
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 20 February 2023
Sec. Cancer Molecular Targets and Therapeutics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1136248
This article is part of the Research TopicWomen in Cancer Molecular Targets and Therapeutics, Volume II: 2022View all 23 articles
 Beatrice Fontana1†
Beatrice Fontana1† Giulia Gallerani1†
Giulia Gallerani1† Irene Salamon1†
Irene Salamon1† Ilaria Pace1
Ilaria Pace1 Roberta Roncarati2*
Roberta Roncarati2* Manuela Ferracin1,3*
Manuela Ferracin1,3*ARID1A belongs to a class of chromatin regulatory proteins that function by maintaining accessibility at most promoters and enhancers, thereby regulating gene expression. The high frequency of ARID1A alterations in human cancers has highlighted its significance in tumorigenesis. The precise role of ARID1A in cancer is highly variable since ARID1A alterations can have a tumor suppressive or oncogenic role, depending on the tumor type and context. ARID1A is mutated in about 10% of all tumor types including endometrial, bladder, gastric, liver, biliopancreatic cancer, some ovarian cancer subtypes, and the extremely aggressive cancers of unknown primary. Its loss is generally associated with disease progression more often than onset. In some cancers, ARID1A loss is associated with worse prognostic features, thus supporting a major tumor suppressive role. However, some exceptions have been reported. Thus, the association of ARID1A genetic alterations with patient prognosis is controversial. However, ARID1A loss of function is considered conducive for the use of inhibitory drugs which are based on synthetic lethality mechanisms. In this review we summarize the current knowledge on the role of ARID1A as tumor suppressor or oncogene in different tumor types and discuss the strategies for treating ARID1A mutated cancers.
Nucleosomes are composed of 147 base-pairs of DNA wrapped around histone octamers (1) and constitute the core unit of chromatin, which is further organized and compacted in higher order structures called topologically associated domains (TADs) in the nucleus (2, 3). The chromatin needs to be dynamically remodeled to guarantee the activation or repression of gene expression during the entire life of the cell, but particularly during the embryonic development (4) and differentiation (5). Remodeling means that regulatory complexes can be “opened” to provide access to the underlying DNA to enable transcription, chromatin assembly, DNA repair, and other processes. Chromatin remodeling is altered in pathological conditions such as cancer and heart failure (6, 7). Different classes of histone modifying enzymes that are involved in the deposition of histone tail modifications, including acetylation, methylation, phosphorylation, SUMOylation and ubiquitination, were identified. Furthermore, there are other ATP-dependent chromatin remodeling complexes, like the SWI/SNF complex, which uses the energy produced from ATP hydrolysis to mobilize and modify the nucleosome chromatin block and recruit the transcriptional machinery to the DNA (8).
The SWI/SNF complex was identified in yeast in 1984 (9) and it was later demonstrated that its structure is highly conserved among species, including mammals (10), suggesting a functional conservation during evolution. The mammalian SWI/SNF complex is comprised of more than 15 subunits (encoded by 29 genes) which assemble into three different complexes: BRG1/BRM-associated factor complex (BAF), polybromo-associated BAF complex (PBAF), and non-canonical BAF (ncBAF) (11). The AT-rich interacting domain containing respectively protein 1A (ARID1A) and 1B (ARID1B) subunits belong to the canonical BAF complex. The main role of ARID1A and B is to link the BAF core module to the subunits with ATPase activity (11). Both proteins are expressed in mammalian cells, and show a specific localization map onto the genome, characterized by a mutually exclusive interaction with the SWI/SNF complex (12).
ARID1A (also known as BAF250a, p270 or SMARCF1) is a key component of the mammalian SWI/SNF protein complex. ARID1A directly binds DNA with low sequence specificity (13), even though recently Rahmanto et al. described some specific DNA binding motifs which were found enriched in ARID1A ChIP-seq peaks in endometrial tumor cells (14). This observation suggests the formation of specific co-regulatory modules in which SWI/SNF complex, and particularly ARID1A subunit, co-localize with different transcription factors (AP-1 (15), FOXA1(16)) to regulate gene transcription in a cell-type specific manner. Moreover, ATAC-seq experiments conducted in colon cancer HCT116 cells proved that the loss of ARID1A profoundly altered chromatin accessibility, revealing a pivotal role of ARID1A in chromatin organization, determining a “gain or loss” of accessibility (15). Kelso et al. also demonstrated that the loss of ARID1 primarily affects enhancer accessibility and active histone marks on these regulatory regions, resulting in a significant alteration of the overall gene expression. Furthermore, ARID1A regulates transcription by modulating the conservative mechanism of RNA polymerase II (RNAPII) pausing which allows rapid and efficient transcription of several loci. This is a mechanism required to maintain cell homeostasis and development (17, 18). The analysis of RNAPII dynamics in ovarian epithelial cells revealed that loss of ARID1A induce changes in pausing versus elongating RNAPII fraction, leading to a significant reduction of transcription on active genes (19). Gao et al. demonstrated in mice that ARID1A deletion on one allele leads to embryonic lethality (20).
In addition, ARID1A can modulate gene transcription in cells, either directly controlling the expression of cancer related genes or indirectly by regulating the recruitment or activity of histone modifier enzymes, which add or remove histone modifications at gene regulatory regions. For example, ARID1A can regulate the immune response by modulating the interferon-responsive gene methylation profile in multiple tumor types (21). ARID1A is also involved in the regulation of Topoisomerase IIa (TOP2A) recruitment on chromatin, which is necessary to resolve R-loop formation during transcription (22).
The immunoprecipitation assay for ATR (ataxia telangiectasia and RAD3-related protein) followed by mass spectrometry, revealed that ARID1A is also an interacting partner and enriched in double strand brake (DSB) sites, further demonstrating that ARID1A is required to create the proper chromatin profile that facilitates the histone variant H2AX phosphorylation, mediated by ATR (23).
Considering the literature findings discussed so far, we can conclude that the role of ARID1A is fundamental in different cellular processes by regulating the transcription of genes involved in cell differentiation and development, although many mechanisms are not completely clarified yet. Several recent studies reported that ARID1A genetic alterations are linked to tumor development. For this reason, we examined the literature describing ARID1A alterations in human cancer, highlighting its double role as a tumor suppressor or oncogene in different tumor types and stages and focusing on its current use in cancer therapy.
One of the most interesting results from the -omics characterization of human cancers was the discovery that chromatin regulation and epigenetic processes are tightly linked to the development of cancer (29). Indeed, nearly all cancers display epigenetic changes that alter DNA expression and chromatin accessibility, and most cancer mutations directly or indirectly affect the epigenome (30, 31). Unexpectedly, many epigenetic-related genetic alterations were attributable to genes encoding subunits of the mammalian ATP-dependent SWI/SNF complex, especially BAF (Brg/Brahma-associated factors) complex (6, 32).
The mammalian SWI/SNF complex (33) is present in multiple forms in mammalian cells, and recent studies demonstrated that the subunit combination determines the functional specificity of the enzyme (34). Overall, the 29 genes of the SWI/SNF complex are mutated in 20% of all cancer types (35); this evidence suggests that the genetic perturbation of SWI/SNF complex is critical for cancer development and can have important oncogenic implications (36, 37). The most frequently mutated gene of the complex is ARID1A (38), which is altered in about 10% of human cancers. In Figure 1A we present the tumor types with frequent ARID1A alterations, according to AACR GENIE project data (39). Genetic alterations are evenly distributed across the gene and comprise missense or truncating mutations that are associated with the loss of function of the protein (Figure 1B).
