 Haoyan Li
Haoyan Li Loraine Gigi
Loraine Gigi Di Zhao
Di Zhao- 1Department of Experimental Radiation Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Texas A&M School of Public Health, Texas A&M University, College Station, TX, United States
Chromatin remodeling proteins contribute to DNA replication, transcription, repair, and recombination. The chromodomain helicase DNA-binding (CHD) family of remodelers plays crucial roles in embryonic development, hematopoiesis, and neurogenesis. As the founding member, CHD1 is capable of assembling nucleosomes, remodeling chromatin structure, and regulating gene transcription. Dysregulation of CHD1 at genetic, epigenetic, and post-translational levels is common in malignancies and other human diseases. Through interacting with different genetic alterations, CHD1 possesses the capabilities to exert oncogenic or tumor-suppressive functions in context-dependent manners. In this Review, we summarize the biochemical properties and dysregulation of CHD1 in cancer cells, and then discuss CHD1’s roles in different contexts of prostate cancer, with an emphasis on its crosstalk with diverse signaling pathways. Furthermore, we highlight the potential therapeutic strategies for cancers with dysregulated CHD1. At last, we discuss current research gaps in understanding CHD1’s biological functions and molecular basis during disease progression, as well as the modeling systems for biology study and therapeutic development.
Introduction
Chromatin remodeling is a major regulator of gene expression. Chromatin remodelers utilize ATP hydrolysis to slide the nucleosomes onto and off the DNA, thereby regulating the accessibility of genes to a range of nuclear factors, including transcriptional factors (1). Chromatin remodeling proteins contribute to DNA recombination, transcription, repair, and replication (2). Based on the similarities and differences in catalytic ATPases and associated subunits, chromatin remodelers can be classified into four subfamilies: Imitation Swtich (ISWI), Chromodomain Helicase DNA-binding (CHD), Switch/sucrose Non-fermentable (SWI/SNF) and INO80 (3). The CHD family comprises nine members and plays crucial roles in embryonic development, hematopoiesis, and neurogenesis (2, 4, 5). Notably, nearly all CHD members are dysregulated and mutated in human malignancies. Increasing evidence points to the roles of CHD members during cancer development and progression. Through promoting the transcription of oncogenes or tumor suppressor genes, some CHD enzymes possess the capability to exert both oncogenic and tumor-suppressive functions in context-dependent manners.
CHD1 is the founding member of the CHD family and is conserved across all eukaryotes (6). CHD1 is capable of assembling nucleosomes, remodeling chromatin structure, modulating histone turnover, and regulating gene transcription (7–9). In embryonic stem cells (ESCs), CHD1 is a key regulator of open/loose chromatin, correlates with a permissive transcriptional state, and directly contributes to developmental pluripotency characteristics (10–13). The induction of CHD1 expression is also essential in the programming of the pluripotent stem cells (5). In the past decade, large-scale cancer genome studies showed recurrent deletions of the CHD1 gene in ~8% of prostate cancer (14–20). In prostate tumors, loss of CHD1 causes DNA repair defects, androgen receptor (AR) redistribution and dysfunction, chromatin instability, and transcriptional plasticity (21–24). However, in PTEN-deficient prostate tumors, the CHD1 protein is stabilized and contributes to cancer progression, tumor microenvironment remodeling, and drug resistance (25–27).
In this Review, we focus on the chromatin remodeler CHD1 that plays multifaceted roles in prostate cancer. We summarize CHD1’s biochemical properties and dysregulation in cancer cells, as well as discuss its biological functions in different contexts of prostate cancer, emphasizing its crosstalk with diverse signaling pathways. In addition, we highlight the differential therapeutic strategies for cancers harboring CHD1 defects or overexpression.
Biochemical and structural properties of CHD1
Compared to other chromatin remodelers, the CHD family is distinguished by two signature motifs: tandem chromodomains located in the N-terminal region and the SNF2-like ATP-dependent helicase domain centered in the middle of the protein (28) (Figure 1). The chromodomains bind to methylation marks on histones, while the SNF2-like ATPase domain confers enzymatic activity and regulates nucleosome remodeling and chromatin conformational change (2, 29). Based on the constituent domains, CHD proteins are classified into three subfamilies: subfamily I (CHD1/2), subfamily II (CHD3–5), and subfamily III (CHD6–9) (Figure 1). In addition to chromodomains and ATPase domain, CHD1 and CHD2 proteins also contain SANT-SLIDE DNA-binding domains located in the C-terminal region (Figure 1), and preferentially bind to AT-rich DNA motifs (28, 30, 31). Although CHD1 and CHD2 are highly homologous to one another, they are significantly divergent in the 3′ regions and may possess distinct functions. In contrast, subfamily II proteins (CHD3–5) are distinguished by N-terminal tandem PHD (plant homeodomain) Zn finger-like domains (Figure 1). They are core components of the nucleosome remodeling and histone deacetylase complex (NuRD) (32). The third subfamily (CHD6–9) is evolutionarily conserved and contains additional featured domains, such as the Brahma and Kismet domain (BRK), the conserved region (CR) domains, and the SANT-SLIDE-like domain (Figure 1).
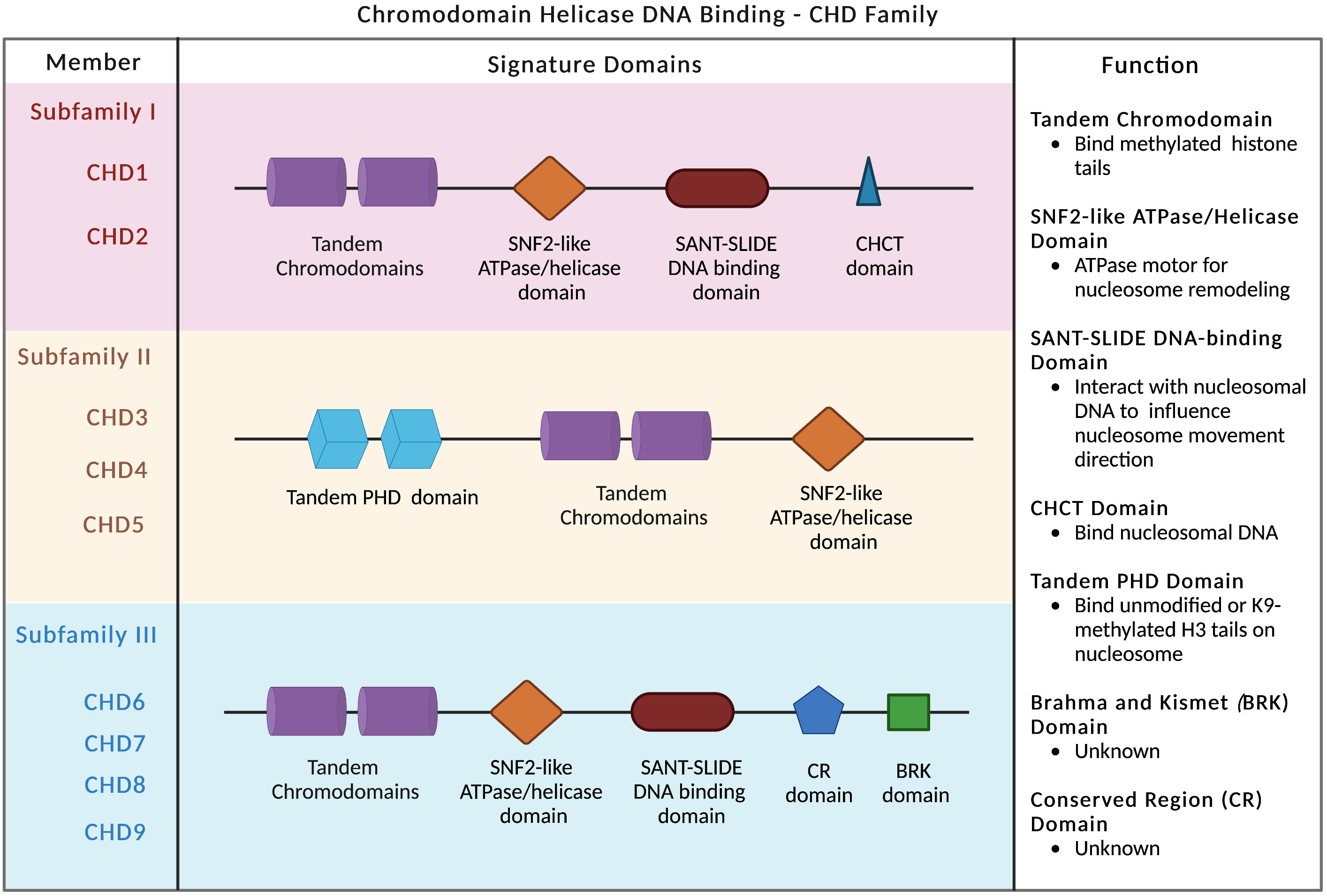
Figure 1 CHD family and signature domains. Three subfamilies of CHD chromatin remodelers are presented with signature domains. The major functions of each domain are listed.
The chromatin association specificity of CHD proteins is largely mediated by interactions with transcription factors, modified histones, and methylated DNA and RNA (4). The tandem chromodomains of human CHD1 protein selectively bind to methylated lysine 4 on the histone H3 tail (H3K4) (33, 34), a hallmark of the transcriptionally active chromatin. The chromodomains target CHD1 to specific areas of chromatin-trimethylated H3K4 marks regions for open chromatin and transcriptional activation (35). Despite the double chromodomains of human CHD2 and yeast CHD1 share significant sequence similarity with human CHD1, they have much lower binding affinity to methylated H3K4 (33). In mice, the chromodomains of CHD1 are also required for proper chromatin localization (36). The SNF2-like ATP-dependent helicase domain shared in the CHD family anchors on the nucleosome and functions as an ATPase motor for the nucleosome remodeling (9, 36, 37). The SANT-SLIDE DNA-binding domains bind to DNA that flanks the nucleosome to increase the nucleosome-binding affinity of CHD1 and influence the direction of the nucleosome movement (12, 38).
CHD1 protein has DNA translocase activity that utilizes the energy of ATP hydrolysis to impel DNA around the octamer and mobilize nucleosomes (4). The CHD1 remodeler is a unique organization of domains on the nucleosome that reveals the direct interdomain communication (12, 37, 39). The chromodomains allow CHD1 to distinguish between nucleosomes and naked DNA by physically gating access to the ATPase motor (37). Disruption of the chromodomain-ATPase interface reduced the reliance on the histone H4 tail for nucleosome sliding (37). Besides, the chromodomains bind to nucleosomal DNA at the superhelical location (SHL) SHL1 site, resulting in ATPase closure; the ATPase motor binding to the SHL2 site is anchored to the N-terminal tail of histone H4 (12, 39). Both pack against the DNA-binding domain on DNA exiting the nucleosome (39). This arrangement spans and bridges two DNA gyres of the nucleosome and enables the ATPase motor to promote the translocation of DNA towards the nucleosome dyad, thereby loosening the first DNA gyre and remodeling the nucleosome (12, 39). The cycles of ATP hydrolysis of the ATPase motor trigger a succession of conformational changes of CHD1, promoting DNA translocation and nucleosome remodeling (4). By the endpoint of the remodeling reaction, the binding affinity of CHD1 for the nucleosome decreases, leading to its release from nucleosome substrates (40).
In addition to the assembly, disruption, and repositioning of nucleosomes, CHD1 is also involved in H3.3 histone variants incorporation and transcription regulation. H3.3 is deposited on gene bodies and regulatory elements marking active transcription, and its levels are constantly high throughout the cell cycle. In Drosophila models, depletion of CHD1 in embryos caused incorrect assembly of H3.3 in the paternal pronucleus chromatin, while CHD1 loss in the adult brain resulted in reduced H3.3 incorporation chromatin, global chromatin perturbation, transcriptional dysregulation, and metabolism reprogramming (41, 42). By disassembling nucleosomes at the promoter region, CHD1 promotes open chromatin and is associated with transcriptionally active locations throughout the genome (10, 43, 44). Deletion of Chd1 resulted in the general downregulation of transcription by RNA polymerases I/II in mouse ESCs (45), and impaired efficient reprogramming of fibroblasts to the pluripotent stem cell state via downregulating the transcriptional factor Oct4 (10). Besides, CHD1 was also found to influence the pre-mRNA splicing, transcription initiation and transcription termination by bridging core factors to H3K4me3 (46–49).
