 Wei Zhang
Wei Zhang Xiaoqiang Zheng
Xiaoqiang Zheng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 04 April 2023
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1122322
This article is part of the Research TopicSelf-assembly Biomaterials as Theragnostic for Injury and DiseaseView all 10 articles
In addition to the rapid development of immune checkpoint inhibitors, there has also been a surge in the development of self-assembly immunotherapy drugs. Based on the immune target, traditional tumor immunotherapy drugs are classified into five categories, namely immune checkpoint inhibitors, direct immune modulators, adoptive cell therapy, oncolytic viruses, and cancer vaccines. Additionally, the emergence of self-assembled drugs with improved precision and environmental sensitivity offers a promising innovation approach to tumor immunotherapy. Despite rapid advances in tumor immunotherapy drug development, all candidate drugs require preclinical evaluation for safety and efficacy, and conventional evaluations are primarily conducted using two-dimensional cell lines and animal models, an approach that may be unsuitable for immunotherapy drugs. The patient-derived xenograft and organoids models, however, maintain the heterogeneity and immunity of the pathological tumor heterogeneity.
Clinical sample sequencing and experiments using animal models have demonstrated that the molecular mechanism of tumorigenesis is due to gene mutations induced by oncogene and anti-oncogene. However, oncogene mutation is not the only factor that eventually causes the development of cancer (1–3). Several preclinical and clinical studies have reported that multiple factors exist between the occurrence of oncogene mutations in cells and the tumors in situ, such as the tumor microenvironment (TME) (4–6).TME as a concept was proposed by Ioannides in 1993 (7). Currently, TME is regarded as the presentation of non-tumor cells and their components in tumors, including the protein molecules produced and released by them(8). Furthermore, the metabolic disorders of TME cells result in the consumption of nutrients, acidification of environmental pH, hypoxia, and the production of regulatory metabolites, thus influencing the immune response to tumors as well as the overexpression of immune checkpoint molecules and tumor metastasis (9–11). The abortive phenomenon of various tumor therapy drugs in previous preclinical and clinical trials has been explained by the discovery of TME.
The concept of immunotherapy was first introduced by William Coley in the 1890s(12). Later, Honjo discovered that programmed death receptor 1 (PD-1) is an inducible gene on activated T lymphocytes, which led to the discovery of cancer immunotherapy through blocking PD-1 (13). Meanwhile, a protein on the molecular surface of immune cells called cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) was discovered by James P. Allison to act as a “molecular brake” that prevents the immune system from responding. Inhibition of CTLA-4 can make T cells proliferate and attack tumor cells (14). The Nobel Prize in Physiology or Medicine was awarded to them in recognition of their contribution to tumor immunotherapy in 2018. Currently, tumor immunotherapy drugs can be mainly divided into five categories: antibody drugs such as immune checkpoint inhibitors (ICIs) (15), direct immune modulators (16–19), chimeric antigen receptor (CAR) -T cells(20), oncolytic viruses (OVs) (21) and cancer vaccines (22).Despite this progress, contributing to off-target toxicity, tissue heterogeneity, poor immunogenicity and tumor infiltration, the clinical use of tumor immunotherapy remains limited to a small subset of cancers. The development of self-assembly nanotechnology provides an opportunity for enhancing the effectiveness and reducing the toxicity of traditional drugs, and a series of nanomaterials were used in the preclinical study of cancer (23). This technology assembles molecules with different functions into highly ordered nanosystems with non-covalent bonds, which is a strategy for building powerful drugs (24).
In recent years, the development of experimental models to accurately replicate the pathophysiology of tumors has become one of the main challenges in the development of new drugs. Researchers emphasize patient-derived tumor xenografts (PDXs) (25)and patient-derived organoids (PDOs) (26) as potential solutions to these problems. PDX preserves the histological structure, degree of differentiation, morphological features, and molecular characteristics of most primary tumors and can better mimic their response to treatment. PDO models and three-dimensional (3D) culture can reproduce TME and biological behavior of tumor cells in vitro by reconstructing 3D communication networks of cell-cell and cell-extracellular matrix (ECM) interactions (16–19). Drug research and development have benefited greatly from the PDX and PDO models (27).
In this review, we discuss the latest advances of the technology in PDX and PDO models for tumor immunotherapy research. We emphasize the use of these preclinical setting to study tumor cell-immune cell interactions and to explore immunotherapeutic drug screens. We also investigate the application of these preclinical models to novel self-assembling drug development and discuss the challenges that need to be overcome to make possible a more widespread and rationalized use of PDX and PDO models. A careful consideration and evaluation must be given to the complexity of humanized PDX and PDO mice and their limitations. As a result, there will be a greater chance of achieving effective research results. In any case, we hope that the optimization of humanized PDX and PDO mouse models will make significant contributions to tumor immunotherapy and personalized medicine for improving the outcome of cancer patients in the future.
Over the years, PDXs have been used to study several aspects of oncological diseases, especially for individualized drug development. It has been proposed that PDX models not only recapitulate key characteristics of human tumors with high fidelity, but also exhibit treatment responses that are concordant with human responses(28–30). In recent years, breakthroughs in tumor immunotherapy have placed increased demands on the development of appropriate preclinical assessment models to evaluate tumor immune responses. Therefore, humanized PDX models have been developed to evaluate the efficacy of immunotherapeutic approaches in cancer. The fundamentals of the humanized PDX model are as follow. In summary, pieces of solid tumors are obtained through surgery or biopsy procedures, and these samples are implanted into the dorsal region or the same organ of immunocompromised mice. To simulate a more realistic state of functional human immune system (HIS), human peripheral blood mononuclear cells (PBMC), CD34+ hematopoietic stem cells (HSC), or other immune cells can be transplanted into immunodeficient mice such as non-obese diabetes (NOD)- severe combined immune deficiency (SCID) gamma(NSG)mice. After human immune reconstruction, patient-derived tumor tissues can be transplanted to create a dual immunogenic model with similar heterogeneity and tumor immune microenvironment (TIME) as patients. This model can not only simulate the growth process of tumors in patients, but also simulate the interaction between a cancer cell and the HIS. The construction process of humanized PDX models is presented in Figure 1.