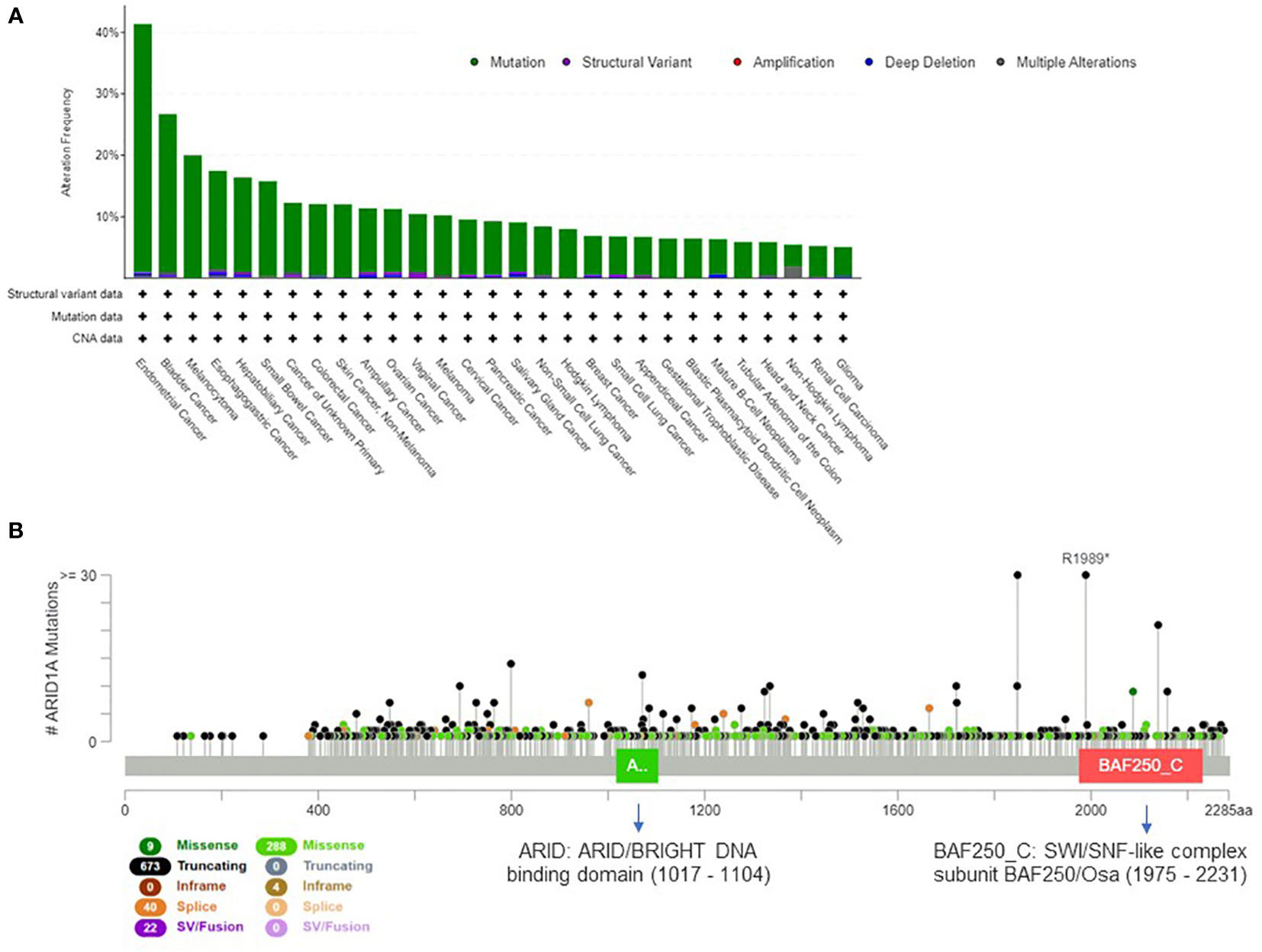
Figure 1 Extent of ARID1A involvement in human cancers. (A) Histogram showing ARID1A alteration frequency in 153.554 samples from the AACR GENIE project (39) and the alteration type across human cancers (minimum frequency cutoff at 5%). (B) Type, frequency and distribution of ARID1A mutations on the gene coding sequence across all AACR GENIE project tumor types.
Some studies report that ARID1A exerts cancer initiation and progression activities in specific cancer types, generally solid tumors, but its function as tumor suppressor or oncogene remains an open question. Mutations in ARID1A gene are usually responsible for its loss of function, thus suggesting a major tumor suppressive role. The survival analysis of ARID1Amut vs. ARID1Awt tumors across TCGA pan-cancer studies and MSK-IMPACT cohorts queried in cBioPortal website (40), generated partially discordant results, and specifically favorable prognosis is reported for ARID1A mutant TCGA cohort (q-value<0.001 for progression-free and disease-free survival, q-value 0.1 for overall survival) and a negative prognosis (p-value 0.007) for the MSK cohort.
In light of the above evidence, ARID1A seems to have a complex role in tumor development, and more studies are required to shed light on ARID1A tumor-specific activity. So far the assessment of its function as a tumor suppressor gene or an oncogene in cancer remains an open question (41, 42).
ARID1A mutations/deletions are documented in up to 80% of clear cell ovarian cancer (CCC), 56% of uterine endometrioid cancer (EC) (43), 40% of endometrial carcinoma and endometroid ovarian cancer and 30% of mucinous ovarian cancer (44), but in 0% of high-grade serous ovarian cancer (44, 45). Wiegand et al. specifically found that 73% of heterozygous ARID1A mutated tumors show a loss of protein expression without loss of heterozygosity, suggesting a haploinsufficiency mechanism (45). In addition, ARID1A mutations were commonly found during the early stages of endometriosis-associated ovarian carcinomas development, thus suggesting a trigger role for ARID1A loss (46). ARID1A mutation seems to be an early event also in endometrial glandular epithelium malignant transformation and ARID1A loss was found in areas with atypical endometriosis (47). These data suggest that ARID1A could be considered a tumor-suppressor gene in ovarian and endometrial cancers.
Gibson et al. analyzed the genomic landscape of endometrial cancer progression and reported the presence of ARID1A mutations since the early stages of tumor development (48). Among the driver genetic alterations, they found mutations in PIK3CA, PTEN, TP53, and PPP2R1A. ARID1A mutations were found to be heterogeneous and subclonal at the early stages, but related to a homogeneous ARID1A protein loss in advanced lesions (48). Reviewing all endometrial cancers in cBioPortal database (40), we observed that ARID1A mutations are mutually exclusive with TP53 mutations and co-occur with PTEN mutations (q-value<0.001).
Bitler et al. demonstrated that ARID1A inactivation upregulates HDAC6 expression, which in turn deacetylates Lys120 of P53 (49). P53K120 acetylation is a pro-apoptotic post-translational modification that selectively regulates apoptosis, without affecting cell cycle regulation (50). Therefore, ARID1A mutations contribute to the final inactivation of the apoptosis-promoting function of P53 by suppressing apoptosis-promoting P53K120Ac. This finding suggests that either the transcriptional repression of oncogenic genes or the transcriptional activation of tumor suppressor genes contribute to the tumor suppressive role of ARID1A.
Unexpectedly, ARID1A inactivation in association with APC and PTEN absence in mouse ovarian cancer models, prompted tumor cells towards epithelial differentiation (51). This observation suggests a context-dependent role for ARID1A in ovarian/endometrial cancer.
Aberrant SWI/SNF mediated chromatin-remodeling can sustain the activity of both oncogenic and tumor suppressive networks, resulting in directionally opposite effects. A double functional role for ARID1A in tumorigenesis has been described in hepatocellular carcinoma (HCC). Indeed, a recent finding by Sun et al. demonstrated that the gain of ARID1A function triggers tumor initiation by enhancing CYP450-mediated oxidative stress, while the loss of ARID1A during the later phases of tumor growth decreases the DNA accessibility and inhibits the transcription of genes associated with migration, invasion, and metastasis (42). In this model, ARID1A haploinsufficiency is enough to drive tumor progression.
