 Junjun Li1
Junjun Li1 Songqing Fan
Songqing Fan Wei Liu
Wei Liu- 1Department of Pathology, Hunan Cancer Hospital, The Affiliated Cancer Hospital of The Xiangya School of Medicine, Central South University, Changsha, China
- 2Department of Pathology, The Second Xiangya Hospital, Central South University, Changsha, China
The development of techniques and immunotherapies are widely applied in cancer treatment such as checkpoint inhibitors, adoptive cell therapy, and cancer vaccines apart from radiation therapy, surgery, and chemotherapy give enduring anti-tumor effects. Minority people utilize single-agent immunotherapy, and most people adopt multiple-agent immunotherapy. The difficulties are resolved by including the biomarkers to choose the non-responders’ and responders’ potentials. The possibility of the potential complications and side effects are examined to improve cancer therapy effects. The Head and Neck Squamous Cell Carcinoma (HNSCC) is analyzed with the help of programmed cell death ligand 1 (PD-L1) and Insulin-like growth factor (IGF). But how IGF and PD-L1 upregulation depends on IL-6, EGFR, and LIN28/Let7-related mechanisms are poorly understood. Briefly, IL-6 stimulates gene expressions of IGF-1/2, and IL-6 cross-activates IGF-1R signaling, NF-κB, and STAT3. NF-κB, up-regulating PD-L1 expressions. IL-6/JAK1 primes PD-L1 for STT3-mediated PD-L1 glycosylation, stabilizes PD-L1 and trafficks it to the cell surface. Moreover, ΔNp63 is predominantly overexpressed over TAp63 in HNSCC, elevates circulating IGF-1 levels by repressing IGFBP3, and activates insulin receptor substrate 1 (IRS1).TP63 and SOX2 form a complex with CCAT1 to promote EGFR expression. EGFR activation through EGF binding extends STAT3 activation, and EGFR and its downstream signaling prolong PD-L1 mRNA half-life. PLC-γ1 binding to a cytoplasmic motif of elevated PD-L1 improves EGF-induced activation of inositol 1,4,5-tri-phosphate (IP3), and diacylglycerol (DAG) subsequently elevates RAC1-GTP. RAC1-GTP was convincingly demonstrated to induce the autocrine production and action of IL-6/IL-6R, forming a feedback loop for IGF and PD-L1 upregulation. Furthermore, the LIN28-Let7 axis mediates the NF-κB-IL-6-STAT3 amplification loop, activated LIN28-Let7 axis up-regulates RAS, AKT, IL-6, IGF-1/2, IGF-1R, Myc, and PD-L1, plays pivotal roles in IGF-1R activation and Myc, NF-κB, STAT3 concomitant activation. Therefore, based on a detailed mechanisms review, our article firstly reveals that IL-6, EGFR, and LIN28/Let7-related mechanisms mediate PD-L1 and IGF upregulation in HNSCC, which comprehensively influences immunity, inflammation, metabolism, and metastasis in the tumor microenvironment, and might be fundamental for overcoming therapy resistance.
1 Introduction
Squamous cell carcinoma is the most common kind of head and neck tumor, and it may develop in several anatomical locations, including the pharynx, oral cavity, and larynx. Over 890,000 new cases and 450,000 deaths were attributed to HNSCC in 2018, making it the sixth most frequent cancer worldwide (1). Long-term smoking, heavy alcohol use, and infection of the human papillomavirus or the Epstein-Barr virus are the main risk factors and oncogenic drivers worldwide (1). There have been improvements in the treatment of HNSCC over the last several decades, but in general, most patients are still given a combination of surgery, radiation, and chemotherapy. About half of all cases are detected at an advanced stage because of the significant difficulties in early diagnosis. Despite developing EGFR-targeted treatment and PD-1/PD-L1 blockade immunotherapy, the 5-year survival rate has remained around 50% in advanced patients (1). To enhance patient care, new treatment methods must be developed. This cannot be done without understanding the particular, detailed processes and co-occurrence of the oncogenic changes of main pathways in the HNSCC microenvironment (2). As a kind of immunotherapy for cancer, targeted antibodies may interfere with cancer cell function and trigger the immune system to target and destroy cancer cells. Some immunotherapy medications, known as immune checkpoint inhibitors, prevent checkpoint proteins from engaging in protein-protein binding. Due to this, the off signal is not sent, and the T cells can effectively eliminate cancer cells. The CTLA-4 checkpoint protein is a target for one such medication. Toxic effects on the immune system or other systems as a consequence of immune system dysfunction are collectively referred to as immunotoxicity.
The Insulin-like development factors system consists of IGF-1 and -2, their receptors (IGF-1R and -2R), and a group of six IGF-binding proteins (IGFBP-1 to -6) (3: 4). Specifically, there is some cross-activation between insulin and IGF-1, IGF-2, and their receptors (5, 6) since IGF1R has a high degree of similarity with the insulin receptor. To interact with their substrates, the IRS and the Src homology 2 domain-containing transforming protein (SH2), IGF-1/2 receptor tyrosine kinase (RTK) activity may be activated by many different stimuli. Phosphorylation of this protein causes it to behave as a docking molecule, activate the RAS/RAF/MEK and PI3K/AKT/mTOR signaling pathways, stimulate cell proliferation, and suppress apoptosis, all of which contribute to carcinogenesis and resistance to numerous therapies (7, 8). PD-L1 overexpression was demonstrated to activate IGF-1R-mediated PI3K/Akt and RAF/MEK/ERK pathways and elevate STAT3, NF-κB, and c-Myc, which are reversible by lycopene in tongue squamous cell carcinoma (9). Moreover, PD-L1 upregulation elicits immune evasion in HNSCC, correlates with Let7 downregulation, PD-L1 expression in HNSCC is heavily glycosylated, Let-7a/let-7b miRNA was shown to promote PD-L1 degradation and inhibit PD-L1 glycosylation (10). IL-6 was found to regulate PD-L1 mutually, and its elevation correlated with disease failure and poor prognosis for HNSCC (11–13). We shall review the oncogenic IL-6/JAK/STAT3, EGFR/RAS/MEK/ERK, PTEN/PI3K/Akt pathways in HNSCC and LIN28/Let7 axis they regulate PD-L1 expression and degradation. Anti-tumor efficacy may be measured in several ways. Still, regardless of the host species, the ultimate objective of any therapy should be to reduce tumor burden, minimize tumor-associated morbidity, enhance the quality of life, and, if feasible, increase the life span. Direct destruction of cancer cells or encouragement of the immune system to fight against the tumor are two possible mechanisms behind its anti-tumor impact. Exposure to potentially toxic compounds, such as chemical warfare agents, may have detrimental consequences on immune function, known as immunotoxicity.
The antitumor immune response is remarkably clinically effective when the PD-L1. programmed cell death receptor-1 (PD-1) signaling axis is inhibited. Low response rates, however, only allow a small subset of patients to benefit from immunotherapy. In recent investigations, the significance of the transmembrane protein PD-L1 in exosomes has been investigated, and it has been discovered that exosomal PD-L1 functions as a mechanism of tumor immune escape and immunotherapy resistance. Exosomal PD-L1 transmits functional PD-L1 across the tumor microenvironment, reduces T cell effector function, and causes systemic immunosuppression (TME). Exosomal PD-L1 has been suggested as a biomarker to predict immunotherapy response and to evaluate treatment efficacy due to its substantial role in immune evasion (14).
The development of several types of solid tumors, including breast cancer (BC) and head and neck squamous cell carcinoma(HNSCC), as well as a number of pathophysiological processes has been linked to the insulin-like growth factor-1 (IGF-1) over the past 20 years, according to numerous research. IGF-1 receptor (R) is overexpressed and hyperphosphorylated in a number of BC subtypes, according to preclinical and clinical studies. This pathway is a key player in the proliferation and spread of tumor cells, making it a crucial therapeutic target. Moreover, the IGF-1 axis has demonstrated a close relationship with the control of estrogen and endocrine therapy, suggesting a potential cure for anti-estrogen resistance. Many clinical trials are currently examining the function of IGF-1R inhibition in regulating resistance mechanisms to target therapies. IGF-1R may potentially interact with other essential therapeutic approaches, such as anti-HER2 medicines and mTOR inhibitors. With an eye on their potential future influence on clinical practice, our goal is to provide a summary of the most current and significant field of application for IGF-1 inhibitors as well as pertinent treatment approaches (1). We will review them in full detail below, emphasizing their connections with PD-L1 and providing possible explanations for PD-L1 overexpression-induced effects and IGF-1R activation (see Figure 1).
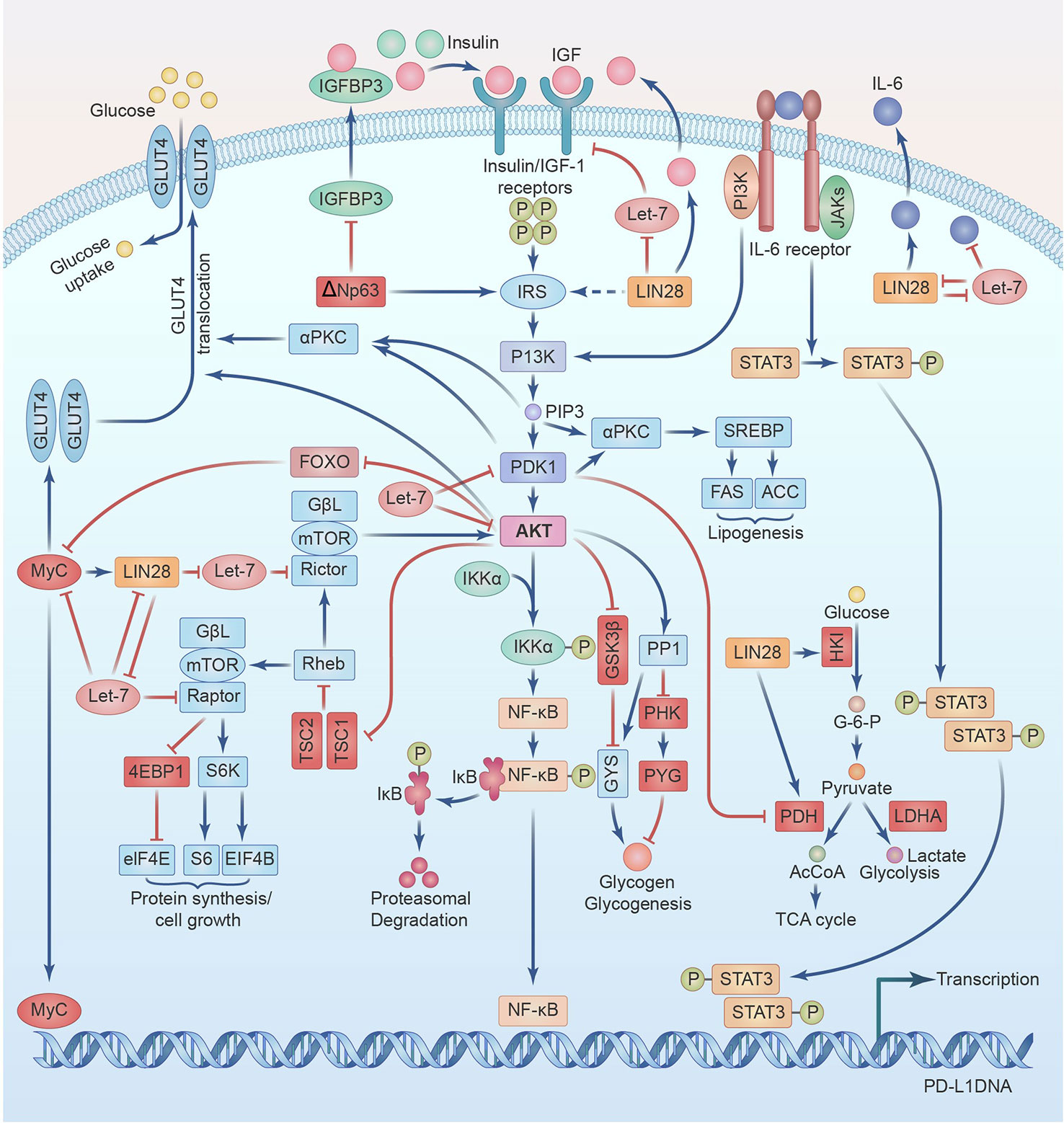
Figure 1 IGF signaling cross-talks with PD-L1 regulation through LIN28-Let7 axis and IL-6 amplification loop in Head and Neck Squamous Cell Cancer.
2 IL-6/JAK and STAT3
2.1 IL-6/JAK
IL-6 is a pleiotropic cytokine involved in many physiological processes and a prominent participant in chronic inflammatory disorders, autoimmune diseases, cytokine storms, and cancer (15). IL-6 was identified in 1986 and is situated on the short arm of chromosome 7 (16). Multiple cell types, including B- and T- lymphocytes, monocytes, fibrocytes, endothelial cells, keratinocytes, adipocytes, and cancer cells, can produce and secrete IL-6 (17). IL-6 is also involved in menstruation and spermatogenesis, bone metabolism through the stimulation of osteoclastogenesis and osteoclast activity (18), liver regeneration (19), and tissue-protective/reparative and inflammatory characteristics of vascular endothelial cells (20).
