 Qingdan Gu
Qingdan Gu Jiazhen Zou2
Jiazhen Zou2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 20 March 2023
Sec. Molecular and Cellular Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1133013
This article is part of the Research TopicInflammation and Chronic DiseaseView all 13 articles
Inflammasomes, composed of the nucleotide-binding oligomerization domain(NOD)-like receptors (NLRs), are immune-functional protein multimers that are closely linked to the host defense mechanism. When NLRs sense pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), they assemble into inflammasomes. Inflammasomes can activate various inflammatory signaling pathways, including nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways, and produce a large number of proinflammatory cytokines, which are closely associated with multiple cancers. They can also accelerate the occurrence and development of cancer by providing suitable tumor microenvironments, promoting tumor cell proliferation, and inhibiting tumor cell apoptosis. Therefore, the exploitation of novel targeted drugs against various inflammasomes and proinflammatory cytokines is a new idea for the treatment of cancer. In recent years, more than 50 natural extracts and synthetic small molecule targeted drugs have been reported to be in the research stage or have been applied to the clinic. Herein, we will overview the mechanisms of inflammasomes in common cancers and discuss the therapeutic prospects of natural extracts and synthetic targeted agents.
Inflammasomes are a critical component of the innate immune system and play an essential role in defending against invasion by external xenobiotics. Unlike adaptive immunity, innate immunity is a nonspecific immunity in which immune cells have pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) from pathogenic microorganisms and damage-associated molecular patterns(DAMPs) from tissue damage to activate the immune response (1, 2). PRRs are distributed not only on the cell membrane but also in the cytoplasm. The membrane PRRs are composed of Toll-like receptors (TLRs) and C-type lectins (CTLs), while the cytoplasmic PRRs consist of the nucleotide-binding oligomerization domain(NOD)-like receptors (NLRs), retinoic acid inducible gene-I (RIG-I)-like receptors (RLRs) and absent-in-melanoma (AIM)-like receptors(ALRs) (3–5). NLRs and ALRs can form inflammasomes, of which, NLRs dominate. Upon recognition of ligands by NLRs, it combined with the apoptosis-associated speck-like protein (ASC) and pro-caspase-1 to form the inflammasome complex. This inflammasome cleaves inactive pro-caspase-1 into active caspase-1. Caspase-1 further promotes the secretion of the pro-inflammatory cytokines IL-1β and IL-18, which trigger inflammation by recruiting immune cells (e.g., macrophages) and inducing programmed cell death (e.g., pyroptosis) (6).
Inflammation is a defensive process of the body against injury and infection, but sustained activation of the inflammatory response may be associated with tumorigenesis and metastasis (7). The research indicated that long-term chronic inflammation is closely associated with growth inhibition resistance, angiogenesis, immune escape, malignant transformation, and metastatic potential acquisition (8). All of these processes are associated with the formation of inflammasomes. Mutations in genes encoding inflammasomes can affect the expression of nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways, which may ultimately contribute to the formation of cancer (9). The occurrence of tumors is related to the ability of cancer cells to capture inflammatory signaling pathways and promote their proliferation, migration and invasion (10). However, the exact molecular basis between inflammation and cancer remains unclear. Due to the different pathogenic mechanisms of inflammasomes in different cancers, inflammasomes and related signaling pathways have become a current research hotspot for molecularly targeted therapies. This review highlights the research progress of NLR inflammasomes in common cancers and outlines the potential of inflammasomes and key molecules in signaling pathways as therapeutic targets.
The NLR proteins consist of three distinct parts, the NACHT domain in the central region, flanked by leucine-rich repeats (LRRs) at the C-terminus, and an effector domain at the N-terminus(Figure 1). The NACHT domain has dNTPase activity and is involved in ATPase-dependent oligomerization (11, 12). LRRs can sense a variety of afferent signals. For example, NOD1 and NOD2 can detect bacterial peptidoglycan (13), NLRC4 and NAIP sense type III secretion system(T3SS) and flagellin (14), NLRP3 can be activated by sensing other molecules, such as ATP, reactive oxygen species (ROS), silica, uric acid crystals and nigericin (15), but the exact mechanisms by which it senses ligands are not yet clarified. The N-terminal effector domain is used to mediate the signal transduction of downstream targets, thereby activating caspases and various signaling pathways (16).
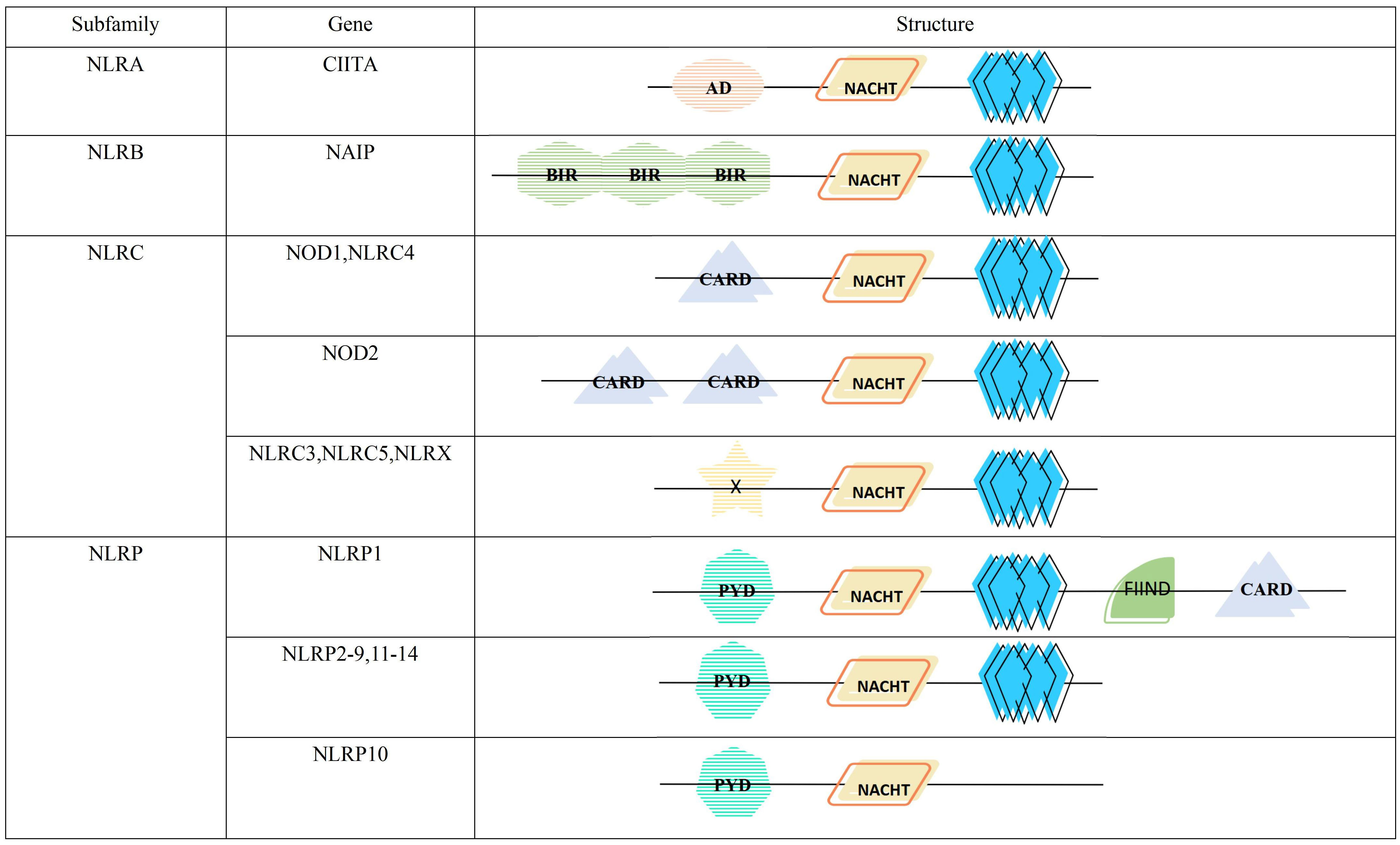
Figure 1 Structure and classification of the NLR family. AD, acidic transactivation domain; NACHT, NACHT domain (consisting of seven distinct con-served motifs, including the ATP/GTPase-specific P-loop, the Mg2+-binding site, and five more-specific motifs); BIR, baculovirus inhibitor of apoptosis repeat; CARD, caspase activation and recruitment domain; X, unidentified; PYD, pyrin domain; FIIND, function to find domain; leucine-rich repeat.
According to the different N-terminal effector domains, NLRs family is divided into 4 subfamilies, containing a total of 22 members (Figure 1). Among them, NLRA subfamily has only one member, the class II major histocompatibility complex transactivator (CIITA), containing the acidic transactivation domain (AD). It can enhance the transcription of MHC class II molecules and inhibits the classical NF-κB pathway (17). NLRB subfamily, characterized by a baculoviral inhibitory repeat-like domain (BIR), is also comprised of a single member, the NLR family apoptosis inhibitory protein(NAIP). It is responsible for preventing apoptosis mainly by inhibiting the activities of caspase-3, caspase-7 and caspase-9 (18). NOD1, NOD2 and NLRC4 with the caspase activation and recruitment domain (CARD), and NLRC3, NLRC5 and NLRX1 with unknown domains all belong to NLRC subfamily. The former can monitor microbial invasion and affect cytokines secretion by upregulating or downregulating inflammatory signaling, while the latter regulates autophagy and cell death by fine-tuning the cascade response of inflammatory signaling and the type I IFN signaling pathway (19). NLRP containing pyrin domain (PYD) has 14 members, namely NLRP1-14, which are mainly responsible for the modulation of inflammatory signaling and apoptosis (20).
Inflammasomes are a set of multi-protein complexes assembled with the participation of natural immune recognition receptors, which function as an inflammatory immune response by secreting inflammatory cytokines and inducing cellular pyroptosis (21). “The canonical inflammasome pathway” was defined when the inflammasome activation was mediated by caspase-1, while “The noncanonical inflammasome pathway” was classified when the inflammasome activation was dependent on human caspase-4/5 or mouse orthologue caspase-11 (22, 23). These two activation pathways and their roles are described in detail below.
