 Yang Wang
Yang Wang Shilei Qin1†
Shilei Qin1† Yanwu Zeng
Yanwu Zeng- 1Department of Thoracic Surgery, Affiliated Hospital of Guilin Medical University, Guilin, China
- 2Operations Department, Shanghai OrigiMed Co., Ltd., Shanghai, China
Background: Lung cancer is the most prevalent cancer worldwide and accounts for approximately 20% of cancer-related death in China every year. High-grade lung cancer poses a significant threat to patients, and developing a novel treatment for these patients requires an understanding of its underlying mechanism.
Methods: Chinese patients with lung cancer were enrolled. The tumor samples were collected by surgery or puncture and applied for next-generation sequencing. A panel of pan-cancer genes was targeted, and the sequencing depth was set to over 1,000 to improve the sensitivity of detecting mutations. Short-length mutations (substitution, insertion, and deletion), copy number variation, and gene fusion were called. Gene mutations were compared between low-grade, middle-grade, and high-grade tumors using Fisher’s exact test. The enriched pathways in each grade of tumors were also inferred.
Results: The study included 173 Chinese patients with non-small cell lung cancer, of whom 98 (56.6%) patients were female and 75 (43.4%) were male, with a mean age of 56.8 years. All patients were microsatellite stable; 66.4% were at the early stages (Stages 0, I, and II) with a tumor mutational burden of approximately 2.5 (confidence interval = [0, 48.3]). Compared to low-grade tumors, high-grade tumors had a significantly higher percentage of mutations in TP53 (75.9% vs 34.4%, p = 1.86e-3) and PIK3CA (24.1% vs. 0%, p = 3.58e-3). Pathway analysis found that high-grade tumors were enriched with mutations in bacterial invasion of epithelial cells (31% vs. 0%, p = 5.8e-4), Epstein–Barr virus infection (79.3% vs. 37.5%, p = 1.72e-3), and the Wnt signaling pathway (75.9% vs. 34.4%, p = 1.91e-3). High-grade tumors had a significantly higher tumor mutational burden than low-grade tumors (p-value = 0.0017). However, actionable mutations with high-level evidence were lower in high-grade tumors.
Conclusion: Patients with high-grade tumors from lung cancer may be more affected by bacteria and Epstein–Barr virus than low-grade tumors. High-grade tumors were specially mutated in TP53 and PIK3CA and may benefit more from immunotherapy. Further research on the underlying mechanism of high-grade lung cancer is necessary to develop new therapeutic options. Lung cancer, tumor grade, genomic mutations, Epstein–Barr virus, pathway analysis
Introduction
Lung cancer is a pervasive form of cancer worldwide, accounting for approximately 20% of cancer-related death in China alone (1). Non-small cell lung cancer (NSCLC) represents approximately ~80% of lung cancer cases (2) and its average overall 5-year survival rate is 32.8%, varying widely from 20.3% to 64.7% among patients (3). Given the diverse feature of NSCLC, further subtyping is crucial for customized treatments.
Tumor grade is a common biomarker used to subtype cancers, and the World Health Organization classification provides a grading system for NSCLC (4). For example, solid or micropapillary predominant adenocarcinoma is classified as high grade, acinar and papillary adenocarcinoma as intermediate grade, and lepidic adenocarcinoma as low grade; large cell neuroendocrine carcinoma is always categorized as high grade. High-grade NSCLC has a median overall survival of 21.6 months (95% CI, 6.8–77.2), which is significantly shorter than other tumors (5). Despite its severity, there are no special treatments for patients with high-grade NSCLC. Targeted therapy and immunotherapy have shown promise for a specific patient group, but knowledge of gene mutations in high-grade NSCLC is limited. Thus, treating patients with high-grade tumors using these techniques remains a challenge, and identifying mutations and understanding their potential mechanisms are crucial.
To address this issue, we conducted a study that enrolled 173 patients with NSCLC, of whom 168 patients had qualified samples for high-depth panel sequencing. We displayed and compared gene mutation profiling between different grades of NSCLC, and functional enrichment analysis revealed the pathway preference of gene mutations for each grade. Additionally, we extracted mutational signatures for each grade of NSCLC and compared them to a known mutational signature database.
Patients and methods
Patients and tumor sample collection
This study was approved by the Ethics Committee of the Affiliated Hospital of Guilin Medical University. The patient inclusion criteria were as follows: (1) patients were diagnosed with NSCLC; (2) patients have qualified paired tumor and blood samples for sequencing; and (3) patients have provided written informed consent. The patient exclusion criteria were as follows: (1) paired samples were successfully sequenced; and (2) the sequencing results had identified mutations. Finally, we enrolled 173 Chinese patients (GMU cohort). The staging system most often used for NSCLC is the 9th edition of the American Joint Committee on Cancer (AJCC) TNM system. Tumor samples were collected by either surgery or puncture and immediately fixed with 4% formalin buffer at 4°C for 24 h. The fixed samples were then embedded in paraffin wax for storage.