Many patients with hematopoietic malignancies or solid tumors have been shown to have elevated levels of IL-6 from tumor cells, stromal cells, and tumor-infiltrating immune cells, which has been linked to poor outcomes (13). Both membrane-bound IL-6R (the classical signalling) and soluble(s) IL-6R (the trans-signalling) are involved in the transmission of IL-6 signals. While IL-6 always works the same way, first binding to the receptor and subsequently to gp130 through cytokine binding domains, the diverse receptor types dictate distinct biological effects (17). Researchers believe only immune cells, liver cells, intestinal epithelial cells, and vascular endothelial cells express IL-6R and benefit from its anti-inflammatory effects (20). The cell surface IL-6R is cleaved into sIL-6R by the proteases disintegrin and ADAM17 and ADAM10(17).
Interestingly, ADAM17 or ADAM10 cleaves PD-L1 off the malignant cells and extracellular vesicle surface to form soluble but still active PD-L1, which impairs the destruction of tumor cells and promotes the death of CD8+ T cells (21). After sIL-6R forms a dimer complex with IL-6, the complex is transported throughout the body. The IL-6-SIL-6R complex forms a heterotrimer, which binds to gp130 to create a hexameric complex with another heterotrimer. The sIL-6R shedding by tumor-infiltrating T cells, neutrophils, and monocytes allows IL-6 to stimulate trans-signaling in tumor and stromal cells, resulting in oncogenic pro-inflammatory responses (17). Since gp130 is ubiquitous, IL-6 can be produced by tumor cells, infiltrating immune cells and stromal cells.
Full activation of JAKs occurs after IL-6-(s) IL-6R complex engagement of gp130, which disrupts JAK homology2 domain (JH2)-mediated regulation of JH1 domain activity by mutual transphosphorylation (17). Since the genes for JAKs, both upstream kinases that control PD-L1 expression, are located on the same chromosome (9p), mutation and amplification of the JAK family lead to increased PD-L1 transcription (22). PD-L1 is protected against GSK3-26S proteasome-driven degradation, improves its association with PD-1 on CD8+ T cells, and mediates EMT, as shown in another study (23). This is accomplished by priming PD-L1 for glycosylation of the catalytic subunit of oligosaccharide transferase STT3. Fully glycosylated PD-L1 has a half-life of 12 h (24), but nonglycosylated PD-L1 has a half-life of 4 h and is quickly proteolyzed. Multiple tyrosines remaining in the cytoplasmic region of gp130 are phosphorylated by active JAK enzymes, transforming them into docking sites that bring proteins into close contact with the enzymes. The proteins are phosphorylated by the active JAK enzymes, which then set off the JAK/STAT3, AKT/PI3K, and RAS/RAF/MEK/MAPK signaling pathway (16). These three signaling pathways, which may also be triggered by EGFR, IGF-1R, and HIF and contribute to PD-L1 regulation, are responsible for the hexameric complex’s ability to elicit anti-inflammatory or pro-inflammatory responses (25).
AP-1, C/EBP, and cAMP-responsive elements are only some transcription factors that bind to specific sites in the IL-6 gene (15). One of the main drivers of IL-6 expression and secretion is NF-κB, which is partially activated via the PI3K/Akt/mTOR pathway. IL-6 induces JAK activation, stimulating STAT3 dimerization via binding to sIL-6R, which subsequently binds to gp130. STAT3’s capacity to increase IL-6 gene expression creates an autocrine feed-forward loop. An amplification loop is formed when IL-6 in the tumor microenvironment activates STAT3 and NF-кB, which then increases the production of IL-6 (17). Perhaps this is the reason why IL-6 levels are so high in HNSCC. The direct effects on tumor cell survival, propagation, and incursion, increased IL-6 levels can also induce the appearance of other growth cytokines or factors significant for HNSCC progression and development, like IGF, GM-CSF, VEGF, monocyte chemoattractant protein (MCP)1, CCL3, CCL2, CCL5, IL-1β, and IL-8, which act in a paracrine or autocrine fashion on both immune and nonimmune cells within the tumor microenvironments (26). Elevated IL-6 levels drive the STAT3, MAPK, PI3K/Akt pathways activation, and NF-κB, which leads to diverse pathological chronic inflammatory diseases like cancer, multiple sclerosis, rheumatoid arthritis, diabetes, Castleman disease, inflammatory bowel disease, Crohn’s and Alzheimer’s disease; the clinical usage of IL-6 signaling inhibitors have proved helpful in the treatment of patients with these disorders (15, 17).
Head and neck cancer and pancreatic cancer cell lines rank the highest according to the cancer inflammation indices, based on IL-6 and PD-L1 genes commonly binding-factors NF-κB, STAT3, and AP-1 (27) and correlated IL-1/NF-κB, IL-6/STAT3, and TNF/AP-1 signaling cascade (28). Together, these constituents provide a much larger positive feedback loop that sustains the altered malignant state (28). An illuminating investigation on non-small-cell lung cancer found an IL-6/STAT3/IGF-1R autocrine loop, in which IL-6 stimulates the gene production of IL-6 itself, as well as IGF-1 and IGF-2, which cross-activate IGF-IR and STAT3, resulting in EMT and an increase in IGF-1R promoter activity (29). IL-6 was also confirmed to stimulate an autocrine or paracrine IGF-1/IGF-1R expression which, through STAT3, stimulated OCT4/NANOG expression in hepatitis B virus-infected hepatocellular carcinoma (30). These findings support that IL-6 mediates PD-L1 and IGF elevation.
2.2 STAT3
The high levels of STAT3 expression in the pericardium, leukocytes, peritoneum, mammary gland, digestive tract, kidney, bladder, lung, bone marrow, and peripheral nervous system all point to critical roles for STAT3 in these organs’ physiological processes (31). The activation of STAT3 is tightly controlled in healthy cells. STAT3 phosphorylation induced by exposure to cytokines peaks within 15–60 minutes and decreases over several hours. Phosphorylated STAT3 returns to the cytoplasm through nuclear pores after being dephosphorylated by nuclear protein-tyrosine phosphatases (PTPs) such as TC-PTP or TC45 (32). Moreover, several other negative protein regulators exist, including suppressor of cytokine signaling proteins (SOCS1-7); the PIAS1-4; several PTPs, including SHP-1, SHP-2, PTPRD, PTPRT, DUSP22, and CD45; and the ubiquitination-dependent proteasomal degradation systems (33). SOCS family negatively regulates the STAT/JAK pathway in terms of preventing the tyrosine kinase receptor and straight binding to JAK or simultaneously binding JAK with gp130 to inhibit JAK kinase activity, (3) targeting SOCS-bound JAK or STAT for proteasomal degradation via polyubiquitination by forming a complex with the elongation protein B/C-cullin5 complex. For example, SOCS3 silencing by CpG islands hypermethylation of the promoter in human lung cancer results in STAT3 hyperactivation and tumor cell proliferation (34). PIAS does not interact with STAT monomers but only inhibits formed STAT dimers after JAK phosphorylation. PIAS mainly regulates transcription by blocking STAT DNA-binding activity, promoting transcript factor sumoylation, and recruiting transcriptional co-repressors such as histone deacetylase (35). PTPs can dephosphorylate STAT and therefore inhibit JAK/STAT signal transduction. SHP1 and SHP2 are examples of tyrosine phosphatases that include the SH2 domain. While SHP2 is expressed in all cell types, SHP1 is mostly found in hematopoietic, smooth muscle, and epithelial cells. Dephosphorylation of active JAKs or STATs occurs when their SH2 domains bind to phospho-tyrosine residues (36). Selected PTPRT mutations correlate with up-regulation of phospho-STAT3 Y705 expression in HNSCC specimens and cell lines, whereas overexpression of wild type reduced STAT3 phosphorylation. Loss of expression of PTPRT and PTPRD occurs due to mutations in these genes in 3.7% and 5.6% of neck and head cancers, respectively, and promoter methylation in 60% of HNSCC cases (17). Cell proliferation is stimulated, and the expression of the inhibitory target phospho-STAT3 Y705 is elevated in response to PTPRD mutations that abolish its action (36). Primary melanoma cells lacking endogenous PTPRD show markedly reduced cell growth and death when treated with exogenous PTPRD (37).
STATs are activated via the tyrosine phosphorylation cascade after binding and stimulation by ligands, including members of the IL-6 cytokine receptor family, such as IL-6, CNTF, LIF, and OSM (15); interferon (IFN-α/β/γ); RTKs, such as EGFRs, VEGFRs, IGFRs, the insulin receptor, c-Kit, and c-Met (38); and nonreceptor tyrosine kinases (NRTKs), such as JAKs and Src family kinases (SFKs). GPCRs, like the sphingosine 1 phosphate receptor 1/2 (S1PR1/2) and angiotensin II (Ang II), and some TLRs, like TLR 2, 3, 4, 7, and 9, can also mediate STAT3 activation (39). STAT3 mutations are rare only in patients with hematological malignancies (17). The hyperactivation of growth factor receptors/NRTKs, including Src/JAKs; the overexpression of stimulatory ligands, such as EGF/IL-6; or the loss of function of negative regulators, such as SOCS/PIAS/PTPs, lead to aberrantly elevated STAT3 activity in over 70% of human cancers (39). STAT3 translocates into the nucleus via an importin/importin 1-dependent process after being phosphorylated at Tyr705 (40). This occurs either through SH2-domain-mediated head-to-tail dimerization or the formation of a dimer with STAT1. The STAT3 dimer binds gamma-activated sites in the promoters of a wide variety of target genes in the nucleus of various cancer cells, including head and neck cancer cells and multiple myeloma (41), influencing the transcription of genes important for cellular proliferation (MYC and cyclin D1), survival (survivin and BCL-xL), angiogenesis (such as VEGF), and maintenance of the immunosuppressive micro-environment. In addition, tumor microenvironmental STAT3 hyperactivation promotes metastasis and cancer progression by elevating the appearance of EMT-related transcription elements such as Twist, Snail, and ZEB1 (42).
STAT3 also underlies resistance to multiple therapies, such as conventional chemotherapy, radiation therapy, and EGFR-targeted therapies; therefore, combination therapy with STAT3 inhibitors may be of clinical benefit (43). Recent elegant research exposed that bone marrow-derived mesenchymal stem cells (BM-MSCs) elicited early-response CD133+/CD83+ lung cancer stem cell (CSC) dissemination and, through LIF/LIFR/pS727-STAT3 signaling, induced mesenchymal-epithelial transition (MET) to establish a macroscopic premetastatic niche. Then, BM-MSCs aided primary cancer cell EMT through IL-6/IL-6R/pS705-STAT3 signaling, leading these cells to acquire CD151+/CD38+ CSC properties and to be attracted to the premetastatic niche. Therefore, the IL-6 cytokine family receptor/JAK/STAT3 axis targeting might also block two-step cancer metastasis and reverse the dissemination of metastasis-related CSCs (44). Microsatellite instability/defective mismatch repair (MSI/dMMR), programmed death ligand 1 (PD-L1), and tumor mutational load are some of the most extensively studied prognostic biomarkers for immunotherapy (TMB). Many preclinical investigations and clinical trials have focused on individuals who have developed a primary or acquired resistance to ICI because of the difficulty of treating this condition. Immune-related adverse events (irAEs) are a distinct spectrum of ICI side effects that mimic autoimmune reactions. The skin, digestive tract, lungs, endocrine, musculoskeletal, and other systems are among the most prevalent sites where irAEs manifest themselves, although they may affect any organ in the body. The current body of data shows that the extreme reserve of TCR inhibitory phosphatases linked with CPI receptors, which are responsible for maintaining peripheral immunological tolerance towards autoantigens, may be the primary cause of irAEs in patients treated with CPI. Active and passive approaches are the two primary categories of cancer immunotherapy to enhance anti-tumor efficacy while abrogating toxicities. Cancer elimination is enhanced by interventions that boost the patient’s immune system, such as vaccination or adjuvant therapy.
