 Sofia Bruni
Sofia Bruni María Florencia Mercogliano
María Florencia Mercogliano Florencia Luciana Mauro
Florencia Luciana Mauro Rosalia Inés Cordo Russo
Rosalia Inés Cordo Russo Roxana Schillaci*
Roxana Schillaci*- Laboratorio de Mecanismos Moleculares de Carcinogénesis. Instituto de Biología y Medicina Experimental (IBYME-CONICET), Buenos Aires, Argentina
Immunotherapy has changed the course of cancer treatment. The initial steps were made through tumor-specific antibodies that guided the setup of an antitumor immune response. A new and successful generation of antibodies are designed to target immune checkpoint molecules aimed to reinvigorate the antitumor immune response. The cellular counterpart is the adoptive cell therapy, where specific immune cells are expanded or engineered to target cancer cells. In all cases, the key for achieving positive clinical resolutions rests upon the access of immune cells to the tumor. In this review, we focus on how the tumor microenvironment architecture, including stromal cells, immunosuppressive cells and extracellular matrix, protects tumor cells from an immune attack leading to immunotherapy resistance, and on the available strategies to tackle immune evasion.
1 Introduction
The tumor microenvironment (TME) is an heterogenous milieu composed not only by cancer cells, but also by different cell types that can contribute to tumoral escape of immune surveillance and dampening of responses to immunotherapy. These cells include myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), tumor infiltrating lymphocytes (TILs), cancer associated fibroblasts (CAFs), blood vessels and the extracellular matrix (ECM), composed of collagen and proteoglycans (1, 2).
Immunotherapy is a field that has been rapidly developing over the past few decades, and immune checkpoint blockade (ICB) has demonstrated a tremendous contribution towards novel therapeutic targets and effective drugs that are now used as standard therapy for several cancers, which show in most cases durable responses and extensions of survival curves. However, it is known that responses are patient-dependent and resistance events hamper their clinical benefit. One of the resistance mechanisms proposed for ICB is the inability of lymphocytes to infiltrate the tumor, also known as immune exclusion. This phenomenon can be due to activated oncogenic pathways (3), hypoxia (4–8), degenerated blood vessels (9, 10), cytokines and chemokines released by tumor (11), stromal cells, immune infiltration of immunosuppressive cells (12) and ECM that limit lymphocyte access to the tumor nest. In this review, we summarize the different mechanisms by which physical barriers may promote immune exclusion and potential therapies to overcome it.
2 Immune exclusion
With the advent of ICB and adoptive cell therapy (ACT), treatment of solid and hematologic malignancies has been revolutionized. However, not all patients benefit from these therapies. It is imperative to elucidate which are the mechanisms of immune resistance that limit the efficacy of immunotherapy (13). A predominant phenomenon observed in cancers that do not respond to immunotherapy is the absence of dialogue between the immune cells and the tumor (14). Immune exclusion is a complex phenomenon in which T cells are recruited to the tumor periphery by chemoattraction and antigenic stimuli that promote their persistence, but existing barriers prevent T cells from infiltrating tumor nests and killing cancer cells. The presence of TILs on the tumor core has been proved to have prognostic value in different cancer types (15–21). Therefore, the impairment of these immune cells in reaching the tumor nests and mounting an efficient immune response against the tumor is an issue of clinical relevance.
2.1 Tumor immunophenotypes
Clinical studies have defined immune profiles that can predict responses to immunotherapy. According to this, three basic immune profiles can be identified: immune inflamed, immune excluded and immune desert phenotypes (14). The immune inflamed tumors are characterized by the presence of CD4+ and CD8+ T cells and myeloid cells in the tumor parenchyma. Immune cells are close to, or in contact with tumor cells. For this reason, they usually respond to ICB therapies. However, not all patients respond, indicating that immune cell infiltration is not sufficient to induce an effective immune response probably caused by T cell exhaustion (22). Therapies with ICB have shown promise in melanoma (23), non-small-cell lung cancer (NSCLC) (22, 24) and urothelial cancer (25, 26), which are examples of the inflamed subtype. The immune-excluded tumors present no infiltration of CD8+ T cells to the tumor core, but have accumulation of them around tumor margins, and are resistant to multiple types of treatment (27). This immunophenotype encompasses the most harmful malignancies, including pancreatic ductal adenocarcinoma (PDAC) (28, 29), breast (30–33) and ovarian cancer (34). The immune desert tumors are characterized by a lack of T cells in the tumor stroma (35–38). Although myeloid cells may be present, few or no CD8+ T cells are present resulting in a non-inflamed TME. As expected, such tumors rarely respond to ICB therapies (37). This immunophenotype likely reflects the absence of pre-existing antitumor immunity, suggesting that tumor-specific T cell generation is the rate-limiting step (14). An example of this immunophenotype is glioblastoma, which is further characterized by a lack of tumor antigens, defects in antigen presentation, and a high accumulation of immunosuppressive cells (39). Both the immune desert and the immune excluded phenotypes can be considered non-inflammatory tumors. ICB therapies have shown promise in immune-inflamed tumors, however, such success is not extended to immune-excluded or desert tumors (40, 41). Therefore, therapies promoting the conversion of these latter two immunophenotypes into inflamed tumors could help reduce resistance to ICBs and provide a new treatment option (27, 42).
2.2 Clinical relevance of TILs
A study carried out by Kather et al. in 177 samples of patients with cancers of different histology, evaluated the topography of immune infiltration using immunohistochemistry. Samples were classified according to the number of cells per mm (2) into three spatial compartments: outer invasive margin (0–500 μm outside the tumor invasion front), inner invasive margin (0–500 μm inside the tumor invasion front), and in the tumor core (>500 μm inside the invasion front). It was found that there was a correlation between the infiltration of the tumor core and the internal invasive margin (43). In this way, different types of tumors were grouped into 3 topographies: high density outside of the tumor with a low density inside the tumor, which can be described as “immune excluded”, low density inside and outside represents “cold” tumors, and high density inside the tumor is classified as “hot” regardless of cell density outside the tumor. A similar approach was carried out by Galon et al. They observed that the infiltration of CD8+ T cells within tumor nests combined with their peri-tumoral presence predicts improved survival of patients with colorectal cancer (CRC) with higher accuracy than the classical TNM staging (44). Moreover, limited observations in head and neck squamous cell carcinoma suggests that immune-excluded cancers are transcriptionally indistinguishable from the immune inflamed ones. Therefore, the distinction between these phenotypes is mainly due to the spatial resolution and localization of the immune cell populations (43, 45).
In large cohorts of early triple-negative breast cancer (TNBC) patients, mostly treated with standard adjuvant therapy, a higher total CD8+ and CD4+ TILs count is significantly associated with favorable outcomes (46–48). However, carrying out a more exhaustive analysis and sub-classifying tumors with different patterns of immune infiltration could be of help when evaluating prognosis. A recent report identified four distinct subtypes of tumors based on their surrounding TME, immunodesert (ID); restricted margin (MR); restricted stroma (RS) and fully inflamed (FI), by integrating gene expression signatures with spatial patterns of CD8+ T cell localization in intratumoral and stromal matched samples of treatment-naive TNBC. Both MR and ID tumors were characterized by low CD8+ infiltration (with the first showing CD8+ accumulation at the margins) and these patients were associated with a poorer disease prognosis. In contrast, FI and SR tumors were characterized by abundance of CD8+ T cells, with SR tumors showing stromal-restricted CD8+ infiltration, and FI showing CD8+ T cell infiltration within the stromal and epithelial compartments. Both FI and SR patients were associated with a more favorable prognosis (49).
While in the previously cited works the authors have focused mainly on making topographic distinctions to classify tumors with different patterns of immune infiltrate, other authors have opted to use gene expression profiles as a prognostic marker that could potentially be included in the clinical practice. ICB effectiveness can be affected by the degree of CD8+ T cell infiltration (50), mutation or neo-antigen load (23), PD-L1 level (51), antigen presentation defects (52), interferon signaling (53), mismatch repair defIciency (54), tumor aneuploidy (55) and intestinal microbiota (56). However, none of these factors are enough to achieve accurate outcome predictions (51). Predicting tumor response to ICB requires an understanding of how tumors escape the immune system. There are two distinct mechanisms of tumor immune escape (38, 57). Some tumors have a high level of CD8+ T cell infiltration, but these cells are dysfunctional. In other tumors, immunosuppressive factors may exclude T cells from infiltrating tumors (58). Jiang et al. developed a computational framework, called Tumor Immune Dysfunction and Exclusion (TIDE), to identify factors that underlie these two mechanisms of tumor immune escape. TIDE integrated and modeled data from 189 human cancer studies and validated an accurate gene signature to model tumor immune escape that could serve as a reliable surrogate biomarker to predict ICB response. TIDE predicted the outcome of melanoma patients treated with first line anti-PD1 or anti-CTLA-4 more accurately than other biomarkers, such as PD-L1 level and tumor mutation burden (59). Finally, other authors have resorted to computational tools to study the TME. In recent years, computational imaging approaches originating from artificial intelligence have achieved success in automatically quantifying radiographic characteristics of tumors (60–62). Radiomics is an emerging field within medical research that aims to use advanced imaging analysis to study tumors and potentially predict treatment outcomes based on radiological features. Jazieh et al. demonstrated that a radiographic image-based biomarker on baseline CT scans is significantly associated with progression-free survival and overall survival (specifically prognostic within individual PD-L1 categories) in patients with NSCLC treated with durvalumab (anti PD-L1 monoclonal antibody) after chemoradiotherapy (CRT) or CRT alone by using radiomic texture patterns within and outside the NSCLC tumor cells (63).
Clinical distinction between the above-mentioned tumor immunophenotypes is highly important for the design of next-generation immunotherapy agents and the possibility of a tailored medicine (64). To determine the prevalent mechanisms of immune exclusion and address them to expand the effectiveness of immunotherapy approaches must be of first priority for researchers and physicians. Among those mechanisms, we can mention physical barriers that will be discussed in the next section of this review, and also metabolic and functional barriers (65) that prevent T cell infiltration to the tumor core and cancer cells elimination (45).
3 Physical barriers within the TME
3.1 Extracellular matrix
It is well known that the ECM plays a crucial role in the interaction of cells within a tissue (66) and, for this reason, it is of high importance during tumor development and progression (67). Interactions between the ECM and the tumor go in both directions: the ECM determines the morphology of the cells, their differentiation and migration, and regulates cell-cell interactions. On the other hand, cells actively remodel the composition and geometry of the ECM in favor of tumor progression (68–71). ECM dysregulation during cancer progression is mainly carried out by stromal cells, including CAFs and immune cells (72, 73). Nevertheless, epithelial cells and mesenchymal stem cells may also be involved at late stages of cancer development (74, 75). How the ECM changes along tumor establishment and progression is well reviewed elsewhere (67, 76). The ECM also has immunostimulants and, hence, potential antitumoral effects. In this sense, it has been reported that ECM components such as biglycan, heparan sulfate or versican can modulate leukocyte (77) and dendritic cells recruitment (78), have an active role in inflammation (79), T cell activation and macrophage recruitment (80), among others. In this review we will focus on the pro-tumoral effects of different components of the ECM and their impact on the efficacy of immunotherapies, since the immunostimulant effects have been reviewed elsewhere (81–84).
The components of the ECM can be divided into two categories: the liquid part, that include cytokines and chemokines, and the polymeric scaffold. The latter contains different types of polymers such as polysaccharides, like hyaluronan and chondroitin sulfate, biglycan and perlecan, and fibrous proteins, like collagen and fibronectin that provide structure to the matrix and participate in cell-cell interactions (67). The ECM plays a key role in the establishment of anti-tumor immune escape mechanisms since it allows the diffusion of signaling (such as cytokines and chemokines), the recruitment of immune cells into the tumors, as well as the interaction between specific ligands and receptors. There are large body of evidence that have described how the ECM components contribute to immune exclusion, like the contribution of chemokines and cytokines, barrier molecules, collagen and hyaluronic acid and the tumor-associated vasculature, that is discussed in the section below and summarized in Figure 1.
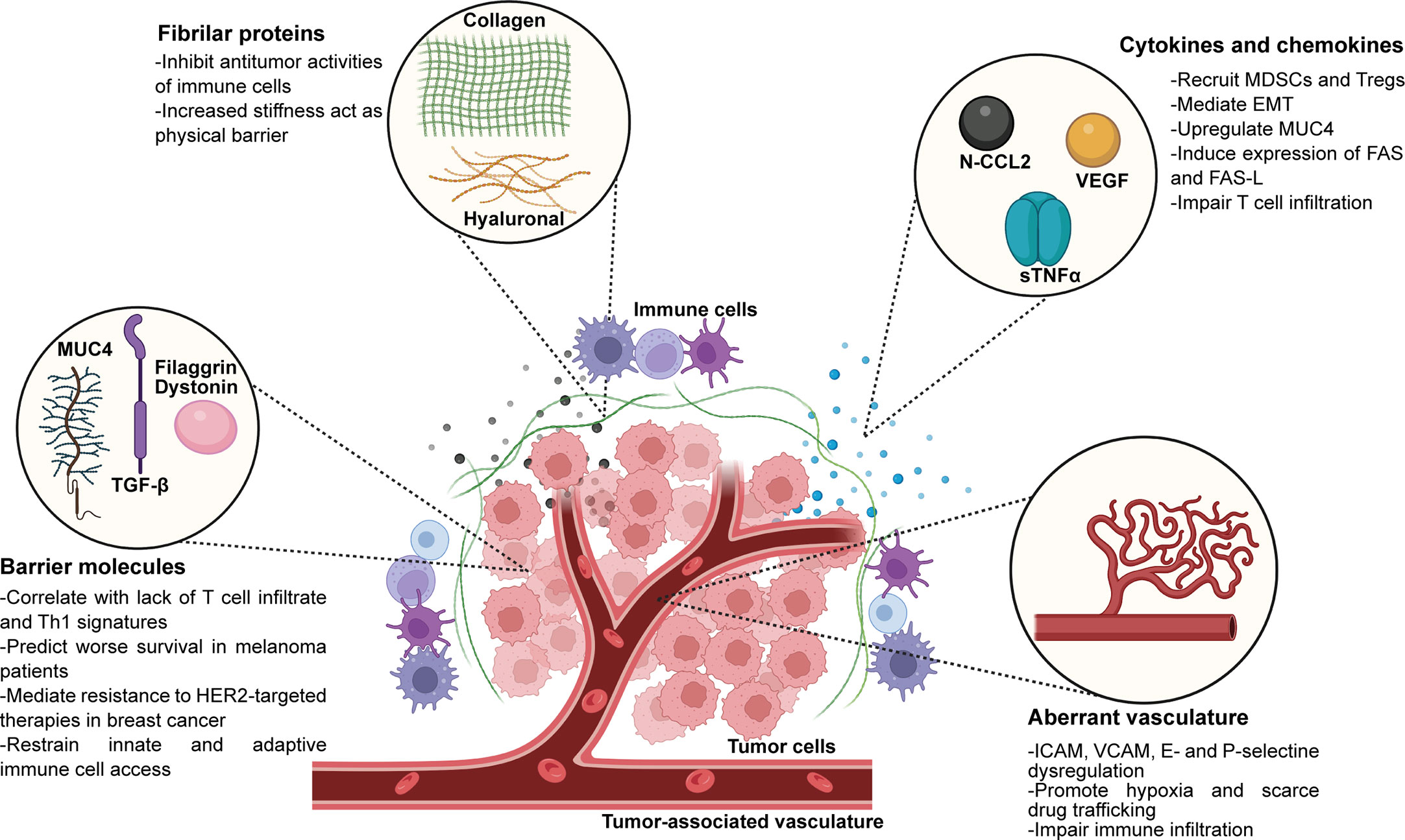
Figure 1 Extracellular matrix components present in the TME that play active roles in immune exclusion and immunosuppression. EMT, epithelial-mesenchimal transition. Created with BioRender.com.
3.1.1 Chemokines, cytokines and barrier molecules
Chemokines released from cells immersed in the TME account for recruitment and extravasation of different immune cell populations. However, the TME can modify chemokines posttranslationally to allow differential cell infiltration and to promote immune exclusion. One example is the production of reactive nitrogen species by MDSCs, which induces nitration of chemokine (C-C motif) ligand 2 (N-CCL2) (85). CCL2 is secreted by different cell types, like T cells, monocytes, and also tumor cells, and works as a chemoattractant for myeloid cells, activated CD4+ and CD8+ T cells, and NK cells (86–89). Moreover, CCL2 has the ability to trigger granule release from both NK and CD8+ T cells (90). The authors prove that nitration of this cytokine results in T cells being trapped within the stroma surrounding human prostate and colon cancer cells (85). In contrast, N-CCL2 attracts MDSCs and TAMs to the tumor core tissue, potentially contributing to the differential recruitment to the TME of these immunosuppressive immune cell types. Interestingly, drugs that inhibit nitration of CCL2 proved to enhance TILs accumulation in preclinical models, and resulted in improved efficacy of adoptive cell therapy. Another important contributor to the composition and abundance of the ECM is the cytokine transforming growth factor-β (TGF-β). This molecule is secreted mainly by CAFs, but it can also be produced by myeloid cells and tumor cells (91–94). TGF-β has several key functions as a major regulator of the ECM homeostasis. For example, it stimulates CAFs to produce structural collagen fibers. Also, this cytokine induces the synthesis of enzymes involved in degradation or crosslinking of collagens (95), thus regulating fibrosis and stiffening of the ECM. Collagen not only acts as a physical barrier to T cell infiltration, but it can also act as a ligand for a vast number of receptors present in tumor and immune cells. One example is the leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1), an immune checkpoint that is expressed on the surface of immune cells. When collagen XVII fibers present in the ECM surrounding the tumor binds to LAIR-1, the signal transduction results in inhibitory signals that cause T cell exhaustion and impairment of NK cell, monocyte and dendritic cell activation and proper function (96). Recently, it has been proved that T cell exclusion and poor response to ICB therapies is associated with a gene signature of TGF-β activation (97–99). Particularly in metastatic urothelial immune-excluded tumors that show no response to atezolizumab, the authors found that there was an enrichment of a fibroblast TGF-β gene signature, and that the expression of this gene signature was directly linked to T cell trapping in the collagen-rich stroma surrounding the tumor (97). Moreover, they demonstrated that the co-inhibition of TGF-β and PD-L1 transformed tumors from excluded to inflamed, adding to the idea that TGF-β signaling restricts T cell infiltration to the TME and hampers efficient antitumor immune responses. Also, TGF-β secreted by stromal cells is able to induce the expression of the vascular endothelial growth factor (VEGF) to promote tumor angiogenesis, and recruit immunosuppressive cells like MDSCs and Tregs, to further enhance immune escape and impair T cell infiltration. Furthermore, Horn and collaborators demonstrated that in two collagen-rich murine carcinomas, MC38 colon and EMT6 breast, the inhibition of LAIR-1 and TGF-β plus anti-PD-L1 therapy was able to control tumor growth and reshape the collagen-rich ECM (100). Authors evidenced that the combined treatment promoted an increased tumor infiltration and activation of CD8+ T cells, and was also able to repolarize protumoral macrophages to the antitumoral subtype. Moreover, they proved that this treatment resulted in high tumor regression rates and long-term protection by tumor-specific immune cells (100). TGF-β also mediates the epithelial-to-mesenchymal transition (EMT) (101, 102), which has been described as another process that raises mechanical barriers for T cell infiltration in various cancers, such as melanoma, ovarian and gastric (103–106). Another cytokine that plays an important role in cancer progression and immune exclusion is the pleiotropic cytokine TNFα. We have previously reviewed the participation of TNFα on the progression and metastasis of different breast cancer subtypes and the antitumor immune response mounted by different therapies (107), and also the mechanisms by which TNFα generates resistance to immunotherapy, such as monoclonal antibodies against cancer cells or immune checkpoints, and adoptive cell therapy elsewhere (108). With regards to immune exclusion, it is known that TNFα may impair the infiltration of effector T cells by negatively regulating the formation of high endothelial venules (HEVs) (109). These HEVs are in close contact with tumor cells and tertiary lymphoid structures (TLS). It has been proved that tumors with high density of HEVs and TLS have enhanced proportion of effector T cells infiltrating the tumor core. Moreover, this has been proved to correlate with favorable clinical outcomes (110, 111). TNFα has also been described to enhance the production of Fas and its ligand FasL on CD8+ T cells (112, 113), which trigger the apoptosis of neighboring CD8+ T cells (114). Our team has demonstrated that soluble TNFα (sTNFα) secretion by HER2+ breast cancer cells induces resistance to the anti-HER2 therapy trastuzumab, by upregulating the expression of the transmembrane glycoprotein mucin 4 (MUC4) through activation of the NF-kB pathway (31, 115). MUC4 belongs to the membrane-bound family of mucins and has two non-covalently associated subunits encoded by a single gene (116). The extracellular subunit, MUC4α, is hyperglycosylated and it favors metastatic dissemination as it confers antiadhesive properties to the tumor cells. The transmembrane subunit MUC4β, contains two EGF-like domains in the extracellular portion that can interact with HER2, preventing its internalization and therefore enhancing its signaling (117). As trastuzumab binds to the juxtamembrane region of HER2 (118), MUC4 with its heavy glycosylation patterns shields trastuzumab epitope on the HER2 molecule hindering its binding and therapeutic effect (119). MUC4 has been proved to interact with HER2 and even to induce its phosphorylation and activation (120). It also promotes HER2 and HER3 translocation to the cell surface, increasing the number of available receptors and keeping them anchored to the membrane during longer periods of time (121). As a consequence, the signaling cascade of the HER2/HER3 heterodimer through activation of PI3K is enhanced (122), causing an increase in cell proliferation and survival in HER2-positive breast cancer (123). In a previous report, we have also demonstrated that MUC4 has a key role in the immune response against the tumors, since it generates an immunosuppressive TME characterized by an increased infiltration of MDSCs, poor NK cells activation and degranulation and an increased proportion of protumoral M2-like macrophages (31). Also, we demonstrated that blocking sTNFα with a dominant negative molecule, INB03, overcomes trastuzumab resistance and remodels the TME into an immunocompetent one, characterized by a decrease in MDSCs proportion, an increase in NK cell activation and degranulation and an enhanced macrophage infiltration and polarization to the antitumor M1-like subtype (31). Moreover, we have proved that MUC4 is an independent biomarker of poor disease-free survival in HER2+ breast cancer patients treated with adjuvant trastuzumab (115). Our studies also focused on the role of MUC4 regarding TILs, since this glycoprotein is heavily glycosylated. We demonstrated that MUC4 acts as a physical barrier for immune exclusion in TNBC and HER2+ breast cancer, since tumor samples from patients with MUC4+ tumors evidenced scarce or null presence of TILs, while patients with MUC4- tumors evidenced abundant TILs on their TME (31, 124). Moreover, mucins can interact with inhibitory receptors like ICAM-1 on T-cells, causing anergy and impaired antigen recognition, and with siglecs on antigen-presenting cells (125, 126). Overexpression of mucins on tumor cells create steric hindrance for therapies, mask the detection of tumor-associated antigens and prevent lysis of the tumor cells by immune cells, generating immune tolerance (126–129). Also, it has been demonstrated that mucins expressed by tumor cells interact with leukocytes in the TME and facilitate the colonization of disseminated cells, since aberrant glycosylation promotes the expression of ligands for selectins expressed by leukocytes and platelets, normally used for adhesion (130). Therefore, mucins form aggregates with these cells and promote metastasis (121, 131). Particularly, it has been described that MUC4 protects disseminated cells from the immune recognition by hiding with its glycosylations the corresponding immunogenic antigens on tumor cells (121), and that, by physically interacting with platelets and macrophages, it able to enhance survival and extravasation (132).
