcidin and ferroportin. Hepcidin agonists (Blue) Hepcidin analogues and other agents that boost expression of the protein hepcidin.
TFRs are known to be involved in the expression of hepcidin. The binding of transferrin and internalization of iron that has been coupled to transferrin is carried out by a class of proteins known as TFR (31). Transferrin is coupled to either monoferric or diferric iron that is absorbed by duodenal enterocytes (32). A cell-surface protein called TFR1 facilitates the uptake of iron through receptor-mediated endocytosis. According to recent studies, iron loading may also impact TFR1’s capacity to influence hepatocytes’ ability to express hepcidin (26). Canali et al. determined that hepatocyte-specific ablation of TFR1 resulted in increased hepcidin compared to the liver’s iron level (26). According to their model, the iron deficit increased TFR1, which bound to HFE and inhibited activation of the HFE-TFR2 complex, which prevented the production of hepcidin from being induced (26, 33). TFR2 is similar to TFR1, except it is produced by hepatocytes and is only found in the liver (34). Although some evidence suggests that HFE’s effects on hepcidin expression may be independent of TFR2, it is widely acknowledged that the interaction between TFR1, TFR2, and HFE is crucial for hepcidin expression in response to circulating iron reserves (34, 35). As previously mentioned, the formation of a multiprotein complex by the HFE-TFR2 complex, which activates SMAD 1/5/8 via phosphorylation by HJV/BMP, leads to an increase in hepcidin synthesis (HFETFR2-HJV) (36–38).
Erythropoiesis is a significant physiological mechanism contributing to the downregulation of hepcidin expression (25, 29). The hormone erythroferrone (ERFE), which is released by erythroblasts in reaction to erythropoietin (EPO), appears to be connected to the mechanism because it directly inhibits hepcidin in the liver (Figure 2) (39). ERFE mediates hepcidin suppression caused by anemia, erythropoiesis, and blood loss; the postulated mechanism is ERFE binding to BMP ligands (39, 40). According to studies, patients with beta-thalassemia and iron deficiency anemia had higher ERFE levels (41, 42). ERFE is a crucial hormone that directly affects the liver to decrease hepcidin expression in reaction to erythropoietic stimulation, according to a significant body of evidence. This is consistent with a logical physiological. However, additional variables are likely to play, such as decreased iron storage due to increased erythropoietic requirement in the bone marrow. Mechanism to increase iron absorption and release when the body needs to increase erythropoiesis. Hypoxia-inducible factors (HIFs) are transcription factors that control how cells react to low oxygen levels (29, 43). Under hypoxic conditions, HIF undergoes post-translational alteration, which results in its translocation to the nucleus and subsequent binding to a hypoxia-responsive element (43). However, it has also been proposed that erythropoiesis-induced activation of ERFE and platelet-derived growth factor-BB and HIF-1 binding to the TMPRSS-6 promoter, may indirectly mediate the effect of hypoxia on hepcidin production (Figure 2) (43, 44).
Hepcidin regulation has been linked to several hormones and growth factors. For example, it is commonly known that male hemochromatosis patients have higher body iron reserves than female ones (27, 45). In addition, other variables, such as testosterone-mediated inhibition of hepcidin synthesis, have been implicated, even though the physiological loss of blood that occurs during menstruation will almost surely have a role (46).
In one study, it was hypothesized that 17 β-estradiol would increase hepcidin expression in HepG2 cells. Also, it was demonstrated that 17 β-estradiol would decrease hepcidin production in human liver HuH7 and HepG2 cells (47). According to reports, progesterone increases hepcidin expression via progesterone receptor membrane component-1 (PGRMC1) (48). The hepatocyte growth factor and epidermal growth factor, which work via the BMP-SMAD pathway, are additional growth factors that have been shown to inhibit hepcidin (49, 50). Hepcidin production in response to infection may be facilitated by activating the toll-like receptor 4 (TLR4) in macrophages (29). Moreover, c-Jun NH2-terminal kinases (JNK) bind to the hepcidin promoter due to LPS-induced TLR4 activation in hepatocytes. TLR4 may also stimulate nuclear factor kappa B (NFkB) signaling, a proposed mechanism by which alcohol inhibits the formation of hepcidin and may explain the elevated hepatic iron reserves typically observed in patients with alcohol-associated liver disease (51, 52).
Furthermore, hypoxia is another factor that has been studied to have a suppressing effect on hepcidin expression. The response to hypoxia is regulated by transcription factors called HIFs (43). Due to posttranslational alteration under hypoxic settings, HIF is translocated to the nucleus and binds to hypoxia-responsive components (43). Despite the possibility that the effect of hypoxia on hepcidin expression may be tangentially induced by activation of ERFE and platelet-derived growth factor-BB as a result of erythropoiesis and by HIF-1 binding to the TMPRSS-6 promoter, hepcidin expression is suppressed in the liver by HIF-2 activation through EPO and possibly through direct binding of HIF-1 to the hepcidin promoter (43, 53). Lastly, furin, a processing enzyme often present in the trans-Golgi network, is another mechanism by which hepcidin production is suppressed during hypoxia. Furin breaks down HJV into its soluble form, which then causes BMP6 to be sequestered and hepcidin expression to be suppressed (54).
Anomalies in iron homeostasis triggered by mutations cause cancer in cells or tissues. Hepcidin, a negative regulator of FPN1 is activated in a number of cancers including breast, colon, and prostate (55–57). Hepcidin dysregulation causes iron homeostasis to be disrupted in cancer (Table 2). Iron mobilization from enterocytes and macrophages into the circulation is inhibited by high plasma hepcidin levels (contributing to cancer anemia). It can lead to iron buildup in tumor cells by degrading FPN-1, which activates signaling pathways including Wnt (61) and NF-β (62), that promote tumor growth (63). Furthermore, hepcidin has been proven in several experiments to increase tumor growth. According to a study by Schwartz et al., compared to their wild-type littermates, mice lacking hepcidin in the colonic tumor epithelium significantly decreased the number, burden, and size of tumors in a sporadic model of colorectal cancer. On the other hand, mice lacking FPN1 developed intracellular iron buildup and increased the likelihood of tumor development (64). SOSTDC1, a BMP4/7 antagonist also regulates hepcidin synthesis, providing a new path for cellular iron deficiency in the thyroid gland through the E4BP4/G9a/SOSTDC1/hepcidin pathway, which can be reduced hepcidin secretion and thyroid cancer cell proliferation (57). Systemic iron anomalies in cancer patients have been documented in addition to the iron effectors produced by cancer cells (65–67). High serum hepcidin levels are found in several diseases, including prostate cancer, multiple myeloma, breast cancer and non-lymphoma Hodgkin’s disease (68, 69). Cancer cells frequently increase iron input while inhibiting outflow, resulting in iron buildup. However, how they would react to the increased iron instability is unclear.

Table 2 Hepcidin role in cancer regulation.
The correlation between hepatic iron buildup and hepatocarcinogenesis has been evident over the past decade because of growing evidence involving molecules controlling iron metabolism or iron-related cell death processes like ferroptosis. Therefore, it is critical to understand the liver’s role in iron homeostasis to understand the association between iron and the development of hepatocellular carcinoma (HCC). First, the liver maintains iron homeostasis by releasing iron into circulation for metabolic needs. Additionally, the liver produces proteins that keep iron equilibrium in the body and also store excess iron (70). Numerous iron-driven pathways can cause HCC or carcinogenesis generally. The most significant is the production of reactive oxygen species (ROS) and the ensuing oxidative stress. The impact of “free” iron, which is present when the iron-binding capabilities of plasma transferrin or intracellular iron-storage protein ferritin are exceeded, has led to the theory that oxidative stress is the fundamental cause of hepatotoxicity under iron-excess circumstances. The toxicity of iron in “Fenton-type” reactions, in which iron promotes the transformation of passive H2O2 into extremely reactive HO, is explained by its redox-active characteristics (71). The iron hormone hepcidin, which upholds systemic iron homeostasis, has a role in HCC pathogenesis. HCC patients may have low amounts of hepcidin, in contrast to tumors with higher hepcidin expression. To find molecular targets for diagnosis, prognosis, and treatments, it is therefore, of enormous clinical benefit to examine the regulation and function of hepcidin in HCC. About 90% of all primary liver cancers are hepatocellular carcinomas (HCC). It is an advanced liver disease and one of the leading causes of cancer-related deaths (72).