Zhao et al. reported that 10–15% of HCCs harbor loss-of-function mutations in this gene and that 83% of HCC show ARID1A mRNA overexpression if compared to adjacent normal tissues (52). According to these observations, the authors proposed two different explanations for ARID1A role in HCC (Figure 2):
1. the change in ARID1A expression could be an early event during the development of HCC, since the silencing of ARID1A enhances cellular proliferation. However, this hypothesis cannot explain the up regulation of ARID1A in most HCC tumors compared with adjacent normal liver tissues.
2. the expression of ARID1A is very low in normal tissues, and at the early stages of the tumorigenesis ARID1A increases to prevent cellular proliferation. Whereas, during the late stages of HCC progression, the ARID1A loss due to acquired mutations, elicits tumor escape and enhance cell proliferation.
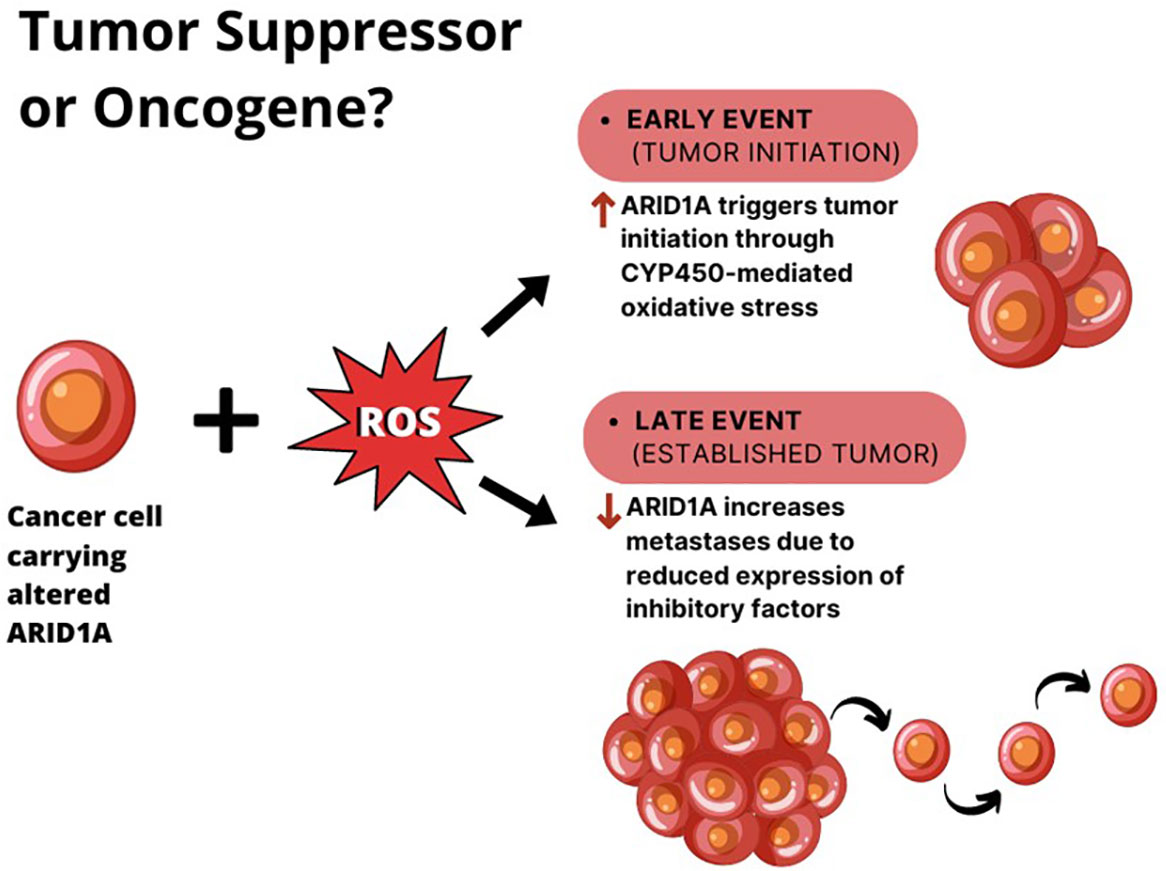
Figure 2 The double role of ARID1A in HCC. In the context of liver cell exposure to reactive oxygen species (ROS), ARID1A is overexpressed in cancer cells during tumor initiation, where it enhances tumor proliferation. When the tumor is established, ARID1A downregulation seems to elicit tumor metastases.
The second hypothesis seems to be more reasonable if we consider that ARID1A expression levels negatively correlated with survival in HCC patients (53).
To test the functional role of ARID1A, Zhang and colleagues used the hepatocellular carcinoma model induced by hydrodynamic transfection of tumor cells with activated AKT/NRAS combined with either ARID1A overexpression or knockdown (54). They observed that ARID1A depletion resulted in accelerated tumor growth and decreased survival in vivo, while ARID1A overexpression had the opposite effect, increasing survival and slowing tumor growth.
Gastric carcinoma (GC) is classified by The Cancer Genome Atlas (TCGA) project into four molecular subtypes: Epstein-Barr virus (EBV) positive with extreme DNA hypermethylation; microsatellite instability (MSI); genomically stable (GS), and chromosomal instability (CIN) (55).
ARID1A genetic alterations in gastric cancer (GC) were first reported by Abe et al. and Wang et al. (56, 57). The authors investigated the role of ARID1A loss in the context of EBV infection and genomic instability. In EBV+ GCs with microsatellite instability (or MHL1-lost), Abe et al. observed a frequent (34%) ARID1A loss of function by using immunohistochemistry (IHC) staining. In the MLH1-lost subgroup, ARID1A loss occurs in the early stages of tumor development, but only in EBV- and MLH1+ GCs the loss of ARID1A expression was associated with prognostic features. The loss of ARID1A expression is a consequence of ARID1A mutations (56, 58), but early stage ARID1A mutations are not always associated with loss of protein. The authors hypothesized that epithelial cells with ARID1A loss can be more easily infected by EBV and this could lately trigger cancer development (56). The transfection of ARID1A gene in gastric cancer cell lines reduced cell proliferation while ARID1A silencing promoted proliferation and migration, thus confirming ARID1A tumor suppressive role in gastric cancer (57).
A systematic meta-analysis of fourteen studies demonstrated that the loss of ARID1A expression predicts poor overall survival in gastric cancer, specifically in Asian populations suggesting a potential role as prognostic biomarker (59). Fitzmaurice et al. showed that PD-L1 is overexpressed in gastric cancer lacking ARID1A expression (60, 61). Hence, gastric cancer lacking ARID1A expression may be more sensitive to PD-1-PD-L1 immune checkpoint therapies.
ARID1A mutations are found in 4% of breast cancers (BCs). ARID1A copy number loss is the most frequent genetic alteration and involves 13–35% of BC cases. In addition, Zhang et al. demonstrated that in a variety of primary invasive BCs, ARID1A expression was epigenetically regulated. (62). Indeed in 86.4% of invasive ductal breast cancers, ARID1A low expression was related to gene promoter hypermethylation (63). Mamo et al. reported a correlation between the absent or decreased expression of the gene and increased tumor aggressiveness (64). Moreover, ARID1A protein expression was demonstrated to be an independent prognostic factor in breast cancer, with higher expression associated with better prognosis (65). Similarly, Takao et al. found that in patients with invasive breast cancer, the partial loss of ARID1A immunoreactivity was associated to a worse prognosis (66).
Among the genes interacting with ARID1A, RAB11FIP1 is overexpressed in breast cancer (67) RAB11FIP is involved in the Rab-11 mediated vesicle recycling, endosomal trafficking and transport between the recycling endosome and the trans-Golgi network, including the trafficking of integrin α5β1, required for cancer cell invasion, metastasis, and resistance to anticancer drugs. In Takao’s study, they found that the downregulation of ARID1A increases RAB11FIP1 expression, resulting in accumulation of integrin α5β1 on breast cancer cell membrane, thus enhancing cancer cell invasion. Specifically, ARID1A decrease alters the three-dimensional structure of the RAB11FIP1 promoter region thus increasing its expression and facilitating invasive breast cancer.