We also downloaded a public cohort (MSK cohort) (6) from cBioportal (http://www.cbioportal.org) for comparison against our cohort. The MSK cohort contained 247 patients with NSCLC.
Pathologic examination
All samples were classified into low, middle, and high grades with histological classification and grade according to the fourth edition of the World Health Organization classification (4) by two expert pathologists.
Library construction and sequencing
FFPE (formalin-fixed paraffin-embedded) tissues were sliced into 4-µm sections. One section of the FFPE sample was dewaxed on the glass slide in a 60°C oven for 1 h and washed with xylene for 15 min. The samples were then processed with gradient concentrations (100%, 95%, 85%, and 75%) of alcohol, cooked with pressure for 4 min, and immersed in cold water for 10 min. These samples were then stained with hematoxylin and eosin. The stained slice underwent a pathologist review to ensure the quality requirements including stained area exceeding 1 cm2, 20% nucleated cellularity, and 20% tumor content. After the sample passed the quality check, 10 unstained FFPE sections were used for sequencing library construction. The library construction and sequencing were performed in the CLIA/CAP-compliant Molecular Diagnostics Service laboratory of Shanghai Origimed Co., Ltd. The detailed procedure had been described in our previous works (7–9). Briefly, typically 50–250 ng of double-stranded DNA was fragmented to ~250 bp by sonication. The KAPA Hyper Prep Kit (KAPA Biosystems) was used for end repair. The following procedures included dA addition, ligation, PCR amplification, and Qubit (Thermofisher Corporation) quality assessment. Samples yielding >40 ng of extracted DNA or 500 ng of sequencing library were further used. A custom-targeted panel of 450 cancer-related genes (Yuansuo®, Origimed incorporation, Shanghai, China) was specifically amplified. Unique molecular identifiers were added to each sonication-shuffled segments before library amplification. Libraries were diluted to 1.05 nM and subsequently sequenced on Illumina NextSeq 500. Paired primers were extended from both ends of targeting sequences for 75 bp. To improve the sensitivity of detecting mutations, the average sequencing depth is over 1000×. For each patient, white blood cells were extracted from paired blood samples. The same library construction and sequencing procedure were applied for white blood cells.
Mutation calling and bioinformatics analysis
The output reads from Illumina NextSeq 500 sequencer were mapped to the human genome (GPRC37) with bwa-mem (version 0.7.9a) and recalibrated with GATK BaseRecalibrator (version 3.8). The mapped reads were deduplicated according to the unique molecular identifiers. Mutect2 (10) and varscan (11) were used to call short nucleotide mutations including single-nucleotide variants and small indels as well. Somatic mutations were annotated and filtered out for possible germline mutations from the known single-nucleotide polymorphism database (ESP6500, 1000 Genomes, gnomAD, and ExAC). Germline mutations were also obtained from sequencing white blood cells for each patient. CNVkit (12) and an in-house pipeline (13) were used to call copy number variation and gene fusion. Tumor mutational burden (TMB) was calculated as the number of mutations dividing the effective coverage length in millions. MutSigCV was used to identify significantly mutated genes, which compares gene mutation frequency by considering mutation biases including gene length and cancer types (14).
Mutational signature analysis was performed for different grades of tumors using the R package “MutationalPatterns” (v3.4.1). First, 96 single-nucleotide mutation contexts were decomposed into components with negative-matrix factorization. The major components were compared to the known COSMIC mutational signature database (https://cancer.sanger.ac.uk/cosmic). The analysis was performed on the R computation platform (v4.05).
A heatmap was generated with the R package “HeatmapComplex” (v2.15.1), bar and scatter plots were generated with the R package “ggpubr” (v0.5.0), and a Sankey plot was generated with the R package “ggalluvial” (v0.12.3).
Statistical analysis
Categorical and scale variables were compared with Fisher’s exact test and Mann–Whitney U test, respectively. The mutation status is defined as “mutated” when a gene or pathway has any mutation in them; otherwise, it is defined as “wild”. The mutation status for each gene and gene pathway was treated as a categorical variable. Benjamin and Hochberg’s method was used to correct the errors from multiple tests. p-values below 0.05 were defined as statistically significant. All the analysis was performed on the R computation platform (v4.05).
Results
Clinical characteristics
This study enrolled 173 Chinese patients (GMU cohort) with lung cancer, of whom 98 (56.6%) patients were female and 75 (43.4%) were male (Table 1). The mean age of these patients was 56.8 years. None of the patients were microsatellite instable. Most of them (66.4%) were at the early stage (Stages 0, I, and II). Patients at advanced stages (Stages III and IV) accounted for 28.3%. The median TMB was 2.5 Mut/Mbp (confidence interval = [0, 48.3]). Of the 173 patients, 29 (16.8%) had high-grade tumors, 34 (19.7%) had middle-grade tumors, and 32 (18.5%) had low-grade tumors.
