 Yuan Sui
Yuan Sui Hanlin Jiang1
Hanlin Jiang1 Collyn M. Kellogg
Collyn M. Kellogg Ralf Janknecht
Ralf Janknecht- 1Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, OK, United States
- 2Department of Cell Biology, University of Oklahoma Health Sciences Center, Oklahoma City, OK, United States
- 3Stephenson Cancer Center, Oklahoma City, OK, United States
BHLHE40 is a transcription factor, whose role in colorectal cancer has remained elusive. We demonstrate that the BHLHE40 gene is upregulated in colorectal tumors. Transcription of BHLHE40 was jointly stimulated by the DNA-binding ETV1 protein and two associated histone demethylases, JMJD1A/KDM3A and JMJD2A/KDM4A, which were shown to also form complexes on their own and whose enzymatic activity was required for BHLHE40 upregulation. Chromatin immunoprecipitation assays revealed that ETV1, JMJD1A and JMJD2A interacted with several regions within the BHLHE40 gene promoter, suggesting that these three factors directly control BHLHE40 transcription. BHLHE40 downregulation suppressed both growth and clonogenic activity of human HCT116 colorectal cancer cells, strongly hinting at a pro-tumorigenic role of BHLHE40. Through RNA sequencing, the transcription factor KLF7 and the metalloproteinase ADAM19 were identified as putative BHLHE40 downstream effectors. Bioinformatic analyses showed that both KLF7 and ADAM19 are upregulated in colorectal tumors as well as associated with worse survival and their downregulation impaired HCT116 clonogenic activity. In addition, ADAM19, but not KLF7, downregulation reduced HCT116 cell growth. Overall, these data have revealed a ETV1/JMJD1A/JMJD2A→BHLHE40 axis that may stimulate colorectal tumorigenesis through upregulation of genes such as KLF7 and ADAM19, suggesting that targeting this axis represents a potential novel therapeutic avenue.
Introduction
The DNA-binding protein ETS variant 1 (ETV1; previously also known as ER81) is a downstream effector of signaling proteins such as RAS and HER2/EGFR2 that induce posttranslational modifications on ETV1 and thereby control its transcriptional activity (1–7). This seems to be important not only for normal development and homeostasis, but also during tumorigenesis (8, 9). The most studied association between ETV1 and malignancy pertains to prostate tumors, where ETV1 can be overexpressed due to chromosomal translocations (10). Mimicking this overexpression in mouse prostates led to neoplastic transformation, proving that ETV1 overexpression can indeed be a driver of tumorigenesis (11–14). Likewise, ETV1 has recently been implicated in colorectal cancer development (15, 16), yet the underlying mechanisms have remained unclear.
Transcriptomic studies on human HCT116 colorectal cancer cells pointed at potential downstream effectors of ETV1, including the basic helix-loop-helix family member E40 (BHLHE40; also known as BHLHB2, DEC1, SHARP2 or STRA13) (16). This DNA-binding transcription factor has pleiotropic functions. For instance, BHLHE40 modulates the circadian rhythm that likely involves its interaction with the seminal clock protein BMAL1 (17–19), affects immune responses and establishment of autoimmune disorders (20), enhances hippocampal synaptic plasticity (21), or attenuates lipid consumption and thereby facilitates high-fat diet-induced obesity (22). However, BHLHE40 is not essential, as respective homozygous knockout mice were viable and fertile and only displayed lymphoproliferative defects at later age with variable penetrance depending on the genetic background (23, 24).
The role of BHLHE40 in cancer is complex. Depending on the tissue and the stage of the disease, BHLHE40 has been implicated to promote or suppress tumorigenesis (25). An earlier study suggested that BHLHE40 is upregulated at both the mRNA and protein level in colon tumors, but this was based on a very small sample number of four (26). A more recent report analyzed BHLHE40 protein levels in ten colorectal tumors by immunohistochemistry and claimed overexpression, but no quantification or statistical analysis was provided (27). Moreover, these authors employed whole-body Bhlhe40 knockout mice and demonstrated that colon carcinogenesis induced by azoxymethane/dextran sulfate was compromised upon BHLHE40 ablation (27). However, chemically induced colon carcinogenesis is highly dependent on inflammation and the immune system. Given that BHLHE40 expression affects the T cell repertoire and thus likely modulates inflammatory responses in colorectal cancer (28), it is possible that BHLHE40 ablation in the immune cells rather than the colon epithelium antagonized azoxymethane/dextran sulfate-induced cell transformation and thereby colon cancer development.
Here, we provide evidence that BHLHE40 is needed for efficient growth and clonogenic activity of colorectal cancer cells. In addition, we demonstrate that BHLHE40 upregulation can be brought about by ETV1 together with two interacting epigenetic regulators, JMJD1A (also known as lysine demethylase (KDM) 3A) and JMJD2A (also known as KDM4A), which are histone demethylases (29–32). Finally, we uncovered how BHLHE40 may promote colon tumorigenesis by regulating the expression of various genes, including KLF7 and ADAM19.
Materials and methods
Virus production and infection
Retrovirus was produced according to standard procedures (33). Viral supernatant was then used to infect 2-3 times HCT116 colorectal cancer cells (CCL-247; American Type Culture Collection), followed by selection with 1-2 µg/ml puromycin (34). To validate effectiveness of viral infection, protein extracts were prepared from stably transduced cells and assessed by Western blotting (35). Rabbit polyclonal antibodies against BHLHE40 [Novus, NB100-1800), ETV1 (#959 generated by our laboratory (36)], JMJD1A (Novus, NB100-77282), JMJD2A (Bethyl, A300-861A) and actin (Sigma, A2066) were used as primary antibodies. The following shRNAs were utilized: shBHLHE40#1 (GGAGACCTACAAATTGCCG), shBHLHE40#2 (GAACCGGATGCAGCTTTCG), shBHLHE40#4 (GAGCTCAGACTCAGAGGAA), shADAM19#2 (GTTGTACTGTGAACTGGAA), shADAM19#3 (GAGTGTGACTGTGGAGAAG), shKLF7#2 (GGAGTCTACAGTCCTTACT) and shKLF7#6 (GCCTTATAAGTGCTCATGG). The shRNA sequences for ETV1, JMJD1A and JMJD2A have been published before (16, 36, 37).
RT-PCR and RNA sequencing
RNA was isolated with Trizol (38) and further purified with the RNeasy Mini kit (Qiagen, #74104). Utilizing random p(dN)6 primers, RNA was reverse transcribed (GoScript Reverse Transcription; Promega, #A5000) and then examined in real-time with the AccuPower 2xGreenstar qPCR kit (Bioneer, #K-6251) in a BioRad light cycler according to the manufacturer’s recommendations. Sequences of RT-PCR primers are provided in Supplementary Table 1. RNA sequencing was performed by Novogene and analysis of data done as described (39).
Chromatin immunoprecipitation assays
HCT116 cells were treated with formaldehyde and chromatin immunoprecipitations then performed as described (40). Antibodies used were control rabbit IgG (Santa Cruz, sc-2027), rabbit polyclonal anti-JMJD1A antibodies (Novus Biologicals, NB100-77282), anti-JMJD2A antibodies (Bethyl, A300-861A) or previously reported (36) anti-ETV1 antibodies. BHLHE40 promoter fragments were amplified with nested PCR (16) and then visualized through ethidium bromide staining (41) after agarose gel electrophoresis (42). Primers used for amplification are listed in Supplementary Table 2.
Luciferase assays
HCT116 cells were grown in 12-wells and transiently transfected with the help of 2 µg polyethylenimine (43). A total of 0.25 µg luciferase reporter plasmid, 0.9 µg pBluescript KS+, and 0.1 µg of empty vector pEV3S or ETV1 expression vector were used for each transfection when comparing pBL2-Basic to BHLHE40-luc, while 0.1 µg BHLHE40 luciferase reporter plasmid, 0.9 µg pBluescript KS+, 80 ng empty vector pEV3S or ETV1 expression vector, and 60 ng of empty vector pEV3S or indicated JMJD expression vector were employed otherwise. Eight hours afterwards, cells were washed with phosphate-buffered saline, and cell extracts were prepared 1.5 days later (44). Luciferase activity was then determined in a luminometer as described (45).
Coimmunoprecipitations
Human embryonic kidney 293T cells (CRL-3216; American Type Culture Collection) were cultured in poly-L-lysine coated 6-cm plates (46). Upon reaching 25% confluency, cells were transiently transfected by the calcium phosphate coprecipitation method (47). Two days after transfection, cell extracts were prepared (48) and immunoprecipitations performed as previously described (49). Precipitated proteins were run on SDS polyacrylamide gels (50), transferred to polyvinylidene fluoride membrane (51), and then challenged with indicated antibodies (52). After incubation with corresponding secondary antibodies coupled to horseradish peroxidase (53), signals were revealed by chemiluminescence (54) and exposure to film (55). Likewise, endogenous proteins were coimmunoprecipitated from human HCT116 colorectal cancer cells, utilizing control rabbit IgG (Santa Cruz, sc-2027) or rabbit polyclonal anti-JMJD1A antibodies (Bethyl, A301-538A) for immunoprecipitation followed by Western blotting with anti-JMJD2A antibodies (Bethyl, A300-861A).
Growth assays
Transduced HCT116 cells were seeded into 96-well plates at a density of 2800 cells/well and their growth measured as described (56). For clonogenic assays, 2000 cells were seeded into 6-well plates, grown for approximately 10 days, and then stained with crystal violet (57).
Results
Regulation of BHLHE40 transcription by ETV1, JMJD1A and JMJD2A
Previously, we performed RNA sequencing on ETV1-depleted human HCT116 colorectal cancer cells and found downregulation of BHLHE40 mRNA (16). To corroborate this finding, we performed Western blotting with HCT116 cells in which ETV1 was downregulated with two different shRNAs. And we expectedly observed that BHLHE40 protein levels became reduced upon ETV1 ablation (Figure 1A). Likewise, we downregulated the histone demethylase JMJD1A, which is capable of binding to ETV1 and also reportedly affects BHLHE40 transcription (16), and observed concurrent decreases of JMJD1A and BHLHE40 protein levels (Figure 1A). Like JMJD1A, the histone demethylase JMJD2A also directly binds to ETV1 (37), prompting us to assess if BHLHE40 would similarly be regulated by JMJD2A. And indeed, BHLHE40 was downregulated at both the protein and mRNA level upon expression of two different JMJD2A shRNAs, and the degree of this downregulation was similar to that observed with ETV1 or JMJD1A shRNAs (Figures 1A, B). These data imply that BHLHE40 gene transcription can be jointly regulated by ETV1, JMJD1A and JMJD2A.