Recent sequencing analyses of PDAC have revealed ARID1A mutations in 6% of the cases (68). PDAC is one of those tumors yet poorly understood. Significant recurrent mutations are found in KRAS, TP53, CDKN2A, SMAD4, RNF43, ARID1A, TGFβR2, GNAS, RREB1 and PBRM1. These mutations are associated with amplification of GATA6 (18q11.2), ERBB2 (17q12), KRAS (12p12.1), AKT2 (19q13) and MYC (8q24.2), and deletion of CDKN2A (9p21.3), SMAD4 (18q21.2), ARID1A (1p36.11) and PTEN (10q23.31) (69).
Birnbaum et al. demonstrated that nine out of ten ARID1A mutated pancreatic cancers carry KRAS hotspot mutation G12D, suggesting that the inactivation of ARID1A may cooperate with KRAS in the early stages of pancreatic cancer formation (70). Li et al. also found that ARID1A deficiency, together with KRAS-G12D mutation, drive the development of pancreatic cancer via miR-503/CDKN2A axis-mediated senescence, although how ARID1A affects miR-503 transcription is not clear (71). These studies prove that ARID1A genetic alteration alone cannot initiate pancreatic cancer but can synergize with other altered genes to promote its pathogenesis.
In the COSMIC database, well-differentiated pancreatic neuroendocrine tumors (with a Ki-67 proliferation rate less than 3%) carry about 20% ARID1A mutations, much higher than 5.35% aggressive PDAC (72).
ARID1A acts as a tumor suppressor gene in renal cell carcinoma (RCC). To define the effects of ARID1A in renal carcinogenesis, Somsuan et al. used a non-malignant kidney epithelial (MDCK) cell line to demonstrate that ARID1A silencing using siRNAs significantly reduced cell death while increasing cell proliferation, with a cell cycle shift from G0/G1 to G2/M phase. In this study, they proved that ARID1A knockdown or deficiency was associated with decreased apoptosis and increased cell proliferation (73). Moreover, the siARID1A-transfected MDCK cells had higher migratory activity and invasive capability, also showing an enlargement of nuclei and multicellular spheroids.
Another study in patients affected by RCC, revealed lower ARID1A protein expression in 67% of samples and decreased ARID1A messenger RNA (mRNA) levels in 68% of samples if compared to normal kidney (74). The loss of ARID1A expression was associated with a larger tumor size, nuclear grade, and higher stage. Furthermore, ARID1A-positive cancers exhibited a longer disease-free and overall survival. Accordingly, Park et al. assessed the clinicopathological correlation and prognostic significance of ARID1A expression by an immunohistochemical study: they proved that low level of ARID1A was significantly correlated with higher nuclear grade, advanced pTNM stage, and shorter cancer-specific and progression-free survival. They proposed ARID1A expression as an independent prognostic factor for progression-free survival in RCC patients (75).
Cancers of unknown primary origin (CUP) comprise newly diagnosed tumors presenting as metastatic cancers, whose primary site cannot be identified after detailed standardized physical examinations, blood analyses, imaging, and immunohistochemical (IHC) testing (76). This tumor type is characterized by an ARID1A mutation frequency of 12-16%.
In a study by Ross et al. ARID1A mutations accounted for 11% of 200 archive CUPs (77). Moreover, in a recent study published by Laprovitera et al. we reported that the intratumor frequency of ARID1A mutation could be associated with CUP progression. Specifically, we longitudinally evaluated the variant allele frequency in circulating cell-free DNA (ccfDNA) samples of a CUP case with ARID1A mutations. The study reports how the fractional abundance of ARID1A mutation (p.R1276_) in ccfDNA decreases during the initial treatment and then increases again during disease worsening, thus suggesting a role in the expansion of the more aggressive subclones (78).
Many genetic alterations occur during the development of melanoma. Thielmann et al. analyzed the clinical pathological features of 116 patients diagnosed with melanoma in association with the most common genetic alterations, including mutations in ARID1A gene (79). They demonstrated that ARID1A mutated melanomas exhibit higher tumor mutational burden (TMB). ARID1A mutations were evenly distributed across the gene without clustering or hotspots. Despite the increased TMB, no statistical significance was noticed in ARID1A mutated patients receiving targeted therapies or immune-checkpoint inhibitors for what concerns progression-free and overall survival. However, ARID1A mutated tumors revealed UV-induced mutation signatures, showing a higher frequency of C>T substitutions in comparison with ARID1A wild-type melanomas. This finding suggests that the impact of ARID1A mutations in immune-checkpoint inhibitors response needs to be better elucidated.
ARID1A expression is progressively lost during colorectal cancer (CRC) development: Wei et al. showed that the loss of ARID1A expression was associated to distant metastasis and late TNM stage of CRC. However, the survival analyses indicated that the loss of ARID1A protein expression was a better prognostic factor for stage IV CRC (80). In line with this observation, Erfani et al. reported no significant association between overall survival and loss of ARID1A expression in CRC (81). Other studies did not find any significant association between loss of ARID1A expression and overall survival; still, they observed that the overall survival was better for patients with no/low ARID1A expression than those with ARID1A expression (82–84). Erfani et al. demonstrated that ARID1A expression is reduced by promoter hypermethylation in CRC and its low expression is associated with lymphatic invasion. These findings suggest that the role of ARID1A in CRC is not completely understood, and possibly different than in other cancer types.
ARID1A mutations are detectable in about 6-7.5% of lung cancers. Hung et al. demonstrated that ARID1A loss-of-function mutations and biallelic inactivation induce the complete loss of ARID1A expression in non-small cell lung cancer (NSCLC) (85). Another study reported that patients with ARID1A loss had a shorter cancer specific survival and a significant association of ARID1A loss to male sex, larger tumor size, smoker status and squamous histology (86). Moreover, ARID1A-loss lung cancers had the worst survival in comparison to ARID1A-positive tumors. Thus, the loss of ARID1A expression might be a valuable prognostic marker in NSCLC (57, 63, 74).
In addition to survival rate, other clinicopathological factors such as lymph node metastasis and tumor infiltration have been positively correlated with loss of ARID1A expression (87). The authors also experimentally verified the impact of ARID1A silencing in lung cancer cell lines, concluding that ARID1A has a tumor suppressive role in this tumor type.
The high frequency of ARID1A mutations among different cancer types, made this gene a very appealing research object for target therapy investigations.
Specifically, as a “care-taker” and “gate-keeper” gene, the mutational status of ARID1A in target therapy was investigated within the context of synthetic lethality. Synthetic lethality is based on essential gene interactions where a genetic alteration, such as a defect in a tumor suppressor gene (genetic context), influence a second gene essential for cell survival (pharmacological target) (88). The use of synthetic lethality as a guidance to develop cancer therapeutics was introduced by Hartwell (89) and Kaelin (90), after the success of PARP inhibitors in BRCA-mutant ovarian cancers (91–93). As for BRCA mutation, ARID1A deficiency in tumors constitutes a promising synthetic lethal phenotype (94) for the use of small inhibitors targeting DNA damage response (DDR), immune-checkpoint blockade (ICB), kinases, and agents leading to a cell-specific cytotoxicity.
Currently there are 23 clinical trials registered in the clinical trials website (https://clinicaltrials.gov/) concerning ARID1A, ranging from phase I to phase II. Table 1 reports a list of concluded clinical trials where ARID1A mutation was considered in outcome evaluations. Since ARID1A deficiency was firstly discovered in gynecological cancers (95); (47, 45), many clinical trials involved uterine/ovarian cancer patients. However, other non-gynecologic clinical trials have been recruiting patients with a wide range of oncologic diseases: bladder cancer, cholangiocarcinoma, pancreatic, colorectal, biliary tract cancer, NSCLC and other solid tumors. In all these clinical trials, ARID1A was considered for synthetic lethal drug screening. The pharmacological targets include molecules directed toward DNA damage response (DDR), immune checkpoint blockade (ICB), kinases, epigenetics effectors.