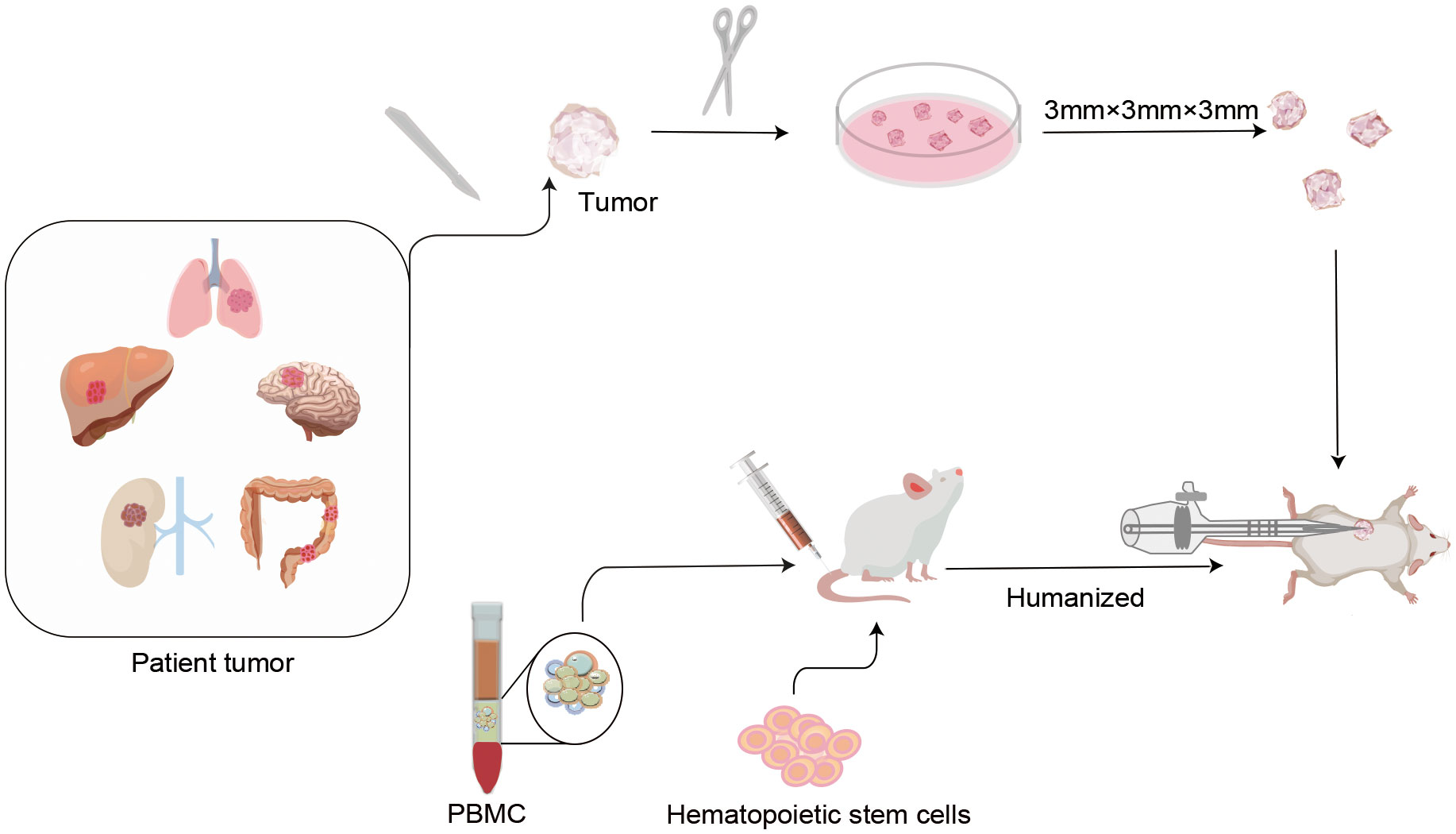
Figure 1 Protocols of humanized patient-derived tumor xenografts (PDX) model construction. In the first step, humanized mice were established by transplanting isolated human peripheral blood mononuclear cells (PBMC) or CD34+ human hematopoietic stem cells (HSC), etc. into severely combined immunodeficient mice. After the human immune system is successfully implanted, a novel humanized PDX model is established by inoculating patient-derived tumor tissues into humanized mice. These types of models not only mimic the phenotypic and molecular characteristics of the original tumor in the patient, but also reproduce the cross-talk between the tumor and the immune system. This is of critical significance for individualized drug marker screening and drug development for tumor immunotherapy.
Humanized PDX models have provided a tremendous boost to the study of tumor pathogenesis and drug development. However, there are still limitations of humanized PDX models: 1) the time period required to build PDX models from patients is long and may take up to 6 months (or longer), 2) the high cost and low throughput, 3) lack of maturation of innate immune cells, coupled with insufficient ability to generate antigen-specific antibodies, 4) limited education of T cells in absence of murine thymus, 5)deficient HLA molecules, and 6) the difficulty in generating lymph node structures and germinal centers(31). These limitations have led to several ongoing efforts to develop novel humanized preclinical models and platforms to develop therapeutic strategies that enhance response to immunotherapy. In general, it is believed that the robustness of drug-screening data will increase when both human-derived immune reconstruction and data analysis become more standardized.
In 2009, Hans Clevers’ team successfully cultured mouse intestinal organoids that self-renew and maintain the villous structure of intestinal gland pits in vitro, bringing new starting for development of cancer therapeutic approaches (32). As an in vitro 3D organ, PDO can not only mimic the cell composition and structure in tumor growth, but also perform specific gene editing, which can satisfy complex tumor microenvironment research and potential drug screening. PDO and organoid-derived PDX(PDOX), as an emerging field, have attracted much attention since they can provide a cancer pre-clinical platform to recapitulate the patient’s tumor and promote translating novel treatments from bench to bedside (33–35). Over the past decades, several PDOs have been successfully cultured, including gastric tumors (36), breast tumors (37), bladder tumors (38), and ovarian tumors (39). The establishment and subsequent screening of PDO/PDOX can generally be completed in a shorter period than for PDX. The model construction process for PDO and PDOX is depicted in Figure 2.
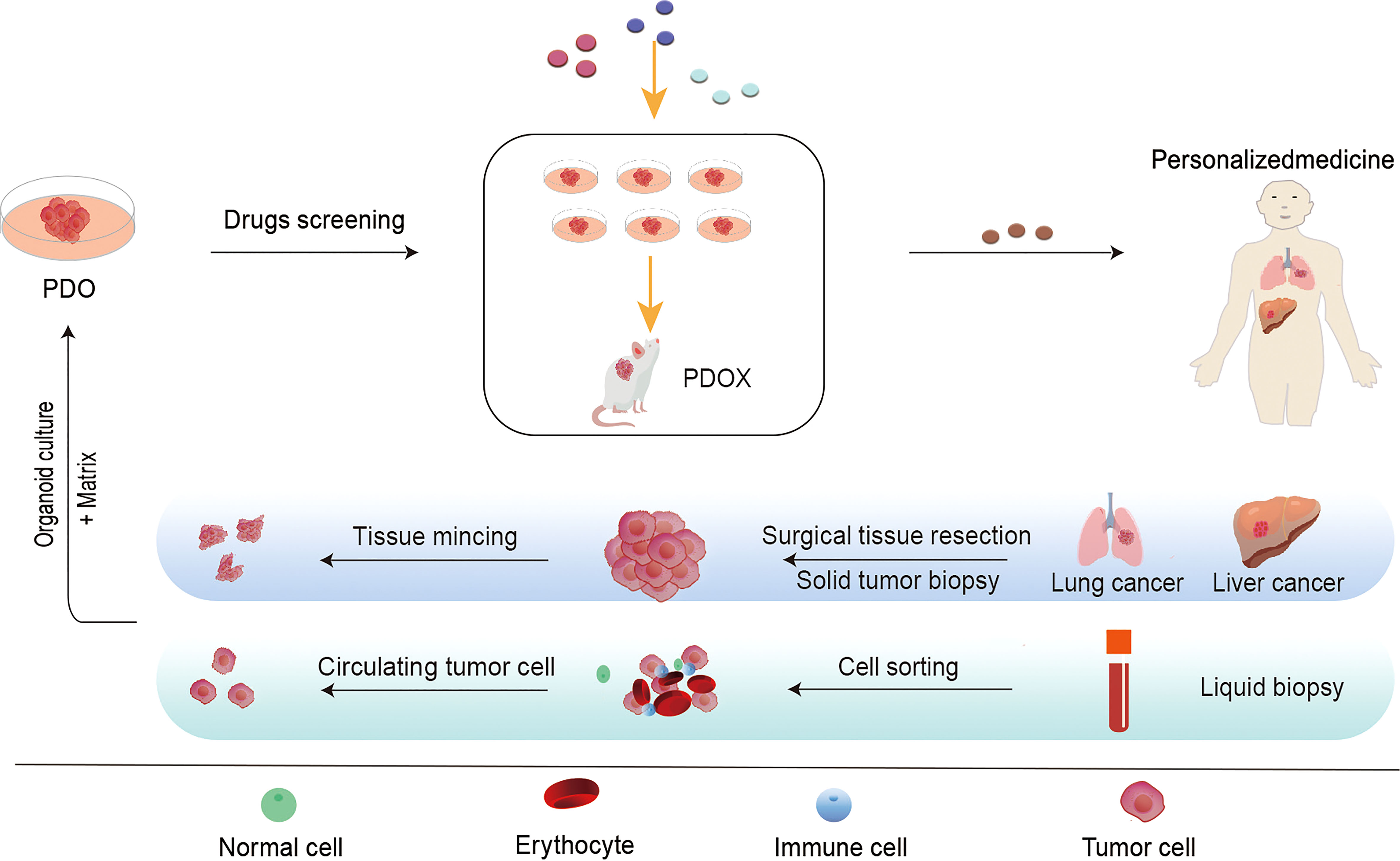
Figure 2 The flow chart of the establishment of patient-derived tumor organoid (PDO) models and the application of PDO in personalized treatment. It is commonly used to obtain primary tumor samples from surgical resections, puncture samples, or circulating tumor cells (CTCs) isolated from blood samples. Patient-derived cancer cells can be propagated in vitro on an enriched Matrigel matrix and cultured into three-dimensional tumor-like organs for in vitro and in vivo applications. These tumor models can be used for drug screening, gene analysis, immunotherapy, and other studies to accurately detect drug efficacy and toxicity. Furthermore, we are able to develop effective individualized treatment strategies for patients.