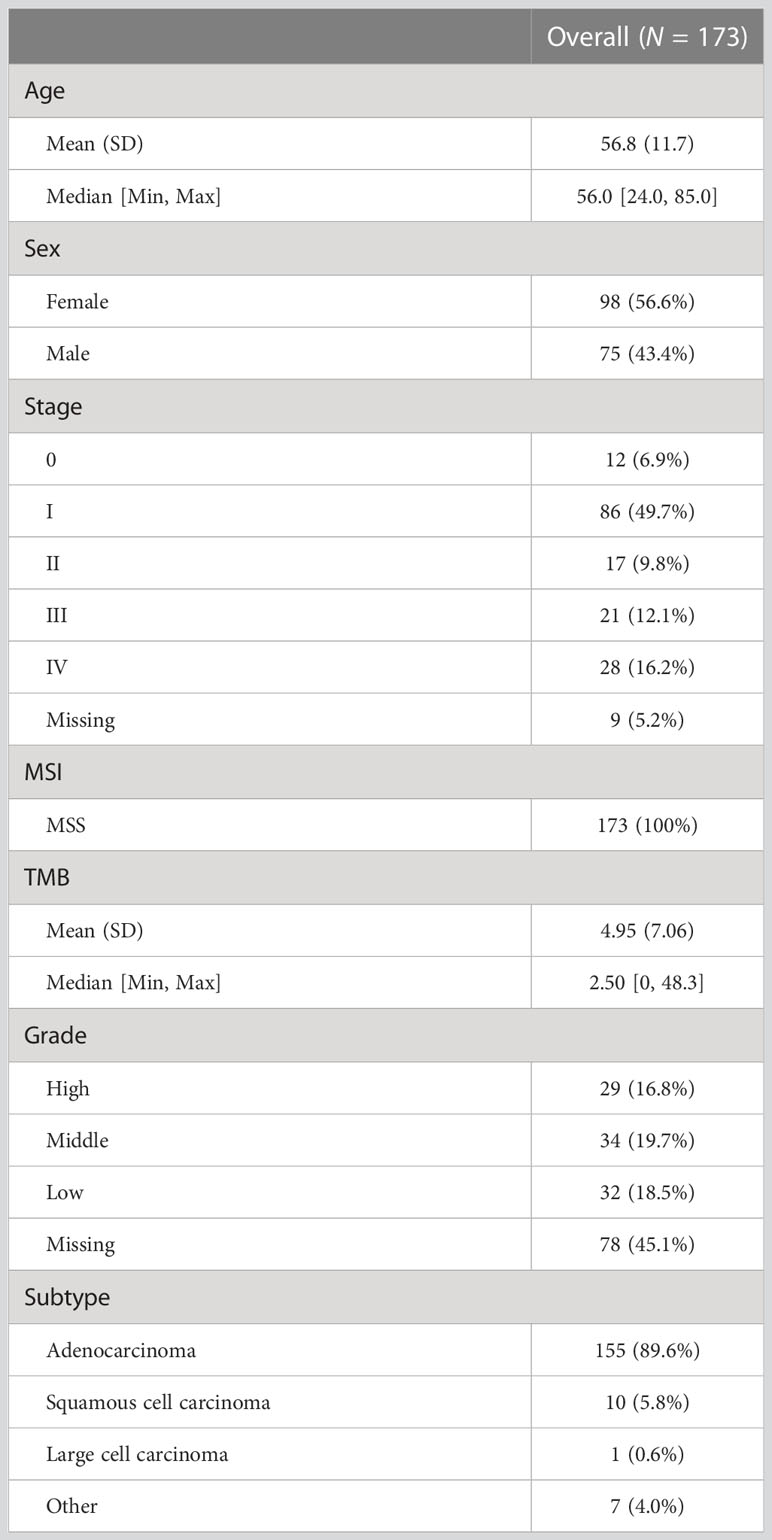
Table 1 Clinical characteristics.
Mutation profiling
Of the 173 patients, 168 had qualified samples for sequencing. As shown in Figure 1, the top 5 mutated genes were EGFR, TP53, KRAS, PIK3CA, and ALK. The mutation frequency of EGFR was 59%. Most of them were substitution or short insert/deletion (indel), and only 11 patients (7%) had EGFR amplification. Pathway enrichment analysis indicated that genes with mutations in more than 1% of patients were mostly related to the regulation of protein phosphorylation, regulation of kinase activity, and regulation of cell proliferation (Supplementary Figure 1A). We also compared the mutation frequency to the MSK cohort. The GMU cohort had a higher percentage of EGFR but a lower percentage of TP53, KRAS, NF1, and STK11 (Supplementary Figure 1B).