Activation of the inflammasomes requires the involvement of “activators”. In the absence of activator stimulation such as DAMPs and PAMPs, LRRs interact with the NACHT domain and inhibit inflammasome formation (24). When LRRs detect DAMPs, PAMPs or environmental stimuli in microbes, the environment or the organism, NLR proteins start recruiting the adaptor protein ASC, which interacts through the Pyrin-Pyrin structural domain (25). Then the effector domain pro-caspase-1 binds to ASC through the CARD-CARD domain to assemble into an inflammasome, and finally, oligomerization to form an inflammasome complex possessing a heptamer (26). Since NLRP1 itself possesses a CARD domain, it can form inflammasomes directly through the interaction of the CARD-CARD domain with pro-caspase-1 without the need for ASC (27). Nonetheless, related studies have shown that ASC can enhance the stability of the CARD-CARD domain between NLRP1 and pro-caspase-1, thereby driving the immune response (28). Thus, ASC is an essential component of the inflammasomes complex assembly process, and when the body is deficient in ASC, it can lead to a variety of diseases (29, 30). NLRC4 does not contain a Pyrin structural domain, and it functions primarily by interacting with NAIP to form NAIP-NLRC4 inflammasomes. First, ligands such as flagellin or bacterial type III secretion system (T3SS) interact with non-activated NAIP, causing a conformational change and thus activation. Then ligands with activated NAIP further cause a conformational change in non-activated NLRC4, and activated NLRC4 can recruit more NLRC4 monomers to form NAIP-NLRC4 inflammasomes (31). Inflammasome assortment can further mediate the formation of active caspase-1 by self-hydrolysis of inactive pro-caspase-1, which can cleave pro-IL-1β and pro-IL-18 to form the mature inflammatory cytokines IL-1β and IL-18 (32). Inflammatory cytokines recruit immune cells, such as T lymphocytes and neutrophils, at the sites of infection and inflammation, thereby regulating the innate and adaptive immune response. They also initiate autocrine and paracrine inflammatory signaling cascades, providing survival signals to normal cells to prolong their lifespan and death signals to abnormal cells to accelerate their death (33). In addition, activated caspase-1 cleaves gasdermin D (GSDMD) and releases its N-terminal domain, which translocates to the cell membrane to form a pore that mediates the release of cellular contents such as IL-1β and IL-18, and induces pyroptosis (34) (Figure 2).
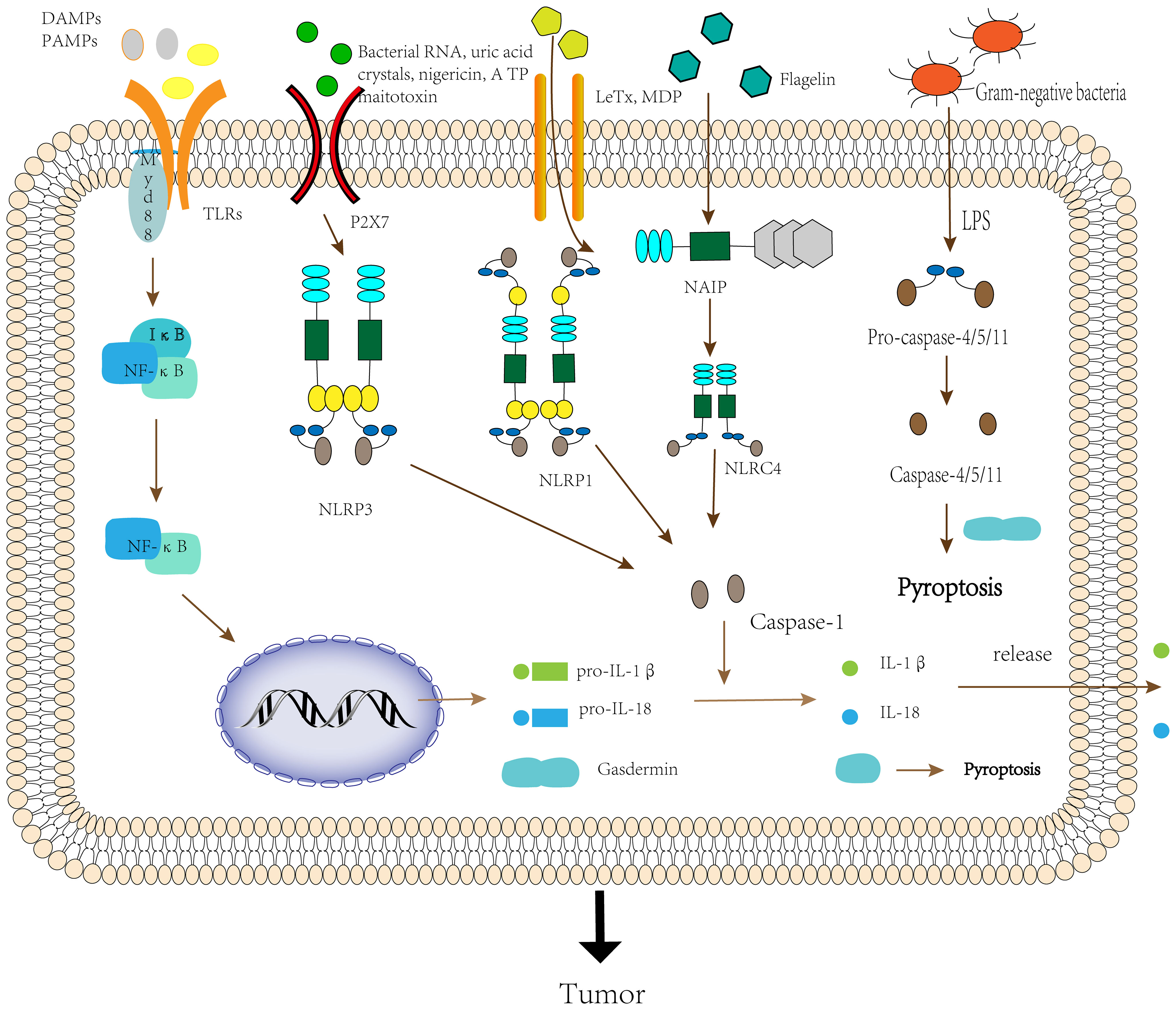
Figure 2 The activation process of the inflammasome.
The noncanonical inflammasome pathway is triggered by caspase-4/5/11. When TLR4 of the organism senses the lipopolysaccharide(LPS) of Gram-negative bacilli, it promotes the transcription of pro-IL-1β and pro-IL-18 by inducing the activation of NF-κB (35). In addition, when signaling to Trif/IRF3 induces the expression of type-I-IFNs, which promotes the secretion of pro-caspase-11. But it remains unknown whether the activation of caspase-11 is due to other additional signals or because pro-caspase-11 is automatically activated after reaching a certain threshold (36). Activated caspase-11 activates the NLRP3 inflammasome to induce caspase-1-dependent maturation and secretion of IL-1β and IL-18, but the activation pathway remains to be determined. In addition, caspase-11 can also cleave GSDMD to induce caspase-1-independent pyroptosis (21). Excessive pyroptosis can lead to a variety of cancer-related diseases, such as gastric cancer, colorectal cancer, and ovarian cancer (37) (Figure 2).
Many studies have demonstrated that abnormal activation of inflammasomes is closely associated with several human diseases, for instance, gout, silicosis, periodic fever syndrome, Crohn’s disease, type 1 diabetes, and so on (6, 38, 39). Currently, the increasing evidence shows that the inflammatory response mediated by NLR inflammasomes is involved in regulating physiological processes such as tumor development and metastasis. When the inflammasomes are abnormally activated, a variety of inflammatory cytokines and chemokines, including TNF-α, IL-1β, IL-6, IL-18, CCL2/MCP-1, are excessively secreted, which affect the development of cancer (40, 41). Among them, TNF-α plays a pro-tumor role by promoting DNA damage and inhibiting DNA repair (42). The expression of IL-1β and IL-18 is significantly elevated in a variety of malignancies, and these cytokines can promote cancer development and distant metastasis by triggering the secretion of VEGF, FGF2 and STAT3 (43, 44). IL-6 can promote the invasive ability of cancer cells, which leads to tumor metastasis. But there are some studies with contradictory findings, probably because inflammasomes not only promote tumor progression, but also correlate with apoptosis of tumor cells (45). The following will review the effects and mechanisms of inflammatory response mediated by NLR inflammasomes on common tumors including colorectal cancer, breast cancer, liver cancer and melanoma.
Colorectal cancer (CRC) is a malignant tumor occurring in intestinal epithelial cells, and the most common pathological carcinoma is adenocarcinoma. The overall incidence rate ranks in the top three among malignant tumors, and the mortality rate ranks the fourth, after lung, liver, and stomach cancers (46, 47). Reducing the prevalence and mortality of CRC has become an urgent issue to be solved. At present, surgical treatment, neoadjuvant chemoradiotherapy and adjuvant chemotherapy are the main clinical treatment methods. With the development of endoscopic technology and various auxiliary technologies, the 5-year survival rate of early-stage CRC can be as high as 92% (48, 49). However, there is still no effective treatment to reduce the prevalence. The search for new treatment methods and new therapeutic targets is currently a hot topic in academic circles.