Collectively, biochemical and structural studies reveal that CHD1 protein predominately interacts with methylated H3K4 histone marks, displays intricate conformational intradomain allosteric regulation, and exhibits nucleosome assembly and remodeling activities. This aligns with its epigenetic functions in chromatin organization, histone variants incorporation, and transcription reprogramming, and provides the mechanistic basis for understanding the phenotypes in animal models and human diseases with dysregulated CHD1.
Dysregulation of CHD1 in human diseases
The tandem chromodomains of CHD1 are highly conserved among species. In yeast, the C-terminal is required for Chd1’s nucleosome-remodeling activity, and the combined mutations in the SANT domain (R1016/K1020) and SLIDE domain (R1255) abolish the binding of Chd1 to DNA and nucleosome and reduce its nucleosome-remodeling activity (50). In Drosophila, the Tryptophans W372/W375 mutants in the first chromodomain or W462 mutant in the second chromodomain impair CHD1’s interaction with trimethylation of H3K4 (H3K4me3) and reduce the assembly of H3.3 into chromatin (44). The flies containing these mutations have decreased viability and fertility (44).
Prior studies have demonstrated the key roles of CHD family remodelers in neurodevelopment in human being (10, 51–54). Large-scale exome sequencing in thousands of autism spectrum disorder cases identified recurrent de novo mutations in CHD2 and CHD8 as genuine autism risk factors (52–54). Pilarowski-Bjornsson syndrome is an autosomal dominant neurodevelopmental disorder characterized by delayed development and intellectual disability, often with autistic features, speech apraxia, and mild dysmorphic features. Several de novo heterozygous missense variants of CHD1 (c.1853G>A, c.5123G>A, c.1379G>A, and c.421A>G) were identified in Pilarowski-Bjornsson syndrome and associated with the closed status of chromatin and the neurodevelopmental disability (51).
Using genome sequencing techniques, many somatic mutations, copy number alterations, and chromosomal rearrangements of chromatin remodelers have been detected in the past decades. Recent cancer genomic studies identified recurrent mutations and deletions of the CHD1 gene in prostate tumors (8-10%), uterine (11%), melanoma (7%), and colorectal cancers (6%) (Figures 2A, B) (55, 56). Mutations are more dominant than deletion in the CHD1 gene in most cancer types, but not in prostate cancer. CHD1 deletion was found in both localized prostate cancer and advanced castration-resistant prostate cancer (CRPC) (14–20). Recent epidemiology and genomics studies of prostate cancer in Asian men uncovered that CHD1 is more often deleted (18%) in the East Asian population with localized prostate cancer than in Western patients (57, 58). Another recent study uncovered that subclonal deletion of CHD1 is about three times more frequent in prostate tumors of African American (AA) men (29.7%) than that of European Ancestry (EA) men (11%) (59). Besides, CHD1 deletion is strongly associated with pathologic stages and rapid biochemical recurrence in AA cases (59).
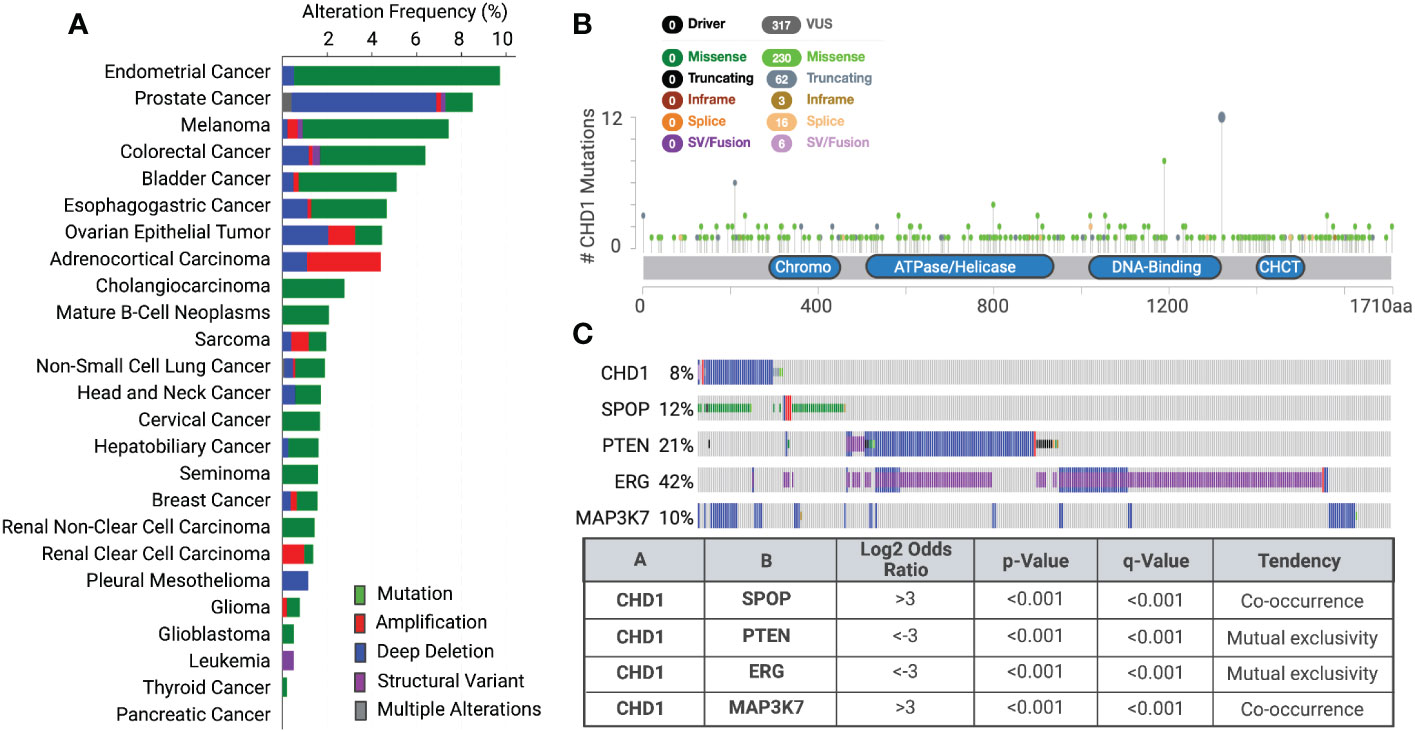
Figure 2 Genetic alterations of CHD1 cross various cancer types (A) Genetic alterations of CHD1 are frequent in human cancers (TCGA datasets). Different types of genetic alterations are highlighted in different colors. Their alteration frequency is presented in each cancer type. (B) Somatic mutations in CHD1 amino acid sequence across human cancers (TCGA databases). The number of single amino acid mutations is shown. Mutation diagram circles are colored with respect to the corresponding mutation types. The functional domains in the CHD1 protein are presented. (C) The co-occurrence and mutual exclusivity of CHD1 deletion and genetic alterations of SPOP, PTEN, ERG, and MAP3K7 in primary prostate tumors are shown (TCGA dataset). The Log2 Odds Ratio, p-value, q-value, and tendency between two genetic events are calculated (cBioportal).
Notably, deletions of CHD1 show distinct patterns of co-occurrence and mutual exclusivity with genetic alterations of some oncogenes and tumor suppressor genes (Figure 2C). CHD1 deletion often co-occurs with missense mutations in SPOP (speckle-type BTB/POZ protein) and defines a new molecular subtype of prostate cancer, characterized by increased DNA methylation and homogeneous gene expression patterns (60). Besides, MAP3K7 and CHD1 were significantly co-deleted in localized prostate tumors and combined loss correlated with poor disease-free survival of patients (20, 61). However, this co-occurrence is rarely found in other cancer types, suggesting their unique functions in prostate cancer development and progression. In contrast, CHD1 deletion is mutually exclusive with PTEN loss or TMPRSS2:ERG fusion in human prostate tumors (14, 16, 25, 62), by crosstalk with key components in PTEN-AKT and AR signaling pathways.
The expression of CHD1 is modulated at both post-transcriptional and post-translational levels, and its dysregulation is associated with cancer development and other human diseases (Table 1). MicroRNAs (miRs) represent a critical class of small, non-coding RNAs and repress target genes either by mRNA degradation or repression of translation. Lifespan-related miRNAs, miR-34a, miR-107, and miR-212-3p, are found preferentially target Chd1 and are associated with high-fat diet and aging (63). In estrogen receptor (ER)+ breast cancer, miR-26 is identified as a microRNA targeting CHD1 and suppresses breast cancer cell proliferation by downregulating the CHD1 expression (67).
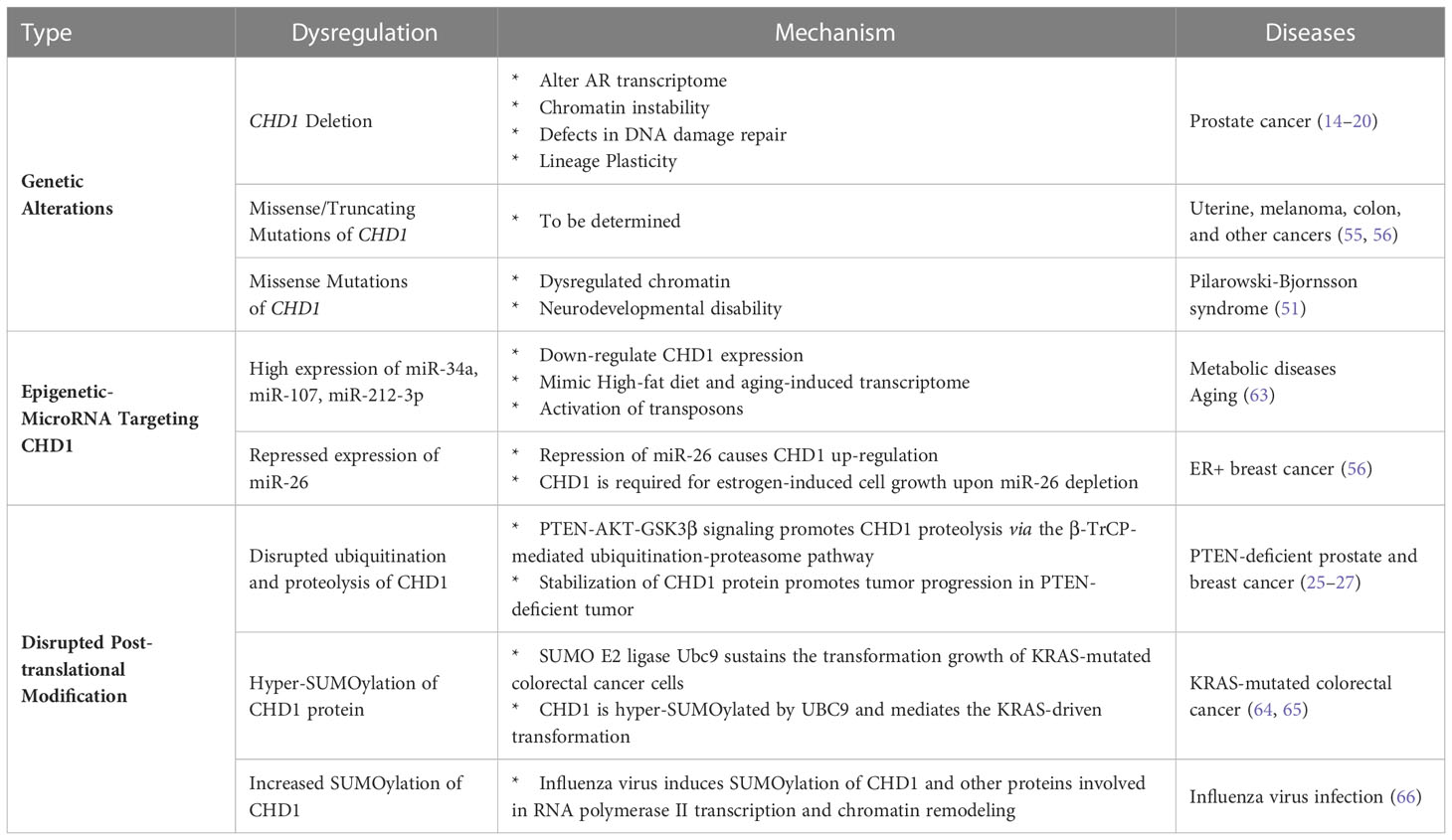
Table 1 Dysregulation of CHD1 in human diseases.