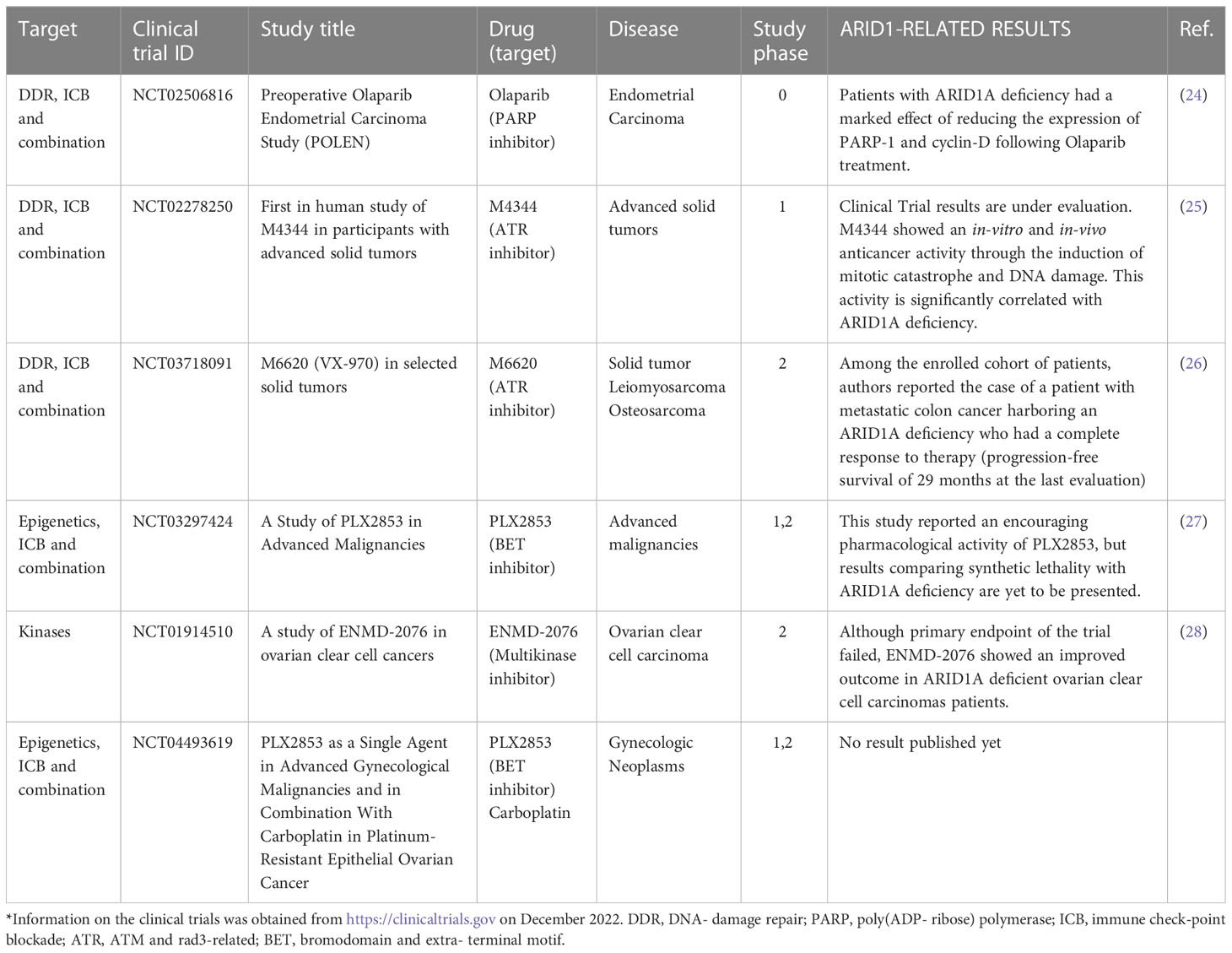
Table 1 Completed clinical trials involving cancers with ARID1A deficiency*.
Genome stability, which essential for cell survival, is compromised in ARID1A-deficient cancer cells. As a result, cancer cells with high levels of replicative stress, become more dependent on compensatory mechanisms such as the activation of ATR signaling (96). This dependency underlies the mechanism of synthetic lethality of ATR inhibitors (ATRi)(97). There are six registered clinical trials using ATRis in patients with ARID1A-deficient solid tumors. The highly potent ATR inhibitor, M4344 (VX-803) is the pharmacological compound used in phase I clinical trial number NCT02278250, which is now concluded, and whose results are under evaluation. M4344 compound demonstrated an anticancer activity both in-vitro and in-vivo by inducing mitotic catastrophe and DNA damage: this effect was significantly correlated with ARID1A deficiency (25). A very interesting result about the synthetic lethality with ATRi in ARID1A-deficient tumors comes from study NCT03718091. Among the results of this study, a patient with metastatic colon cancer harboring an ARID1A deficiency had a complete response to therapy (progression-free survival of 29 months at the last evaluation) (98).
Another synthetic lethality relationship currently exploited is between DNA damage response (DDR) deficiency and PARP inhibitors. In this context Shen et al. demonstrated both in-vitro and in-vivo that PARP inhibitors are selectively active towards ARID1A-deficient cells, thus providing a novel approach for stratifying patients for clinical trials of targeted therapy with PARP inhibitors (23). The POLEN study was a window-of-opportunity trial where PARP inhibitor Olaparib was administered as neoadjuvant therapy in patients with early-stage endometrial carcinoma before surgery (24). Authors showed that treatment with Olaparib reduced the expression of PARP-1 and cyclin-D, this effect was more prominent in patients with ARID1A deficiency.
Another PARP inhibitor, niraparib, was used in a phase II clinical trial in metastatic solid tumors (99). Patients were stratified for the presence of mutations in genes involved in DNA damage repair including ARID1A (court B). Preliminary results of the study were presented at ASCO 2022 and described that in the court B patients who had stable disease were ARID1A mutated (99). Moreover, there are preclinical studies associating the loss of ARID1A function with synthetic lethality based on inhibitors of the bromodomain and extra-terminal family, BET (BETi) (100); (16, 101). This led to the development of clinical trials involving BET inhibitors as single agents or in combination with existing treatment options in multiple human cancers bearing ARID1A deficiency. On this basis, PLX2853, a potent BET inhibitor, was used in two completed phase I/II clinical trials (NCT03297424, NCT04493619). Gordon et al, presented early results from the “PLX2853 in Advanced Malignancies” study reporting an encouraging pharmacological activity, but results comparing synthetic lethality with ARID1A deficiency are yet to be present (27). Targeting Aurora kinase A (AURKA) is a further synthetic lethality interaction in ARID1A defective tumors. Starting from the finding of Wu et al. unveiling the direct repression of AURKA by ARID1A (102), studies have been conducted with pan-aurora kinase inhibitors in colon and ovarian cancer cells with ARID1A deficiency, causing chromosomal abnormalities leading to synthetic lethality (103).
Finally, a phase 2 clinical trial to assess the activity of a strong selective inhibitor for AURKA, ENMD- 2076, in treating patients with ovarian clear cell carcinomas was recently concluded (NCT01914510). Despite ENMD- 2076 did not meet the efficacy bar set in this trial, this AURKA inhibitor has been reported as potentially advantageous for ovarian clear cell carcinomas patients with ARID1A deficiency (28).