3 EGFR/PD-L1
EGFR is a 170 kDa transmembrane glycoprotein and RTK that is an associate of the ErbB/HER family with ErbB3 (HER3), ErbB2 (HER2/neu), and ErbB4 (HER4). Except for HER2/neu, ligands have been identified for all HER family members. Seven ligands for EGFR are known, including the four high-affinity ligands EGF, heparin-binding EGF, TGF-α, and amphiregulin, and the three low-affinity ligands epiregulin, betacellulin, and epigen. When a ligand hits EGFR, it stimulates homo- or heterodimerization of EGFR with additional HER2, HER3 receptors or other RTKs, such as IGF-1R or MET (45). Dimerization of the extracellular region of the receptor induces intracellular conformation changes, which releases intramolecular cis-autoinhibition, resulting in the transphosphorylation of key tyrosine residues, which allows the kinase to assume an active conformation (46). EGFR activates four downstream signaling cascades: the MAPK, PI3K/AKT/mTOR, JAK/STAT, and PLCγ/PKC pathways. Dysregulation of these pathways is commonly observed in the occurrence and metastasis of many cancer types, especially HNSCC (47). A study in SkHep-1 cells revealed that EGF, TGF-α, HB-EGF, and beta-cellulin stimulation, but not amphiregulin or epiregulin, caused dose-dependent EGFR accumulation in the nucleus. Stimulation with the former four ligands also increased the phosphorylation of EGFR tyrosine residues and wound closure rate more than stimulation with the latter two ligands. However, a single ligand stimulation failed to promote proliferation (48). An informative study revealed that unlike new PD-L1 transcription mediated by interferon regulatory factor 1 interaction with the PD-L1 promoter succeeding IFN-γ activates JAK/STAT axis, the binding EGF to EGFR elevates PD-L1 transcript levels by prolonging the mRNA half-life. PD-L1 overexpression in HeLa cells promoted migration and invasion; doubled the thymidine incorporation into DNA; and facilitated EMT by increasing SNAIL, SLUG, and ZEB1 levels, and H1975 cells overexpressing PD-L1 displayed more robust tumorigenesis in athymic mice. PD-L1 deletion or PD-L1 antibodies abrogated EGF and TGF-β -inducible migration, tumorigenesis, and metastasis (49). Most interestingly, the authors created an EGF gradient and measured directionality that connects the linear distance between the starting point and endpoint (D) to the total distance (T) traveled. PD-L1 deficiency or application of the EGFR kinase inhibitors significantly reduced persistence. As actin filaments and Rho family GTPases control the switch from random to persistent migration, PD-L1 ablation caused a reduction in RAC1-GTP and CDC42-GTP levels but elevated RHOA-GTP levels with visually severe loss of actin cables (49). Most importantly, RAC1-GTP was convincingly demonstrated to induce the autocrine production and action of IL-6/IL-6R, leading to STAT3 activation, which might induce PD-L1 expression and EMT, linking PD-L1 signaling with regulation (50–52). The cytoplasmic tail of PD-L1 was cloned into a yeast 2-hybrid bait construct, and >50 million connections have been examined to discover direct PD-L1 binders. The breakthrough occurred when filamin A and PLC-γ1 were identified as the two PD-L1 interaction partners with the maximum confidence scores (49). Each monomer of the F-actin crosslinking protein Filamin A consists of an actin-binding domain and a rod segment. Filamin A dimers crosslink actin filaments to create networks orthogonal to parallel bundles, while the two hinges in the rod section allow for the inherent flexibility of actin networks. Briefly, the distinctive structure of filamin A and its specific interaction with F-actin confer dynamic mechanical features on the cytoskeleton (51). The binding of PLC-γ1 to a conserved cytoplasmic domain of PD-L1 promotes EGF-induced activation, resulting in the production of DAG and IP3. The former concentration-dependently activates PKC-, while the latter increases cytosolic calcium ion levels and regulates actin-myosin contractions. Notably, PLC-γ1 was not activated when EGFR was inactive, or PD-L1 was scarce; this process is known as the EGFR and PD-L1 two-hit model of PLC- activation. The attachment of PLC-γ1 to the cytoplasmic region of PD-L1 has also been associated with protection against IFN-γ cytotoxicity (49).
A previous report revealed that the genomic binding sites of master transcription factors TP63 and SOX2 in SCCs overlap. An epigenomic profiling of various SCCs, including HNSCC, demonstrated that SOX2 and TP63 cooperatively bind and activate the super-enhancers and promoter of the long non-coding RNA (lncRNA) CCAT1 to regulate its expression. ChIP analysis showed that oncogenic CCAT1 forms a multifaceted with SOX2 and TP63, which binds the super-enhancers of EGFR to regulate EGFR expression, thereby activating both PD-L1-related MEK/ERK1/2 and PI3K/AKT signaling pathways (53). In light of TP63, ΔNp63, which is predominantly overexpressed over TAp63 in HNSCC (27), has been shown to elevate circulating IGF-1 levels by repressing IGFBP3 and increasing intracellular MEK/ERK1/2 and PI3K/AKT/mTOR signaling by activating IRS1 expression, ultimately promoting the growth and survival of SCC cells (54). SOX2 was found to bind a proximal site in the Lin28 promoter region and positively regulate Lin28 expression in neural precursor cells (55). Lin28 elevates IGF-2 and IL-6, activates IGF-1R and Myc, and enables the activation of NF-kB, STAT3, and c-Myc to elevate PD-L1 expression (56). EGFR mutations were demonstrated to upregulation PD-L1 expression via the IL-6/STAT3/JAK pathway in non-small cell lung cancer, mediating tyrosine kinase inhibitors (TKIs) resistance (57). PD-L1 recruits phospholipase C-γ1 to its cytoplasmic motif, which enhances phospholipase C-γ1 activation by EGFR and tumorigenicity of EGFR-mutant lung tumors (49). Matrine and Polyphyllin I overcomes EMT-associated resistance to TKI via IL-6/STAT3 pathway inhibition in lung cancer (58, 59). Non-coding RNA miR-146b concurrently blocked both the IL-6-STAT3 and EGFR pathways in vitro and might be of clinical benefit in ovarian cancer (60). Reports that lycopene reverses PD-L1 expression and signaling in HNSCC and that tomato lectin specifically blocks EGFR signaling-mediated poly-LacNAc glycosylation necessary for the PD-L1/PD-1 interaction in triple-negative breast cancer (TNBC) might provide some hope (58; 61). These results identified an SCC-specific DNA/RNA/protein multifaceted that stimulates IGF signaling and upregulates PD-L1, progressing our understanding of metastasis and immunity in cancer biology.
EGFR is a crucial switch in the activation of STAT3 by IL-6. SOCS3 may directly block the kinase activity of receptor-associated JAK proteins, preventing subsequent activation of STAT3. EGFR activation can inhibit SOCS3 and nullify the negative control of JAK kinase activity (62). IL-6 stimulates the shedding of EGFR ligands through ADAM10 and ADAM17, so it transactivates EGFR in normal prostate epithelial cells and promotes cell proliferation (63). IGFBP2 is a secreted protein that controls the bioavailability and localization of IGF-1 and IGF-2 by binding to them. IGFBP2 was a possible biomarker for PI3K/Akt pathway activation and PTEN status, and it was demonstrated to upregulate PD-L1 expression in glioma, prostate, breast, colorectal, and HNSCC (64). IGFBP2 and EGFR nuclear colocalization were reported in glioblastoma and astrocytoma cells. Additional research demonstrated that IGFBP2 enhances the EGFR nuclear accumulation and the activation of the EGFR/STAT3/PD-L1 signaling pathway in melanoma cells (65). IGFBP2 nuclear translocation in cancer cells is based on a typical importin-α-dependent mechanism mediated by a nuclear localization signal (179PKKLRPP185) in the linker domain of IGFBP2 (64).
The RAS/RAF/MEK/ERK signal transduction pathway activates numerous critical cellular mediators, leading to proliferation, cell growth, differentiation, invasion, migration, and survival (64). HRAS, KRAS, and NRAS are the three members of the Ras gene family. Ras is a GTPase localized on the intracellular side of the plasma membrane that RTKs, including EGFR, activate. Guanine nucleotide exchange factors (GEFs), which permit GTP loading, influence the transition of Ras from an inactive to an active state. GTP hydrolysis mediates the reverse switch, which is mediated by GTPase-activating proteins (GAPs) (66). Once Ras is active, it stimulates the RAF (RAF1) and PI3K pathways (45). RAF has three variants: ARAF, BRAF, and CRAF (RAF1). GTP-bound RAS interacts with and recruits RAF, accumulating RAF at the plasma membrane and subsequent dimerization, activating RAF kinases. Although all three isoforms activate MEK, data suggest that oncogenic RAS may activate them differently. Activated RAF phosphorylates MEK1 and MEK2 kinases, activating the MAP kinases ERK1 and ERK2. These MAP kinases may either phosphorylate cytoplasmic substrates or translocate into the nucleus to target genes that govern cell growth, proliferation, and survival (66). Mutated RAS phosphorylation increases hyperactive MEK signaling and inhibits tristetraprolin, which usually prohibits PD-L1 and LIN28 mRNA by binding AU-rich regions, thereby extending their half-lives (67) (see Figure 2).
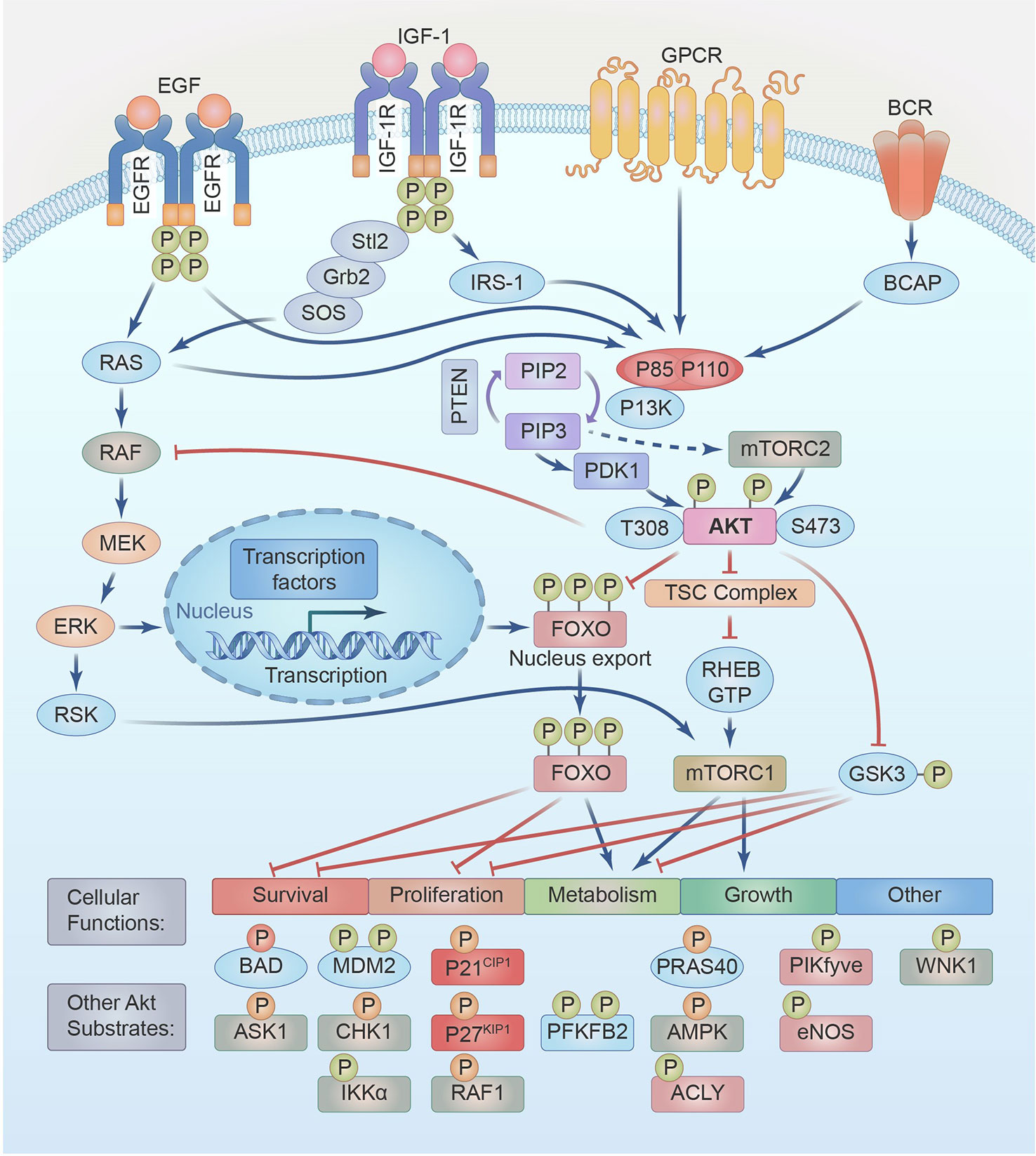
Figure 2 IGF/IGFR,EGF/EGFR,GPCR,BCR signal through PI3K-AKT-mTOR and RAS-RAF-MEK-ERK pathways. A partial list of their functions and substrates are shown.