Our prior studies in prostate cancer demonstrated that PTEN-AKT-GSK3β signaling promotes CHD1 protein degradation via the β-TrCP-mediated ubiquitination-proteasome pathway (25). β-TrCP is an F-box protein that acts as the substrate-recognition subunit for the SCFβ-TrCP (Skp1–Cullin1–F-box protein) E3 ubiquitin ligases. We found that β-TrCP E3 ligase directly interacts with CHD1 protein, induces its poly-ubiquitination, and promotes the proteolysis of CHD1 (25). Through E3 ligase consensus-sequence scanning, we also identified two evolutionarily conserved putative β-TrCP consensus-binding motifs (DSGXXS) at the N terminus of CHD1 (25). Another study also reported that the N-terminal serine-rich region (SRR) of CHD1 is modified by phosphorylation and depletion of SRR impaired differentiation of the ESCs (68). Systematic mass spectrometric analysis and consensus site prediction also showed that PGK and GSK3 kinases might be involved in the phosphorylation of CHD1 (69). Notably, β-TrCP E3 ligase recognizes and interacts specifically with phosphorylated substrates, and importantly, β-TrCP-binding motifs in CHD1 protein contain GSK3β consensus sequences (SXXXS). Further biochemical and molecular biological studies established that GSK3β serves as a kinase of CHD1 and mediates its recognition and interaction with β-TrCP E3 ligase, resulting in CHD1 protein ubiquitination and degradation (25). In PTEN-deficient cancers, AKT activation-induced GSK3β suppression results in the disruption of CHD1 proteolysis and aberrant accumulation of the CHD1 protein (25–27), which contributes to tumor development and tumor microenvironment (TME) remodeling.
Like ubiquitination, SUMOylation is a post-translational modification that regulates protein stability, activity, localization, and interactome. SUMOylation involves various cellular processes, such as transcription, chromatin remodeling, DNA damage repair, cell cycle progression, ribosome biogenesis, and mitochondrial dynamics (70–72). In KRAS-mutated colorectal cancer, CHD1 protein is hyper-SUMOylated by the SUMO E2 ligase UBC9, and depletion of CHD1 impairs the KRAS-driven transformation (64, 65). Besides, influenza virus infection was also found to induce the SUMOylation of CHD1 and other proteins involved in RNA polymerase II transcription and chromatin remodeling (66).
As a chromatin remodeler, CHD1 dysregulation is associated with malignancies and other human diseases. Diverse mechanisms, including genetic alterations, epigenetic regulations, and post-translational modifications, lead to the dysregulation of CHD1 in context-dependent manners (Table 1). It is equally important to understand the biological functions of CHD1 in different contexts, which will uncover the therapeutic vulnerabilities of diseases with dysregulated CHD1.
Multifaceted roles of CHD1 in prostate cancer
Genetic studies in yeast, fruit flies, zebrafish, and mice underscore the roles of CHD family enzymes in regulating cellular fate and identity, embryonic development, stem cell maintenance, and neuronal development and pathologies. These studies have been summarized and discussed in several comprehensive review articles (2, 4, 5). The increasing evidence documented individual CHD remodelers function as context-dependent oncogenes or tumor suppressors in human malignancies. For instance, CHD4, as a crucial subunit of the NuRD complex, promotes tumorigenesis by epigenetic silencing tumor suppressor genes or serving as a coactivator of HIF in colorectal, breast, and endometrial cancers (73–75). In contrast, CHD5 was identified as a tumor suppressor gene in gliomas, breast, colon, lung, ovarian, and prostate cancers (76, 77). Given the frequent alterations and dysregulation of CHD1 in prostate tumors, in this section, we review CHD1’s biological functions in prostate cancer with an emphasis on its crosstalk with different genetic alterations and diverse signaling pathways.
Prostate tumorigenesis
As noted earlier, CHD1 is homozygously deleted in 8~18% of prostate cancer, supporting the hypothesis that CHD1 is a tumor suppressor in prostate cancer. Earlier in vitro studies using siRNA showed that downregulation of CHD1 in nontumorigenic prostate epithelial cells promoted cell invasiveness and enhanced cell clonogenicity, but had no impact on cell growth (17, 18). To obtain the genetic evidence, our and other independent groups established prostate-specific Chd1 deletion genetically engineered mouse (GEM) models, in which conditional Chd1 alleles deleted by a Probasin (Pb) promoter-driven Cre recombinase (Pb-Cre; Chd1L/L) (21, 23, 27). Homozygous deletion of Chd1 in prostate glands showed no observed differences in cell proliferation, cell survival, androgen receptor (AR) expression, or glandular structure (21, 23, 27). No invasive adenocarcinoma was observed in mice up to 1 year of age, as characterized by well-maintained smooth muscle actin structures (21). This genetic evidence suggests that Chd1 loss alone is insufficient to drive tumorigenesis in the prostate.
Notably, CHD1-depleted tumors often harbor additional genetic alterations, including SPOP mutations and MAP3K7 deletion, but also show mutual exclusivity with PTEN loss or ERG translocation (Figure 2C) (14, 16, 17, 20, 25, 26, 62). CHD1 depletion reduced cell proliferation, invasiveness, and tumor growth of PTEN-deficient cancer cells (14, 20, 25, 26); while loss of MAP3K7 and CHD1 coordinates to promote aggressive prostate cancer (20, 61). These observations seem paradoxical at first glance; however, they established the context-dependent roles of CHD1 in prostate cancer (Figure 3). Importantly, CHD1’s distinct roles in different contexts are largely mediated by the crosstalk with diverse signaling pathways, which will be introduced individually in the following subsections.
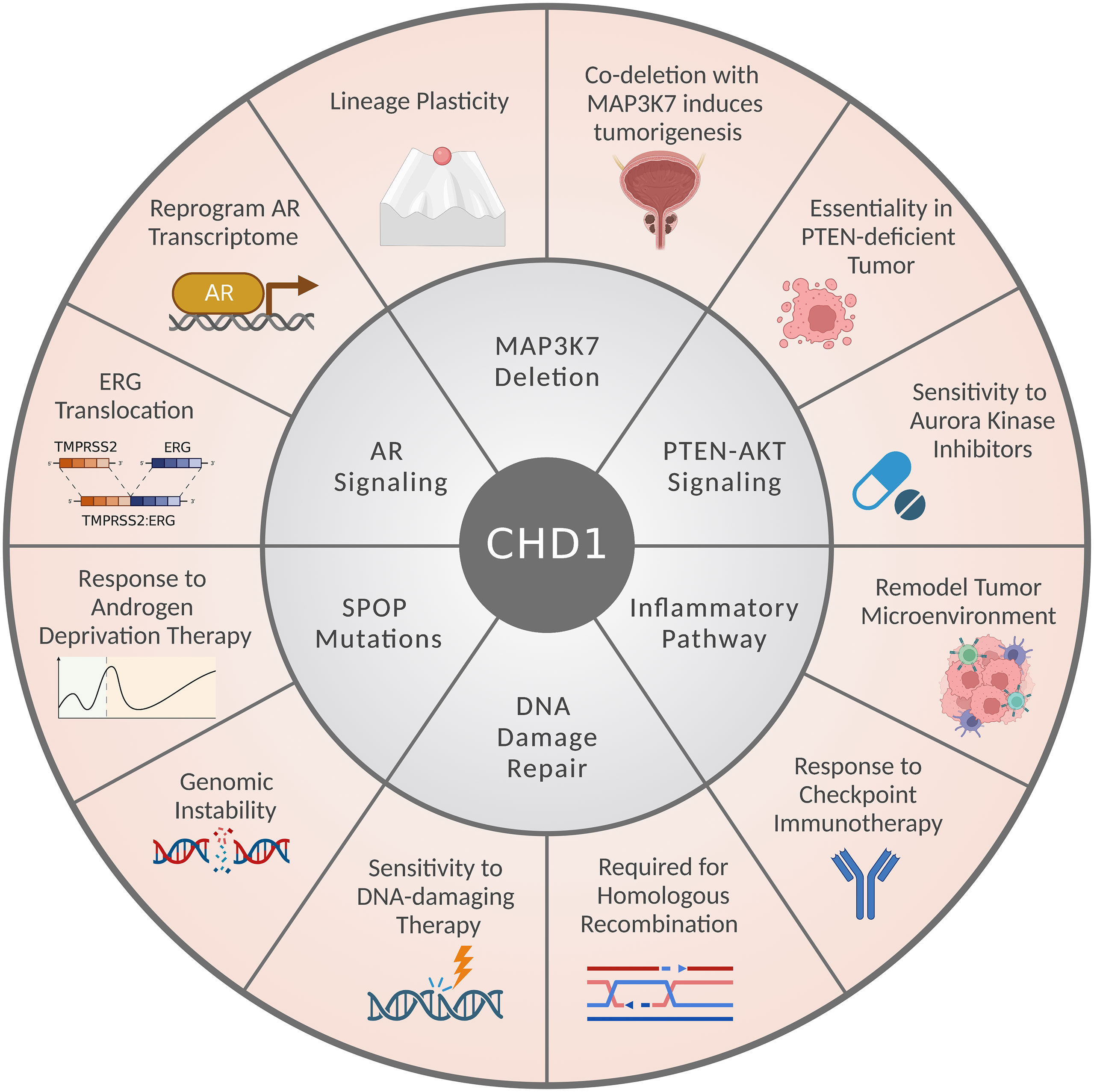
Figure 3 CHD1’s multifaceted roles and crosstalk with signaling pathways in prostate cancer. The CHD1-associated signaling pathways or genetic alterations are presented in the middle circle. CHD1’s biological functions and therapeutic implications in prostate cancers are listed in the outer circle.
AR signaling
Prostate cancer is largely driven by androgen receptor (AR) signaling. Androgen deprivation therapy (ADT) and AR inhibition are the main strategies for prostate cancer treatment (78). Although CHD1 protein does not directly bind to AR (14, 16, 23), loss of CHD1 caused the transcriptome reprogramming of AR signaling and is strongly associated with the ERG translocation (14, 16, 22, 23). By performing the chromatin-bound interactome analysis, Augello et al. uncovered that CHD1 interacts with the cofactors of AR and other nuclear receptors (23). Chromatin immunoprecipitation (ChIP) sequencing showed that CHD1 colocalizes to gene enhancers enriched for AR and its cofactors, such as HOXB13, ETV1, and FOXA1 (23). Specifically, they found that CHD1 localizes to chromatin-containing canonical AR binding sites, but CHD1 loss causes AR to redistribute to HOXB13-enriched sites, which drives a unique AR transcriptome that contributes to the tumor formation (23).
In prostate cancer, the most common genetic rearrangement involves the fusion of the androgen-regulated gene TMPRSS2 with the ETS transcription factor ERG (79). The fusion joins the 5′-UTR of TMPRSS2 (21q22) with the 3′-end of ERG (21q22) and leads to the TMPRSS2:ERG mRNA fusion transcript, which is induced by androgen. Using whole exome sequencing, FISH, or confocal microscopy, several groups showed the mutual exclusivity of CHD1 deletion with ERG fusion in human prostate tumors (14, 16, 62). CHD1 deletion is also strongly associated with early PSA recurrence (14, 59). Using a doxorubicin/dihydrotestosterone-induced DNA double-strand breaks system, Burkhardt and colleagues showed that CHD1 depletion prevents the formation of ERG rearrangements. Mechanistically, they found that CHD1 is required to recruit AR to responsive promoters and regulates the expression of AR-responsive genes, such as NKX3-1, FOXO1, and PPARγ (14). Given that AR-dependent transcription is a prerequisite for ERG translocation, these studies concluded that a functional CHD1 supports AR signaling transcriptome and ERG fusion development in prostate cancer.
Lysine-specific demethylase 1 (KDM1A/LSD1) removes the mono- and di-methylation from H3K4 and H3K9, and plays an important role in regulating AR-dependent gene expression in prostate cancer (80, 81). A prior study by the Schule group reported that the LSD1 protein is modified by di-methylation at K114 (K114me2) (82). By solving the cocrystal structure, they identified CHD1 as an LSD1-K114me2 reader and uncovered that chromatin colocalization of CHD1 and LSD1-K114me2 drive AR-dependent transcription and TMPRSS2-ERG translocation (82). This structural study provides additional evidence and mechanistic insight into CHD1’s roles in modulating AR signaling and ERG fusions during prostate cancer evolution.