ARID1A acts predominantly as a tumor suppressor gene in many solid tumors, although its functional role seems to be stage and tumor type dependent. For example, ARID1A expression is initially upregulated in HCC and then lost by acquisitions of truncating/missense mutations. The loss of ARID1A expression in human cancers is generally associated with negative prognostic features, tumor progression and increased tumor growth and invasion. The lack of ARID1A in tumors is associated with an increased tumor mutational burden and genomic instability, which is currently pharmacologically exploited according to synthetic lethality principles. Although the exact mechanisms are still elusive, clinical trials with small inhibitors directed toward DNA damage response (DDR), immune checkpoint blockade (ICB), kinase inhibitors and epigenetics effectors are showing promising results and are worth investigating in future studies.
MF contributed to conception and design of the study. BF, IS, GG, and RR wrote the first draft of the manuscript. IP wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
The research leading to these results has received funding from AIRC under IG 2021 - ID. 25789 project – P.I. Ferracin Manuela.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 a resolution. Nature (1997) 389:251–60. doi: 10.1038/38444
2. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature (2012) 485:376–80. doi: 10.1038/nature11082
3. Bonev B, Cavalli G. Organization and function of the 3D genome. Nat Rev Genet (2016) 17:661–78. doi: 10.1038/nrg.2016.112
4. Cenik BK, Shilatifard A. COMPASS and SWI/SNF complexes in development and disease. Nat Rev Genet (2021) 22:38–58. doi: 10.1038/s41576-020-0278-0
5. Padeken J, Methot SP, Gasser SM. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat Rev Mol Cell Biol (2022) 23:623–40. doi: 10.1038/s41580-022-00483-w
6. Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv (2015) 1:e1500447. doi: 10.1126/sciadv.1500447
7. Papait A, Vertua E, Magatti M, Ceccariglia S, De Munari S, Silini AR, et al. Mesenchymal stromal cells from fetal and maternal placenta possess key similarities and differences: Potential implications for their applications in regenerative medicine. Cells (2020) 9:127. doi: 10.3390/cells9010127
8. Tang L, Nogales E, Ciferri C. Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog Biophys Mol Biol (2010) 102:122–8. doi: 10.1016/j.pbiomolbio.2010.05.001
9. Neigeborn L, Carlson M. Genes affecting the regulation of SUC2 gene expression by glucose repression in saccharomyces cerevisiae. Genetics (1984) 108:845–58. doi: 10.1093/genetics/108.4.845
10. Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev (1996) 10:2117–30. doi: 10.1101/gad.10.17.2117
11. Mashtalir N, D'avino AR, Michel BC, Luo J, Pan J, Otto JE, et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell (2018) 175:1272–88:e1220. doi: 10.1016/j.cell.2018.09.032
12. Raab JR, Resnick S, Magnuson T. Genome-wide transcriptional regulation mediated by biochemically distinct SWI/SNF complexes. PLos Genet (2015) 11:e1005748. doi: 10.1371/journal.pgen.1005748
13. Wilsker D, Patsialou A, Zumbrun SD, Kim S, Chen Y, Dallas PB, et al. The DNA-binding properties of the ARID-containing subunits of yeast and mammalian SWI/SNF complexes. Nucleic Acids Res (2004) 32:1345–53. doi: 10.1093/nar/gkh277
14. Suryo Rahmanto Y, Shen W, Shi X, Chen X, Yu Y, Yu ZC, et al. Inactivation of Arid1a in the endometrium is associated with endometrioid tumorigenesis through transcriptional reprogramming. Nat Commun (2020) 11:2717. doi: 10.1038/s41467-020-16416-0
15. Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, Hargreaves DC. Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. Elife (2017) 6:e30506. doi: 10.7554/eLife.30506.054
16. Nagarajan S, Rao SV, Sutton J, Cheeseman D, Dunn S, Papachristou EK, et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat Genet (2020) 52:187–97. doi: 10.1038/s41588-019-0541-5
17. Levine M. Paused RNA polymerase II as a developmental checkpoint. Cell (2011) 145:502–11. doi: 10.1016/j.cell.2011.04.021
18. Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: Emerging roles in metazoans. Nat Rev Genet (2012) 13:720–31. doi: 10.1038/nrg3293
19. Trizzino M, Kapusta A, Brown CD. Transposable elements generate regulatory novelty in a tissue-specific fashion. BMC Genomics (2018) 19:468. doi: 10.1186/s12864-018-4850-3
20. Gao X, Tate P, Hu P, Tjian R, Skarnes WC, Wang Z. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci U.S.A. (2008) 105:6656–61. doi: 10.1073/pnas.0801802105
21. Li J, Wang W, Zhang Y, Cieslik M, Guo J, Tan M, et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest (2020) 130:2712–26. doi: 10.1172/JCI134402
22. Tsai S, Fournier LA, Chang EY, Wells JP, Minaker SW, Zhu YD, et al. ARID1A regulates r-loop associated DNA replication stress. PLos Genet (2021) 17:e1009238. doi: 10.1371/journal.pgen.1009238
23. Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discovery (2015) 5:752–67. doi: 10.1158/2159-8290.CD-14-0849
24. Romero I, Rubio MJ, Medina M, Matias-Guiu X, Santacana M, Schoenenberger JA, et al. An olaparib window-of-opportunity trial in patients with early-stage endometrial carcinoma: POLEN study. Gynecol Oncol (2020) 159:721–31. doi: 10.1016/j.ygyno.2020.09.013
25. Jo U, Senatorov IS, Zimmermann A, Saha LK, Murai Y, Kim SH, et al. Novel and highly potent ATR inhibitor M4344 kills cancer cells with replication stress, and enhances the chemotherapeutic activity of widely used DNA damaging agents. Mol Cancer Ther (2021) 20:1431–41. doi: 10.1158/1535-7163.MCT-20-1026
26. Yap TA, O'carrigan B, Penney MS, Lim JS, Brown JS, De Miguel Luken MJ, et al. Phase I trial of first-in-Class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol (2020) 38:3195–204. doi: 10.1200/JCO.19.02404
27. Gordon MS, Carvajal RD, Spira AI, Huang M, Watkins P, Powell B, et al. Phase 1b/2a study of PLX2853, a small molecule BET inhibitor, in subjects with advanced solid tumors and lymphoma. J Clin Oncol (2021) 39:3018–8. doi: 10.1200/JCO.2021.39.15_suppl.3018
28. Lheureux S, Tinker A, Clarke B, Ghatage P, Welch S, Weberpals JI, et al. A clinical and molecular phase II trial of oral ENMD-2076 in ovarian clear cell carcinoma (OCCC): A study of the princess Margaret phase II consortium. Clin Cancer Res (2018) 24:6168–74. doi: 10.1158/1078-0432.CCR-18-1244
29. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer (2011) 11:726–34. doi: 10.1038/nrc3130
30. Dawson MA, Kouzarides T. Cancer epigenetics: From mechanism to therapy. Cell (2012) 150:12–27. doi: 10.1016/j.cell.2012.06.013
31. Morgan MA, Shilatifard A. Chromatin signatures of cancer. Genes Dev (2015) 29:238–49. doi: 10.1101/gad.255182.114
32. Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell (2018) 173:371–385.e318. doi: 10.1016/j.cell.2018.02.060