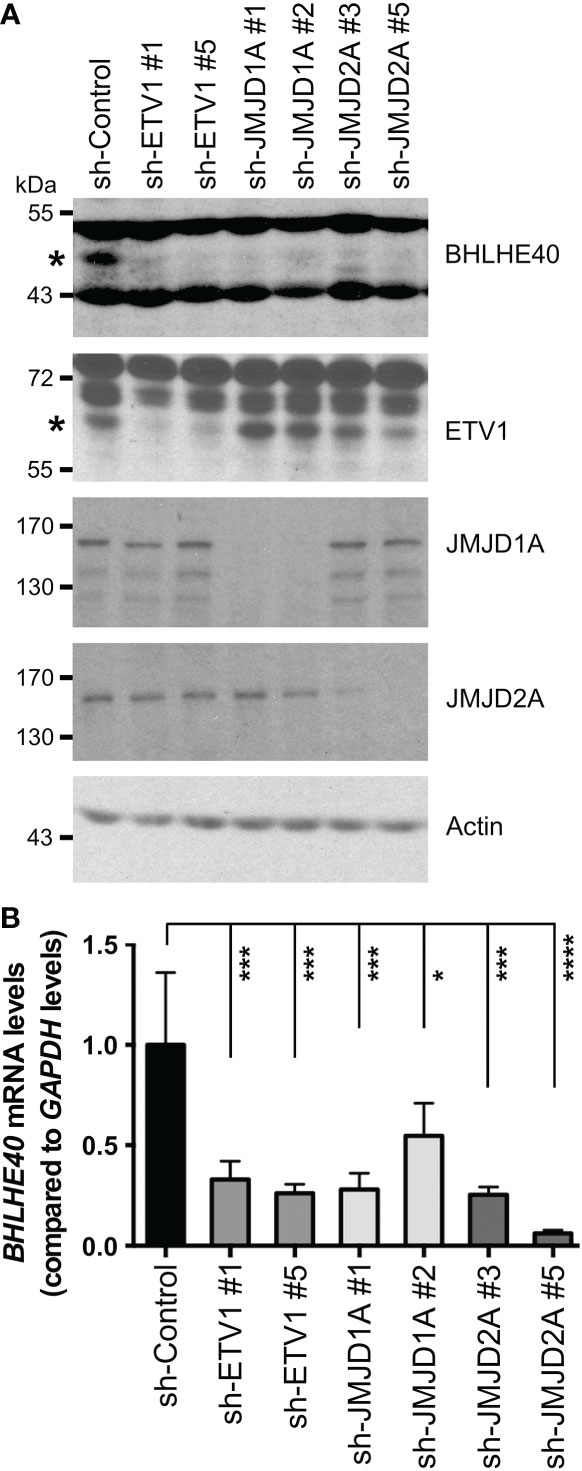
Figure 1 Regulation of BHLHE40 expression by ETV1 and associated JMJD cofactors. (A) Downregulation of ETV1, JMJD1A or JMJD2A with respective shRNAs in HCT116 cells reduced BHLHE40 protein levels. Asterisks mark BHLHE40 or ETV1, while other bands are unspecific in the respective blots. (B) Corresponding quantitative RT-PCR examining BHLHE40 mRNA levels. Shown are means with standard deviations (n=3). One-way ANOVA with post hoc Dunnett’s multiple comparisons test. *P < 0.05; ***P < 0.001; ****P < 0.0001.
Consistently, chromatin immunoprecipitation assays revealed that all three of these proteins bound to several regions within the human BHLHE40 gene promoter (Figure 2A). Notably, the BHLHE40 promoter contains numerous potential ETV1 binding sites throughout its length (Supplementary Figure 1), providing a possible explanation why ETV1 bound to all four tested regions of the BHLHE40 promoter. Further, we cloned the BHLHE40 promoter in front of a luciferase gene and then determined that ETV1 was capable of activating this reporter construct (Figure 2B). In contrast, neither ectopic JMJD1A nor ectopic JMJD2A alone activated the BHLHE40 luciferase reporter gene, but either one of these histone demethylases cooperated with ETV1 in doing so (Figure 2C). In contrast, catalytically inactive mutants of JMJD1A and JMJD2A were unable to cooperate with ETV1, and their expression alone even suppressed the BHLHE40 luciferase reporter gene (Figure 2C), possibly by competing with and thus precluding endogenous JMJD1A and JMJD2A to cooperate with endogenous ETV1. Overall, these data suggest that ETV1, JMJD1A and JMJD2A are jointly involved in the upregulation of the BHLHE40 gene promoter. In further support of this notion, BHLHE40 mRNA levels are positively correlated with ETV1, JMJD1A and JMJD2A in the 592 colorectal adenocarcinomas represented in The Cancer Genome Atlas (TCGA; Supplementary Figure 2).
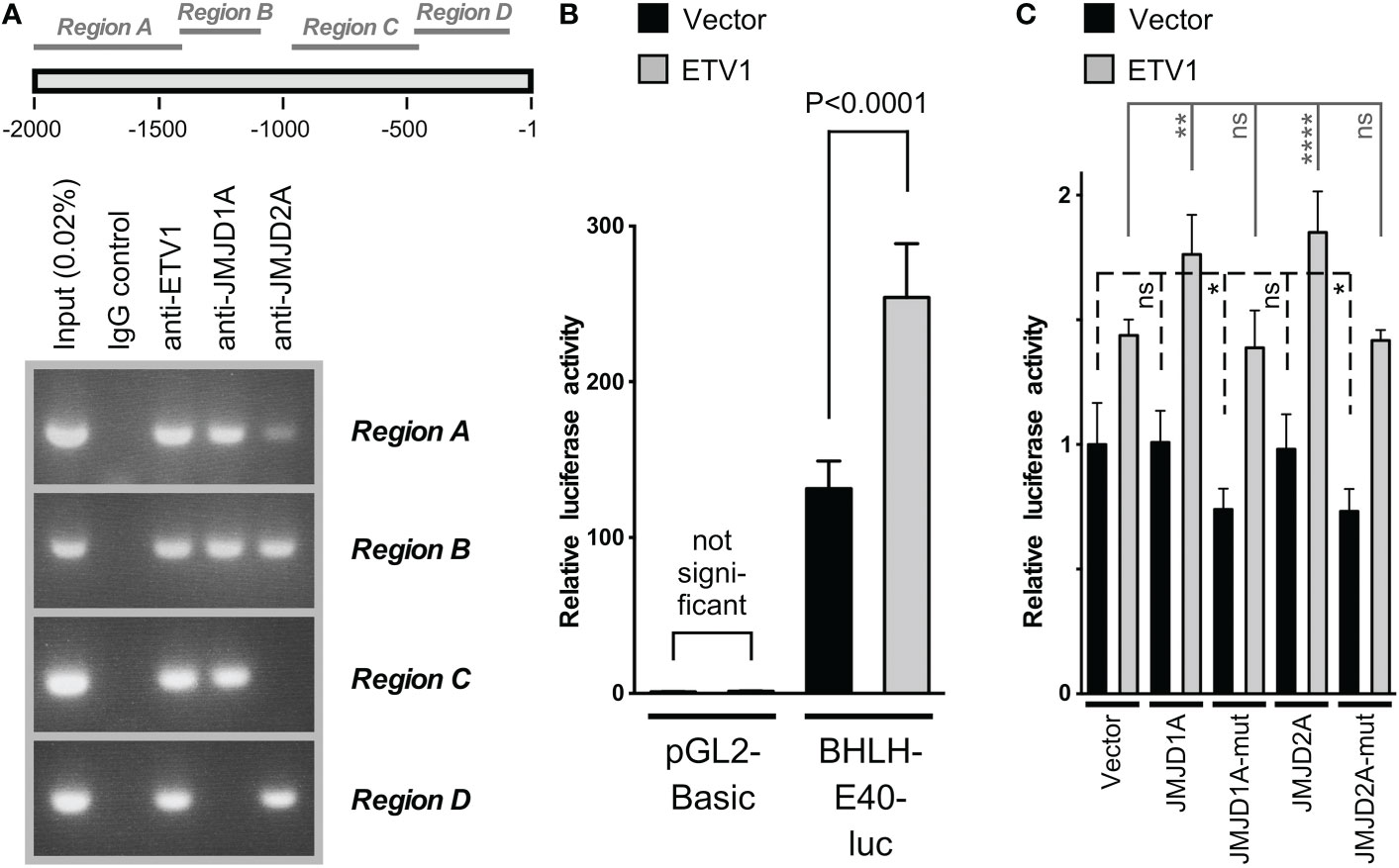
Figure 2 Analysis of the BHLHE40 gene promoter in HCT116 cells. (A) Chromatin immunoprecipitation assay. Four different regions (see scheme on top) of the BHLHE40 promoter were amplified after immunoprecipitation with indicated antibodies. (B) Activation of the (-2000/+50) BHLHE40 promoter luciferase reporter construct or the parental vector pGL2-Basic upon ETV1 overexpression. Shown are means with standard deviations (n=4). One-way ANOVA with post hoc Tukey’s multiple comparisons test. (C) Similar, joint stimulation of the (-2000/+50) BHLHE40 promoter luciferase activity by ETV1 and either JMJD1A or JMJD2A; “mut” indicates catalytically inactive mutants of JMJD1A (H1120A/D1122G) or JMJD2A (H188A). Shown are means with standard deviations (n=6). Statistical significance was determined by one-way ANOVA with post hoc Tukey’s multiple comparisons test. *P < 0.05; **P < 0.01; ****P < 0.0001; ns, not significant.
Because ETV1 can bind to both JMJD1A and JMJD2A (16, 37), their joint regulation of BHLHE40 transcription may involve the formation of a ternary complex of all these three factors. This would be consistent with the fact that all of these three transcription factors interacted with the same regions A and B of the BHLHE40 gene promoter in our chromatin immunoprecipitation experiments (Figure 2A). This additionally prompted us to examine if JMJD1A and JMJD2A would also form complexes with each other and not only with ETV1. To this end, we coexpressed Flag-tagged JMJD1A and Myc-tagged JMJD2A, performed immunoprecipitations with anti-Flag antibodies and probed with anti-Myc antibodies if JMJD2A would coimmunoprecipitate. And this was the case (Figure 3A), and this interaction between JMJD1A and JMJD2A could be confirmed by switching the tags in another coimmunoprecipitation experiment (Figure 3B) as well as upon assessing the complex formation between endogenous JMJD1A and endogenous JMJD2A in HCT116 cells (Figure 3C). We then determined that the N-terminal half of JMJD1A (amino acids 2-650), which does not contain the catalytic JmjC domain nor a Zn-finger also required for catalytic activity, primarily mediated the interaction with JMJD2A (Figure 3D). Deletion of the N-terminal JmjN domain that is required for the catalytic activity of JMJD2A did not abolish binding to JMJD1A (see 61-1064 in Figure 3E), but further deletion of the catalytic core JmjC domain did (see 301-1064). While this shows that the JmjC domain of JMJD2A is required for binding to JMJD1A, it is not sufficient since JMJD2A amino acids 2-350 did not bind to JMJD1A (Figure 3E). But JMJD2A amino acids 2-703 interacted with JMJD1A, indicating that the JMJD2A JmjC domain together with about 400 amino acids C-terminal of it jointly mediate the interaction with JMJD1A.