Lineage plasticity
Transcriptomic and epigenetic profiling studies in ESCs and cancer cells showed that CHD1 is required for sustaining the opening of chromatin and the global transcription (10, 22, 23, 25, 27, 83). CHD1 deficiency causes the accumulation of heterochromatin, diminishing the pluripotency of ESCs (10). In prostate cancer, CHD1 co-localizes with H3K4me3 to the promoters of actively transcribed genes, while CHD1 depletion reduces H3K4me3 marked genes, alters the chromatin assembly across the genome, and reprograms the global transcription (22, 23, 25). Lineage plasticity of cancer cells has been proposed as a source of intratumoral heterogeneity and resistance to targeted anticancer treatments (84). In prostate cancer, the histological transformation from AR-dependent adenocarcinoma to AR-indifferent neuroendocrine or small-cell carcinoma is a well-known pathway of lineage plasticity, which might occur as a consequence of ADT (85, 86). In addition to the deregulation of AR signaling, CHD1 loss is linked to lineage plasticity by inducing a lineage-specific transcriptome (20, 22).
Cramer’s group initially proposed this hypothesis. They found MAP3K7 and CHD1 were significantly co-deleted in localized prostate tumors and combined loss correlated with poor disease-free survival of patients (20, 61). CHD1 knockdown reduced cell proliferation, impaired tumor growth, and prolonged the overall survival of mice in PTEN-deficient LNCaP-derived xenograft models. However, additional MAP3K7 loss completely rescued this effect and promoted prostate cancer progression (20, 61). Co-suppression of MAP3K7 and CHD1 induces androgen-independent growth and causes resistance to AR inhibitors, such as enzalutamide (61). Combining mouse prostate epithelial progenitor/stem cells (PrP/SC) and tissue recombination model, they found that CHD1-depleted PrP/SCs grafts are mostly benign, characterized by intact p63+ basal layer (20). This is consistent with the phenotypes observed in Pb-Cre; Chd1L/L GEM model (21, 23, 27). In contrast, MAP3K7-depleted grafts displayed a mixture of benign, high-grade prostatic intraepithelial neoplasia (PIN), and carcinoma phenotypes. Strikingly, dual MAP3K7–CHD1 loss grafts displayed high-grade PIN and invasive carcinoma phenotypes (20). Compared to MAP3K7 or CHD1 depletion alone, dual depletion caused lineage switching, characterized by loss of AR and epithelial markers (CK5, p63, CK14, and CK18) along with the upregulation of neuroendocrine differentiation markers (SYP and Nestin) and mucin production (20). It remains unclear if MAP3K7/CHD1 double-depletion affects metastatic progression. Nevertheless, better understanding their interactions and underlying mechanisms might provide novel therapeutic strategies for MAP3K7/CHD1 loss prostate cancer.
Recently, Zhang et al. showed that CHD1 loss renders prostate cancer cells more resistant to AR inhibition via inducing lineage plasticity (22). They showed that loss of CHD1 induces the transcription factors of GR, BRN2, TBX2, and NR2F1, which are required to promote tumor heterogeneity and resistance to AR inhibitors in CHD1-deficient tumors (22). They also found that enzalutamide-resistant xenograft tumors with CHD1 depletion and high expression of those transcription factors, lost luminal lineage identities (AR, CK8, and CK18), but displayed increased basal markers (CK5 and p63) and epithelial to mesenchymal transition genes (SNAI2, TWIST1, SNAI1, and ZEB1) (22). These non-luminal lineage programs and plastic chromatin landscape induced by CHD1 loss may serve as mechanisms to enable heterogeneous subclones less dependent on AR.
DNA damage repair
Endogenous cell metabolism and environmental factors often cause DNA double-strand break (DSB). Homologous recombination (HR) and non-homologous end joining (NHEJ) are two major repair mechanisms in response to DSB (87). Using prostate cancer cell lines and GEM models, several studies reported that CHD1 loss causes defects in HR-mediated DNA damage repair (DDR) and increases sensitivity to DNA-damaging therapies (21, 24, 88, 89). Besides, recent studies in metastatic prostate cancer patients showed that CHD1 deletion is associated with HR deficiency-related mutational signatures (59, 90).
Mechanistically, CHD1 accumulates at the DNA damage sites, maintains the open status of chromatin, and co-localizes with γH2AX in response to DNA damage (24). CHD1 interacts with and recruits DDR factors, such as CtIP, 53BP1, RIF1, and KU70, to the DNA damage sites (21, 24). CtIP is a key player in HR by resecting DSB ends. CHD1 loss impairs the recruitment of CtIP to DNA damage sites and suppresses the initiation of HR (24, 88). As a key DDR protein, 53BP1 maintains the balance of repair pathway choices and genomic stability. Shenoy et al. found that CHD1 forms a complex with NHEJ components and negatively regulates the protein stability of 53BP1 (21). CHD1 loss stabilizes 53BP1 protein and causes the switch from HR to NHEJ pathway for DSB repair. Although AR signaling is known to regulate the expression of DDR-related genes and promotes NHEJ repair, the role of CHD1 in modulating DDR is independent of the AR pathway (21). CHD1 loss is also associated with chromosomal and genomic instability in prostate cancers (21, 22), and DDR defects may serve as one of the mechanisms.
When CHD1 loss meets SPOP mutations
Recurrent missense mutations in SPOP (speckle-type BTB/POZ protein) occur in 10-15% of localized prostate tumors and metastatic CRPC (60, 91–93). In occurrence with CHD1 deletion (Figure 2C), SPOP mutations define a distinct prostate cancer subtype, characterized by genomic instability, increased AR transcriptional activity, absence of ERG rearrangements, and increased DNA methylation (60, 91, 92, 94). SPOP protein is a substrate adaptor for the Cullin3-RING-based BCR E3 ligase complex (CUL3-SPOP), which mediates the ubiquitination and proteasomal degradation of target proteins. In prostate cancer, hotspot SPOP mutations are only observed in the MATH domain that is responsible for substrate recognition and recruitment. The mutant reduces the substrate-binding affinity and results in the aberrant accumulation of substrates (95).
Several oncogenic proteins in AR signaling were identified as substrates of CUL3-SPOP, such as AR (96), SRC3 (97), and ERG (98, 99). CUL3-SPOP complex mediates the ubiquitination-degradation of AR by binding to the 645ASSTT649 Motif in the hinge domain of AR. Prostate cancer-associated SPOP mutants (Y87C, Y87N, F102C, S119N, F125V, W131G, F133L, and F133V) fail to bind AR protein, thereby increasing the protein stability and activity of AR during tumorigenesis (96, 100). By establishing a tissue-specific SPOP-F133V overexpressing GEM model, Blattner and colleagues reported that SPOP mutation promotes prostate tumorigenesis through coordinate regulation of PI3K/mTOR and AR signaling (101). Clinical trials in men with metastatic prostate cancer found that SPOP mutations are associated with improved survival outcomes after ADT (93, 102, 103). Although it remains unclear whether SPOP mutations crosstalk with CHD1 loss when regulating AR signaling, a clinical study in metastatic CRPC showed that SPOP mutations and CHD1 loss are associated with a higher response rate to abiraterone (inhibitor of androgen biosynthesis) and a longer time on the abiraterone treatment (93).
In addition to modulating AR signaling, coordinate CHD1 deletion and SPOP mutations are also involved in DNA damage response. Phenocopying CHD1 loss, SPOP mutations also cause genomic instability and impaired HR DSB repair, as well as promote the sensitivity of prostate tumors to DNA-damaging therapeutic agents, such as PARP inhibitors (94, 104, 105). Mechanistically, SPOP is accumulated at DNA double-strand break sites, where it interacts with ATM kinase and plays an essential for DDR (94, 105). Depletion or mutations of SPOP inhibits HR and promotes NHEJ by downregulating DNA repair factors (RAD51, BRCA2, CHK1, and ATR), reducing RAD51 foci formation, and stabilizing 53BP1 (94, 106). Recent studies found that SPOP mutations and CHD1 deletion sensitize prostate cancer cells to DNA damage inducers and show synergistic effects on the DNA damage repair (59, 89). By generating prostate-specific Chd1 and/or Spop deletion GEM models, Zhu and colleagues found that co-deletion of Chd1 and Spop in the prostate synergistically induces the response to DNA DSBs, characterized by increased γH2AX staining (89). Besides, they showed that the combination of CHD1 depletion and SPOP mutations significantly augmented the DNA damage response and sensitized human prostate cells to DNA-damaging agents (89). Another study in AA men revealed that, compared to cases with either alteration alone, prostate tumors with both CHD1 deletion and SPOP mutations showed significantly higher levels of HR deficiency-associated signatures and large-scale structural rearrangements (59). These studies demonstrated the synergistic effects of CHD1 loss and SPOP mutations in modulating AR signaling and DDR pathways, providing insights into the molecular basis of their frequent co-occurrence in prostate cancers.
Essentiality in PTEN-deficient cancers
Tumor suppressor PTEN is frequently altered in prostate and other cancer types. As a dual lipid and protein phosphatase, PTEN dephosphorylates PIP3 and suppresses the activation of AKT, leading to a hyperactive PI3K signaling (107, 108). PTEN/AKT pathway is critical for cellular processes, such as metabolism and cell proliferation (109). Genetic deletion and mutations of PTEN occur in ~20% of localized prostate tumors and are further enriched in ~40% of CRPC with strong associations with metastatic disease and poor overall outcome (60, 110). In prior studies, we found that CHD1 deletions show a mutually exclusive pattern with PTEN loss in prostate tumors (Figure 2C), and CHD1 negatively correlates with PTEN expression at protein levels (25). Mechanistically, PTEN loss stabilizes CHD1 protein in cancer cells and prostate tumors by disrupting CHD1’s ubiquitination and degradation (25–27), as described above. Functionally, we identified CHD1 as a synthetic essential gene in cancers containing PTEN deficiency (25–27). CHD1 depletion significantly suppressed tumor growth in PTEN-deficient xenograft models (25), consistent with earlier observations in LNCaP xenograft tumors (20). However, CHD1 knockdown showed little effect on benign prostatic hyperplasia cells or PTEN-intact tumors (25, 26).
In GEM models, Pb-Cre-driven Pten loss (Pb-Cre; PtenL/L) in the prostate triggers non-lethal invasive tumors after a long latency (111). By crossing a Chd1 conditional knockout allele into this GEM model, Augello et al. reported that CHD1 loss promotes prostate tumor progression (23). The limitations of this study include the small animal cohort (n = 5), low frequency of tumor progression (1 in 5 mice), and lack of survival data. In contrast, we established prostate-specific Chd1 deletion in two well-established PTEN-deficient GEM models, Pb-Cre; PtenL/L and Pb-Cre; PtenL/L; Smad4L/L (112), and then determined the impact of Chd1 deletion with much larger cohorts (n = 22 or 18) (27). In both models, we found that CHD1 depletion significantly delayed the development and progression of PTEN-deficient prostate tumors and prolonged the survival of mice, providing genetic evidence supporting the essential roles of CHD1 in the context of PTEN defects (27). Given that CHD1-null prostates are phenotypically normal (21, 23, 27), these studies revealed the therapeutic potential of targeting CHD1 in PTEN-deficient tumors with an acceptable therapeutic window. Despite these encouraging factors, it is worth noting that tumor progression was rarely observed in some Pten/Chd1 double-knockout mice. Although these cases appear to result from clonal expansion of prostate cancer cells undergoing incomplete Chd1 deletion, future study is needed to identify potential second-site suppression events that may underlie CHD1 bypass. It will also provide rational combinatorial strategies targeting CHD1 in PTEN-deficient tumors.
Tumor microenvironment remodeling
Tumor development and progression are largely driven by interactions between cancer cells, extracellular matrix, stromal cells, and immune cells in the tumor microenvironment (TME) (78). Prostate cancer has a TME characterized in part by a relative paucity of infiltrating T cells and a high proportion of immunosuppressive myeloid cells, including myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) (78, 113). MDSCs are a heterogeneous group of myeloid cells that play immunosuppressive roles via interaction with T and NK cells (114). Our prior studies using multiple GEM models demonstrated that CHD1 is involved in the inflammatory response and plays a crucial role in modulating the TME via promoting MDSC infiltration and suppressing tumor-infiltrating lymphocytes (TILs) (27). In PTEN-deficient prostate tumors, CHD1 deletion caused reduced MDSC infiltration and increased CD8+ T cells (27). Transcriptional and epigenetic profiling analyses revealed that CHD1 cooperates with NF-κB, the central player of inflammation, to regulate the transcription of inflammatory genes (25, 27). Besides, we identified IL-6 as a direct target of CHD1 and mediates the recruitment and activation of MDSC, which contributes to T cell suppression in the prostate tumors (27). In addition to NF-κB and IL-6/Stat3 signaling, CHD1 modulates several other TME-related pathways, such as inflammatory response, interferon alpha and gamma pathways, and angiogenesis (23, 27).