33. Euskirchen G, Auerbach RK, Snyder M. SWI/SNF chromatin-remodeling factors: multiscale analyses and diverse functions. J Biol Chem (2012) 287:30897–905. doi: 10.1074/jbc.R111.309302
34. Wu JI. Diverse functions of ATP-dependent chromatin remodeling complexes in development and cancer. Acta Biochim Biophys Sin (Shanghai) (2012) 44:54–69. doi: 10.1093/abbs/gmr099
35. Helming KC, Wang X, Roberts CWM. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell (2014) 26:309–17. doi: 10.1016/j.ccr.2014.07.018
36. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet (2013) 45:592–601. doi: 10.1038/ng.2628
37. Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLos One (2013) 8:e55119. doi: 10.1371/journal.pone.0055119
38. Jiang T, Chen X, Su C, Ren S, Zhou C. Pan-cancer analysis of ARID1A alterations as biomarkers for immunotherapy outcomes. J Cancer (2020) 11:776–80. doi: 10.7150/jca.41296
39. Consortium APG. AACR project GENIE: Powering precision medicine through an international consortium. Cancer Discovery (2017) 7:818–31. doi: 10.1158/2159-8290.CD-17-0151
40. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6:pl1. doi: 10.1126/scisignal.2004088
41. Hohmann AF, Vakoc CR. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet (2014) 30:356–63. doi: 10.1016/j.tig.2014.05.001
42. Sun X, Wang SC, Wei Y, Luo X, Jia Y, Li L, et al. Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell (2017) 32574-589.e576. doi: 10.1016/j.ccell.2017.10.007
43. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med (2017) 23:703–13. doi: 10.1038/nm.4333
44. Kuroda Y, Chiyoda T, Kawaida M, Nakamura K, Aimono E, Yoshimura T, et al. ARID1A mutation/ARID1A loss is associated with a high immunogenic profile in clear cell ovarian cancer. Gynecol Oncol (2021) 162:679–85. doi: 10.1016/j.ygyno.2021.07.005
45. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med (2010) 363:1532–43. doi: 10.1056/NEJMoa1008433
46. Cornen S, Adelaide J, Bertucci F, Finetti P, Guille A, Birnbaum DJ, et al. Mutations and deletions of ARID1A in breast tumors. Oncogene (2012) 31:4255–6. doi: 10.1038/onc.2011.598
47. Wiegand KC, Lee AF, Al-Agha OM, Chow C, Kalloger SE, Scott DW, et al. Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. J Pathol (2011) 224:328–33. doi: 10.1002/path.2911
48. Gibson WJ, Hoivik EA, Halle MK, Taylor-Weiner A, Cherniack AD, Berg A, et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat Genet (2016) 48:848–55. doi: 10.1038/ng.3602
49. Bitler BG, Wu S, Park PH, Hai Y, Aird KM, Wang Y, et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat Cell Biol (2017) 19:962–73. doi: 10.1038/ncb3582
50. Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell (2006) 24:841–51. doi: 10.1016/j.molcel.2006.11.026
51. Zhai Y, Kuick R, Tipton C, Wu R, Sessine M, Wang Z, et al. Arid1a inactivation in an apc- and pten-defective mouse ovarian cancer model enhances epithelial differentiation and prolongs survival. J Pathol (2016) 238:21–30. doi: 10.1002/path.4599
52. Zhao J, Chen J, Lin H, Jin R, Liu J, Liu X, et al. The clinicopathologic significance of BAF250a (ARID1A) expression in hepatocellular carcinoma. Pathol Oncol Res (2016) 22:453–9. doi: 10.1007/s12253-015-0022-9
53. Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science (2017) 357. doi: 10.1126/science.aan2507
54. Zhang S, Zhou YF, Cao J, Burley SK, Wang HY, Zheng XFS. mTORC1 promotes ARID1A degradation and oncogenic chromatin remodeling in hepatocellular carcinoma. Cancer Res (2021) 81:5652–65. doi: 10.1158/0008-5472.CAN-21-0206
55. Sohn BH, Hwang JE, Jang HJ, Lee HS, Oh SC, Shim JJ, et al. Clinical significance of four molecular subtypes of gastric cancer identified by the cancer genome atlas project. Clin Cancer Res (2017) 23:4441–9. doi: 10.1158/1078-0432.CCR-16-2211
56. Abe H, Maeda D, Hino R, Otake Y, Isogai M, Ushiku AS, et al. ARID1A expression loss in gastric cancer: Pathway-dependent roles with and without Epstein-Barr virus infection and microsatellite instability. Virchows Arch (2012) 461:367–77. doi: 10.1007/s00428-012-1303-2
57. Wang DD, Chen YB, Pan K, Wang W, Chen SP, Chen JG, et al. Decreased expression of the ARID1A gene is associated with poor prognosis in primary gastric cancer. PLos One (2012) 7:e40364. doi: 10.1371/journal.pone.0040364
58. Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet (2011) 43:1219–23. doi: 10.1038/ng.982
59. Yang L, Wei S, Zhao R, Wu Y, Qiu H, Xiong H. Loss of ARID1A expression predicts poor survival prognosis in gastric cancer: A systematic meta-analysis from 14 studies. Sci Rep (2016) 6:28919. doi: 10.1038/srep28919
60. Global Burden of Disease Cancer, C, Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, et al. The global burden of cancer 2013. JAMA Oncol (2015) 1:505–27. doi: 10.1001/jamaoncol.2015.0735
61. Pan QX, Su ZJ, Zhang JH, Wang CR, Ke SY. A comparison of the prognostic value of preoperative inflammation-based scores and TNM stage in patients with gastric cancer. Onco Targets Ther (2015) 8:1375–85. doi: 10.2147/OTT.S82437
62. Zhang X, Sun Q, Shan M, Niu M, Liu T, Xia B, et al. Promoter hypermethylation of ARID1A gene is responsible for its low mRNA expression in many invasive breast cancers. PLos One (2013) 8:e53931. doi: 10.1371/journal.pone.0053931
63. Cho HD, Lee JE, Jung HY, Oh MH, Lee JH, Jang SH, et al. Loss of tumor suppressor ARID1A protein expression correlates with poor prognosis in patients with primary breast cancer. J Breast Cancer (2015) 18:339–46. doi: 10.4048/jbc.2015.18.4.339
64. Mamo A, Cavallone L, Tuzmen S, Chabot C, Ferrario C, Hassan S, et al. An integrated genomic approach identifies ARID1A as a candidate tumor-suppressor gene in breast cancer. Oncogene (2012) 31:2090–100. doi: 10.1038/onc.2011.386
65. Zhao J, Liu C, Zhao Z. ARID1A: A7 potential prognostic factor for breast cancer. Tumour Biol (2014) 35:4813–9. doi: 10.1007/s13277-014-1632-7
66. Takao C, Morikawa A, Ohkubo H, Kito Y, Saigo C, Sakuratani T, et al. Downregulation of ARID1A, a component of the SWI/SNF chromatin remodeling complex, in breast cancer. J Cancer (2017) 8:1–8. doi: 10.7150/jca.16602
67. Wang B, Yang Z, Wang H, Cao Z, Zhao Y, Gong C, et al. MicroRNA-320a inhibits proliferation and invasion of breast cancer cells by targeting RAB11A. Am J Cancer Res (2015) 5:2719–29. doi: 10.1158/1538-7445.AM2015-2719
68. Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev (2016) 30:355–85. doi: 10.1101/gad.275776.115
69. Cancer Genome Atlas Research Network, Electronic Address, A.a.D.H.E, Cancer Genome Atlas Research, N Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell (2017) 32:185–203.e113. doi: 10.1016/j.ccell.2017.07.007
70. Birnbaum DJ, Adelaide J, Mamessier E, Finetti P, Lagarde A, Monges G, et al. Genome profiling of pancreatic adenocarcinoma. Genes Chromosomes Cancer (2011) 50:456–65. doi: 10.1002/gcc.20870