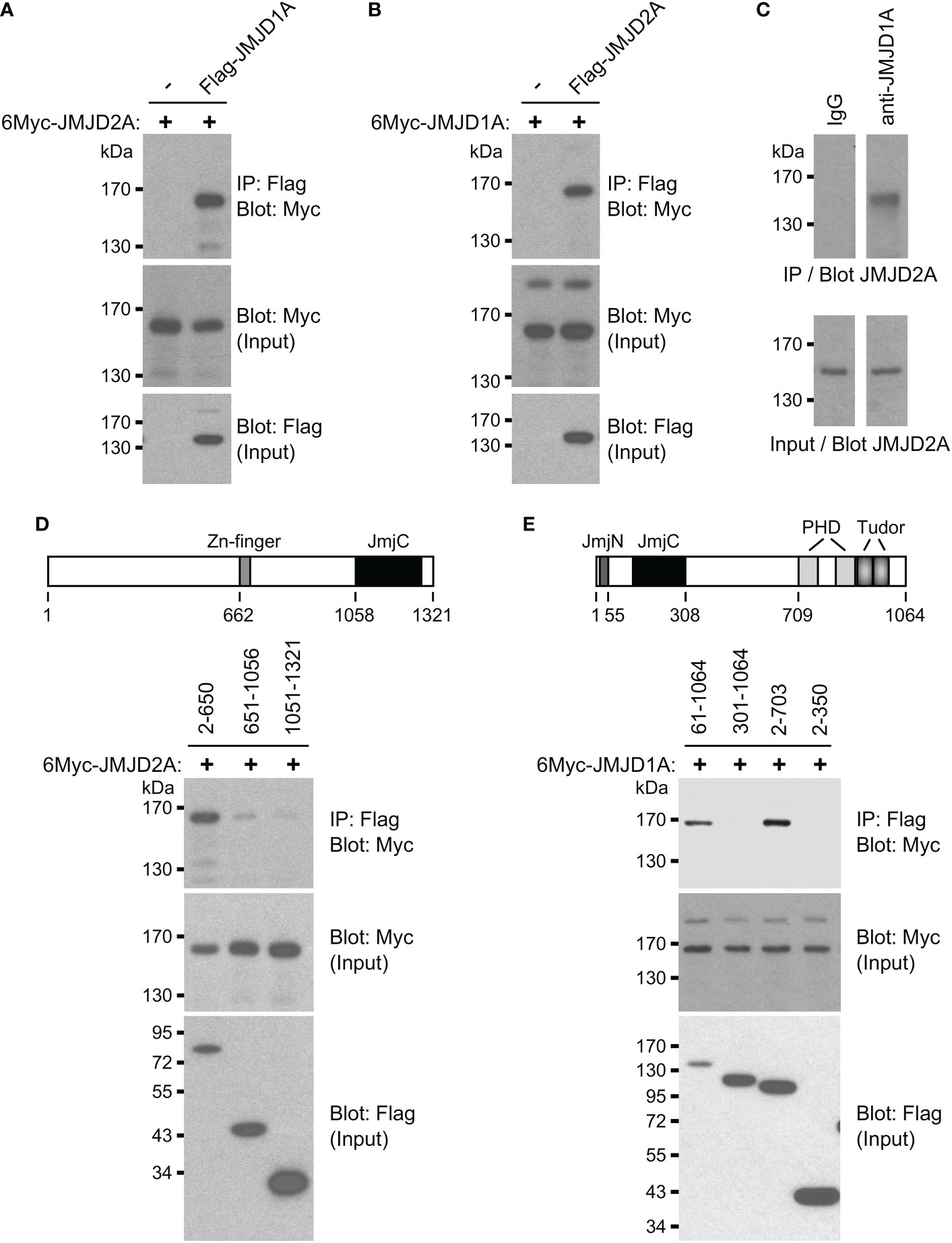
Figure 3 Complex formation between JMJD1A and JMJD2A. (A) Coimmunoprecipitation experiment with 6Myc-JMJD2A and Flag-JMJD1A in 293T cells. The top panel shows immunoprecipitation (IP) with anti-Flag antibodies followed by anti-Myc Western blotting, while the bottom two panels show input levels for 6Myc-JMJD2A and Flag-JMJD1A. (B) Likewise, interaction between 6Myc-JMJD1A and Flag-JMJD2A. (C) Endogenous coimmunoprecipitation of JMJD2A with JMJD1A in HCT116 cells. Either control IgG or anti-JMJD1A antibody was used for immunoprecipitation. (D) Coimmunoprecipitations of indicated Flag-tagged JMJD1A amino acids with 6Myc-JMJD2A. Top shows a sketch of JMJD1A. (E) Coimmunoprecipitation of 6Myc-JMJD1A with indicated Flag-tagged JMJD2A amino acids. Top shows a sketch of JMJD2A.
Stimulation of HCT116 cells by BHLHE40
Ablation of ETV1 (16), JMJD1A (58–60) or JMJD2A (61) alone reportedly suppressed growth or clonogenic activity of HCT116 cells, indicating that all three factors are required for the full oncogenic potential of these colorectal cancer cells. Hence, we tested whether their joint target gene, BHLHE40, would also promote the growth and/or clonogenic activity of HCT116 cells. To this end, we downregulated BHLHE40 with three different shRNAs and observed that this resulted in both reduced cell growth and clonogenic activity (Figure 4). While this manuscript was under preparation, another study similarly showed that a BHLHE40 siRNA was capable of reducing HCT116 cell growth (62). These data implicate that BHLHE40 in its own right is a tumor promoter in colorectal cancer. In agreement with this, analysis of published microarray data (63, 64) revealed that BHLHE40 mRNA levels are upregulated in colorectal tumors (Figure 5A and Supplementary Figures 3A-D) and may be a predictor of disease recurrence (Figure 5B and Supplementary Figures 3E, F) and disease-free survival (Figure 5C). Altogether, these data indicate that upregulation of BHLHE40 in human colorectal tumors may promote their growth and aggressiveness.
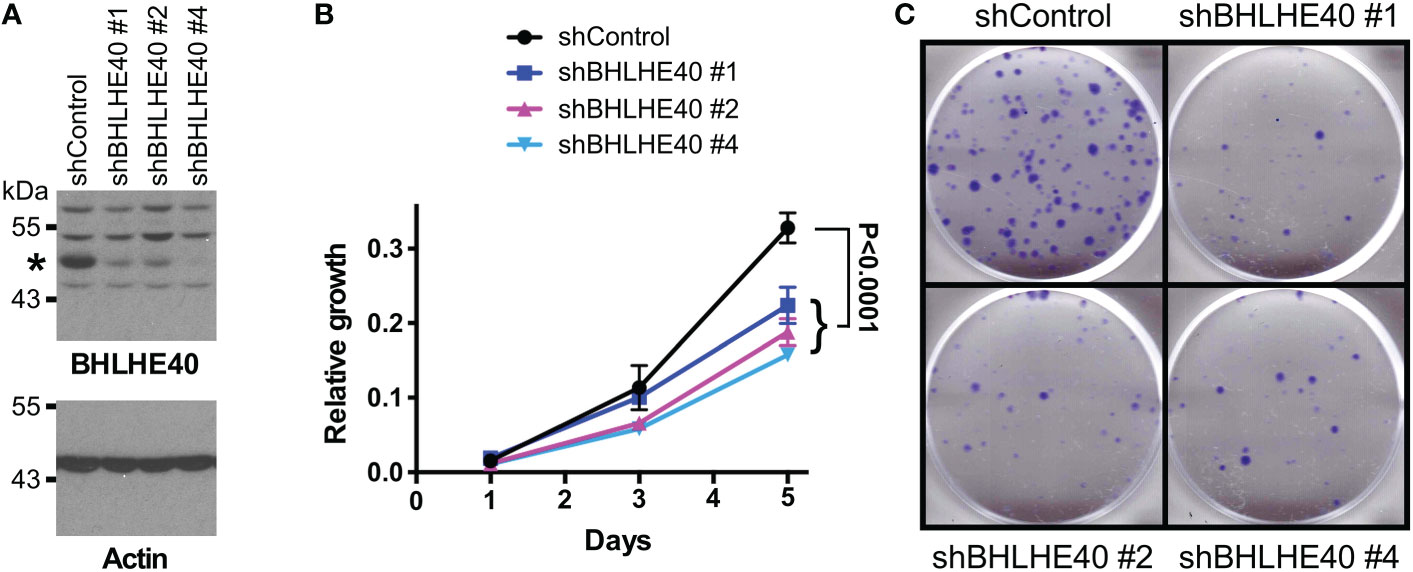
Figure 4 BHLHE40 promotes HCT116 growth. (A) Downregulation of BHLHE40 with three different shRNAs. Shown are BHLHE40 and actin Western blots. Asterisk marks BHLHE40. (B) Cell growth assay. Shown are means with standard deviations (n=3). Two-way ANOVA with post hoc Tukey’s multiple comparisons test. (C) Corresponding clonogenic assays with representative images.