EGFR is overexpressed in 90% of HNSCC cases with pathway genetic alteration frequency at 42% correlates with poor prognosis and treatment outcomes (2). Malignant tumors are often addicted to EGFR signaling for sustained survival and proliferation, even after acquiring resistance to multiple therapies (68). Strategies targeting EGFR for the treatment of HNSCC have been widely evaluated. Their mechanisms are oral TKIs bind the intracellular tyrosine kinase domain of EGFR and prevent downstream signaling, intravenously administered antibodies bind the extracellular domain of EGFR to promote its internalization, also facilitate ADCC, which is mediated by NK cells, and antigen-presenting cells give rise to innate and adaptive antitumor immune responses via EGFR-specific T cells (68).
4 PTEN/PI3K/AKT/mTOR
6% of HNSCC tumors have reduced expression of PTEN or the loss of its function due to genetic and epigenetic alterations (2). PTEN was identified as a tumor suppressor for the first time in 1997, and its expression is usually diminished due to homozygous deletion in advanced cancers (69). Subsequent research identified PTEN as a strong adverse regulator of significant cell growth and survival signaling pathways, notably the PI3K/AKT/mTOR signaling pathways (70). Recent investigations have shown that PTEN also works as a key molecular switch that controls the reprogramming of cancer cell metabolism and autophagy (71). PTEN phosphatase decreases the activity of PI3K, hence impeding insulin/IGF-1 pathway signaling. Loss of PTEN function consistently underlying the pathogenesis of HNSCC by activating the PI3K/Akt/mTOR pathway, which increases PD-L1 protein translation ratios, as observed for glioma (72).
PTEN is a dual lipid and protein phosphatase, and its biological effects are driven by its capacity to dephosphorylate PIP3. In contrast, PTEN’s potential substrates include focal adhesion kinase (FAK), IRS1, c-SRC, and PTEN itself (73). When the p85α regulatory subunit of PI3K loses affinity for the p110α catalytic subunit, it’s binding to PTEN enhances its steadiness and phosphatase action (74). PI3K effector substrates, particularly AKT, become spontaneously activated without external oncogenic activation when PTEN is mutated or otherwise inactive (75).
PTEN regulates metabolic processes, including gluconeogenesis, glycolysis, glycogen synthesis, mitochondrial metabolism, and lipid metabolism (76). Cancer cells with inadequate PTEN expression manifest a glycolytic phenotype, termed the Warburg effect (77), by which metabolism is reprogrammed to match cell proliferation. In contrast, PTEN accumulation represses glycolysis and enhances oxidative phosphorylation by inhibiting the PI3K/Akt pathway (78, 79). The PTEN/PI3K/AKT axis regulates de novo lipogenesis by controlling sterol response element-binding protein (SREBP) expression and inhibiting the forkhead transcription factor FOXO1, which negatively regulates SREBP transcription and vice versa (80, 81). Notably, SREBP was also confirmed to be involved in PD-L1-mediated EMT, and overexpression of PD-L1 enhanced SREBP-1c promoter activity, which might result from the inhibition of FOXO1 (81–83).
PTEN also has important tumor-suppressor functions in the nucleus, mainly via its participation in various processes, including genome maintenance, DNA replication, DNA repair, cell cycle control, and gene expression (78). In prostate cancer, it was discovered that the X-linked tumor suppressor USP11 regulates protein stability and PTEN polyubiquitination in both the cytoplasm and nucleus (84). Mechanistically, PTEN inhibits PI3K/AKT, leading to FOXO1 dephosphorylation and nuclear translocation, activating the binding of FOXO1 to the USP11 promoter, which increases USP11 protein and mRNA levels to increase PTEN stability. This mechanism is exploited by mouse embryonic fibroblasts cultured at a high density to upregulate USP11 expression to increase PTEN protein levels for contact inhibition without mRNA changes. USP11 downregulation in cancer patients correlates with FOXO nuclear localization and PTEN expression. The PTEN-PI3K-FOXO-USP11 feed-forward loop improves the tumor-suppressive activity and stability of PTEN (84).
Overexpression of interleukin-8 (IL-8) correlates with poor outcomes in various cancers, including HNSCC, by activating the STAT3 pathway, which a CXCR1/2 inhibitor can block. More importantly, the binding of IL-8 to CXCR1/2 facilitates the phosphorylation and inactivation of PTEN, which functions as an important switch in the IL-8/STAT3 pathway, elevating snail expression and the autocrine/paracrine activity of IL-8 to form a novel positive feedback loop that might drive malignant progression (85). PTEN is also transcriptionally repressed by two PD-L1-related zinc finger-like transcription factors, SNAIL and SLUG, which compete with p53 for PTEN promoter binding (86). PTEN is regulated by non-coding RNAs and is important in adaptive and innate immunity; this is beyond the scope of this review and has been well-reviewed in the past (87, 88).
The PI3K/AKT/mTOR pathway is active in over 80% of HNSCC tumors because of EGFR activation (47%), PIK3CA mutations or amplifications (25%), PIK3R1 overexpression (2%) or PTEN mutation (6%) (2, 89). In HNSCC with wild-type PIKCA expression, prolonged tyrosine phosphorylation of PI3K and HER3 recruitment resulted in abnormal PI3K/AKT/mTOR signaling and immune suppression, as shown by an independent kinome-wide siRNA screen. HER3 antibody inhibition decreases PI3K/AKT/mTOR signaling, slows tumor development, and reverses the immunosuppressive tumor microenvironment (90). Activating the PI3K axis in HNSCC cells induces excessive proliferation, enhanced cancer cell survival, and resistance to several anticancer treatments (91). Recent studies have revealed that oncogenes may be activated by class I PI3K, AKT, and mTOR, contributing to the inhibition of autophagy and the promotion of cancer (92). PI3K axis activation is responsible for the immortal nature of HNSCC and might be conveniently exploited as an “Achilles heel” to improve therapeutic responses (93). In addition to EGFR, IGF1-R, HER2/neu, HER3, G protein-coupled receptors (GPCRs), and Ras GTPase, other RTKs can activate the PI3K/Akt signaling axis (91, 92). Different ligands, such as cytokines, homologous growth factors, and hormones, bind to the 58 RTKs from 20 different classes discovered in cells. Phosphorylation of RTK YXXM motifs upon ligand binding initiates signaling either directly through binding a regulatory component of PI3K (p85) or indirectly through adaptor proteins like IRS-1 (91). This interaction abrogates p85’s inhibition of p110, allowing PI3K to be activated. PIP2 is phosphorylated into PIP3 when PI3K is bound to the plasma membrane and activated (94). GPCRs are a huge family of cell surface receptors. Approximately 800 GPCRs and 35 G protein subunits transmit different signaling cascades and play crucial roles in several cellular and physiological functions (95). GPCRs may either straight activate PI3K by enhancing the kinase activity of p110β or circuitously by phosphorylating RasGEF and RasGRP4. The Gβγ subunits of GPCR bind with great affinity to a specific area in the C2-helical domain linker of p110β (96). RAS is another PI3K/Akt axis upstream activator (45). RAC1 and CDC42 binds directly to the N-terminal RAS-binding domain (RBD) of p110β and cooperates with RTKs to exert lipid kinase activity. After RTK activation, PIP3 propagates the PI3K/Akt signaling axes, activating the Rho complex to activate the PI3K enzyme through Ras-dependent p110β activation (97).
Once activated by the PIP3 binding to the PH domain, which modifies its configuration, AKT is recruited to the plasma membrane, where its phosphorylation is done at Thr308 by phosphoinositide-dependent protein kinase 1 (PDK1) (98). PIP3 also activates the mTORC2 complex by interacting with the PH domain within the SIN1 element and activated mTORC2 phosphorylates AKT at Ser 473 in the C-terminal hydrophobic motif, which is vital for high activation (99). IRS-1 and PI3K are recruited and activated by IGF-1-IGF-1R interaction. Activated PI3K transforms PIP2 to PIP3 as described above to activate AKT, and the dephosphorylation of AKT at Thr308 and Ser473 by protein phosphatase 2A (PP2A) modulates the activation state of AKT and its downstream effects (73). Activated AKT regulates multiple cellular processes by phosphorylating various targets, including mTORC1, TSC2, GSK3, FOXO, RAF1, IKKα, MDM2, CHK1, ASK1, p27, p21, BAD, PRAS40, eNOS, AMPK, and WNK1. These downstream effectors control important cellular processes, such as proliferation, transcription, metabolism, protein synthesis, cell survival, apoptosis, autophagy, angiogenesis, and EMT (see Figure 2). Phosphorylated AKT can activate mTORC1 either by direct phosphorylation at Ser2448 or by indirect phosphorylation and inhibition of TSC1/2 at Ser939 and Thr1462 (100), increasing protein, lipid, and nucleotide synthesis while downregulating autophagy, which supports cell survival, growth, and proliferation (91). Over C-terminal domains and GTPase-activating protein domains, TSC1 and TSC2 inhibit mTORC1 activity by converting Ras-related Rheb-GTP, strong mTORC1 activators, to Rheb-GDP. Inactivation of TSC2 maintains Rheb in its GTP-bound state, thereby augmenting mTORC1 activation (101). Activated Akt also inhibits the activity of both GSK-3α and GSK-3β by phosphorylating GSK-3α at Ser21 or GSK-3β at Ser9 in their N-terminal regulatory domains, which leads to different changes in transcription factors and upregulation of the anti-apoptotic BCL-2 family member MCL-1, transcription factors c-Myc, SREBP1c, β-catenin, HIF1α, glycogen synthase, and glucose transporter protein, etc. (100). Without inhibition by active AKT or p-EGFR, GSK-3β-mediated phosphorylation PD-L1 at T180 and S184 induces its relationship with the E3 ligase beta-transducin repeats-containing protein (b-TrCP), leading to PD-L1 degradation in the cytoplasm (102).
Other potent substrates of AKT are the Forkhead Box O transcription elements (FoxO1, 3, 4, and 6), which are generally considered to be tumor suppressors with established functions in DNA damage repair, the scavenging of reactive oxygen species, cell cycle arrest, apoptosis, metabolism, cell migration and angiogenesis (103). High nutrient availability or oncogenic activation of PI3K promotes Akt-mediated FOXO1 (Ser319, Thr24, and Ser256), FOXO3 (Ser315, Thr32, and Ser253), FOXO4 (Ser258, Thr28, and Ser193) and FOXO6 (Ser184 and Thr26) phosphorylation. This phosphorylation facilitates 14-3-3 chaperone protein binding, promoting FOXO nuclear export to the cytoplasm and preventing FOXO re-entry, blocking FOXO transcriptional activity. FOXOs accumulated in the cytosol can be polyubiquitinated and degraded via ubiquitin–proteasome pathway (103). Conversely, dephosphorylation mediated by PP2A rescues active AKT-phosphorylated FOXO1 and FOXO3 from nuclear exclusion and degradation (104). Extreme oxidative stress activates upstream c-Jun N-terminal kinase (JNK) and mammalian sterile 20-like kinase (MST1), JNK-mediated phosphorylation of FOXO4 (Thr447, Thr451) and FOXO3 (Ser574) and MST1-mediated phosphorylation of FOXO3 (Ser207) and FOXO1 (Ser212); promotes the capacity of FOXO proteins to control the expressions of target genes convoluted in survival (BIM and PUMA), cell growths (Sestrin3, MAP1LCB, and BNIP3), proliferation (p21 and p27), and metabolism (G6PC and PEPCK) is modulated (105,91) (see Figure 2).
Some studies have indicated that AKT signaling can phosphorylate the STAT3 protein, although it is sometimes unclear whether AKT phosphorylates STAT3 directly or indirectly through activating PKM2 or mTOR (105). Interestingly, IGF-1R signaling can activate STAT3 through interferon-induced transmembrane protein 2 (IFITM2) in gastric cancer (106), through the Stat3-Midkine-Stat3 loop in hepatocellular carcinoma (107), through the NF-κB-IL-6-STAT3 loop in non-small cell lung cancer (29), through IGFBP2-MDA-9 in prostate cancer (108), and RACK1 in ovarian cancer (109), suggesting the general need for an adaptor to mediate the activation of STAT3 by IGF-1R signaling. Determining whether this is the case in HNSCC needs careful investigation. Still, regardless, cytokine-mediated activation of STAT3 induces the expression of IGF-1R, so activation of the JAK/STAT3 pathway can cause insulin/IGF-1 signaling sensitization.