Numerous studies in the past decades have demonstrated how using organoids enhances the accuracy of the drug screening system(37, 40). These PDOs have been widely employed in the research of anti-tumor drugs. There are many advantages associated with organoids, primarily in the realization of individualized precision medicine, the reduction of modeling time, high throughput screening, genomic screening, and drug screening(41, 42). Unfortunately, no single mouse model can capture every aspect of the parent tumor and immune landscape. Some major drawbacks should be considered. Organoids cannot perfectly replicate the microenvironment in vivo, they lack tumor blood vessels and immune cells, and the co-culture system with other cell types is not yet well established. There are difficulties in studying the role of other systems and organs within the body. A global standard for organoids establishment and quality control does not yet exist. Despite its limitations, it still provides an extremely valuable contribution to the research and development of cancer drugs(43). In any case, to maximize the potential for translational research, it is imperative to select the most suitable humanized mouse model (44).
Immune surveillance is a vital tool for inhibiting tumorigenesis and maintaining the body’s internal environment’s homeostasis. A tumor cell can self-modify or release factors that influence TIME, such as engaging immune checkpoint pathways, to evade immune surveillance. Several immune checkpoints have been identified, including PD-1 and its ligand (PD-L1), which regulate the activity of T cells and cancer growth (45). Since the development and clinical application of ICIs, cancer immunotherapy has significantly expanded our toolkit for fighting the disease. At present, ICIs primarily consist of antibodies against CTLA-4 (ipilimumab), PD-1 (nivolumab, pembrolizumab, cemiplimab) and PD-L1 (atezolizumab, durvalumab, avelumab) (15, 46, 47).
Researchers recently established a humanized mouse NPC-PDX model by engrafting nasopharyngeal carcinoma (NPC) biopsies in NSG mice. This model was used to investigate the anti-tumor efficacy of nivolumab and ipilimumab (48). A study published in 2019 evaluated the efficacy of nivolumab against colorectal cancer (CRC) in a hematopoietic humanized PDX mouse model (hu-CB-BRGS). It was observed that PD-1 blockade therapy induced the immune system to kill tumors in this mode of action (49). Kleinmanns et al. established a HIS-PDX model of ovarian carcinoma in situ of NSG mice, which were injected with CD34+HSC via the vein beforehand. They further investigated the change in immune cells by flow cytometry after the animal was treated with nivolumab. The results indicated that the overall response to monotherapy was modest, and the combination therapy might be more effective (50). Here, we summarized the conditions in which PDX models were used in the preclinical evaluation of immune checkpoint mAb drugs that are currently available, to better understand the utilization of the PDX model in drug development (Table 1).
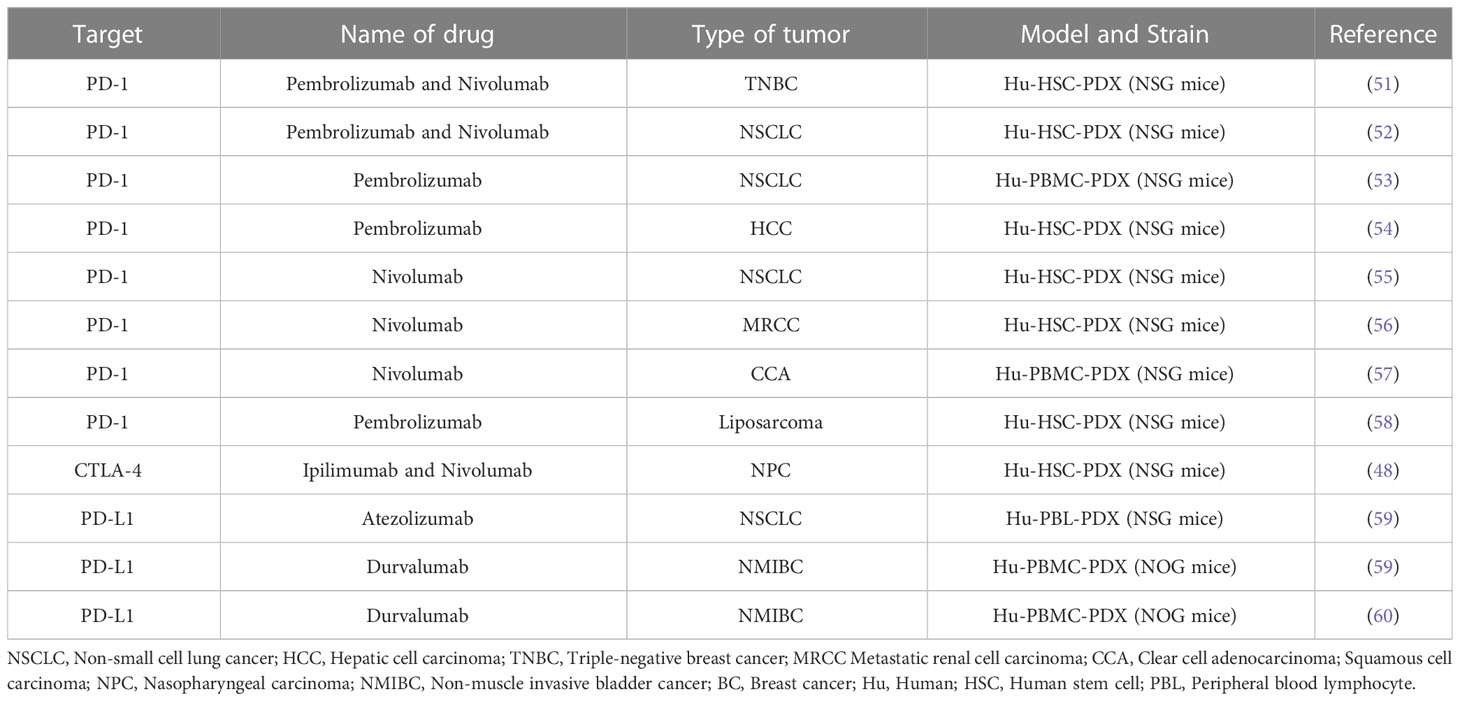
Table 1 Patient-derived tumor xenografts (PDX) models in preclinical evaluation of immune checkpoint monoclonal antibody drugs.