71. Li ZY, Zhu SS, Chen XJ, Zhu J, Chen Q, Zhang YQ, et al. ARID1A suppresses malignant transformation of human pancreatic cells via mediating senescence-associated miR-503/CDKN2A regulatory axis. Biochem Biophys Res Commun (2017) 493:1018–25. doi: 10.1016/j.bbrc.2017.09.099
72. De Wilde RF, Edil BH, Hruban RH, Maitra A. Well-differentiated pancreatic neuroendocrine tumors: from genetics to therapy. Nat Rev Gastroenterol Hepatol (2012) 9:199–208. doi: 10.1038/nrgastro.2012.9
73. Somsuan K, Peerapen P, Boonmark W, Plumworasawat S, Samol R, Sakulsak N, et al. ARID1A knockdown triggers epithelial-mesenchymal transition and carcinogenesis features of renal cells: Role in renal cell carcinoma. FASEB J (2019) 33:12226–39. doi: 10.1096/fj.201802720RR
74. Lichner Z, Scorilas A, White NM, Girgis AH, Rotstein L, Wiegand KC, et al. The chromatin remodeling gene ARID1A is a new prognostic marker in clear cell renal cell carcinoma. Am J Pathol (2013) 182:1163–70. doi: 10.1016/j.ajpath.2013.01.007
75. Park JH, Lee C, Suh JH, Chae JY, Kim HW, Moon KC. Decreased ARID1A expression correlates with poor prognosis of clear cell renal cell carcinoma. Hum Pathol (2015) 46:454–60. doi: 10.1016/j.humpath.2014.12.002
76. Fizazi K, Greco FA, Pavlidis N, Daugaard G, Oien K, Pentheroudakis G, et al. Cancers of unknown primary site: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2015) 26 Suppl 5:v133–138. doi: 10.1093/annonc/mdv305
77. Ross JS, Wang K, Gay L, Otto GA, White E, Iwanik K, et al. Comprehensive genomic profiling of carcinoma of unknown primary site: New routes to targeted therapies. JAMA Oncol (2015) 1:40–9. doi: 10.1001/jamaoncol.2014.216
78. Laprovitera N, Salamon I, Gelsomino F, Porcellini E, Riefolo M, Garonzi M, et al. Genetic characterization of cancer of unknown primary using liquid biopsy approaches. Front Cell Dev Biol (2021) 9:666156. doi: 10.3389/fcell.2021.666156
79. Thielmann CM, Matull J, Roth S, Placke JM, Chorti E, Zaremba A, et al. Genetic and clinical characteristics of ARID1A mutated melanoma reveal high tumor mutational load without implications on patient survival. Cancers (Basel) (2022) 14. doi: 10.3390/cancers14092090
80. Wei XL, Wang DS, Xi SY, Wu WJ, Chen DL, Zeng ZL, et al. Clinicopathologic and prognostic relevance of ARID1A protein loss in colorectal cancer. World J Gastroenterol (2014) 20:18404–12. doi: 10.3748/wjg.v20.i48.18404
81. Erfani M, Hosseini SV, Mokhtari M, Zamani M, Tahmasebi K, Alizadeh Naini M, et al. Altered ARID1A expression in colorectal cancer. BMC Cancer (2020) 20:350. doi: 10.1186/s12885-020-6706-x
82. Chou A, Toon CW, Clarkson A, Sioson L, Houang M, Watson N, et al. Loss of ARID1A expression in colorectal carcinoma is strongly associated with mismatch repair deficiency. Hum Pathol (2014) 45:1697–703. doi: 10.1016/j.humpath.2014.04.009
83. Lee SY, Kim DW, Lee HS, Ihn MH, Oh HK, Park DJ, et al. Loss of AT-rich interactive domain 1A expression in gastrointestinal malignancies. Oncology (2015) 88:234–40. doi: 10.1159/000369140
84. Lee LH, Sadot E, Ivelja S, Vakiani E, Hechtman JF, Sevinsky CJ, et al. ARID1A expression in early stage colorectal adenocarcinoma: An exploration of its prognostic significance. Hum Pathol (2016) 53:97–104. doi: 10.1016/j.humpath.2016.02.004
85. Hung YP, Redig A, Hornick JL, Sholl LM. ARID1A mutations and expression loss in non-small cell lung carcinomas: Clinicopathologic and molecular analysis. Mod Pathol (2020) 33:2256–68. doi: 10.1038/s41379-020-0592-2
86. Jang SH, Lee JH, Lee HJ, Cho H, Ahn H, Song IH, et al. Loss of ARID1A expression is associated with poor prognosis in non-small cell lung cancer. Pathol Res Pract (2020) 216:153156. doi: 10.1016/j.prp.2020.153156
87. Zhang Y, Xu X, Zhang M, Bai X, Li H, Kan L, et al. ARID1A is downregulated in non-small cell lung cancer and regulates cell proliferation and apoptosis. Tumour Biol (2014) 35:5701–7. doi: 10.1007/s13277-014-1755-x
88. Rancati G, Moffat J, Typas A, Pavelka N. Emerging and evolving concepts in gene essentiality. Nat Rev Genet (2018) 19:34–49. doi: 10.1038/nrg.2017.74
89. Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science (1997) 278:1064–8. doi: 10.1126/science.278.5340.1064
90. Kaelin WG Jr. Choosing anticancer drug targets in the postgenomic era. J Clin Invest (1999) 104:1503–6. doi: 10.1172/JCI8888
91. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature (2005) 434:913–7. doi: 10.1038/nature03443
92. Farmer H, Mccabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434:917–21. doi: 10.1038/nature03445
93. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med (2009) 361:123–34. doi: 10.1056/NEJMoa0900212
94. Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A, et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun (2016) 7:13837. doi: 10.1038/ncomms13837
95. Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science (2010) 330:228–31. doi: 10.1126/science.1196333
96. Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol (2017) 18:622–36. doi: 10.1038/nrm.2017.67
97. Karnitz LM, Zou L. Molecular pathways: Targeting ATR in cancer therapy. Clin Cancer Res (2015) 21:4780–5. doi: 10.1158/1078-0432.CCR-15-0479
98. Ngoi NYL, Pham MM, Tan DSP, Yap TA. Targeting the replication stress response through synthetic lethal strategies in cancer medicine. Trends Cancer (2021) 7:930–57. doi: 10.1016/j.trecan.2021.06.002
99. George TJ, Lee J-H, Hosein PJ, Deremer DL, Chatzkel JA, Ramnaraign BH, et al. Results of a phase II trial of the PARP inhibitor, niraparib, in BAP1 and other DNA damage response pathway deficient neoplasms. J Clin Oncol (2022) 40:3122–2. doi: 10.1200/JCO.2022.40.16_suppl.3122
100. Caumanns JJ, Wisman GBA, Berns K, van der Zee AGJ, De Jong S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim Biophys Acta Rev Cancer (2018) 1870:176–84. doi: 10.1016/j.bbcan.2018.07.005
101. Wanior M, Kramer A, Knapp S, Joerger AC. Exploiting vulnerabilities of SWI/SNF chromatin remodelling complexes for cancer therapy. Oncogene (2021) 40:3637–54. doi: 10.1038/s41388-021-01781-x
102. Wu C, Lyu J, Yang EJ, Liu Y, Zhang B, Shim JS. Targeting AURKA-CDC25C axis to induce synthetic lethality in ARID1A-deficient colorectal cancer cells. Nat Commun (2018) 9:3212. doi: 10.1038/s41467-018-05694-4
Keywords: ARID1A, tumor suppressor gene, oncogene, solid tumors, SWI/SNF complex, synthetic lethality
Citation: Fontana B, Gallerani G, Salamon I, Pace I, Roncarati R and Ferracin M (2023) ARID1A in cancer: Friend or foe? Front. Oncol. 13:1136248. doi: 10.3389/fonc.2023.1136248
Received: 02 January 2023; Accepted: 06 February 2023;
Published: 20 February 2023.
Edited by:
Antonis Kirmizis, University of Cyprus, CyprusReviewed by:
Ivana De La Serna, University of Toledo Medical Center, United StatesCopyright © 2023 Fontana, Gallerani, Salamon, Pace, Roncarati and Ferracin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuela Ferracin, bWFudWVsYS5mZXJyYWNpbkB1bmliby5pdA==; Roberta Roncarati, cm9iZXJ0YS5yb25jYXJhdGlAY25yLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.