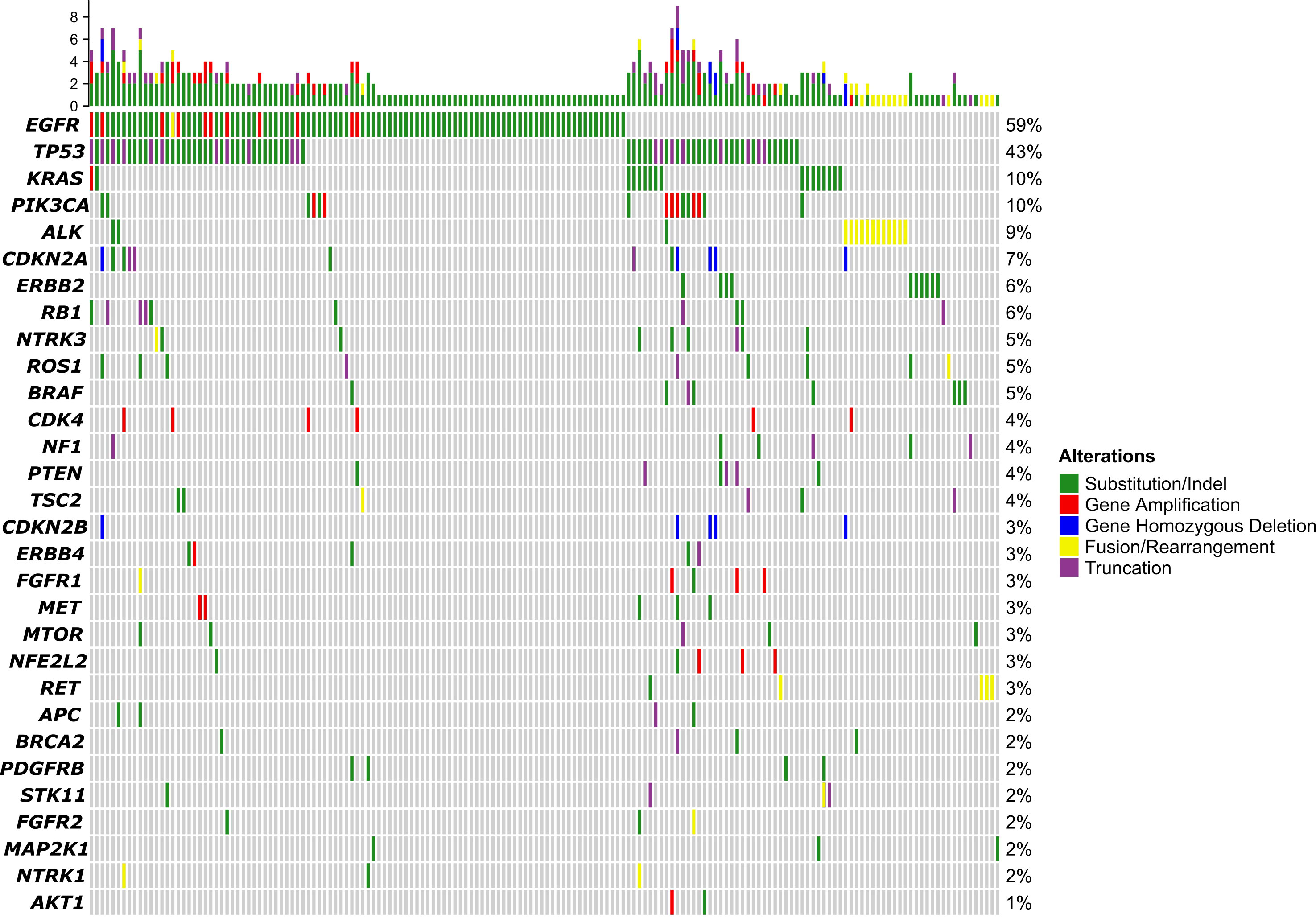
Figure 1 Gene mutation profiling.
Driver genes were associated with tumor grades
To find out significantly mutated genes, MutSigCV was used. It found 23 and 15 significantly mutated genes in the GMU and MSK cohorts, respectively. The median allele frequency in GMU and MSK cohorts is plotted in Figure 2A. Eleven genes were both significantly mutated in the GMU and MSK cohorts. The shared significant genes (RB1, TP53, EGFR, BRAF, PIK3CA, etc.) had a high allele frequency in both cohorts. ALK, PDGFRA, ERBB2, RET, and MET had higher alleles in both cohorts but were only significantly mutated in the MSK cohort. RB1, TP53, and PIK3CA were most mutated in high-grade tumors and EGFR was most mutated in middle- and low-grade tumors (Figure 2B).