Moreover, proteins with known barrier functions have been described to promote immune exclusion (103). Examples of these proteins are filaggrin, TACSTD2 and desmosomal proteins like desmocollin 3, dystonin, desmoplakin, periplakin, plakophilin 3 and junction plakoglobin. In healthy tissues, these proteins are usually expressed in the outer layer of the skin and play a crucial role as mechanical barriers that protect our body from pathogens. However, Salerno and collaborators found that a subset of metastatic melanomas and ovarian carcinomas express high levels of genes encoding these proteins. Moreover, they prove that the expression of genes encoding proteins with barrier functions correlated with a lack of T cell infiltrates and immune related signatures in multiple data sets. What is more, overexpression of the barrier molecules was shown to predict worse survival for melanoma and ovarian cancer patients across various clinical subsets (103). Melanoma patients that received adjuvant therapy and did not have overexpression of barrier molecules showed improved survival, which may suggest a potential predictive value for this subset of proteins. Particularly, Chen and collaborators described by means of cellular experiments that filaggrin is an oncogene in bladder urothelial carcinoma (BLCA), and that a knockdown of this barrier protein suppressed BLCA cell proliferation and promoted apoptosis (133). Furthermore, based on TCGA data sets the authors proved that BLCA patients expressing the wild-type isoform of filaggrin presented less overall survival than patients expressing mutated forms of the barrier protein, consistent with the fact that higher tumor mutation burden favors responses to immunotherapy (133). T cell infiltration to the tumor bed was modulated by filaggrin mutational state, since patients with wild-type filaggrin showed significant down-regulation of CD4+ naïve T cells, central memory T cells, and natural killer T cells, while Th1 cells were significantly upregulated. Also, these patients showed poorer response to ICB (133). Another recent study evaluated multiplex immunofluorescence histology of tumor samples from 65 advanced melanoma patients, and proved that the expression of another barrier molecule periplakin negatively correlates with response to pembrolizumab (134). Decreased expression of periplakin and increased proportion of CD103+ CD8+ T cell infiltration correlated with improved survival in these patients, so authors propose periplakin as a predictive biomarker to ICB. Moreover, Pai and collaborators proved that the expression of another barrier molecule, the desmosomal protein dystonin, was inversely correlated with the Th1-like immune signature in patients with metastatic melanoma and ovarian cancer (45), and was associated with worse prognosis in patients with melanoma (103). Particularly, dystonin expression allowed the authors to identify tumors with null expression of the Th1-like immune signature, suggesting no interaction between these immune cells and cancer cells. Interestingly, dystonin expression identified a subset of melanoma patients with no CD8+ gene signatures. However, this was not associated with decreased patient survival, which adds to the idea that the presence of T cells within a given TME is not enough to predict prognosis, and that a better approach could be to study the ratio between lymphoid and myeloid or other cellular infiltrate to better define prognostic significance (135–137). These findings raise the attention to barrier molecules as future potential biomarkers for defining T cell accessibility to the tumor core and, consequently, ICB responses.
3.1.2 Collagen and hyaluronic acid
Another important component of the ECM is collagen, which distributes between the cells as fibers. Historically, collagen was demonstrated to be a passive barrier to resist tumor cell infiltration and establishment. However, the role of collagen in cancer progression, immune escape and poorer patient outcome is now widely studied (138–141). Collagen I, III and IV are the predominant types in forming the scaffolds of the TME (142–144). Upon tumor progression, the ECM remodeling of collagen fibers which can be degraded (145) and later re-depositioned (143, 146), can promote tumor infiltration, angiogenesis, invasion and migration (70, 71, 146, 147). How collagen can be a double-edged sword in tumor progression, both inhibiting and promoting tumor progression at different stages of cancer development has been exhaustively reviewed elsewhere (148). In this section, we will focus on the role of collagen fibers as a contributor to immune exclusion.
A recent article from Kuo-Sheng Hsu and collaborators has proven that the survival of cancer cells depend on the uptake of collagen I into their associated stroma (149). Also, evidence indicates that breast tumors with dense collagen depositions correlate with a worse patient’s outcome (150). Moreover, it has been shown that collagen XIII expression is increased in human breast cancer samples compared to normal mammary gland tissue, and increased levels of collagen XIII mRNA correlate with short distance recurrence free survival (151). One of the mechanisms contributing to collagen I-mediated tumor growth promotion is its ability to mediate immune cell exclusion (152). It has been demonstrated that interaction of collagen fibers and immune cells account for inhibition of their antitumor activities (153), and Salmon et al. revealed that the stromal ECM, particularly fibronectin and collagen fibers, influences antitumor immunity against human lung tumors by controlling the localization and migration of T cells (154). Movement of cells towards and within the ECM is prevented through an unusually dense and stiff composition. Salmon stated that it was the higher density of the ECM in the tumor islets that caused T cells to preferentially migrate to the tumor stroma rather than infiltrate the islets. Other researchers also highlight that the remodeling of the ECM that increases tumor stiffness can mechanically activate pathways that lead to tumor progression and dampen T cell infiltration to the tumor islets (155–158). Particularly, studies have demonstrated that T cells are impaired to infiltrate from the stroma to the tumor core of PDAC when the ECM density is high (159, 160). Moreover, it has been stated that tumor fibrosis (accumulation of collagen type I and III fibers in the tumor ECM) is associated with a variety of malignancies (142, 143, 161) and particularly correlates with worse prognosis in breast cancer (162). Also, tumor fibrosis not only increases metstasis risks in different tumor types (163, 164), but it is also necessary for the successful establishment of metastasis foci (165, 166). When fibrosis is highly extensive, for example in PDAC, the scar-like ECM acts as a physical barrier to cytotoxic T-cell infiltration into tumors (167). Collagen type I is the most abundant type of collagen fiber present in the ECM (168). Researchers proved that loose areas of fibronectin and collagen usually facilitate T cell infiltration, while dense and stiff ECM reduce T cell velocity and migration (167). In line with this, it has been proved that central fibrosis in tumors exclude immune cells from CRC metastases (169). One of the collagen receptors, the discoidin domain receptor 1 (DDR1), has also been studied in this regard. The different roles of DDR1 in cancer promotion are summarized in Table 1. This receptor has an intracellular domain with tyrosine kinase activity (170), which is triggered by collagen binding and participates in different downstream signal transduction (170, 171), but has no impact on tumor growth (152). In contrast, researchers have found that it is the extracellular DDR1 collagen-binding domain (ECD), the one required for tumor growth in immunocompetent hosts (152). As it happens with collagen, DDR1 also has paradoxical and context-depending roles in cancer. Some authors have described DDR1 as a tumor suppressor, as it induces apoptosis of basal-like breast cancer cells in 3D collagen I matrices (172–174) and in luminal breast cancer cells in young collagen 1-enriched ECM (175). However, when collagen 1 ages, DDR1 no longer promotes apoptosis and the tumor keeps growing. On the other hand, several publications suggest that DDR1 overexpression at protein levels has shown to be associated with cancer progression (176–179) and metastases (180), and the increase of DDR1 mRNA levels was proved to be associated with worse overall survival in all subtypes of breast cancer patients, and particularly in TNBC (152). Regarding invasion, Juin et al. demonstrated that DDR1 plays a key role in the formation and matrix-degradation ability of F-actin invadosomes, promoted by collagen I fibrils (181). Bravo-Cordero and colleagues proved that the DDR1/STAT1 axis favors dormancy by triggering collagen III expression. However, changes in the alignment of collagen III fibers to a more linear orientation may awake tumor cells from dormancy and reactivate proliferation and metastasis (182). Sun and collaborators demonstrated that DDR1 knockout (KO) tumors were impaired to grow in immunocompetent mice, but were capable of growing in immunodeficient hosts. However, depleting CD8+ T cells allowed DDR1 KO tumors to grow at the same rate as wild-type ones in immunocompetent mice (152). Moreover, the authors proved that DDR1 KO tumors exhibited an increase in CD4+ and CD8+ activated T cells, compared to DDR1 wild-type tumors. What is more, when representative tumor samples from each experimental group were analyzed by multiplex immunofluorescence staining, authors found that CD8+ T cells were largely restricted to the tumor margins of DDR1 wild-type tumors, a phenomenon that was also evident by other researchers in similar mouse models of TNBC (183). Strikingly, DDR1 KO tumors showed an increased infiltration of CD8+ T cells on the tumor core, suggesting that DDR1 is a key factor of the TME that physically restricts T cell infiltration. Adding to this, it has been proved that both mRNA and protein levels of DDR1 negatively correlate with genes that define anti-tumour immunity, a gene expression signature for intratumoral T cell accumulation, CD8+ T cell signature scores, and the cytolytic effector pathway (59, 184). The authors found that in TNBC patient samples which have never received any treatment, the percentage of DDR1 positive cells in DDR1 high tumors inversely correlated with the abundance of infiltrating CD8+ T cells, which were circumscribed to the tumor margin rather than present in the tumor core. In contrast, tumors classified as DDR1 low did not show evident differences between the abundance of CD8+ T cells in the tumor core or at the margins. Moreover, when tumor samples were stratified according to the immune phenotype, all tumor samples classified as immune-excluded were DDR1 high and most tumors classified as non-excluded were DDR1 low, confirming the role of tumoral DDR1 in immune exclusion. Finally, with respect to the role of DDR1 in the ECM, it was proved that DDR1-ECD promotes the alignment of collagen fibers, reinforcing the defenses of tumors against immune infiltration (152).

Table 1 Roles of DDR1 in promoting cancer.
ECM alignment also plays a key role in controlling immune cell migration (154, 185, 186). Particularly, a collagen-alignment signature could be a prognostic factor for the survival of patients with breast cancer (187). The migratory capacity of T cells was impaired in fresh human tumor explants due to the architecture of collagen fibrils that act both as a guiding path to lead T cells out of the tumor core and as a physical barrier that prevents their entrance, leading to a T cell excluded profile (154, 188). Moreover, it has been recently demonstrated that T cell infiltration inversely correlates with stiffening and high density of ECM in different preclinical murine models of pancreatic, breast, and bile duct carcinomas (189). The authors proved that ECM stiffness measurements correlated with tumor growth and ECM crosslinking of collagen fibers. They showed that interfering with collagen crosslinking into fibers by inhibition of the lysyl oxidase (LOX) enzyme reduces ECM content and tumor stiffness, improving T cell migration and improving the efficacy of anti-PD-1 blockade. Combination of anti-PD-1 and LOX inhibition resulted in an increased accumulation of effector CD8+ T cells in the tumor and significant delays on tumor progression in a pancreatic cancer mouse model (189).
Hyaluronan (HA) is a linear glycosaminoglycan, composed of repeating disaccharide units of D-glucuronic acid and N-acetyl-D-glucosamine (190). HA is a conspicuous component of the ECM, where it possesses several functions both in physiological and pathological conditions such as morphogenesis, tissue repair, inflammation and tumorigenesis (191–194). In healthy tissues, HA presents high molecular weight (HMW-HA) (>1,000 kDa) and has a structural function. HA fragmentation is critical during inflammation, tissue-remodeling processes, and cancer. HA fragments of 200-1000 KDa are called low molecular weight HA (LMW-HA), and fragments <10 kDa are HA oligosaccharides (195). HA levels and molecular size mainly depend on a fine tune between the expression of the enzymes involved in its synthesis (hyaluronan synthases) and its degradation (hyaluronidases) (196). Hyaluronan levels are increased in different solid tumors and associated with tumor aggressiveness and progression, reviewed elsewhere (197, 198). HA forms a hydrogel-like matrix surrounding the tumor cells acting as an exclusion barrier and, thereby, inhibiting the permeation of different drugs (such as chemotherapeutics or monoclonal antibodies) as well as limiting the accessibility of cells from the immune system. A HA-rich matrix confers resistance to anti-HER2 therapy with trastuzumab in breast cancer cells due to HA-induced masking of HER2 and to inhibition of NK cells access to the tumors. Depletion of HA levels by treatment with a pegylated version of hyaluronidase, PEGPH20, resulted in increased NK cell access and enhanced ADCC (199–201). The HA role in the immune exclusion of T cells has been reported in PDAC, which are characterized by HA accumulation. Indeed, depletion of HA by PEGPH20 in combination with a whole-cell PDAC vaccine modulated myeloid cell function by inhibiting the CXCR4 immunosuppressive signaling axis and lead to enhanced T cell infiltration with a rise of intratumoral effector memory T cells (202). Another study demonstrated that treatment with the inhibitor of hyaluronan synthesis 4-methylumbelliferone, promoted infiltration of inoculated γδT cells into tumor tissue and, consequently, suppressed the growth of PDAC (203). In both studies, PDAC stroma remodeling by HA depletion presented an effective immunosensitizer effect. Despite HA function as an exclusion barrier, HA exerts immunomodulatory actions by interacting with receptors including CD44, RHAMM, or TLR4. HA can modulate both the innate and the adaptive immune responses and its final effects depend mainly on the molecular weight of HA, the immune cell type, and the receptor involved. While native HA appears to be anti-inflammatory and anti-tumorigenic, HA fragments and oligomers are pro-inflammatory and pro-tumorigenic (204). The effects of both HMW-HA and HA fragments on TAM function has been extensively reviewed elsewhere (205, 206). While the role of HA on TILs remains to be explored, HA is an important mediator of T cell trafficking. HA/CD44 interaction is known to facilitate the rolling and extravasation of T cells to inflammatory sites (207). Furthermore, HMW-HA, but not LMW-HA, induced the immunosuppressive actions of regulatory T cells. Indeed, HMW-HA enhanced Foxp3 expression and IL-10 production by Tregs (208, 209). Altogether, these reports point out a crucial role of HA in the regulation of T cell recruitment and function in the TME. Further studies aimed to investigate the levels and the molecular weight of HA polymers within the TME will help to elucidate their specific cellular functions in vivo.
3.1.3 Aberrant vasculature
Tumor vasculature density and molecular signature has also been shown to have a role in T cell infiltration (210–212). The whole process of T cell migration into target tissues is reviewed elsewhere (213, 214); the last stage of leukocyte infiltration is called transendothelial migration and extravasation. This process usually takes place in HEVs, and it has been reported that tumors with higher amounts of HEVs will promote increased infiltration than tumors with aberrant vasculature formation (210, 211). Tumor-associated endothelial cells play key roles during tumor development such as angiogenesis, regulating vessel permeability, transportation of the immune cells, intravasation and extravasation of tumor cells during dissemination (12). Leukocyte attachment, rolling and transmigration into tissues is regulated by endothelial cells, through the expression of a series of adhesion molecules, for example the intracellular adhesion molecule (ICAM), the vascular cell adhesion molecule (VCAM) as well as E- and P-selectin (12). However, TILs infiltration to the tumor core is affected by a dysregulation of these receptors on the tumor-associated vasculature. Aberrant tumor vasculature is characterized by collapsed blood vessels that are unevenly formed and are often leaky. These vessels usually induce hypoxia, scarce drug trafficking and immune infiltration to the tumor core (215). Also, the endothelium expresses molecules that act as ligands for T cells, which can either inhibit or stimulate immune cell extravasation (216). One example of molecules that stimulate cell extravasation is the extracellular superoxide dismutase 3 (SOD3), an enzyme expressed within the TME that improves the aberrant functions of tumor-associated vasculature by stabilizing hypoxia-inducible factor 2 (HIF2α) that, in turn, induces the expression of vascular endothelial cadherin (212, 217). This signal transduction cascade causes a reduction in vascular leakage and increases blood flow towards the tumor (217). However, SOD3 expression is downregulated in several tumors, including pancreatic, colorectal, lung and breast cancer (218–220). Mira and collaborators have demonstrated that SOD3 enhances effector T cell transmigration and extravasation through the upregulation of WNT ligands, induced by HIF2α. WNT ligands cause the increase of laminin subunit alpha-4 (LAMA4) expression in endothelial cells, a laminin that participates in the maturation of microvessels (221) and gives permissive signals in favor of the transendothelial T cell extravasation (44). Interestingly, SOD3 does not trigger permissive signals for migration of Tregs or myeloid cells. Since SOD3 expression is associated with an increased infiltration of CD8+ T cells and improved outcomes in patients with stage II CRC, the authors claim that this regulatory mechanism of vascular normalization could have clinical implications (217). On the other hand, there are negative signals that inhibit effector T cell adherence to the tumor endothelium and impair T cell recruitment and extravasation to tumor sites. This phenomenon is known as endothelial cell anergy. For example, endothelin B receptor (ETBR) synthesis has been proved to interfere with the adhesion of T cells to the endothelium and, consequently, a successful transendothelial migration and extravasation process. ETBR expression has been proved to be increased in the endothelium of human ovarian cancer and inhibits T cell infiltration to the tumor core. Authors have demonstrated that this inhibition on T cell infiltration can be reversed through treatment with the ETBR neutralizing agent BQ-788 (222). Another overexpressed factor on the tumor-associated vasculature that has been implicated in TILs exclusion is Fas ligand (FasL), produced by murine and human tumors, like ovarian, colon, prostate, breast, bladder, and renal cancer (223). T cells express Fas receptor on their membrane and, upon FasL binding, CD8+ T cells undergo apoptosis. However, this effect has not been observed in Tregs, since they also express c-FLIP on their cell membrane, a protein capable of inhibiting apoptosis. As a consequence, if the tumor-associated vasculature expresses high levels of FasL, although CD8+ T cells may be recruited to the tumor core they are not able to penetrate successfully, and Tregs are preferentially recruited instead (223). Authors also analyzed cohorts of ovarian, colon, bladder, prostate, and renal cancer patients and found that intraepithelial CD3+ or CD8+ cells correlated with significant increases in overall survival, and that high percentages of tumor vessels with FasL+ inversely correlated with intraepithelial CD3+ cells presence. This was further analyzed and authors found that the lack of CD3+ cells was mostly due to a lack in intraepithelial CD8+ TILs, since tumors with FasL+ vessels exhibited abundance of intraepithelial FoxP3+ cells. This data indicates that tumor endothelium induces FasL expression and selectively increases Treg:effector T cell ratio on the tumor core, allowing for CD8+ T cell exclusion in several solid tumor types (223). In line with this, in animal models the inhibition of FasL increased significantly the infiltration of effector T cells and resulted in tumor growth suppression. The authors described that FasL expression is regulated by VEGF, prostaglandin E2 (PGE2), and IL-10. They claim that blocking tumor angiogenesis promotes effector CD8+ T cell infiltration by limiting their tumor vasculature FasL-mediated apoptosis. This is relevant particularly for ex vivo activated T cells used for ACT or endogenous activated T cells for cancer vaccines, as activated T cells are prone to undergo apoptosis mediated by FasL. It is known that VEGF and fibroblast growth factor 2 (FGF2) can cause endothelial cell anergy by downregulating the expression of ICAM1 and VCAM1 on the endothelium, two key adhesion molecules for T cell infiltration (224). Moreover, increased expression of some growth factors like VEGF, platelet-derived growth factor (PDGFC) and placental growth factor are able to promote tumor angiogenesis (225, 226). Moreover, it was proved that VEGF can inhibit endothelium activation induced by NF-κB signaling, and impair the T cell infiltration, by blocking the production of chemokines CXCL10 and CXCL11 secreted by tumor and stromal cells (227). These chemokines correlate to T cell abundance in the tumor core of lung (228, 229), colorectal (230) and melanoma (228) patients. Another factor that can influence T cell infiltration into the tumor nest is the integrin-selectin signature expressed in the tumor vasculature, since they are key players in the stages of leukocyte rolling to achieve successful extravasation into target tissues (231). In this regard, researchers have determined the role of the ECM, particularly integrins, in the preferential localization, migration and activation of T cells in lung adenocarcinomas, TNBC and HER2+ breast cancer tumors (154, 232). Particularly, integrin subunit alpha 5 (ITGA5) has a crucial role in promoting cancer cell invasion, metastasis and it was recently proved to be overexpressed in gastrointestinal tumors and was associated with worse overall survival and disease-free survival in colorectal, pancreatic, gastric and liver cancers (233). What is more, expression of ITGA5 was associated with an increased infiltration to the tumor core of CD4 + T cells, macrophages, neutrophils and dendritic cells. Also, ITGA5 expression measured by immunohistochemistry in gastrointestinal tumor samples was proved to be associated with gene expression signatures related to immunotolerant populations, such as TAMs, Th2 cells and protumoral M2-like cells. Authors claim that ITGA5 may have a potential role in polarizing macrophages to the M2 subtype and promoting the activation of tolerogenic lymphocytes (233). ITGA5 is not only expressed in tumor cells, but it has also been reported to be expressed in CAFs (234), TAMs (235) and chimeric antigen-receptor expressing T cells (233). CAFs have a clear role in regulating the recruitment of immune cells and their functions with regards to their anti-tumor responses (167, 236), which will be discussed afterwards in this review. ITGA5 expressed in the surface of TAMs and T cells can directly regulate recruitment and alternative activation of immune cells, and its interaction with other integrins is crucial for the integrin-mediated signaling pathways, leukocyte migration and cell-matrix adhesion (237). This data proves, once more, the importance of the interplay between vasculature and the ECM in immune exclusion and response to therapy.
3.2 Immunosuppressive cells
Numerous preclinical studies have proposed several stromal cell types of the TME, like CAFs, MDSCs and TAMs as the key players for restricting infiltration of T cells in the tumor core (135, 238–243). In this section we discuss the most recent evidence on the mechanisms by which these cells promote immune exclusion and therapy failure. The main roles of immunosuppressive cells in immune exclusion are summarized in Figure 2.