In response to tissue iron overload and inflammation, BMP6 and IL6 are two of the main stimulators of hepcidin synthesis in hepatocytes (73). The major cell signaling pathway that controls hepatic hepcidin production in response to iron is activated by the BMPs, which act as ligands (74). BMP6 is the potent and prevalent hepcidin inducer in response to elevated iron. Other BMPs, such as BMP9, BMP4, and BMP2, can activate the transcription of hepcidin in primary human hepatocytes, mouse models, and HCC cell lines (75). Notably, BMPs 6 and 4 are upregulated in HCC patients’ livers, and BMP4 and BMP9 are both triggered by hypoxia in HCC tissues where they are highly expressed (76). Therefore, it would seem plausible to anticipate that HCC would express hepcidin more highly due to the high levels of these BMPs. Nevertheless, this contrasts with what is seen in HCC, which mostly has reduced hepcidin expression (77).
Similarly, IL6 is another intriguing factor that deviates from the usual in the setting of hepcidin in HCC. Hepcidin is categorized as an acute-phase protein and is the primary mediator of the acute-phase response, which IL6 mediates (78). Hepcidin is typically brought on by inflammation, specifically IL6 through the JAK-STAT3 pathway and the non-canonical BMP pathway. Higher serum IL6 levels are associated with a higher risk of developing HCC, and higher serum IL6 levels were found in HCC patients. Similarly, the JAK/STAT pathway is constitutively active in HCC, which promotes tumor proliferation, invasion, and metastasis (79). The hepcidin gene itself being downregulated may be one factor causing low levels of hepcidin in HCC. The DNA on the HAMP promoter region is more heavily methylated in HCC tissues. This reduces the production of hepcidin in HCC by suppressing HAMP transcription (80). Interestingly, this downregulation happens despite normal serum iron levels (131.4 ± 23.4 mg/dL) and normal or high ferritin levels (414.4 (328.2-1121) ng/mL) in some HCC patients, as reported in one study (81) or elevated levels of iron, ferritin, and transferrin saturation in the sera of HCC patients compared to control patients in another study (76). Shen et al. demonstrated that the knockdown of HAMP elevated cellular iron levels, migratory cell ability, and proliferation in human liver cancer cell lines. The first discovery was confirmed in mice, where tumor weights were larger in groups with lower Hamp expression than in controls (82).
In summary, hepcidin downregulation can exacerbate HCC pathophysiology and promote cancer growth. One of the HCC diagnostic markers could be hepcidin. Hepcidin is presented as a promising prognostic marker due to its downregulation, which predicts poor patient outcomes. Hepcidin regulation may be a means of improving current HCC therapy approaches and changing HCC pathophysiology.
Breast cancer is the most prevalent form of cancer affecting women. More than 2.26 million women were diagnosed with breast cancer in 2020. One of the most crucial objectives of biomedical research is to understand the molecular pathways involved in breast cancer incidence and development due to the intricacy of breast cancer studies and their life-threatening relevance (83). Hepcidin significantly influences the growth of breast tumors. Although ferroportin expression is downregulated in breast cancer, its expression in hepcidin is elevated (55). Hepcidin released by breast cancer cells binds to ferroportin, which causes ferroportin breakdown by preventing iron outflow and increasing iron retention (55, 84). In an attempt to decipher the mechanisms that control hepcidin expression in breast cancer cells, Blanchette-Farra et al. presented a new model for tumor-mediated regulation of iron via hepcidin by tumor architecture and the microenvironment of breast tumors (83). The authors investigated the potential function of cancer-associated fibroblasts in controlling breast tumor pathways by iron and hepcidin using a 3D cell culture technique. Accordingly, Blanchette-Farra et al. indicated that multiple molecules from the TGF-β superfamily, including BMP and growth differentiation factor-15 (GDF-15), were responsible for the metabolism of hepcidin in breast cancer. They emphasize that GDF-15 expression was substantially correlated with hepcidin levels in patient samples, both at the mRNA and protein levels, indicating a function for GDF-15 in controlling hepcidin in breast tumors (83). Regarding the relationship between the expression of hepcidin and BMPs, they noted that BMPs, specifically BMP6, were crucial mediators for synthesizing hepcidin in breast cancer cells. Additionally, the proliferation of tumor-associated fibroblasts in breast cancer spheroids causes IL-6, one of the most significant pro-inflammatory cytokines to be secreted, further promoting hepcidin (83). More importantly, to reinforce Blanchette’s findings, another study from a clinical standpoint revealed that the combined expression profile of hepcidin and ferroportin serves as a potent predictor for survival following mastectomy for women with breast cancer (55).
In men, prostate cancer (PCa) is a form of malignancy with a high incidence of morbidity and mortality (85). Studies have indicated that prostate cancer cells metastasis, proliferation and angiogenesis have all been linked to intracellular iron excess (60). A prostate cancer-specific tumor biomarker is the prostate-specific antigen (PSA), while an indicator of intracellular iron concentration and storage is the soluble transferrin receptor (sTfR). Hepcidin expression can influence PSA and sTfR levels in prostate cancer cells, which can then influence the proliferation of tumor cells (39). Following these considerations, Wang et al. explored how cell proliferation, migration, and apoptosis are related to hepcidin and iron metabolism in prostate cancer cells (86). They assessed prostate cancer cell lines, subjecting them to overexpression and low expression groups, and determined PSA, sTfR, and ferroportin levels. They showed that prostate cancer cells have high hepcidin levels, which regulate cell growth, migration, and death by enhancing intracellular iron transportation (86).
Moreover, the case for iron dysmetabolism in prostate cancer has lately gained further support from a growing body of studies. It is distinguished by various iron-related proteins expressed differently from normal cells (87). Similar to other malignancies, PCa cells require iron to survive. Iron is also required for the activity of enzymes that regulate the transcriptional activity of the androgen receptor, a known PCa promoter (88). Also, iron is required by PCa cells to “reconfigure” internal enzyme activity to boost energy production and extracellular matrix disintegration (89). According to clinical data, iron sequestration in PCa cancer cells is elevated, although it is low in normal cells next to PCa cells (89). Research has shown that senescent prostate cells have higher iron loads due to altered protein expression that controls cellular iron transport (90). Interestingly and incredibly similar to PCa cells, iron dysregulation is seen in experimental models with senescent prostate epithelial cells. It is characterized by the upregulation of TFR1, IRP2, and ferritin, while FPN is upregulated but predominantly localized intracellularly, preventing it from taking part in iron export (90). Senescent cells experience these alterations due to compromised ferritinophagy, which results in increased iron absorption into ferritin. Senescent cells would receive this signal as a deficiency of intracellular iron, enhancing iron import through activation of TFR1. Ferritinophagy is induced chemically to inhibit this process (90). The need for ferritinophagy and its impact on PCa is crucial in investigating this potential disturbance. Tesfay et al. also agree that it increases intracellular iron retention and promotes the survival of prostate cancer cells. Prostatic hepcidin acts as an autocrine hormone by reducing the iron exporter ferroportin on the cell surface (60). To investigate the expression of hepcidin in regulating ferroportin in prostrate cells, they tested expression in prostate cancer cells and non-malignant prostate epithelial cells and also examined cancer cells whose proliferation is either sensitive or insensitive to androgen. They also evaluated the role of prostatic hepcidin in regulating ferroportin in an autocrine fashion.
A significant finding from their work confirms that hepcidin is produced by normal prostate cells and that hepcidin synthesis is significantly increased in prostate cancer cells and tissue. In detail, hepcidin affects both healthy and cancerous prostate cells significantly and functions in an autocrine manner. It enhances metabolically accessible iron, decreases ferroportin levels and maintains viability. Since hepcidin activity is both a source and a local target in prostate cells, this autocrine regulatory axis may support healthy prostate biology. Additionally, these findings show that local synthesis of hepcidin by peripheral tissues also has significant impacts, even though hepcidin has primarily been researched to regulate systemic iron intake and recycling (60).