Colorectal cancer model is the most studied cancer model, and a number of studies have shown that a variety of NLR inflammasomes are closely related to it, among which NLRC3, NLRC4, NOD2, NLRP1, NLRP6, NLRP12 inflammasomes have protective effects on colorectal cancer, while the effects of NLRP3 and NOD1 inflammasomes are still controversial (50). Rajendra’s team (51) found that NLRC3 inflammasomes modulated the multiplication and differentiation of stem cells and could promote apoptosis. What’s more, the upregulation of Nlrc3 was negatively correlated with the occurrence of tumors. In contrast, in Nlrc3 downregulated enterocytes, cell proliferation was not controlled due to the inability to inhibit the activation of the P13K-mTOR signaling axis, which may lead to tumorigenesis (52). Similarly, in a mouse model of colorectal cancer, the expression of Nlrc4 mRNA was diminished (50). Compared with wild-type mice, Nlrc4-/- mice could promote the proliferation of colonic epithelial cells and inhibit apoptosis of tumor cells, thereby promoting tumor formation, suggesting a protective effect of NLRC4 inflammasomes on colorectal carcinogenesis (53). In addition, Allam’s team (54) concluded that the higher incidence of colorectal cancer in Naip-/- mice may be related to the inability of Naip-/- mice to suppress the overactivation of STAT3, a transcription factor that promotes tumor growth. STAT3 hyperactivation was not observed in Nlrc4-/- mice, demonstrating that the protective effect of NAIP inflammasomes may be independent of NLRC4 inflammasomes. Couturier et al. (55) found an increased distal colon tumor load in Nod2-/- mice by comparison with wild-type mice. This phenomenon may be due to an imbalance of pro- and anti-inflammatory cytokines and a loss of autophagy and apoptosis, leading to chronic inflammation as well as an increased risk of cancer (56). Nashir (57) and his team concluded that NOD2 inflammasomes could inhibit CRC by blocking IRF4, thereby downregulating NF-κB and MAPK signaling pathways. NLRP1 and NLRP6 inflammasomes prevent CRC by mediating the production of IL-1β and IL-18 through caspase-1 (58–60). In addition, it has been shown that CCL5-induced inflammation can increase the number of tumors in Nlrp6-/- mice (61). The team of Zaki (62) found that NF-κB, ERK, and STAT3 signaling pathways were highly activated and that tumor incidence was increased in the colon of Nlrp12-/- mice. The ERK pathway activates the oncogenic transcription factor cMyc, which activates multiple oncogenic factors including COX. STAT3 regulates the pro-inflammatory cytokines IL-17 and IL-23, the anti-apoptotic protein Bcl-xL, and a variety of growth factors. Allen (11) also found that classical and non-classical NF-κB signaling pathways were activated and the body was highly susceptible to colorectal cancer in Nlrp12-/- mouse models. Among them, NLRP12 interacted with NIK in the non-classical NF-κB pathway and inhibited the production of p52, making this pathway dysregulated. Compared to the non-classical NF-κB pathway, the classical pathway has a relatively weak effect. In addition, NLRP12 inflammasomes also was able to regulate MAPK and AKT pathways, resulting in reduced tumorigenesis in mouse models (62). Stefanie’s group (63) found an interesting phenomenon in that NLRP5 inflammasomes expression was not detected in normal intestinal tissues, but it expressed in tissues with intestinal cancer, which indicated that NLRP5 inflammasomes expression was associated with tumorigenesis. However, the specific mechanism of action between NLRP5 inflammasomes and CRC needs to be further explored. The occurrence of CRC is probably related to immunological factors such as the activation of immune response and recruitment of immune cells. NLRP3 was found to be expressed in both immune cells and epithelial cells of colon cancer, manifesting that NLRP3 inflammasomes are involved in the formation of CRC (64). However, this aspect of research is still controversial: some studies have shown a positive correlation between NLRP3 inflammasomes and CRC, but a part of the research holds the opposite view, they suggest that NLRP3 inflammasomes may inhibit tumor development (65). Zaki et al. (66) found that NLRP3 inflammasomes lacking the adaptor proteins ASC and caspase-1 played a protective role in CRC compared to wild-type mice. Another study demonstrated that the use of NLRP3 inflammasomes small molecule inhibitors reduced the incidence of CRC (67). In addition, it has been reported that NOD1 inflammasomes deficiency caused damage to the intestinal epithelial barrier, increased apoptosis, and release of inflammatory cytokines in mouse models, leading to the development and progression of CRC (68, 69). Conversely, some other authors have argued that NOD1 is highly expressed in CRC mice and patients, and increased NOD1 expression would decrease the long-term survival of patients. NOD1 inflammasomes enhances tumor cell adhesion, migration and metastasis by activating MAPK signaling pathway (70). Tumorigenesis and metastasis require an appropriate microenvironment, and Charles et al. (71) suggested that NOD1 expression on myeloid cells formed a microenvironment conducive to tumor growth and may contribute to the development of CRC (Table 1).

Table 1 Effects of NLR inflammasomes in cancer.
Among female malignancies, breast cancer ranks first and has become one of the major public health problems in the world (89, 90). With the increasing improvement of breast cancer treatment methods, in addition to traditional surgery, adjuvant chemoradiotherapy and hormone therapy, targeted gene therapy and immunotherapy have also been used in clinical practice. Although the 5-year survival rate of patients has improved significantly, the mortality rate is still high (91, 92). It is of great clinical importance to find new therapeutic targets and develop new preventive and therapeutic measures to reduce the morbidity and mortality of breast cancer.
There have been many studies showing that abnormal expression of multiple NLR inflammasomes are closely connected with breast cancer. The NLRC4/IL-1β signaling pathway has been shown to promote the progression of breast cancer. In adipocytes, NLRC4 inflammasomes induce tumor infiltration into myeloid cells, and IL-1β promotes vascular endothelial growth factor A (VEGFA) secretion and angiogenesis, thereby driving disease progression (72). A study showed that NAIP was expressed in breast cancer and at significantly higher levels than in tumor control groups. In addition, NAIP was overexpressed in patients with poor outcomes and poor prognoses. These results suggest an aggravating role for NAIP inflammasomes in breast cancer (73). Nour (75) et al. proposed that the NLRP3/IL-1β pathway was strongly connected with the development and metastasis of breast cancer. Compared with normal breast tissue, the expression of Nlrp3, caspase-1 and IL-1β genes in the NLRP3 inflammatory pathway was significantly upregulated in mouse and human mammary tumor stroma. Among them, NLRP3 inflammasomes could mediate infiltration of CD11b+Gr1+ immune cells into mammary tumor tissues, and IL-1β could promote mammary tumor progression and tumor cell metastasis to the lung by upregulating adhesion molecules expression in primary tumors and metastatic sites. Guo et al. (76) also made similar findings that IL-1β secretion and tumor cell metastasis to the lung were reduced in caspase-1-/- and Nlrp3-/- mice compared to wild-type mice. What’s more, blocking IL-1R with inhibitors inhibited tumor growth and metastasis. The authors posited that this may be because activation of inflammasomes and production of IL-1β could recruit myeloid-derived suppressor cells (MDSC) and tumor-associated macrophages to tumor tissues, creating a favorable microenvironment for tumor metastasis. However, François et al. (74) held the opposite view, arguing that NLRP3 inflammasomes exerted anti-tumor functions by stimulating IL-1β release from dendritic cells. It has been proposed that NLRP1 inflammasomes promotes proliferation, migration and invasion of the breast cancer cell line MCF-7 compared to normal breast tissue. IL-1β, IL-18 and ASC, the pivotal elements of the inflammatory signaling pathway, are upregulated in breast cancer cells (77), but the specific cancer-promoting mechanism of NLRP1 inflammasomes needs to be further verified. Some investigators suggested that NOD1 and NOD2 inflammasomes may also be relevant to the negative regulation of breast cancer. In a study of estrogen receptor(ER)-negative Hs578T cell line, the investigators found that the proliferation rate of cells overexpressing NOD1 was reduced in vitro, and a similar phenomenon was seen in NOD2 overexpressed cells, even more significant than the former, which may be the result of activation of MAPK pathway signaling (93). Another in vitro cellular experiment showed that in the ER-positive MCF7 breast cancer cell line, NOD1 upregulation promoted RIP2 and caspase-8 mediated apoptosis and decreased estrogen-induced cell proliferation response (94). In addition, NOD1 downregulation enhanced the multiplication of breast cancer cells (80). Therefore, NOD1 inflammasomes was considered to be a tumor suppressor in ER-positive breast cancer cells (Table 1).
Liver cancer is the fifth most common cancer, with a mortality rate of 9.1% of all cancers. It is estimated that there were about 780,000 new cases of liver cancer and nearly 740,000 deaths in 2012 (78). Hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC) are the two main histological types of liver cancer, of which 70%-85% are HCC and 10% - 20% are ICC (79, 95). There are many causes of hepatocellular carcinomas, such as immune activation due to chronic inflammation, bacterial translocation of intestinal flora and damage to hepatocytes by endotoxins secreted by them, etc. (96, 97). Most researchers now believe that certain genetic mutations, upregulation or downregulation of gene expression due to chronic inflammation caused by hepatitis viruses, such as hepatitis B virus (HBV) or hepatitis C virus (HCV), may further contribute to the development of cancer, which is considered a potential risk factor for hepatocellular carcinoma (98). Significant genetic variants in NLRs were identified in a genomic sequence study, among which the gene mutations of NLRP3, NLRC5, NLRP12 and activation of their signaling pathways were widely noted (99, 100) (Table 1).
Raised levels of NLRP3 inflammasomes were observed in patients with chronic HBV infection, indicating that NLRP3 inflammasomes is an important intracellular receptor for HBV infection and that it may induce inflammatory responses by stimulating the secretion of IL-1β and IL-18 (101). In patients suffering HCV, the NLRP3 inflammatory signaling pathway has also been implicated as a factor stimulating elevated serum IL-1β levels (100). In a clinical study, NLRP3 expression was found to be upregulated in hepatitis and cirrhosis but downregulated in HCC (102). Another study found that human stanniocalcin-1 could reduce the volume of HCC tumor tissue by upregulating NLRP3 signaling pathway (103). This implies that NLRP3 inflammasomes are negatively associated with HCC. NLRC5 inflammasomes can promote HCC, it has been shown that knocking out Nlrp5 can suppress tumor growth and metastasis by targeting the Wnt/β-catenin signaling pathway (104). In addition, NLRP12 inflammasomes also influence the progression of HCC. A proto-oncogene cJun exists in hepatocytes, and JNK, a cJun N-terminal kinase, is prominently activated in hepatocellular carcinoma. NLRP12 inflammasomes can inhibit HCC by suppressing the JNK signaling pathway. Additionally, they can mediate NF-κB downregulation and ERK activation, which has negative regulatory effects on HCC (81). Hepatocyte-specific cytokines include chemokines such as CXCL1, CXCL2 and CCL2, which enhance inflammatory cell infiltration, thereby increasing inflammation in the tumor environment. It has been found that NLRP12 inflammasomes can induce the downregulation of hepatocyte-specific cytokines and exert an inhibitory effect on HCC (82) (Table 1).