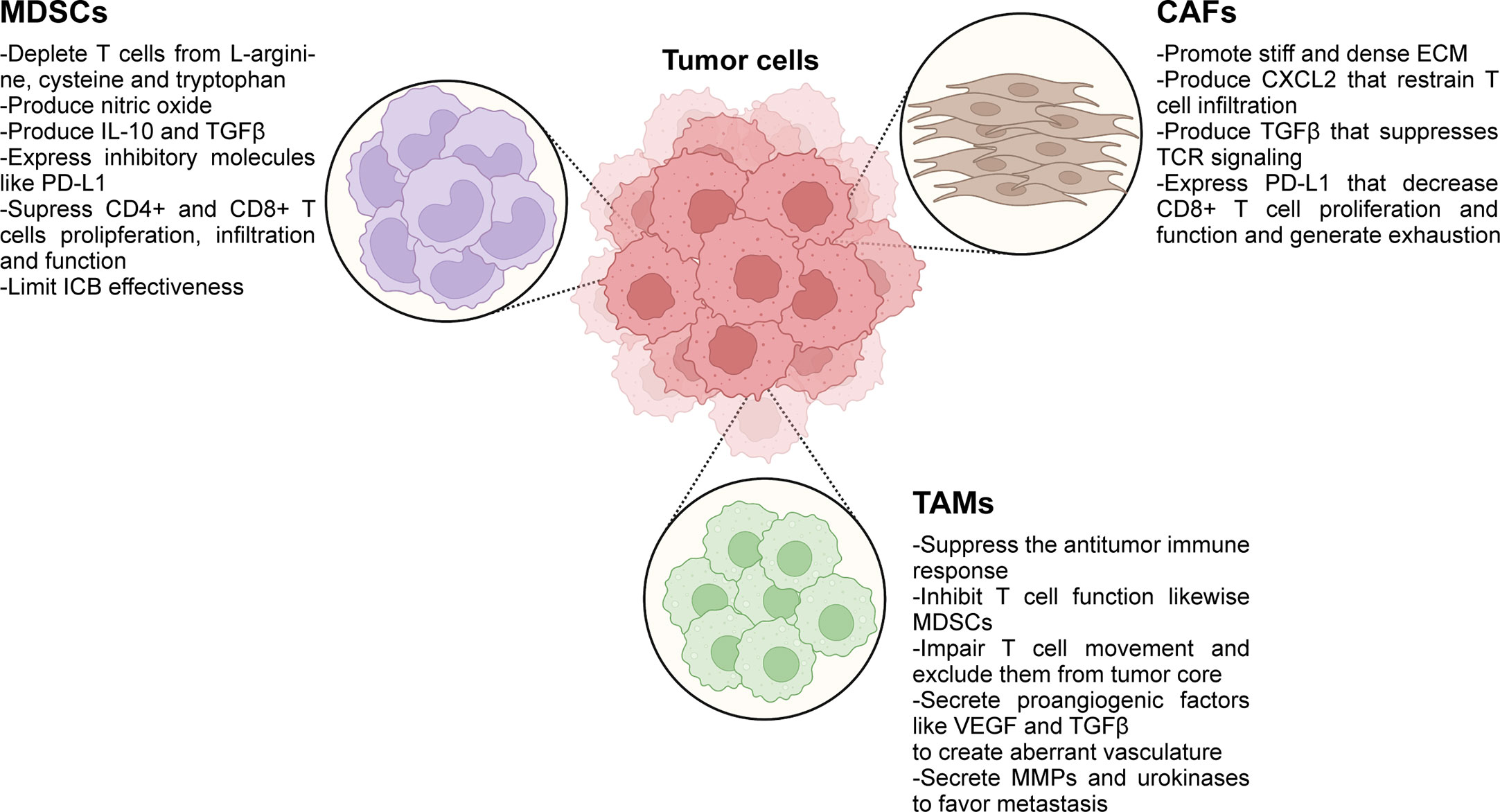
Figure 2 Immunosuppressive cells present in the TME play active roles in immune exclusion and immunosuppression. ICB, immune checkpoint blockade; ECM, extracellular matrix; MDSCs, myeloid-derived suppressor cells; TAMs, tumor-associated macrophages; CAFs, cancer-associated fibroblasts; MMPs, matrix metalloproteinases. Created with BioRender.com.
3.2.1 Myeloid cell barrier
Thanks to their plasticity, myeloid cells are able to modify their differentiation conditioned by tumor factors to achieve an immunosuppressive phenotype that fosters immune evasion. MDSCs are produced by an abnormal myeloid progenitor development in the bone marrow induced by high levels of factors produced by tumors and their stroma, such as G-CSF, GM-CSF, IL-6, and VEGF, which activate Stat3 and Stat5 favoring their differentiation to the detriment of normal monocytes, macrophages and dendritic cells (244–247). The relationship between cytokines and immunosuppressive cells was clearly depicted in pancreatic cancer and liver metastasis, as tumor-GM-CSF induces the activation and expansion of MDSCs, which in turn, suppress T cell function (242, 248). MDSCs enter the circulation from the bone marrow and infiltrate the primary or metastatic tumor site in response to gradients of chemokines such as IL-8, CCL2, and CXCL12 (249–251). Once in the TME, different factors such as hypoxia, adenosine, the activation of the endoplasmic reticulum stress response pathway, among others, increase the immunosuppressive activities of MDSCs (252–254). Two major subpopulations have been described in humans and mice: the polymorphonuclear MDSC (PMN-MDSC) and the monocytic MDSC (M-MDSC). The former is derived from granulocytic precursors and are similar to neutrophils, while the latter are derived from monocytic precursors and are similar to monocytes. In this complex scenario, Long et al. described that erythroid progenitor cells can switch to the myeloid lineage and differentiate to erythroid-differentiated myeloid cells (EDMC), which suppress T cell function and promote anemia (255). EDMC in the TME together with anemia, are described as biomarkers of poor response to ICB therapy in several cancer types (255). Recently, immunotherapy has burst in the oncology arena as a promising approach to exploit the immune system to combat cancer, focusing mainly on the cytotoxic capacity of CD8+T cells. On the one hand, the ICB treatment is prone to induce an active immune response, reinvigorating in vivo exhausted endogenous T cells. On the other hand, other therapies rely on a passive strategy by transferring autologous T cells that were genetically engineered to express chimeric antigen receptors. MDSCs have been recognized as a barrier that limits ICB effectiveness (256, 257). MDSCs manage to suppress CD8+ and CD4+ T cell proliferation and function by depleting nutrients, such as L-arginin, cysteine and tryptophan, by inducing the production of nitric oxide (NO) by the inducible nitrous oxide synthase (iNOS), the production of the immunosuppressive cytokines IL-10 and TGF-β, and by expressing the inhibitor molecule PD-L1 (258–262). In addition, in a mouse melanoma model it was demonstrated that MDSCs impair the infiltration of activated CD8+ T cells (240). In a meta-analysis, elevated numbers of MDSCs in the circulation were found to be an independent indicator of poor outcome in patients with solid tumors (263). In melanoma patients subjected to ICB treatment, a basal higher level of PMN-MDSC was proved to be present in progressive disease vs. patients that evidence clinical benefit. The patients that responded to therapy exhibited an increase in circulating MDSCs after ICB treatment, suggesting that MDSCs count could be considered as a prognostic biomarker of ICB effectiveness (264).
The other myeloid cells in the TME scenario are macrophages. Macrophages are present in every tissue to maintain their homeostasis. The tissue-resident macrophages originate from monocytes from the bone marrow but are also derived from precursors that seed the tissues upon different waves of embryonic hematopoiesis and are locally proliferating (265). In particular, a glioblastoma can have 30% of its tumor mass composed of bone marrow–derived macrophages and its abundance is related to tumor grade (266). In a scenario of acute or chronic inflammation (this latter case, includes cancer) bone marrow-derived monocytes migrate to the inflamed tissue attracted by CCL2 and CSF 1, and differentiate to macrophages (267). The macrophages present in tumors are defined as tumor-associated macrophages (TAMs) and can also derive from M-MDSCs as precursor cells (268). Indeed, the cancer-related inflammation is promoted by TNFα secreted by TAMs that induces constitutive activation of NF-κB in tumor cells, which promotes their survival and invasion (269). The ability of the TME to polarize the recruited monocytes to TAMs instead of to dendritic cells has been recently uncovered and is due to the production of retinoic acid by tumor cells (270). Macrophages are plastic and signals from the TME, derived from tumor cells, lymphocytes and stroma cells, condition their fate to acquire either immunosuppressive or immunostimulant function. The “classically activated”, M1-like or antitumoral macrophages are originated by interferon (IFN)-γ or microbial stimuli and produce IL-12, which polarize CD4+T cells to a type I response. In addition, they have cytotoxic activity against microorganisms and tumor cells. On the other hand, the “alternatively activated” macrophages, M2-like or protumoral macrophages differentiate after IL-4/IL-13/IL-10/TGFβ stimuli, have high IL-10 production, downregulate M1-like macrophages´ function and are associated with repair of wound tissues (271, 272). Macrophage polarization conditioned by TME has been recently reviewed (273). It is generally accepted that TAMs have an M2-like phenotype that can promote cancer initiation, progression and favor metastasis while suppressing the antitumor immune response (274–276). In particular, TAMs inhibit CD8+ T cell activation and function in a similar way to that was described for MDSCs: TAMs promote L-arginine depletion in the TME, secrete immunosuppressive cytokines, including IL-10 and TGF-β, and express the inhibitory molecules PD-L1, PD-L2, CD80 and CD86. This profile hijacks the anti-PD-1/PD-L1 therapies, as has thoroughly been reviewed elsewhere (277). TAMs also act as a trap for CD8+T cells. In human lung squamous-cell carcinoma it has been demonstrated that the exclusion of CD8+T cells from the tumor nest was not only due to the density of the ECM but also induced by a long-lasting interaction between TAMs and CD8 T cells which impairs T cell movement (278). In addition, TAMs secrete C-chemokine ligand (CCL)-2, CCL-3, and other members of the CCL family that promote the recruitment of regulatory T cells (Treg) and type 2 conventional dendritic cell (cDC2) HLA-DRlow which, upon their interaction in hypoxic conditions, results in a loss of antigen-presenting capacity of cDC2 (279). TAMs also secrete angiogenic cytokines such as VEGF-A, TGF-α, TGF-β, EGF, and PDGF that allow aberrant tumor-associated vasculature (274), which has been described to play a key role in immune exclusion, as reviewed above. Metastasis is a multistep process that allows tumor cells to survive and proliferate in distant organs. In this respect, TAMs secrete matrix metalloproteinases and urokinase that remodel the TME allowing local invasion of cancer cells that form adherens junctions to the ECM. Moreover, the M2-like macrophages are an important component of the tumor microenviromment of metastasis (TMEM) doorways. This structure is composed of perivascular M2-like macrophages, endothelial cells and tumor cells expressing Mammalian enabled actin-regulatory protein (MENA). The tumor cell interaction with M2-like macrophages flow through the tumor cells via the TMEM doorway releasing them to the bloodstream to accomplish another step of the metastatic cascade (280–282). These niches, where the dissemination trajectories of tumor cells occur, are immune desert and enable the successful migration of tumor cells to disseminate through the host (283). Another layer of complexity of TAM impact on the TME relies on their ability to remodel tissue by interacting with cancer-associated fibroblasts (CAFs) promoting collagen production and fibrosis, fostering the exclusion of T cells and causing failure of ICB (284). As CAFs secrete high levels of IL-6, it induces Stat3 activation and macrophage polarization to M2-like increasing the immunosuppressive tumor milieu (285).
3.2.2 CAFs and the ECM
The major producer of ECM in solid tumors are the CAFs (286), which are mainly co-opted resident (287) or recruited fibroblast-like cells that undergo reprogramming by the tumor (288). In several tumors, an increased proportion of CAFs on the TME has been associated with poor prognosis (289), but lately a wider and more specific classification of CAFs subpopulations highlights the idea that not all CAFs are biomarkers of poor outcome (290, 291), and that different CAF subtypes may have different contributions to cancer progression. This is due to the heterogeneity of CAFs, as they can be immunosuppressive or immunostimulatory for T cells. As PDAC is characterized to be surrounded by abundant desmoplasia, it has been crucial to study CAFs subpopulations. Öhlund et al. revealed that pancreatic stellate cells are a main source of CAFs, since they can either differentiate to inflammatory CAFs (iCAFs) or myofibroblastic CAF (myoCAF). These subpopulations were defined based on their protein expression: iCAFs secrete IL-6 and pro-inflammatory factors, while myCAFs have elevated expression of α-smooth muscle actin (αSMA). By RNAseq, authors identified that the proinflammatory genes as Il6, Il11, and Lif, and chemokines, such as Cxcl1 and Cxcl2 were upregulated in the iCAFs, while a TGFβ-response genes were characteristic of myCAFs. In addition, a spatial separation between iCAFs and myCAFs was reported: myCAFs are in contact with tumor cells, while iCAFs are situated far from the neoplastic nest (290). These two subpopulations were confirmed by single-cell RNA sequencing in human PDAC (292). Nevertheless, another subtype of CAFs emerged from scRNA in human and mouse PDAC, the so-called antigen-presenting CAFs (apCAFs). These cells have high levels of major histocompatibility complex (MHC) class II family members and, in a mouse model, showed capacity to activate CD4+T cells (291).
CAFs have shown to have different strategies to restrain T cells to the stroma and prevent them from penetrating the tumor core. One of these strategies is their contribution to the production of a stiff and dense ECM, since it has been demonstrated by live imaging of lung tumor tissue slices that T cells move actively in regions of loose fibronectin and collagen fibers, while dense ECM surrounding the tumor core presented a clear impairment to T cell mobility (154). Interestingly, when collagenase was used to reduce the rigidity of the ECM around tumor islets, an enhanced movement of T cells was observed from the stroma and into the tumor core, in close contact with tumor cells. Another strategy by which CAFs can control T cell infiltration is by means of the production of the cytokine CXCL12. In a preclinical mouse model of PDAC, it was demonstrated that CAFs that express the fibroblast activation protein (FAP) within the ECM produce and secrete CXCL12, which in turn binds to the PDAC tumor cells, coating them (241). This phenomenon has also been reported for colorectal and ovarian cancer (293, 294). Feig and collaborators found that T cells were absent from tumor regions containing cancer cells, which were coated with CXCL12. Moreover, they proved that the main source of CXCL12 production in the TME were the FAP+ CAFs. The administration of AMD3100, an inhibitor of the CXCL12 receptor (CXCR4), highly increased T cell infiltration among cancer cells. What is more, authors claim that AMD3100 synergized with anti-PD-L1 therapy to diminish the proportion of cancer cells within the lesion and to arrest tumor growth (241). CD8+T cells functionality is hijacked by CAFs at different levels. TCR signaling can be suppress by secretion of TGFb produced by CAFs (295). CAFs also express PD-L1 and PD-L1 that decrease CD8+ T cell proliferation, function and probably contributes to T cell exhaustion (296, 297).
4 Clinical impact
Given the importance of the TME in treatment response, in particular immunotherapies, in this section we will address the current state-of-the-art of targeting the above-mentioned physical barriers as novel treatments in combination with standard of care therapies and their clinical implications.
4.1 Targeting MDSCs
MDSCs are a subpopulation of immature myeloid cells that expand massively during tumor progression and invasion and have immunosuppressive effects and promote tumor immune escape. There are several reports of MDSCs involvement in chemo-, radio- and immunotherapy (257, 298–303), which poses this cell population as an attractive therapeutic target. Numerous strategies are being tested in clinical trials: promoting maturation, differentiation or depletion of MDSCs, prevention of expansion and recruitment, inhibition of their function or their metabolism (299, 304–306), among others.
4.1.1 Maturation, differentiation or depletion
Vitamin D3 showed to stimulate MDSCs maturation and improved the antitumor immune response in patients with head and neck squamous cell carcinoma extending their progression-free survival (307, 308). All trans retinoic acid in combination with immunotherapy was proven to induce MDSCs differentiation into macrophages, granulocytes, and dendritic cells (309, 310) which not only abolishes the immunosuppressive effects but also can reduce the MDSCs number from the blood (311, 312). In respect to MDSCs depletion, chemotherapeutic agents can diminish their number, but it is not specific for this population (313). Therefore, a monoclonal antibody against CD33 conjugated with a toxin was tested in acute myeloid leukemia for depletion of MDSCs in a phase II clinical trial (314).
4.1.2 Prevention of expansion and recruitment
Stat3 activation is crucial for MDSCs expansion and function and its inhibition with an antisense oligonucleotide called AZD9150 reduced MDSCs circulation in peripheral blood of patients with diffuse large B-cell lymphoma (315), and is currently being tested in combination with ICB in solid tumors (NCT02499328). MDSCs are regulated by the same colony stimulating factors (CSF) that modulate normal myelopoiesis (316). It has been demonstrated in preclinical models for different cancers that blockade of GM-CSF/G-CSF can inhibit the accumulation of MDSCs and exert an antitumor immune response while restraining polarization of the macrophages to the M2-like phenotype (317–320). Moreover, combination of GM-CSF/G-CSF blocking agents with ICB and chemotherapy have given promising results (321) which led to clinical trials that test these combinations (322, 323) in prostate (NCT02961257, NCT01499043), melanoma (NCT02071940, NCT02975700), glioblastoma (NCT01499043) and breast (NCT02265536) among others. Naturally, targeting the complex cytokine and chemokine network or their receptors which modulate MDSCs is expected to have great clinical implications. For example, propagermanium (a CCL2 inhibitor) was shown to be safely tolerated by patients with primary breast cancer and an antimetastatic effect was observed (324). On the other hand, a monoclonal antibody against CCL2 showed no antitumor effect as a single agent in patients with metastatic castration-resistant prostate cancer (325). A small molecule inhibitor of CCR2 combined with FOLFIRINOX, the standard of care for patients with PDAC, had promising results, decreasing TAMs and migration of MDSCs (326, 327). In spite of these results, most of the approaches targeting the CCL2/CCR2 axis have disappointing results, and this could be due to the impediment for long term blocking (328). Targeting CXCR1/2 in combination with chemotherapies showed a potent antitumor response, and enhances the efficacy of chemotherapy in an animal (329) model of gastric cancer, and furthermore have shown to improve ICBs therapy in head and neck tumors and (330)oral and lung carcinoma (331, 332),. Numerous CXCR1/2 inhibitors have been tested in clinical trials for cancer patients and are extensively described elsewhere (330). Another key player in MDSCs recruitment and mobilization is VEGF, which is also produced by them to promote metastasis and angiogenesis (333). VEGF expression in ovarian cancer correlated with a poorer prognosis and MDSCs infiltration (334). Different studies have approached the use of anti-VEGF/VEGFR therapies in patients with NSCLC (335), glioblastoma (336) and colorectal (337) cancer and presented good outcomes. Sunitinib, a tyrosine kinase inhibitor that targets PDGFRs and VEGFRs, has shown to inhibit MDSCs immunosuppressive effect in patients with various cancer types (338). Another study in renal cancer patients showed that sunitinib decreases circulating MDSCs (339) by inhibiting VEGF and STAT3 activation (338). To achieve effectiveness, combination treatments that target several of these factors are being tested in preclinical models, for example in a preclinical breast cancer model all trans retinoic acid administration together with VEGFR2 inhibitors and chemotherapy have been tested and not only they diminished MDSCs infiltration, but also exhibited hindered tumor growth (340).
4.1.3 Inhibition of function
These treatments aim to inhibit pathways related with MDSCs immunosuppressive mechanisms. Inhibition of STAT3 through different strategies in preclinical models and clinical trials (316, 341, 342) have been tested in solid tumors with limited effectiveness and unwarranted toxicities (343). The COX2/PGE2 axis is another interesting target since it has been shown to be involved in tumor promotion and invasion in ovarian cancer (344), tumor evasion in colorectal adenoma (345), among others (346). Inhibition of said signaling pathway showed to improve immune response and decreased arginase expression (347, 348) and exhibited great inhibitory effect over the MDSCs population in several cancers (349–351). Combination with immunotherapy has been already reviewed elsewhere (352). Inhibitors of the histone deacetylase have also shown to reduce COX2, arginase and iNOS levels, resulting in an inhibition of the immunosuppressive function of MDSCs. These drugs have been tested in preclinical models (352, 353) and in clinical trials resulting in sustained survival, retarded tumor growth and a positive immunostimulatory effect over CD8+ T cells (354, 355). Phosphodiesterase inhibitors can also alter MDSCs function downregulating iNOS and arginase stimulating antitumor immunity in mice (356) and in patients with HNSCC (357) and melanoma (358).
4.2 Targeting TAMs
TAMs account for 30-50% of the TME (359) and promote tumor progression (360, 361) and an immunosuppressive TME depending on their polarization (362, 363). These characteristics make them an appealing target to modulate the TME and obtain tumor regression or increase efficacy of current therapies. Depletion of TAMs, stimulation of their phagocytosis, reprogramming and inhibition of their recruitment are some of the proposed targeted therapies.
Paralleling CAR-T cells, recently, chimeric antigen receptor macrophages (CAR-M) have been developed. CAR-M have shown to polarize macrophages to the M1 antitumoral subtype and to enhance the phagocytosis of cancer cells (364–367), and are also capable of transforming the TME into a pro-inflammatory one, which promotes antitumoral effects in preclinical models (366). CD47 (don’t eat me signal) is a crucial molecule expressed by tumors that impedes macrophage recognition of the tumoral cells, which results in a decreased phagocytosis and in tumor evasion (368). Several works have studied potential therapies directed against CD47 and have shown favorable results (369–372). Regarding TAMs depletion, bisphosphonates have been shown to be effective and they also inhibit M2-like TAMs proliferation and migration, but they do not achieve a durable response (373–377).
Another interesting strategy is TAMs reprogramming. Macrophages can shift between M1-like antitumoral or M2-like pro-tumoral phenotypes, depending on the cytokines present in the microenvironment (378). As it was described for MDSCs, recruitment of M1-like macrophages to the tumor core is an interesting therapeutic idea and given both cells belong to the myeloid population, some of the therapies used for MDSCs also affect TAMs. In line with this, TLR agonists are currently being tested in the clinical setting to repolarize M2-like TAMs to the M1-like subtype (379–384). Moreover, CSF-1/CSFR-1 blockade inhibits TAMs recruitment to the tumor bed and has antitumoral effects in mice and in patients (385–388). The CCL2/CCR2 axis is also important for TAMs recruitment and therapies that target these proteins have shown to be effective in this sense and they also reduce tumor growth (326, 389, 390). Inhibitors of CSF-1/CSFR-1 not only affect TAMs recruitment but also have a reprogramming effect (322, 385, 391, 392). Given the rise of immunotherapies, several strategies that target TAMs are now being tested in combination with ICBs and have shown to improve their effectiveness (375, 393–399).