Research has shown that a distinct regulatory cascade regulates hepcidin synthesis in prostate cancer. A unique confluence of pathways, including BMP4/7, IL6, Wnt, and the dual BMP and Wnt antagonist SOSTDC1, regulates the synthesis of hepcidin in prostate cancer (60, 91). Furthermore, prostate cancer patients exhibit a faster rate of disease progression due to enhanced epigenetic methylation-mediated silencing of SOSTDC1 (60). The findings imply a new connection between iron metabolism and prostate cancer, and prostatic dysregulation of hepcidin encourages the development and progression of prostate cancer.
Lung cancer is the second most prevalent cancer and the main factor in cancer-related deaths globally (92). Several malignant malignancies, particularly lung cancer, are closely associated with iron dysregulation at both the onset and growth stages. There is growing evidence that iron plays a significant role in lung cancer. Iron is also significant for creating potential lung cancer treatment plans because it participates prominently in numerous types of cell death. IL-6 is said to be increased in lung cancer patients and linked to lung carcinogenesis and poor patient survival (93, 94). It is known to upregulate hepcidin, which reduces iron efflux from cells and results in cancer-related anemia. According to reports, 88% and 62% of non-small cell lung cancer (NSCLC) patients have high ferritin levels and TFR1 expression. In individuals with NSCLC and small-cell lung cancer (SCLC), the serum ferritin level was shown to be increased (95). Research shows that EGFR regulates iron homeostasis by redistributing TFR1, which boosts cellular iron import and encourages the growth of lung cancer. In NSCLC, the expression of membrane TFR1 and iron levels are significantly linked with EGFR activation (96).
Given the close relationship between iron regulation and its influence on lung cancer development, hepcidin evaluation is the key factor to a broader understanding to decipher the mechanism involved. Therefore, fan et al. looked into how hepcidin is engaged in lung cancer metastasis and immune infiltration to understand better its involvement in this process and its molecular regulation (97). They used the Tumor Immune Estimation Resource (TIMER) online database to analyze the mRNA expression of hepcidin in a number of human cancers, and they discovered increased expressions of hepcidin in colon adenocarcinoma, invasive breast carcinoma, esophageal carcinoma, and other cancer cell types compared to their matched normal tissues.
They also discovered that hepcidin mRNA was expressed more strongly in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) tissues than in healthy lung tissues, according to analyses from the gene expression profiling interactive analysis (GEPIA) and UALCAN databases, a web-based tool that provides in-depth analyses of transcriptome data from The Cancer Genome Atlas (TCGA) and MET500 data (97). They specifically found that hepcidin expression was significantly higher in lung cancer tissues when compared to non-tumor tissues. These findings confirmed an earlier study and revealed that hepcidin might act as an oncogene by promoting lung cancer development and spread (98). The authors then considered the hepcidin gene’s prognostic potential because the level of hepcidin expression is tightly linked to lung cancer growth and metastasis. Following the Kaplan-Meier plotter database, they discovered lung cancer patients with increased hepcidin gene expression had worse general and progression-free survival but not post-progression survival (97). This suggests that hepcidin is closely linked to the prognosis of lung cancer. The scientists also examined the link between hepcidin expression and six other invading immune cells, such as CD4+T cells, CD8+T cells, dendritic cells, neutrophils, and macrophages, to understand better how these immune cells are related to hepcidin expression. A high positive correlation was found between B cells, CD4+T cells, macrophages, neutrophils, and dendritic cells but not between hepcidin expression levels and CD8+T cells in LUAD (97). Additionally, LUSC immune cell infiltration by all six different kinds was positively and strongly correlated with hepcidin expression. These findings highlight hepcidin’s prominent role in carcinogenesis and suggest that hepcidin may be crucial in controlling immune cell infiltration in lung cancer (97).
The kidney is the organ responsible for the reabsorption of iron. In essence, iron that is both transferrin-bound and non-transferrin-bound is capable of entering the glomerular filtrate. The tubular epithelia reabsorb a significant portion of this iron. Numerous transporters, such as the multiligand megalin-cubilin receptor complex, transferrin receptor 1, divalent metal transporter 1, zinc transporter ZIP8, and zinc transporter ZIP14, have been connected to this reuptake (73, 97, 99). In a study, it was noted that mice lacking megalin had significantly higher levels of urine hepcidin without concurrent glomerular filtration impairment (100). Van Swelm et al. validated the megalin-induced reabsorption of hepcidin in the proximal tubules. However, their research suggests that some of the primary hepcidin isoform, hepcidin-25, is degraded into hepcidin-22 and hepcidin-20, which can maintain some of hepcidin-25’s pharmacological effects (101).
Hepcidin expression in the kidney is relatively low compared to the liver, which is known to be the ultimate source of hepcidin (8, 102). Kulaksiz et al. claim that renal hepcidin is primarily found in the connecting tubules and the cortical thick ascending limb of nephron tubules (103). Also, Veuthey et al. hypothesized that the proximal tubule produces prohepcidin. However, it should be noted that the authors of this study measured the immunohistochemical expression of this peptide rather than its mRNA expression, which may indicate that the peptide’s presence inside proximal tubular cells is caused by absorption from the blood filtrate, which contains the peptide’s systemic form (103, 104). However, hepcidin expression is not restricted to tubular cells, as there is evidence that it can also be found in the leukocytes of the renal interstitium when the kidney is inflamed (105). IL-6 and interferon-alpha may cause interstitial renal leukocytes to increase the expression of hepcidin, with interferon-alpha having a more substantial effect on renal monocytes than hepatocytes (106).
Additionally, it has been demonstrated that increased oxidative stress caused by acute kidney injury (AKI) results in an upregulation of renal hepcidin expression (107). Studies have also shown that hepcidin may impact renal iron metabolism separately from ferroportin (101, 108). On the other hand, the BMP6/SMAD pathway may stimulate renal hepcidin, much as it was possible with liver hepcidin (60). Finally, it is worth noting that the ablation of renal BMP6 is associated with gradual renal injury spurred on by a considerable rise in iron load and oxidative stress (109).
Having looked at the expression and regulation of hepcidin in the kidney, it is essential to see how its impact influences cancer development in the kidney. Renal cell carcinoma (RCC) is the most frequent malignant kidney tumor and the second most prevalent urological malignancy (110). Therefore, Kamai et al., interested in investigating whether hepcidin is involved in renal cell carcinoma, examined serum hepcidin-25 and compared hepcidin mRNA expression between RCC tissues and non-neoplastic tissues from the same resected specimens. They further examined the possibility of using hepcidin to assess patients with RCC’s prognosis (111). In their findings, they realized that compared to individuals without metastatic tumors, those with metastatic RCC had serum levels of hepcidin-25 that were higher. Also, hepcidin mRNA expression was higher in metastatic RCCs compared to non-metastatic RCCs. They concluded that poorer overall survival in our RCC patients was associated with increased tumor expression of hepcidin mRNA when assessing patients’ prognoses (111). This study demonstrated that an increased serum level of hepcidin-25 is a marker for the metastatic condition. In contrast, an increased amount of hepcidin mRNA in tumor tissue is linked to the tumor progression of RCC. Similarly, Traeger et al. investigated the viability of serum hepcidin in the quest to find a suitable biomarker to correlate patient’s survival in urinary tract urothelial carcinomas (UUTUC) and RCC as a potential biomarker in UUTUC and RCC (59). They also concluded that serum levels of hepcidin were associated with metastases in both UUTUC and RCC, making serum hepcidin a prognostic marker in RCC and UUTUC (59).