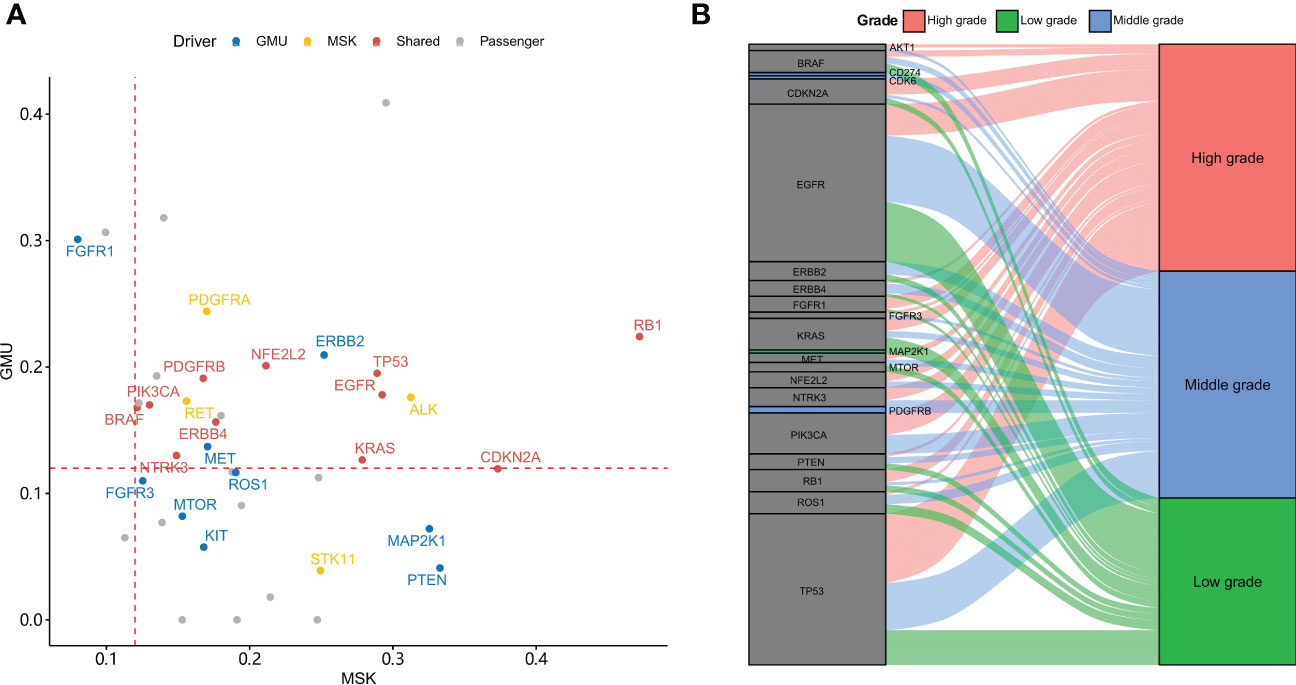
Figure 2 Driver genes in the GMU and MSK cohorts. (A) The allele frequency of driver gene mutation in both MSK and GMU cohorts is compared. (B) Driver genes preferentially mutate in different tumor grades.
Grade-associated gene mutations and pathway
Of the 168 samples sequenced, 95 samples have histological grade records. Gene mutations within low-grade, middle-grade, and high-grade tumors were compared with Fisher’s exact test. The enriched pathways in each grade of tumors were also inferred. When comparing mutation frequency between histological grades (Supplementary Figures 2A–C), we found that high-grade tumors had an elevated percentage of mutations in TP53 (75.9% vs 34.4%, p = 1.86e-3) and PIK3CA (24.1% vs. 0%, p = 3.58e-3) compared to low-grade tumors (Supplementary Figure 2C). Pathway analysis found that high-grade tumors were enriched with mutations in bacterial invasion of epithelial cells (31% vs. 0%, p = 5.8e-4), Epstein–Barr virus (EBV) infection (79.3% vs. 37.5%, p = 1.72e-3), and the Wnt signaling pathway (75.9% vs. 34.4%, p = 1.91e-3) (Figure 3). No gene was significantly mutated between low- and middle-grade tumors and between middle- and high-grade tumors (Supplementary Figures 2A, B).