4.3 Targeting CAFs
Lately, CAFs have emerged as an important factor in the regulation of the TME and immunotherapy response. Fibroblast activating protein (FAP) is one of the main targets for depleting CAFs (400), and inhibition of said protein showed good outcomes in mice models (401, 402). However, a monoclonal antibody targeting FAP did not show the expected promising results in a phase II clinical trial of patients with metastatic colorectal cancer (403). Bispecific antibodies of FAP and IL-2 receptor (RO6874281) or 1BBL (RG7826) are being tested in combination with ICBs on clinical trials and have shown to be safe, and preclinical models encourage the advancement due to promising results such as tumor regression, accumulation of CD8+ T and NK cells. Other approaches aim to inhibit pathways related to CAFs activation, for example, focal adhesion kinase (FAK) inhibitors combined with anti-PD-1 had a good effect in preclinical and clinical models (159, 404) (NCT03727880, NCT02758587, NCT02546531). In this sense, inhibition of the platelet derived growth factor (PDGF) (405) and FGFR (406) pathways have shown positive outcomes for the patients but the adverse effects and resistant events remain to be elucidated. On the other hand, depletion of α-SMA+ myCAF induces reduced survival of animals with PDAC and scarce myCAF in human tumor samples was correlated with poor overall survival (407). Therefore, a deep insight of CAFs subtype and their relationship with tumor types should be unraveled before their approval in patients. Recent review highlights the challenges that targeting CAFs has to overcome to reach clinical practice (408, 409).
Some downstream effectors of CAFs have been studied as potential therapeutic targets such as CXCR4/CXCL-12 (410) (NCT02907099, NCT02826486) or TGF-β (36, 97, 411–413). Interestingly, some of them are also being proposed as targets of other cell populations as mentioned above, meaning these therapies affect multiple cell types. Given the recent description and implications of CAFs subsets in the TME remodeling and immunotherapy response, future studies should disclose the role of said subsets and new therapeutic targets will arise.
4.4 Targeting collagen
Collagen is a central element of the ECM, and therefore for the TME and immune (414) exclusion, has implications in tumor progression, metastasis, and conditions response to treatment. However, collagen has also shown to exhibit antitumor activity (415). Collagen-binding domains (CBD) are present in collagen and mediate interactions with other proteins (416). Given this, CBD-engineered biomolecules could deliver drugs or monoclonal antibodies to the tumor site and their effect is limited to the ECM, augmenting the therapeutic outcomes and decreasing off-target effects and toxicity (417). Among these examples, CBD fused with the EGFR binding portion of cetuximab showed antitumor activity in vitro and longer retention times and enrichment in tumor human epidermoid squamous carcinoma xenografts (418, 419). In the same manner, CBD carrying IL-2 or IL-12 in combination with ICB treatment have been tested, exhibiting promising results in breast cancer (420) and melanoma (421). In another approach, collagen-binding albumin, a protein that accumulates in the TME and is used as an energy source for cancer growth, was conjugated to chemotherapy in combination with ICB in a colon carcinoma preclinical model (422). The treatment not only caused accumulation of the drug on the TME but also generated complete tumor inhibition in breast and colorectal cancer models (423). Recently, a fusion protein of CBD and the Fc of SIRPα (signal regulatory protein α) (423), which targets the SIRPα-CD47 axis, showed accumulation in the tumor tissue, more antitumor activity and an increase infiltration of M1-like macrophages than SIRPα Fc alone, in a xenograft model of NSCLC (424). Collagenase treatment loosens the ECM, allows an improved drug penetration and delivery and provides an antitumor effect (425–427). Alternatives to achieve collagen degradation include activators of matrix metalloproteinases (428, 429), engineered bacteria (430) and armed oncolytic virus expressing ECM components (431–434).
Another strategy is not to deplete collagen, but to modulate its production by interfering with collagen crosslinking. The LOX family of enzymes is critical in collagen crosslinking and therefore ECM stiffness. In fact, it has been shown that aberrant LOX expression is implicated in tumor progression (435) and therapy resistance (436). Therapies targeting LOX have been developed and tested in preclinical models in various cancers showing good results (437, 438). Inhibition of collagen synthesis by antifibrotic drugs loosens ECM architecture allowing greater immune cell infiltration and drug delivery in PDAC (439, 440), NSCLC (441), melanoma (442), among others. There are several clinical trials testing antifibrotic drugs that could potentially be used in cancer treatment in the future (443). Clinical trials testing therapies targeted collagen have been reviewed elsewhere (138).
Recently, it was demonstrated that an antibody against the collagen receptor discoidin (DDR1) disrupts collagen alignment and inhibits tumor growth in TNBC (152). Finally, given that CAFs are the main producers of collagen, therapies targeting this cell population can achieve the above-mentioned collagen-related effects.
4.5 Targeting HA
As mentioned above, HA can act as a physical barrier causing both the exclusion of immune cells and of drugs (such as chemotherapeutics and monoclonal antibodies), thus leading to failure of treatment. Finding efficient tools to remodel the HA matrix within tumors is complex and constitutes a matter of intense research nowadays. One strategy proposes to target the hyaluronidase activity with O-sulfated HA, and showed a pronounced effect over invasion in PDAC (444) and prostate cancer (445).
The application of hyaluronidases to degrade HA has also been investigated as a possible anticancer drug alone or in combination with other drugs. Indeed, intravenous hyaluronidase treatment of mice implanted with human breast cancer cells significantly reduced tumor growth (446). Intravenous trastuzumab constitutes the main standard of care for HER2-positive BC since 1998. As an alternative, a subcutaneous trastuzumab formulation was developed containing the recombinant human hyaluronidase, which degrades interstitial HA, enables the administration of large drug volumes subcutaneously, and favors trastuzumab delivery to the circulation (447, 448). After a phase III trial confirmed the comparable efficacy and safety of subcutaneous and intravenous trastuzumab (449), subcutaneous trastuzumab was approved for HER2-positive early BC. This trastuzumab formulation presents several advantages compared with the intravenous trastuzumab including shorter administration times and increased convenience and preference for patients (450). Preclinical studies in pancreatic cancer models demonstrated that the hyaluronidase PEGPH20, besides increasing CD8+ T cell infiltration as stated before, increases chemotherapy effectiveness (451). Based on this, PEGPH20 treatment was assessed in clinical trials for PDAC, where it showed an increase in perfusion and chemotherapy delivery (452). Although early phase studies showed promising results, further clinical studies demonstrated that addition of PEGPH20 to chemotherapy either increased toxicity or failed to achieve an improvement in the progression-free survival or overall survival, so the studies were discontinued for PDAC (452–454). It is worth noting that although HA may be a physical barrier hampering drug delivery, it may also have signaling properties. Therefore, hyaluronidases may also generate HA fragments, which could have pro-tumorigenic (pro-angiogenic) actions and may explain the failure of PEGPH20 treatment in PDAC. Blocking peptides for LMW HA is another interesting strategy to prevent HA to bind its receptors and downregulate signaling, but to date there is not extensive work approaching this task. However, it has been demonstrated that HA-binding peptides can inhibit survival and invasion of breast and prostate cancer cell lines in vitro and in vivo (455–457).
Another therapeutic strategy proposes to target HA synthesis by treatment with 4-metylumbilliferone (4-MU), a natural compound that inhibits the expression of HA synthases (152, 458) and therefore downregulates HA signaling. Preclinical studies have shown that 4-MU has antitumoral effects affecting progression, migration, and invasion in several cancers, such as colorectal, pancreatic, prostate, ovarian, breast, melanoma, hepatocellular cancers (459). 4-MU also has shown to reduce metastasis in breast (460), ovarian (461) and skin (462) cancers. Since 4-MU is already approved in Europe for biliary spasms treatment, and given that it decreases HA levels in human participants (463), 4-MU constitutes a promising anticancer agent. Recently, another inhibitor of HA synthesis, the thymidine analog 5′-Deoxy-5′-(1,3-Diphenyl-2-Imidazolidinyl)-Thymidine (DDIT), demonstrated more potent anti-tumorigenic properties than 4-MU in breast cancer (464), further supporting targeting HA synthesis as an antitumoral therapeutic strategy. Further studies investigating the impact of HA-targeting strategies on HA levels, HA molecular weight, and the activation of HA receptors will help to improve current and develop new therapeutic tools aiming HA to tailor HA metabolism in cancer.
4.6 Targeting MUC4
MUC4 expression was reported to be higher in various cancer types such as breast, pancreatic, lung, ovarian, bladder and cervical cancer (465). Our group reported that TNFα-induced MUC4 is involved in HER2-targeted therapy resistance in vivo and in vitro, and that it is an independent predictor of trastuzumab response in HER2-positive breast cancer patients (115). We have disclosed that a dominant-negative protein (INB03) that selectively blocks sTNFα downregulates MUC4 and its administration with trastuzumab turns the immunosuppressive TME into a immunostimulatory one through polarization to M1-like macrophages and favoring the crosstalk between macrophages and NK cells in preclinical models (466). Furthermore, MUC4-positive breast tumors are associated with an immunologically cold phenotype, suggesting that INB03 could be used in combination with trastuzumab and ICBs in these tumors. MUC4 has also been proved to be a biomarker for pancreatic cancer and drives cell tumor proliferation and invasion in PDAC (467, 468). In ovarian cancer MUC4 downregulation potentiates the antitumoral effect of auranofin (469). Moreover, MUC4 expression has been linked to the metastatic potential of the cancer cells (470). Given the aforementioned information and the lack of therapies targeting MUC4, we proposed that TNFα-blocking agents or MUC4-targeted therapies in combination with immunotherapy and the standard of care for each cancer type should be further investigated in the clinical setting.
5 Closing remarks
It is clear that the phenomenon of immune exclusion has strong clinical relevance for different tumor types, but has not yet been fully elucidated. We envision that understanding the immune-excluding mechanisms of action of the different components of the TME will open up new avenues of therapeutic approaches. However, a full comprehension of the TME of each tumor type, and even in each patient should be taken into consideration in the clinical setting. In the near future, diagnosis would be performed by promising non-invasive techniques that will help to characterize the TME and select a proper treatment. Moreover, the plasticity of myeloid cells, the depletion of suppressive cells and the reduction of the TME stiffness are different strategies that are now being explored to reshape the immunosuppressive tumor milieu into an immunocompetent one that will render the success of the present IBC.
Author contributions
SB and RS contributed to the conception of the manuscript. SB, MFM, FLM, RIRC and RS wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
Agencia Nacional de Promoción Científica y Tecnológica PICT 2017-1517, PICT 2018-2086, PICT aplicación intensiva 2021-023 and PICT 2020-0026 awarded to RS and PICT 2017-0419, PICT 2020-2315 awarded to RICR. Fundacion Williams and Fundacion Rene Baron for institutional support.
Conflict of interest
RS is a consultant for and a research grant from INmune Bio.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi: 10.1038/nm.3394
2. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21(3):309–22. doi: 10.1016/j.ccr.2012.02.022
3. Aguilera TA, Giaccia AJ. Molecular pathways: oncologic pathways and their role in T-cell exclusion and immune evasion-a new role for the AXL receptor tyrosine kinase. Clin Cancer Res (2017) 23(12):2928–33. doi: 10.1158/1078-0432.CCR-17-0189
4. Hatfield SM, Kjaergaard J, Lukashev D, Belikoff B, Schreiber TH, Sethumadhavan S, et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J Mol Med (2014) 92(12):1283–92. doi: 10.1007/s00109-014-1189-3
5. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med (2015) 7(277):277ra30. doi: 10.1126/scitranslmed.aaa1260
6. Hatfield SM, Sitkovsky M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1α driven immunosuppression and improve immunotherapies of cancer. Curr Opin Pharmacol (2016) 29:90–6. doi: 10.1016/j.coph.2016.06.009
7. Daniel SK, Sullivan KM, Labadie KP, Pillarisetty VG. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin Transl Med (2019) 8(1):10. doi: 10.1186/s40169-019-0226-9
8. Hatfield S, Veszeleiova K, Steingold J, Sethuraman J, Sitkovsky M. Mechanistic justifications of systemic therapeutic oxygenation of tumors to weaken the hypoxia inducible factor 1α-mediated immunosuppression. Adv Exp Med Biol (2019) 1136:113–21. doi: 10.1007/978-3-030-12734-3_8
9. Li YL, Zhao H, Ren XB. Relationship of VEGF/VEGFR with immune and cancer cells: staggering or forward? Cancer Biol Med (2016) 13(2):206–14. doi: 10.20892/j.issn.2095-3941.2015.0070
10. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol (2018) 9:978. doi: 10.3389/fimmu.2018.00978
11. Zhang Y, Guan XY, Jiang P. Cytokine and chemokine signals of T-cell exclusion in tumors. Front Immunol (2020) 11:594609. doi: 10.3389/fimmu.2020.594609
12. Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The tumor microenvironment: a milieu hindering and obstructing antitumor immune responses. Front Immunol (2020) 11:940. doi: 10.3389/fimmu.2020.00940
13. Bedognetti D, Ceccarelli M, Galluzzi L, Lu R, Palucka K, Samayoa J, et al. Toward a comprehensive view of cancer immune responsiveness: a synopsis from the SITC workshop. J Immunother Cancer (2019) 7(1):131. doi: 10.1186/s40425-019-0602-4
14. Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature (2017) 541(7637):321–30. doi: 10.1038/nature21349
15. He J, Fu F, Wang W, Xi G, Guo W, Zheng L, et al. Prognostic value of tumour-infiltrating lymphocytes based on the evaluation of frequency in patients with oestrogen receptor–positive breast cancer. Eur J Cancer (2021) 1:154:217–26. doi: 10.1016/j.ejca.2021.06.011
16. Orhan A, Vogelsang RP, Andersen MB, Madsen MT, Hölmich ER, Raskov H, et al. The prognostic value of tumour-infiltrating lymphocytes (TILs) in pancreatic cancer: a systematic review and meta-analysis. Ann Oncol (2019) 30:xi2–3. doi: 10.1093/annonc/mdz447.006
17. Moore MR, Friesner ID, Rizk EM, Fullerton BT, Mondal M, Trager MH, et al. Automated digital TIL analysis (ADTA) adds prognostic value to standard assessment of depth and ulceration in primary melanoma. Sci Rep (2021) 11(1):2809. doi: 10.1038/s41598-021-82305-1
18. Ropponen KM, Eskelinen MJ, Lipponen PK, Alhava E, Kosma VM. Prognostic value of tumour-infiltrating lymphocytes (TILs) in colorectal cancer [Internet]. Vol 182 J Pathol (1997) 182(3)318–24. doi: 10.1002/(sici)1096-9896(199707)182:3<318::aid-path862>3.0.co;2-6
19. Gao G, Wang Z, Qu X, Zhang Z. Prognostic value of tumor-infiltrating lymphocytes in patients with triple-negative breast cancer: a systematic review and meta-analysis. BMC Cancer (2020) 20(1):179. doi: 10.1186/s12885-020-6668-z
20. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med (2003) 348(3):203–13. doi: 10.1056/NEJMoa020177
21. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA (2005) 102(51):18538–43. doi: 10.1073/pnas.0509182102
22. Indini A, Massi D, Pirro M, Roila F, Grossi F, Sahebkar A, et al. Targeting inflamed and non-inflamed melanomas: biological background and clinical challenges. Semin Cancer Biol (2022) 86(Pt 2):477–90. doi: 10.1016/j.semcancer.2022.06.005
23. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371(23):2189–99. doi: 10.1056/NEJMoa1406498
24. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet (2016) 387(10030):1837–46. doi: 10.1016/S0140-6736(16)00587-0
25. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet (2016) 387(10031):1909–20. doi: 10.1016/S0140-6736(16)00561-4
26. Chen X, Chen H, Lin R, Li Y, Guo Y, Chen Q, et al. Correlation between PD-L1 expression of the tumour cells and lymphocytes infiltration in the invasive front of urothelial carcinoma. J Clin Pathol (2022). doi: 10.1136/jcp-2021-207795
27. Huang Y, Chen Y, Zhou S, Chen L, Wang J, Pei Y, et al. Dual-mechanism based CTLs infiltration enhancement initiated by nano-sapper potentiates immunotherapy against immune-excluded tumors. Nat Commun (2020) 11(1):622. doi: 10.1038/s41467-020-14425-7
28. Kim JE, Kim H, Kim B, Chung HG, Chung HH, Kim KM, et al. Comprehensive immunoprofile analysis of prognostic markers in pancreaticobiliary tract cancers. Cancer Med (2023) 12(7):7748–61. doi: 10.1002/cam4.5530
29. Schalck A, Sakellariou-Thompson D, Forget MA, Sei E, Hughes TG, Reuben A, et al. Single-cell sequencing reveals trajectory of tumor-infiltrating lymphocyte states in pancreatic cancer. Cancer Discovery (2022) 12(10):2330–49. doi: 10.1158/2159-8290.CD-21-1248
30. Ren X, Song Y, Pang J, Chen L, Zhou L, Liang Z, et al. Prognostic value of various immune cells and immunoscore in triple-negative breast cancer. Front Immunol (2023) 14:1137561. doi: 10.3389/fimmu.2023.1137561
31. Bruni S, Mauro FL, Proietti CJ, Cordo-Russo RI, Rivas MA, Inurrigarro G, et al. Blocking soluble TNFα sensitizes HER2-positive breast cancer to trastuzumab through MUC4 downregulation and subverts immunosuppression. J Immunother Cancer (2023) 11(3):e005325. doi: 10.1136/jitc-2022-005325
32. Carter JM, Chumsri S, Hinerfeld DA, Ma Y, Wang X, Zahrieh D, et al. Distinct spatial immune microlandscapes are independently associated with outcomes in triple-negative breast cancer. Nat Commun (2023) 14(1):2215. doi: 10.1038/s41467-023-37806-0
33. Laenkholm AV, Callagy G, Balancin M, Bartlett JMS, Sotiriou C, Marchio C, et al. Incorporation of TILs in daily breast cancer care: how much evidence can we bear? Virchows Arch (2022) 480(1):147–62. doi: 10.1007/s00428-022-03276-w
34. Ghisoni E, Imbimbo M, Zimmermann S, Valabrega G. Ovarian cancer immunotherapy: turning up the heat. Int J Mol Sci (2019) 20(12):2927. doi: 10.3390/ijms20122927
35. Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol (2016) 27(8):1492–504. doi: 10.1093/annonc/mdw217
36. Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res (2016) 22(8):1865–74. doi: 10.1158/1078-0432.CCR-15-1507
37. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515(7528):563–7. doi: 10.1038/nature14011
38. Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol (2013) 25(2):268–76. doi: 10.1016/j.coi.2013.02.009
39. Wu W, Klockow JL, Zhang M, Lafortune F, Chang E, Jin L, et al. Glioblastoma multiforme (GBM): an overview of current therapies and mechanisms of resistance. Pharmacol Res (2021) 171:105780. doi: 10.1016/j.phrs.2021.105780
40. Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother (2010) 33(8):828–33. doi: 10.1097/CJI.0b013e3181eec14c
41. Brahmer JR, Tykodi SS, Chow LQM, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
42. Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics (2021) 11(11):5365–86. doi: 10.7150/thno.58390
43. Kather JN, Suarez-Carmona M, Charoentong P, Weis CA, Hirsch D, Bankhead P, et al. Topography of cancer-associated immune cells in human solid tumors. Elife (2018) 4:7. doi: 10.7554/eLife.36967
44. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi: 10.1126/science.1129139
45. Pai SI, Cesano A, Marincola FM. The paradox of cancer immune exclusion: immune oncology next frontier. Cancer Treat Res (2020) 180:173–95. doi: 10.1007/978-3-030-38862-1_6
46. Mahmoud SMA, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AHS, et al. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol (2011) 29(15):1949–55. doi: 10.1200/JCO.2010.30.5037
47. Liu S, Lachapelle J, Leung S, Gao D, Foulkes WD, Nielsen TO. CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res (2012) 14(2):R48. doi: 10.1186/bcr3148
48. Schmidt M, Weyer-Elberich V, Hengstler JG, Heimes AS, Almstedt K, Gerhold-Ay A, et al. Prognostic impact of CD4-positive T cell subsets in early breast cancer: a study based on the FinHer trial patient population. Breast Cancer Res (2018) 20(1):15. doi: 10.1186/s13058-018-0942-x
49. Gruosso T, Gigoux M, Manem VSK, Bertos N, Zuo D, Perlitch I, et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J Clin Invest (2019) 129(4):1785–800. doi: 10.1172/JCI96313
50. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science (2015) 350(6257):207–11. doi: 10.1126/science.aad0095
51. Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol (2017) 14(11):655–68. doi: 10.1038/nrclinonc.2017.88
52. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med (2016) 375(9):819–29. doi: 10.1056/NEJMoa1604958
53. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest (2017) 127(8):2930–40. doi: 10.1172/JCI91190
54. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMoa1500596
55. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science (2017) 355(6322):eaaf8399. doi: 10.1126/science.aaf8399
56. Kroemer G, Zitvogel L. The breakthrough of the microbiota. Nat Rev Immunol (2018) 18(2):87–8. doi: 10.1038/nri.2018.4
57. Joyce JA, Fearon DT. T Cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348(6230):74–80. doi: 10.1126/science.aaa6204
58. Spranger S, Gajewski TF. Tumor-intrinsic oncogene pathways mediating immune avoidance. Oncoimmunology (2016) 5(3):e1086862. doi: 10.1080/2162402X.2015.1086862
59. Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med (2018) 24(10):1550–8. doi: 10.1038/s41591-018-0136-1
60. Gillies RJ, Kinahan PE, Hricak H. Radiomics: images are more than pictures, they are data. Radiology (2016) 278(2):563–77. doi: 10.1148/radiol.2015151169
61. Doi K. Computer-aided diagnosis in medical imaging: historical review, current status and future potential. Comput Med Imaging Graph (2007) 31(4-5):198–211. doi: 10.1016/j.compmedimag.2007.02.002
62. Lambin P, Leijenaar RTH, Deist TM, Peerlings J, de Jong EEC, van Timmeren J, et al. Radiomics: the bridge between medical imaging and personalized medicine. Nat Rev Clin Oncol (2017) 14(12):749–62. doi: 10.1038/nrclinonc.2017.141
63. Jazieh K, Khorrami M, Saad A, Gad M, Gupta A, Patil P, et al. Novel imaging biomarkers predict outcomes in stage III unresectable non-small cell lung cancer treated with chemoradiation and durvalumab. J Immunother Cancer (2022) 10(3):e003778. doi: 10.1136/jitc-2021-003778
64. Tiwari A, Oravecz T, Dillon LA, Italiano A, Audoly L, Fridman WH, et al. Towards a consensus definition of immune exclusion in cancer. Front Immunol (2023) 14:1084887. doi: 10.3389/fimmu.2023.1084887
65. Pietrobon V, Cesano A, Marincola F, Kather JN. Next generation imaging techniques to define immune topographies in solid tumors. Front Immunol (2020) 11:604967. doi: 10.3389/fimmu.2020.604967
66. Cox TR. The matrix in cancer. Nat Rev Cancer (2021) 21(4):217–38. doi: 10.1038/s41568-020-00329-7
67. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol (2012) 196(4):395–406. doi: 10.1083/jcb.201102147
68. Bissell MJ, Aggeler J. Dynamic reciprocity: how do extracellular matrix and hormones direct gene expression? Prog Clin Biol Res (1987) 249:251–62.