Brain tumor cells have also been discovered to regulate hepcidin distinctively. Some tumors have low local hepcidin levels, whereas in others, the concentrations are similar to those of healthy brain tissue (112). Distinct cell types in the brain regulate iron metabolism differently. For instance, tumor stem cells in glioblastoma extract iron from the surrounding tissue more efficiently than other tumor cells (113). It is also possible that the distinctive hepcidin regulation in brain tumors is due to the significant roles played by pericytes and glial cells in the modulation of hepcidin in brain tissue (114). In any event, similar to how it functions in other tissues, hepcidin found in brain tissue can inhibit FPN (115). Additionally, it has been demonstrated that this hepcidin-induced action in neurons and vascular endothelial cells is associated with the downregulation of TFR1 and the lowering of transferrin-bound iron from the periphery into the brain (115, 116). Additionally, astrocytes’ intriguing phenomenon of hepcidin-induced downregulation of TFR1 was reported (117). This is crucial because TFR1 overexpression promotes the survival of brain cancer cells. Since there are contradictory data about the effect of hepcidin in lowering the iron burden in brain cells, how these hepcidin-induced effects might influence the brain’s iron metabolism is currently unknown (115). Because studies have demonstrated that reducing the iron availability to brain tumor cells might have significant antitumor effects, it is crucial to understand how hepcidin affects the iron metabolism of these tumors (118, 119).
More consideration must be given to measuring hepcidin levels in cancer patients. The dynamics between local and systemic hepcidin levels are probably more important than the absolute serum hepcidin levels. Plasma iron concentrations are primarily regulated by hepcidin. A study by Rivera et al. indicates that within 1 hour after hepcidin injection, mice’s serum iron levels dropped significantly (120). Although hepcidin is quickly degraded from the plasma, the effects of a single dose were felt for up to 72 h. This is likely because it takes time to synthesize enough ferroportin (the hepcidin receptor) again.
Several transcription-inhibition techniques are now being tested to reduce superfluous hepcidin synthesis. Similarly, by disrupting the interaction between hepcidin and ferroportin, hepcidin function can be suppressed (Figure 3) (121). Most models suggest that relative to healthy neighboring tissue (122), cancer tissues express more hepcidin and less FPN (60, 84). A high hepcidin/low FPN model associated with high TFR1 expression provides additional evidence that enhanced iron supply is the norm in cancer tissue, promoting cellular proliferation in cancer tissues (58, 122). Cancer cell growth is inhibited by suppressing liver hepcidin, and a similar effect can be obtained by knocking out tumor hepcidin (84). Anti-HJV antibodies can stop hepcidin expression from providing a comparable outcome (123). Anti-hepcidin antibodies may be used as part of a local anti-hepcidin strategy to inhibit the activity of the local hepcidin. Anti-hepcidin antibodies treat prostate cancer to restore FPN expression and stop cancer growth. A different tactic would involve preventing local hepcidin from acting on FPN in cancer cells (60). This might be accomplished by transforming cancer cells’ FPN genes, resulting in an FPN mutant that is resistant to hepcidin’s activities. Hepcidin’s use as a therapeutic target has historically coincided with the production of hepatic hepcidin agonists and antagonists. One strategy to combat hepcidin is to use RNA interference-causing agents like short interfering RNA (124, 125). Hepcidin expression in the liver has been demonstrated to be blocked by small interfering RNAs. Still, no hepcidin-specific small interfering RNA has yet been created that has been shown to be effective in human trials (124, 125). However, some hepcidin antibodies, such as LY2787106, have already been tested in human clinical research to manage cancer-related anemia. Although the preliminary findings indicated that patients tolerated the medication well, the observed efficacy of this treatment was regrettably transient. This was likely because of the homeostatic reparative output of hepcidin, which should be considered when blocking liver hepcidin with particular antibodies (126). Inhibiting the activity of hepcidin expression regulators like BMP molecules, including BMPR, HJV, and MT2 is another aspect of hepcidin antagonism (Figure 3) (124).
Additionally, pharmacotherapeutic drugs may be used to manipulate hepcidin. For example, iron-chelation therapy has been proven to improve the anticancer effects of medications like sorafenib (127). Interestingly, sorafenib can inhibit the Ras/MAPK pathway, leading to the induction of hepcidin expression. However, it is yet unknown whether this means that sorafenib’s prohepcidin action enhances its anticancer effects in hepatocellular carcinoma (HCC) (128).
Heparins effectively decrease the expression of hepcidin by acting on the BMP/SMAD pathway (125, 128). Furthermore, heparins can bind a variety of BMPs, which explains how they can decrease hepcidin even in BMP6 knockout models (125, 128, 129). Chemically synthesized heparins with non-anticoagulant characteristics, including SST0001, have demonstrated effectiveness in multiple myeloma (MM) and sarcoma mice by suppressing tumor development alone or in combination with conventional chemotherapy (130). However, the processes underlying SST001’s anticancer effects are complex, and it is currently unclear if they are connected to hepcidin suppression.
The only accepted rationale for the use of hepcidin treatments in cancer up until this point has been anemia of cancer (124, 125). High blood hepcidin levels characterize this disease, which also has low hemoglobin and serum iron levels. The primary source of iron for plasma is hepcidin, which elevates FPN downregulation and cellular iron sequestration in enterocytes and macrophages (123, 126, 131). Hepcidin is the main supply of iron for plasma. The ability of local or systemic hepcidin treatment to affect cancer cell proliferation is uncertain.
Although iron is required for the proper functioning of cellular metabolism, it can also produce active oxygen because it is a redox-active metal., which has the potential to be hazardous. As a result, the amount of attention dedicated to investigating iron homeostasis in healthy cells and anomalies in iron metabolism in cancer has increased dramatically over the past few years. While the levels of numerous proteins and the activity of many enzymes are altered in most tumors, cancer cells generally maintain iron metabolism pathways similar to those of normal cells. This shows that altering iron metabolism is essential to maintaining tumor cells.
In cancer cells, elevated iron levels promote the activation of iron-dependent proteins, guard against the adverse effects of excess iron, and achieve “adjusted iron homeostasis” in sync with tumor metabolism. Although the particular mechanism is still not entirely understood, there are still some unanswered problems, such as whether the iron metabolism of different tumor cell types is consistent. How can iron levels be monitored in a way that selectively damages tumor cells while protecting healthy cells?
An essential peptide for maintaining cellular iron homeostasis is hepcidin. This is significant because the amount of iron deposited influences the cellular redox state. One of the harmful factors connected to several diseases is oxidative. Therefore, it should not be surprising that hepcidin dysregulation has been linked to conditions like liver cirrhosis diabetes, and heart disease. A hepcidin imbalance may also be common in cancer, although the involvement of hepcidin in cancer has received less attention. Hepcidin levels are generally higher in cancers, except for HCC and some brain tumors, where they are lower. Hepcidin dysregulation in cancer is significant because it gives tumors the iron they need to survive. The precise linkages between activated leukocytes and tumor cells in terms of the interplay of iron metabolism in the cancer environment will need to be addressed in studies on hepcidin aberrations in cancer. Additionally, research should clarify the precise mechanisms by which malignant cells manipulate the expression of hepcidin and other iron proteins to attack non-cancerous cells. In some malignancies, preventing the activity of local hepcidin can slow tumor growth. For instance, in breast cancer inhibiting liver hepcidin along with local hepcidin suppression has been demonstrated to inhibit tumor proliferation. Hepcidin suppression in MM would be more successful by direct actions on hepcidin, as evidenced by the inconsistent outcomes of blocking hepcidin in MM using anti-IL-6 antibodies. Although these results are quite encouraging, similar research from other tumors will help us understand the function of hepcidin treatment in cancer.
When blocking liver hepcidin, we must exercise caution because doing so may have unintended consequences. Hepcidin levels are low in other malignancies, such as HCC; thus, the anticancer approach aims to augment the levels of hepcidin. The situation is more complicated with brain cancer since many brain tumors have low hepcidin levels, except that this observation does not apply to all brain cancers. Hepcidin expression is also regulated differently by various types of brain cells. The findings that suggest that hepcidin therapy may be significant as a safeguard for brain tissue seem to be compatible with the minuscule quantities of hepcidin found in brain tumors. The ability to stop the formation of brain tumors through hepcidin modification is still an open subject.