Melanoma is one of the fastest growing cancers in the world and the most aggressive of all skin cancers, with an increasing incidence worldwide in recent years (83, 84). It is a malignant tumor derived from the malignant transformation of melanocytes and usually occurs in the skin and mucous membranes. Surgery is the radical treatment for most early melanoma, and the treatment of metastatic melanoma includes molecularly targeted therapy and immunosuppressive therapy. However, when it comes to patients who do not respond to immunotherapy, who do not have appropriately targeted drugs, who have relapsed, or who have failed to exhaust their available treatment options, their mortality rate is very high (105). Therefore, finding new therapeutic targets is of great importance to prolong the long-term survival of patients.
NLRP3 and NLRP1 inflammasomes polymorphisms were associated with susceptibility to melanoma in a Swedish case-control study (106). Zhai (107) mentioned in his paper that NLRP1 expression was upregulated in melanoma cells. Moreover, in Nlrp1-/- metastatic melanoma cells, decreased inflammatory cytokines secretion and NF-κB activity were observed, while increased caspase-2/-9 activity and promoted apoptosis. These phenomena indicated that NLRP1 inflammasomes promote melanoma growth. Similarly, NLRP3 inflammasomes promote melanoma growth by activating caspase-1 and producing IL-1β, which leads to suppression of anti-tumor immunity generated by NK cells and T cells (9, 108). The role of NLRC4 inflammasomes in melanoma is controversial. A study showed that subcutaneous injection of B16F10 melanoma in Nlrc4-/- mice accelerated tumor growth (85). The authors also observed that the production of inflammatory factors, chemokines and IFN-γ was reduced in the Nlrc4-/- model, which could be responsible for the accelerated growth of the tumors. Interestingly, another study used the same mouse model, but there was no discrepancy in neoplasm incidence between wild-type and Nlrc4-/- mice from the same litter (86). This may be due to the fact that littermate mice largely eliminate the effects of confounding factors such as gut microbiota and genetic differences (53) (Table 1).
The excessive activation of the inflammasome has been shown to be directly related to a variety of autoimmune diseases and cancers. Therefore, the development of targeted therapeutic drugs targeting the inflammasomes and key molecules in their signaling pathways is very promising. More than 50 therapeutic agents targeting inflammasomes and key molecules in their signaling pathways are reported to be still in development or already on the market (87). Most of them are inhibitors of NLRP3 and IL-1. Currently, there are two main classes of drugs targeting inflammasomes, namely natural extracts and synthetic small molecule agents.
Sulforaphane, a compound extracted from cruciferous vegetables, exerts biological effects by inhibiting the activation of NLRC4 and NLRP3 inflammasomes and the secretion of IL-1β (88). Due to its anti-inflammatory and anti-tumor properties, it is currently in clinical trials for the treatment of prostate cancer (109) and breast cancer (88). It has been shown that in 20 patients of recurrent prostate cancer treated with sulforaphane, the time to Prostate-specific antigen (PSA) doubling was longer than that before treatment (6.1 months before treatment vs. 9.6 months during treatment (p = 0.044) (109), indicating that the progression of the disease was slowed down. Andrographolide is a natural diterpene compound isolated from Andrographis paniculate. It inhibits the activation of inflammasomes mediated by NF-κB, TNF-α and mitochondrial autophagy, resulting in reduced secretion of IL-1β and IL-6, thereby delaying the progression of colon cancer and reducing the tumor load (67, 110). GL-V9 is a derivative of baicalein, which degrades NLRP3 inflammasomes by inducing autophagy, and can also delay colon cancer progression and tumorigenesis (111). Fumigaclavine C is a natural toxin derived from the marine-derived Aspergillus fumigatus. Guo (112) et al. suggested that it could reduce the occurrence of colitis and delay the progression of colon cancer in mice by inhibiting the NLRP3 signaling pathway and downregulating TNF-α, IL-1β and IL-17A. Li et al. (113) mentioned that Fumigaclavine C could significantly downregulate the expression of NF-κB, inhibit the MAPK signaling pathway and induce caspases-3, -8 and -9-mediated apoptosis to inhibit breast cancer development, thus it may be used as a targeted therapy for breast cancer. In addition, Caffeic acid phenethylester, a natural component isolated from propolis, has been proposed as a potential therapeutic component. It further inhibits caspase-1 activation and IL-1β production by inhibiting NLRP3, thereby suppressing the development of inflammation (114). Besides the above drugs, many botanical drugs also target NLRP3 inflammasomes, such as Withaferin A (115) and Mangiferin (116). Beyond that, there are a lot of plant extracts that target other NLRs inflammasomes, such as apigenin, a flavonoid, that protects against colitis in mice by inhibiting the NLRP6 signaling pathway, which is currently in clinical trials for CRC and breast cancer (117) (Table 2).
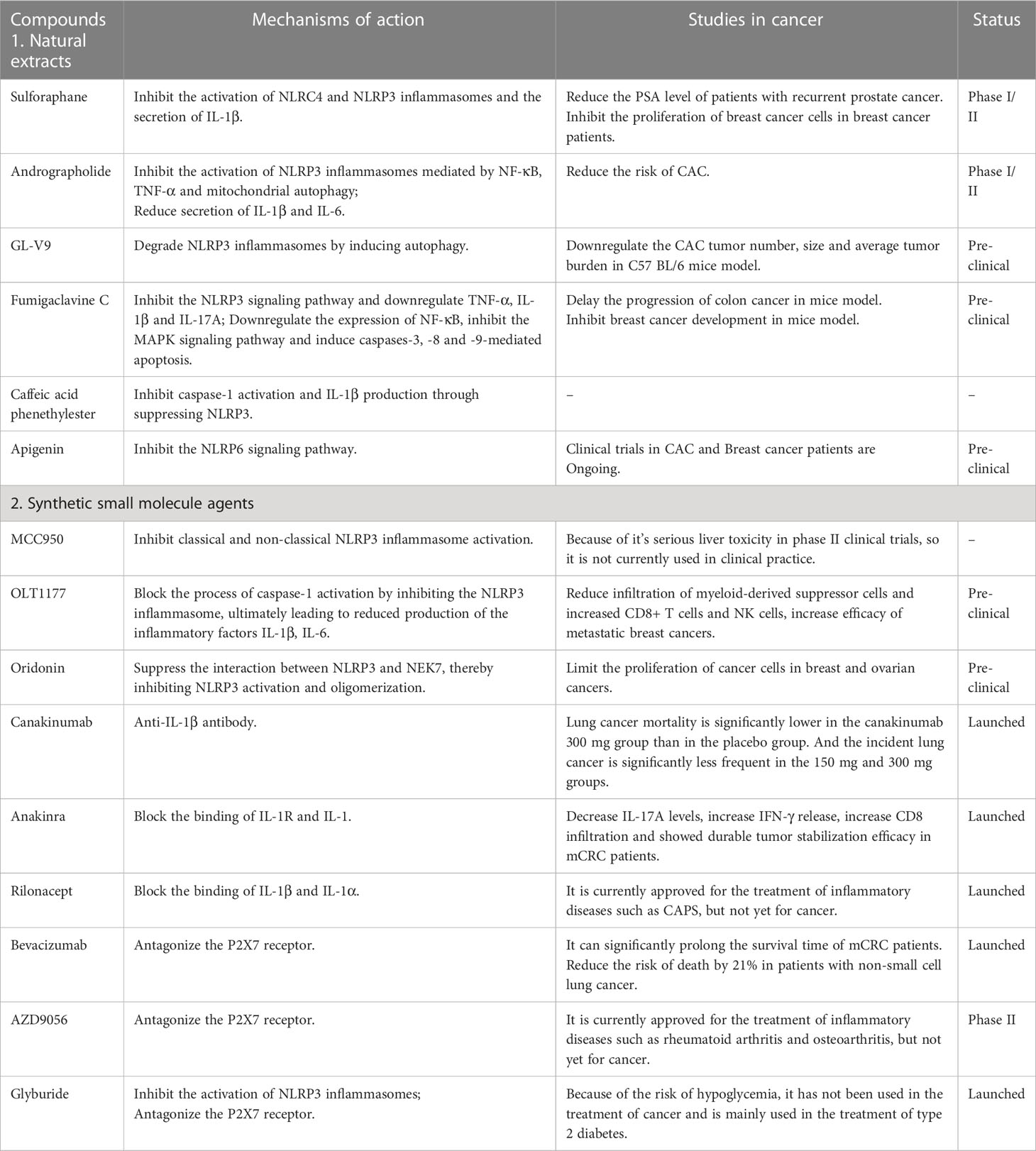
Table 2 The agents targeting inflammasomes for cancer treatment.
In recent years, several small molecules targeted agents have been identified that act directly on inflammasomes with the advantages of greater specificity, lower cost and less invasiveness, and have been of increasing interest to researchers. Here, several NLRP3 inflammasomes inhibitors are introduced. MCC950, was found to specifically inhibit classical and non-classical NLRP3 inflammasome activation pathways in macrophages in vitro (118). However, it has been found to have serious liver toxicity in phase II clinical trials, so it is not currently used in clinical practice (119). OLT1177, another NLRP3 inflammasome inhibitor based on MCC950, not only avoids the occurrence of liver injury mentioned above, but also can treat inflammation-related diseases by inhibiting NLRP3 inflammasome activation (120). It has been shown that patients with Cryopyrin-associated periodic syndromes (CAPS) treated with high concentrations of OLT1177 for 8 consecutive days have no liver injury and a significant reduction in blood inflammatory factors (120). OLT1177 not only plays a role in the treatment of inflammation-related diseases, but also has a promising future in the field of tumor. Inhibition of the NLRP3 inflammasomes blocked the process of caspase-1 activation, ultimately leading to reduced production of the inflammatory factors IL-1β, IL-6. This process has been shown to be related to the reduction of tumor cell proliferation (121). Oridonin is a small molecule derived from Rabdosia rubescens, and current studies have revealed that it can suppress the interaction between NLRP3 and NEK7, thereby inhibiting NLRP3 inflammasomes activation and oligomerization, but it does not affect AIM2 and NLRC4 (122). It has been shown that Oridonin can limit the proliferation of cancer cells in breast and ovarian cancers. Besides, Bay 11-7082 and Tranilast are also currently developed specific NLRP3 inflammasomes targeting inhibitors (123, 124) (Table 2).