69. Yu H, Mouw JK, Weaver VM. Forcing form and function: biomechanical regulation of tumor evolution. Trends Cell Biol (2011) 21(1):47–56. doi: 10.1016/j.tcb.2010.08.015
70. Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia (2004) 9(4):325–42. doi: 10.1007/s10911-004-1404-x
71. Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell (2005) 8(3):241–54. doi: 10.1016/j.ccr.2005.08.010
72. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature (2004) 432(7015):332–7. doi: 10.1038/nature03096
73. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell (2005) 121(3):335–48. doi: 10.1016/j.cell.2005.02.034
74. Quante M, Tu SP, Tomita H, Gonda T, Wang SSW, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell (2011) 19(2):257–72. doi: 10.1016/j.ccr.2011.01.020
75. Singer NG, Caplan AI. Mesenchymal stem cells: mechanisms of inflammation. Annu Rev Pathol (2011) 6:457–78. doi: 10.1146/annurev-pathol-011110-130230
76. Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech (2011) 4(2):165–78. doi: 10.1242/dmm.004077
77. Kumar AV, Katakam SK, Urbanowitz AK, Gotte M. Heparan sulphate as a regulator of leukocyte recruitment in inflammation. Curr Protein Pept Sci (2015) 16(1):77–86. doi: 10.2174/1573402111666150213165054
78. El Ghazal R, Yin X, Johns SC, Swanson L, Macal M, Ghosh P, et al. Glycan sulfation modulates dendritic cell biology and tumor growth. Neoplasia (2016) 18(5):294–306. doi: 10.1016/j.neo.2016.04.004
79. Zeng-Brouwers J, Beckmann J, Nastase MV, Iozzo RV, Schaefer L. De novo expression of circulating biglycan evokes an innate inflammatory tissue response via MyD88/TRIF pathways. Matrix Biol (2014) 35:132–42. doi: 10.1016/j.matbio.2013.12.003
80. He Y, Liu T, Dai S, Xu Z, Wang L, Luo F. Tumor-associated extracellular matrix: how to be a potential aide to anti-tumor immunotherapy? Front Cell Dev Biol (2021) 9:739161. doi: 10.3389/fcell.2021.739161
81. Neill T, Schaefer L, Iozzo RV. Decoding the matrix: instructive roles of proteoglycan receptors. Biochemistry (2015) 54(30):4583–98. doi: 10.1021/acs.biochem.5b00653
82. Du H, Bartleson JM, Butenko S, Alonso V, Liu WF, Winer DA, et al. Tuning immunity through tissue mechanotransduction. Nat Rev Immunol (2023) 23(3):174–88. doi: 10.1038/s41577-022-00761-w
83. García-García A, Martin I. Extracellular matrices to modulate the innate immune response and enhance bone healing. Front Immunol (2019) 10:2256. doi: 10.3389/fimmu.2019.02256
84. Gordon-Weeks A, Yuzhalin AE. Cancer extracellular matrix proteins regulate tumour immunity. Cancers (2020) 12(11):3331. doi: 10.3390/cancers12113331
85. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208(10):1949–62. doi: 10.1084/jem.20101956
86. Allavena P, Bianchi G, Zhou D, van Damme J, Jílek P, Sozzani S, et al. Induction of natural killer cell migration by monocyte chemotactic protein-1, -2 and -3. Eur J Immunol (1994) 24(12):3233–6. doi: 10.1002/eji.1830241249
87. Hao Q, Vadgama JV, Wang P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun Signal (2020) 18(1):82. doi: 10.1186/s12964-020-00589-8
88. Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med (1998) 187(1):129–34. doi: 10.1084/jem.187.1.129
89. Nansen A, Marker O, Bartholdy C, Thomsen AR. CCR2+ and CCR5+ CD8+ T cells increase during viral infection and migrate to sites of infection. Eur J Immunol (2000) 30(7):1797–806. doi: 10.1002/1521-4141(200007)30:7<1797::AID-IMMU1797>3.0.CO;2-B
90. Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B. Activation of NK cells by CC chemokines. chemotaxis, Ca2+ mobilization, and enzyme release. J Immunol (1996) 156(1):322–7.
91. Erdogan B, Webb DJ. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans (2017) 45(1):229–36. doi: 10.1042/BST20160387
92. Liu Z, Kuang W, Zhou Q, Zhang Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int J Mol Med (2018) 42(6):3395–403. doi: 10.3892/ijmm.2018.3923
93. Rodón L, Gonzàlez-Juncà A, Inda M del M, Sala-Hojman A, Martínez-Sáez E, Seoane J. Active CREB1 promotes a malignant TGFβ2 autocrine loop in glioblastoma. Cancer Discovery (2014) 4(10):1230–41. doi: 10.1158/2159-8290.CD-14-0275
94. Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer (2013) 110(3):724–32. doi: 10.1038/bjc.2013.768b
95. Budi EH, Schaub JR, Decaris M, Turner S, Derynck R. TGF-β as a driver of fibrosis: physiological roles and therapeutic opportunities. J Pathol (2021) 254(4):358–73. doi: 10.1002/path.5680
96. Carvalheiro T, Garcia S, Pascoal Ramos MI, Giovannone B, Radstake TRDJ, Marut W, et al. Leukocyte associated immunoglobulin like receptor 1 regulation and function on monocytes and dendritic cells during inflammation. Front Immunol (2020) 11:1793. doi: 10.3389/fimmu.2020.01793
97. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
98. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
99. Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun (2018) 9(1):4692. doi: 10.1038/s41467-018-06654-8
100. Horn LA, Chariou PL, Gameiro SR, Qin H, Iida M, Fousek K, et al. Remodeling the tumor microenvironment via blockade of LAIR-1 and TGF-β signaling enables PD-L1-mediated tumor eradication. J Clin Invest (2022) 132(8):e155148. doi: 10.1172/JCI155148
101. Katsuno Y, Lamouille S, Derynck R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol (2013) 25(1):76–84. doi: 10.1097/CCO.0b013e32835b6371
102. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal (2014) 7(344):re8. doi: 10.1126/scisignal.2005189
103. Salerno EP, Bedognetti D, Mauldin IS, Deacon DH, Shea SM, Pinczewski J, et al. Human melanomas and ovarian cancers overexpressing mechanical barrier molecule genes lack immune signatures and have increased patient mortality risk. Oncoimmunology (2016) 5(12):e1240857. doi: 10.1080/2162402X.2016.1240857
104. Fintha A, Gasparics Á, Rosivall L, Sebe A. Therapeutic targeting of fibrotic epithelial-mesenchymal transition-an outstanding challenge. Front Pharmacol (2019) 10:388. doi: 10.3389/fphar.2019.00388
105. Runyan RB, Savagner P. Epithelial-mesenchymal transition and plasticity in the developmental basis of cancer and fibrosis. Dev Dyn (2018) 247(3):330–1. doi: 10.1002/dvdy.24620
106. Kurata T, Fushida S, Kinoshita J, Oyama K, Yamaguchi T, Okazaki M, et al. Low-dose eribulin mesylate exerts antitumor effects in gastric cancer by inhibiting fibrosis via the suppression of epithelial-mesenchymal transition and acts synergistically with 5-fluorouracil. Cancer Manag Res (2018) 10:2729–42. doi: 10.2147/CMAR.S167846
107. Mercogliano MF, Bruni S, Elizalde PV, Schillaci R. Tumor necrosis factor α blockade: an opportunity to tackle breast cancer. Front Oncol (2020) 10:584. doi: 10.3389/fonc.2020.00584
108. Mercogliano MF, Bruni S, Mauro F, Elizalde PV, Schillaci R. Harnessing tumor necrosis factor alpha to achieve effective cancer immunotherapy. Cancers (2021) 13(3):564. doi: 10.3390/cancers13030564
109. Bertrand F, Rochotte J, Colacios C, Montfort A, Tilkin-Mariamé AF, Touriol C, et al. Blocking tumor necrosis factor α enhances CD8 T-cell-Dependent immunity in experimental melanoma. Cancer Res (2015) 75(13):2619–28. doi: 10.1158/0008-5472.CAN-14-2524
110. Avram G, Sánchez-Sendra B, Martín JM, Terrádez L, Ramos D, Monteagudo C. The density and type of MECA-79-positive high endothelial venules correlate with lymphocytic infiltration and tumour regression in primary cutaneous melanoma. Histopathology (2013) 63(6):852–61. doi: 10.1111/his.12235
111. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, et al. Human solid tumors contain high endothelial venules: association with T- and b-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res (2011) 71(17):5678–87. doi: 10.1158/0008-5472.CAN-11-0431
112. Choi C, Park JY, Lee J, Lim JH, Shin EC, Ahn YS, et al. Fas ligand and fas are expressed constitutively in human astrocytes and the expression increases with IL-1, IL-6, TNF-alpha, or IFN-gamma. J Immunol (1999) 162(4):1889–95. doi: 10.4049/jimmunol.162.4.1889
113. Lee WH, Seo D, Lim SG, Suk K. Reverse signaling of tumor necrosis factor superfamily proteins in macrophages and microglia: superfamily portrait in the neuroimmune interface. Front Immunol (2019) 10:262. doi: 10.3389/fimmu.2019.00262
114. Alderson MR, Tough TW, Davis-Smith T, Braddy S, Falk B, Schooley KA, et al. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med (1995) 181(1):71–7. doi: 10.1084/jem.181.1.71
115. Mercogliano MF, De Martino M, Venturutti L, Rivas MA, Proietti CJ, Inurrigarro G, et al. TNFα-induced mucin 4 expression elicits trastuzumab resistance in HER2-positive breast cancer. Clin Cancer Res (2017) 23(3):636–48. doi: 10.1158/1078-0432.CCR-16-0970
116. Carraway KL, Price-Schiavi SA, Komatsu M, Jepson S, Perez A, Carraway CA. Muc4/sialomucin complex in the mammary gland and breast cancer. J Mammary Gland Biol Neoplasia (2001) 6(3):323–37. doi: 10.1023/A:1011327708973
117. Carraway KL, Rossi EA, Komatsu M, Price-Schiavi SA, Huang D, Guy PM, et al. An intramembrane modulator of the ErbB2 receptor tyrosine kinase that potentiates neuregulin signaling. J Biol Chem (1999) 274:5263–6. doi: 10.1074/jbc.274.9.5263
118. Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, et al. Structure of the extracellular region of HER2 alone and in complex with the herceptin fab. Nature (2003) 421:756–60. doi: 10.1038/nature01392
119. Price-Schiavi SA, Jepson S, Li P, Arango M, Rudland PS, Yee L, et al. Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int J Cancer (2002) 99(6):783–91. doi: 10.1002/ijc.10410
120. Workman HC, Sweeney C, Carraway KL 3rd. The membrane mucin Muc4 inhibits apoptosis induced by multiple insults via ErbB2-dependent and ErbB2-independent mechanisms. Cancer Res (2009) 69(7):2845–52. doi: 10.1158/0008-5472.CAN-08-2089
121. Chaturvedi P, Singh AP, Chakraborty S, Chauhan SC, Bafna S, Meza JL, et al. MUC4 mucin interacts with and stabilizes the HER2 oncoprotein in human pancreatic cancer cells. Cancer Res (2008) 68(7):2065–70. doi: 10.1158/0008-5472.CAN-07-6041
122. Funes M, Miller JK, Lai C, Carraway KL 3rd, Sweeney C. The mucin Muc4 potentiates neuregulin signaling by increasing the cell-surface populations of ErbB2 and ErbB3. J Biol Chem (2006) 281(28):19310–9. doi: 10.1074/jbc.M603225200
123. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA (2003) 100(15):8933–8. doi: 10.1073/pnas.1537685100
124. Schillaci R, Bruni S, Mauro F, Mercogliano MF, Roldan-Deamicis A, Proietti CJ, et al. Abstract P5-13-32: mucin 4 expression in high risk breast cancer: predicting and overcoming resistance to immunotherapy. Cancer Res (2022) 82:P5–13. doi: 10.1158/1538-7445.sabcs21-p5-13-32
125. Cornelissen LAM, Van Vliet SJ. A bitter sweet symphony: immune responses to altered O-glycan epitopes in cancer. Biomolecules (2016) 6(2):26. doi: 10.3390/biom6020026
126. Agrawal B, Krantz MJ, Reddish MA, Longenecker BM. Cancer-associated MUC1 mucin inhibits human T-cell proliferation, which is reversible by IL-2. Nat Med (1998) 4(1):43–9. doi: 10.1038/nm0198-043
127. Chauhan SC, Kumar D, Jaggi M. Mucins in ovarian cancer diagnosis and therapy. J Ovarian Res (2009) 2:21. doi: 10.1186/1757-2215-2-21
128. Komatsu M, Yee L, Carraway KL. Overexpression of sialomucin complex, a rat homologue of MUC4, inhibits tumor killing by lymphokine-activated killer cells. Cancer Res (1999) 59(9):2229–36.
129. van de Wiel-van Kemenade E, Ligtenberg MJ, de Boer AJ, Buijs F, Vos HL, Melief CJ, et al. Episialin (MUC1) inhibits cytotoxic lymphocyte-target cell interaction. J Immunol (1993) 151(2):767–76. doi: 10.4049/jimmunol.151.2.767
130. McEver RP. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res (2015) 107(3):331–9. doi: 10.1093/cvr/cvv154
131. Baldus SE, Mönig SP, Hanisch FG, Zirbes TK, Flucke U, Oelert S, et al. Comparative evaluation of the prognostic value of MUC1, MUC2, sialyl-lewis(a) and sialyl-lewis(x) antigens in colorectal adenocarcinoma. Histopathology (2002) 40(5):440–9. doi: 10.1046/j.1365-2559.2002.01389.x
132. Rowson-Hodel AR, Wald JH, Hatakeyama J, O’Neal WK, Stonebraker JR, VanderVorst K, et al. Membrane mucin Muc4 promotes blood cell association with tumor cells and mediates efficient metastasis in a mouse model of breast cancer. Oncogene (2018) 37(2):197–207. doi: 10.1038/onc.2017.327
133. Chen L, Huang X, Xiong L, Chen W, An L, Wang H, et al. Analysis of prognostic oncogene filaggrin (FLG) wild-type subtype and its implications for immune checkpoint blockade therapy in bladder urothelial carcinoma. Transl Androl Urol (2022) 11(10):1419–32. doi: 10.21037/tau-22-573
134. Edmonds NL, Flores SE, Mahmutovic A, Young SJ, Mauldin IS, Slingluff CL Jr. CD103 and periplakin are potential biomarkers for response of metastatic melanoma to pembrolizumab. Melanoma Res (2022) 32(6):440–50. doi: 10.1097/CMR.0000000000000855
135. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discovery (2011) 1(1):54–67. doi: 10.1158/2159-8274.CD-10-0028
136. Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep (2017) 19(1):203–17. doi: 10.1016/j.celrep.2017.03.037
137. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell (2015) 27(4):462–72. doi: 10.1016/j.ccell.2015.02.015
138. Xu S, Xu H, Wang W, Li S, Li H, Li T, et al. The role of collagen in cancer: from bench to bedside. J Transl Med (2019) 17(1):309. doi: 10.1186/s12967-019-2058-1
139. Shields MA, Dangi-Garimella S, Redig AJ, Munshi HG. Biochemical role of the collagen-rich tumour microenvironment in pancreatic cancer progression. Biochem J (2012) 441(2):541–52. doi: 10.1042/BJ20111240
140. Paolillo M, Schinelli S. Extracellular matrix alterations in metastatic processes. Int J Mol Sci (2019) 20(19):4947. doi: 10.3390/ijms20194947
141. Nissen NI, Karsdal M, Willumsen N. Collagens and cancer associated fibroblasts in the reactive stroma and its relation to cancer biology. J Exp Clin Cancer Res (2019) 38(1):115. doi: 10.1186/s13046-019-1110-6
142. Zhu GG, Risteli L, Mäkinen M, Risteli J, Kauppila A, Stenbäck F. Immunohistochemical study of type I collagen and type I pN-collagen in benign and malignant ovarian neoplasms. Cancer (1995) 75(4):1010–7. doi: 10.1002/1097-0142(19950215)75:4<1010::AID-CNCR2820750417>3.0.CO;2-O
143. Huijbers IJ, Iravani M, Popov S, Robertson D, Al-Sarraj S, Jones C, et al. A role for fibrillar collagen deposition and the collagen internalization receptor endo180 in glioma invasion. PloS One (2010) 5(3):e9808. doi: 10.1371/journal.pone.0009808
144. Yamauchi M, Barker TH, Gibbons DL, Kurie JM. The fibrotic tumor stroma. J Clin Invest (2018) 128(1):16–25. doi: 10.1172/JCI93554
145. Arnold SA, Rivera LB, Miller AF, Carbon JG, Dineen SP, Xie Y, et al. Lack of host SPARC enhances vascular function and tumor spread in an orthotopic murine model of pancreatic carcinoma. Dis Model Mech (2010) 3(1-2):57–72. doi: 10.1242/dmm.003228
146. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell (2009) 139(5):891–906. doi: 10.1016/j.cell.2009.10.027
147. Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res (2007) 67(6):2649–56. doi: 10.1158/0008-5472.CAN-06-1823
148. Fang M, Yuan J, Peng C, Li Y. Collagen as a double-edged sword in tumor progression. Tumour Biol (2014) 35(4):2871–82. doi: 10.1007/s13277-013-1511-7
149. Hsu KS, Dunleavey JM, Szot C, Yang L, Hilton MB, Morris K, et al. Cancer cell survival depends on collagen uptake into tumor-associated stroma. Nat Commun (2022) 13(1):7078. doi: 10.1038/s41467-022-34643-5
150. Boyd NF, Guo H, Martin LJ, Sun L, Stone J, Fishell E, et al. Mammographic density and the risk and detection of breast cancer. N Engl J Med (2007) 356(3):227–36. doi: 10.1056/NEJMoa062790
151. Zhang H, Fredericks T, Xiong G, Qi Y, Rychahou PG, Li JD, et al. Membrane associated collagen XIII promotes cancer metastasis and enhances anoikis resistance. Breast Cancer Res (2018) 20(1):116. doi: 10.1186/s13058-018-1030-y
152. Sun X, Wu B, Chiang HC, Deng H, Zhang X, Xiong W, et al. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature (2021) 599:673–8. doi: 10.1038/s41586-021-04057-2
153. Meyaard L. The inhibitory collagen receptor LAIR-1 (CD305). J Leukoc Biol (2008) 83(4):799–803. doi: 10.1189/jlb.0907609
154. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest (2012) 122(3):899–910. doi: 10.1172/JCI45817
155. Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol (2014) 15(12):802–12. doi: 10.1038/nrm3896
156. Datar I, Schalper KA. Epithelial-mesenchymal transition and immune evasion during lung cancer progression: the chicken or the egg? Clin Cancer Res (2016) 22(14):3422–4. doi: 10.1158/1078-0432.CCR-16-0336
157. Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP, et al. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis (2017) 6(7):e352. doi: 10.1038/oncsis.2017.54
158. Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol (2017) 19(5):518–29. doi: 10.1038/ncb3513
159. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med (2016) 22(8):851–60. doi: 10.1038/nm.4123
160. Hartmann N, Giese NA, Giese T, Poschke I, Offringa R, Werner J, et al. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin Cancer Res (2014) 20(13):3422–33. doi: 10.1158/1078-0432.CCR-13-2972
161. Kauppila S, Stenbäck F, Risteli J, Jukkola A, Risteli L. Aberrant type I and type III collagen gene expression in human breast cancer in vivo. J Pathol (1998) 186(3):262–8. doi: 10.1002/(SICI)1096-9896(1998110)186:3<262::AID-PATH191>3.0.CO;2-3
162. Hasebe T, Sasaki S, Imoto S, Mukai K, Yokose T, Ochiai A. Prognostic significance of fibrotic focus in invasive ductal carcinoma of the breast: a prospective observational study. Modern Pathol (2002) 15:502–16. doi: 10.1038/modpathol.3880555
163. Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors [Internet]. Nat Genet (2003) 33:49–54. doi: 10.1038/ng1060
164. Tavazoie SF, Alarcón C, Oskarsson T, Padua D, Wang Q, Bos PD, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature (2008) 451(7175):147–52. doi: 10.1038/nature06487
165. Erler JT, Weaver VM. Three-dimensional context regulation of metastasis. Clin Exp Metastasis (2009) 26(1):35–49. doi: 10.1007/s10585-008-9209-8
166. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature (2005) 438(7069):820–7. doi: 10.1038/nature04186
167. Jiang H, Hegde S, DeNardo DG. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol Immunother (2017) 66(8):1037–48. doi: 10.1007/s00262-017-2003-1
168. Boot-Handford RP, Tuckwell DS. Fibrillar collagen: the key to vertebrate evolution? a tale of molecular incest. BioEssays (2003) 25:142–51. doi: 10.1002/bies.10230
169. Wang E, Shibutani M, Nagahara H, Fukuoka T, Iseki Y, Okazaki Y, et al. Abundant intratumoral fibrosis prevents lymphocyte infiltration into peritoneal metastases of colorectal cancer. PloS One (2021) 16(7):e0255049. doi: 10.1371/journal.pone.0255049
170. Leitinger B. Discoidin domain receptor functions in physiological and pathological conditions. Int Rev Cell Mol Biol (2014) 310:39–87. doi: 10.1016/b978-0-12-800180-6.00002-5
171. Vogel WF, Abdulhussein R, Ford CE. Sensing extracellular matrix: an update on discoidin domain receptor function. Cell Signal (2006) 18:1108–16. doi: 10.1016/j.cellsig.2006.02.012
172. Takai K, Drain AP, Lawson DA, Littlepage LE, Karpuj M, Kessenbrock K, et al. Discoidin domain receptor 1 (DDR1) ablation promotes tissue fibrosis and hypoxia to induce aggressive basal-like breast cancers. Genes Dev (2018) 32(3-4):244–57. doi: 10.1101/gad.301366.117
173. Saby C, Collin G, Sinane M, Buache E, Van Gulick L, Saltel F, et al. DDR1 and MT1-MMP expression levels are determinant for triggering BIK-mediated apoptosis by 3D type I collagen matrix in invasive basal-like breast carcinoma cells. Front Pharmacol (2019) 10:462. doi: 10.3389/fphar.2019.00462
174. Assent D, Bourgot I, Hennuy B, Geurts P, Noël A, Foidart JM, et al. A membrane-type-1 matrix metalloproteinase (MT1-MMP)-discoidin domain receptor 1 axis regulates collagen-induced apoptosis in breast cancer cells. PloS One (2015) 10(3):e0116006. doi: 10.1371/journal.pone.0116006
175. Saby C, Maquoi E, Saltel F, Morjani H. Collagen and discoidin domain receptor 1 partnership: a multifaceted role in the regulation of breast carcinoma cell phenotype. Front Cell Dev Biol (2021) 9:808625. doi: 10.3389/fcell.2021.808625
176. Hidalgo-Carcedo C, Hooper S, Chaudhry SI, Williamson P, Harrington K, Leitinger B, et al. Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat Cell Biol (2011) 13(1):49–58. doi: 10.1038/ncb2133
177. Valiathan RR, Marco M, Leitinger B, Kleer CG, Fridman R. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev (2012) 31(1-2):295–321. doi: 10.1007/s10555-012-9346-z
178. Ruggeri JM, Franco-Barraza J, Sohail A, Zhang Y, Long D, Pasca di Magliano M, et al. Discoidin domain receptor 1 (DDR1) is necessary for tissue homeostasis in pancreatic injury and pathogenesis of pancreatic ductal adenocarcinoma. Am J Pathol (2020) 190(8):1735–51. doi: 10.1016/j.ajpath.2020.03.020
179. Ambrogio C, Gómez-López G, Falcone M, Vidal A, Nadal E, Crosetto N, et al. Combined inhibition of DDR1 and notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med (2016) 22(3):270–7. doi: 10.1038/nm.4041
180. Gao H, Chakraborty G, Zhang Z, Akalay I, Gadiya M, Gao Y, et al. Multi-organ site metastatic reactivation mediated by non-canonical discoidin domain receptor 1 signaling. Cell (2016) 166(1):47–62. doi: 10.1016/j.cell.2016.06.009
181. Juin A, Di Martino J, Leitinger B, Henriet E, Gary AS, Paysan L, et al. Discoidin domain receptor 1 controls linear invadosome formation via a Cdc42-tuba pathway. J Cell Biol (2014) 207(4):517–33. doi: 10.1083/jcb.201404079
182. Di Martino JS, Nobre AR, Mondal C, Taha I, Farias EF, Fertig EJ, et al. A tumor-derived type III collagen-rich ECM niche regulates tumor cell dormancy. Nat Cancer (2022) 3(1):90–107. doi: 10.1038/s43018-021-00291-9
183. Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest (2021) 131(4):e140100. doi: 10.1172/JCI140100
184. Jiménez-Sánchez A, Cast O, Miller ML. Comprehensive benchmarking and integration of tumor microenvironment cell estimation methods. Cancer Res (2019) 79:6238–46. doi: 10.1158/0008-5472.can-18-3560
185. Kaur A, Ecker BL, Douglass SM, Kugel CH 3rd, Webster MR, Almeida FV, et al. Remodeling of the collagen matrix in aging skin promotes melanoma metastasis and affects immune cell motility. Cancer Discovery (2019) 9(1):64–81. doi: 10.1158/2159-8290.CD-18-0193
186. Ray A, Provenzano PP. Aligned forces: origins and mechanisms of cancer dissemination guided by extracellular matrix architecture. Curr Opin Cell Biol (2021) 72:63–71. doi: 10.1016/j.ceb.2021.05.004
187. Tomko LA, Hill RC, Barrett A, Szulczewski JM, Conklin MW, Eliceiri KW, et al. Targeted matrisome analysis identifies thrombospondin-2 and tenascin-c in aligned collagen stroma from invasive breast carcinoma. Sci Rep (2018) 8(1):12941. doi: 10.1038/s41598-018-31126-w
188. Peranzoni E, Rivas-Caicedo A, Bougherara H, Salmon H, Donnadieu E. Positive and negative influence of the matrix architecture on antitumor immune surveillance. Cell Mol Life Sci (2013) 70(23):4431–48. doi: 10.1007/s00018-013-1339-8
189. Nicolas-Boluda A, Vaquero J, Vimeux L, Guilbert T, Barrin S, Kantari-Mimoun C, et al. Tumor stiffening reversion through collagen crosslinking inhibition improves T cell migration and anti-PD-1 treatment. Elife (2021) 10:e58688. doi: 10.7554/eLife.58688
190. Laurent TC, Fraser JR. Hyaluronan. FASEB J (1992) 6(7):2397–404. doi: 10.1096/fasebj.6.7.1563592
191. Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol (2007) 23:435–61. doi: 10.1146/annurev.cellbio.23.090506.123337
192. Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer (2004) 4(7):528–39. doi: 10.1038/nrc1391
193. Johnson P, Ruffell B. CD44 and its role in inflammation and inflammatory diseases. Inflammation Allergy - Drug Targets (2009) 8:208–20. doi: 10.2174/187152809788680994
194. Spicer AP, Tien JYL. Hyaluronan and morphogenesis. Birth Defects Res Part C: Embryo Today: Rev (2004) 72:89–108. doi: 10.1002/bdrc.20006
195. Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front Immunol (2015) 6:201. doi: 10.3389/fimmu.2015.00201
196. Vigetti D, Passi A. Chapter four - hyaluronan synthases posttranslational regulation in cancer. In: Simpson MA, Heldin P, editors. Advances in cancer research. Academic Press (2014). p. 95–119.