FL and AT wrote the original manuscript. YZ and HZ conceptualized the idea. GH, FP, and AH reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
This project was funded by the Zhejiang Science and Technology Major Program on Aquatic New Variety Breeding (2021C02069-8), the Key R&D Program of Zhejiang (2022C02028) and the major Project of Science and Technology of Zhejiang Province (2018C02019).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mleczko-Sanecka K, Silvestri L. Cell-type-specific insights into iron regulatory processes. Am J Hematol (2021) 96:110–27. doi: 10.1002/ajh.26001
2. Pantopoulos K. Inherited disorders of iron overload. Front Nutr (2018) 5:103. doi: 10.3389/fnut.2018.00103
3. Asperti M, Cantamessa L, Ghidinelli S, Gryzik M, Denardo A, Giacomini A, et al. The antitumor didox acts as an iron chelator in hepatocellular carcinoma cells. Pharmaceuticals (2019) 12:129. doi: 10.3390/ph12030129
4. Morales M, Xue X. Targeting iron metabolism in cancer therapy. Theranostics (2021) 11:8412–29. doi: 10.7150/thno.59092
5. Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta - Mol Cell Res (2012) 1823:1434–43. doi: 10.1016/J.BBAMCR.2012.01.014
6. Billesbølle CB, Azumaya CM, Kretsch RC, Powers AS, Gonen S, Schneider S, et al. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature (2020) 586:807. doi: 10.1038/S41586-020-2668-Z
7. Xu Y, Alfaro-Magallanes VM, Babitt JL. Physiological and pathophysiological mechanisms of hepcidin regulation: Clinical implications for iron disorders. Br J Haematology (2021) 193:882–93. doi: 10.1111/BJH.17252
8. Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem (2001) 276:7811–19. doi: 10.1074/JBC.M008923200
9. Daher R, Lefebvre T, Puy H, Karim Z. Extrahepatic hepcidin production: The intriguing outcomes of recent years. World J Clin cases (2019) 7:1926–26. doi: 10.12998/WJCC.V7.I15.1926
10. Agarwal AK, Yee J. Hepcidin. Adv Chronic Kidney Dis (2019) 26:298–305. doi: 10.1053/J.ACKD.2019.04.005
12. Vela D, Vela-Gaxha Z. Differential regulation of hepcidin in cancer and non-cancer tissues and its clinical implications. Exp Mol Med (2018) 50:e436. doi: 10.1038/EMM.2017.273
13. Gupta R, Musallam KM, Taher AT, Rivella S. Ineffective erythropoiesis: anemia and iron overload. Hematology/Oncology Clinics North America (2018) 32:213–21. doi: 10.1016/J.HOC.2017.11.009
14. Murray SC, Serra Barros A, Brown DA, Dudek P, Ayling J. Mellor j. a pre-initiation complex at the 3'-end of genes drives antisense transcription independent of divergent sense transcription. Nucleic Acids Res (2012) 40:2432–44. doi: 10.1093/nar/gkr1121
15. Theurl I, Aigner E, Theurl M, Nairz M, Seifert M, Schroll A, et al. Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: Diagnostic and therapeutic implications. Blood (2009) 113:5277–86. doi: 10.1182/BLOOD-2008-12-195651
16. Finberg KE, Heeney MM, Campagna DR, Aydinok Y, Pearson HA, Hartman KR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat Genet (2008) 40:569–71. doi: 10.1038/NG.130
17. Cui Y, Wu Q, Zhou Y. Iron-refractory iron deficiency anemia: New molecular mechanisms. Kidney Int (2009) 76:1137–41. doi: 10.1038/KI.2009.357
18. Al-Jamea LH, Woodman A, Heiba NM, Elshazly SA, Khalaf NB, Fathallah DM, et al. Genetic analysis of TMPRSS6 gene in Saudi female patients with iron deficiency anemia. Hematology/Oncology Stem Cell Ther (2021) 14:41–50. doi: 10.1016/J.HEMONC.2020.04.007
19. Vela D. Low hepcidin in liver fibrosis and cirrhosis; a tale of progressive disorder and a case for a new biochemical marker. Mol Med (2018) 24:5. doi: 10.1186/S10020-018-0008-7
20. Han CY, Koo JH, Kim SH, Gardenghi S, Rivella S, Strnad P, et al. Hepcidin inhibits Smad3 phosphorylation in hepatic stellate cells by impeding ferroportin-mediated regulation of akt. Nat Commun (2016) 7:13817. doi: 10.1038/ncomms13817
21. Arlet JB, Hermine O, Darnige L, Ostland V, Westerman M, Badoual C, et al. Iron-deficiency anemia in castleman disease: Implication of the interleukin 6/hepcidin pathway. Pediatrics (2010) 126:e1608–12. doi: 10.1542/peds.2010-1123
22. Song SNJ, Tomosugi N, Kawabata H, Ishikawa T, Nishikawa T, Yoshizaki K. Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric castleman disease. Blood (2010) 116:3627–34. doi: 10.1182/BLOOD-2010-03-271791
23. Joachim JH, Mehta KJ. Hepcidin in hepatocellular carcinoma. Br J Cancer (2022) 127:185–92. doi: 10.1038/s41416-022-01753-2
24. Li Y, Miller I, Prasad P, George NA, Parrow NL, Fleming RE. Effects of exogenous transferrin on the regulation of iron metabolism and erythropoiesis in iron deficiency with or without anemia. Front Physiol (2022) 13:893149. doi: 10.3389/fphys.2022.893149
25. Xiao X, Alfaro-Magallanes VM, Babitt JL. Bone morphogenic proteins in iron homeostasis. Bone (2020) 138:115495. doi: 10.1016/j.bone.2020.115495
26. Canali S, Zumbrennen-Bullough KB, Core AB, Wang CY, Nairz M, Bouley R, et al. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood (2017) 129:405–14. doi: 10.1182/blood-2016-06-721571
27. Parrow NL, Fleming RE. Liver sinusoidal endothelial cells as iron sensors. Blood (2017) 129:397–98. doi: 10.1182/BLOOD-2016-12-754499
28. Radhakrishnan K, Kim YH, Jung YS, Kim J, Kim DK, Cho SJ, et al. Orphan nuclear receptor ERRgamma is a novel transcriptional regulator of IL-6 mediated hepatic BMP6 gene expression in mice. Int J Mol Sci (2020) 21:7148. doi: 10.3390/ijms21197148
29. Rishi G, Subramaniam VN. Signaling pathways regulating hepcidin. Vitamins Hormones (2019) 110:47–70. doi: 10.1016/BS.VH.2019.01.003
30. Piperno A, Pelucchi S, Mariani R. Inherited iron overload disorders. Trans Gastroenterol Hepatol (2020) 5:25. doi: 10.21037/TGH.2019.11.15
31. Hamdi-Rozé H, Ben Ali Z, Ropert M, Detivaud L, Aggoune S, Simon D, et al. Variable expressivity of HJV related hemochromatosis: "Juvenile" hemochromatosis? Blood Cells Molecules Dis (2019) 74:30–3. doi: 10.1016/J.BCMD.2018.10.006
32. Anderson GJ, Frazer DM. Current understanding of iron homeostasis. Am J Clin Nutr (2017) 106:1559S–66S. doi: 10.3945/AJCN.117.155804
33. Gammella E, Buratti P, Cairo G, Recalcati S. The transferrin receptor: The cellular iron gate. Metallomics (2017) 9:1367–75. doi: 10.1039/C7MT00143F
34. Fillebeen C, Charlebois E, Wagner J, Katsarou A, Mui J, Vali H, et al. Transferrin receptor 1 controls systemic iron homeostasis by fine-tuning hepcidin expression to hepatocellular iron load. Blood (2019) 133:344–55. doi: 10.1182/BLOOD-2018-05-850404