Currently, three drugs, canakinumab, anakinra and rilonacept, are approved by the US Food and Drug Administration as targeted therapy for IL-1 (125). Among them, canakinumab is an IL-1β antibody, which has been reported to significantly reduce lethality in lung cancer patients (126). In addition, clinical efforts are also underway to study the efficacy of canakinumab in other cancers, such as non-small cell lung cancer (NSCLC) and triple-negative breast cancer (TNBC) (127). Anakinra is a recombinant IL-1 receptor antagonist that exerts biological effects by directly blocking the binding of IL-1R and IL-1. At present, it has been used to treat a variety of diseases such as myeloma and mCRC (128, 129). Furthermore, various clinical trials have proven that its use in combination with other chemotherapeutic agents can significantly improve patient prognosis and increase survival rates (130). Moreover, rilonacept is a blocking receptor that binds both IL-1β and IL-1α, which is currently approved for the treatment of CAPS (131). Given the preliminary therapeutic effects of these approved biologics on NLRP3-related inflammatory diseases, clinical studies and drug development of the aforementioned IL-1 blockers are steadily progressing (Table 2).
P2X7 receptor plays an important role in the NLRP3/caspase-1 cascade, which can promote tumor proliferation, migration and invasion (132). Bevacizumab, a P2X7 receptor antagonist, has an inhibitory effect on tumor growth (133). As early as 2004, it has been approved by the United States for the treatment of metastatic CRC(mCRC). At present, it has been approved for a variety of solid tumors, such as non-small cell lung cancer, renal cell carcinoma, and glioblastoma. Studies have shown that compared with chemotherapy agents (irinotecan, fluorouracil and leucovorin) alone, Patients with mCRC treated with bevacizumab combined with chemotherapy had significantly longer survival time (6.2 months vs10.6 months; p<0.001) (134). Another study showed that the combination of bevacizumab and carboplatin plus paclitaxel in patients with non-small cell lung cancer reduced the risk of death by 21%, as compared with chemotherapy alone (135). What’s more, AZD9056 and Glyburide are also the P2X7 receptor antagonist. The former is used to treat autoimmune diseases such as rheumatoid arthritis and osteoarthritis, and the latter is used to treat type 2 diabetes (136) (Table 2).
This review focuses on the biological effect of inflammasomes and the targeted therapeutic agents that aim at inflammatory signaling pathways in various cancers. In recent years, we have gained a deeper insight into the role of NLR inflammasomes, which go far beyond the simple recognition of and fight against pathogens outside the organism, and have demonstrated that NLR inflammasomes-mediated inflammatory signaling pathways are closely associated with all stages of cancer development and metastasis. Sustained activation of inflammasomes can promote the secretion of inflammatory cytokines, leading to increased immune cell infiltration, thereby changing the tumor microenvironment. In addition, inflammasomes can inhibit T-cell and NK cell-mediated antitumor activity, all of which can accelerate cancer progression. It has been found that inflammasomes also have vital roles in inhibiting tumorigenesis development, which provides a theoretical basis for the development of novel anticancer drugs.
Since the intricate relationship between inflammasomes and various types of cancer is difficult to elucidate, the mechanism of inflammasomes in cancer and whether they can be potential therapeutic targets for cancer have attracted extensive attention. Currently, a variety of natural plants targeting key molecules in the NLR inflammasomes signaling pathway have been developed for the treatment of inflammatory diseases (115, 116, 137, 138). Nevertheless, their exact mechanisms in the activation of inflammasomes and their anti-cancer effects still need further investigation. What’s more, a number of synthetic inflammasomes antagonists and monoclonal antibodies are used to modulate inflammasomes activity, thereby inhibiting tumor development and metastasis (139, 140). One of them, targeting effector cytokines, proved to be less effective therapeutically. It has been found that this approach may affect other key functions of inflammatory cytokines, for example, patients on long-term IL-1β inhibitors may be more susceptible to infections (141). In addition, we need to be alert to the fact that inappropriate molecular therapy may not only lead to an increased susceptibility of the body to infectious and autoimmune diseases but also aggravate the disease by diminishing the body’s anti-tumor immune response.
In clinical practice, resistance to immunotherapy is considered to be a great challenge. In-depth exploration of the mechanism of inflammasomes and immunotherapy drugs may help to solve this mystery. Currently, immune checkpoint inhibitors (ICIs) have been identified as effective anti-cancer therapies, such as anti-PD-1, in which the role of inflammasomes cannot be ignored. In the study on diffuse large B-cell lymphoma (DLBCL), Lu et al. found that NLRP3 inflammasome activation and significantly elevated IL-18 levels in DLBCL tissues promoted CD8+ T cell apoptosis by up-regulating programmed death ligand-1 (PD-L1), thereby exerting immunosuppressive effects (142). This process, to some extent, inhibits the tumor suppressive effect of anti-PD-1 drugs. Another showed that inhibition of the NLRP3 inflammasome significantly enhanced the efficacy of anti-PD-1 drugs (143). Huseni et al. found in a clinical study that interleukin-6 (IL-6) production due to inflammasome activation was associated with anti-PD-L1 resistance and that, compared with Atezolizumab(anti-PD-L1) alone, combined blockade of PD-L1 and IL-6 receptor (IL6R) significantly reduced drug resistance (144). In addition, the activation of NLRC4 inflammasome can exert immunosuppressive effects by promoting the expression of PD-L1 (145). These studies demonstrate a correlation between NLR inflammasome and adaptive resistance to anti-PD-1 checkpoint inhibitor immunotherapy in the treatment of cancer. Therefore, targeted antagonists against inflammasomes have the potential to become a new immunotherapy strategy.
Although many achievements have been made in the study of inflammasomes, many challenges remain, for example: (1) The activators of multiple inflammasomes are still unknown, and the signaling mechanisms activated by different inflammasomes need to be further explored (146). (2) Different inflammasomes play different roles in the same tissue, and the same inflammasomes play very distinct roles in different tissues (147), making the exploration of mechanisms extremely complicated. (3) Studies on the relevance of inflammasomes to tumors are limited and mostly focused on mouse models. (4) Some diseases involve the regulation of multiple NLR inflammasomes, and whether the effect of the combination of these inflammasomes-targeted drugs is superior to the use of a single targeted drug, etc. These mysteries need more researchers to collaborate to unravel them one by one. Exploring the specificity of NLR inflammasomes in tissues or cells, thoroughly uncovering their biological roles and their pathogenic mechanisms in various stages of tumors, and finding possible therapeutic targets will contribute to the development of novel anti-cancer drugs.
QD, QG, JZ, and ZY make substantial contributions to the conception or design of the work. JZ, and ZY selected extracted relevant papers of this manuscript. QG wrote the manuscript. QD had primary responsibility for final content. All authors contributed to the article and approved the submitted version.
Guangdong basic and Applied Basic Research Foundation (2020A1515010179). Shenzhen Longhua District Science and Technology Bureau Project (2021010). Shenzhen Longhua District Science and Technology Innovation Special Fund Project (11501A20220923BE863B1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454(7203):428–35. doi: 10.1038/nature07201
2. Jeong E, Lee JY. Intrinsic and extrinsic regulation of innate immune receptors. Yonsei Med J (2011) 52(3):379–92. doi: 10.3349/ymj.2011.52.3.379
3. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
4. Huysamen C, Brown GD. The fungal pattern recognition receptor, dectin-1, and the associated cluster of c-type lectin-like receptors. FEMS Microbiol Lett (2009) 290(2):121–8. doi: 10.1111/j.1574-6968.2008.01418.x
5. Yoneyama M, Fujita T. Rig-I family rna helicases: Cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev (2007) 18(5-6):545–51. doi: 10.1016/j.cytogfr.2007.06.023
6. Kim YK, Shin JS, Nahm MH. Nod-like receptors in infection, immunity, and diseases. Yonsei Med J (2016) 57(1):5–14. doi: 10.3349/ymj.2016.57.1.5
7. Greten FR, Grivennikov SI. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity (2019) 51(1):27–41. doi: 10.1016/j.immuni.2019.06.025
8. Shalapour S, Karin M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J Clin Invest (2015) 125(9):3347–55. doi: 10.1172/JCI80007
9. Karki R, Man SM, Kanneganti TD. Inflammasomes and cancer. Cancer Immunol Res (2017) 5(2):94–9. doi: 10.1158/2326-6066.CIR-16-0269