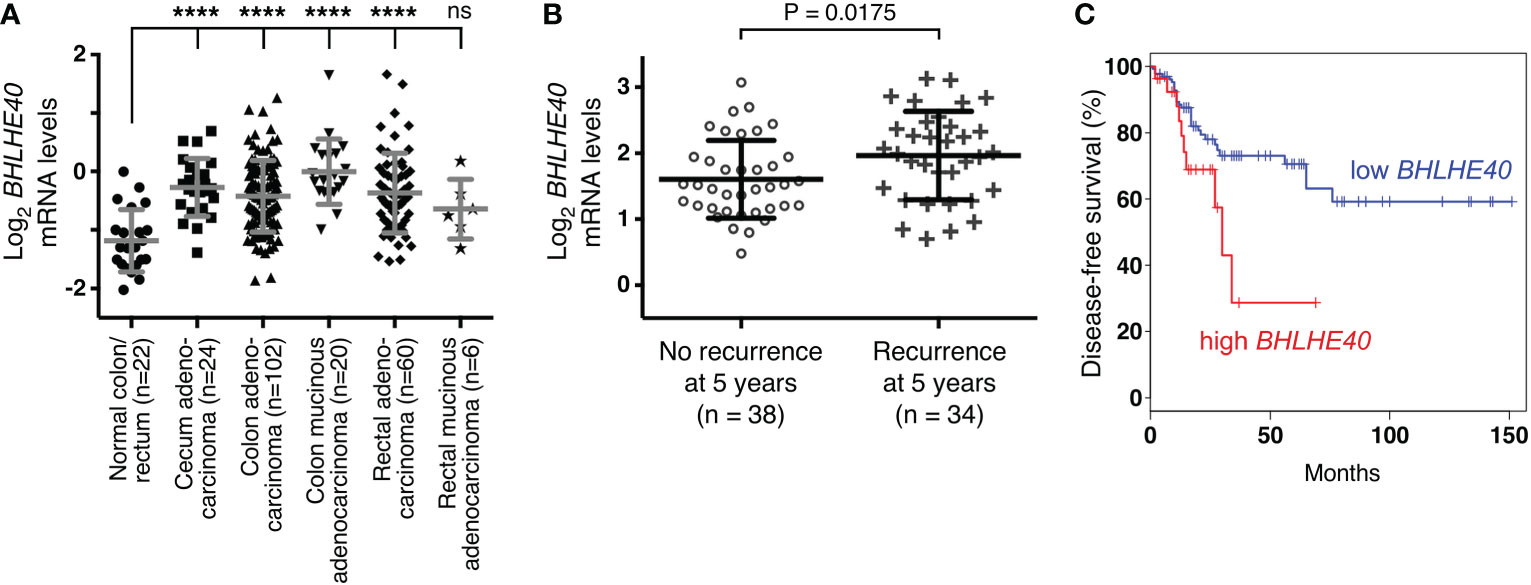
Figure 5 Expression of BHLHE40 in colorectal tumors. (A) BHLHE40 mRNA levels (reporter A_23_P57838; median-centered ratio) in TCGA microarray experiments (63). One-way ANOVA (Dunnett’s multiple comparisons test); ****P < 0.0001; ns, not significant. (B) Association of BHLHE40 mRNA levels with recurrence of colorectal carcinomas 5 years after diagnosis (data from reference (64); reporter 201169_s_at; median-centered ratio); unpaired, two-tailed t test. (C) Kaplan-Meier survival curve for patients with colorectal adenocarcinoma comparing high (90% cutoff, n=27) with low (50% cutoff, n=135) BHLHE40 mRNA expression. Analysis was done with GEPIA (http://gepia.cancer-pku.cn/index.html). P=0.028 (Logrank test).
Influence of BHLHE40 on the transcriptome
To gain mechanistic insights how BHLHE40 could affect colon cancer cells, we performed RNA sequencing on HCT116 cells that expressed three different BHLHE40 shRNAs. We found that this caused the down-/upregulation of 275/146 genes (Figures 6A, B). The fact that only 1.4% of all transcripts were significantly altered implies that BHLHE40 has a strong selectivity in its function as a transcriptional regulator. We then validated the RNA sequencing data by quantitative RT-PCR with a small selection of the differentially expressed genes (all downregulated in RNA sequencing data; Figure 6C). Amongst those, we selected ADAM19, which encodes for a metalloproteinase, and KLF7, which encodes for a DNA-binding transcription factor, for further analysis, because their expression was positively correlated with BHLHE40 in 592 colon adenocarcinomas (Figures 6D, E) and little was known about their role in colon cancer.
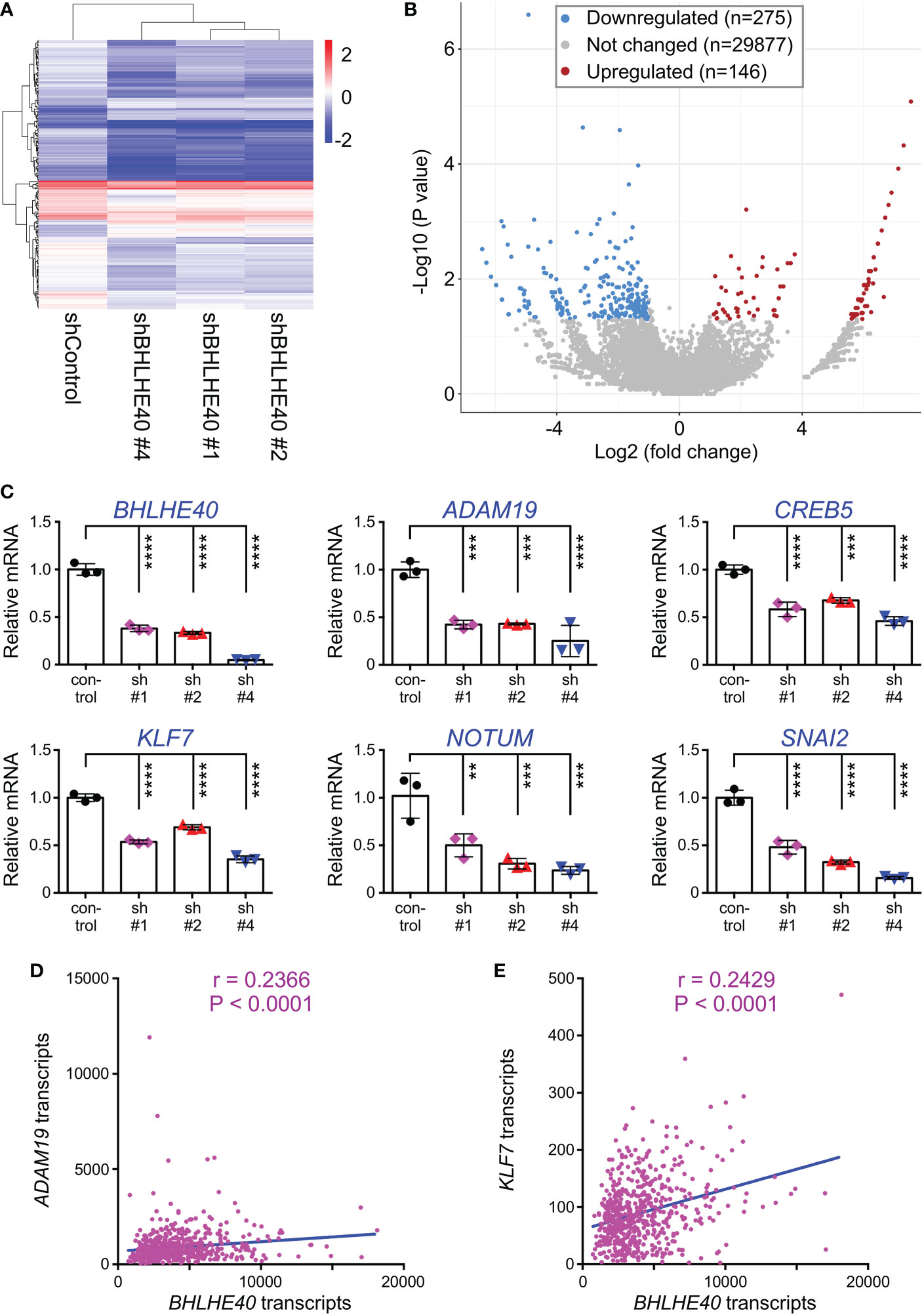
Figure 6 Transcriptome changes upon BHLHE40 downregulation. (A) Heat map of transcript changes between control HCT116 cells and those with downregulation of BHLHE40. (B) Volcano plot showing average P values versus average fold changes. Only genes with absolute transcript level changes of greater than 2 and P<0.05 were considered up- or downregulated. (C) Validation by quantitative RT-PCR for indicated transcripts that were found downregulated upon expression of BHLHE40 shRNAs. All mRNA levels were normalized to GAPDH. One-way ANOVA (Dunnett’s multiple comparisons test; n=3); **P < 0.01; ***P < 0.001; ****P < 0.0001. (D) Correlation of BHLHE40 mRNA levels with those of ADAM19 in 592 colorectal adenocarcinomas. Data were derived from the TCGA PanCancer Atlas (RSEM values; batch normalized from Illumina HiSeq_RNASeqV2); r=Spearman correlation coefficient. (E) Analogous for KLF7.
Role of KLF7 and ADAM19 in colorectal cancer cells
First, we determined in published (63) microarray data that KLF7 mRNA is significantly upregulated in various subtypes of colorectal cancer (Figure 7A). Likewise, analysis of public RNA sequencing data revealed upregulation of KLF7 mRNA in colorectal adenocarcinomas at the primary tumor site (Figure 7B). Moreover, we found a significant association of KLF7 mRNA with disease recurrence in another published (65) microarray data set (Figure 7C). Further, high KLF7 levels are associated with reduced disease-free survival (Figure 7D). Altogether, these data suggest that KLF7 may promote colon tumorigenesis and may be a predictor of worse outcome. Similarly, we found that ADAM19 is significantly overexpressed in colorectal adenocarcinomas (Figure 7E), corroborating a previous study comparing a small number (n=14) of colorectal tumors to normal tissue (66). And high ADAM19 expression appears to also be predictive of worse disease-free survival, although this does not reach statistical significance (Figure 7F), suggesting that ADAM19 exerts pro-tumorigenic functions in colon cancer.