5 STAT3/NF-kB/c-Myc and PD-L1
Head and neck and pancreatic cancer cell lines rank the highest according to the cancer inflammation indices, which are based on IL-6 and PD-L1 genes commonly binding-factors NF-κB, STAT3, and AP-1 and their correlated IL-1/NF-κB, IL-6/STAT3, and TNF/AP-1 signaling cascades (28). The coactivation of NF-κB, STAT3, and AP-1 was confirmed in an updated TCGA report of HNSCC (27). Importantly, apart from mediating STAT3 activation via autocrine production and action of IL-6/IL-6R, activated Rac1 was reported to promote the deprivation of IκBα and the nuclear translocation of STAT3-NFκB multiplexes in starved cancer cells (110). Interestingly, cigarette smoke or cigarette smoke extracts (CSE) exposure was found to dose- and time-dependently elevate RAC1 expression and activity in mice or human bronchial epithelial cells under IL-8 and IL-6 expressions. CSE dose- and time-dependently persuaded the phosphorylation of STAT3, Erk1/2, and ERK1/2 or STAT3 inhibitor markedly reduced level of IL-8 and IL-6 after CSE exposure, inhibition of activity or expression of Rac1 blocked cigarette smoke or CSE-induced Erk1/2 and STAT3 phosphorylation in vivo or in vitro (111). Notably, oscillatory (1Hz) shear stress (10 dynes/cm2) activated RAC1 and CDC42 to stimulate the translocation of β-catenin to the nucleus and increase TCF/LEF activity in osteoblasts (112). STAT3 directly binds the IL-6 promoter to persuade IL-6 expressions. HPV was inveterate to drive the Rac1-NFκB-IL-6-STAT3 axis in cervical tumor to elevate IL-6 and STAT3, which correlates with cervical disease progression (113). NF-κB elevates LIN28 levels, inhibits Let-7 expression, and increases its target gene IL-6 expression, which activates NF-κB and STAT3, thereby forming positive feedback loops (56). This loop is reinforced by STAT3 activation, miR-181b, and miR-21, which are detected in the plasma of HNSCC patients (114) and can be up-regulated by STAT3 through direct binding to the promoter regions. miR-181b directly inhibits cylindromatosis (CYLD), a tumor suppressor and negative modulator of NF-κB, while miR-21 directly inhibits PTEN to activate PI3K-AKT- IKK-IκBs pathway. Both lead to NF-κB activation and act as a part of the epigenetic switch through the LIN28-Let-7 axis linking inflammation to cancer (56). PDLIM2, which is downregulated in HNSCC by systematic analysis of TACGA data, is a gene essential for NF-κB and STAT3 deprivation; normally, it improves the expression of proteins included in antigen presentation and endorses T-cell activation while inhibiting multidrug resistance genes, thereby renders mutated cells susceptible to immune observation and cytotoxicity (115). miR-221 subsidizes the constitutive activation of NF-κB and STAT3 by directly targeting and inhibiting PDLIM2 expression (116). Taken together, NF-κB and STAT3 co-activation occur in HNSCC through common induction by RAC1 and IL-6, common inhibition of degradation by PDLIM2, STAT3 indirectly reinforces NF-κB by inhibiting its negative modulator, and finally, co-binding to their common target genes.
The RNA-binding protein Lin28 is present as two homologs, Lin28A and Lin28B. Lin28A, first identified in 1997 in research on C. elegans, is commonly present in mammals (117, 118), while Lin28B was first identified in hepatocellular carcinoma; therefore, both are involved in the development and cancer (119, 120). LIN28 homologs are typically expressed in undifferentiated and pluripotent cells, and their expression is then reduced in response to development and differentiation. Let7 was first identified in C. elegans in 2001 as a typical miRNA 19-22 nucleotide in length that binds the 3’UTRs of target mRNAs, including Lin28 and PD-L1, facilitating mRNA decay and translation inhibition (121). Members of the let7 family perform vital roles in regulating cell differentiation, metabolism, and the progression of certain diseases, including tumorigenesis (122). Lin28A/B binding to pre-let-7 or pri-let-7 not only suppresses let-7 precursor processing by Dicer and Drosha into its mature form but also induces oligo-uridylation at the 3′ terminus of pre-let-7, rendering it susceptible to degradation by the 3’-5’ exonuclease Dis3L2. Thus, the Lin28-Let-7 axis is considered a double-negative feedback loop involved in regulating various biological functions; normally, most LIN28-binding sites are non-miRNA transcript, such as ribosomal and coding RNA, whose binding sequesters the LIN28 protein and competitively inhibits LIN28-repression on cellular miRNAs (123). LIN28-mediated mechanisms that are either Let-7-dependent or Let-7-independent regulate the hallmarks of cancer, such as proliferation, metabolism, the evasion of immune destruction, tumor-associated inflammation, cancer cell death, and genome instability (124) (see Figure 3; Table 1).
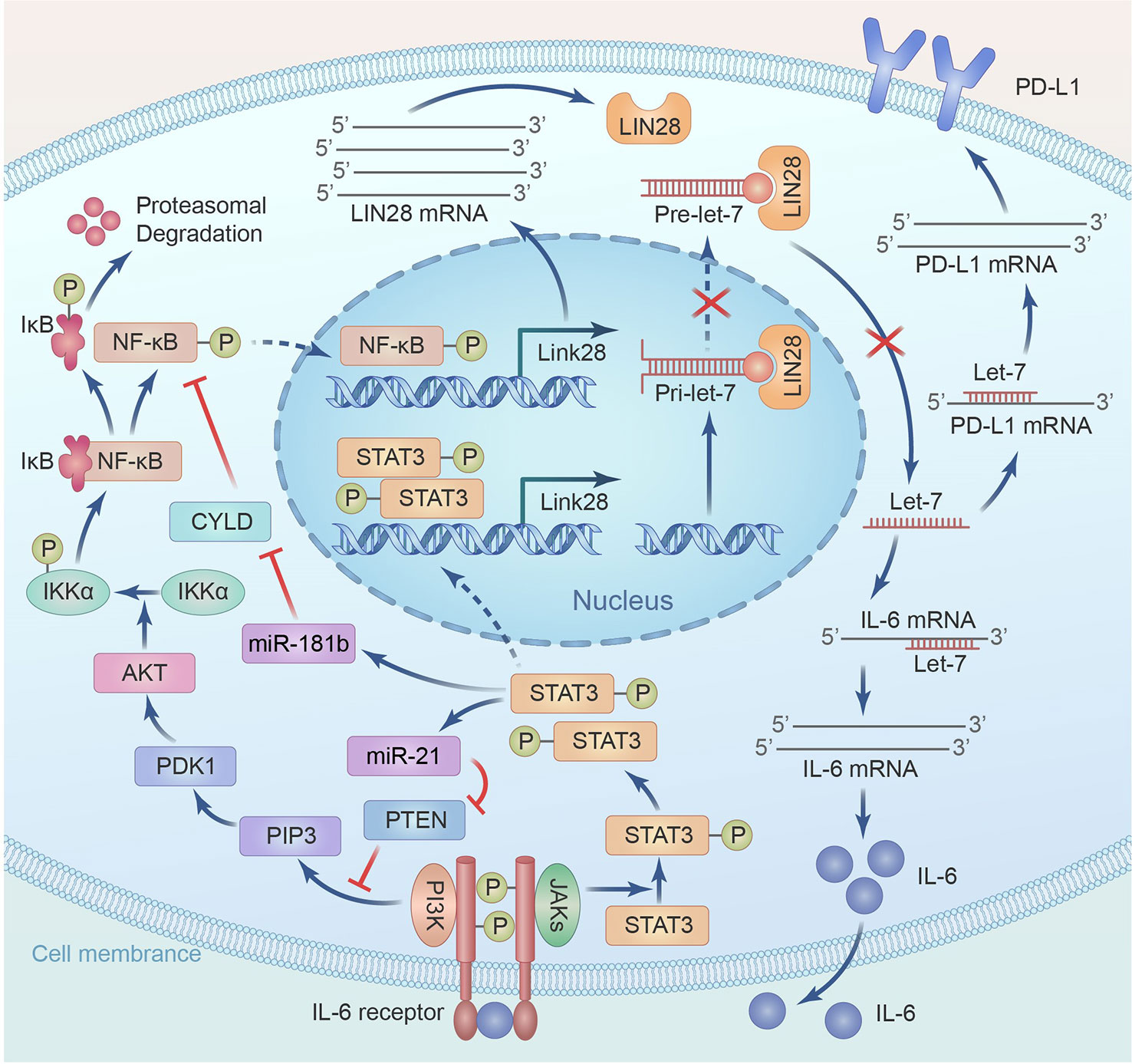
Figure 3 A positive feed-back between LIN28/Let7 axis and NF-κB/STAT3 mediates up-regulation of IL-6 and PD-L1.

Table 1 Lin28 regulates Let7 related and Let7 independent targets in cancer.
Lin28 overexpression and let-7 inhibition increase IL-6 expression and subsequently activate NF-κB and STAT3, but accumulating evidence in various cancers, including HNSCC, has revealed that Lin28 overexpression and let-7 inhibition also leads to Myc overexpression. In contrast, the induction of let-7a expression significantly reduces Myc expression. The binding of Let-7 miRNA represses Myc mRNA, and Lin28 elevates Myc expression by inhibiting Let-7 miRNA binding. Moreover, Myc can increase Lin28 expression by occupying the Lin28a and Lin28b promoters, so a reciprocal positive feedback loop between Lin28 and Myc exists (124). c-Myc binds directly to the E-box component in the CCAT-1 promoter area and increases CCAT-1 expression (125). CCAT-1-L resides inside a super-enhancer region and promotes c-Myc production by facilitating chromatin looping connections among the c-Myc promoter and its enhancers via its interaction with CTCF (126). In addition, CCAT-1 may increase c-Myc expression by acting as a sponge for tumor-suppressor microRNAs that straightly target c-Myc (127). Therefore, a positive feedback loop among CCAT-1 and c-Myc boosts cancer cell aggressiveness by increasing the expression of each gene. Myc gene amplification is common in many solid tumor types, while chromosomal translocation is common in B-cell/T-cell leukemias and lymphomas; Myc is activated in about 70% of HNSCC via amplification or pathway activation (128). The detailed mechanisms of Myc activation by related signaling pathways and the multiple crosstalk mechanisms with the hallmarks of cancer are presented in Figure 4.
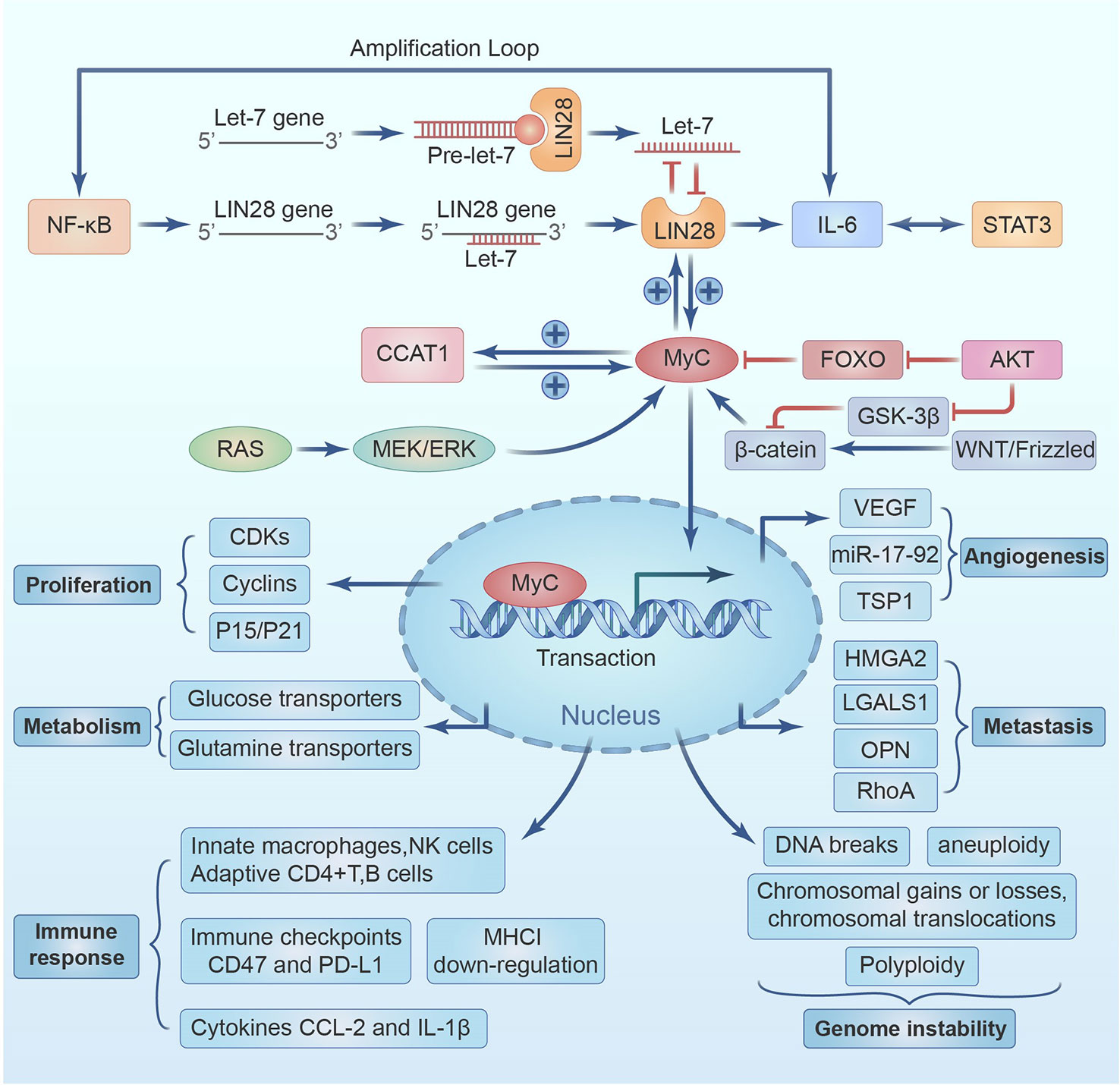
Figure 4 Mechanisms of Myc activation and cross-talk with the hallmarks of cancer.