197. Itano N, Kimata K. Altered hyaluronan biosynthesis in cancer progression. Semin Cancer Biol (2008) 18:268–74. doi: 10.1016/j.semcancer.2008.03.006
198. Heldin P, Karousou E, Skandalis SS. CHAPTER 3 - growth factor regulation of hyaluronan deposition in malignancies. In: Stern R, editor. Hyaluronan in cancer biology. San Diego: Academic Press (2009). p. 37–50.
199. Singha NC, Nekoroski T, Zhao C, Symons R, Jiang P, Frost GI, et al. Tumor-associated hyaluronan limits efficacy of monoclonal antibody therapy. Mol Cancer Ther (2015) 14:523–32. doi: 10.1158/1535-7163.mct-14-0580
200. Varadi T, Mocanu MM, Mersich T, Auvinen P, Tammi R, Tammi M, et al. 5027 POSTER binding of trastuzumab to ErbB2 is inhibited by a high local density of hyaluronan. Eur J Cancer (2011) 47:S339. doi: 10.1016/s0959-8049(11)71469-1
201. Pályi-Krekk Z, Barok M, Isola J, Tammi M, Szöllo˝si J, Nagy P. Hyaluronan-induced masking of ErbB2 and CD44-enhanced trastuzumab internalisation in trastuzumab resistant breast cancer. Eur J Cancer (2007) 43:2423–33. doi: 10.1016/j.ejca.2007.08.018
202. Blair AB, Kim VM, Muth ST, Saung MT, Lokker N, Blouw B, et al. Dissecting the stromal signaling and regulation of myeloid cells and memory effector T cells in pancreatic cancer. Clin Cancer Res (2019) 25(17):5351–63. doi: 10.1158/1078-0432.CCR-18-4192
203. Suto A, Kudo D, Yoshida E, Nagase H, Suto S, Mimura J, et al. Increase of tumor infiltrating γδ T-cells in pancreatic ductal adenocarcinoma through remodeling of the extracellular matrix by a hyaluronan synthesis suppressor, 4-methylumbelliferone. Pancreas (2019) 48(2):292–8. doi: 10.1097/MPA.0000000000001211
204. Schwertfeger KL, Cowman MK, Telmer PG, Turley EA, McCarthy JB. Hyaluronan, inflammation, and breast cancer progression. Front Immunol (2015) 6:236. doi: 10.3389/fimmu.2015.00236
205. Spinelli FM, Vitale DL, Sevic I, Alaniz L. Hyaluronan in the tumor microenvironment. In: Birbrair A, editor. Tumor microenvironment: extracellular matrix components – part a. Cham: Springer International Publishing (2020). p. 67–83.
206. Caon I, Bartolini B, Parnigoni A, Caravà E, Moretto P, Viola M, et al. Revisiting the hallmarks of cancer: the role of hyaluronan. Semin Cancer Biol (2020) 62:9–19. doi: 10.1016/j.semcancer.2019.07.007
207. Bonder CS, Clark SR, Norman MU, Johnson P, Kubes P. Use of CD44 by CD4+ Th1 and Th2 lymphocytes to roll and adhere. Blood (2006) 107(12):4798–806. doi: 10.1182/blood-2005-09-3581
208. Bollyky PL, Wu RP, Falk BA, Lord JD, Long SA, Preisinger A, et al. ECM components guide IL-10 producing regulatory T-cell (TR1) induction from effector memory T-cell precursors. Proc Natl Acad Sci USA (2011) 108(19):7938–43. doi: 10.1073/pnas.1017360108
209. Bollyky PL, Falk BA, Long SA, Preisinger A, Braun KR, Wu RP, et al. CD44 costimulation promotes FoxP3+ regulatory T cell persistence and function via production of IL-2, IL-10, and TGF-beta. J Immunol (2009) 183(4):2232–41. doi: 10.4049/jimmunol.0900191
210. Park HS, Kim YM, Kim S, Lee WS, Kong SJ, Yang H, et al. High endothelial venule is a surrogate biomarker for T-cell inflamed tumor microenvironment and prognosis in gastric cancer. J Immunother Cancer (2021) 9(10):e003353. doi: 10.1136/jitc-2021-003353
211. Martinet L, Le Guellec S, Filleron T, Lamant L, Meyer N, Rochaix P, et al. High endothelial venules (HEVs) in human melanoma lesions. OncoImmunology (2012) 1:829–39. doi: 10.4161/onci.20492
212. Carmona-Rodríguez L, Martínez-Rey D, Fernández-Aceñero MJ, González-Martín A, Paz-Cabezas M, Rodríguez-Rodríguez N, et al. SOD3 induces a HIF-2α-dependent program in endothelial cells that provides a selective signal for tumor infiltration by T cells. J ImmunoTher Cancer (2020) 8:e000432. doi: 10.1136/jitc-2019-000432
213. von Andrian UH, von Andrian UH, Mackay CR. T-Cell function and migration {{/amp]]mdash; two sides of the same coin. New Engl J Med (2000) 343:1020–34. doi: 10.1056/nejm200010053431407
214. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7:678–89. doi: 10.1038/nri2156
215. Viallard C, Larrivée B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis (2017) 20(4):409–26. doi: 10.1007/s10456-017-9562-9
216. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest (2019) 129(10):4350–64. doi: 10.1172/JCI125413
217. Mira E, Carmona-Rodríguez L, Pérez-Villamil B, Casas J, Fernández-Aceñero MJ, Martínez-Rey D, et al. SOD3 improves the tumor response to chemotherapy by stabilizing endothelial HIF-2α. Nat Commun (2018) 9(1):575. doi: 10.1038/s41467-018-03079-1
218. Teoh-Fitzgerald MLT, Fitzgerald MP, Jensen TJ, Futscher BW, Domann FE. Genetic and epigenetic inactivation of extracellular superoxide dismutase promotes an invasive phenotype in human lung cancer by disrupting ECM homeostasis. Mol Cancer Res (2012) 10:40–51. doi: 10.1158/1541-7786.mcr-11-0501
219. Teoh-Fitzgerald ML, Fitzgerald MP, Zhong W, Askeland RW, Domann FE. Epigenetic reprogramming governs EcSOD expression during human mammary epithelial cell differentiation, tumorigenesis and metastasis. Oncogene (2014) 33:358–68. doi: 10.1038/onc.2012.582
220. O’Leary BR, Fath MA, Bellizzi AM, Hrabe JE, Button AM, Allen BG, et al. Loss of SOD3 (EcSOD) expression promotes an aggressive phenotype in human pancreatic ductal adenocarcinoma. Clin Cancer Res (2015) 21(7):1741–51. doi: 10.1158/1078-0432.CCR-14-1959
221. Thyboll J, Kortesmaa J, Cao R, Soininen R, Wang L, Iivanainen A, et al. Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol Cell Biol (2002) 22(4):1194–202. doi: 10.1128/MCB.22.4.1194-1202.2002
222. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin b receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med (2008) 14(1):28–36. doi: 10.1038/nm1699
223. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med (2014) 20(6):607–15. doi: 10.1038/nm.3541
224. Bellone M, Calcinotto A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front Oncol (2013) 3:231. doi: 10.3389/fonc.2013.00231
225. Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, et al. PDGF-c mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell (2009) 15(1):21–34. doi: 10.1016/j.ccr.2008.12.004
226. Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell (2011) 19(1):31–44. doi: 10.1016/j.ccr.2010.11.009
227. Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J (2015) 29(1):227–38. doi: 10.1096/fj.14-250985
228. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res (2009) 69(7):3077–85. doi: 10.1158/0008-5472.CAN-08-2281
229. Andersson Å, Yang SC, Huang M, Zhu L, Kar UK, Batra RK, et al. IL-7 promotes CXCR3 ligand-dependent T cell antitumor reactivity in lung cancer. J Immunol (2009) 182:6951–8. doi: 10.4049/jimmunol.0803340
230. Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, et al. Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology (2010) 138(4):1429–40. doi: 10.1053/j.gastro.2009.10.057
231. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer (2010) 10(1):9–22. doi: 10.1038/nrc2748
232. Rojas K, Baliu-Piqué M, Manzano A, Saiz-Ladera C, García-Barberán V, Cimas FJ, et al. In silico transcriptomic mapping of integrins and immune activation in basal-like and HER2+ breast cancer. Cell Oncol (2021) 44(3):569–80. doi: 10.1007/s13402-020-00583-9
233. Zhu H, Wang G, Zhu H, Xu A. ITGA5 is a prognostic biomarker and correlated with immune infiltration in gastrointestinal tumors. BMC Cancer (2021) 21(1):269. doi: 10.1186/s12885-021-07996-1
234. Lu L, Xie R, Wei R, Cai C, Bi D, Yin D, et al. Integrin α5 subunit is required for the tumor supportive role of fibroblasts in colorectal adenocarcinoma and serves as a potential stroma prognostic marker. Mol Oncol (2019) 13:2697–714. doi: 10.1002/1878-0261.12583
235. Xu M, Zhang S, Jia L, Wang S, Liu J, Ma X, et al. E-m, an engineered endostatin with high ATPase activity, inhibits the recruitment and alternative activation of macrophages in non-small cell lung cancer. Front Pharmacol (2017) 8:532. doi: 10.3389/fphar.2017.00532
236. Harper J, Sainson RCA. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin Cancer Biol (2014) 25:69–77. doi: 10.1016/j.semcancer.2013.12.005
237. Kuninty PR, Bansal R, De Geus SWL, Mardhian DF, Schnittert J, van Baarlen J, et al. ITGA5 inhibition in pancreatic stellate cells attenuates desmoplasia and potentiates efficacy of chemotherapy in pancreatic cancer. Sci Adv (2019) 5(9):eaax2770. doi: 10.1126/sciadv.aax2770
238. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell (2012) 21:822–35. doi: 10.1016/j.ccr.2012.04.025
239. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8 T cells. OncoImmunology (2013) 2:e26968. doi: 10.4161/onci.26968
240. Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res (2012) 72(4):876–86. doi: 10.1158/0008-5472.CAN-11-1792
241. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
242. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell (2012) 21(6):836–47. doi: 10.1016/j.ccr.2012.04.024
243. Garcia AJ, Ruscetti M, Arenzana TL, Tran LM, Bianci-Frias D, Sybert E, et al. Pten null prostate epithelium promotes localized myeloid-derived suppressor cell expansion and immune suppression during tumor initiation and progression. Mol Cell Biol (2014) 34(11):2017–28. doi: 10.1128/MCB.00090-14
244. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
245. Mastio J, Condamine T, Dominguez G, Kossenkov AV, Donthireddy L, Veglia F, et al. Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J Exp Med (2019) 216(9):2150–69. doi: 10.1084/jem.20181952
246. Casbon AJ, Reynaud D, Park C, Khuc E, Gan DD, Schepers K, et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci USA (2015) 112(6):E566–75. doi: 10.1073/pnas.1424927112
247. Porembka MR, Mitchem JB, Belt BA, Hsieh CS, Lee HM, Herndon J, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother (2012) 61(9):1373–85. doi: 10.1007/s00262-011-1178-0
248. Thorn M, Guha P, Cunetta M, Espat NJ, Miller G, Junghans RP, et al. Tumor-associated GM-CSF overexpression induces immunoinhibitory molecules via STAT3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther (2016) 23(6):188–98. doi: 10.1038/cgt.2016.19
249. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol (2018) 19(2):108–19. doi: 10.1038/s41590-017-0022-x
250. Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol (2018) 200(2):422–31. doi: 10.4049/jimmunol.1701019
251. Colligan SH, Tzetzo SL, Abrams SI. Myeloid-driven mechanisms as barriers to antitumor CD8+ T cell activity. Mol Immunol (2020) 118:165–73. doi: 10.1016/j.molimm.2019.12.012
252. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer (2017) 17(12):709–24. doi: 10.1038/nrc.2017.86
253. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207(11):2439–53. doi: 10.1084/jem.20100587
254. Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, et al. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity (2014) 41(3):389–401. doi: 10.1016/j.immuni.2014.08.015
255. Long H, Jia Q, Wang L, Fang W, Wang Z, Jiang T, et al. Tumor-induced erythroid precursor-differentiated myeloid cells mediate immunosuppression and curtail anti-PD-1/PD-L1 treatment efficacy. Cancer Cell (2022) 40(6):674–93. doi: 10.1016/j.ccell.2022.04.018
256. Sade-Feldman M, Kanterman J, Klieger Y, Ish-Shalom E, Olga M, Saragovi A, et al. Clinical significance of circulating CD33+CD11b+HLA-DR- myeloid cells in patients with stage IV melanoma treated with ipilimumab. Clin Cancer Res (2016) 22(23):5661–72. doi: 10.1158/1078-0432.CCR-15-3104
257. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
258. Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, et al. L-arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res (2015) 75(2):275–83. doi: 10.1158/0008-5472.CAN-14-1491
259. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res (2010) 70(1):68–77. doi: 10.1158/0008-5472.CAN-09-2587
260. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111(8):4233–44. doi: 10.1182/blood-2007-07-099226
261. Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol (2006) 16(1):53–65. doi: 10.1016/j.semcancer.2005.07.005
262. Ballbach M, Dannert A, Singh A, Siegmund DM, Handgretinger R, Piali L, et al. Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol Lett (2017) 192:1–6. doi: 10.1016/j.imlet.2017.10.001
263. Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The role of myeloid-derived suppressor cells in patients with solid tumors: a meta-analysis. PloS One (2016) 11:e0164514. doi: 10.1371/journal.pone.0164514
264. Sun SH, Benner B, Savardekar H, Lapurga G, Good L, Abood D, et al. Effect of immune checkpoint blockade on myeloid-derived suppressor cell populations in patients with melanoma. Front Immunol (2021) 12:740890. doi: 10.3389/fimmu.2021.740890
265. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity (2016) 44(3):439–49. doi: 10.1016/j.immuni.2016.02.024
266. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci (2016) 19(1):20–7. doi: 10.1038/nn.4185
267. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475(7355):222–5. doi: 10.1038/nature10138
268. Ribechini E, Hutchinson JA, Hergovits S, Heuer M, Lucas J, Schleicher U, et al. Novel GM-CSF signals via IFN-γR/IRF-1 and AKT/mTOR license monocytes for suppressor function. Blood Adv (2017) 1(14):947–60. doi: 10.1182/bloodadvances.2017006858
269. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
270. Devalaraja S, To TKJ, Folkert IW, Natesan R, Alam MZ, Li M, et al. Tumor-derived retinoic acid regulates intratumoral monocyte differentiation to promote immune suppression. Cell (2020) 180(6):1098–114.e16. doi: 10.1016/j.cell.2020.02.042
271. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi: 10.1016/j.cell.2010.01.025
272. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Letters (2008) 267:204–15. doi: 10.1016/j.canlet.2008.03.028
273. Kerneur C, Cano CE, Olive D. Major pathways involved in macrophage polarization in cancer. Front Immunol (2022) 13:1026954. doi: 10.3389/fimmu.2022.1026954
274. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi: 10.1016/j.cell.2010.03.014
275. Liu Y, Cao X. The origin and function of tumor-associated macrophages. Cell Mol Immunol (2015) 12(1):1–4. doi: 10.1038/cmi.2014.83
276. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer (2004) 4:71–8. doi: 10.1038/nrc1256
277. Pu Y, Ji Q. Tumor-associated macrophages regulate PD-1/PD-L1 immunosuppression. Front Immunol (2022) 13:874589. doi: 10.3389/fimmu.2022.874589
278. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci USA (2018) 115(17):E4041–50. doi: 10.1073/pnas.1720948115
279. Suthen S, Lim CJ, Nguyen PHD, Dutertre CA, Lai HLH, Wasser M, et al. Hypoxia-driven immunosuppression by treg and type-2 conventional dendritic cells in HCC. Hepatology (2022) 76(5):1329–44. doi: 10.1002/hep.32419
280. Oktay MH, Jones JG. TMEM: a novel breast cancer dissemination marker for the assessment of metastatic risk. biomark Med (2015) 9(2):81–4. doi: 10.2217/bmm.14.104
281. Robinson BD, Sica GL, Liu YF, Rohan TE, Gertler FB, Condeelis JS, et al. Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clin Cancer Res (2009) 15(7):2433–41. doi: 10.1158/1078-0432.CCR-08-2179
282. Rohan TE, Xue X, Lin HM, D’Alfonso TM, Ginter PS, Oktay MH, et al. Tumor microenvironment of metastasis and risk of distant metastasis of breast cancer. J Natl Cancer Inst (2014) 106(8):dju136. doi: 10.1093/jnci/dju136
283. Asiry S, Kim G, Filippou PS, Sanchez LR, Entenberg D, Marks DK, et al. The cancer cell dissemination machinery as an immunosuppressive niche: a new obstacle towards the era of cancer immunotherapy. Front Immunol (2021) 12:654877. doi: 10.3389/fimmu.2021.654877
284. Hurkmans DP, Jensen C, Koolen SLW, Aerts J, Karsdal MA, Mathijssen RHJ, et al. Blood-based extracellular matrix biomarkers are correlated with clinical outcome after PD-1 inhibition in patients with metastatic melanoma. J Immunother Cancer (2020) 8(2):e001193. doi: 10.1136/jitc-2020-001193
285. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res (2013) 73(10):3007–18. doi: 10.1158/0008-5472.CAN-12-4601
286. Liu T, Zhou L, Li D, Andl T, Zhang Y. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front Cell Dev Biol (2019) 7:60. doi: 10.3389/fcell.2019.00060
287. Arina A, Idel C, Hyjek EM, Alegre ML, Wang Y, Bindokas VP, et al. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc Natl Acad Sci USA (2016) 113(27):7551–6. doi: 10.1073/pnas.1600363113
288. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer (2016) 16(9):582–98. doi: 10.1038/nrc.2016.73
289. Liu L, Liu L, Yao HH, Zhu ZQ, Ning ZL, Huang Q. Stromal myofibroblasts are associated with poor prognosis in solid cancers: a meta-analysis of published studies. PloS One (2016) 11(7):e0159947. doi: 10.1371/journal.pone.0159947
290. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214(3):579–96. doi: 10.1084/jem.20162024
291. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discovery (2019) 9(8):1102–23. doi: 10.1158/2159-8290.CD-19-0094
292. Bernard V, Semaan A, Huang J, San Lucas FA, Mulu FC, Stephens BM, et al. Single-cell transcriptomics of pancreatic cancer precursors demonstrates epithelial and microenvironmental heterogeneity as an early event in neoplastic progression. Clin Cancer Res (2019) 25(7):2194–205. doi: 10.1158/1078-0432.CCR-18-1955
293. Scotton CJ, Wilson JL, Scott K, Stamp G, Wilbanks GD, Fricker S, et al. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res (2002) 62(20):5930–8.