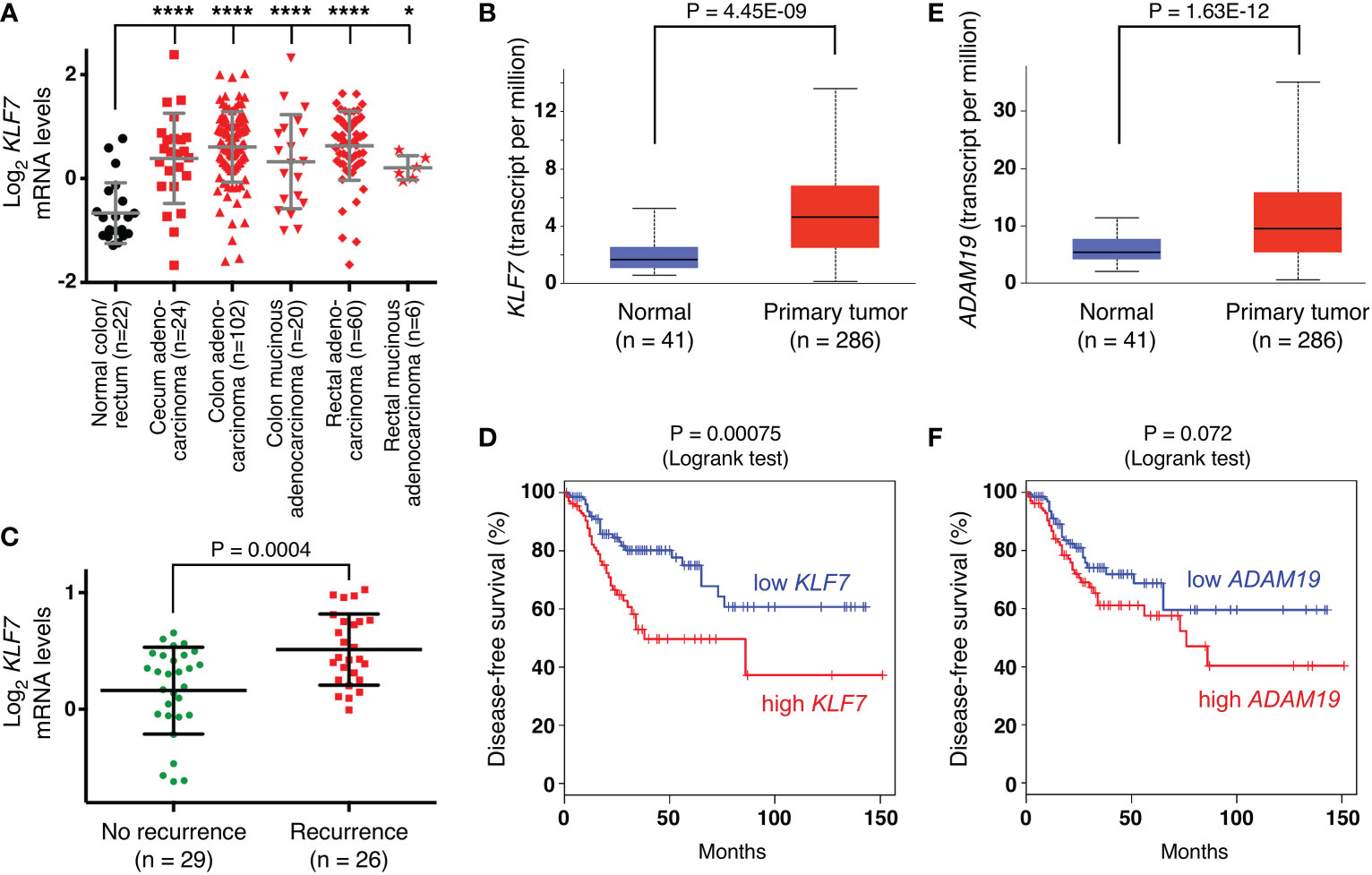
Figure 7 Expression of KLF7 and ADAM19 in colorectal tumors. (A) KLF7 mRNA levels (reporter A_23_P67978; median-centered ratio) in TCGA microarray experiments (63). One-way ANOVA (Dunnett’s multiple comparisons test); *P < 0.05; ****P < 0.0001. (B) KLF7 transcripts determined by RNA sequencing in TCGA colorectal adenocarcinomas (box plot; unpaired, two-tailed t test). Analysis was done with UALCAN (http://ualcan.path.uab.edu). (C) Association of KLF7 mRNA levels with recurrence of colorectal carcinomas 5 years after diagnosis (data from reference (65); reporter 204334_at; median-centered ratio); unpaired, two-tailed t test. (D) Disease-free survival for patients with colorectal adenocarcinoma comparing high (median cutoff, n=135) with low (median cutoff, n=135) KLF7 mRNA expression. Analysis was done with GEPIA (http://gepia.cancer-pku.cn/index.html). (E) As in panel (B) for ADAM19. (F) As in panel D for ADAM19.
We then tested how downregulation of KLF7 or ADAM19 would affect HCT116 cells. Using two independent shRNAs, we achieved efficient ADAM19 downregulation (Figure 8A). Both of these ADAM19 shRNAs caused a significant decrease in HCT116 cell growth and clonogenic activity (Figures 8B, C), which is similar to what we observed upon BHLHE40 downregulation (see Figure 4). In contrast, downregulation of KLF7 did not affect HCT116 cell growth, but still led to a reduction in clonogenic activity (Figures 8D-F), albeit less so than compared to ADAM19 downregulation. These data suggest that KLF7 and especially ADAM19 are downstream effectors of BHLHE40.
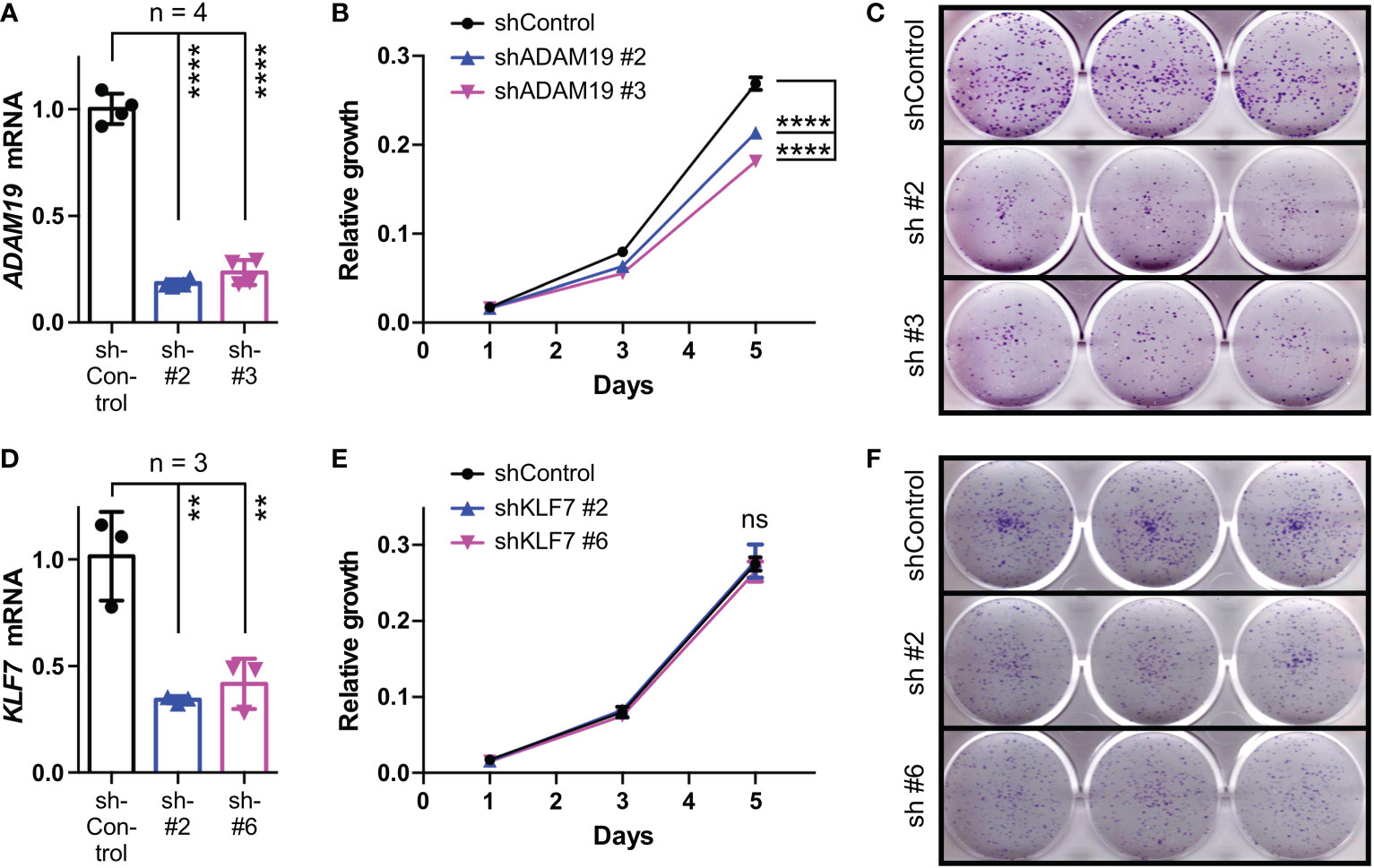
Figure 8 Role of KLF7 and ADAM19 for HCT116 cell growth and clonogenicity. (A) Downregulation of ADAM19 with two different shRNAs. Shown are ADAM19 mRNA levels normalized to GAPDH levels (means with standard deviations). One-way ANOVA with post hoc Tukey’s multiple comparisons test. (B) Impact of ADAM19 downregulation on cell growth. Shown are means with standard deviations (n=3). Two-way ANOVA with post hoc Tukey’s multiple comparisons test. (C) Clonogenic assays upon ADAM19 downregulation; shown are representative images. (D-F) Analogous to panels A-C for KLF7. **P < 0.01; ****P < 0.0001; ns, not significant.
Discussion
In the present study, we have discovered that BHLHE40 transcription is jointly stimulated by ETV1, JMJD1A and JMJD2A, that BHLHE40 can stimulate growth and clonogenic activity of HCT116 colorectal cancer cells, that KLF7 and ADAM19 are regulated by BHLHE40, and that KLF7 and ADAM19 are in their own right potential promoters of colorectal tumors.
Corroborating previous studies (26, 27), we found that BHLHE40 is upregulated in colorectal tumors. Importantly, our results demonstrated that high BHLHE40 levels are correlated with more disease recurrence and reduced survival, implying that BHLHE40 levels can predict disease aggressiveness. Why BHLHE40 is upregulated in colorectal cancer has remained unknown, but our study strongly indicates that this could be due to enhanced activity of ETV1, JMJD1A and/or JMJD2A. Both ETV1 and JMJD1A are reportedly overexpressed in colorectal tumors (15, 16, 58–60), providing one plausible explanation for BHLHE40 upregulation in this cancer. Since ETV1, JMJD1A and JMJD2A are implicated in many different types of malignancies (8, 29–32), BHLHE40 upregulation by these three proteins is likely not limited to colorectal tumors. Notably, results from our chromatin immunoprecipitation experiments support the notion that a ternary complex of ETV1, JMJD1A and JMJD2A may bind to the BHLHE40 promoter in regions A and B (see Figure 2A); however, it appears that binary complexes of ETV1/JMJD1A and ETV1/JMJD2A are also involved in BHLHE40 upregulation, as exclusive binding of respective protein pairs was found in regions C and D, respectively, of the BHLHE40 gene promoter (see Figure 2A). Further, since we demonstrated that JMJD1A and JMJD2A formed complexes by themselves, these two proteins could be recruited together by a DNA-binding transcription factor(s) other than ETV1 to various gene promoters. The joint recruitment of JMJD1A and JMJD2A may be very effective to remove the repressive H3K9me3/2 epigenetic promoter marks, because JMJD2A preferentially demethylates H3K9me3 to H3K9me2, which can then be converted to H3K9me1 by JMJD1A (29–32). And the cooperation between JMJD1A and JMJD2A in regulating BHLHE40 transcription may be relevant to diseases other than cancer, including cardiac hypertrophy and fibrosis that are dependent on BHLHE40 (67, 68) and promoted by JMJD1A and JMJD2A overexpression (69, 70).