A recent immunogenicity study in localized prostate cancer provides additional evidence. Using multiplex immunofluorescence, Calagua and colleagues identified the genomic alterations associated with immunogenic (PD-L1 ≥5% and extensive TILs) and nonimmunogenic (PD-L1 negative and no TILs) tumor foci (115). They found that deep deletions of CHD1 are strongly associated with dendritic cell signatures and immunogenic phenotype, characterized by enriched T cell infiltration (115). The regulatory axis of CHD1/IL-6/MDSC may serve as one mechanism by which CHD1 loss drives immunogenicity. Besides, immunogenic localized prostate cancer shows high rates of genomic instability and variable tumor mutational burden (TMB) (115), suggesting chromatin instability and DDR defects induced by CHD1 loss may also contribute to immunogenic features.
Therapeutic strategies targeting CHD1 dysregulation
In the past decade, we have gained a better understanding of CHD1 biology and how its dysregulation impacts cancer development and progression. This knowledge lays an important foundation for developing effective therapeutics targeting the dysregulated CHD1 in cancers and using CHD1 as a biomarker for predicting the response to therapies. In this section, we highlight the response of CHD1-deficient tumors to DNA-damaging and antiandrogen therapies. Given that CHD1 is upregulated and plays an essential role in PTEN-deficient cancers, we also discuss the therapeutic potential of targeting CHD1 and its downstream effectors in tumors containing PTEN deficiency.
DNA-damaging therapy
As noted above, CHD1 plays a key role in DNA damage response and modulates the choice between HR and NHEJ DDR pathways. Several preclinical studies using prostate cancer cell lines, PDX models, and GEM models demonstrated that CHD1 loss leads to hypersensitivity to ionizing radiation (IR), PARP inhibition, and DNA-damaging agents, such as mitomycin C, carboplatin, irinotecan, and camptothecin (21, 24, 88–90).
By comparing the response of wildtype and Chd1-null (Pb-Cre; Chd1L/L) mice to a single dose of 10 Gy of IR, Shenoy and colleagues found that Chd1 deleted prostate tissues and ESCs are more sensitive to IR, as evidenced by increased γH2AX and phosphorylation of H2A and p53 (21). Similar phenotypes were also observed in CHD1-depleted prostate cell lines (21, 24). PARP (Poly-ADP-ribose polymerase) detects and initiates single-strand DNA breaks (SSB) DNA damage repair. Prior studies uncovered that PARP inhibitors have synthetic lethal effects in cells with HR defects, such as BRCA1 and BRCA2 loss (116). PARP inhibitors have been clinically tested in CRPC, and genetic alterations in DDR pathways are associated with better responses (117, 118). Preclinical studies showed that CHD1 loss-induced HR defects sensitize prostate tumors to PARP inhibitors, Olaparib and Talazoparib, both in vitro and in vivo (21, 24, 59, 88), suggesting CHD1 might be a predictive biomarker. The second-generation platinum agent, carboplatin, also showed a good response in a metastatic CRPC patient with homozygous CHD1 loss (21).
Notably, SPOP depletion also sensitizes cancer cells to IR and PARP inhibitors (94, 104, 105). A recent study demonstrated that SPOP mutations and CHD1 loss synergistically promote sensitivity to camptothecin, an inducer of double-strand breaks (89). Given that co-occurrence of SPOP mutations and CHD1 deletion define a distinct molecular subtype of prostate cancer, further studies are needed to assess if they have synergistic effects in response to DNA-damaging therapies. Their potential as biomarkers for predicting the response to radiotherapy, PARP inhibitors, and DNA-damaging agents in advanced prostate cancers remains to be determined.
Antiandrogen therapy
In 1941, Huggins and Hodges reported that castration led to tumor regression in prostate cancer patients, first recognizing hormone responsiveness as a central feature of prostate cancer (119). Androgen deprivation by castration or agents that block the androgen pathway is the standard of care for prostate cancer. Resistance to ADT facilitates the development of CRPC with high rates of metastasis and mortality (120). Given the important role of CHD1 in AR signaling, preclinical and clinical studies have been conducted to determine the impact of CHD1 loss on response to antiandrogen therapies using different model systems (14, 16, 22, 23, 61).
Using an androgen-driven regrowth model, Augello et al. showed castrated Chd1-deficient mice (Pb-Cre; Chd1L/L) showed increased proliferation in regenerated epithelium upon androgen re-stimulation, suggesting Chd1 deletion may render the prostate tissue more dependent to androgen (23). However, Zhang et al. used AR-overexpressing LNCaP models and showed that CHD1 loss renders human prostate cancer cells more resistant to AR inhibitors in vitro and in vivo in castrated mice (22). They also found that low expression of CHD1 is associated with shorter clinical response to next-generation antiandrogen therapies (enzalutamide or abiraterone) in CRPC patients (22). Along the same line, Jillson and colleagues showed that co-suppression of MAP3K7 and CHD1 causes androgen-independent growth of prostate cancer cells and promotes resistance to AR inhibitor enzalutamide (61).
In prostate cancer patients, CHD1 loss was associated with a shorter time to PSA recurrence, suggesting its potential as a prognostic biomarker (14, 59, 61, 121). However, recent clinical trials in men with metastatic prostate cancer found that SPOP mutations are associated with improved survival outcomes after ADT (93, 102, 103). When considered as an individual variable, CHD1 loss is associated with a higher response rate to abiraterone (OR, 7.30, P= 0.08) and a longer time on abiraterone (HR, 0.50, P = 0.06) in metastatic CRPC patients (93). Prospective clinical trials are needed to validate the impact of CHD1 deletion on response to castration, abiraterone, enzalutamide, and other antiandrogen drugs in both hormone-sensitive and -resistant prostate cancers. Given the context-dependent role of CHD1 in prostate tumors, genes showing co-occurrence (SPOP or MAP3K7) or mutual exclusivity (ERG and PTEN) should also be considered as influence factors in these clinical studies.
Notably, the upregulation of transcription factors of GR, BRN2, TBX2, and NR2F1 was found to mediate the resistance to enzalutamide in CHD1-deficient prostate cancer, since inhibition of each factor re-sensitizes CHD1 loss prostate tumors to AR inhibitor (22). This offers new insights into synthetic lethal interactions with CHD1 and potential therapeutic vulnerabilities in prostate cancers containing CHD1 deficiency. Given that GR (Glucocorticoid Receptor) inhibition has been tested in clinical studies of CRPC (NCT02012296), future biomarker studies are needed to assess if GR inhibition is more effective in CRPC patients harboring CHD1 loss.
Targeting CHD1 in PTEN-deficient cancers
Our prior studies in xenograft and GEMM models established CHD1 as a synthetic essential gene and potential therapeutic target in prostate cancers containing PTEN defects (25–27). Several independent groups are dedicated to developing small-molecule inhibitors targeting CHD1, and the efficacies of top hits will be tested in cancer cell lines and diverse preclinical models. We expect that these drugs have better therapeutic effects on PTEN-deficient tumors but may have modest effects on PTEN-intact tumors. When some of them enter the early clinical phase, it is important to use PTEN as a biomarker for patient selection. Given that CHD1 inhibition sensitizes tumor cells to DNA-damaging agents, the combination of CHD1 inhibitors and DNA-damaging therapies should be tested in preclinical and clinical studies as well. It is also worth determining if CHD1 inhibitors synergize with AR or GR inhibitors in suppressing CRPC tumor growth and progression. However, caution should be taken when pharmacologically inhibiting CHD1 in prostate cancer with SPOP or MAP3K7 deletions, reasoning that CHD1 inhibition may play tumor-promoting roles in these contexts.
Aurora kinase inhibitors
Combining high-throughput epigenetic screening and pan-cancer drug sensitivity analyses, we reported that CHD1 promotes the susceptibility of cancer cells to inhibitors targeting Aurora kinases (26). Aurora kinases are key players in mitotic control. Among three mammalian paralogues, Aurora A is required for centrosome maturation and mitotic spindle assembly (122–124). Several small-molecule inhibitors targeting Aurora kinases have been tested in clinical trials, and subsets of patients showed significant clinical benefits from the single agent or in combination with other agents (125–131).
In our recent study, we uncovered that CHD1 loss impaired the in vitro and in vivo efficacy of Aurora kinase inhibitors, while high expression of CHD1 is associated with increased sensitivity in a pan-cancer manner (26). Prior studies demonstrated that the activity of Aurora A is largely modulated by the autophosphorylation and interaction with the co-activator TPX2 (132–135). Mechanistic studies revealed that the regulatory axis of CHD1-KPNA2 suppressed the interaction between Aurora A and TPX2, thereby rendering cancer cells more vulnerable to Aurora A inhibition (26). Furthermore, our studies in GEM models, patient-derived organoids, and patient samples showed that PTEN defects are associated with a better response to Aurora A inhibition in advanced prostate cancer by inducing CHD1 protein stabilization (26). This study establishes the important role of CHD1 in modulating Aurora kinases and provides insights for using PTEN and CHD1 as predictive biomarkers to improve patient selections in clinical trials of Aurora A inhibitors.
Checkpoint immunotherapy
Immunotherapy has shown only modest activity in advanced prostate cancer, partially due to low tumor mutation burden (TMB), lack of infiltrating T cells, and immunosuppressive TME (78, 113). Immune checkpoint inhibitors that target cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed death 1 (PD-1), and its ligand (PD-L1) display minimal or no activity as single agents or in combination with AR inhibitors in advanced prostate cancers (136–141). As noted above, CHD1 contributes to immunosuppressive TME by promoting MDSCs and suppressing tumor-killing T cells (27). Our recent studies in GEM and syngeneic models revealed that depletion of CHD1 reverses the immunosuppressive TME and sensitizes prostate tumors to the checkpoint immunotherapy (27). As a direct target gene of CHD1, IL-6 mediates the recruitment and activation of MDSCs in prostate tumors. Phenocopying CHD1 depletion, pharmacological inhibition of IL-6 and dual blockade of PD-1/CTLA-4 showed synergistic effects in preclinical models of PTEN-deficient prostate cancer (27). Notably, IL-6 inhibition was found to reduce immune-related adverse events in patients by de-coupling autoimmunity from antitumor immunity induced by immune checkpoint blockade (142). Further clinical studies are needed to test the above combinations in CRPC patients, particularly in PTEN-loss/CHD1-high tumors.
Conclusion and perspective
CHD1 was discovered over two decades ago, and significant progress has been made in understanding CHD1 biology. However, many questions remain to be answered, regarding CHD1’s context-dependent roles and the molecular basis in human diseases, as well as the modeling systems for studying CHD1 biology and therapeutics development.
It remains a debate on whether CHD1 is a tumor suppressor or an oncogene during tumorigenesis and cancer progression. Prior studies in cell lines and GEM models showed that CHD1 deletion alone is insufficient to drive prostate tumorigenesis (21, 23, 27). Functionally, CHD1 is required for conventional AR signaling and transcriptome (14, 16, 22, 23), which plays a key role in prostate cancer development and progression. However, CHD1 loss causes chromatin instability and lineage plasticity, resulting in the androgen-independent growth of prostate tumors and less sensitivity to antiandrogen therapy (14, 16, 22, 23, 61). As discussed above, the impact of CHD1 loss may vary when combined with different genetic alterations in prostate cancer. In the context of PTEN deficiency, CHD1 is essential for tumor growth and the immunosuppressive TME (25, 27); in contrast, CHD1 deletion augments the tumor-promoting role of MAP3K7 loss (20, 61). Hence, through interacting with different genetic events and altering the transcription of distinct pathways, CHD1 possesses the capability to exert both oncogenic and tumor-suppressive functions in context-dependent manners.
Most CHD family members are components of large multi-subunit complexes, however, CHD1 remodeler exists predominantly as a monomer or dimer (9, 143). The epigenetic machinery and interactome of CHD1 have been reported in different species. The yeast Chd1 was identified as a component of SAGA (Spt-Ada-Gcn5 acetyltransferase) and SLIK (SAGA-like) complexes, two highly homologous and conserved histone acetyltransferase complexes (144). Besides, yeast Chd1 forms complexes with RNA polymerase II and elongation factors Spt5 and Pob3 for the gene transcription (49, 145). Drosophila Chd1 was found to interact with SSRP1, a nuclear protein involved in the transcription regulation (36). Despite no direct binding to AR, mouse and human CHD1 proteins form complexes with AR cofactors, such as NCoR (146), HOXB13, ETV1, and FOXA1 (23), which mediate AR transcriptome changes upon CHD1 loss. In addition, both mouse and human CHD1 proteins interact with NF-κB, resulting in the activation of inflammatory pathways (25, 27). Given the context-dependent role of CHD1, it is crucial to identify the interactome of CHD1 in different genetic and molecular subsets of prostate cancer. Combined with high-throughput transcriptome and epigenetic profiling, these studies will uncover the molecular basis of CHD1 during cancer development, progression, and response to therapies.