294. Akishima-Fukasawa Y, Nakanishi Y, Ino Y, Moriya Y, Kanai Y, Hirohashi S. Prognostic significance of CXCL12 expression in patients with colorectal carcinoma. Am J Clin Pathol (2009) 132(2):202–10. doi: 10.1309/AJCPK35VZJEWCUTL
295. Goehrig D, Nigri J, Samain R, Wu Z, Cappello P, Gabiane G, et al. Stromal protein βig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut (2019) 68:693–707. doi: 10.1136/gutjnl-2018-317570
296. Nazareth MR, Broderick L, Simpson-Abelson MR, Kelleher RJ Jr, Yokota SJ, Bankert RB. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J Immunol (2007) 178(9):5552–62. doi: 10.4049/jimmunol.178.9.5552
297. Gorchs L, Fernández Moro C, Bankhead P, Kern KP, Sadeak I, Meng Q, et al. Human pancreatic carcinoma-associated fibroblasts promote expression of Co-inhibitory markers on CD4+ and CD8+ T-cells. Front Immunol (2019) 10:847. doi: 10.3389/fimmu.2019.00847
298. Li T, Liu T, Zhu W, Xie S, Zhao Z, Feng B, et al. Targeting MDSC for immune-checkpoint blockade in cancer immunotherapy: current progress and new prospects. Clin Med Insights Oncol (2021) 15:11795549211035540. doi: 10.1177/11795549211035540
299. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther (2021) 6(1):362. doi: 10.1038/s41392-021-00670-9
300. Kang C, Jeong SY, Song SY, Choi EK. The emerging role of myeloid-derived suppressor cells in radiotherapy. Radiat Oncol J (2020) 38(1):1–10. doi: 10.3857/roj.2019.00640
301. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. OncoImmunology (2017) 6:e1320011. doi: 10.1080/2162402x.2017.1320011
302. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer (2019) 120:16–25. doi: 10.1038/s41416-018-0333-1
303. De Cicco P, Ercolano G, Ianaro A. The new era of cancer immunotherapy: targeting myeloid-derived suppressor cells to overcome immune evasion. Front Immunol (2020) 11:1680. doi: 10.3389/fimmu.2020.01680
304. Hofer F, Di Sario G, Musiu C, Sartoris S, De Sanctis F, Ugel S. A complex metabolic network confers immunosuppressive functions to myeloid-derived suppressor cells (MDSCs) within the tumour microenvironment. Cells (2021) 10(10):2700. doi: 10.3390/cells10102700
305. Li J, Bolyard C, Xin G, Li Z. Targeting metabolic pathways of myeloid cells improves cancer immunotherapy. Front Cell Dev Biol (2021) 9:747863. doi: 10.3389/fcell.2021.747863
306. Hu C, Pang B, Lin G, Zhen Y, Yi H. Energy metabolism manipulates the fate and function of tumour myeloid-derived suppressor cells. Br J Cancer (2020) 122(1):23–9. doi: 10.1038/s41416-019-0644-x
307. Fleet JC, Burcham GN, Calvert RD, Elzey BD, Ratliff TL. 1α, 25 dihydroxyvitamin d (1,25(OH)2D) inhibits the T cell suppressive function of myeloid derived suppressor cells (MDSC). J Steroid Biochem Mol Biol (2020) 198:105557. doi: 10.1016/j.jsbmb.2019.105557
308. Lathers DMR, Clark JI, Achille NJ, Young MRI. Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamin D3. Cancer Immunol Immunother (2004) 53(5):422–30. doi: 10.1007/s00262-003-0459-7
309. Ni X, Hu G, Cai X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit Rev Food Sci Nutr (2019) 59(sup1):S71–80. doi: 10.1080/10408398.2018.1509201
310. Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res (2006) 66(18):9299–307. doi: 10.1158/0008-5472.CAN-06-1690
311. Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with ipilimumab. Int Immunopharmacol (2018) 63:282–91. doi: 10.1016/j.intimp.2018.08.007
312. Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother (2013) 62(5):909–18. doi: 10.1007/s00262-013-1396-8
313. Welters MJ, van der Sluis TC, van Meir H, Loof NM, van Ham VJ, van Duikeren S, et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci Trans Med (2016) 8:334ra52. doi: 10.1126/scitranslmed.aad8307
314. Fournier E, Duployez N, Ducourneau B, Raffoux E, Turlure P, Caillot D, et al. Mutational profile and benefit of gemtuzumab ozogamicin in acute myeloid leukemia. Blood (2020) 135(8):542–6. doi: 10.1182/blood.2019003471
315. Reilley MJ, McCoon P, Cook C, Lyne P, Kurzrock R, Kim Y, et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial. J ImmunoTher Cancer (2018) 6:119. doi: 10.1186/s40425-018-0436-5
316. Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget (2017) 8(2):3649–65. doi: 10.18632/oncotarget.12278
317. Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine (2016) 6:50–8. doi: 10.1016/j.ebiom.2016.02.024
318. Li W, Zhang X, Chen Y, Xie Y, Liu J, Feng Q, et al. G-CSF is a key modulator of MDSC and could be a potential therapeutic target in colitis-associated colorectal cancers. Protein Cell (2016) 7(2):130–40. doi: 10.1007/s13238-015-0237-2
319. Ma N, Liu Q, Hou L, Wang Y, Liu Z. MDSCs are involved in the protumorigenic potentials of GM-CSF in colitis-associated cancer. Int J Immunopathol Pharmacol (2017) 30(2):152–62. doi: 10.1177/0394632017711055
320. Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood (2010) 115(7):1461–71. doi: 10.1182/blood-2009-08-237412
321. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res (2014) 74(18):5057–69. doi: 10.1158/0008-5472.CAN-13-3723
322. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J ImmunoTher Cancer (2017) 5:53. doi: 10.1186/s40425-017-0257-y
323. Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells (2020) 9(3):561. doi: 10.3390/cells9030561
324. Masuda T, Noda M, Kogawa T, Kitagawa D, Hayashi N, Jomori T, et al. Phase I dose-escalation trial to repurpose propagermanium, an oral CCL2 inhibitor, in patients with breast cancer. Cancer Sci (2020) 111(3):924–31. doi: 10.1111/cas.14306
325. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs (2013) 31(3):760–8. doi: 10.1007/s10637-012-9869-8
326. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol (2016) 17(5):651–62. doi: 10.1016/S1470-2045(16)00078-4
327. Noel MS, Hezel AF, Linehan D, Wang-Gillam A, Eskens F, Sleijfer S, et al. Orally administered CCR2 selective inhibitor CCX872-b clinical trial in pancreatic cancer. J Clin Oncol (2017) 35:276–6. doi: 10.1200/jco.2017.35.4_suppl.276
328. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature (2014) 515(7525):130–3. doi: 10.1038/nature13862
329. Wang J, Hu W, Wang K, Yu J, Luo B, Luo G, et al. Repertaxin, an inhibitor of the chemokine receptors CXCR1 and CXCR2, inhibits malignant behavior of human gastric cancer MKN45 cells in vitro and in vivo and enhances efficacy of 5-fluorouracil. Int J Oncol (2016) 48(4):1341–52. doi: 10.3892/ijo.2016.3371
330. Che J, Song R, Chen B, Dong X. Targeting CXCR1/2: the medicinal potential as cancer immunotherapy agents, antagonists research highlights and challenges ahead. Eur J Med Chem (2020) 185:111853. doi: 10.1016/j.ejmech.2019.111853
331. Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight (2019) 4(7):e126853. doi: 10.1172/jci.insight.126853
332. Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-cell immunotherapy in head and neck cancer models. Clin Cancer Res (2020) 26(6):1420–31. doi: 10.1158/1078-0432.CCR-19-2625
333. Rivera LB, Bergers G. Intertwined regulation of angiogenesis and immunity by myeloid cells. Trends Immunol (2015) 36(4):240–9. doi: 10.1016/j.it.2015.02.005
334. Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin Cancer Res (2017) 23(2):587–99. doi: 10.1158/1078-0432.CCR-16-0387
335. Koinis F, Vetsika EK, Aggouraki D, Skalidaki E, Koutoulaki A, Gkioulmpasani M, et al. Effect of first-line treatment on myeloid-derived suppressor cells’ subpopulations in the peripheral blood of patients with non-small cell lung cancer. J Thorac Oncol (2016) 11(8):1263–72. doi: 10.1016/j.jtho.2016.04.026
336. Peereboom DM, Alban TJ, Grabowski MM, Alvarado AG, Otvos B, Bayik D, et al. Metronomic capecitabine as an immune modulator in glioblastoma patients reduces myeloid-derived suppressor cells. JCI Insight (2019) 4(22):e130748. doi: 10.1172/jci.insight.130748
337. Limagne E, Euvrard R, Thibaudin M, Rébé C, Derangère V, Chevriaux A, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX–bevacizumab drug treatment regimen. Cancer Res (2016) 76:5241–52. doi: 10.1158/0008-5472.can-15-3164
338. Chen HM, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, et al. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin Cancer Res (2015) 21(18):4073–85. doi: 10.1158/1078-0432.CCR-14-2742
339. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res (2009) 15(6):2148–57. doi: 10.1158/1078-0432.CCR-08-1332
340. Bauer R, Udonta F, Wroblewski M, Ben-Batalla I, Santos IM, Taverna F, et al. Blockade of myeloid-derived suppressor cell expansion with all- retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res (2018) 78(12):3220–32. doi: 10.1158/0008-5472.CAN-17-3415
341. Tcyganov E, Mastio J, Chen E, Gabrilovich DI. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol (2018) 51:76–82. doi: 10.1016/j.coi.2018.03.009
342. Bitsch R, Kurzay A, Özbay Kurt F, de la Torre C, Lasser S, Lepper A, et al. STAT3 inhibitor napabucasin abrogates MDSC immunosuppressive capacity and prolongs survival of melanoma-bearing mice. J Immunother Cancer (2022) 10(3):e004384. doi: 10.1136/jitc-2021-004384
343. Wong ALA, Hirpara JL, Pervaiz S, Eu JQ, Sethi G, Goh BC. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin Investig Drugs (2017) 26:883–7. doi: 10.1080/13543784.2017.1351941
344. Zhang X, Yan K, Deng L, Liang J, Liang H, Feng D, et al. Cyclooxygenase 2 promotes proliferation and invasion in ovarian cancer cells via the PGE2/NF-κB pathway. Cell Transplant (2019) 28(1_suppl):1S–13S. doi: 10.1177/0963689719890597
345. Wei J, Zhang J, Wang D, Cen B, Lang JD, DuBois RN. The COX-2–PGE2 pathway promotes tumor evasion in colorectal adenomas. Cancer Prev Res (2022) 15:285–96. doi: 10.1158/1940-6207.capr-21-0572
346. Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis (2009) 30:377–86. doi: 10.1093/carcin/bgp014
347. Eruslanov E, Daurkin I, Ortiz J, Vieweg J, Kusmartsev S. Pivotal advance: tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE2 catabolism in myeloid cells. J Leukoc Biol (2010) 88(5):839–48. doi: 10.1189/jlb.1209821
348. Veltman JD, Lambers MEH, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JGJV, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. celecoxib influences MDSC function. BMC Cancer (2010) 10:464. doi: 10.1186/1471-2407-10-464
349. Hou W, Sampath P, Rojas JJ, Thorne SH. Oncolytic virus-mediated targeting of PGE2 in the tumor alters the immune status and sensitizes established and resistant tumors to immunotherapy. Cancer Cell (2016) 30(1):108–19. doi: 10.1016/j.ccell.2016.05.012
350. Wang SJ, Khullar K, Kim S, Yegya-Raman N, Malhotra J, Groisberg R, et al. Effect of cyclo-oxygenase inhibitor use during checkpoint blockade immunotherapy in patients with metastatic melanoma and non-small cell lung cancer. J Immunother Cancer (2020) 8(2):e000889. doi: 10.1136/jitc-2020-000889
351. Li S, Jiang M, Wang L, Yu S. Combined chemotherapy with cyclooxygenase-2 (COX-2) inhibitors in treating human cancers: recent advancement. BioMed Pharmacother (2020) 129:110389. doi: 10.1016/j.biopha.2020.110389
352. Pu D, Yin L, Huang L, Qin C, Zhou Y, Wu Q, et al. Cyclooxygenase-2 inhibitor: a potential combination strategy with immunotherapy in cancer. Front Oncol (2021) 11:637504. doi: 10.3389/fonc.2021.637504
353. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci USA (2014) 111(32):11774–9. doi: 10.1073/pnas.1410626111
354. Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, et al. Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res (2018) 6(12):1561–77. doi: 10.1158/2326-6066.CIR-18-0070
355. Hashimoto A, Fukumoto T, Zhang R, Gabrilovich D. Selective targeting of different populations of myeloid-derived suppressor cells by histone deacetylase inhibitors. Cancer Immunol Immunother (2020) 69(9):1929–36. doi: 10.1007/s00262-020-02588-7
356. Lin S, Wang J, Wang L, Wen J, Guo Y, Qiao W, et al. Erratum: phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am J Cancer Res (2018) 8(12):2590.
357. Califano JA, Khan Z, Noonan KA, Rudraraju L, Zhang Z, Wang H, et al. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res (2015) 21(1):30–8. doi: 10.1158/1078-0432.CCR-14-1716
358. Hassel JC, Jiang H, Bender C, Winkler J, Sevko A, Shevchenko I, et al. Tadalafil has biologic activity in human melanoma. results of a pilot trial with tadalafil in patients with metastatic melanoma (TaMe). Oncoimmunology (2017) 6(9):e1326440. doi: 10.1080/2162402X.2017.1326440
359. Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol (2019) 40(4):310–27. doi: 10.1016/j.it.2019.02.003
360. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J BioMed Sci (2019) 26(1):78. doi: 10.1186/s12929-019-0568-z
361. Dallavalasa S, Beeraka NM, Basavaraju CG, Tulimilli SV, Sadhu SP, Rajesh K, et al. The role of tumor associated macrophages (TAMs) in cancer progression, chemoresistance, angiogenesis and metastasis - current status. Curr Med Chem (2021) 28(39):8203–36. doi: 10.2174/0929867328666210720143721
362. Wang H, Yung MMH, Ngan HYS, Chan KKL, Chan DW. The impact of the tumor microenvironment on macrophage polarization in cancer metastatic progression. Int J Mol Sci (2021) 22(12):6560. doi: 10.3390/ijms22126560
363. Gao J, Liang Y, Wang L. Shaping polarization of tumor-associated macrophages in cancer immunotherapy. Front Immunol (2022) 13:888713. doi: 10.3389/fimmu.2022.888713
364. Zhang L, Tian L, Dai X, Yu H, Wang J, Lei A, et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J Hematol Oncol (2020) 13(1):153. doi: 10.1186/s13045-020-00983-2
365. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Cancer Res (2021) 81(5):1201–8. doi: 10.1158/0008-5472.CAN-20-2990
366. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol (2020) 38(8):947–53. doi: 10.1038/s41587-020-0462-y
367. Wang S, Yang Y, Ma P, Zha Y, Zhang J, Lei A, et al. CAR-macrophage: an extensive immune enhancer to fight cancer. EBioMedicine (2022) 76:103873. doi: 10.1016/j.ebiom.2022.103873
368. Giatromanolaki A, Mitrakas A, Anestopoulos I, Kontosis A, Koukourakis IM, Pappa A, et al. Expression of CD47 and SIRPα macrophage immune-checkpoint pathway in non-Small-Cell lung cancer. Cancers (2022) 14(7):1801. doi: 10.3390/cancers14071801
369. Veillette A, Tang Z. Signaling regulatory protein (SIRP)α-CD47 blockade joins the ranks of immune checkpoint inhibition. J Clin Oncol (2019) 37(12):1012–4. doi: 10.1200/JCO.19.00121
370. Wang H, Sun Y, Zhou X, Chen C, Jiao L, Li W, et al. CD47/SIRPα blocking peptide identification and synergistic effect with irradiation for cancer immunotherapy. J Immunother Cancer (2020) 8(2):e000905. doi: 10.1136/jitc-2020-000905
371. Lin F, Xiong M, Hao W, Song Y, Liu R, Yang Y, et al. A novel blockade CD47 antibody with therapeutic potential for cancer. Front Oncol (2020) 10:615534. doi: 10.3389/fonc.2020.615534
372. Gu S, Ni T, Wang J, Liu Y, Fan Q, Wang Y, et al. CD47 blockade inhibits tumor progression through promoting phagocytosis of tumor cells by M2 polarized macrophages in endometrial cancer. J Immunol Res (2018) 2018:6156757. doi: 10.1155/2018/6156757
373. Junankar S, Shay G, Jurczyluk J, Ali N, Down J, Pocock N, et al. Real-time intravital imaging establishes tumor-associated macrophages as the extraskeletal target of bisphosphonate action in cancer. Cancer Discovery (2015) 5(1):35–42. doi: 10.1158/2159-8290.CD-14-0621
374. Rogers TL, Holen I. Tumour macrophages as potential targets of bisphosphonates. J Trans Med (2011) 9:177. doi: 10.1186/1479-5876-9-177
375. Anfray C, Ummarino A, Andón FT, Allavena P. Current strategies to target tumor-Associated-Macrophages to improve anti-tumor immune responses. Cells (2019) 9(1):46. doi: 10.3390/cells9010046
376. Zang X, Zhang X, Hu H, Qiao M, Zhao X, Deng Y, et al. Targeted delivery of zoledronate to tumor-associated macrophages for cancer immunotherapy. Mol Pharm (2019) 16(5):2249–58. doi: 10.1021/acs.molpharmaceut.9b00261
377. Lv J, Chen FK, Liu C, Liu PJ, Feng ZP, Jia L, et al. Zoledronic acid inhibits thyroid cancer stemness and metastasis by repressing M2-like tumor-associated macrophages induced wnt/β-catenin pathway. Life Sci (2020) 256:117925. doi: 10.1016/j.lfs.2020.117925
378. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol (2020) 877:173090. doi: 10.1016/j.ejphar.2020.173090
379. Liu Z, Xie Y, Xiong Y, Liu S, Qiu C, Zhu Z, et al. TLR 7/8 agonist reverses oxaliplatin resistance in colorectal cancer via directing the myeloid-derived suppressor cells to tumoricidal M1-macrophages. Cancer Lett (2020) 469:173–85. doi: 10.1016/j.canlet.2019.10.020
380. Molgora M, Colonna M. Turning enemies into allies–reprogramming tumor-associated macrophages for cancer therapy. Med (2021) 2:666–81. doi: 10.1016/j.medj.2021.05.001
381. Genard G, Lucas S, Michiels C. Reprogramming of tumor-associated macrophages with anticancer therapies: radiotherapy versus chemo- and immunotherapies. Front Immunol (2017) 8:828. doi: 10.3389/fimmu.2017.00828
382. Kapp K, Volz B, Oswald D, Wittig B, Baumann M, Schmidt M. Beneficial modulation of the tumor microenvironment and generation of anti-tumor responses by TLR9 agonist lefitolimod alone and in combination with checkpoint inhibitors. Oncoimmunology (2019) 8(12):e1659096. doi: 10.1080/2162402X.2019.1659096
383. Thomas M, Ponce-Aix S, Navarro A, Riera-Knorrenschild J, Schmidt M, Wiegert E, et al. Immunotherapeutic maintenance treatment with toll-like receptor 9 agonist lefitolimod in patients with extensive-stage small-cell lung cancer: results from the exploratory, controlled, randomized, international phase II IMPULSE study. Ann Oncol (2018) 29(10):2076–84. doi: 10.1093/annonc/mdy326
384. Belgiovine C, Digifico E, Anfray C, Ummarino A, Andón FT. Targeting tumor-associated macrophages in anti-cancer therapies: convincing the traitors to do the right thing. J Clin Med (2020) 9:3226. doi: 10.3390/jcm9103226
385. Wesolowski R, Sharma N, Reebel L, Rodal MB, Peck A, West BL, et al. Phase ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther Adv Med Oncol (2019) 11:1758835919854238. doi: 10.1177/1758835919854238
386. Tap WD, Gelderblom H, Palmerini E, Desai J, Bauer S, Blay JY, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet (2019) 394(10197):478–87. doi: 10.1016/S0140-6736(19)30764-0
387. Liu YQ, Wang YN, Lu XY, Tong LJ, Li Y, Zhang T, et al. Identification of compound D2923 as a novel anti-tumor agent targeting CSF1R. Acta Pharmacol Sin (2018) 39(11):1768–76. doi: 10.1038/s41401-018-0056-0