To enable PDOs reproduce the TIME, researchers have developed a number of novel platforms to evaluate the efficacy of tumor immunotherapy in recent years. For example, researchers have constructed a complex air-liquid interface approach PDO from different cancer types, allowing in vitro preservation of the tumor epithelium and its stromal microenvironment, and even immunologically active CD8+ T cells, NK cells, etc. Using this model, it is possible to simulate the biological behavior and therapeutic response of tumors during anti-PD-1 therapy (61). By combining PDO and humanized mouse techniques, the investigators constructed a new model of spontaneous multi-organ metastasis from microsatellite instability-H CRC and also provided empirical evidence for a key role of B cells in generating site-dependent anti-tumor immunity after anti-CTLA-4 treatment(62). Researchers also demonstrated using a patient-derived organotypic tumor spheroids (PDOTS) and a matched PDO drug screening platform that inhibition of innate immune kinase TANK-binding kinase 1 coupled with PD-1 blockade was an effective strategy for overcoming tumor immunotherapy resistance (63). In addition, the investigators established a glioblastoma (GBO)-related organoid biobank for individualized therapeutic screening. This is a PDO model with significant clinical translational potential to simulate tumor response to CAR-T cell immunotherapy (64). These studies demonstrate that immuno-oncology studies can be successfully conducted using organoid models that may facilitate personalized immunotherapy testing. In order to better understand the advantages and disadvantages of the PDOs model. We also summarize the studies with the PDO model to evaluate ICIs drugs briefly (Table 2).
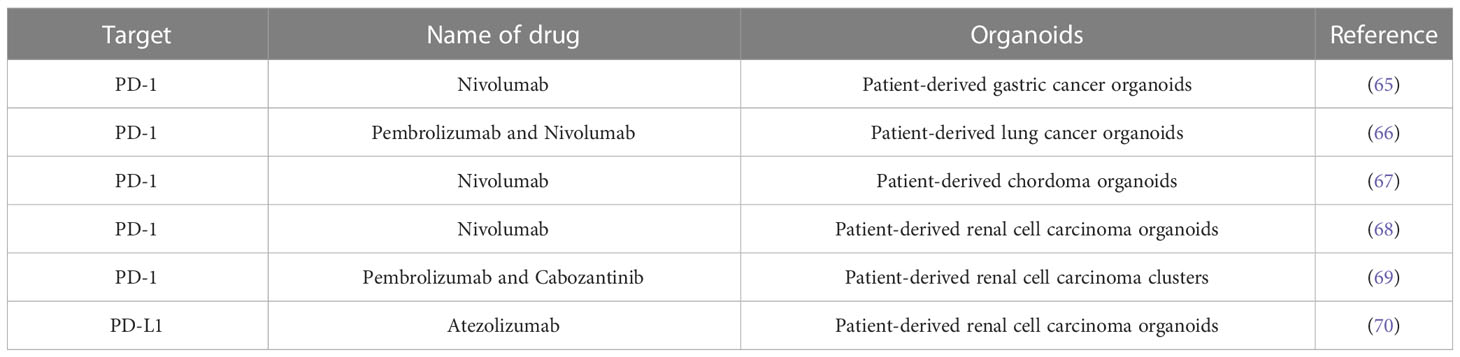
Table 2 Patient-derived organoids (PDOs) models in preclinical evaluation of immune checkpoint monoclonal antibody drugs.
Immunosuppressive cells (such as myeloid-derived suppressor cells and regulatory T cells) can release inhibitory cytokines in the TME to evade the immune system (71). Cytokines, such as interferon (INF)-alpha and interleukin (IL)-2, also play a crucial role in tumor immunotherapy. In 1986, the FDA approved INF-α as a cancer therapy drug for the treatment of leukemia. Currently, IFN-α and IL-2 have become the most widely used drugs in tumor immunotherapy strategies, however, several other cytokines are currently under clinical investigation (72, 73). Aside from cytokines, non-specific immune drugs also include target natural killer (NK) cells, macrophages, and immunomodulators. Pexidartinib, the first macrophage-targeting medicine approved by the FDA, is recommended for adult patients with symptomatic giant cell tumors of tenosynovitis because it restricts macrophage proliferation by blocking CSF cytokines (74). In a recent study on pexidartinib, researchers evaluated its impact on PDX and observed that pexidartinib can significantly inhibit osteosarcoma tumor growth (75). Immunotherapy with IL-2 and GM-CSF has significantly improved survival in children with high-risk neuroblastoma (76). Treatment failure and IL2-related toxicity, however, pose significant challenges to the treatment of one third of these patients. There has been evidence in recent clinical trials that NK cells hyperproliferate and acquire an activated phenotype in patients receiving recombinant human IL-15, resulting in NK cell expansion in vivo and tumor shrinkage in two patients. As a result, scholars validated the tumor suppressive effect of IL-15 on PDX models, and they demonstrated that the replacement of IL-2 with IL-15 was associated with significant tumor regression in vivo, supporting clinical trials of IL-15 for pediatric neuroblastoma (77). Additionally, related study has also demonstrated that IL-15 enhanced the anti-tumor activity of γδ T cells, and effectively suppressed tumor growth, and prolonged the survival of renal cancer-bearing PDX mice (78). Taking these results into consideration, it appears that cytokines might be able to have significant clinical implications in the future.
CAR- T cell therapy, as a novel approach in anticancer therapy, in which T cells are retargeted against the tumor cell following the engineered expression of CARs (79). Currently, two CAR-T cell products have been used for the treatment of lymphoblastic leukemia and lymphoma (80). Besides, it has been reported that CAR-T cells engineered to simultaneously produce interleukin (IL)-7 and chemokine (C–C motif) ligand 19 (CCL19) were effective against solid tumors in pancreatic cancer (PC) PDX model (81). Additionally, other researchers have also verified CAR-T cells anti-tumor immunotherapy effects on triple-negative breast cancer(TNBC) (82). Other studies have found that in a patient with late-stage HCC, anti-GPC3 IL-7/CCL19 CAR-T therapy resulted in complete tumor disappearance 30 days post-intra-tumor injection. And in a patient with advanced PC, anti-MSLN-IL-7/CCL19 CAR-T cellular therapy resulted in almost complete tumor disappearance 240 days post-intravenous infusion (83). Both preclinical and clinical studies suggest that novel CAR-T cells have significant potential for the treatment of solid tumors.
In 2010, Zhao Y et al., reported that they developed a PDX model to evaluate CAR-T therapy (84). Jiang Z et al., reported that CAR T cells demonstrated a positive therapeutic effect on liver cancer in a PDX mouse model. They concluded that the growth of the tumor in the PDX model could be inhibited following CAR-T cells therapy (85). The investigators developed a highly specific SynNotch-CAR-T cells, which was validated using the PDX model to target gliomas and exert anti-tumor effects with reduced off-target toxicity (86).The emergence of adaptive therapy has stimulated the development of new CAR-NK cells therapy techniques (20). In 2021, Cao B and his team developed mesothelin (MSLN)-CAR NK cells, which were evaluated using PDX (NSG mice). According to the findings, these cells demonstrated strong anti-tumor properties and offer a promising treatment for gastric cancer (87). Although CAR-NK cells have obvious advantages in tumor therapy, the short life cycle of NK cells in vivo and the immunosuppression of the TME limit the clinical transformation of CAR-NK cells.
Ding S et al., generated thousands of micro-organ spheres from patient tissues and assessed tumor drug response (88). The establishment of an organoid biobank, as mentioned earlier, is a valuable platform for evaluating tumor treatment strategies such as CAR-T cell therapy (89). In addition, combining organoid and 3D imaging technologies, the investigators have provided a platform to reveal the mode of action of cellular anti-cancer immunotherapies in a patient-specific manner and apply them to develop multiple engineered T cell products(90). PDOs are ideal for short drug screening cycles and convenient sampling of the model, which can be achieved through several methods, including surgery, biopsy, urine, and lung lavage fluid (88, 91). The development of PDOs will greatly shorten preclinical study time and facilitate drug development.