BHLHE40 is capable of binding to specific DNA sites and this often leads to transcriptional repression, possibly through recruitment of a histone deacetylase (25, 71–73). However, BHLHE40 can also stimulate gene transcription, but this may in part be through recruitment by other DNA-binding proteins such as STAT3 or SP1 (74, 75). Furthermore, BHLHE40 may influence gene transcription by modulating the formation of chromatin loops through binding CTCF (76). Our transcriptomic analyses in HCT116 colorectal cancer cells showed that downregulation of BHLHE40 led more often to a reduction than an increase of gene transcription, suggesting that BHLHE40 is mostly stimulating transcription of genes, including KLF7 and ADAM19. If this is through direct DNA binding at respective gene promoters or other means remains to be studied.
The transcription factor KLF7 plays important roles in neuronal morphogenesis, which is why its knockout in mice led to neonatal death (77, 78). It also plays important roles in tumorigenesis. For instance, KLF7 has been implicated as a tumor promoter in oral squamous carcinoma (79), pancreatic cancer (80), hepatocellular carcinoma (81) or gastric cancer (82), but its role in colorectal cancer has been elusive. Our data indicate that KLF7 may promote the clonogenic activity of colorectal cancer cells and that KLF7 overexpression may even have prognostic value. Interestingly, KLF7 has been shown to modulate stem cells and lineage differentiation (83, 84), but if KLF7 likewise influences cancer stem cells and how this may relate to colon tumorigenesis remains to be studied.
ADAM19 belongs to a family of metalloproteinases that have various functions. Their proteolytic activity can activate growth factors and cytokines, lead to the shedding of ectodomains of membrane-bound receptors, influence integrin signaling and degrade extracellular matrix components. As such, ADAM proteins can have pleiotropic effects, including regulating cell proliferation, migration and invasion in normal and cancer cells, and are therefore potential targets in cancer therapy (85, 86). This pertains also to ADAM19, which has previously been shown to be overexpressed in brain tumors and implicated as a promoter of invasion (87, 88). Likewise, a previous study (66) and our data strongly suggest that ADAM19 is overexpressed in colorectal cancer and may be a predictor of survival. Overexpression of ADAM19 in colorectal cancer may in part be due to the downregulation of microRNAs targeting ADAM19 (89, 90). Interestingly, overexpression of ADAM10, ADAM17 and ADAM19 together resulted in increased migration and invasion of HCT116 cells, but it remained undefined which of the three ADAMs was responsible (66). Our data show that ADAM19 downregulation decreased HCT116 cell growth and clonogenic activity, which hints at a pro-tumorigenic function of ADAM19 in colorectal cancer.
In our RNA sequencing study, we found that many genes besides KLF7 and ADAM19 were, directly or indirectly, regulated by BHLHE40 and might therefore also (partially) mediate BHLHE40’s tumorigenic functions. For instance, the CREB5 transcription factor gene was downregulated in the BHLHE40 shRNA-treated HCT116 cells, suggesting that BHLHE40 stimulates CREB5 expression. And CREB5 is overexpressed in colorectal tumors and capable of enhancing proliferation, invasion and metastasis of colorectal cancer cells (91, 92). Similarly, SNAI2, which encodes for a zinc-finger transcription factor regulating epithelial-mesenchymal transition and is overexpressed in colorectal tumors (93), was positively regulated by BHLHE40, and SNAI2 is capable of stimulating colorectal cancer cell invasion and tumor formation (94–96). NOTUM, which encodes for a palmitoleoyl-protein carboxylesterase being overexpressed in colon tumors and supporting tumor initiation and proliferation (97–100), was also found to be downregulated upon BHLHE40 ablation. This suggests that BHLHE40 acts through multiple effectors to facilitate tumorigenesis.
In conclusion, our data have uncovered a ETV1/JMJD1A/JMJD2A→BHLHE40 axis in colon cancer cells. Antagonizing this axis may potentially suppress colorectal tumor formation, and likewise the inhibition of BHLHE40-regulated genes such as ADAM19 or KLF7 may have therapeutic value.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA859517.
Author contributions
YS, HJ, CK and RJ conceived, designed and performed experiments. YS, HJ, CK, SO and RJ analyzed and interpreted data as well as contributed to the writing of this manuscript. RJ supervised this study. All authors contributed to the article and approved the submitted version.
Funding
This work was in part funded by grant R01 CA154745 (to RJ) from the National Cancer Institute of the USA. RJ has also been supported in part by the Oklahoma Tobacco Settlement Endowment Trust through an award made to the University of Oklahoma/Stephenson Cancer Center. Additional assistance was provided by the Stephenson Cancer Center Molecular Biology Core that was supported by National Institutes of Health grants P30 CA225520 and P20 GM103639. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the granting agencies.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1122238/full#supplementary-material
References
1. Janknecht R. Analysis of the ERK-stimulated ETS transcription factor ER81. Mol Cell Biol (1996) 16:1550–6. doi: 10.1128/MCB.16.4.1550
2. Bosc DG, Goueli BS, Janknecht R. HER2/Neu-mediated activation of the ETS transcription factor ER81 and its target gene MMP-1. Oncogene (2001) 20:6215–24. doi: 10.1038/sj.onc.1204820
3. Bosc DG, Janknecht R. Regulation of HER2/Neu promoter activity by the ETS transcription factor, ER81. J Cell Biochem (2002) 86:174–83. doi: 10.1002/jcb.10205
4. Wu J, Janknecht R. Regulation of the ETS transcription factor ER81 by the 90-kDa ribosomal S6 kinase 1 and protein kinase a. J Biol Chem (2002) 277:42669–79. doi: 10.1074/jbc.M205501200
5. Goel A, Janknecht R. Acetylation-mediated transcriptional activation of the ETS protein ER81 by p300, P/CAF, and HER2/Neu. Mol Cell Biol (2003) 23:6243–54. doi: 10.1128/MCB.23.17.6243-6254.2003
6. Janknecht R. Regulation of the ER81 transcription factor and its coactivators by mitogen- and stress-activated protein kinase 1 (MSK1). Oncogene (2003) 22:746–55. doi: 10.1038/sj.onc.1206185
7. Goueli BS, Janknecht R. Upregulation of the catalytic telomerase subunit by the transcription factor ER81 and oncogenic HER2/Neu, ras, or raf. Mol Cell Biol (2004) 24:25–35. doi: 10.1128/MCB.24.1.25-35.2004
8. Oh S, Shin S, Janknecht R. ETV1, 4 and 5: An oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta (2012) 1826:1–12. doi: 10.1016/j.bbcan.2012.02.002
9. Nicholas TR, Strittmatter BG, Hollenhorst PC. Oncogenic ETS factors in prostate cancer. Adv Exp Med Biol (2019) 1210:409–36. doi: 10.1007/978-3-030-32656-2_18
10. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science (2005) 310:644–8. doi: 10.1126/science.1117679
11. Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature (2007) 448:595–9. doi: 10.1038/nature06024
12. Shin S, Kim TD, Jin F, van Deursen JM, Dehm SM, Tindall DJ, et al. Induction of prostatic intraepithelial neoplasia and modulation of androgen receptor by ETS variant 1/ETS-related protein 81. Cancer Res (2009) 69:8102–10. doi: 10.1158/0008-5472.CAN-09-0941
13. Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y, et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev (2013) 27:683–98. doi: 10.1101/gad.211011.112
14. Oh S, Shin S, Song H, Grande JP, Janknecht R. Relationship between ETS transcription factor ETV1 and TGF-beta-regulated SMAD proteins in prostate cancer. Sci Rep (2019) 9:8186. doi: 10.1038/s41598-019-44685-3
15. Orlando G, Law PJ, Cornish AJ, Dobbins SE, Chubb D, Broderick P, et al. Promoter capture Hi-c-based identification of recurrent noncoding mutations in colorectal cancer. Nat Genet (2018) 50:1375–80. doi: 10.1038/s41588-018-0211-z
16. Oh S, Song H, Freeman WM, Shin S, Janknecht R. Cooperation between ETS transcription factor ETV1 and histone demethylase JMJD1A in colorectal cancer. Int J Oncol (2020) 57:1319–32. doi: 10.3892/ijo.2020.5133
17. Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M, et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature (2002) 419:841–4. doi: 10.1038/nature01123
18. Sato F, Kawamoto T, Fujimoto K, Noshiro M, Honda KK, Honma S, et al. Functional analysis of the basic helix-loop-helix transcription factor DEC1 in circadian regulation. interaction with BMAL1. Eur J Biochem (2004) 271:4409–19. doi: 10.1111/j.1432-1033.2004.04379.x
19. Nakashima A, Kawamoto T, Honda KK, Ueshima T, Noshiro M, Iwata T, et al. DEC1 modulates the circadian phase of clock gene expression. Mol Cell Biol (2008) 28:4080–92. doi: 10.1128/MCB.02168-07
20. Cook ME, Jarjour NN, Lin CC, Edelson BT. Transcription factor Bhlhe40 in immunity and autoimmunity. Trends Immunol (2020) 41:1023–36. doi: 10.1016/j.it.2020.09.002
21. Hamilton KA, Wang Y, Raefsky SM, Berkowitz S, Spangler R, Suire CN, et al. Mice lacking the transcriptional regulator Bhlhe40 have enhanced neuronal excitability and impaired synaptic plasticity in the hippocampus. PloS One (2018) 13:e0196223. doi: 10.1371/journal.pone.0196223
22. Noshiro M, Kawamoto T, Nakashima A, Ozaki N, Ueno T, Saeki M, et al. Deficiency of the basic helix-loop-helix transcription factor DEC1 prevents obesity induced by a high-fat diet in mice. Genes Cells (2018) 23:658–69. doi: 10.1111/gtc.12607
23. Sun H, Lu B, Li RQ, Flavell RA, Taneja R. Defective T cell activation and autoimmune disorder in Stra13-deficient mice. Nat Immunol (2001) 2:1040–7. doi: 10.1038/ni721
24. Miyazaki K, Miyazaki M, Guo Y, Yamasaki N, Kanno M, Honda Z, et al. The role of the basic helix-loop-helix transcription factor Dec1 in the regulatory T cells. J Immunol (2010) 185:7330–9. doi: 10.4049/jimmunol.1001381
25. Kiss Z, Mudryj M, Ghosh PM. Non-circadian aspects of BHLHE40 cellular function in cancer. Genes Cancer (2020) 11:1–19. doi: 10.18632/genesandcancer.201
26. Li Y, Zhang H, Xie M, Hu M, Ge S, Yang D, et al. Abundant expression of Dec1/stra13/sharp2 in colon carcinoma: Its antagonizing role in serum deprivation-induced apoptosis and selective inhibition of procaspase activation. Biochem J (2002) 367:413–22. doi: 10.1042/bj20020514
27. Shan E, Huo Y, Wang H, Zhang Z, Hu J, Wang G, et al. Differentiated embryonic chondrocyte expressed gene-1 (DEC1) enhances the development of colorectal cancer with an involvement of the STAT3 signaling. Neoplasia (2022) 27:100783. doi: 10.1016/j.neo.2022.100783
28. Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature (2018) 564:268–72. doi: 10.1038/s41586-018-0694-x
29. Yoo J, Jeon YH, Cho HY, Lee SW, Kim GW, Lee DH, et al. Advances in histone demethylase KDM3A as a cancer therapeutic target. Cancers (Basel) (2020) 12:1098. doi: 10.3390/cancers12051098
30. Sui Y, Gu R, Janknecht R. Crucial functions of the JMJD1/KDM3 epigenetic regulators in cancer. Mol Cancer Res (2020) 18:1615–25. doi: 10.1158/1541-7786.MCR-20-0404
31. Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: Epigenetic regulators in cancer cells. Cancer Res (2013) 73:2936–42. doi: 10.1158/0008-5472.CAN-12-4300
32. Lee DH, Kim GW, Jeon YH, Yoo J, Lee SW, Kwon SH. Advances in histone demethylase KDM4 as cancer therapeutic targets. FASEB J (2020) 34:3461–84. doi: 10.1096/fj.201902584R
33. Kim TD, Oh S, Lightfoot SA, Shin S, Wren JD, Janknecht R. Upregulation of PSMD10 caused by the JMJD2A histone demethylase. Int J Clin Exp Med (2016) 9:10123–34.