Based on mechanisms that contribute to concomitant activation, the master transcription factors NF-κB, STAT3, FOXO, AKT and Myc bind the promoter of LIN28 and PD-L1 and upregulate their expressions. Similarly, the RAS and MEK/ERK also bind with the promoter of Myc and eventually upregulate the expression of LIN28. The β-catein, GSK-3 β and WNT/Frizzled also affect the expression of Myc and LIN28. The expression of Myc leads to the activation of CDKs, Cyclins and P15/P21 which causes cell proliferation. The LIN28-Let7 axis underpins the whole IGF and PD-L1 upregulation network. Furthermore, the expression of Myc is associated with VEGF, and miR-17-92 and TSP1 that causes angiogenesis. Myc is also associated with HMGA2, LGALS1, OPN and RhoA linked with the metastasis. Let-7a downregulation is negatively linked with higher CCAT1, c-Myc, and PD-L1 expression levels in TNBC, confirming this hypothesis. Let-7a overexpression or CCAT1 inhibition dramatically suppressed PD-L1 expression in MDA-MB-231 cells and TNBC reducing cell survival and colony formation (129). Interestingly, metformin and the Lin28A inhibitor C1632 exhibited synergistic antitumor effects in oral squamous cell carcinoma via increasing mature Let-7 and downregulated PD-L1. In addition to the ability of metformin to decrease c-myc to induce tristetraprolin expression in breast cancer (7), it stimulates Dicer through the AMP-activated protein kinase pathways to permit the maturation of let-7 (121). Let-7 overexpression was reported to reduce PD-L1 glycosylation and endorse its deprivation in HNSCC, and the specific mechanism needs clarification (130). In addition, C1632 therapy may inhibit PD-L1 in antigen-presenting THP-1 macrophage, and the increased release of interferon and TNFα boosts T-cell-mediated anti-tumor activity (131). Despite the discovery of T helper 17 (Th17) cells in tumor tissues, their role in cancer immunity remains unknown. Mice lacking the immune cell-signaling molecule interleukin-17A (IL-17A) are more likely to develop malignant lung cancers. Instead, adoptive T-cell treatment with tumor-specific Th17 cells successfully suppressed tumor growth. Th17 cell treatment was thought to have little impact on tumor growth, dramatically stimulated the production of tumor-specific CD8+ T cells, which were crucial to the therapeutic outcome. Tumor-infiltrating dendritic cells (DCs) and CD8(+) DCs carrying tumor antigens were both enhanced in number by Th17 cells draining to lymph nodes. Th17 cells boosted the production of the chemokine CCL20 in tumor tissues. Because of this, tumor-specific CD8+ T cell activation is facilitated by the inflammation induced by Th17 cells. These results may have major consequences for anticancer immunotherapies (132). In cancer immunoediting, tumors may evade the immune system by eliminating primary histocompatibility compound class I (MHC-I) molecules. The vast majority of natural killer (NK) cells in advanced malignancies have been shown in recent research to be faulty, allowing malignant MHC-I-deficient tumors to escape the regulation of the immune system. Here, we demonstrate that a vaccination based on antigen-presenting cells (APCs) expressing tumor antigens loaded with natural killer T (NKT)-cell ligands are very effective against these late-stage tumors. They discover that MHC-I downregulation in tumors is intimately linked to developing NK-cell fatigue in tumor-bearing mice and human cancer patients. This co-expression characterizes functionally tired NK cells in advanced tumors. The anti-tumor benefits of the vaccination against advanced tumors depend partly on IL-21’s ability to restore NK-cell activity. These findings provide light on the mechanism behind the generation of NK-cell dysfunction in advanced tumors and may help guide the design of immunotherapeutic approaches to this disease. The methodologies for studying linked biomarkers, like high-throughput sequencing of T-cell receptors (TCRs), are discussed, as are the bioinformatics involved in designing personalized, neoantigen-based vaccines, including detection of mutations and the prediction of potential epitopes, as well as data analysis and the bioinformatics quantitative of immune cell infiltration in tumor tissues. (133).
6 Conclusions
These findings show that mutually connected IL-6, EGFR, and LIN28/Let7 axis-related mechanisms modulate IGF and PD-L1 upregulation in the HNSCC microenvironment, which comprehensively influences immunity, inflammation, metabolism, and metastasis and might be fundamental for overcoming therapy resistance. IGF-1/2 gene expression is stimulated by IL-6, which also cross-activates the IGF-1R signaling pathway, NF-κB, and STAT3 genes to promote their expression. PD-L1 is stabilized and transported to the cell surface through STT3-mediated PD-L1 glycosylation, which is facilitated by IL-6/JAK1. It has been concluded that PD-L1 and IGF overexpression in HNSCC are mediated by IL-6, EGFR, and LIN28/Let7-related pathways. This extensively affects immunity, inflammation, metabolism, and metastasis in the tumor microenvironment and may be crucial for overcoming therapeutic resistance.
Author contributions
WL proposed the idea. SF and HY affirmed its value. WL developed the train of thought and wrote the article. JL, XJ and YX took part in the discussion. SF revised the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Research Climb Plan of Hunan Cancer Hospital (Wei Liu 20201228-1001).
Acknowledgments
We want to express our special thanks to Huayue Yu and Qiuqiu Song, only for their kind encouragement and nurturing tolerance, we have travelled the long way from initial conception to final composition of this review article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Ianza A, Sirico M, Bernocchi O, Generali D. Role of the IGF-1 axis in overcoming resistance in breast cancer. Front Cell Dev Biol (2021) 9:641449. doi: 10.3389/fcell.2021.641449
2. Li H, Batth IS, Qu X, Xu L, Song N, Wang R, et al. IGF-IR signaling in epithelial to mesenchymal transition and targeting IGF-IR therapy: overview and new insights. Mol Cancer (2017) 16(1). doi: 10.1186/s12943-016-0576-5
3. Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer (2012) 12(3):159–69. doi: 10.1038/nrc3215
4. Simpson A, Petnga W, Macaulay VM, Weyer-Czernilofsky U, Bogenrieder T. Insulin-like growth factor (IGF) pathway targeting in cancer: Role of the IGF axis and opportunities for future combination studies. Targeted Oncol (2017) 12(5):571–97. doi: 10.1007/s11523-017-0514-5
5. Radhakrishnan Y, Shen X, Maile LA, Xi G, Clemmons DR. IGF-I stimulates cooperative interaction between the IGF-I receptor and CSK homologous kinase that regulates SHPS-1 phosphorylation in vascular smooth muscle cells. Mol Endocrinol (2011) 25(9):1636–49. doi: 10.1210/me.2011-0035
6. Sbuquois B, Carré N, Burnol A-F. Regulation of insulin and type 1 insulin-like growth factor signaling and action by the Grb10/14 and SH2B1/B2 adaptor proteins. FEBS J (2013) 280(3):794–816. doi: 10.1111/febs.12080
7. Yu D, Liu X, Han G, Liu Y, Zhao X, Wang D, et al. The let-7 family of microRNAs suppresses immune evasion in head and neck squamous cell carcinoma by promoting PD-L1 degradation. Cell Communication Signaling (2019) 17(1). doi: 10.1186/s12964-019-0490-8
8. Peng M, Fan S, Li J, Zhou X, Liao Q, Tang F, et al. Programmed death-ligand 1 signaling and expression are reversible by lycopene via PI3K/AKT and Raf/MEK/ERK pathways in tongue squamous cell carcinoma. Genes Nutr (2022) 17(1). doi: 10.1186/s12263-022-00705-y
9. Li J, Xu J, Yan X, Jin K, Li W, Zhang R. Targeting interleukin-6 (IL-6) sensitizes anti-PD-L1 treatment in a colorectal cancer preclinical model. Med Sci Monitor (2018) 24:5501–8. doi: 10.12659/msm.907439
10. Tsukamoto H, Fujieda K, Miyashita A, Fukushima S, Ikeda T, Kubo Y, et al. Combined blockade of IL6 and PD-1/PD-L1 signaling abrogates mutual regulation of their immunosuppressive effects in the tumor microenvironment. Cancer Res (2018) 78(17):5011–22. doi: 10.1158/0008-5472.can-18-0118
11. Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, et al. Complementary DNA for a novel human interleukin (BSF-2) that induces b lymphocytes to produce immunoglobulin. Nature (1986) 324(6092):73–6. doi: 10.1038/324073a0
12. Kumari N, Dwarakanath BS, Das A, Bhatt AN. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour biol: J Int Soc Oncodevelopmental Biol Med (2016) 37(9):11553–72. doi: 10.1007/s13277-016-5098-7
13. Hirano T. IL-6 in inflammation, autoimmunity and cancer. Int Immunol (2020) 33(3):127–48. doi: 10.1093/intimm/dxaa078
14. Ayala-Mar S, Donoso-Quezada J, González-Valdez J. Clinical implications of exosomal PD-L1 in cancer immunotherapy. J Immunol Res (2021) 2021:8839978. doi: 10.1155/2021/8839978 eCollection 2021.
15. Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (2012) 122(4):143–59. doi: 10.1042/cs20110340
16. Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol (2018) 15(4):234–48. doi: 10.1038/nrclinonc.2018.8
17. Naseem S, Hussain T, Manzoor S. Interleukin-6: A promising cytokine to support liver regeneration and adaptive immunity in liver pathologies. Cytokine Growth Factor Rev (2018) 39:36–45. doi: 10.1016/j.cytogfr.2018.01.002
18. Zegeye MM, Lindkvist M, Fälker K, Kumawat AK, Paramel G, Grenegård M, et al. Activation of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL-6 trans-signaling-mediated pro-inflammatory response in human vascular endothelial cells. Cell Communication Signaling (2018) 16(1). doi: 10.1186/s12964-018-0268-4
19. Orme JJ, Jazieh KA, Xie T, Harrington S, Liu X, Ball M, et al. ADAM10 and ADAM17 cleave PD-L1 to mediate PD-(L)1 inhibitor resistance. OncoImmunology (2020) 9(1). doi: 10.1080/2162402x.2020.1744980
20. Prestipino A, Emhardt AJ, Aumann K, O’Sullivan D, Gorantla SP, Duquesne S, et al. Oncogenic JAK2 V617F causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci Trans Med (2018) 10(429):eaam7729. doi: 10.1126/scitranslmed.aam7729
21. Hsu J-M, Xia W, Hsu Y-H, Chan L-C, Yu W-H, Cha J-H, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun (2018) 9(1):1908. doi: 10.1038/s41467-018-04313-6
22. Cha J-H, Chan L-C, Li C-W, Hsu JL, Hung M-C. Mechanisms controlling PD-L1 expression in cancer. Mol Cell (2019) 76(3):359–70. doi: 10.1016/j.molcel.2019.09.030
23. Zerdes I, Matikas A, Bergh J, Rassidakis GZ, Foukakis T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: biology and clinical correlations. Oncogene (2018) 37(34):4639–61. doi: 10.1038/s41388-018-0303-3
24. Wang Y, Shen Y, Wang S, Shen Q, Zhou X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett (2018) 415:117–28. doi: 10.1016/j.canlet.2017.12.003
25. Campbell JD, Yau C, Bowlby R, Liu Y, Brennan K, Fan H, et al. Genomic, pathway network, and immunologic features distinguishing squamous carcinomas. Cell Rep (2018) 23(1):194–212.e6. doi: 10.1016/j.celrep.2018.03.063
26. Ji Z, He L, Regev A, Struhl K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc Natl Acad Sci (2019), 201821068. doi: 10.1073/pnas.1821068116
27. Zheng X, Lu G, Yao Y, Gu W. An autocrine IL-6/IGF-1R loop mediates EMT and promotes tumor growth in non-small cell lung cancer. Int J Biol Sci (2019) 15(9):1882–91. doi: 10.7150/ijbs.31999
28. Chang T-S, Wu Y-C, Chi C-C, Su W-C, Chang P-J, Lee K-F, et al. Activation of IL6/IGFIR confers poor prognosis of HBV-related hepatocellular carcinoma through induction of OCT4/NANOG expression. Clin Cancer Res (2015) 21(1):201–10. doi: 10.1158/1078-0432.ccr-13-3274
29. Bharadwaj U, Kasembeli MM, Robinson P, Tweardy DJ. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: Rationale, progress, and caution. Pharmacol Rev (2020) 72(2):486–526. doi: 10.1124/pr.119.018440
30. Böhmer F-D, Friedrich K. Protein tyrosine phosphatases as wardens of STAT signaling. JAK-STAT (2014) 3(1):e28087. doi: 10.4161/jkst.28087
31. Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in cancer immunotherapy. Mol Cancer (2020) 19(1):145. doi: 10.1186/s12943-020-01258-7
32. Yuan J, Zhang F, Niu R. Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci Rep (2015) 5:17663. doi: 10.1038/srep17663
33. Kim SJ, Yoon S. Activated Rac1 regulates the degradation of IκBα and the nuclear translocation of STAT3-NFκB complexes in starved cancer cells. Exp Mol Med (2016) 48(5):e231. doi: 10.1038/emm.2016.17
34. Peyser ND, Du Y, Li H, Lui V, Xiao X, Chan TA, et al. Loss-of-Function PTPRD mutations lead to increased STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. PloS One (2015) 10(8):e0135750. doi: 10.1371/journal.pone.0135750
35. Solomon DA, Kim J-S, Cronin JC, Sibenaller Z, Ryken T, Rosenberg SA, et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res (2008) 68(24):10300–6. doi: 10.1158/0008-5472.can-08-3272
36. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer (2014) 14(11):736–46. doi: 10.1038/nrc3818
37. Cimica V, Chen H-C, Iyer JK, Reich NC. Dynamics of the STAT3 transcription factor: Nuclear import dependent on ran and importin-β1. PloS One (2011) 6(5):e20188. doi: 10.1371/journal.pone.0020188
38. Naseem S, Hussain T, Manzoor S. Interleukin-6: A promising cytokine to support liver regeneration and adaptive immunity in liver pathologies. Cytokine Growth Factor Rev (2018) 39:36–45. doi: 10.1016/j.cytogfr.2018.01.002
39. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9(11):798–809. doi: 10.1038/nrc2734
40. Sadrkhanloo M, Entezari M, Orouei S, Ghollasi M, Rezaei S, Hejazi ES, et al. STAT3-EMT axis in tumors: Modulation of cancer metastasis, stemness and therapy response. Pharmacol Res (2022) 182:106311. doi: 10.1016/j.phrs.2022.106311
41. Tan F, Putoczki T, Stylli S, Luwor R. The role of STAT3 signaling in mediating tumor resistance to cancer therapy. Curr Drug Targets (2014) 15(14):1341–53. doi: 10.2174/1389450115666141120104146
42. Lin W-H, Chang Y-W, Hong M-X, Hsu T-C, Lee K-C, Lin C, et al. STAT3 phosphorylation at Ser727 and Tyr705 differentially regulates the EMT–MET switch and cancer metastasis. Oncogene (2020) 40(4):791–805. doi: 10.1038/s41388-020-01566-8
43. Yotsumoto F, Sanui A, Fukami T, Shirota K, Horiuchi S, Tsujioka H, et al. Efficacy of ligand-based targeting for the EGF system in cancer. Anticancer Res (2009) 29(11):4879–85. doi: 10.1038/s41388-020-01566-8
44. Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer (2018) 17(1):58. doi: 10.1186/s12943-018-0782-4
45. Ortiz-Cuaran S, Bouaoud J, Karabajakian A, Fayette J, Saintigny P. Precision medicine approaches to overcome resistance to therapy in head and neck cancers. Front Oncol (2021) 11:614332. doi: 10.3389/fonc.2021.614332
46. Faria JAQA, de Andrade C, Goes AM, Rodrigues MA, Gomes DA. Effects of different ligands on epidermal growth factor receptor (EGFR) nuclear translocation. Biochem Biophys Res Commun (2016) 478(1):39–45. doi: 10.1016/j.bbrc.2016.07.097
47. Ghosh S, Nataraj NB, Noronha A, Patkar S, Sekar A, Mukherjee S, et al. PD-L1 recruits phospholipase c and enhances tumorigenicity of lung tumors harboring mutant forms of EGFR. Cell Rep (2021) 35(8):109181. doi: 10.1016/j.celrep.2021.109181
48. Faruqi TR, Gomez D, Bustelo XR, Bar-Sagi D, Reich NC. Rac1 mediates STAT3 activation by autocrine IL-6. Proc Natl Acad Sci (2001) 98(16):9014–9. doi: 10.1073/pnas.161281298
49. Zhu P, Kang G, Jiao Y, Gui C, Fan H, Li X, et al. The α5-nAChR/PD-L1 axis facilitates lung adenocarcinoma cell migration and invasion. Hum Cell (2022) 35(4):1207–18. doi: 10.1007/s13577-022-00709-1
50. Shao Q-Q, Zhang T-P, Zhao W-J, Liu Z-W, You L, Zhou L, et al. Filamin a: Insights into its exact role in cancers. Pathol Oncol research: POR (2016) 22(2):245–52. doi: 10.1007/s12253-015-9980-1
51. Frezza V, Fierro C, Gatti E, Peschiaroli A, Lena AM, Petruzzelli MA, et al. ΔNp63 promotes IGF1 signalling through IRS1 in squamous cell carcinoma. Aging (2018) 10(12):4224–40. doi: 10.18632/aging.101725
52. Jiang Y, Jiang Y-Y, Xie J-J, Mayakonda A, Hazawa M, Chen L, et al. Co-Activation of super-enhancer-driven CCAT1 by TP63 and SOX2 promotes squamous cancer progression. Nat Commun (2018) 9(1):3619. doi: 10.1038/s41467-018-06081-9
53. Cimadamore F, Amador-Arjona A, Chen C, Huang C-T, Terskikh AV. SOX2-LIN28/let-7 pathway regulates proliferation and neurogenesis in neural precursors. Proc Natl Acad Sci (2013) 110(32):E3017–26. doi: 10.1073/pnas.1220176110
54. Wang H, Zhao Q, Deng K, Guo X, Xia J. Lin28: an emerging important oncogene connecting several aspects of cancer. Tumor Biol (2016) 37(3):2841–8. doi: 10.1007/s13277-015-4759-2
55. Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu Z, et al. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol (2016) 49(4):1360–8. doi: 10.3892/ijo.2016.3632
56. Chen S-F, Zhang Z-Y, Zhang J-L. Matrine increases the inhibitory effects of afatinib on H1975 cells via the IL−6/JAK1/STAT3 signaling pathway. Mol Med Rep (2017) 16(3):2733–9. doi: 10.3892/mmr.2017.6865
57. Lou W, Chen Y, Zhu K-Y, Deng H, Wu T, Wang J. Polyphyllin I overcomes EMT-associated resistance to erlotinib in lung cancer cells via IL-6/STAT3 pathway inhibition. Biol Pharm Bull (2017) 40(8):1306–13. doi: 10.1248/bpb.b17-00271
58. Chan L-C, Li C-W, Xia W, Hsu J-M, Lee H-H, Cha J-H, et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Invest (2019) 129(8):3324–38. doi: 10.1172/JCI126022
59. Yan M, Han M, Yang X, Shen R, Wang H, Zhang L, et al. Dual inhibition of EGFR and IL-6-STAT3 signalling by miR-146b: a potential targeted therapy for epithelial ovarian cancer. J Enzyme Inhibition Medicinal Chem (2021) 36(1):1905–15. doi: 10.1080/14756366.2021.1963240
60. Wang Y, van Boxel-Dezaire AHH, Cheon H, Yang J, Stark GR. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc Natl Acad Sci (2013) 110(42):16975–80. doi: 10.1073/pnas.1315862110
61. Poncet N, Guillaume J, Mouchiroud G. Epidermal growth factor receptor transactivation is implicated in IL-6-induced proliferation and ERK1/2 activation in non-transformed prostate epithelial cells. Cell Signalling (2011) 23(3):572–8. doi: 10.1016/j.cellsig.2010.11.009
62. Yau SW, Azar WJ, Sabin MA, Werther GA, Russo VC. IGFBP-2 - taking the lead in growth, metabolism and cancer. J Cell Communication Signaling (2015) 9(2):125–42. doi: 10.1007/s12079-015-0261-2
63. Li T, Zhang C, Zhao G, Zhang X, Hao M, Hassan S, et al. IGFBP2 regulates PD-L1 expression by activating the EGFR-STAT3 signaling pathway in malignant melanoma. Cancer Lett (2020) 477:19–30. doi: 10.1016/j.canlet.2020.02.036
64. Degirmenci U, Wang M, Hu J. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells (2020) 9(1):198. doi: 10.3390/cells9010198
65. Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase direct target of ras. Nature (1994) 370(6490):527–32. doi: 10.1038/370527a0
66. Coelho MA, de Carné Trécesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity (2017) 47(6):1083–1099.e6. doi: 10.1016/j.immuni.2017.11.016
67. Steck PA, Pershouse MA, Jasser SA, Yung WKA, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet (1997) 15(4):356–62. doi: 10.1038/ng0497-356
68. Squarize CH, Castilho RM, Abrahao AC, Molinolo A, Lingen MW, Gutkind JS. PTEN deficiency contributes to the development and progression of head and neck cancer. Neoplasia (2013) 15(5):461–71. doi: 10.1593/neo.121024
69. Aquila S, Santoro M, Caputo A, Panno ML, Pezzi V, De Amicis F. The tumor suppressor PTEN as molecular switch node regulating cell metabolism and autophagy: Implications in immune system and tumor microenvironment. Cells (2020) 9(7):1725. doi: 10.3390/cells9071725
70. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med (2006) 13(1):84–8. doi: 10.1038/nm1517
71. Haddadi N, Lin Y, Travis G, Simpson AM, Nassif NT, McGowan EM. PTEN/PTENP1: ‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol Cancer (2018) 17(1):37. doi: 10.1186/s12943-018-0803-3
72. Jiang N, Dai Q, Su X, Fu J, Feng X, Peng J. Role of PI3K/AKT pathway in cancer: the framework of malignant behavior. Mol Biol Rep (2020) 47(6):4587–629. doi: 10.1007/s11033-020-05435-1
73. He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW, et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduction Targeted Ther (2021) 6(1):1–17. doi: 10.1038/s41392-021-00828-5
74. Chen C-Y, Chen J, He L, Stiles BL. PTEN: Tumor suppressor and metabolic regulator. Front Endocrinol (2018) 9:338. doi: 10.3389/fendo.2018.00338
75. Cordero-Espinoza L, Hagen T. Increased concentrations of fructose 2,6-bisphosphate contribute to the warburg effect in phosphatase and tensin homolog (PTEN)-deficient cells. J Biol Chem (2013) 288(50):36020–8. doi: 10.1074/jbc.M113.510289
76. Lee Y-R, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol (2018) 19(9):547–62. doi: 10.1038/s41580-018-0015-0
77. Kanagasabai T, Li G, Shen TH, Gladoun N, Castillo-Martin M, Celada SI, et al. MicroRNA-21 deficiency suppresses prostate cancer progression through downregulation of the IRS1-SREBP-1 signaling pathway. Cancer Lett (2022) 525:46–54. doi: 10.1016/j.canlet.2021.09.041