More than a century ago, a phenomenon was observed in clinical therapy, that is some patients with cancer experience the cancer regression if they were infected with certain viruses (92). Based on this case, OVs therapy was further developed to advance cancer biological therapy. OVs possess excellent safety in clinical trials, which greatly promotes their research and development. A novel OV (OAd-MUC16-BiTE) with better anti-tumor characteristics was developed for treating ovarian cancer in PDX mice models (93). Other study evaluated the anticancer efficacy of VG161, a herpes virus type 1 (HSV-1), in HLA-matched CD34+ humanized PDX model. It was found that VG161 significantly inhibited tumor growth and would realize enhancement of OV-induced antitumor immunity for long-term maintenance treatment (94). In research by Quinn CH et al., oncolytic herpes simplex viruses (oHSVs) were demonstrated to be effective in treating high-risk neuroblastoma in PDX mice (NOD-SCID) (95). OVs therapy has the advantages of excellent replication efficiency, a potent killing effect, fewer adverse reactions, and inexpensive cost, making it one of the most promising tumor immunotherapy methods in the future (21, 33). In addition, exploring the anticancer activity of OVs based on pancreatic PDOs proved to be an effective predictive tool (96). However, the delivery of OVs was by intertumoral injection, which limited its clinical use. Therefore, how to deliver these OVs to the tumor tissue more effectively and how to improve the potential of these viruses to disseminate within the tumor tissue site may be the future focus of this therapy.
Tumor immunotherapy has changed the treatment of advanced tumors, however, the proportion of patients responding to immunotherapies remains low. In recent years, supramolecular chemistry and self-assembled systems have been extensively investigated in the field of cancer therapy and hold great promise for improving immunotherapeutic outcomes in tumor patients (97, 98). Unlike conventional cancer immunotherapy, rationally designed nano-self-assembled drugs can trigger specific tumoricidal effects, thereby improving infiltration of TIME such as killer immune T lymphocytes, optimizing antigen presentation, and inducing durable immune responses (23). The development of nanotechnology provides an opportunity for enhancing the effectiveness and reducing the toxicity of traditional drugs, and a series of nanomaterials were used in the preclinical study of cancer (24). In conclusion, self-assembled drugs have a broad potential for application in tumor immunotherapy, especially in refractory and recurrent cancers.
The self-assembled peptides can respond to various environmental conditions, such as pH, temperature, and molecular interactions, while also possessing high biocompatibility and drug loading capabilities(99, 100). According to current research, self-assembly peptides can be classified into two main categories for tumor immunotherapy research: 1) Self-assembly into nanodrugs using their drug loading capacity, delivering molecules such as peptides or siRNA, inhibiting specific proteins or genes in tumor cells to enhance tumor immunotherapy response. 2) Using peptide self-assembly to simulate tumor antigens as tumor vaccines to stimulate the body to produce anti-tumor antibodies(101, 102). In short, self-assembling peptide drugs may improve the treatment of tumors immune therapy significantly in future.
Self-assembled nanomedicines have received significant attention due to their excellent biocompatibility, high modification versatility and ease of synthesis, controllable and adaptive nanostructures(103). Recently, a study pointed out that through targeted inhibition of MDM2, p53 can be activated, the tumor immune microenvironment can be reprogramed, and immunotherapy resistance can be overcome (104). Researchers created TPA, a combined targeted peptide that inhibited the PD-1/PD-L1 axis, activated p53, and showed tumor killing and immunotherapeutic sensitization effects on a humanized PBMC-engrafted PDX model. There is now a potential pathway for the development of self-assembled peptide drugs for cancer therapy (105). For tumor targeting, the researchers synthesized size-tunable nanostructures with a spherical morphology by combining partially reductive HSA with hydrophobic Fluvastatin, known as AB-Flu. According to the study, these nanodrugs effectively enhanced the potency of Anti-PD1 antibodies against colon cancer in a humanized CRC-PDX mouse model while maintaining acceptable levels of safety (100). Generally, self-assembled drugs have unique anti-tumor effects and are low in toxicity. Through blocking the supply of tumor nutrients, improving drug targeting, and even recruiting multiple immune cells, they can achieve tumor therapy. Therefore, development and research into self-assembled drugs is warranted. Anti-tumor potential of self-assembled drugs creates new hope for tumor treatment, and the PDX and PDO models facilitate clinical transformation as well.
The rapid development of immunotherapy drugs brings hope to clinical patients with cancer (100, 106–108). However, the preclinical evaluation of drugs still restricts the development of drug research. Although the drug evaluation system has advanced from a 2D cell line evaluation system to PDX/PDO system and even developed a PDX model with human immune cells to more accurately simulate the immune environment in vivo (109, 110), there are still limitations. The establishing cycle of PDX/humanized PDX is long, the technology is challenging, and it cannot completely simulate the TME. Although the modeling cycle of PDOs is short, but it is still necessary to investigate whether the medium possesses antigenicity because the composition of the medium is unknown. In addition, despite the extensive genetic heterogeneity of tumors in vivo, it is unknown whether tumor organoids can capture the entire range of heterogeneity that originates from the primary tumor (111). Despite the complexity of cancer, there are still several unresolved issues, including those related to its pathogenesis, mechanism of metastasis, patient response to treatment, or mechanism of drug resistance. Further studies are required to constantly improve the simulation of TME to create PDX/PDO that is more similar to the primary tumor characteristics, to better serve drug development.
WZ wrote the manuscript. XZ reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Van Dyke T, Jacks T. Cancer modeling in the modern era: progress and challenges. Cell (2002) 108(2):135–44. doi: 10.1016/s0092-8674(02)00621-9
2. He W, Wang S, Yan J, Qu Y, Jin L, Sui F, et al. Self-assembly of therapeutic peptide into stimuli-responsive clustered nanohybrids for cancer-targeted therapy. Advanced Funct Mater (2019) 29(10):1807736. doi: 10.1002/adfm.201807736
3. Martinez-Jimenez F, Muinos F, Sentis I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, et al. A compendium of mutational cancer driver genes. Nat Rev Cancer (2020) 20(10):555–72. doi: 10.1038/s41568-020-0290-x
4. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi: 10.1038/nm.3394
5. Yan S, Yan J, Liu D, Li X, Kang Q, You W, et al. A nano-predator of pathological MDMX construct by clearable supramolecular gold(I)-thiol-peptide complexes achieves safe and potent anti-tumor activity. Theranostics (2021) 11(14):6833–46. doi: 10.7150/thno.59020
6. Bilotta MT, Antignani A, Fitzgerald DJ. Managing the TME to improve the efficacy of cancer therapy. Front Immunol (2022) 13:954992. doi: 10.3389/fimmu.2022.954992
7. Ioannides CG, Whiteside TL. T Cell recognition of human tumors: implications for molecular immunotherapy of cancer. Clin Immunol Immunopathol (1993) 66(2):91–106. doi: 10.1006/clin.1993.1012
8. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
9. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity (2015) 43(3):435–49. doi: 10.1016/j.immuni.2015.09.001
10. Domblides C, Lartigue L, Faustin B. Control of the antitumor immune response by cancer metabolism. Cells (2019) 8(2):104. doi: 10.3390/cells8020104