Last but not least, better cancer model systems are needed for studying CHD1’s biology and its impact on drug responsiveness. Unlike other common cancer types, only a small number of human prostate cancer cell lines are available for preclinical studies. They are insufficient to recapitulate the diversity of molecular subtypes and genetic features in human disease. Although CHD1 loss are frequently found in primary or castration-resistant prostate tumors, none of those prostate cancer cell lines contains homogeneous deletions of CHD1. In the past decade, hundreds of patient-derived organoids and xenograft (PDX) models have been generated by multiple institutes and widely used in the prostate cancer research (147, 148). With high fidelity of histopathologic, genomic, and molecular characteristics, they capture the diverse molecular landscape of naïve prostate cancer or CRPC and enable the development and evaluation of biomarker-driven therapy. However, CHD1 loss or SPOP mutations rarely exist in prostate cancer PDX models. De Sarkar et al. recently identified two PDX models, LuCaP78 and LuCaP78CR, lack transcript and protein of CHD1 (90). Both lines, originating from the same patient, contain a combination of monoallelic genomic loss and epigenetic silencing of the remaining allele, show homology-directed DNA repair deficiency features, and are sensitive to IR and carboplatin treatment (90). It remains unclear why prostate tumors with CHD1 loss and/or SPOP mutation have a lower engraftment rate when generating PDX lines, but it is important to establish additional PDX models to mimic this distinct molecular subtype for biology studies and the development of effective therapeutics.
Several GEM models containing prostate-specific Chd1 deletion have been generated, and provide important tools for investigating CHD1 biology. However, none of them fully recapitulate the genetic and molecular features of prostate cancers with CHD1 deletion. Conditional knockout of Pten is the most used allele when generating GEM models of prostate cancer, and that’s why most CHD1 loss GEM models contain PTEN co-deletion (23, 27). These models provide good tools to study the roles of CHD1 in PTEN-deficient tumors, but they showed benign or less aggressive phenotypes due to CHD1’s essentiality in this context. Given the mutual exclusivity between CHD1 deletion and PTEN loss in prostate cancer patients, the co-deletion GEM models couldn’t represent genetic features in human diseases. Efforts have been made to generate GEM models to mimic the molecular subtype of CHD1 deletion and SPOP mutations, but the Chd1/Spop double-knockout mice displayed prostatic intraepithelial neoplasia at 12 months of age and failed to generate prostate adenocarcinoma (89). Using mouse prostate epithelial progenitor/stem cells (PrP/SC) graft model, Cramer’s group showed that co-suppression of CHD1 and MAP3K7 led to high-grade PIN and invasive carcinoma phenotypes (20). It is worth testing whether this combination drives tumorigenesis and progression in Pb-Cre-driven GEM models. Nevertheless, combining the next-generation CHD1 deletion GEM models with cutting-edge single-cell transcriptome profiling will help us fully understand the impact of CHD1 on disease progression, lineage plasticity, response to therapy, and the crosstalk between cancer cells and diverse immune components in the TME. Importantly, the knowledge obtained in prostate cancer will also inform the studies of CHD1 and other CHD remodelers in other cancer types.
Author contributions
HL and DZ designed the framework of the review. HL, LG, and DZ wrote the manuscript. LG and DZ drew the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported, for LG, in part by NIH 1R25CA240137-01A1 UPWARDS Training Program (Underrepresented Minorities Working Towards Research Diversity in Science) and CPRIT training Award RP210028 (to LG). This work was supported, for DZ, in part by CPRIT Recruitment of First-Time Tenure-Track Faculty Award RR190021 (to DZ, a CPRIT Scholar in Cancer Research), NIH/NCI R01 CA275990 (to DZ), and Prostate Cancer Foundation Challenge Award FP00016492 (to DZ).
Conflict of interest
DZ reports grants from NIH/NCI, CPRIT, and Prostate Cancer Foundation during the conduct of the study.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lu Y, Tan F, Zhao Y, Zhou S, Chen X, Hu Y, et al. A chromodomain-Helicase-DNA-Binding factor functions in chromatin modification and gene regulation. Plant Physiol (2020) 183(3):1035–46. doi: 10.1104/pp.20.00453
2. Marfella CG, Imbalzano AN. The chd family of chromatin remodelers. Mutat Res (2007) 618(1-2):30–40. doi: 10.1016/j.mrfmmm.2006.07.012
3. Clapier CR, Iwasa J, Cairns BR, Peterson CL. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol (2017) 18(7):407–22. doi: 10.1038/nrm.2017.26
4. Alendar A, Berns A. Sentinels of chromatin: chromodomain helicase DNA-binding proteins in development and disease. Genes Dev (2021) 35(21-22):1403–30. doi: 10.1101/gad.348897.121
5. Liu C, Kang N, Guo Y, Gong P. Advances in chromodomain helicase DNA-binding (CHD) proteins regulating stem cell differentiation and human diseases. Front Cell Dev Biol (2021) 9:710203. doi: 10.3389/fcell.2021.710203
6. Flaus A, Martin DM, Barton GJ, Owen-Hughes T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res (2006) 34(10):2887–905. doi: 10.1093/nar/gkl295
7. Smolle M, Venkatesh S, Gogol MM, Li H, Zhang Y, Florens L, et al. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol (2012) 19(9):884–92. doi: 10.1038/nsmb.2312
8. Stockdale C, Flaus A, Ferreira H, Owen-Hughes T. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J Biol Chem (2006) 281(24):16279–88. doi: 10.1074/jbc.M600682200
9. Lusser A, Urwin DL, Kadonaga JT. Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat Struct Mol Biol (2005) 12(2):160–6. doi: 10.1038/nsmb884
10. Gaspar-Maia A, Alajem A, Polesso F, Sridharan R, Mason MJ, Heidersbach A, et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature (2009) 460(7257):863–8. doi: 10.1038/nature08212
11. Baumgart SJ, Najafova Z, Hossan T, Xie W, Nagarajan S, Kari V, et al. CHD1 regulates cell fate determination by activation of differentiation-induced genes. Nucleic Acids Res (2017) 45(13):7722–35. doi: 10.1093/nar/gkx377
12. Farnung L, Vos SM, Wigge C, Cramer P. Nucleosome-Chd1 structure and implications for chromatin remodelling. Nature (2017) 550(7677):539–42. doi: 10.1038/nature24046
13. Tsukiyama T. The in vivo functions of ATP-dependent chromatin-remodelling factors. Nat Rev Mol Cell Biol (2002) 3(6):422–9. doi: 10.1038/nrm828
14. Burkhardt L, Fuchs S, Krohn A, Masser S, Mader M, Kluth M, et al. CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res (2013) 73(9):2795–805. doi: 10.1158/0008-5472.CAN-12-1342
15. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, et al. The genomic complexity of primary human prostate cancer. Nature (2011) 470(7333):214–20. doi: 10.1038/nature09744
16. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature (2012) 487(7406):239–43. doi: 10.1038/nature11125
17. Huang S, Gulzar ZG, Salari K, Lapointe J, Brooks JD, Pollack JR. Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene (2012) 31(37):4164–70. doi: 10.1038/onc.2011.590
18. Liu W, Lindberg J, Sui G, Luo J, Egevad L, Li T, et al. Identification of novel CHD1-associated collaborative alterations of genomic structure and functional assessment of CHD1 in prostate cancer. Oncogene (2012) 31(35):3939–48. doi: 10.1038/onc.2011.554
19. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell (2013) 153(3):666–77. doi: 10.1016/j.cell.2013.03.021
20. Rodrigues LU, Rider L, Nieto C, Romero L, Karimpour-Fard A, Loda M, et al. Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res (2015) 75(6):1021–34. doi: 10.1158/0008-5472.CAN-14-1596
21. Shenoy TR, Boysen G, Wang MY, Xu QZ, Guo W, Koh FM, et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann Oncol (2017) 28(7):1495–507. doi: 10.1093/annonc/mdx165
22. Zhang Z, Zhou C, Li X, Barnes SD, Deng S, Hoover E, et al. Loss of CHD1 promotes heterogeneous mechanisms of resistance to AR-targeted therapy via chromatin dysregulation. Cancer Cell (2020) 37(4):584–598 e11. doi: 10.1016/j.ccell.2020.03.001
23. Augello MA, Liu D, Deonarine LD, Robinson BD, Huang D, Stelloo S, et al. CHD1 loss alters AR binding at lineage-specific enhancers and modulates distinct transcriptional programs to drive prostate tumorigenesis. Cancer Cell (2019) 35(4):603–617 e8. doi: 10.1016/j.ccell.2019.03.001
24. Kari V, Mansour WY, Raul SK, Baumgart SJ, Mund A, Grade M, et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep (2016) 17(11):1609–23. doi: 10.15252/embr.201642352
25. Zhao D, Lu X, Wang G, Lan Z, Liao W, Li J, et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature (2017) 542(7642):484–8. doi: 10.1038/nature21357
26. Li H, Wang Y, Lin K, Venkadakrishnan VB, Bakht M, Shi W, et al. CHD1 promotes sensitivity to aurora kinase inhibitors by suppressing interaction of AURKA with its coactivator TPX2. Cancer Res (2022) 82(17):3088–101. doi: 10.1158/0008-5472.CAN-22-0631
27. Zhao D, Cai L, Lu X, Liang X, Li J, Chen P, et al. Chromatin regulator CHD1 remodels the immunosuppressive tumor microenvironment in PTEN-deficient prostate cancer. Cancer Discovery (2020) 10(9):1374–87. doi: 10.1158/2159-8290.CD-19-1352
28. Woodage T, Basrai MA, Baxevanis AD, Hieter P, Collins FS. Characterization of the CHD family of proteins. Proc Natl Acad Sci U.S.A. (1997) 94(21):11472–7. doi: 10.1073/pnas.94.21.11472
29. Yasin H ZF. Chromodomain helicase DNA-binding proteins and neurodevelopmental disorders. J Trans Genet Genomics (2020) 4):307–19. doi: 10.20517/jtgg.2020.30
30. Stokes DG, Perry RP. DNA-Binding and chromatin localization properties of CHD1. Mol Cell Biol (1995) 15(5):2745–53. doi: 10.1128/MCB.15.5.2745
31. Delmas V, Stokes DG, Perry RP. A mammalian DNA-binding protein that contains a chromodomain and an SNF2/SWI2-like helicase domain. Proc Natl Acad Sci U.S.A. (1993) 90(6):2414–8. doi: 10.1073/pnas.90.6.2414
32. Mansfield RE, Musselman CA, Kwan AH, Oliver SS, Garske AL, Davrazou F, et al. Plant homeodomain (PHD) fingers of CHD4 are histone H3-binding modules with preference for unmodified H3K4 and methylated H3K9. J Biol Chem (2011) 286(13):11779–91. doi: 10.1074/jbc.M110.208207
33. Flanagan JF, Blus BJ, Kim D, Clines KL, Rastinejad F, Khorasanizadeh S. Molecular implications of evolutionary differences in CHD double chromodomains. J Mol Biol (2007) 369(2):334–42. doi: 10.1016/j.jmb.2007.03.024
34. Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, et al. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature (2005) 438(7071):1181–5. doi: 10.1038/nature04290
35. Persson J, Ekwall K. Chd1 remodelers maintain open chromatin and regulate the epigenetics of differentiation. Exp Cell Res (2010) 316(8):1316–23. doi: 10.1016/j.yexcr.2010.02.029
36. Kelley DE, Stokes DG, Perry RP. CHD1 interacts with SSRP1 and depends on both its chromodomain and its ATPase/helicase-like domain for proper association with chromatin. Chromosoma (1999) 108(1):10–25. doi: 10.1007/s004120050347