34. Kim TD, Fuchs JR, Schwartz E, Abdelhamid D, Etter J, Berry WL, et al. Pro-growth role of the JMJD2C histone demethylase in HCT-116 colon cancer cells and identification of curcuminoids as JMJD2 inhibitors. Am J Transl Res (2014) 6:236–47.
35. Shin S, Bosc DG, Ingle JN, Spelsberg TC, Janknecht R. Rcl is a novel ETV1/ER81 target gene upregulated in breast tumors. J Cell Biochem (2008) 105:866–74. doi: 10.1002/jcb.21884
36. Oh S, Shin S, Lightfoot SA, Janknecht R. 14-3-3 proteins modulate the ETS transcription factor ETV1 in prostate cancer. Cancer Res (2013) 73:5110–9. doi: 10.1158/0008-5472.CAN-13-0578
37. Kim TD, Jin F, Shin S, Oh S, Lightfoot SA, Grande JP, et al. Histone demethylase JMJD2A drives prostate tumorigenesis through transcription factor ETV1. J Clin Invest (2016) 126:706–20. doi: 10.1172/JCI78132
38. DiTacchio L, Bowles J, Shin S, Lim DS, Koopman P, Janknecht R. Transcription factors ER71/ETV2 and SOX9 participate in a positive feedback loop in fetal and adult mouse testis. J Biol Chem (2012) 287:23657–66. doi: 10.1074/jbc.M111.320101
39. Sui Y, Li X, Oh S, Zhang B, Freeman WM, Shin S, et al. Opposite roles of the JMJD1A interaction partners MDFI and MDFIC in colorectal cancer. Sci Rep (2020) 10:8710. doi: 10.1038/s41598-020-65536-6
40. Kim TD, Shin S, Janknecht R. ETS transcription factor ERG cooperates with histone demethylase KDM4A. Oncol Rep (2016) 35:3679–88. doi: 10.3892/or.2016.4747
41. Oh S, Janknecht R. Histone demethylase JMJD5 is essential for embryonic development. Biochem Biophys Res Commun (2012) 420:61–5. doi: 10.1016/j.bbrc.2012.02.115
42. Shin S, Oh S, An S, Janknecht R. ETS variant 1 regulates matrix metalloproteinase-7 transcription in LNCaP prostate cancer cells. Oncol Rep (2013) 29:306–14. doi: 10.3892/or.2012.2079
43. Li X, Moon G, Shin S, Zhang B, Janknecht R. Cooperation between ETS variant 2 and jumonji domain-containing 2 histone demethylases. Mol Med Rep (2018) 17:5518–27. doi: 10.3892/mmr.2018.8507
44. Mooney SM, Goel A, D’Assoro AB, Salisbury JL, Janknecht R. Pleiotropic effects of p300-mediated acetylation on p68 and p72 RNA helicase. J Biol Chem (2010) 285:30443–52. doi: 10.1074/jbc.M110.143792
45. Knebel J, De Haro L, Janknecht R. Repression of transcription by TSGA/Jmjd1a, a novel interaction partner of the ETS protein ER71. J Cell Biochem (2006) 99:319–29. doi: 10.1002/jcb.20945
46. Berry WL, Shin S, Lightfoot SA, Janknecht R. Oncogenic features of the JMJD2A histone demethylase in breast cancer. Int J Oncol (2012) 41:1701–6. doi: 10.3892/ijo.2012.1618
47. De Haro L, Janknecht R. Functional analysis of the transcription factor ER71 and its activation of the matrix metalloproteinase-1 promoter. Nucleic Acids Res (2002) 30:2972–9. doi: 10.1093/nar/gkf390
48. Dowdy SC, Mariani A, Janknecht R. HER2/Neu- and TAK1-mediated up-regulation of the transforming growth factor beta inhibitor Smad7 via the ETS protein ER81. J Biol Chem (2003) 278:44377–84. doi: 10.1074/jbc.M307202200
49. Kim J, Shin S, Subramaniam M, Bruinsma E, Kim TD, Hawse JR, et al. Histone demethylase JARID1B/KDM5B is a corepressor of TIEG1/KLF10. Biochem Biophys Res Commun (2010) 401:412–6. doi: 10.1016/j.bbrc.2010.09.068
50. Kim TD, Shin S, Janknecht R. Repression of Smad3 activity by histone demethylase SMCX/JARID1C. Biochem Biophys Res Commun (2008) 366:563–7. doi: 10.1016/j.bbrc.2007.12.013
51. Papoutsopoulou S, Janknecht R. Phosphorylation of ETS transcription factor ER81 in a complex with its coactivators CREB-binding protein and p300. Mol Cell Biol (2000) 20:7300–10. doi: 10.1128/MCB.20.19.7300-7310.2000
52. Goel A, Janknecht R. Concerted activation of ETS protein ER81 by p160 coactivators, the acetyltransferase p300 and the receptor tyrosine kinase HER2/Neu. J Biol Chem (2004) 279:14909–16. doi: 10.1074/jbc.M400036200
53. Rossow KL, Janknecht R. The ewing’s sarcoma gene product functions as a transcriptional activator. Cancer Res (2001) 61:2690–5.
54. Mooney SM, Grande JP, Salisbury JL, Janknecht R. Sumoylation of p68 and p72 RNA helicases affects protein stability and transactivation potential. Biochemistry (2010) 49:1–10. doi: 10.1021/bi901263m
55. Oh S, Shin S, Janknecht R. Sumoylation of transcription factor ETV1 modulates its oncogenic potential in prostate cancer. Int J Clin Exp Pathol (2021) 14:795–810.