78. Deng X, Zhang W, O-Sullivan I, Williams JB, Dong Q, Park EA, et al. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J Biol Chem (2012) 287(24):20132–43. doi: 10.1074/jbc.m112.347211
79. Hu Y, Zai H, Jiang W, Ou Z, Yao Y, Zhu Q. The mutual inhibition of FoxO1 and SREBP-1c regulated the progression of hepatoblastoma by regulating fatty acid metabolism. Mediators Inflammation (2021) 2021:5754592. doi: 10.1155/2021/5754592
80. Wang Y, Wang H, Zhao Q, Xia Y, Hu X, Guo J. PD-L1 induces epithelial-to-mesenchymal transition via activating SREBP-1c in renal cell carcinoma. Med Oncol (Northwood London England) (2015) 32(8):212. doi: 10.1007/s12032-015-0655-2
81. Xu G-L, Ni C-F, Liang H-S, Xu Y-H, Wang W-S, Shen J, et al. Upregulation of PD-L1 expression promotes epithelial-to-mesenchymal transition in sorafenib-resistant hepatocellular carcinoma cells. Gastroenterol Rep (2020) 8(5):390–8. doi: 10.1093/gastro/goaa049
82. Park MK, Yao Y, Xia W, Setijono SR, Kim JH, Vila IK, et al. PTEN self-regulates through USP11 via the PI3K-FOXO pathway to stabilize tumor suppression. Nat Commun (2019) 10(1):636. doi: 10.1038/s41467-019-08481-x
83. He L, Xu K, Niu L, Lin L. Astragalus polysaccharide (APS) attenuated PD-L1-mediated immunosuppression via the miR-133a-3p/MSN axis in HCC. Pharm Biol (2022) 60(1):1710–20. doi: 10.1080/13880209.2022.2112963
84. Uygur B, Abramo K, Leikina E, Vary C, Liaw L, Wu W-S. SLUG is a direct transcriptional repressor of PTEN tumor suppressor. Prostate (2015) 75(9):907–16. doi: 10.1002/pros.22974
85. Ghafouri-Fard S, Abak A, Shoorei H, Mohaqiq M, Majidpoor J, Sayad A, et al. Regulatory role of microRNAs on PTEN signaling. Biomed Pharmacother (2021) 133:110986. doi: 10.1016/j.biopha.2020.110986
86. Taylor H, Laurence ADJ, Uhlig HH. The role of PTEN in innate and adaptive immunity. Cold Spring Harbor Perspect Med (2019) 9(12):a036996. doi: 10.1101/cshperspect.a036996
87. Marquard FE, Jücker M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem Pharmacol (2020) 172:113729. doi: 10.1016/j.bcp.2019.113729
88. Wang Z, Goto Y, Allevato MM, Wu VH, Saddawi-Konefka R, Gilardi M, et al. Disruption of the HER3-PI3K-mTOR oncogenic signaling axis and PD-1 blockade as a multimodal precision immunotherapy in head and neck cancer. Nat Commun (2021) 12(1):2383. doi: 10.1038/s41467-021-22619-w
89. Akbari Dilmaghani N, Safaroghli-Azar A, Pourbagheri-Sigaroodi A, Bashash D. The PI3K/Akt/mTORC signaling axis in head and neck squamous cell carcinoma: Possibilities for therapeutic interventions either as single agents or in combination with conventional therapies. IUBMB Life (2021) 73(4):618–42. doi: 10.1002/iub.2446
90. Xu Z, Han X, Ou D, Liu T, Li Z, Jiang G, et al. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol (2019) 104(2):575–87. doi: 10.1007/s00253-019-10257-8
91. Manning BD, Toker A. AKT/PKB signaling: Navigating the network. Cell (2017) 169(3):381–405. doi: 10.1016/j.cell.2017.04.001
92. Milligan G, Kostenis E. Heterotrimeric G-proteins: a short history. Br J Pharmacol (2009) 147(S1):S46–55. doi: 10.1038/sj.bjp.0706405
93. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers (2020) 6(1):92. doi: 10.1038/s41572-020-00224-3
94. Kurosu H, Maehama T, Okada T, Yamamoto T, Hoshino S, Fukui Y, et al. Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110beta is synergistically activated by the betagamma subunits of G proteins and phosphotyrosyl peptide. J Biol Chem (1997) 272(39):24252–6. doi: 10.1074/jbc.272.39.24252
95. Schiller MR. Coupling receptor tyrosine kinases to rho GTPases–GEFs what’s the link. Cell Signalling (2006) 18(11):1834–43. doi: 10.1016/j.cellsig.2006.01.022
96. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase bα. Curr Biol (1997) 7(4):261–9. doi: 10.1016/s0960-9822(06)00122-9
97. Sarbassov DD. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307(5712):1098–101. doi: 10.1126/science.1106148
98. Meier R. Inactivation and dephosphorylation of protein kinase balpha (PKBalpha) promoted by hyperosmotic stress. EMBO J (1998) 17(24):7294–303. doi: 10.1093/emboj/17.24.7294
99. Hoxhaj G, Hughes-Hallett J, Timson RC, Ilagan E, Yuan M, Asara JM, et al. The mTORC1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep (2017) 21(5):1331–46. doi: 10.1016/j.celrep.2017.10.029
100. Jiramongkol Y, Lam EW-F. FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev (2020) 39(3):681–709. doi: 10.1007/s10555-020-09883-w
101. Li C-W, Lim S-O, Xia W, Lee H-H, Chan L-C, Kuo C-W, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun (2016) 7(1):12632. doi: 10.1038/ncomms12632
102. Singh A, Ye M, Bucur O, Zhu S, Tanya Santos M, Rabinovitz I, et al. Protein phosphatase 2A reactivates FOXO3a through a dynamic interplay with 14-3-3 and AKT. Mol Biol Cell (2010) 21(6):1140–52. doi: 10.1091/mbc.e09-09-0795
103. Salminen A, Kaarniranta K, Kauppinen A. Insulin/IGF-1 signaling promotes immunosuppression via the STAT3 pathway: impact on the aging process and age-related diseases. Inflammation Res (2021) 70(10-12):1043–61. doi: 10.1007/s00011-021-01498-3
104. Xu L, Zhou R, Yuan L, Wang S, Li X, Ma H, et al. IGF1/IGF1R/STAT3 signaling-inducible IFITM2 promotes gastric cancer growth and metastasis. Cancer Lett (2017) 393:76–85. doi: 10.1016/j.canlet.2017.02.014
105. Bie C, Chen Y, Tang H, Li Q, Zhong L, Peng X, et al. Insulin-like growth factor 1 receptor drives hepatocellular carcinoma growth and invasion by activating Stat3-Midkine-Stat3 loop. Digestive Dis Sci (2022) 67(2):569–84. doi: 10.1007/s10620-021-06862-1
106. Das SK, Pradhan AK, Bhoopathi P, Talukdar S, Shen X-N, Sarkar D, et al. The MDA-9/Syntenin/IGF1R/STAT3 axis directs prostate cancer invasion. Cancer Res (2018) 78(11):2852–63. doi: 10.1158/0008-5472.CAN-17-2992
107. Zhang W, Zong CS, Hermanto U, Lopez-Bergami P, Ronai Z, Wang L-H. RACK1 recruits STAT3 specifically to insulin and insulin-like growth factor 1 receptors for activation, which is important for regulating anchorage-independent growth. Mol Cell Biol (2006) 26(2):413–24. doi: 10.1128/mcb.26.2.413-424.2006
108. Kim SJ, Yoon S. Activated Rac1 regulates the degradation of IκBα and the nuclear translocation of STAT3–NFκB complexes in starved cancer cells. Exp Mol Med (2016) 48(5):e231–1. doi: 10.1038/emm.2016.17
109. Jiang J-X, Zhang S-J, Shen H-J, Guan Y, Liu Q, Zhao W, et al. Rac1 signaling regulates cigarette smoke-induced inflammation in the lung via the Erk1/2 MAPK and STAT3 pathways. Biochim Et Biophys Acta Mol Basis Dis (2017) 1863(7):1778–88. doi: 10.1016/j.bbadis.2017.04.013
110. Wan Q, Cho E, Yokota H, Na S. Rac1 and Cdc42 GTPases regulate shear stress-driven β-catenin signaling in osteoblasts. Biochem Biophys Res Commun (2013) 433(4):502–7. doi: 10.1016/j.bbrc.2013.03.020
111. Morgan EL, Macdonald A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1–NFκB—IL-6 signalling axis. PloS Pathog (2019) 15(6):e1007835. doi: 10.1371/journal.ppat.1007835
112. de Paula Silva E, Marti LC, Andreghetto FM, de Sales RO, Hoberman M, Dos Santos Dias B, et al. Extracellular vesicles cargo from head and neck cancer cell lines disrupt dendritic cells function and match plasma microRNAs. Sci Rep (2021) 11(1):18534. doi: 10.1038/s41598-021-97753-y
113. Zeng Y, Lin D, Gao M, Du G, Cai Y. Systematic evaluation of the prognostic and immunological role of PDLIM2 across 33 cancer types. Sci Rep (2022) 12(1):1933. doi: 10.1038/s41598-022-05987-1
114. Liu S, Sun X, Wang M, Hou Y, Zhan Y, Jiang Y, et al. A microRNA 221– and 222–mediated feedback loop maintains constitutive activation of NFκB and STAT3 in colorectal cancer cells. Gastroenterology (2014) 147(4):847–859.e11. doi: 10.1053/j.gastro.2014.06.006
115. Horvitz HR, Sulston JE. ISOLATION AND GENETIC CHARACTERIZATION OF CELL-LINEAGE MUTANTS OF THE NEMATODE CAENORHABDITIS ELEGANS. Genetics (1980) 96(2):435–54. doi: 10.1093/genetics/96.2.435
116. Guo Y, Chen Y, Ito H, Watanabe A, Ge X, Kodama T, et al. Identification and characterization of lin-28 homolog b (LIN28B) in human hepatocellular carcinoma. Gene (2006) 384:51–61. doi: 10.1016/j.gene.2006.07.011
117. Chen Y, Xie C, Zheng X, Nie X, Wang Z, Liu H, et al. LIN28/let-7/PD-L1 pathway as a target for cancer immunotherapy. Cancer Immunol Res (2019) 7(3):487–97. doi: 10.1158/2326-6066.cir-18-0331
118. Parisi S, Castaldo D, Piscitelli S, D’Ambrosio C, Divisato G, Passaro F, et al. Identification of RNA-binding proteins that partner with Lin28a to regulate Dnmt3a expression. Sci Rep (2021) 11(1):2345. doi: 10.1038/s41598-021-81429-8
119. Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol (2008) 18(10):505–16. doi: 10.1016/j.tcb.2008.07.007
120. Krsnik D, Marić T, Bulić-Jakuš F, Sinčić N, Bojanac AK. LIN28 family in testis: Control of cell renewal, maturation, fertility and aging. Int J Mol Sci (2022) 23(13):7245. doi: 10.3390/ijms23137245
121. Tan FE, Sathe S, Wheeler EC, Yeo GW. Non-microRNA binding competitively inhibits LIN28 regulation. Cell Rep (2021) 36(6):109517. doi: 10.1016/j.celrep.2021.109517
122. Wang T, Wang G, Hao D, Liu X, Wang D, Ning N, et al. Aberrant regulation of the LIN28A/LIN28B and let-7 loop in human malignant tumors and its effects on the hallmarks of cancer. Mol Cancer (2015) 14(1):125. doi: 10.1186/s12943-015-0402-5
123. He X, Tan X, Wang X, Jin H, Liu L, Ma L, et al. C-myc-activated long noncoding RNA CCAT1 promotes colon cancer cell proliferation and invasion. Tumor Biol (2014) 35(12):12181–8. doi: 10.1007/s13277-014-2526-4
124. Xiang J-F, Yin Q-F, Chen T, Zhang Y, Zhang X-O, Wu Z, et al. Human colorectal cancer-specific CCAT1-l lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res (2014) 24(5):513–31. doi: 10.1038/cr.2014.35
125. Wang N, Yu Y, Xu B, Zhang M, Li Q, Miao L. Pivotal prognostic and diagnostic role of the long non−coding RNA colon cancer−associated transcript 1 expression in human cancer (Review). Mol Med Rep (2019) 19(2):771–82. doi: 10.3892/mmr.2018.9721
126. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell (2018) 173(2):321–337.e10. doi: 10.1016/j.cell.2018.03.035
127. Dhanasekaran R, Deutzmann A, Mahauad-Fernandez WD, Hansen AS, Gouw AM, Felsher DW. The MYC oncogene — the grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol (2021) 19(1):23–36. doi: 10.1038/s41571-021-00549-2
128. Selem N, Nafae H, Youness RA, Gad MZ. 32P immunoregulatory loop between let-7a and CCAT1 lncRNA coordinated by c-myc underlies the PD-1/PD-L1 immunoresistance in triple negative breast cancer patients. Ann Oncol (2021) 32:S1355. doi: 10.1016/j.annonc.2021.08.2028
129. Pandiri I, Chen Y, Joe Y, Kim HJ, Park J, Chung HT, et al. Tristetraprolin mediates the anti-proliferative effects of metformin in breast cancer cells. Breast Cancer Res Treat (2016) 156(1):57–64. doi: 10.1007/s10549-016-3742-y
130. Chen H, Sa G, Li L, He S, Wu T. In vitro and in vivo synergistic anti-tumor effect of LIN28 inhibitor and metformin in oral squamous cell carcinoma. Eur J Pharmacol (2021) 891:173757. doi: 10.1016/j.ejphar.2020.173757
131. Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T Helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity (2009) 31(5):787–98. doi: 10.1016/j.immuni.2009.09.014
132. Seo H, Jeon I, Kim B-S, Park M, Bae E-A, Song B, et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat Commun (2017) 8:15776. doi: 10.1038/ncomms15776
Keywords: immunotherapy, insulin-like growth factor, metabolism, immune checkpoint inhibitors, IL-6, EGFR, LIN28-let7 axis, HNSCC
Citation: Li J, Xiao Y, Yu H, Jin X, Fan S and Liu W (2023) Mutual connected IL-6, EGFR and LIN28/Let7-related mechanisms modulate PD-L1 and IGF upregulation in HNSCC using immunotherapy. Front. Oncol. 13:1140133. doi: 10.3389/fonc.2023.1140133
Received: 15 January 2023; Accepted: 28 March 2023;
Published: 12 April 2023.
Edited by:
Da Liu, Changchun University of Chinese Medicine, ChinaReviewed by:
Kailin Yang, Cleveland Clinic, United StatesNerina Denaro, IRCCS Ca ‘Granda Foundation Maggiore Policlinico Hospital, Italy
Copyright © 2023 Li, Xiao, Yu, Jin, Fan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Liu, bHc5OTcwMEAxNjMuY29t