35. Rishi G, Crampton EM, Wallace DF, Subramaniam VN. In situ proximity ligation assays indicate that hemochromatosis proteins hfe and transferrin receptor 2 (Tfr2) do not interact. PloS One (2013) 8:e77267. doi: 10.1371/JOURNAL.PONE.0077267
36. Andriopoulos B, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet (2009) 41:482–87. doi: 10.1038/NG.335
37. Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet (2009) 41:478–81. doi: 10.1038/NG.320
38. Corradini E, Schmidt PJ, Meynard D, Garuti C, Montosi G, Chen S, et al. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in hfe knockout mice. Gastroenterology (2010) 139:1721–29. doi: 10.1053/j.gastro.2010.07.044
39. Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Author correction: Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet (2020) 52:463. doi: 10.1038/S41588-019-0548-Y
40. Ganz T, Jung G, Naeim A, Ginzburg Y, Pakbaz Z, Walter PB, et al. Immunoassay for human serum erythroferrone. Blood (2017) 130:1243–46. doi: 10.1182/BLOOD-2017-04-777987
41. Xu LH, Zhang Y, Wang Y, Hu D, Xu JH. Hepcidin and erythroferrone levels in child-bearing women with iron deficiency anemia. Zhongguo Shi Yan xue Ye Xue Za Zhi (2021) 29:213–16. doi: 10.19746/j.cnki.issn.1009-2137.2021.01.034
42. El Gendy FM, El-Hawy MA, Shehata AMF, Osheba HE. Erythroferrone and iron status parameters levels in pediatric patients with iron deficiency anemia. Eur J Haematology (2018) 100:356–60. doi: 10.1111/EJH.13021
43. Renassia C, Peyssonnaux C. New insights into the links between hypoxia and iron homeostasis. Curr Opin Hematol (2019) 26:125–30. doi: 10.1097/MOH.0000000000000494
44. Ravasi G, Pelucchi S, Buoli Comani G, Greni F, Mariani R, Pelloni I, et al. Hepcidin regulation in a mouse model of acute hypoxia. Eur J Haematology (2018) 100:636–43. doi: 10.1111/EJH.13062
45. Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet (2016) 388:706–16. doi: 10.1016/S0140-6736(15)01315-X
46. Guo W, Schmidt PJ, Fleming MD, Bhasin S. Hepcidin is not essential for mediating testosterone's effects on erythropoiesis. Andrology (2020) 8:82–90. doi: 10.1111/andr.12622
47. Ikeda Y, Tajima S, Izawa-Ishizawa Y, Kihira Y, Ishizawa K, Tomita S, et al. Estrogen regulates hepcidin expression via GPR30-BMP6-dependent signaling in hepatocytes. PloS One (2012) 7:e40465. doi: 10.1371/JOURNAL.PONE.0040465
48. Li X, K.Rhee D, Malhotra R, Mayeur C, Hurst LA, Ager E, et al. Progesterone receptor membrane component-1 regulates hepcidin biosynthesis. J Clin Invest (2016) 126:389–401. doi: 10.1172/JCI83831
49. Goodnough JB, Ramos E, Nemeth E, Ganz T. Inhibition of hepcidin transcription by growth factors. Hepatology (2012) 56:291–91. doi: 10.1002/HEP.25615
50. Silvestri L, Nai A, Dulja A, Pagani A. Hepcidin and the BMP-SMAD pathway: An unexpected liaison. Vitamin hormones (2019) 110:71–99. doi: 10.1016/bs.vh.2019.01.004
51. Lee P, Peng H, Gelbart T, Wang L, Beutler E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc Natl Acad Sci U.S.A. (2005) 102:1906–10. doi: 10.1073/PNAS.0409808102
52. Zinatizadeh MR, Schock B, Chalbatani GM, Zarandi PK, Jalali SA, Miri SR. The nuclear factor kappa b (NF-kB) signaling in cancer development and immune diseases. Genes Dis (2021) 8:287–97. doi: 10.1016/j.gendis.2020.06.005
53. Kowdley KV, Gochanour EM, Sundaram V, Shah RA, Handa P. Hepcidin signaling in health and disease: ironing out the details. Hepatol Commun (2021) 5:723–35. doi: 10.1002/HEP4.1717
54. Silvestri L, Pagani A, Camaschella C. Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood (2008) 111:924–31. doi: 10.1182/BLOOD-2007-07-100677
55. Pinnix ZK, Miller LD, Wang W, D'Agostino R, Kute T, Willingham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Trans Med (2010) 2:43ra56. doi: 10.1126/SCISIGNAL.3001127
56. Sornjai W, Nguyen Van Long F, Pion N, Pasquer A, Saurin JC, Marcel V, et al. Iron and hepcidin mediate human colorectal cancer cell growth. Chemico-Biological Interact (2020) 319. doi: 10.1016/J.CBI.2020.109021
57. Zhou Q, Chen J, Feng J, Wang J. E4BP4 promotes thyroid cancer proliferation by modulating iron homeostasis through repression of hepcidin. Cell Death Dis (2018) 9:987. doi: 10.1038/s41419-018-1001-3
58. Kovac S, Böser P, Cui Y, Ferring-Appel D, Casarrubea D, Huang L, et al. Anti-hemojuvelin antibody corrects anemia caused by inappropriately high hepcidin levels. Haematologica (2016) 101:e173–e76. doi: 10.3324/HAEMATOL.2015.140772
59. Hänninen MM, Haapasalo J, Haapasalo H, Fleming RE, Britton RS, Bacon BR, et al. Expression of iron-related genes in human brain and brain tumors. BMC Neurosci (2009) 10:36. doi: 10.1186/1471-2202-10-36
60. Wang J, Liu W, Bai R, Li JC, Li M, Zhu R. Hepcidin downregulation correlates with disease aggressiveness and immune infiltration in liver cancers. Frontier Oncol (2021) 11:714756. doi: 10.3389/FONC.2021.714756
61. Brookes MJ, Boult J, Roberts K, Cooper BT, Hotchin NA, Matthews G, et al. A role for iron in wnt signalling. Oncogene (2008) 27:966–75. doi: 10.1038/SJ.ONC.1210711
62. Xiong S, She H, Takeuchi H, Han B, Engelhardt JF, Barton CH, et al. Signaling role of intracellular iron in NF-κb activation. J Biol Chem (2003) 278:17646–54. doi: 10.1074/JBC.M210905200
63. Chua ACG, Klopcic B, Lawrance IC, Olynyk JK, Trinder D. Iron: An emerging factor in colorectal carcinogenesis. World J Gastroenterol (2010) 16:663–72. doi: 10.3748/WJG.V16.I6.663
64. Schwartz AJ, Goyert JW, Solanki S, Kerk SA, Chen B, Castillo C, et al. Hepcidin sequesters iron to sustain nucleotide metabolism and mitochondrial function in colorectal cancer epithelial cells. Nat Metab (2021) 3:969–82. doi: 10.1038/S42255-021-00406-7
65. Guo Q, Li L, Hou S, Yuan Z, Li C, Zhang W, et al. The role of iron in cancer progression. Front Oncol (2021) 11:778492. doi: 10.3389/fonc.2021.778492
66. Abdel-Razeq H, Hashem H. Recent update in the pathogenesis and treatment of chemotherapy and cancer induced anemia. Crit Rev Oncology/Hematology (2020) 145:102837. doi: 10.1016/j.critrevonc.2019.102837
67. Torti SV, Torti FM. Iron and cancer: 2020 vision. Cancer Res (2020) 80:5435–48. doi: 10.1158/0008-5472.CAN-20-2017
68. Zhao B, Li R, Cheng G, Li Z, Zhang Z, Li J, et al. Role of hepcidin and iron metabolism in the onset of prostate cancer. Oncol Lett (2018) 15:9953–58. doi: 10.3892/OL.2018.8544
69. El-Mahdy RI, Zakhary MM, Maximous DW, Mokhtar AA, El Dosoky MI. Circulating osteocyte-related biomarkers (vitamin d, sclerostin, dickkopf-1), hepcidin, and oxidative stress markers in early breast cancer: Their impact in disease progression and outcome. J Steroid Biochem Mol Biol (2020) 204:105773. doi: 10.1016/j.jsbmb.2020.105773