56. Kim TD, Oh S, Shin S, Janknecht R. Regulation of tumor suppressor p53 and HCT116 cell physiology by histone demethylase JMJD2D/KDM4D. PloS One (2012) 7:e34618. doi: 10.1371/journal.pone.0034618
57. Berry WL, Kim TD, Janknecht R. Stimulation of beta-catenin and colon cancer cell growth by the KDM4B histone demethylase. Int J Oncol (2014) 44:1341–8. doi: 10.3892/ijo.2014.2279
58. Uemura M, Yamamoto H, Takemasa I, Mimori K, Hemmi H, Mizushima T, et al. Jumonji domain containing 1A is a novel prognostic marker for colorectal cancer: In vivo identification from hypoxic tumor cells. Clin Cancer Res (2010) 16:4636–46. doi: 10.1158/1078-0432.CCR-10-0407
59. Peng K, Su G, Ji J, Yang X, Miao M, Mo P, et al. Histone demethylase JMJD1A promotes colorectal cancer growth and metastasis by enhancing wnt/beta-catenin signaling. J Biol Chem (2018) 293:10606–19. doi: 10.1074/jbc.RA118.001730
60. Li X, Oh S, Song H, Shin S, Zhang B, Freeman WM, et al. A potential common role of the jumonji c domain-containing 1A histone demethylase and chromatin remodeler ATRX in promoting colon cancer. Oncol Lett (2018) 16:6652–62. doi: 10.3892/ol.2018.9487
61. Kim TD, Shin S, Berry WL, Oh S, Janknecht R. The JMJD2A demethylase regulates apoptosis and proliferation in colon cancer cells. J Cell Biochem (2012) 113:1368–76. doi: 10.1002/jcb.24009
62. Wang J, Li B, Yang S, Ma C, Liu K, Chen X, et al. Upregulation of INHBA mediated by the transcription factor BHLHE40 promotes colon cancer cell proliferation and migration. J Clin Lab Anal (2022) 36:e24539. doi: 10.1002/jcla.24539
63. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature (2012) 487:330–7. doi: 10.1038/nature11252
64. Smith JJ, Deane NG, Wu F, Merchant NB, Zhang B, Jiang A, et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology (2010) 138:958–68. doi: 10.1053/j.gastro.2009.11.005
65. Lin YH, Friederichs J, Black MA, Mages J, Rosenberg R, Guilford PJ, et al. Multiple gene expression classifiers from different array platforms predict poor prognosis of colorectal cancer. Clin Cancer Res (2007) 13:498–507. doi: 10.1158/1078-0432.CCR-05-2734
66. Sun L, Chen B, Wu J, Jiang C, Fan Z, Feng Y, et al. Epigenetic regulation of a disintegrin and metalloproteinase (ADAM) transcription in colorectal cancer cells: Involvement of beta-catenin, BRG1, and KDM4. Front Cell Dev Biol (2020) 8:581692. doi: 10.3389/fcell.2020.581692
67. Le HT, Sato F, Kohsaka A, Bhawal UK, Nakao T, Muragaki Y, et al. Dec1 deficiency suppresses cardiac perivascular fibrosis induced by transverse aortic constriction. Int J Mol Sci (2019) 20:4967. doi: 10.3390/ijms20194967
68. Li X, Le HT, Sato F, Kang TH, Makishima M, Zhong L, et al. Dec1 deficiency protects the heart from fibrosis, inflammation, and myocardial cell apoptosis in a mouse model of cardiac hypertrophy. Biochem Biophys Res Commun (2020) 532:513–9. doi: 10.1016/j.bbrc.2020.08.058
69. Zhang QJ, Chen HZ, Wang L, Liu DP, Hill JA, Liu ZP. The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J Clin Invest (2011) 121:2447–56. doi: 10.1172/JCI46277
70. Zhang QJ, Tran TAT, Wang M, Ranek MJ, Kokkonen-Simon KM, Gao J, et al. Histone lysine dimethyl-demethylase KDM3A controls pathological cardiac hypertrophy and fibrosis. Nat Commun (2018) 9:5230. doi: 10.1038/s41467-018-07173-2
71. Sun H, Taneja R. Stra13 expression is associated with growth arrest and represses transcription through histone deacetylase (HDAC)-dependent and HDAC-independent mechanisms. Proc Natl Acad Sci USA (2000) 97:4058–63. doi: 10.1073/pnas.070526297
72. Zawel L, Yu J, Torrance CJ, Markowitz S, Kinzler KW, Vogelstein B, et al. DEC1 is a downstream target of TGF-beta with sequence-specific transcriptional repressor activities. Proc Natl Acad Sci USA (2002) 99:2848–53. doi: 10.1073/pnas.261714999
73. St-Pierre B, Flock G, Zacksenhaus E, Egan SE. Stra13 homodimers repress transcription through class b e-box elements. J Biol Chem (2002) 277:46544–51. doi: 10.1074/jbc.M111652200
74. Ivanova AV, Ivanov SV, Zhang X, Ivanov VN, Timofeeva OA, Lerman MI. STRA13 interacts with STAT3 and modulates transcription of STAT3-dependent targets. J Mol Biol (2004) 340:641–53. doi: 10.1016/j.jmb.2004.05.025
75. Li Y, Xie M, Yang J, Yang D, Deng R, Wan Y, et al. The expression of antiapoptotic protein survivin is transcriptionally upregulated by DEC1 primarily through multiple sp1 binding sites in the proximal promoter. Oncogene (2006) 25:3296–306. doi: 10.1038/sj.onc.1209363
76. Hu G, Dong X, Gong S, Song Y, Hutchins AP, Yao H. Systematic screening of CTCF binding partners identifies that BHLHE40 regulates CTCF genome-wide distribution and long-range chromatin interactions. Nucleic Acids Res (2020) 48:9606–20. doi: 10.1093/nar/gkaa705
77. Laub F, Lei L, Sumiyoshi H, Kajimura D, Dragomir C, Smaldone S, et al. Transcription factor KLF7 is important for neuronal morphogenesis in selected regions of the nervous system. Mol Cell Biol (2005) 25:5699–711. doi: 10.1128/MCB.25.13.5699-5711.2005
78. Blackmore MG, Wang Z, Lerch JK, Motti D, Zhang YP, Shields CB, et al. Krüppel-like factor 7 engineered for transcriptional activation promotes axon regeneration in the adult corticospinal tract. Proc Natl Acad Sci USA (2012) 109:7517–22. doi: 10.1073/pnas.1120684109
79. Ding X, Wang X, Gong Y, Ruan H, Sun Y, Yu Y. KLF7 overexpression in human oral squamous cell carcinoma promotes migration and epithelial-mesenchymal transition. Oncol Lett (2017) 13:2281–9. doi: 10.3892/ol.2017.5734
80. Gupta R, Malvi P, Parajuli KR, Janostiak R, Bugide S, Cai G, et al. KLF7 promotes pancreatic cancer growth and metastasis by up-regulating ISG expression and maintaining golgi complex integrity. Proc Natl Acad Sci USA (2020) 117:12341–51. doi: 10.1073/pnas.2005156117
81. Guo Y, Chai B, Jia J, Yang M, Li Y, Zhang R, et al. KLF7/VPS35 axis contributes to hepatocellular carcinoma progression through CCDC85C-activated beta-catenin pathway. Cell Biosci (2021) 11:73. doi: 10.1186/s13578-021-00585-6
82. Li Y, Wang Q, Wang D, Fu W. KLF7 promotes gastric carcinogenesis through regulation of ANTXR1. Cancer Manag Res (2021) 13:5547–57. doi: 10.2147/CMAR.S308071
83. Caiazzo M, Colucci-D’Amato L, Esposito MT, Parisi S, Stifani S, Ramirez F, et al. Transcription factor KLF7 regulates differentiation of neuroectodermal and mesodermal cell lineages. Exp Cell Res (2010) 316:2365–76. doi: 10.1016/j.yexcr.2010.05.021
84. Schuettpelz LG, Gopalan PK, Giuste FO, Romine MP, van Os R, Link DC. Kruppel-like factor 7 overexpression suppresses hematopoietic stem and progenitor cell function. Blood (2012) 120:2981–9. doi: 10.1182/blood-2012-02-409839
85. Mochizuki S, Okada Y. ADAMs in cancer cell proliferation and progression. Cancer Sci (2007) 98:621–8. doi: 10.1111/j.1349-7006.2007.00434.x
86. Raeeszadeh-Sarmazdeh M, Do LD, Hritz BG. Metalloproteinases and their inhibitors: Potential for the development of new therapeutics. Cells (2020) 9:1313. doi: 10.3390/cells9051313
87. Wildeboer D, Naus S, Amy Sang QX, Bartsch JW, Pagenstecher A. Metalloproteinase disintegrins ADAM8 and ADAM19 are highly regulated in human primary brain tumors and their expression levels and activities are associated with invasiveness. J Neuropathol Exp Neurol (2006) 65:516–27. doi: 10.1097/01.jnen.0000229240.51490.d3
88. Qi B, Newcomer RG, Sang QX. ADAM19/adamalysin 19 structure, function, and role as a putative target in tumors and inflammatory diseases. Curr Pharm Des (2009) 15:2336–48. doi: 10.2174/138161209788682352
89. Zhang Q, Yu L, Qin D, Huang R, Jiang X, Zou C, et al. Role of microRNA-30c targeting ADAM19 in colorectal cancer. PloS One (2015) 10:e0120698. doi: 10.1371/journal.pone.0120698
90. Li Y, Hu J, Wang M, Yuan Y, Zhou F, Zhao H, et al. Exosomal circPABPC1 promotes colorectal cancer liver metastases by regulating HMGA2 in the nucleus and BMP4/ADAM19 in the cytoplasm. Cell Death Discovery (2022) 8:335. doi: 10.1038/s41420-022-01124-z
91. Zhang M, Li Y, Wang H, Yu W, Lin S, Guo J. LncRNA SNHG5 affects cell proliferation, metastasis and migration of colorectal cancer through regulating miR-132-3p/CREB5. Cancer Biol Ther (2019) 20:524–36. doi: 10.1080/15384047.2018.1537579
92. Wang S, Qiu J, Liu L, Su C, Qi L, Huang C, et al. CREB5 promotes invasiveness and metastasis in colorectal cancer by directly activating MET. J Exp Clin Cancer Res (2020) 39:168. doi: 10.1186/s13046-020-01673-0
93. Boufraqech M, Zhang L, Nilubol N, Sadowski SM, Kotian S, Quezado M, et al. Lysyl oxidase (LOX) transcriptionally regulates SNAI2 expression and TIMP4 secretion in human cancers. Clin Cancer Res (2016) 22:4491–504. doi: 10.1158/1078-0432.CCR-15-2461
94. Camp ER, Findlay VJ, Vaena SG, Walsh J, Lewin DN, Turner DP, et al. Slug expression enhances tumor formation in a noninvasive rectal cancer model. J Surg Res (2011) 170:56–63. doi: 10.1016/j.jss.2011.02.012
95. Li Y, Zhao Z, Xu C, Zhou Z, Zhu Z, You T. HMGA2 induces transcription factor slug expression to promote epithelial-to-mesenchymal transition and contributes to colon cancer progression. Cancer Lett (2014) 355:130–40. doi: 10.1016/j.canlet.2014.09.007
96. Yao C, Su L, Shan J, Zhu C, Liu L, Liu C, et al. IGF/STAT3/NANOG/Slug signaling axis simultaneously controls epithelial-mesenchymal transition and stemness maintenance in colorectal cancer. Stem Cells (2016) 34:820–31. doi: 10.1002/stem.2320
97. De Robertis M, Arigoni M, Loiacono L, Riccardo F, Calogero RA, Feodorova Y, et al. Novel insights into notum and glypicans regulation in colorectal cancer. Oncotarget (2015) 6:41237–57. doi: 10.18632/oncotarget.5652
98. Yoon JH, Kim D, Kim J, Lee H, Ghim J, Kang BJ, et al. NOTUM is involved in the progression of colorectal cancer. Cancer Genomics Proteomics (2018) 15:485–97. doi: 10.21873/cgp.20107
99. Flanagan DJ, Pentinmikko N, Luopajarvi K, Willis NJ, Gilroy K, Raven AP, et al. NOTUM from apc-mutant cells biases clonal competition to initiate cancer. Nature (2021) 594:430–5. doi: 10.1038/s41586-021-03525-z
Keywords: colorectal cancer, gene transcription, ADAM19, KLF7, BHLHE40, RNA sequencing, histone demethylase
Citation: Sui Y, Jiang H, Kellogg CM, Oh S and Janknecht R (2023) Promotion of colorectal cancer by transcription factor BHLHE40 involves upregulation of ADAM19 and KLF7. Front. Oncol. 13:1122238. doi: 10.3389/fonc.2023.1122238
Received: 12 December 2022; Accepted: 30 January 2023;
Published: 20 February 2023.
Edited by:
Zhaohui Huang, Affiliated Hospital of Jiangnan University, ChinaReviewed by:
Xiuping Yu, Louisiana State University Health Shreveport, United StatesShangze Li, Chongqing University, China
Copyright © 2023 Sui, Jiang, Kellogg, Oh and Janknecht. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf Janknecht, cmFsZi1qYW5rbmVjaHRAb3Voc2MuZWR1