11. Ren M, Zheng X, Gao H, Jiang A, Yao Y, He W. Nanomedicines targeting metabolism in the tumor microenvironment. Front Bioeng Biotechnol (2022) 10:943906. doi: 10.3389/fbioe.2022.943906
12. Oiseth S, Aziz M. Cancer immunotherapy: a brief review of the history, possibilities, and challenges ahead. J Cancer Metastasis Treat (2017) 3:250–61. doi: 10.20517/2394-4722.2017.41
13. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11(11):3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
14. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271(5256):1734–6. doi: 10.1126/science.271.5256.1734
15. Ai L, Xu A, Xu J. Roles of PD-1/PD-L1 pathway: Signaling, cancer, and beyond. Adv Exp Med Biol (2020) 1248:33–59. doi: 10.1007/978-981-15-3266-5_3
16. Hausser HJ, Brenner RE. Phenotypic instability of saos-2 cells in long-term culture. Biochem Biophys Res Commun (2005) 333(1):216–22. doi: 10.1016/j.bbrc.2005.05.097
17. Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res (2013) 73(14):4372–82. doi: 10.1158/0008-5472.CAN-12-3342
18. Tsai S, McOlash L, Palen K, Johnson B, Duris C, Yang Q, et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer (2018) 18(1):335. doi: 10.1186/s12885-018-4238-4
19. Ho T, Msallam R. Tissues and tumor microenvironment (TME) in 3D: Models to shed light on immunosuppression in cancer. Cells (2021) 10(4):831. doi: 10.3390/cells10040831
20. Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine (2020) 59:102975. doi: 10.1016/j.ebiom.2020.102975
21. Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol (2020) 13(1):84. doi: 10.1186/s13045-020-00922-1
22. Song Q, Zhang CD, Wu XH. Therapeutic cancer vaccines: From initial findings to prospects. Immunol Lett (2018) 196:11–21. doi: 10.1016/j.imlet.2018.01.011
23. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer (2017) 17(1):20–37. doi: 10.1038/nrc.2016.108
24. Muntimadugu E, Kommineni N, Khan W. Exploring the potential of nanotherapeutics in targeting tumor microenvironment for cancer therapy. Pharmacol Res (2017) 126:109–22. doi: 10.1016/j.phrs.2017.05.010
25. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol (2012) 9(6):338–50. doi: 10.1038/nrclinonc.2012.61
26. Qu J, Kalyani FS, Liu L, Cheng T, Chen L. Tumor organoids: synergistic applications, current challenges, and future prospects in cancer therapy. Cancer Commun (Lond) (2021) 41(12):1331–53. doi: 10.1002/cac2.12224
27. Yan J, Zheng X, You W, He W, Xu GK. A bionic-homodimerization strategy for optimizing modulators of protein-protein interactions: From statistical mechanics theory to potential clinical translation. Adv Sci (Weinh) (2022) 9(11):e2105179. doi: 10.1002/advs.202105179
28. Rygaard J, Povlsen CO. Heterotransplantation of a human malignant tumour to "Nude" mice. Acta Pathol Microbiol Scand (1969) 77(4):758–60. doi: 10.1111/j.1699-0463.1969.tb04520.x
29. Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discovery (2014) 4(9):998–1013. doi: 10.1158/2159-8290.Cd-14-0001
30. Woo XY, Giordano J, Srivastava A, Zhao ZM, Lloyd MW, de Bruijn R, et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat Genet (2021) 53(1):86–99. doi: 10.1038/s41588-020-00750-6
31. Chuprin J, Buettner H, Seedhom MO, Greiner DL, Keck JG, Ishikawa F, et al. Humanized mouse models for immuno-oncology research. Nat Rev Clin Oncol (2023) 20(3):192–206. doi: 10.1038/s41571-022-00721-2
32. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature (2009) 459(7244):262–5. doi: 10.1038/nature07935
33. Russell L, Peng KW, Russell SJ, Diaz RM. Oncolytic viruses: Priming time for cancer immunotherapy. BioDrugs (2019) 33(5):485–501. doi: 10.1007/s40259-019-00367-0
34. Tuveson D, Clevers H. Cancer modeling meets human organoid technology. Science (2019) 364(6444):952–5. doi: 10.1126/science.aaw6985
35. Zhou Z, Cong L, Cong X. Patient-derived organoids in precision medicine: Drug screening, organoid-on-a-Chip and living organoid biobank. Front Oncol (2021) 11:762184. doi: 10.3389/fonc.2021.762184
36. Nanki K, Toshimitsu K, Takano A, Fujii M, Shimokawa M, Ohta Y, et al. Divergent routes toward wnt and r-spondin niche independency during human gastric carcinogenesis. Cell (2018) 174(4):856–869.e817. doi: 10.1016/j.cell.2018.07.027
37. Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell (2018) 172(1-2):373–386.e310. doi: 10.1016/j.cell.2017.11.010
38. Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, et al. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell (2018) 173(2):515–528.e517. doi: 10.1016/j.cell.2018.03.017
39. Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ Jr., et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discovery (2018) 8(11):1404–21. doi: 10.1158/2159-8290.CD-18-0474
40. Boucherit N, Gorvel L, Olive D. 3D tumor models and their use for the testing of immunotherapies. Front Immunol (2020) 11:603640. doi: 10.3389/fimmu.2020.603640
41. Weeber F, Ooft SN, Dijkstra KK, Voest EE. Tumor organoids as a pre-clinical cancer model for drug discovery. Cell Chem Biol (2017) 24(9):1092–100. doi: 10.1016/j.chembiol.2017.06.012
42. Guillen KP, Fujita M, Butterfield AJ, Scherer SD, Bailey MH, Chu Z, et al. A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology. Nat Cancer (2022) 3(2):232–50. doi: 10.1038/s43018-022-00337-6
43. Bar-Ephraim YE, Kretzschmar K, Clevers H. Organoids in immunological research. Nat Rev Immunol (2020) 20(5):279–93. doi: 10.1038/s41577-019-0248-y
44. Wu W, Li X, Yu S. Patient-derived tumour organoids: A bridge between cancer biology and personalised therapy. Acta Biomater (2022) 146:23–36. doi: 10.1016/j.actbio.2022.04.050
45. Khan E, Shrestha AK, Elkhooly M, Wilson H, Ebbert M, Srivastava S, et al. CNS and PNS manifestation in immune checkpoint inhibitors: A systematic review. J Neurol Sci (2022) 432:120089. doi: 10.1016/j.jns.2021.120089
46. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
47. Dyck L, Mills KHG. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol (2017) 47(5):765–79. doi: 10.1002/eji.201646875
48. Liu WN, Fong SY, Tan WWS, Tan SY, Liu M, Cheng JY, et al. Establishment and characterization of humanized mouse NPC-PDX model for testing immunotherapy. Cancers (Basel) (2020) 12(4):1025. doi: 10.3390/cancers12041025
49. Capasso A, Lang J, Pitts TM, Jordan KR, Lieu CH, Davis SL, et al. Characterization of immune responses to anti-PD-1 mono and combination immunotherapy in hematopoietic humanized mice implanted with tumor xenografts. J Immunother Cancer (2019) 7(1):37. doi: 10.1186/s40425-019-0518-z
50. Kleinmanns K, Gullaksen SE, Bredholt G, Davidson B, Torkildsen CF, Grindheim S, et al. Humanized ovarian cancer patient-derived xenografts for improved preclinical evaluation of immunotherapies. Cancers (Basel) (2022) 14(13):3092. doi: 10.3390/cancers14133092
51. Rosato RR, Davila-Gonzalez D, Choi DS, Qian W, Chen W, Kozielski AJ, et al. Evaluation of anti-PD-1-based therapy against triple-negative breast cancer patient-derived xenograft tumors engrafted in humanized mouse models. Breast Cancer Res (2018) 20(1):108. doi: 10.1186/s13058-018-1037-4
52. Meraz IM, Majidi M, Meng F, Shao R, Ha MJ, Neri S, et al. An improved patient-derived xenograft humanized mouse model for evaluation of lung cancer immune responses. Cancer Immunol Res (2019) 7(8):1267–79. doi: 10.1158/2326-6066.CIR-18-0874
53. Lin S, Huang G, Cheng L, Li Z, Xiao Y, Deng Q, et al. Establishment of peripheral blood mononuclear cell-derived humanized lung cancer mouse models for studying efficacy of PD-L1/PD-1 targeted immunotherapy. MAbs (2018) 10(8):1301–11. doi: 10.1080/19420862.2018.1518948
54. Zhao Y, Wang J, Liu WN, Fong SY, Shuen TWH, Liu M, et al. Analysis and validation of human targets and treatments using a hepatocellular carcinoma-immune humanized mouse model. Hepatology (2021) 74(3):1395–410. doi: 10.1002/hep.31812
55. Martin-Ruiz A, Fiuza-Luces C, Rincon-Castanedo C, Fernandez-Moreno D, Galvez BG, Martinez-Martinez E, et al. Benefits of exercise and immunotherapy in a murine model of human non-small-cell lung carcinoma. Exerc Immunol Rev (2020) 26:100–15.