70. Hino K, Yanatori I, Hara Y, Nishina S. Iron and liver cancer: An inseparable connection. FEBS J (2022) 289:7810–29. doi: 10.1111/febs.16208
71. Kim E, Viatour P. Hepatocellular carcinoma: Old friends and new tricks. Exp Mol Medicin (2020) 52:1898–907. doi: 10.1038/s12276-020-00527-1
72. Nemeth E, Ganz T. Hepcidin-ferroportin interaction controls systemic iron homeostasis. Int J Mol Sci (2021) 22:6493. doi: 10.3390/ijms22126493
73. Rishi G, Wallace DF, Subramaniam VN. Hepcidin: Regulation of the master iron regulator. Bioscience Rep (2015) 35:e00192. doi: 10.1042/BSR20150014
74. Truksa J, Peng H, Lee P, Beutler E. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc Natl Acad Sci U.S.A. (2006) 103:10289–93. doi: 10.1073/pnas.0603124103
75. Maegdefrau U, Arndt S, Kivorski G, Hellerbrand C, Bosserhoff AK. Downregulation of hemojuvelin prevents inhibitory effects of bone morphogenetic proteins on iron metabolism in hepatocellular carcinoma. Lab Invest (2011) 91:1615–23. doi: 10.1038/labinvest.2011.123
76. Maegdefrau U, Amann T, Winklmeier A, Braig S, Schubert T, Weiss TS, et al. Bone morphogenetic protein 4 is induced in hepatocellular carcinoma by hypoxia and promotes tumour progression. J Pathol (2009) 218:520–9. doi: 10.1002/path.2563
77. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood (2003) 101:2461–3. doi: 10.1182/blood-2002-10-3235
78. Hin Tang JJ, Hao Thng DK, Lim JJ, Toh TB. JAK/STAT signaling in hepatocellular carcinoma. Hepat Oncol (2020) 7:HEP18. doi: 10.2217/hep-2020-0001
79. Udali S, Castagna A, Corbella M, Ruzzenente A, Moruzzi S, Mazzi F, et al. Hepcidin and DNA promoter methylation in hepatocellular carcinoma. Eur J Clin Invest (2018) 48:e12870. doi: 10.1111/eci.12870
80. Kijima H, Sawada T, Tomosugi N, Kubota K. Expression of hepcidin mRNA is uniformly suppressed in hepatocellular carcinoma. BMC Cancer (2008) 8:167. doi: 10.1186/1471-2407-8-167
81. Shen Y, Li X, Su Y, Badshah SA, Zhang B, Xue Y, et al. HAMP downregulation contributes to aggressive hepatocellular carcinoma via mechanism mediated by Cyclin4-dependent kinase-1/STAT3 pathway. Diagnostics (2019) 9:48. doi: 10.3390/diagnostics9020048
82. Blanchette-Farra N, Kita D, Konstorum A, Tesfay L, Lemler D, Hegde P, et al. Contribution of three-dimensional architecture and tumor-associated fibroblasts to hepcidin regulation in breast cancer. Oncogene 2018 37:29 (2018) 37:4013–32. doi: 10.1038/s41388-018-0243-y
83. Zhang S, Chen Y, Guo W, Yuan L, Zhang D, Xu Y, et al. Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell Signalling (2014) 26:2539–50. doi: 10.1016/J.CELLSIG.2014.07.029
84. Tadayon F, Arezegar HR, Khorrami MH, Hashemi Juzdani R, Shahdoost AA, Mellat M. Evaluation of prostatic cancer prevalence in patients with prostatic-specific antigen between 4 and 10 and normal digital rectal examination. Advanced Biomed Res (2016) 5:112. doi: 10.4103/2277-9175.184298
85. Tesfay L, Clausen KA, Kim JW, Hegde P, Wang X, Miller LD, et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res (2015) 75:2254–63. doi: 10.1158/0008-5472.CAN-14-2465
86. De Berardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv (2016) 2:e1600200. doi: 10.1126/sciadv.1600200
87. Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci U.S.A. (2007) 104:19226–31. doi: 10.1073/PNAS.0700735104
88. Ornstein DL, Zacharski LR. Iron stimulates urokinase plasminogen activator expression and activates NF-kappa b in human prostate cancer cells. Nutr Cancer (2007) 58:115–26. doi: 10.1080/01635580701308265
89. Masaldan S, Clatworthy SAS, Gamell C, Meggyesy PM, Rigopoulos AT, Haupt S, et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol (2018) 14:100–15. doi: 10.1016/J.REDOX.2017.08.015
90. Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci (2020) 21:1–47. doi: 10.3390/IJMS21124507
91. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA: A Cancer J Clin (2022) 72:7–33. doi: 10.3322/CAAC.21708
92. Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest (2007) 117:3846–56. doi: 10.1172/JCI31871
93. Brooks GD, McLeod L, Alhayyani S, Miller A, Russell PA, Ferlin W, et al. IL6 trans-signaling promotes KRAS-driven lung carcinogenesis. Cancer Res (2016) 76:866–76. doi: 10.1158/0008-5472.CAN-15-2388
94. Yildirim A, Meral M, Kaynar H, Polat H, Ucar EY. Relationship between serum levels of some acute-phase proteins and stage of disease and performance status in patients with lung cancer. Med Sci Monitor (2007) 13:195–200.
95. Wang B, Zhang J, Song F, Tian M, Shi B, Jiang H, et al. EGFR regulates iron homeostasis to promote cancer growth through redistribution of transferrin receptor 1. Cancer Lett (2016) 381:331–40. doi: 10.1016/J.CANLET.2016.08.006
96. Fan Y, Liu B, Chen F, Song Z, Han B, Meng Y, et al. Hepcidin upregulation in lung cancer: A potential therapeutic target associated with immune infiltration. Front Immunol (2021) 12:612144/FULL. doi: 10.3389/FIMMU.2021.612144/FULL
97. Chen Q, Wang L, Ma Y, Wu X, Jin L, Yu F. Increased hepcidin expression in non-small cell lung cancer tissue and serum is associated with clinical stage. Thorac Cancer (2014) 5:14. doi: 10.1111/1759-7714.12046
98. Mohammad G, Matakidou A, Robbins PA, Lakhal-Littleton S. The kidney hepcidin/ferroportin axis controls iron reabsorption and determines the magnitude of kidney and systemic iron overload. Kidney Int (2021) 100:559–69. doi: 10.1016/J.KINT.2021.04.034
99. Peters HPE, Laarakkers CMM, Pickkers P, Masereeuw R, Boerman OC, Eek A, et al. Tubular reabsorption and local production of urine hepcidin-25. BMC Nephrol (2013) 14:70. doi: 10.1186/1471-2369-14-70
100. Van Swelm RPL, Wetzels JFM, Verweij VGM, Laarakkers CMM, Pertijs JCLM, van der Wijst J, et al. Renal handling of circulating and renal-synthesized hepcidin and its protective effects against hemoglobin-mediated kidney injury. J Am Soc Nephrol (2016) 27:2720–32. doi: 10.1681/ASN.2015040461
101. Krause A, Neitz S, Mägert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett (2000) 480:147–50. doi: 10.1016/S0014-5793(00)01920-7
102. Kulaksiz H, Theilig F, Bachmann S, Gehrke SG, Rost D, Janetzko A, et al. The iron-regulatory peptide hormone hepcidin: Expression and cellular localization in the mammalian kidney. J Endocrinol (2005) 184:361–70. doi: 10.1677/JOE.1.05729
103. Vela D. Systemic and local hepcidin as emerging and important peptides in renal homeostasis and pathology. BioFactors (2019) 45:118–34. doi: 10.1002/BIOF.1468
104. Rovin BH, Zhang X. Biomarkers for lupus nephritis: The quest continues. Clin J Am Soc Nephrol (2009) 4:1858–65. doi: 10.2215/CJN.03530509