10. Liu P, Lu Z, Liu L, Li R, Liang Z, Shen M, et al. Nod-like receptor signaling in inflammation-associated cancers: From functions to targeted therapies. Phytomedicine (2019) 64:152925. doi: 10.1016/j.phymed.2019.152925
11. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. Nlrp12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical nf-kappab signaling. Immunity (2012) 36(5):742–54. doi: 10.1016/j.immuni.2012.03.012
12. Danis J, Mellett M. Nod-like receptors in host defence and disease at the epidermal barrier. Int J Mol Sci (2021) 22(9):4677. doi: 10.3390/ijms22094677
13. Caruso R, Warner N, Inohara N, Nunez G. Nod1 and Nod2: Signaling, host defense, and inflammatory disease. Immunity (2014) 41(6):898–908. doi: 10.1016/j.immuni.2014.12.010
14. West TE, Myers ND, Chantratita N, Chierakul W, Limmathurotsakul D, Wuthiekanun V, et al. Nlrc4 and Tlr5 each contribute to host defense in respiratory melioidosis. PloS Negl Trop Dis (2014) 8(9):e3178. doi: 10.1371/journal.pntd.0003178
15. Broz P, Dixit VM. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
16. Oviedo-Boyso J, Bravo-Patino A, Baizabal-Aguirre VM. Collaborative action of toll-like and nod-like receptors as modulators of the inflammatory response to pathogenic bacteria. Mediators Inflammation (2014) 2014:432785. doi: 10.1155/2014/432785
17. Velloso FJ, Trombetta-Lima M, Anschau V, Sogayar MC, Correa RG. Nod-like receptors: Major players (and targets) in the interface between innate immunity and cancer. Biosci Rep (2019) 39(4). doi: 10.1042/BSR20181709
18. Hausmann A, Bock D, Geiser P, Berthold DL, Fattinger SA, Furter M, et al. Intestinal epithelial Naip/Nlrc4 restricts systemic dissemination of the adapted pathogen salmonella typhimurium due to site-specific bacterial pamp expression. Mucosal Immunol (2020) 13(3):530–44. doi: 10.1038/s41385-019-0247-0
19. Fekete T, Bencze D, Biro E, Benko S, Pazmandi K. Focusing on the cell type specific regulatory actions of Nlrx1. Int J Mol Sci (2021) 22(3):1316. doi: 10.3390/ijms22031316
20. Hayward JA, Mathur A, Ngo C, Man SM. Cytosolic recognition of microbes and pathogens: Inflammasomes in action. Microbiol Mol Biol Rev (2018) 82(4):e00015-18. doi: 10.1128/MMBR.00015-18
21. Ding J, Shao F. Snapshot: The noncanonical inflammasome. Cell (2017) 168(3):544–e1. doi: 10.1016/j.cell.2017.01.008
22. Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: A supramolecular assembly of asc dimers mediating inflammatory cell death Via caspase-1 activation. Cell Death Differ (2007) 14(9):1590–604. doi: 10.1038/sj.cdd.4402194
23. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
24. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. Nlrp3 inflammasome and its inhibitors: A review. Front Pharmacol (2015) 6:262. doi: 10.3389/fphar.2015.00262
25. Dick MS, Sborgi L, Ruhl S, Hiller S, Broz P. Asc filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun (2016) 7:11929. doi: 10.1038/ncomms11929
26. Wang L, Hauenstein AV. The Nlrp3 inflammasome: Mechanism of action, role in disease and therapies. Mol Aspects Med (2020) 76:100889. doi: 10.1016/j.mam.2020.100889
27. Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted Nalp1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell (2007) 25(5):713–24. doi: 10.1016/j.molcel.2007.01.032
28. Platnich JM, Muruve DA. Nod-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys (2019) 670:4–14. doi: 10.1016/j.abb.2019.02.008
29. de Torre-Minguela C, Mesa Del Castillo P, Pelegrin P. The Nlrp3 and pyrin inflammasomes: Implications in the pathophysiology of autoinflammatory diseases. Front Immunol (2017) 8:43. doi: 10.3389/fimmu.2017.00043
30. He Y, Hara H, Nunez G. Mechanism and regulation of Nlrp3 inflammasome activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
31. Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y, et al. Structural and biochemical basis for induced self-propagation of Nlrc4. Science (2015) 350(6259):399–404. doi: 10.1126/science.aac5489
32. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol (2012) 28:137–61. doi: 10.1146/annurev-cellbio-101011-155745
33. van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA. Inflammasome activation and il-1beta and il-18 processing during infection. Trends Immunol (2011) 32(3):110–6. doi: 10.1016/j.it.2011.01.003
34. Sharma BR, Kanneganti TD. Inflammasome signaling in colorectal cancer. Transl Res (2022) 252:45–52. doi: 10.1016/j.trsl.2022.09.002
35. Broz P, Monack DM. Noncanonical inflammasomes: Caspase-11 activation and effector mechanisms. PloS Pathog (2013) 9(2):e1003144. doi: 10.1371/journal.ppat.1003144
36. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 increases susceptibility to salmonella infection in the absence of caspase-1. Nature (2012) 490(7419):288–91. doi: 10.1038/nature11419
37. Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: A new frontier in cancer. BioMed Pharmacother (2020) 121:109595. doi: 10.1016/j.biopha.2019.109595
38. Lamkanfi M, Vande Walle L, Kanneganti TD. Deregulated inflammasome signaling in disease. Immunol Rev (2011) 243(1):163–73. doi: 10.1111/j.1600-065X.2011.01042.x
39. Hoffman HM, Brydges SD. Genetic and molecular basis of inflammasome-mediated disease. J Biol Chem (2011) 286(13):10889–96. doi: 10.1074/jbc.R110.135491
40. Hu Z, Chai J. Structural mechanisms in nlr inflammasome assembly and signaling. Curr Top Microbiol Immunol (2016) 397:23–42. doi: 10.1007/978-3-319-41171-2_2
41. Sauter KA, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1beta through activation of the Nlrp3 inflammasome. Cancer Biol Ther (2011) 11(12):1008–16. doi: 10.4161/cbt.11.12.15540
42. Cruceriu D, Baldasici O, Balacescu O, Berindan-Neagoe I. The dual role of tumor necrosis factor-alpha (Tnf-alpha) in breast cancer: Molecular insights and therapeutic approaches. Cell Oncol (Dordr) (2020) 43(1):1–18. doi: 10.1007/s13402-019-00489-1
43. Tas F, Tilgen Yasasever C, Karabulut S, Tastekin D, Duranyildiz D. Clinical significance of serum interleukin-18 (Il-18) levels in patients with gastric cancer. BioMed Pharmacother (2015) 70:19–23. doi: 10.1016/j.biopha.2014.12.040
44. Fabbi M, Carbotti G, Ferrini S. Context-dependent role of il-18 in cancer biology and counter-regulation by il-18bp. J Leukoc Biol (2015) 97(4):665–75. doi: 10.1189/jlb.5RU0714-360RR
45. Zhiyu W, Wang N, Wang Q, Peng C, Zhang J, Liu P, et al. The inflammasome: An emerging therapeutic oncotarget for cancer prevention. Oncotarget (2016) 7(31):50766–80. doi: 10.18632/oncotarget.9391
46. Uchino Y, Goto Y, Konishi Y, Tanabe K, Toda H, Wada M, et al. Colorectal cancer patients have four specific bacterial species in oral and gut microbiota in common-a metagenomic comparison with healthy subjects. Cancers (Basel) (2021) 13(13):3332. doi: 10.3390/cancers13133332
48. Fisher DA, Princic N, Miller-Wilson LA, Wilson K, Fendrick AM, Limburg P. Utilization of a colorectal cancer screening test among individuals with average risk. JAMA Netw Open (2021) 4(9):e2122269. doi: 10.1001/jamanetworkopen.2021.22269
49. McFerran E, O'Mahony JF, Fallis R, McVicar D, Zauber AG, Kee F. Evaluation of the effectiveness and cost-effectiveness of personalized surveillance after colorectal adenomatous polypectomy. Epidemiol Rev (2017) 39(1):148–60. doi: 10.1093/epirev/mxx002
50. Liu R, Truax AD, Chen L, Hu P, Li Z, Chen J, et al. Expression profile of innate immune receptors, nlrs and Aim2, in human colorectal cancer: Correlation with cancer stages and inflammasome components. Oncotarget (2015) 6(32):33456–69. doi: 10.18632/oncotarget.5587
51. Karki R, Malireddi RKS, Zhu Q, Kanneganti TD. Nlrc3 regulates cellular proliferation and apoptosis to attenuate the development of colorectal cancer. Cell Cycle (2017) 16(13):1243–51. doi: 10.1080/15384101.2017.1317414
52. Karki R, Man SM, Malireddi RKS, Kesavardhana S, Zhu Q, Burton AR, et al. Nlrc3 is an inhibitory sensor of Pi3k-mtor pathways in cancer. Nature (2016) 540(7634):583–7. doi: 10.1038/nature20597
53. Kay C, Wang R, Kirkby M, Man SM. Molecular mechanisms activating the naip-Nlrc4 inflammasome: Implications in infectious disease, autoinflammation, and cancer. Immunol Rev (2020) 297(1):67–82. doi: 10.1111/imr.12906
54. Allam R, Maillard MH, Tardivel A, Chennupati V, Bega H, Yu CW, et al. Epithelial naips protect against colonic tumorigenesis. J Exp Med (2015) 212(3):369–83. doi: 10.1084/jem.20140474
55. Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, et al. Nod2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest (2013) 123(2):700–11. doi: 10.1172/JCI62236
56. Branquinho D, Freire P, Sofia C. Nod2 mutations and colorectal cancer - where do we stand? World J Gastrointest Surg (2016) 8(4):284–93. doi: 10.4240/wjgs.v8.i4.284
57. Udden SMN, Peng L, Gan JL, Shelton JM, Malter JS, Hooper LV, et al. Nod2 suppresses colorectal tumorigenesis Via downregulation of the tlr pathways. Cell Rep (2017) 19(13):2756–70. doi: 10.1016/j.celrep.2017.05.084
58. Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, et al. Nod-like receptor pyrin domain-containing protein 6 (Nlrp6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U.S.A. (2011) 108(23):9601–6. doi: 10.1073/pnas.1100981108
59. Chen GY, Liu M, Wang F, Bertin J, Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol (2011) 186(12):7187–94. doi: 10.4049/jimmunol.1100412
60. Williams TM, Leeth RA, Rothschild DE, Coutermarsh-Ott SL, McDaniel DK, Simmons AE, et al. The Nlrp1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J Immunol (2015) 194(7):3369–80. doi: 10.4049/jimmunol.1402098
61. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. Nlrp6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145(5):745–57. doi: 10.1016/j.cell.2011.04.022
62. Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, et al. The nod-like receptor Nlrp12 attenuates colon inflammation and tumorigenesis. Cancer Cell (2011) 20(5):649–60. doi: 10.1016/j.ccr.2011.10.022
63. Huhn S, da Silva Filho MI, Sanmuganantham T, Pichulik T, Catalano C, Pardini B, et al. Coding variants in nod-like receptors: An association study on risk and survival of colorectal cancer. PloS One (2018) 13(6):e0199350. doi: 10.1371/journal.pone.0199350
64. Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, et al. Inflammasome components nalp 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem (2007) 55(5):443–52. doi: 10.1369/jhc.6A7101.2006
65. Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, et al. The Nlrp3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med (2010) 207(5):1045–56. doi: 10.1084/jem.20100050
66. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The Nlrp3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32(3):379–91. doi: 10.1016/j.immuni.2010.03.003
67. Guo W, Sun Y, Liu W, Wu X, Guo L, Cai P, et al. Small molecule-driven mitophagy-mediated Nlrp3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy (2014) 10(6):972–85. doi: 10.4161/auto.28374
68. Guo H, Gibson SA, Ting JPY. Gut microbiota, nlr proteins, and intestinal homeostasis. J Exp Med (2020) 217(10):e20181832. doi: 10.1084/jem.20181832
69. Chen GY, Shaw MH, Redondo G, Nunez G. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res (2008) 68(24):10060–7. doi: 10.1158/0008-5472.CAN-08-2061
70. Jiang HY, Najmeh S, Martel G, MacFadden-Murphy E, Farias R, Savage P, et al. Activation of the pattern recognition receptor Nod1 augments colon cancer metastasis. Protein Cell (2020) 11(3):187–201. doi: 10.1007/s13238-019-00687-5
71. Maisonneuve C, Tsang DKL, Foerster EG, Robert LM, Mukherjee T, Prescott D, et al. Nod1 promotes colorectal carcinogenesis by regulating the immunosuppressive functions of tumor-infiltrating myeloid cells. Cell Rep (2021) 34(4):108677. doi: 10.1016/j.celrep.2020.108677
72. Kolb R, Phan L, Borcherding N, Liu Y, Yuan F, Janowski AM, et al. Obesity-associated Nlrc4 inflammasome activation drives breast cancer progression. Nat Commun (2016) 7:13007. doi: 10.1038/ncomms13007
73. Choi J, Hwang YK, Choi YJ, Yoo KE, Kim JH, Nam SJ, et al. Neuronal apoptosis inhibitory protein is overexpressed in patients with unfavorable prognostic factors in breast cancer. J Korean Med Sci (2007) 22(Suppl):S17–23. doi: 10.3346/jkms.2007.22.S.S17
74. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the Nlrp3 inflammasome in dendritic cells induces il-1beta-Dependent adaptive immunity against tumors. Nat Med (2009) 15(10):1170–8. doi: 10.1038/nm.2028
75. Ershaid N, Sharon Y, Doron H, Raz Y, Shani O, Cohen N, et al. Nlrp3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat Commun (2019) 10(1):4375. doi: 10.1038/s41467-019-12370-8
76. Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting Inflammasome/Il-1 pathways for cancer immunotherapy. Sci Rep (2016) 6:36107. doi: 10.1038/srep36107
77. Wei Y, Huang H, Qiu Z, Li H, Tan J, Ren G, et al. Nlrp1 overexpression is correlated with the tumorigenesis and proliferation of human breast tumor. BioMed Res Int (2017) 2017:4938473. doi: 10.1155/2017/4938473
78. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
79. Massarweh NN, El-Serag HB. Epidemiology of hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Cancer Control (2017) 24(3):1073274817729245. doi: 10.1177/1073274817729245
80. da Silva Correia J, Miranda Y, Austin-Brown N, Hsu J, Mathison J, Xiang R, et al. Nod1-dependent control of tumor growth. Proc Natl Acad Sci U.S.A. (2006) 103(6):1840–5. doi: 10.1073/pnas.0509228103
81. Udden SN, Kwak YT, Godfrey V, Khan MAW, Khan S, Loof N, et al. Nlrp12 suppresses hepatocellular carcinoma via downregulation of cjun n-terminal kinase activation in the hepatocyte. Elife (2019) 8:e40396. doi: 10.7554/eLife.40396
82. Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, et al. Hepatocyte necrosis induced by oxidative stress and il-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell (2008) 14(2):156–65. doi: 10.1016/j.ccr.2008.06.016
83. Sasse AD, Sasse EC, Clark LG, Clark OAC. Withdrawn: Chemoimmunotherapy versus chemotherapy for metastatic malignant melanoma. Cochrane Database Syst Rev (2018) 2:CD005413. doi: 10.1002/14651858.CD005413.pub3
84. Donley GM, Liu WT, Pfeiffer RM, McDonald EC, Peters KO, Tucker MA, et al. Reproductive factors, exogenous hormone use and incidence of melanoma among women in the united states. Br J Cancer (2019) 120(7):754–60. doi: 10.1038/s41416-019-0411-z
85. Janowski AM, Colegio OR, Hornick EE, McNiff JM, Martin MD, Badovinac VP, et al. Nlrc4 suppresses melanoma tumor progression independently of inflammasome activation. J Clin Invest (2016) 126(10):3917–28. doi: 10.1172/JCI86953
86. Tenthorey JL, Chavez RA, Thompson TW, Deets KA, Vance RE, Rauch I. Nlrc4 inflammasome activation is Nlrp3- and phosphorylation-independent during infection and does not protect from melanoma. J Exp Med (2020) 217(7):e20191736. doi: 10.1084/jem.20191736
87. Baldwin AG, Brough D, Freeman S. Inhibiting the inflammasome: A chemical perspective. J Med Chem (2016) 59(5):1691–710. doi: 10.1021/acs.jmedchem.5b01091
88. Zhang Z, Atwell LL, Farris PE, Ho E, Shannon J. Associations between cruciferous vegetable intake and selected biomarkers among women scheduled for breast biopsies. Public Health Nutr (2016) 19(7):1288–95. doi: 10.1017/S136898001500244X
89. Zidi O, Souai N, Raies H, Ben Ayed F, Mezlini A, Mezrioui S, et al. Fecal metabolic profiling of breast cancer patients during neoadjuvant chemotherapy reveals potential biomarkers. Molecules (2021) 26(8):2266. doi: 10.3390/molecules26082266
90. Malvezzi M, Carioli G, Bertuccio P, Boffetta P, Levi F, La Vecchia C, et al. European Cancer mortality predictions for the year 2019 with focus on breast cancer. Ann Oncol (2019) 30(5):781–7. doi: 10.1093/annonc/mdz051
91. Austreid E, Lonning PE, Eikesdal HP. The emergence of targeted drugs in breast cancer to prevent resistance to endocrine treatment and chemotherapy. Expert Opin Pharmacother (2014) 15(5):681–700. doi: 10.1517/14656566.2014.885952
92. Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol (2019) 21(9):1113–26. doi: 10.1038/s41556-019-0373-7
93. Velloso FJ, Sogayar MC, Correa RG. Expression and in vitro assessment of tumorigenicity for Nod1 and Nod2 receptors in breast cancer cell lines. BMC Res Notes (2018) 11(1):222. doi: 10.1186/s13104-018-3335-4
94. da Silva Correia J, Miranda Y, Leonard N, Hsu J, Ulevitch RJ. Regulation of Nod1-mediated signaling pathways. Cell Death Differ (2007) 14(4):830–9. doi: 10.1038/sj.cdd.4402070
95. Sia D, Villanueva A, Friedman SL, Llovet JM. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology (2017) 152(4):745–61. doi: 10.1053/j.gastro.2016.11.048
96. Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and Tlr4. Cancer Cell (2012) 21(4):504–16. doi: 10.1016/j.ccr.2012.02.007
97. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature (2013) 499(7456):97–101. doi: 10.1038/nature12347
98. de Martel C, Maucort-Boulch D, Plummer M, Franceschi S. World-wide relative contribution of hepatitis b and c viruses in hepatocellular carcinoma. Hepatology (2015) 62(4):1190–200. doi: 10.1002/hep.27969
99. Everson GT, Terrault NA, Lok AS, Rodrigo del R, Brown RS Jr., Saab S, et al. A randomized controlled trial of pretransplant antiviral therapy to prevent recurrence of hepatitis c after liver transplantation. Hepatology (2013) 57(5):1752–62. doi: 10.1002/hep.25976
100. Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, et al. Il-1beta production through the Nlrp3 inflammasome by hepatic macrophages links hepatitis c virus infection with liver inflammation and disease. PloS Pathog (2013) 9(4):e1003330. doi: 10.1371/journal.ppat.1003330
101. Askari A, Nosratabadi R, Khaleghinia M, Zainodini N, Kennedy D, Shabani Z, et al. Evaluation of Nlrc4, Nlrp1, and Nlrp3, as components of inflammasomes, in chronic hepatitis b virus-infected patients. Viral Immunol (2016) 29(9):496–501. doi: 10.1089/vim.2016.0045
102. Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, et al. Deregulation of the Nlrp3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest (2014) 94(1):52–62. doi: 10.1038/labinvest.2013.126
103. Yeung BH, Shek FH, Lee NP, Wong CK. Stanniocalcin-1 reduces tumor size in human hepatocellular carcinoma. PloS One (2015) 10(10):e0139977. doi: 10.1371/journal.pone.0139977
104. Peng YY, He YH, Chen C, Xu T, Li L, Ni MM, et al. Nlrc5 regulates cell proliferation, migration and invasion in hepatocellular carcinoma by targeting the Wnt/Beta-catenin signaling pathway. Cancer Lett (2016) 376(1):10–21. doi: 10.1016/j.canlet.2016.03.006
105. Mukherjee N, Skees J, Todd KJ, West DA, Lambert KA, Robinson WA, et al. Mcl1 inhibitors S63845/Mik665 plus navitoclax synergistically kill difficult-to-Treat melanoma cells. Cell Death Dis (2020) 11(6):443. doi: 10.1038/s41419-020-2646-2
106. Verma D, Bivik C, Farahani E, Synnerstad I, Fredrikson M, Enerback C, et al. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res (2012) 25(4):506–13. doi: 10.1111/j.1755-148X.2012.01008.x