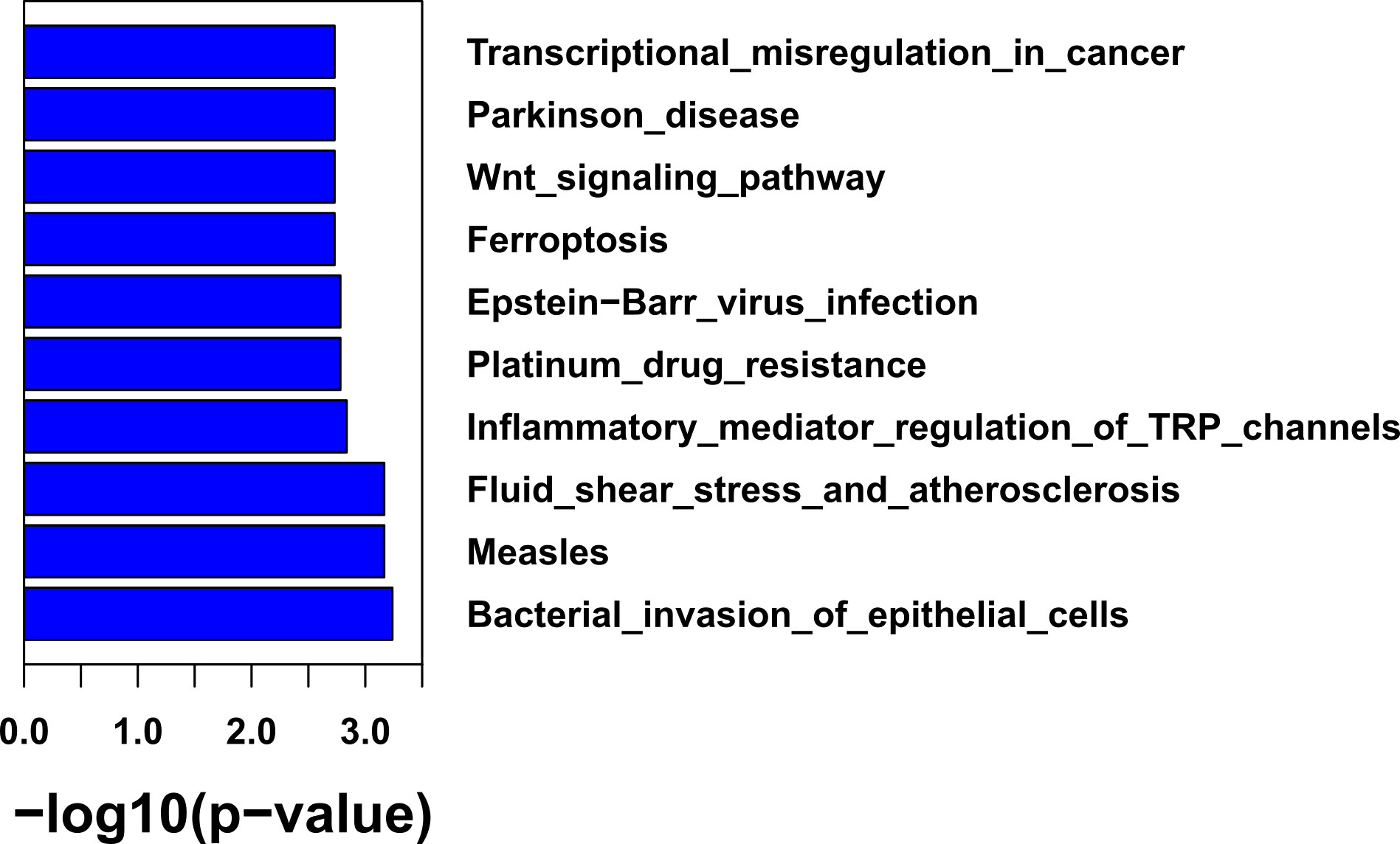
Figure 3 The functional enrichment of genes with mutations in high-grade tumors.
High-grade tumors were associated with Epstein–Barr virus-related gene mutations
Mutational signature analysis indicated that high-grade tumors only had one mutational signature, which is like SBS40 (cosine similarity = 0.78, Figure 4A). Middle-grade and low-grade tumors each had a mutational signature like SBS33 with cosine similarities of 0.69 and 0.64, respectively (Figures 4B, C). Genes with mutations in the EBV infection pathway had a similar mutation profile to COSMIC SBS40 (cosine similarity = 0.81) (Figure 4D). We also compared the mutation profile of genes with mutations in the EBV infection pathway for the MSK cohort to the COSMIC signature database. Those tumors also presented mutational signatures like SBS40 (cosine similarity = 0.78) (Supplementary Figure 3A). The mutational signature similarity between GMU and MSK cohorts was 0.76. SBS40 was considered related with age in the COSMIC database. We failed to find an association between age and tumor grade (Supplementary Figure 3B).