105. Zhang X, Rovin BH. Hepcidin expression by human monocytes in response to adhesion and pro-inflammatory cytokines. Biochim Biophys Acta (2010) 1800:1262–67. doi: 10.1016/J.BBAGEN.2010.08.005
106. Scindia Y, Dey P, Thirunagari A, Liping H, Rosin DL, Floris M, et al. Hepcidin mitigates renal ischemia-reperfusion injury by modulating systemic iron homeostasis. J Am Soc Nephrol (2015) 26:2800–14. doi: 10.1681/ASN.2014101037
107. Van Swelm RPL, Vos M, Verhoeven F, Thévenod F, Swinkels DW. Endogenous hepcidin synthesis protects the distal nephron against hemin and hemoglobin mediated necroptosis. Cell Death Dis (2018) 9:550. doi: 10.1038/S41419-018-0568-Z
108. Dendooven A, Van Oostrom O, van der Giezen DM, Leeuwis JW, Snijckers C, Joles JA, et al. Loss of endogenous bone morphogenetic protein-6 aggravates renal fibrosis. Am J Pathol (2011) 178:1069–79. doi: 10.1016/J.AJPATH.2010.12.005
109. Huang S, Luo Q, Huang J, Wei J, Wang S, Hong C, et al. A cluster of metabolic-related genes serve as potential prognostic biomarkers for renal cell carcinoma. Front Genet (2022) 13:902064. doi: 10.3389/FGENE.2022.902064
110. Kamai T, Tomosugi N, Abe H, Arai K, Yoshida KI. Increased serum hepcidin-25 level and increased tumor expression of hepcidin mRNA are associated with metastasis of renal cell carcinoma. BMC Cancer (2009) 9:270. doi: 10.1186/1471-2407-9-270
111. Traeger L, Ellermann I, Wiethoff H, Ihbe J, Gallitz I, Eveslage M, et al. Serum hepcidin and GDF-15 levels as prognostic markers in urothelial carcinoma of the upper urinary tract and renal cell carcinoma. BMC Cancer (2019) 19:74. doi: 10.1186/S12885-019-5278-0
112. Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, et al. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell (2015) 28:441–55. doi: 10.1016/J.CCELL.2015.09.002
113. McCarthy RC, Kosman DJ. Glial cell ceruloplasmin and hepcidin differentially regulate iron efflux from brain microvascular endothelial cells. PloS One (2014) 9:e89003. doi: 10.1371/JOURNAL.PONE.0089003
114. Zhou YF, Zhang C, Yang G, Qian ZM, Zhang MW, Ma J, et al. Hepcidin protects neuron from hemin-mediated injury by reducing iron. Front Physiol (2017) 8:332. doi: 10.3389/FPHYS.2017.00332
115. Du F, Qian ZM, Luo Q, Yung WH, Ke Y. Hepcidin suppresses brain iron accumulation by downregulating iron transport proteins in iron-overloaded rats. Mol Neurobiol (2015) 52:101–14. doi: 10.1007/S12035-014-8847-X
116. Du F, Qian C, Ming Qian Z, Wu XM, Xie H, Yung WH, et al. Hepcidin directly inhibits transferrin receptor 1 expression in astrocytes via a cyclic AMP-protein kinase a pathway. Glia (2011) 59:936–45. doi: 10.1002/GLIA.21166
117. Valdés PA, Samkoe K, O'Hara JA, Roberts DW, Paulsen KD, Pogue BW. Deferoxamine iron chelation increases delta-aminolevulinic acid induced protoporphyrin IX in xenograft glioma model. Photochem Photobiol (2010) 86:471–75. doi: 10.1111/J.1751-1097.2009.00664.X
118. Dayani PN, Bishop MC, Black K, Zeltzer PM. Desferoxamine (DFO)–mediated iron chelation: Rationale for a novel approach to therapy for brain cancer. J Neuro-oncology (2004) 67:367–77. doi: 10.1023/B:NEON.0000024238.21349.37
119. Rivera S, Nemeth E, Gabayan V, Lopez MA, Farshidi D, Ganz T. Synthetic hepcidin causes rapid dose-dependent hypoferremia and is concentrated in ferroportin-containing organs. Blood (2005) 106:2196–9. doi: 10.1182/blood-2005-04-1766
120. Vyoral D, Jiri P. Therapeutic potential of hepcidin - the master regulator of iron metabolism. Pharmacol Res (2017) 115:242–54. doi: 10.1016/j.phrs.2016.11.010
121. Gu Z, Wang H, Xia J, Yang Y, Jin Z, Xu H, et al. Decreased ferroportin promotes myeloma cell growth and osteoclast differentiation. Cancer Res (2015) 75:2211–21. doi: 10.1158/0008-5472.CAN-14-3804
122. Tuhkanen H, Hartikainen JM, Soini Y, Velasco G, Sironen R, Nykopp TK, et al. Matriptase-2 gene (TMPRSS6) variants associate with breast cancer survival, and reduced expression is related to triple-negative breast cancer. Int J Cancer (2013) 133:2334–40. doi: 10.1002/IJC.28254
123. Crielaard BJ, Lammers T, Rivella S. Targeting iron metabolism in drug discovery and delivery. Nat Rev Drug Discovery (2017) 16:400–23. doi: 10.1038/NRD.2016.248
124. Poli M, Asperti M, Ruzzenenti P, Regoni M, Arosio P. Hepcidin antagonists for potential treatments of disorders with hepcidin excess. Front Pharmacol (2014) 5:86. doi: 10.3389/FPHAR.2014.00086
125. Vadhan-Raj S, Abonour R, Goldman JW, Smith DA, Slapak CA, Ilaria RL, et al. A first-in-human phase 1 study of a hepcidin monoclonal antibody, LY2787106, in cancer-associated anemia. J Hematol Oncol (2017) 10:73. doi: 10.1186/S13045-017-0427-X
126. Mleczko-Sanecka K, Roche F, Da Silva AR, Call D, D'Alessio F, Ragab A, et al. Unbiased RNAi screen for hepcidin regulators links hepcidin suppression to proliferative Ras/RAF and nutrient-dependent mTOR signaling. Blood (2014) 123:1574–85. doi: 10.1182/BLOOD-2013-07-515957
127. Poli M, Girelli D, Campostrini N, Maccarinelli F, Finazzi D, Luscieti S, et al. Heparin: A potent inhibitor of hepcidin expression. Vitro vivo. Blood (2011) 117:997–1004. doi: 10.1182/blood-2010-06-289082
128. Poli M, Asperti M, Naggi A, Campostrini N, Girelli D, Corbella M, et al. Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression. Vitro vivo. Blood (2014) 123:1564–73. doi: 10.1182/BLOOD-2013-07-515221
129. Galli M, Chatterjee M, Grasso M, Specchia G, Magen H, Einsele H, et al. Phase I study of the heparanase inhibitor roneparstat: An innovative approach for ultiple myeloma therapy. Haematologica (2018) 103:e469–e72. doi: 10.3324/HAEMATOL.2017.182865
130. Tisi MC, Bozzoli V, Giachelia M, Massini G, Ricerca BM, Maiolo E, et al. Anemia in diffuse large b-cell non-Hodgkin lymphoma: The role of interleukin-6, hepcidin and erythropoietin. Leukemia lymphoma (2014) 55:270–75. doi: 10.3109/10428194.2013.802314
Keywords: hepcidin, cancer, iron, metabolism, therapeutics, homeostasis
Citation: Lin F, Tuffour A, Hao G, Peprah FA, Huang A, Zhou Y and Zhang H (2023) Distinctive modulation of hepcidin in cancer and its therapeutic relevance. Front. Oncol. 13:1141603. doi: 10.3389/fonc.2023.1141603
Received: 10 January 2023; Accepted: 08 February 2023;
Published: 21 February 2023.
Edited by:
Pierre-Jean Lamy, Groupe Inovie, FranceReviewed by:
Wang Liu, University of Kansas Medical Center, United StatesCopyright © 2023 Lin, Tuffour, Hao, Peprah, Huang, Zhou and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiqi Zhang, emhhbmdocUB6amZpc2guY29tLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.