388. Ho WJ, Jaffee EM. Macrophage-targeting by CSF1/1R blockade in pancreatic cancers. Cancer Res (2021) 81(24):6071–3. doi: 10.1158/0008-5472.CAN-21-3603
389. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut (2017) 66(1):157–67. doi: 10.1136/gutjnl-2015-310514
390. Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc Natl Acad Sci USA (2020) 117(2):1129–38. doi: 10.1073/pnas.1910856117
391. Fujiwara T, Yakoub MA, Chandler A, Christ AB, Yang G, Ouerfelli O, et al. CSF1/CSF1R signaling inhibitor pexidartinib (PLX3397) reprograms tumor-associated macrophages and stimulates T-cell infiltration in the sarcoma microenvironment. Mol Cancer Ther (2021) 20(8):1388–99. doi: 10.1158/1535-7163.MCT-20-0591
392. Wang Q, Lu Y, Li R, Jiang Y, Zheng Y, Qian J, et al. Therapeutic effects of CSF1R-blocking antibodies in multiple myeloma. Leukemia (2018) 32(1):176–83. doi: 10.1038/leu.2017.193
393. Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer (2020) 19(1):41. doi: 10.1186/s12943-020-01165-x
394. Wu X, Singh R, Hsu DK, Zhou Y, Yu S, Han D, et al. A small molecule CCR2 antagonist depletes tumor macrophages and synergizes with anti–PD-1 in a murine model of cutaneous T-cell lymphoma (CTCL). J Invest Dermatol (2020) 140:1390–400.e4. doi: 10.1016/j.jid.2019.11.018
395. Li Z, Ding Y, Liu J, Wang J, Mo F, Wang Y, et al. Depletion of tumor associated macrophages enhances local and systemic platelet-mediated anti-PD-1 delivery for post-surgery tumor recurrence treatment. Nat Commun (2022) 13(1):1845. doi: 10.1038/s41467-022-29388-0
396. Li M, He L, Zhu J, Zhang P, Liang S. Targeting tumor-associated macrophages for cancer treatment. Cell Biosci (2022) 12:85. doi: 10.1186/s13578-022-00823-5
397. Datta M, Coussens LM, Nishikawa H, Hodi FS, Jain RK. Reprogramming the tumor microenvironment to improve immunotherapy: emerging strategies and combination therapies. Am Soc Clin Oncol Educ Book. (2019) 39:165–74. doi: 10.1200/EDBK_237987
398. Chamseddine AN, Assi T, Mir O, Chouaib S. Modulating tumor-associated macrophages to enhance the efficacy of immune checkpoint inhibitors: a TAM-pting approach. Pharmacol Ther (2022) 231:107986. doi: 10.1016/j.pharmthera.2021.107986
399. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther (2021) 6:75. doi: 10.1038/s41392-021-00484-9
400. Xin L, Gao J, Zheng Z, Chen Y, Lv S, Zhao Z, et al. Fibroblast activation protein-α as a target in the bench-to-Bedside diagnosis and treatment of tumors: a narrative review. Front Oncol (2021) 11:648187. doi: 10.3389/fonc.2021.648187
401. Santos AM, Jung J, Aziz N, Kissil JL, Puré E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest (2009) 119(12):3613–25. doi: 10.1172/JCI38988
402. Gunderson AJ, Yamazaki T, McCarty K, Phillips M, Alice A, Bambina S, et al. Blockade of fibroblast activation protein in combination with radiation treatment in murine models of pancreatic adenocarcinoma. PloS One (2019) 14(2):e0211117. doi: 10.1371/journal.pone.0211117
403. Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jäger D, Renner C, et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie (2003) 26(1):44–8. doi: 10.1159/000069863
404. Mohanty A, Pharaon RR, Nam A, Salgia S, Kulkarni P, Massarelli E. FAK-targeted and combination therapies for the treatment of cancer: an overview of phase I and II clinical trials. Expert Opin Investig Drugs (2020) 29(4):399–409. doi: 10.1080/13543784.2020.1740680
405. Kim S, You D, Jeong Y, Yoon SY, Kim SA, Lee JE. Inhibition of platelet-derived growth factor c and their receptors additionally increases doxorubicin effects in triple-negative breast cancer cells. Eur J Pharmacol (2021) 895:173868. doi: 10.1016/j.ejphar.2021.173868
406. Kommalapati A, Tella SH, Borad M, Javle M, Mahipal A. FGFR inhibitors in oncology: insight on the management of toxicities in clinical practice. Cancers (2021) 13(12):2968. doi: 10.3390/cancers13122968
407. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2015) 28(6):831–3. doi: 10.1016/j.ccell.2015.11.002
408. Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol (2021) 18(12):792–804. doi: 10.1038/s41571-021-00546-5
409. Brewer G, Fortier AM, Park M, Moraes C. The case for cancer-associated fibroblasts: essential elements in cancer drug discovery? Future Drug Discovery (2022) 4(1):FDD71. doi: 10.4155/fdd-2021-0004
410. Lefort S, Thuleau A, Kieffer Y, Sirven P, Bieche I, Marangoni E, et al. CXCR4 inhibitors could benefit to HER2 but not to triple-negative breast cancer patients. Oncogene (2017) 36:1211–22. doi: 10.1038/onc.2016.284
411. Teicher BA. TGFβ-directed therapeutics: 2020. Pharmacol Ther (2021) 217:107666. doi: 10.1016/j.pharmthera.2020.107666
412. Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci Transl Med (2018) 10(424):eaan5488. doi: 10.1126/scitranslmed.aan5488
413. Martin CJ, Datta A, Littlefield C, Kalra A, Chapron C, Wawersik S, et al. Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci Transl Med (2020) 12(536):eaay8456. doi: 10.1126/scitranslmed.aay8456
414. Collagen receptor implicated in immune exclusion. Cancer Discovery (2022) 12(1):6. doi: 10.1158/2159-8290.CD-NB2021-0397
415. Shi R, Zhang Z, Zhu A, Xiong X, Zhang J, Xu J, et al. Targeting type I collagen for cancer treatment. Int J Cancer (2022) 151(5):665–83. doi: 10.1002/ijc.33985
416. Pareti FI, Fujimura Y, Dent JA, Holland LZ, Zimmerman TS, Ruggeri ZM. Isolation and characterization of a collagen binding domain in human von willebrand factor. J Biol Chem (1986) 261(32):15310–5. doi: 10.1016/S0021-9258(18)66869-3
417. Wahyudi H, Reynolds AA, Li Y, Owen SC, Michael Yu S. Targeting collagen for diagnostic imaging and therapeutic delivery. J Controlled Release (2016) 240:323–31. doi: 10.1016/j.jconrel.2016.01.007
418. Liang H, Li X, Wang B, Chen B, Zhao Y, Sun J, et al. A collagen-binding EGFR antibody fragment targeting tumors with a collagen-rich extracellular matrix. Sci Rep (2016) 6:18205. doi: 10.1038/srep18205
419. Liang H, Li X, Chen B, Wang B, Zhao Y, Zhuang Y, et al. A collagen-binding EGFR single-chain fv antibody fragment for the targeted cancer therapy. J Control Release (2015) 209:101–9. doi: 10.1016/j.jconrel.2015.04.029
420. Ishihara J, Ishihara A, Sasaki K, Lee SSY, Williford JM, Yasui M, et al. Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci Transl Med (2019) 11(487):eaau3259. doi: 10.1126/scitranslmed.aau3259
421. Mansurov A, Ishihara J, Hosseinchi P, Potin L, Marchell TM, Ishihara A, et al. Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat BioMed Eng (2020) 4(5):531–43. doi: 10.1038/s41551-020-0549-2
422. Cho H, Jeon SI, Ahn CH, Shim MK, Kim K. Emerging albumin-binding anticancer drugs for tumor-targeted drug delivery: current understandings and clinical translation. Pharmaceutics (2022) 14(4):728. doi: 10.3390/pharmaceutics14040728
423. Sasaki K, Ishihara J, Ishihara A, Miura R, Mansurov A, Fukunaga K, et al. Engineered collagen-binding serum albumin as a drug conjugate carrier for cancer therapy. Sci Adv (2019) 5(8):eaaw6081. doi: 10.1126/sciadv.aaw6081
424. Liu J, Meng Z, Xu T, Kuerban K, Wang S, Zhang X, et al. A SIRPαFc fusion protein conjugated with the collagen-binding domain for targeted immunotherapy of non-small cell lung cancer. Front Immunol (2022) 13:845217. doi: 10.3389/fimmu.2022.845217
425. García-Olmo D, Villarejo Campos P, Barambio J, Garcia Gomez-Heras S, Vega-Clemente L, Olmedillas-Lopez S, et al. Intraperitoneal collagenase as a novel therapeutic approach in an experimental model of colorectal peritoneal carcinomatosis. Sci Rep (2021) 11:503. doi: 10.1038/s41598-020-79721-0
426. Dolor A, Szoka FC Jr. Digesting a path forward: the utility of collagenase tumor treatment for improved drug delivery. Mol Pharm (2018) 15(6):2069–83. doi: 10.1021/acs.molpharmaceut.8b00319
427. Pan A, Wang Z, Chen B, Dai W, Zhang H, He B, et al. Localized co-delivery of collagenase and trastuzumab by thermosensitive hydrogels for enhanced antitumor efficacy in human breast xenograft. Drug Deliv (2018) 25(1):1495–503. doi: 10.1080/10717544.2018.1474971
428. Dong X, Liu HJ, Feng HY, Yang SC, Liu XL, Lai X, et al. Enhanced drug delivery by nanoscale integration of a nitric oxide donor to induce tumor collagen depletion. Nano Lett (2019) 19(2):997–1008. doi: 10.1021/acs.nanolett.8b04236
429. Zhang W, Liu L, Su H, Liu Q, Shen J, Dai H, et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br J Cancer (2019) 121(10):837–45. doi: 10.1038/s41416-019-0578-3
430. Ebelt ND, Zamloot V, Zuniga E, Passi KB, Sobocinski LJ, Young CA, et al. Collagenase-expressing salmonella targets major collagens in pancreatic cancer leading to reductions in immunosuppressive subsets and tumor growth. Cancers (2021) 13:3565. doi: 10.3390/cancers13143565
431. Zhang W, Zhang C, Tian W, Qin J, Chen J, Zhang Q, et al. Efficacy of an oncolytic adenovirus driven by a chimeric promoter and armed with decorin against renal cell carcinoma. Hum Gene Ther (2020) 31(11-12):651–63. doi: 10.1089/hum.2019.352
432. Jung KH, Choi IK, Lee HS, Yan HH, Son MK, Ahn HM, et al. Oncolytic adenovirus expressing relaxin (YDC002) enhances therapeutic efficacy of gemcitabine against pancreatic cancer. Cancer Lett (2017) 396:155–66. doi: 10.1016/j.canlet.2017.03.009
433. Wan PKT, Ryan AJ, Seymour LW. Beyond cancer cells: targeting the tumor microenvironment with gene therapy and armed oncolytic virus. Mol Ther (2021) 29(5):1668–82. doi: 10.1016/j.ymthe.2021.04.015
434. Moaven O, Mangieri CW, Stauffer JA, Anastasiadis PZ, Borad MJ. Strategies to develop potent oncolytic viruses and enhance their therapeutic efficacy. JCO Precis Oncol (2021) 5:PO.21.00003. doi: 10.1200/PO.21.00003
435. Wang TH, Hsia SM, Shieh TM. Lysyl oxidase and the tumor microenvironment. Int J Mol Sci (2016) 18:62. doi: 10.3390/ijms18010062
436. Koorman T, Jansen KA, Khalil A, Haughton PD, Visser D, Rätze MAK, et al. Spatial collagen stiffening promotes collective breast cancer cell invasion by reinforcing extracellular matrix alignment. Oncogene (2022) 41(17):2458–69. doi: 10.1038/s41388-022-02258-1
437. Saatci O, Kaymak A, Raza U, Ersan PG, Akbulut O, Banister CE, et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat Commun (2020) 11(1):2416. doi: 10.1038/s41467-020-16199-4
438. Setargew YFI, Wyllie K, Grant RD, Chitty JL, Cox TR. Targeting lysyl oxidase family meditated matrix cross-linking as an anti-stromal therapy in solid tumours. Cancers (2021) 13(3):491. doi: 10.3390/cancers13030491
439. Elahi-Gedwillo KY, Carlson M, Zettervall J, Provenzano PP. Antifibrotic therapy disrupts stromal barriers and modulates the immune landscape in pancreatic ductal adenocarcinoma. Cancer Res (2019) 79(2):372–86. doi: 10.1158/0008-5472.CAN-18-1334
440. Kim JH, Shin BC, Park WS, Lee J, Kuh HJ. Antifibrotic effects of pentoxifylline improve the efficacy of gemcitabine in human pancreatic tumor xenografts. Cancer Sci (2017) 108(12):2470–7. doi: 10.1111/cas.13405
441. Fujiwara A, Funaki S, Fukui E, Kimura K, Kanou T, Ose N, et al. Effects of pirfenidone targeting the tumor microenvironment and tumor-stroma interaction as a novel treatment for non-small cell lung cancer. Sci Rep (2020) 10(1):10900. doi: 10.1038/s41598-020-67904-8
442. Diazzi S, Tartare-Deckert S, Deckert M. Bad neighborhood: fibrotic stroma as a new player in melanoma resistance to targeted therapies. Cancers (2020) 12(6):1364. doi: 10.3390/cancers12061364
443. Zhao M, Wang L, Wang M, Zhou S, Lu Y, Cui H, et al. Targeting fibrosis: mechanisms and clinical trials. Signal Transduct Target Ther (2022) 7:206. doi: 10.1038/s41392-022-01070-3
444. Sato N, Cheng XB, Kohi S, Koga A, Hirata K. Targeting hyaluronan for the treatment of pancreatic ductal adenocarcinoma. Acta Pharm Sin B (2016) 6:101–5. doi: 10.1016/j.apsb.2016.01.002
445. Benitez A, Yates TJ, Lopez LE, Cerwinka WH, Bakkar A, Lokeshwar VB. Targeting hyaluronidase for cancer therapy: antitumor activity of sulfated hyaluronic acid in prostate cancer cells. Cancer Res (2011) 71(12):4085–95. doi: 10.1158/0008-5472.CAN-10-4610
446. Shuster S, Frost GI, Csoka AB, Formby B, Stern R. Hyaluronidase reduces human breast cancer xenografts in SCID mice. Int J Cancer (2002) 102(2):192–7. doi: 10.1002/ijc.10668
447. Frost GI. Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert Opin Drug Deliv (2007) 4(4):427–40. doi: 10.1517/17425247.4.4.427
448. Bookbinder LH, Hofer A, Haller MF, Zepeda ML, Keller GA, Lim JE, et al. A recombinant human enzyme for enhanced interstitial transport of therapeutics. J Control Release (2006) 114(2):230–41. doi: 10.1016/j.jconrel.2006.05.027
449. Jackisch C, Stroyakovskiy D, Pivot X, Ahn JS, Melichar B, Chen SC, et al. Subcutaneous vs intravenous trastuzumab for patients with ERBB2-positive early breast cancer: final analysis of the HannaH phase 3 randomized clinical trial. JAMA Oncol (2019) 5(5):e190339. doi: 10.1001/jamaoncol.2019.0339
450. O’Shaughnessy J, Sousa S, Cruz J, Fallowfield L, Auvinen P, Pulido C, et al. Preference for the fixed-dose combination of pertuzumab and trastuzumab for subcutaneous injection in patients with HER2-positive early breast cancer (PHranceSCa): a randomised, open-label phase II study. Eur J Cancer (2021) 152:223–32. doi: 10.1016/j.ejca.2021.03.047
451. Morosi L, Meroni M, Ubezio P, Fuso Nerini I, Minoli L, Porcu L, et al. PEGylated recombinant human hyaluronidase (PEGPH20) pre-treatment improves intra-tumour distribution and efficacy of paclitaxel in preclinical models. J Exp Clin Cancer Res (2021) 40(1):286. doi: 10.1186/s13046-021-02070-x
452. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21(3):418–29. doi: 10.1016/j.ccr.2012.01.007
453. Van Cutsem E, Tempero MA, Sigal D, Oh DY, Fazio N, Macarulla T, et al. Randomized phase III trial of pegvorhyaluronidase Alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J Clin Oncol (2020) 38(27):3185–94. doi: 10.1200/JCO.20.00590
454. Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, et al. Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J Clin Oncol (2019) 37(13):1062–9. doi: 10.1200/JCO.18.01295
455. Hauser-Kawaguchi A, Luyt LG, Turley E. Design of peptide mimetics to block pro-inflammatory functions of HA fragments. Matrix Biol (2019) 78-79:346–56. doi: 10.1016/j.matbio.2018.01.021
456. Liu N, Xu XM, Chen J, Wang L, Yang S, Underhill CB, et al. Hyaluronan-binding peptide can inhibit tumor growth by interacting with bcl-2. Int J Cancer (2004) 109(1):49–57. doi: 10.1002/ijc.11636
457. Ikemoto H, Lingasamy P, Anton Willmore AM, Hunt H, Kurm K, Tammik O, et al. Hyaluronan-binding peptide for targeting peritoneal carcinomatosis. Tumour Biol (2017) 39(5):1010428317701628. doi: 10.1177/1010428317701628
458. Kakizaki I, Kojima K, Takagaki K, Endo M, Kannagi R, Ito M, et al. A novel mechanism for the inhibition of hyaluronan biosynthesis by 4-methylumbelliferone. J Biol Chem (2004) 279(32):33281–9. doi: 10.1074/jbc.M405918200
459. Vitale DL, Icardi A, Rosales P, Spinelli FM, Sevic I, Alaniz LD. Targeting the tumor extracellular matrix by the natural molecule 4-methylumbelliferone: a complementary and alternative cancer therapeutic strategy. Front Oncol (2021) 11:710061. doi: 10.3389/fonc.2021.710061
460. Urakawa H, Nishida Y, Knudson W, Knudson CB, Arai E, Kozawa E, et al. Therapeutic potential of hyaluronan oligosaccharides for bone metastasis of breast cancer. J Orthop Res (2012) 30(4):662–72. doi: 10.1002/jor.21557
461. Lokman NA, Price ZK, Hawkins EK, Macpherson AM, Oehler MK, Ricciardelli C. 4-methylumbelliferone inhibits cancer stem cell activation and overcomes chemoresistance in ovarian cancer. Cancers (2019) 11(8):1187. doi: 10.3390/cancers11081187
462. Bhattacharyya SS, Paul S, Mandal SK, Banerjee A, Boujedaini N, Khuda-Bukhsh AR. A synthetic coumarin (4-methyl-7 hydroxy coumarin) has anti-cancer potentials against DMBA-induced skin cancer in mice. Eur J Pharmacol (2009) 614(1-3):128–36. doi: 10.1016/j.ejphar.2009.04.015
463. Rosser JI, Nagy N, Goel R, Kaber G, Demirdjian S, Saxena J, et al. Oral hymecromone decreases hyaluronan in human study participants. J Clin Invest (2022) 132(9):e157983. doi: 10.1172/JCI157983
464. Karalis T, Shiau AK, Gahman TC, Skandalis SS, Heldin CH, Heldin P. Identification of a small molecule inhibitor of hyaluronan synthesis, DDIT, targeting breast cancer cells. Cancers (2022) 14(23):5800. doi: 10.3390/cancers14235800
465. Gao XP, Dong JJ, Xie T, Guan X. Integrative analysis of MUC4 to prognosis and immune infiltration in pan-cancer: friend or foe? Front Cell Dev Biol (2021) 9:695544. doi: 10.3389/fcell.2021.695544
466. Bruni S, Mauro FL, Mercogliano MF, Proietti CJ, Adami C, Dupont A, et al. Abstract 2047: MUC4 enables immune tumor evasion in HER2 breast cancer. Cancer Res (2022) 82:2047–7. doi: 10.1158/1538-7445.am2022-2047
467. Stoup N, Liberelle M, Schulz C, Cavdarli S, Vasseur R, Magnez R, et al. The EGF domains of MUC4 oncomucin mediate HER2 binding affinity and promote pancreatic cancer cell tumorigenesis. Cancers (2021) 13(22):5746. doi: 10.3390/cancers13225746
468. Gautam SK, Kumar S, Dam V, Ghersi D, Jain M, Batra SK. MUCIN-4 (MUC4) is a novel tumor antigen in pancreatic cancer immunotherapy. Semin Immunol (2020) 47:101391. doi: 10.1016/j.smim.2020.101391
469. Bae JS, Lee J, Park Y, Park K, Kim JR, Cho DH, et al. Attenuation of MUC4 potentiates the anticancer activity of auranofin via regulation of the Her2/Akt/FOXO3 pathway in ovarian cancer cells. Oncol Rep (2017) 38(4):2417–25. doi: 10.3892/or.2017.5853
Keywords: tumor microenvironment, immune exclusion, physical barrier, myeloid cells, extracellular matrix, tumor infiltrating lymphocytes (TILs), tumor-associated vasculature, tumor-associated macrophage (TAMs)
Citation: Bruni S, Mercogliano MF, Mauro FL, Cordo Russo RI and Schillaci R (2023) Cancer immune exclusion: breaking the barricade for a successful immunotherapy. Front. Oncol. 13:1135456. doi: 10.3389/fonc.2023.1135456
Received: 31 December 2022; Accepted: 10 May 2023;
Published: 22 May 2023.
Edited by:
Jing Hong Wang, University of Pittsburgh Medical Center, United StatesReviewed by:
Rebecca Kesselring, University of Freiburg Medical Center, GermanyHamid Morjani, Université de Reims Champagne-Ardenne, France
Copyright © 2023 Bruni, Mercogliano, Mauro, Cordo Russo and Schillaci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roxana Schillaci, cm94YW5hc2NoaWxsYWNpQGdtYWlsLmNvbQ==; cnNjaGlsbGFjaUBpYnltZS5jb25pY2V0Lmdvdi5hcg==