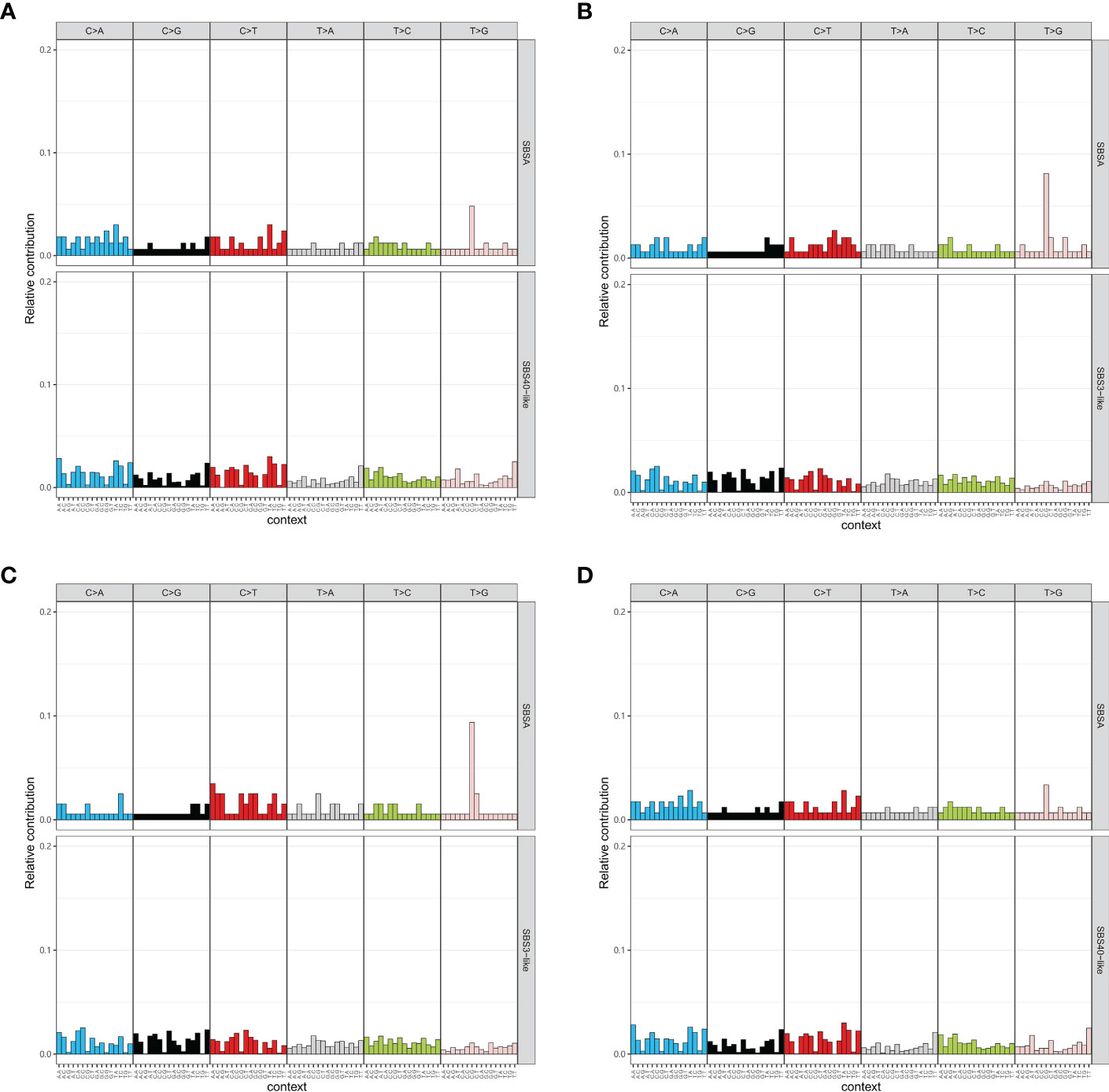
Figure 4 Mutational signature of tumors with different grades. The mutational signatures of high-grade tumors (A), middle-grade tumors (B), and low-grade tumors (C) are compared to COSMIC signatures. (D) Tumors with mutations in the Epstein–Barr virus-related pathway have a signature similar to COSMIC SBS40.
Actionability and tumor mutational burden
Gene mutations are regularly used as biomarkers for targeted therapy and immunotherapy. We further compared the actionability among different grades of tumors (Figure 5A). High-grade tumors had a lower percentage of high actionability evidence (Level_1). High-grade tumors had fewer EGFR actionable mutations than the other tumors (Supplementary Figures 4A–C).
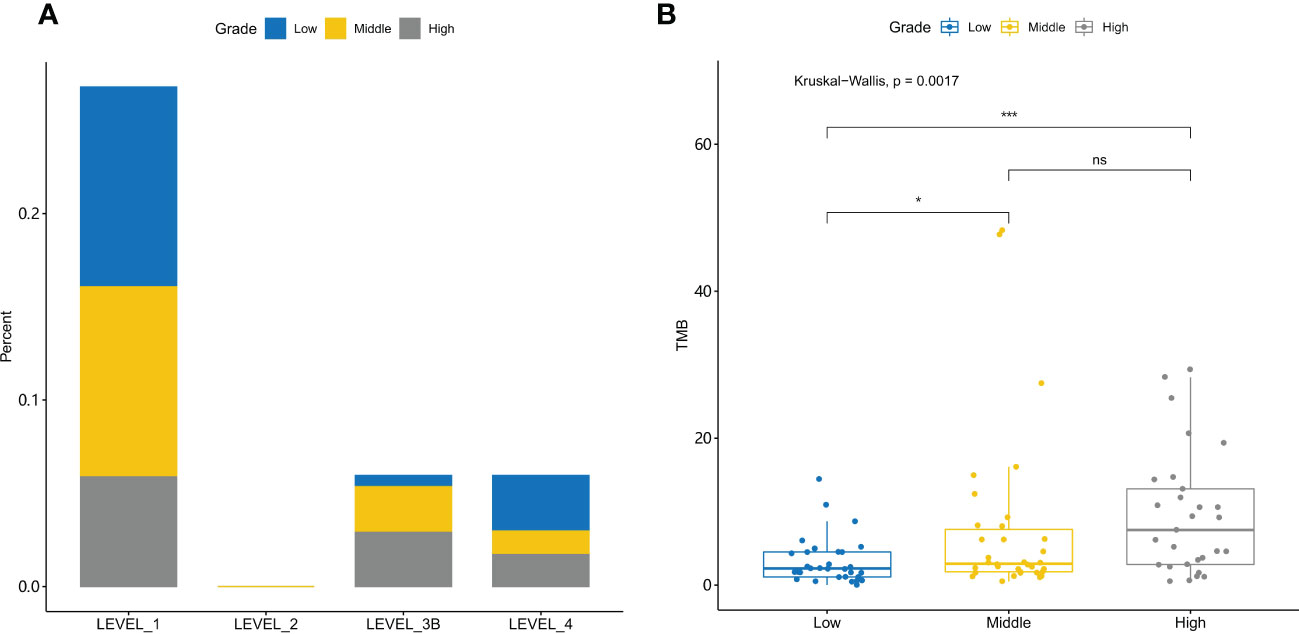
Figure 5 Actionability and tumor mutational burden are different for low-, middle-, and high-grade tumors (A) The evidence levels of actionability in lung cancers are shown for different grades of tumors. (B) Tumor mutational burden (TMB) is significantly different among different grades of tumors. *, p-value<0.05; ***, p-value<0.001; ns, Non-significant.