37. Hauk G, McKnight JN, Nodelman IM, Bowman GD. The chromodomains of the Chd1 chromatin remodeler regulate DNA access to the ATPase motor. Mol Cell (2010) 39(5):711–23. doi: 10.1016/j.molcel.2010.08.012
38. McKnight JN, Jenkins KR, Nodelman IM, Escobar T, Bowman GD. Extranucleosomal DNA binding directs nucleosome sliding by Chd1. Mol Cell Biol (2011) 31(23):4746–59. doi: 10.1128/MCB.05735-11
39. Nodelman IM, Bleichert F, Patel A, Ren R, Horvath KC, Berger JM, et al. Interdomain communication of the Chd1 chromatin remodeler across the DNA gyres of the nucleosome. Mol Cell (2017) 65(3):447–459 e6. doi: 10.1016/j.molcel.2016.12.011
40. Langst G, Manelyte L. Chromatin remodelers: From function to dysfunction. Genes (Basel) (2015) 6(2):299–324. doi: 10.3390/genes6020299
41. Schoberleitner I, Bauer I, Huang A, Andreyeva EN, Sebald J, Pascher K, et al. CHD1 controls H3.3 incorporation in adult brain chromatin to maintain metabolic homeostasis and normal lifespan. Cell Rep (2021) 37(1):109769. doi: 10.1016/j.celrep.2021.109769
42. Konev AY, Tribus M, Park SY, Podhraski V, Lim CY, Emelyanov AV, et al. CHD1 motor protein is required for deposition of histone variant h3.3 into chromatin in vivo. Science (2007) 317(5841):1087–90. doi: 10.1126/science.1145339
43. Lin JJ, Lehmann LW, Bonora G, Sridharan R, Vashisht AA, Tran N, et al. Mediator coordinates PIC assembly with recruitment of CHD1. Genes Dev (2011) 25(20):2198–209. doi: 10.1101/gad.17554711
44. Morettini S, Tribus M, Zeilner A, Sebald J, Campo-Fernandez B, Scheran G, et al. The chromodomains of CHD1 are critical for enzymatic activity but less important for chromatin localization. Nucleic Acids Res (2011) 39(8):3103–15. doi: 10.1093/nar/gkq1298
45. Guzman-Ayala M, Sachs M, Koh FM, Onodera C, Bulut-Karslioglu A, Lin CJ, et al. Chd1 is essential for the high transcriptional output and rapid growth of the mouse epiblast. Development (2015) 142(1):118–27. doi: 10.1242/dev.114843
46. Lee Y, Park D, Iyer VR. The ATP-dependent chromatin remodeler Chd1 is recruited by transcription elongation factors and maintains H3K4me3/H3K36me3 domains at actively transcribed and spliced genes. Nucleic Acids Res (2017) 45(12):7180–90. doi: 10.1093/nar/gkx321
47. Sims RJ 3rd, Millhouse S, Chen CF, Lewis BA, Erdjument-Bromage H, Tempst P, et al. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell (2007) 28(4):665–76. doi: 10.1016/j.molcel.2007.11.010
48. Radman-Livaja M, Quan TK, Valenzuela L, Armstrong JA, van Welsem T, Kim T, et al. A key role for Chd1 in histone H3 dynamics at the 3 ‘ ends of long genes in yeast. PloS Genet (2012) 8(7). doi: 10.1371/journal.pgen.1002811
49. Alén C, Kent NA, Jones HS, O'Sullivan J, Aranda A, Proudfoot NJ. A role for chromatin remodeling in transcriptional termination by RNA polymerase II. Mol Cell (2002) 10(6):1441–52. doi: 10.1016/S1097-2765(02)00778-5
50. Ryan DP, Sundaramoorthy R, Martin D, Singh V, Owen-Hughes T. The DNA-binding domain of the Chd1 chromatin-remodelling enzyme contains SANT and SLIDE domains. EMBO J (2011) 30(13):2596–609. doi: 10.1038/emboj.2011.166
51. Pilarowski GO, Vernon HJ, Applegate CD, Boukas L, Cho MT, Gurnett CA, et al. Missense variants in the chromatin remodeler CHD1 are associated with neurodevelopmental disability. J Med Genet (2018) 55(8):561–6. doi: 10.1136/jmedgenet-2017-104759
52. O'Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun (2014) 5:5595. doi: 10.1038/ncomms6595
53. De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature (2014) 515(7526):209–15. doi: 10.1038/nature13772
54. Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature (2012) 485(7397):242–5. doi: 10.1038/nature11011
55. Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell (2018) 173(2):291–304 e6. doi: 10.1016/j.cell.2018.03.022
56. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell (2018) 173(2):321–337 e10. doi: 10.1016/j.cell.2018.03.035
57. Zhu Y, Mo M, Wei Y, Wu J, Pan J, Freedland SJ, et al. Epidemiology and genomics of prostate cancer in Asian men. Nat Rev Urol (2021) 18(5):282–301. doi: 10.1038/s41585-021-00442-8
58. Li J, Xu C, Lee HJ, Ren S, Zi X, Zhang Z, et al. A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature (2020) 580(7801):93–9. doi: 10.1038/s41586-020-2135-x
59. Miklos Diossy VT, Li H, Zhou J, Sztupinszki Z, Young D, Nousome D, et al. Increased frequency of CHD1 deletions in prostate cancers of African American men is associated with distinct homologous recombination deficiency associated DNA aberration profiles. medRxiv (2021). doi: 10.1101/2021.02.08.21251199
60. Cancer Genome Atlas Research, N. The molecular taxonomy of primary prostate cancer. Cell (2015) 163(4):1011–25. doi: 10.1016/j.cell.2015.10.025
61. Jillson LK, Rider LC, Rodrigues LU, Romero L, Karimpour-Fard A, Nieto C, et al. MAP3K7 loss drives enhanced androgen signaling and independently confers risk of recurrence in prostate cancer with joint loss of CHD1. Mol Cancer Res (2021) 19(7):1123–36. doi: 10.1158/1541-7786.MCR-20-0913
62. Tereshchenko IV, Zhong H, Chekmareva MA, Kane-Goldsmith N, Santanam U, Petrosky W, et al. ERG and CHD1 heterogeneity in prostate cancer: use of confocal microscopy in assessment of microscopic foci. Prostate (2014) 74(15):1551–9. doi: 10.1002/pros.22873
63. Green CD, Huang Y, Dou X, Yang L, Liu Y, Han JJ. Impact of dietary interventions on noncoding RNA networks and mRNAs encoding chromatin-related factors. Cell Rep (2017) 18(12):2957–68. doi: 10.1016/j.celrep.2017.03.001
64. Licciardello MP, Kubicek S. Pharmacological treats for SUMO addicts. Pharmacol Res (2016) 107:390–7. doi: 10.1016/j.phrs.2016.01.004
65. Yu B, Swatkoski S, Holly A, Lee LC, Giroux V, Lee CS, et al. Oncogenesis driven by the Ras/Raf pathway requires the SUMO E2 ligase Ubc9. Proc Natl Acad Sci U.S.A. (2015) 112(14):E1724–33. doi: 10.1073/pnas.1415569112
66. Domingues P, Golebiowski F, Tatham MH, Lopes AM, Taggart A, Hay RT, et al. Global reprogramming of host SUMOylation during influenza virus infection. Cell Rep (2015) 13(7):1467–80. doi: 10.1016/j.celrep.2015.10.001
67. Tan S, Ding K, Li R, Zhang W, Li G, Kong X, et al. Identification of miR-26 as a key mediator of estrogen stimulated cell proliferation by targeting CHD1, GREB1 and KPNA2. Breast Cancer Res (2014) 16(2):R40. doi: 10.1186/bcr3644
68. Piatti P, Lim CY, Nat R, Villunger A, Geley S, Shue YT, et al. Embryonic stem cell differentiation requires full length Chd1. Sci Rep (2015) 5:8007. doi: 10.1038/srep08007
69. Dirksen EH, Pinkse MW, Rijkers DT, Cloos J, Liskamp RM, Slijper M, et al. Investigating the dynamic nature of the interactions between nuclear proteins and histones upon DNA damage using an immobilized peptide chemical proteomics approach. J Proteome Res (2006) 5(9):2380–8. doi: 10.1021/pr060278b
70. Hickey CM, Wilson NR, Hochstrasser M. Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol (2012) 13(12):755–66. doi: 10.1038/nrm3478
71. Jalal D, Chalissery J, Hassan AH. Genome maintenance in saccharomyces cerevisiae: the role of SUMO and SUMO-targeted ubiquitin ligases. Nucleic Acids Res (2017) 45(5):2242–61. doi: 10.1093/nar/gkw1369
72. Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol (2007) 8(12):947–56. doi: 10.1038/nrm2293
73. Xia L, Huang W, Bellani M, Seidman MM, Wu K, Fan D, et al. CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell (2017) 31(5):653–668 e7. doi: 10.1016/j.ccell.2017.04.005
74. Cai Y, Geutjes EJ, de Lint K, Roepman P, Bruurs L, Yu LR, et al. The NuRD complex cooperates with DNMTs to maintain silencing of key colorectal tumor suppressor genes. Oncogene (2014) 33(17):2157–68. doi: 10.1038/onc.2013.178
75. Wang Y, Chen Y, Bao L, Zhang B, Wang JE, Kumar A, et al. CHD4 promotes breast cancer progression as a coactivator of hypoxia-inducible factors. Cancer Res (2020) 80(18):3880–91. doi: 10.1158/0008-5472.CAN-20-1049
76. Bagchi A, Papazoglu C, Wu Y, Capurso D, Brodt M, Francis D, et al. CHD5 is a tumor suppressor at human 1p36. Cell (2007) 128(3):459–75. doi: 10.1016/j.cell.2006.11.052
77. Kolla V, Zhuang T, Higashi M, Naraparaju K, Brodeur GM. Role of CHD5 in human cancers: 10 years later. Cancer Res (2014) 74(3):652–8. doi: 10.1158/0008-5472.CAN-13-3056
78. Wang G, Zhao D, Spring DJ, DePinho RA. Genetics and biology of prostate cancer. Genes Dev (2018) 32(17-18):1105–40. doi: 10.1101/gad.315739.118
79. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science (2005) 310(5748):644–8. doi: 10.1126/science.1117679
80. Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell (2011) 20(4):457–71. doi: 10.1016/j.ccr.2011.09.001
81. Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature (2005) 437(7057):436–9. doi: 10.1038/nature04020
82. Metzger E, Willmann D, McMillan J, Forne I, Metzger P, Gerhardt S, et al. Assembly of methylated KDM1A and CHD1 drives androgen receptor-dependent transcription and translocation. Nat Struct Mol Biol (2016) 23(2):132–9. doi: 10.1038/nsmb.3153
83. Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell (2006) 10(1):105–16. doi: 10.1016/j.devcel.2005.10.017
84. Quintanal-Villalonga Á, Chan JM, Yu HA, Pe'er D, Sawyers CL, Sen T, et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol (2020) 17(6):360–71. doi: 10.1038/s41571-020-0340-z
85. Antonarakis ES. Targeting lineage plasticity in prostate cancer. Lancet Oncol (2019) 20(10):1338–40. doi: 10.1016/S1470-2045(19)30497-8
86. Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol (2018) 15(5):271–86. doi: 10.1038/nrurol.2018.22
87. Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell (2012) 47(4):497–510. doi: 10.1016/j.molcel.2012.07.029
88. Zhou J, Li J, Serafim RB, Ketchum S, Ferreira CG, Liu JC, et al. Human CHD1 is required for early DNA-damage signaling and is uniquely regulated by its n terminus. Nucleic Acids Res (2018) 46(8):3891–905. doi: 10.1093/nar/gky128
89. Zhu Y, Wen J, Huang G, Mittlesteadt J, Wen X, Lu X. CHD1 and SPOP synergistically protect prostate epithelial cells from DNA damage. Prostate (2021) 81(1):81–8. doi: 10.1002/pros.24080
90. De Sarkar N, Dasgupta S, Chatterjee P, Coleman I, Ha G, Ang LS, et al. Genomic attributes of homology-directed DNA repair deficiency in metastatic prostate cancer. JCI Insight (2021) 6(23). doi: 10.1172/jci.insight.152789
91. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet (2012) 44(6):685–9. doi: 10.1038/ng.2279
92. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell (2015) 162(2):454. doi: 10.1016/j.cell.2015.05.001
93. Boysen G, Rodrigues DN, Rescigno P, Seed G, Dolling D, Riisnaes R, et al. SPOP-Mutated/CHD1-Deleted lethal prostate cancer and abiraterone sensitivity. Clin Cancer Res (2018) 24(22):5585–93. doi: 10.1158/1078-0432.CCR-18-0937
94. Boysen G, Barbieri CE, Prandi D, Blattner M, Chae SS, Dahija A, et al. SPOP mutation leads to genomic instability in prostate cancer. Elife (2015) 4. doi: 10.7554/eLife.09207
95. Wang Z, Song Y, Ye M, Dai X, Zhu X, Wei W. The diverse roles of SPOP in prostate cancer and kidney cancer. Nat Rev Urol (2020) 17(6):339–50. doi: 10.1038/s41585-020-0314-z
96. Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res (2014) 74(19):5631–43. doi: 10.1158/0008-5472.CAN-14-0476
97. Li C, Ao J, Fu J, Lee DF, Xu J, Lonard D, et al. Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene (2011) 30(42):4350–64. doi: 10.1038/onc.2011.151
98. Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, Liu P, et al. SPOP promotes ubiquitination and degradation of the ERG oncoprotein to suppress prostate cancer progression. Mol Cell (2015) 59(6):917–30. doi: 10.1016/j.molcel.2015.07.026
99. An J, Ren S, Murphy SJ, Dalangood S, Chang C, Pang X, et al. Truncated ERG oncoproteins from TMPRSS2-ERG fusions are resistant to SPOP-mediated proteasome degradation. Mol Cell (2015) 59(6):904–16. doi: 10.1016/j.molcel.2015.07.025
100. An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep (2014) 6(4):657–69. doi: 10.1016/j.celrep.2014.01.013
101. Blattner M, Liu D, Robinson BD, Huang D, Poliakov A, Gao D, et al. SPOP mutation drives prostate tumorigenesis in vivo through coordinate regulation of PI3K/mTOR and AR signaling. Cancer Cell (2017) 31(3):436–51. doi: 10.1016/j.ccell.2017.02.004
102. Stopsack KH, Nandakumar S, Wibmer AG, Haywood S, Weg ES, Barnett ES, et al. Oncogenic genomic alterations, clinical phenotypes, and outcomes in metastatic castration-sensitive prostate cancer. Clin Cancer Res (2020) 26(13):3230–8. doi: 10.1158/1078-0432.CCR-20-0168
103. Swami U, Isaacsson Velho P, Nussenzveig R, Chipman J, Sacristan Santos V, Erickson S, et al. Association of SPOP mutations with outcomes in men with de Novo metastatic castration-sensitive prostate cancer. Eur Urol (2020) 78(5):652–6. doi: 10.1016/j.eururo.2020.06.033
104. Xiao M, Fried JS, Ma J, Su Y, Boohaker RJ, Zeng Q, et al. A disease-relevant mutation of SPOP highlights functional significance of ATM-mediated DNA damage response. Signal Transduct Target Ther (2021) 6(1):17. doi: 10.1038/s41392-020-00381-7
105. Zhang D, Wang H, Sun M, Yang J, Zhang W, Han S, et al. Speckle-type POZ protein, SPOP, is involved in the DNA damage response. Carcinogenesis (2014) 35(8):1691–7. doi: 10.1093/carcin/bgu022
106. Hjorth-Jensen K, Maya-Mendoza A, Dalgaard N, Sigurðsson JO, Bartek J, Iglesias-Gato D, et al. SPOP promotes transcriptional expression of DNA repair and replication factors to prevent replication stress and genomic instability. Nucleic Acids Res (2018) 46(18):9891. doi: 10.1093/nar/gky719
107. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem (1998) 273(22):13375–8. doi: 10.1074/jbc.273.22.13375
108. Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell (1998) 95(1):29–39. doi: 10.1016/S0092-8674(00)81780-8
109. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell (2008) 133(3):403–14. doi: 10.1016/j.cell.2008.04.013
110. Aparicio AM, Shen L, Tapia EL, Lu JF, Chen HC, Zhang J, et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin Cancer Res (2016) 22(6):1520–30. doi: 10.1158/1078-0432.CCR-15-1259
111. Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell (2003) 4(3):209–21. doi: 10.1016/S1535-6108(03)00215-0
112. Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature (2011) 470(7333):269–73. doi: 10.1038/nature09677
113. Gerritsen WR, Sharma P. Current and emerging treatment options for castration-resistant prostate cancer: a focus on immunotherapy. J Clin Immunol (2012) 32(1):25–35. doi: 10.1007/s10875-011-9595-6
114. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21(8):485–98. doi: 10.1038/s41577-020-00490-y
115. Calagua C, Ficial M, Jansen CS, Hirz T, Del Balzo L, Wilkinson S, et al. A subset of localized prostate cancer displays an immunogenic phenotype associated with losses of key tumor suppressor genes. Clin Cancer Res (2021) 27(17):4836–47. doi: 10.1158/1078-0432.CCR-21-0121
116. Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science (2017) 355(6330):1152–8. doi: 10.1126/science.aam7344
117. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair defects and olaparib in metastatic prostate cancer. N Engl J Med (2015) 373(18):1697–708. doi: 10.1056/NEJMoa1506859
118. Schiewer MJ, Knudsen KE. DNA Damage response in prostate cancer. Cold Spring Harb Perspect Med (2019) 9(1). doi: 10.1101/cshperspect.a030486
119. Huggins C, Hodges CV. Studies on prostatic cancer - I the effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res (1941) 1(4):293–7. doi: 10.3322/canjclin.22.4.232
120. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer (2015) 15(12):701–11. doi: 10.1038/nrc4016
121. Hernández-Llodrà S, Segalés L, Juanpere N, Marta Lorenzo T, Salido M, Nonell L, et al. SPOP and CHD1 alterations in prostate cancer: Relationship with PTEN loss, tumor grade, perineural infiltration, and PSA recurrence. Prostate (2021) 81(16):1267–77. doi: 10.1002/pros.24218
122. Hannak E, Kirkham M, Hyman AA, Oegema K. Aurora-a kinase is required for centrosome maturation in caenorhabditis elegans. J Cell Biol (2001) 155(7):1109–15. doi: 10.1083/jcb.200108051
123. Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell (1995) 81(1):95–105. doi: 10.1016/0092-8674(95)90374-7
124. Berdnik D, Knoblich JA. Drosophila aurora-a is required for centrosome maturation and actin-dependent asymmetric protein localization during mitosis. Curr Biol (2002) 12(8):640–7. doi: 10.1016/S0960-9822(02)00766-2
125. Mehra R, Serebriiskii IG, Burtness B, Astsaturov I, Golemis EA. Aurora kinases in head and neck cancer. Lancet Oncol (2013) 14(10):E425–35. doi: 10.1016/S1470-2045(13)70128-1
126. Pitts TM, Bradshaw-Pierce EL, Bagby SM, Hyatt SL, Selby HM, Spreafico A, et al. Antitumor activity of the aurora a selective kinase inhibitor, alisertib, against preclinical models of colorectal cancer. Oncotarget (2016) 7(31):50290–301. doi: 10.18632/oncotarget.10366
127. Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of n-myc in childhood neuroblastoma. Cancer Cell (2013) 24(1):75–89. doi: 10.1016/j.ccr.2013.05.005
128. Falchook G, Coleman RL, Roszak A, Behbakht K, Matulonis U, Ray-Coquard I, et al. Alisertib in combination with weekly paclitaxel in patients with advanced breast cancer or recurrent ovarian cancer: A randomized clinical trial. JAMA Oncol (2019) 5(1):e183773. doi: 10.1001/jamaoncol.2018.3773
129. Melichar B, Adenis A, Lockhart AC, Bennouna J, Dees EC, Kayaleh O, et al. Safety and activity of alisertib, an investigational aurora kinase a inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol (2015) 16(4):395–405. doi: 10.1016/S1470-2045(15)70051-3
130. DuBois SG, Mosse YP, Fox E, Kudgus RA, Reid JM, McGovern R, et al. Phase II trial of alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma. Clin Cancer Res (2018) 24(24):6142–9. doi: 10.1158/1078-0432.CCR-18-1381
131. Beltran H, Oromendia C, Danila DC, Montgomery B, Hoimes C, Szmulewitz RZ, et al. A phase II trial of the aurora kinase a inhibitor alisertib for patients with castration-resistant and neuroendocrine prostate cancer: Efficacy and biomarkers. Clin Cancer Res (2019) 25(1):43–51. doi: 10.1158/1078-0432.CCR-18-1912
132. Littlepage LE, Wu H, Andresson T, Deanehan JK, Amundadottir LT, Ruderman JV. Identification of phosphorylated residues that affect the activity of the mitotic kinase aurora-a. Proc Natl Acad Sci United States America (2002) 99(24):15440–5. doi: 10.1073/pnas.202606599
133. Walter AO, Seghezzi W, Korver W, Sheung J, Lees E. The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene (2000) 19(42):4906–16. doi: 10.1038/sj.onc.1203847
134. Bayliss R, Sardon T, Vernos I, Conti E. Structural basis of aurora-a activation by TPX2 at the mitotic spindle. Mol Cell (2003) 12(4):851–62. doi: 10.1016/S1097-2765(03)00392-7
135. Kufer TA, Silljé HH, Körner R, Gruss OJ, Meraldi P, Nigg EA. Human TPX2 is required for targeting aurora-a kinase to the spindle. Cell Motil Cytoskeleton (2003) 54(2):179–9. doi: 10.1083/jcb.200204155
136. Maia MC, Hansen AR. A comprehensive review of immunotherapies in prostate cancer. Crit Rev Oncol Hematol (2017) 113:292–303. doi: 10.1016/j.critrevonc.2017.02.026
137. Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol (2017) 35(1):40–7. doi: 10.1200/JCO.2016.69.1584
138. Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol (2014) 15(7):700–12. doi: 10.1016/S1470-2045(14)70189-5
139. Graff JN, Alumkal JJ, Drake CG, Thomas GV, Redmond WL, Farhad M, et al. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget (2016) 7(33):52810–7. doi: 10.18632/oncotarget.10547
140. Powles T, Yuen KC, Gillessen S, Kadel EE 3rd, Rathkopf D, Matsubara N, et al. Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: a randomized phase 3 trial. Nat Med (2022) 28(1):144–53. doi: 10.1038/s41591-021-01600-6
141. Subudhi SK, Siddiqui BA, Aparicio AM, Yadav SS, Basu S, Chen H, et al. Combined CTLA-4 and PD-L1 blockade in patients with chemotherapy-naive metastatic castration-resistant prostate cancer is associated with increased myeloid and neutrophil immune subsets in the bone microenvironment. J Immunother Cancer (2021) 9(10). doi: 10.1136/jitc-2021-002919
142. Hailemichael Y, Johnson DH, Abdel-Wahab N, Foo WC, Bentebibel SE, Daher M, et al. Interleukin-6 blockade abrogates immunotherapy toxicity and promotes tumor immunity. Cancer Cell (2022) 40(5):509–523 e6. doi: 10.1016/j.ccell.2022.04.004
143. Tran HG, Steger DJ, Iyer VR, Johnson AD. The chromo domain protein chd1p from budding yeast is an ATP-dependent chromatin-modifying factor. EMBO J (2000) 19(10):2323–31. doi: 10.1093/emboj/19.10.2323
144. Pray-Grant MG, Daniel JA, Schieltz D, Yates JR 3rd, Grant PA. Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature (2005) 433(7024):434–8. doi: 10.1038/nature03242
145. Simic R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, et al. Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J (2003) 22(8):1846–56. doi: 10.1093/emboj/cdg179
146. Tai HH, Geisterfer M, Bell JC, Moniwa M, Davie JR, Boucher L, et al. CHD1 associates with NCoR and histone deacetylase as well as with RNA splicing proteins. Biochem Biophys Res Commun (2003) 308(1):170–6. doi: 10.1016/S0006-291X(03)01354-8
147. Palanisamy N, Yang J, Shepherd PDA, Li-Ning-Tapia EM, Labanca E, Manyam GC, et al. The MD Anderson prostate cancer patient-derived xenograft series (MDA PCa PDX) captures the molecular landscape of prostate cancer and facilitates marker-driven therapy development. Clin Cancer Res (2020) 26(18):4933–46. doi: 10.1158/1078-0432.CCR-20-0479
Keywords: CHD1, prostate cancer, epigenetic remodeler, dysregulation, therapeutic strategy
Citation: Li H, Gigi L and Zhao D (2023) CHD1, a multifaceted epigenetic remodeler in prostate cancer. Front. Oncol. 13:1123362. doi: 10.3389/fonc.2023.1123362
Received: 13 December 2022; Accepted: 11 January 2023;
Published: 26 January 2023.
Edited by:
Shuai Gao, New York Medical College, United StatesReviewed by:
Changmeng Cai, University of Massachusetts Boston, United StatesDaisuke Obinata, Nihon University, Japan
Copyright © 2023 Li, Gigi and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Di Zhao, ZHpoYW8yQG1kYW5kZXJzb24ub3Jn