56. Kang Y, Armstrong AJ, Hsu DS. An autologous humanized patient-derived xenograft (PDX) model for evaluation of nivolumab immunotherapy in renal cell cancer: a case report. Stem Cell Investig (2022) 9:8. doi: 10.21037/sci-2022-029
57. Vargas R, Gopal P, Kuzmishin GB, DeBernardo R, Koyfman SA, Jha BK, et al. Case study: patient-derived clear cell adenocarcinoma xenograft model longitudinally predicts treatment response. NPJ Precis Oncol (2018) 2:14. doi: 10.1038/s41698-018-0060-3
58. Choi B, Lee JS, Kim SJ, Hong D, Park JB, Lee KY. Anti-tumor effects of anti-PD-1 antibody, pembrolizumab, in humanized NSG PDX mice xenografted with dedifferentiated liposarcoma. Cancer Lett (2020) 478:56–69. doi: 10.1016/j.canlet.2020.02.042
59. Chen X, Shen C, Wei Z, Zhang R, Wang Y, Jiang L, et al. Patient-derived non-small cell lung cancer xenograft mirrors complex tumor heterogeneity. Cancer Biol Med (2021) 18(1):184–98. doi: 10.20892/j.issn.2095-3941.2020.0012
60. Blinova E, Samishina E, Deryabina O, Blinov D, Roshchin D, Shich E, et al. Expression of p53 protein associates with anti-PD-L1 treatment response on human-derived xenograft model of GATA3/CR5/6-negative recurrent nonmuscular invasive bladder urothelial carcinoma. Int J Mol Sci (2021) 22(18):9856. doi: 10.3390/ijms22189856
61. Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid modeling of the tumor immune microenvironment. Cell (2018) 175(7):1972–1988.e1916. doi: 10.1016/j.cell.2018.11.021
62. Küçükköse E, Heesters BA, Villaudy J, Verheem A, Cercel M, van Hal S, et al. Modeling resistance of colorectal peritoneal metastases to immune checkpoint blockade in humanized mice. J Immunother Cancer (2022) 10(12):e005345. doi: 10.1136/jitc-2022-005345
63. Sun Y, Revach OY, Anderson S, Kessler EA, Wolfe CH, Jenney A, et al. Targeting TBK1 to overcome resistance to cancer immunotherapy. Nature (2023) 615:158–67. doi: 10.1038/s41586-023-05704-6
64. Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discovery (2018) 8(2):196–215. doi: 10.1158/2159-8290.Cd-17-0833
65. Chakrabarti J, Koh V, Steele N, Hawkins J, Ito Y, Merchant JL, et al. Disruption of Her2-induced PD-L1 inhibits tumor cell immune evasion in patient-derived gastric cancer organoids. Cancers (Basel) (2021) 13(24):6158. doi: 10.3390/cancers13246158
66. Takahashi N, Hoshi H, Higa A, Hiyama G, Tamura H, Ogawa M, et al. An In vitro system for evaluating molecular targeted drugs using lung patient-derived tumor organoids. Cells (2019) 8(5):481. doi: 10.3390/cells8050481
67. Scognamiglio G, De Chiara A, Parafioriti A, Armiraglio E, Fazioli F, Gallo M, et al. Patient-derived organoids as a potential model to predict response to PD-1/PD-L1 checkpoint inhibitors. Br J Cancer (2019) 121(11):979–82. doi: 10.1038/s41416-019-0616-1
68. Esser LK, Branchi V, Leonardelli S, Pelusi N, Simon AG, Klümper N, et al. Cultivation of clear cell renal cell carcinoma patient-derived organoids in an air-liquid interface system as a tool for studying individualized therapy. Front Oncol (2020) 10:1775. doi: 10.3389/fonc.2020.01775
69. Ao Z, Wu Z, Cai H, Hu L, Li X, Kaurich C, et al. Rapid profiling of tumor-immune interaction using acoustically assembled patient-derived cell clusters. Adv Sci (Weinh) (2022) 9(22):e2201478. doi: 10.1002/advs.202201478
70. Hamdan F, Ylösmäki E, Chiaro J, Giannoula Y, Long M, Fusciello M, et al. Novel oncolytic adenovirus expressing enhanced cross-hybrid IgGA fc PD-L1 inhibitor activates multiple immune effector populations leading to enhanced tumor killing in vitro, in vivo and with patient-derived tumor organoids. J Immunother Cancer (2021) 9(8):e003000. doi: 10.1136/jitc-2021-003000
71. Klener P Jr., Otahal P, Lateckova L, Klener P. Immunotherapy approaches in cancer treatment. Curr Pharm Biotechnol (2015) 16(9):771–81. doi: 10.2174/1389201016666150619114554
72. Mitchell MS. Immunotherapy as part of combinations for the treatment of cancer. Int Immunopharmacol (2003) 3(8):1051–9. doi: 10.1016/s1567-5769(03)00019-5
73. Wrangle JM, Patterson A, Johnson CB, Neitzke DJ, Mehrotra S, Denlinger CE, et al. IL-2 and beyond in cancer immunotherapy. J Interferon Cytokine Res (2018) 38(2):45–68. doi: 10.1089/jir.2017.0101
74. Monestime S, Lazaridis D. Pexidartinib (TURALIO): The first FDA-indicated systemic treatment for tenosynovial giant cell tumor. Drugs R D (2020) 20(3):189–95. doi: 10.1007/s40268-020-00314-3
75. Smeester BA, Slipek NJ, Pomeroy EJ, Laoharawee K, Osum SH, Larsson AT, et al. PLX3397 treatment inhibits constitutive CSF1R-induced oncogenic ERK signaling, reduces tumor growth, and metastatic burden in osteosarcoma. Bone (2020) 136:115353. doi: 10.1016/j.bone.2020.115353
76. Furman WL, McCarville B, Shulkin BL, Davidoff A, Krasin M, Hsu CW, et al. Improved outcome in children with newly diagnosed high-risk neuroblastoma treated with chemoimmunotherapy: Updated results of a phase II study using hu14.18K322A. J Clin Oncol (2022) 40(4):335–44. doi: 10.1200/jco.21.01375
77. Nguyen R, Moustaki A, Norrie JL, Brown S, Akers WJ, Shirinifard A, et al. Interleukin-15 enhances anti-GD2 antibody-mediated cytotoxicity in an orthotopic PDX model of neuroblastoma. Clin Cancer Res (2019) 25(24):7554–64. doi: 10.1158/1078-0432.Ccr-19-1045
78. Zhang B, Li H, Liu W, Tian H, Li L, Gao C, et al. Adoptive cell therapy of patient-derived renal cell carcinoma xenograft model with IL-15-induced γδT cells. Med Oncol (2021) 38(3):30. doi: 10.1007/s12032-021-01474-1
79. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nat Rev Drug Discovery (2020) 19(3):185–99. doi: 10.1038/s41573-019-0051-2
80. Fournier C, Martin F, Zitvogel L, Kroemer G, Galluzzi L, Apetoh L. Trial watch: Adoptively transferred cells for anticancer immunotherapy. Oncoimmunology (2017) 6(11):e1363139. doi: 10.1080/2162402X.2017.1363139
81. Goto S, Sakoda Y, Adachi K, Sekido Y, Yano S, Eto M, et al. Enhanced anti-tumor efficacy of IL-7/CCL19-producing human CAR-T cells in orthotopic and patient-derived xenograft tumor models. Cancer Immunol Immunother (2021) 70(9):2503–15. doi: 10.1007/s00262-021-02853-3
82. Ye X, Deng X, Wen J, Li Y, Zhang M, Cai Z, et al. Folate receptor-alpha targeted 7x19 CAR-γδT suppressed triple-negative breast cancer xenograft model in mice. J Oncol (2022) 2022:2112898. doi: 10.1155/2022/2112898
83. Pang N, Shi J, Qin L, Chen A, Tang Y, Yang H, et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J Hematol Oncol (2021) 14(1):118. doi: 10.1186/s13045-021-01128-9
84. Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res (2010) 70(22):9053–61. doi: 10.1158/0008-5472.CAN-10-2880
85. Jiang Z, Jiang X, Chen S, Lai Y, Wei X, Li B, et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front Immunol (2016) 7:690. doi: 10.3389/fimmu.2016.00690
86. Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med (2021) 13(591):eabe7378. doi: 10.1126/scitranslmed.abe7378
87. Cao B, Liu M, Huang J, Zhou J, Li J, Lian H, et al. Development of mesothelin-specific CAR NK-92 cells for the treatment of gastric cancer. Int J Biol Sci (2021) 17(14):3850–61. doi: 10.7150/ijbs.64630
88. Ding S, Hsu C, Wang Z, Natesh NR, Millen R, Negrete M, et al. Patient-derived micro-organospheres enable clinical precision oncology. Cell Stem Cell (2022) 29(6):905–917.e906. doi: 10.1016/j.stem.2022.04.006
89. Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH, et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell (2020) 180(1):188–204.e122. doi: 10.1016/j.cell.2019.11.036
90. Dekkers JF, Alieva M, Cleven A, Keramati F, Wezenaar AKL, van Vliet EJ, et al. Uncovering the mode of action of engineered T cells in patient cancer organoids. Nat Biotechnol (2023) 41(1):60–9. doi: 10.1038/s41587-022-01397-w
91. Driehuis E, Kolders S, Spelier S, Lõhmussaar K, Willems SM, Devriese LA, et al. Oral mucosal organoids as a potential platform for personalized cancer therapy. Cancer Discovery (2019) 9(7):852–71. doi: 10.1158/2159-8290.Cd-18-1522
92. Mondal M, Guo J, He P, Zhou D. Recent advances of oncolytic virus in cancer therapy. Hum Vaccin Immunother (2020) 16(10):2389–402. doi: 10.1080/21645515.2020.1723363
93. Wang Q, Ma X, Wu H, Zhao C, Chen J, Li R, et al. Oncolytic adenovirus with MUC16-BiTE shows enhanced antitumor immune response by reversing the tumor microenvironment in PDX model of ovarian cancer. Oncoimmunology (2022) 11(1):2096362. doi: 10.1080/2162402x.2022.2096362
94. Ding J, Murad YM, Sun Y, Lee IF, Samudio I, Liu X, et al. Pre-existing HSV-1 immunity enhances anticancer efficacy of a novel immune-stimulating oncolytic virus. Viruses (2022) 14(11):2327. doi: 10.3390/v14112327
95. Quinn CH, Beierle AM, Hutchins SC, Marayati R, Bownes LV, Stewart JE, et al. Targeting high-risk neuroblastoma patient-derived xenografts with oncolytic virotherapy. Cancers (Basel) (2022) 14(3):762. doi: 10.3390/cancers14030762
96. Raimondi G, Mato-Berciano A, Pascual-Sabater S, Rovira-Rigau M, Cuatrecasas M, Fondevila C, et al. Patient-derived pancreatic tumour organoids identify therapeutic responses to oncolytic adenoviruses. EBioMedicine (2020) 56:102786. doi: 10.1016/j.ebiom.2020.102786
97. Li L-L, Qiao S-L, Liu W-J, Ma Y, Wan D, Pan J, et al. Intracellular construction of topology-controlled polypeptide nanostructures with diverse biological functions. Nat Commun (2017) 8(1):1276. doi: 10.1038/s41467-017-01296-8
98. Nasser TA, Adel R, Badr A, Teleb M, Bekhit AA, Elkhodairy KA, et al. Combined cancer immunotheranostic nanomedicines: Delivery technologies and therapeutic outcomes. ACS Omega (2023) 8(5):4491–507. doi: 10.1021/acsomega.2c05986
99. Fisher E, Pavlenko K, Vlasov A, Ramenskaya G. Peptide-based therapeutics for oncology. Pharm Med (2019) 33(1):9–20. doi: 10.1007/s40290-018-0261-7
100. Ma F, Liu T, Yang W, You W, He W, Yan J, et al. Turning fluvastatin into a supramolecular immuno-sensitizer towards augmented tumor immunotherapy. Chem Eng J (2022) 437:135310. doi: 10.1016/j.cej.2022.135310
101. Xiao YF, Jie MM, Li BS, Hu CJ, Xie R, Tang B, et al. Peptide-based treatment: A promising cancer therapy. J Immunol Res (2015) 2015:761820. doi: 10.1155/2015/761820
102. Qiu F, Becker KW, Knight FC, Baljon JJ, Sevimli S, Shae D, et al. Poly(propylacrylic acid)-peptide nanoplexes as a platform for enhancing the immunogenicity of neoantigen cancer vaccines. Biomaterials (2018) 182:82–91. doi: 10.1016/j.biomaterials.2018.07.052
103. Wang F, Xu D, Su H, Zhang W, Sun X, Monroe MK, et al. Supramolecular prodrug hydrogelator as an immune booster for checkpoint blocker-based immunotherapy. Sci Adv (2020) 6(18):eaaz8985. doi: 10.1126/sciadv.aaz8985
104. Zhou X, Singh M, Sanz Santos G, Guerlavais V, Carvajal LA, Aivado M, et al. Pharmacologic activation of p53 triggers viral mimicry response thereby abolishing tumor immune evasion and promoting antitumor immunity. Cancer Discovery (2021) 11(12):3090–105. doi: 10.1158/2159-8290.Cd-20-1741
105. Zhang L, Jiang Z, Yang X, Qian Y, Wang M, Wu S, et al. A totipotent "All-In-One" peptide sequentially blocks immune checkpoint and reverses the immunosuppressive tumor microenvironment. Adv Mater (2023) 35(2):e2207330. doi: 10.1002/adma.202207330
106. She J, Li Y, Yan S, Yan Y, Liu D, Li S, et al. De novo supraparticle construction by a self-assembled janus cyclopeptide to tame hydrophilic microRNA and hydrophobic molecule for anti-tumor cocktail therapy and augmented immunity. Chem Eng J (2020) 401:126080. doi: 10.1016/j.cej.2020.126080
107. Shields C, Wang LL, Evans MA, Mitragotri S. Materials for immunotherapy. Adv Mater (2020) 32(13):e1901633. doi: 10.1002/adma.201901633
108. Yan J, He W, Li X, You W, Liu X, Lin S, et al. Carnosic acid-induced co-self-assembly of metal-peptide complexes into a nanocluster-based framework with tumor-specific accumulation for augmented immunotherapy. Chem Eng J (2021) 416:129141. doi: 10.1016/j.cej.2021.129141
109. Olson B, Li Y, Lin Y, Liu ET, Patnaik A. Mouse models for cancer immunotherapy research. Cancer Discovery (2018) 8(11):1358–65. doi: 10.1158/2159-8290.Cd-18-0044
110. Yan J, Yao Y, Yan S, Gao R, Lu W, He W. Chiral protein supraparticles for tumor suppression and synergistic immunotherapy: An enabling strategy for bioactive supramolecular chirality construction. Nano Lett (2020) 20(8):5844–52. doi: 10.1021/acs.nanolett.0c01757
Keywords: drug discovery, PDO, PDX, tumor immunotherapy, tumor microenvironment
Citation: Zhang W and Zheng X (2023) Patient-derived xenografts or organoids in the discovery of traditional and self-assembled drug for tumor immunotherapy. Front. Oncol. 13:1122322. doi: 10.3389/fonc.2023.1122322
Received: 23 December 2022; Accepted: 24 March 2023;
Published: 04 April 2023.
Edited by:
Sen Hou, Beihang University, ChinaReviewed by:
Yumeng Xue, Northwestern Polytechnical University, ChinaCopyright © 2023 Zhang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqiang Zheng, emhlbmd4aWFvcWlhbmdAeGp0dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.