107. Zhai Z, Liu W, Kaur M, Luo Y, Domenico J, Samson JM, et al. Nlrp1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene (2017) 36(27):3820–30. doi: 10.1038/onc.2017.26
108. Okamoto M, Liu W, Luo Y, Tanaka A, Cai X, Norris DA, et al. Constitutively active inflammasome in human melanoma cells mediating autoinflammation Via caspase-1 processing and secretion of interleukin-1beta. J Biol Chem (2010) 285(9):6477–88. doi: 10.1074/jbc.M109.064907
109. Alumkal JJ, Slottke R, Schwartzman J, Cherala G, Munar M, Graff JN, et al. A phase ii study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Invest New Drugs (2015) 33(2):480–9. doi: 10.1007/s10637-014-0189-z
110. Bertoglio JC, Baumgartner M, Palma R, Ciampi E, Carcamo C, Caceres DD, et al. Andrographis paniculata decreases fatigue in patients with relapsing-remitting multiple sclerosis: A 12-month double-blind placebo-controlled pilot study. BMC Neurol (2016) 16:77. doi: 10.1186/s12883-016-0595-2
111. Zhao Y, Guo Q, Zhao K, Zhou Y, Li W, Pan C, et al. Small molecule gl-V9 protects against colitis-associated colorectal cancer by limiting Nlrp3 inflammasome through autophagy. Oncoimmunology (2017) 7(1):e1375640. doi: 10.1080/2162402X.2017.1375640
112. Guo W, Hu S, Elgehama A, Shao F, Ren R, Liu W, et al. Fumigaclavine c ameliorates dextran sulfate sodium-induced murine experimental colitis Via Nlrp3 inflammasome inhibition. J Pharmacol Sci (2015) 129(2):101–6. doi: 10.1016/j.jphs.2015.05.003
113. Li YX, Himaya SW, Dewapriya P, Zhang C, Kim SK. Fumigaclavine c from a marine-derived fungus aspergillus fumigatus induces apoptosis in mcf-7 breast cancer cells. Mar Drugs (2013) 11(12):5063–86. doi: 10.3390/md11125063
114. Lee HE, Yang G, Kim ND, Jeong S, Jung Y, Choi JY, et al. Targeting asc in Nlrp3 inflammasome by caffeic acid phenethyl ester: A novel strategy to treat acute gout. Sci Rep (2016) 6:38622. doi: 10.1038/srep38622
115. Kanak MA, Shahbazov R, Yoshimatsu G, Levy MF, Lawrence MC, Naziruddin B. A small molecule inhibitor of nfkappab blocks er stress and the Nlrp3 inflammasome and prevents progression of pancreatitis. J Gastroenterol (2017) 52(3):352–65. doi: 10.1007/s00535-016-1238-5
116. Qu S, Wang W, Li D, Li S, Zhang L, Fu Y, et al. Mangiferin inhibits mastitis induced by lps Via suppressing nf-kb and Nlrp3 signaling pathways. Int Immunopharmacol (2017) 43:85–90. doi: 10.1016/j.intimp.2016.11.036
117. Radulovic K, Normand S, Rehman A, Delanoye-Crespin A, Chatagnon J, Delacre M, et al. A dietary flavone confers communicable protection against colitis through Nlrp6 signaling independently of inflammasome activation. Mucosal Immunol (2018) 11(3):811–9. doi: 10.1038/mi.2017.87
118. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the Nlrp3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21(3):248–55. doi: 10.1038/nm.3806
119. Wu D, Chen Y, Sun Y, Gao Q, Li H, Yang Z, et al. Target of Mcc950 in inhibition of Nlrp3 inflammasome activation: A literature review. Inflammation (2020) 43(1):17–23. doi: 10.1007/s10753-019-01098-8
120. Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. Olt1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the Nlrp3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U.S.A. (2018) 115(7):E1530–E9. doi: 10.1073/pnas.1716095115
121. Tricot GJ. New insights into role of microenvironment in multiple myeloma. Int J Hematol (2002) 76(Suppl 1):334–6. doi: 10.1007/BF03165279
122. He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, et al. Oridonin is a covalent Nlrp3 inhibitor with strong anti-inflammasome activity. Nat Commun (2018) 9(1):2550. doi: 10.1038/s41467-018-04947-6
123. Irrera N, Vaccaro M, Bitto A, Pallio G, Pizzino G, Lentini M, et al. Bay 11-7082 inhibits the nf-kappab and Nlrp3 inflammasome pathways and protects against imq-induced psoriasis. Clin Sci (Lond) (2017) 131(6):487–98. doi: 10.1042/CS20160645
124. Darakhshan S, Pour AB. Tranilast: A review of its therapeutic applications. Pharmacol Res (2015) 91:15–28. doi: 10.1016/j.phrs.2014.10.009
125. Ozaki E, Campbell M, Doyle SL. Targeting the Nlrp3 inflammasome in chronic inflammatory diseases: Current perspectives. J Inflammation Res (2015) 8:15–27. doi: 10.2147/JIR.S51250
126. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet (2017) 390(10105):1833–42. doi: 10.1016/S0140-6736(17)32247-X
127. Hamarsheh S, Zeiser R. Nlrp3 inflammasome activation in cancer: A double-edged sword. Front Immunol (2020) 11:1444. doi: 10.3389/fimmu.2020.01444
128. Lee C, Do HTT, Her J, Kim Y, Seo D, Rhee I. Inflammasome as a promising therapeutic target for cancer. Life Sci (2019) 231:116593. doi: 10.1016/j.lfs.2019.116593
129. Isambert N, Hervieu A, Rebe C, Hennequin A, Borg C, Zanetta S, et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (Irafu): A single-arm phase 2 study. Oncoimmunology (2018) 7(9):e1474319. doi: 10.1080/2162402X.2018.1474319
130. Lust JA, Lacy MQ, Zeldenrust SR, Witzig TE, Moon-Tasson LL, Dinarello CA, et al. Reduction in c-reactive protein indicates successful targeting of the il-1/Il-6 axis resulting in improved survival in early stage multiple myeloma. Am J Hematol (2016) 91(6):571–4. doi: 10.1002/ajh.24352
131. Stahl N, Radin A, Mellis S. Rilonacept–caps and beyond. Ann N Y Acad Sci (2009) 1182:124–34. doi: 10.1111/j.1749-6632.2009.05074.x
132. Bianchi G, Vuerich M, Pellegatti P, Marimpietri D, Emionite L, Marigo I, et al. Atp/P2x7 axis modulates myeloid-derived suppressor cell functions in neuroblastoma microenvironment. Cell Death Dis (2014) 5(3):e1135. doi: 10.1038/cddis.2014.109
133. Adinolfi E, Raffaghello L, Giuliani AL, Cavazzini L, Capece M, Chiozzi P, et al. Expression of P2x7 receptor increases in vivo tumor growth. Cancer Res (2012) 72(12):2957–69. doi: 10.1158/0008-5472.CAN-11-1947
134. Hurwitz HI, Fehrenbacher L, Hainsworth JD, Heim W, Berlin J, Holmgren E, et al. Bevacizumab in combination with fluorouracil and leucovorin: An active regimen for first-line metastatic colorectal cancer. J Clin Oncol (2005) 23(15):3502–8. doi: 10.1200/JCO.2005.10.017
135. Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-Small-Cell lung cancer. N Engl J Med (2006) 355(24):2542–50. doi: 10.1056/NEJMoa061884
136. Keystone EC, Wang MM, Layton M, Hollis S, McInnes IB, Team DCS. Clinical evaluation of the efficacy of the P2x7 purinergic receptor antagonist Azd9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann Rheum Dis (2012) 71(10):1630–5. doi: 10.1136/annrheumdis-2011-143578
137. Lee J, Ahn H, Hong EJ, An BS, Jeung EB, Lee GS. Sulforaphane attenuates activation of Nlrp3 and Nlrc4 inflammasomes but not Aim2 inflammasome. Cell Immunol (2016) 306-307:53–60. doi: 10.1016/j.cellimm.2016.07.007
138. Wu D, Wu K, Zhu Q, Xiao W, Shan Q, Yan Z, et al. Formononetin administration ameliorates dextran sulfate sodium-induced acute colitis by inhibiting Nlrp3 inflammasome signaling pathway. Mediators Inflammation (2018) 2018:3048532. doi: 10.1155/2018/3048532
139. He A, Shao J, Zhang Y, Lu H, Wu Z, Xu Y. Cd200fc reduces lps-induced il-1beta activation in human cervical cancer cells by modulating Tlr4-Nf-Kappab and Nlrp3 inflammasome pathway. Oncotarget (2017) 8(20):33214–24. doi: 10.18632/oncotarget.16596
140. Li S, Liang X, Ma L, Shen L, Li T, Zheng L, et al. Mir-22 sustains Nlrp3 expression and attenuates h. pylori-induced gastric carcinogenesis. Oncogene (2018) 37(7):884–96. doi: 10.1038/onc.2017.381
141. Fleischmann RM, Schechtman J, Bennett R, Handel ML, Burmester GR, Tesser J, et al. Anakinra, a recombinant human interleukin-1 receptor antagonist (R-Methuil-1ra), in patients with rheumatoid arthritis: A Large, international, multicenter, placebo-controlled trial. Arthritis Rheum (2003) 48(4):927–34. doi: 10.1002/art.10870
142. Lu F, Zhao Y, Pang Y, Ji M, Sun Y, Wang H, et al. Nlrp3 inflammasome upregulates pd-L1 expression and contributes to immune suppression in lymphoma. Cancer Lett (2021) 497:178–89. doi: 10.1016/j.canlet.2020.10.024
143. Theivanthiran B, Evans KS, DeVito NC, Plebanek M, Sturdivant M, Wachsmuth LP, et al. A tumor-intrinsic pd-L1/Nlrp3 inflammasome signaling pathway drives resistance to anti-Pd-1 immunotherapy. J Clin Invest (2020) 130(5):2570–86. doi: 10.1172/JCI133055
144. Huseni MA, Wang L, Klementowicz JE, Yuen K, Breart B, Orr C, et al. Cd8(+) T cell-intrinsic il-6 signaling promotes resistance to anti-Pd-L1 immunotherapy. Cell Rep Med (2023) 4(1):100878. doi: 10.1016/j.xcrm.2022.100878
145. Akkaya I, Oylumlu E, Ozel I, Uzel G, Durmus L, Ciraci C. Nlrc4 inflammasome-mediated regulation of eosinophilic functions. Immune Netw (2021) 21(6):e42. doi: 10.4110/in.2021.21.e42
146. He Q, Fu Y, Tian D, Yan W. The contrasting roles of inflammasomes in cancer. Am J Cancer Res (2018) 8(4):566–83.
Keywords: inflammasomes, NOD-like receptors (NLRs), cancer, targeted therapeutics, natural extracts, synthetic small molecule targeted drugs
Citation: Gu Q, Zou J, Zhou Y and Deng Q (2023) Mechanism of inflammasomes in cancer and targeted therapies. Front. Oncol. 13:1133013. doi: 10.3389/fonc.2023.1133013
Received: 28 December 2022; Accepted: 06 March 2023;
Published: 20 March 2023.
Edited by:
Nan-Shan Chang, National Cheng Kung University, TaiwanReviewed by:
Haiwei Mou, Wistar Institute, United StatesCopyright © 2023 Gu, Zou, Zhou and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiuchan Deng, Z3JhY2U4MTlAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.