In high-grade tumors, tumors with FGFR1 or TP53 mutations had higher TMB than those without, and tumors with EGFR mutations had lower TMB (Supplementary Figure 4D). In middle-grade tumors, PDGFRB- and PIK3CA-mutated tumors had a higher TMB, and EGFR-mutated tumors had a lower TMB (Supplementary Figure 4E). In low-grade tumors, TP53-mutated tumors had a higher TMB (Supplementary Figure 4F).
TMB is a biomarker for immunotherapy. TMB was significantly different among different grade tumors (Kruskal–Wallis test, p-value = 1.7e-3) (Figure 5B). Further post-hoc comparison confirmed that TMB in high- and middle-grade NSCLC tumors is significantly higher than that in low-grade tumors with p-value = 6.6e-4 and 4.2e-2, respectively.
Discussion
In this study, we investigated gene mutations in NSCLC. Our analysis of the gene mutation profile in the GMU cohort revealed a different pattern compared to the MSK cohort. Specifically, we found that EGFR mutations were more frequent in the GMU cohort (59% vs. 10%) than in the MSK cohort (10%), whereas TP53 (43% vs. 66%) and KRAS (10% vs. 41%) mutations were less common. Our results are consistent with a previous study on NSCLC patients in a Chinese population, which reports a higher percentage of EGFR mutations (56%) and a lower percentage of KRAS mutations (12%) (15).
Histological grade is an independent prognostic factor for resectable NSCLC (16) and high-grade NSCLC is more dangerous than the low-grade one (17). Understanding the mechanism of high-grade NSCLC could help develop novel treatments for patients. To identify candidate driver genes, MutSigCV was used, and 20 genes were found to be significantly mutated in the GMU cohort. Of those, 11 genes were also significantly mutated in the MSK cohort, including RB1, EGFR (15, 18), TP53, and PIK3CA (19). All the sharing candidate driver genes had a higher variant allele frequency in both cohorts, and RB1, TP53, and PIK3CA mutations were more commonly detected in high-grade NSCLC. TP53 mutations were found to occur more frequently in high-grade ovarian (20) and endometrial cancers (21), but not in breast cancer (22). RB1 mutations were found to be prone to high-grade bladder cancer (23), astrocytic gliomas (24), and Merkel cell carcinoma (25), and they could function by activating the NF-κB pathway and exerting anti-inflammatory effects (26). Furthermore, EGFR-resistant lung adenocarcinoma with RB1 mutations could transform into a more aggressive small-cell lung cancer (27). Similarly, loss of RB1 and TP53 in NSCLC could also lead to the transformation of NSCLC into small-cell lung cancer (28). The association of RB1, TP53, and PIK3CA mutations with histological grade in NSCLC could be meaningful for predicting histological grade in certain scenarios, such as using ctDNA for gene mutation detection and histological grade prediction before surgery.
Further pathway enrichment analysis of differentially mutated genes revealed that the bacterial invasion of epithelial cells pathway and EBV infection pathway were significantly mutated in high-grade NSCLC. EBV has been shown to promote tumor development in many cancers, including lung cancer (29, 30), breast cancer (31), gastric cancer (32), and nasopharyngeal carcinoma (33). In NSCLC, EBV infection has been closely linked to pulmonary lymphoepithelioma-like carcinomas (a subtype of large cell carcinoma) (29, 30), lung squamous-cell carcinomas (34), and lung adenocarcinomas (35). EBV has also been associated with a higher mutation rate in the NF-κB pathway, including TRAF3 and NFKBIA (30, 36). Notably, gene mutations in the EBV infection response pathway, such as TRAF3 (37), NFKBIA (30), RB1 (38), and PIK3CA (39), have been reported to play an important role during cancer development. Mutations in the EBV infection pathway could potentially be a mechanism by which EBV evades the immune response in hosts.
Previous studies have reported an association between EBV infection and histological grade in lymphoma (40) and breast cancer (41), but this association has not been explored in lung cancer. To further investigate, mutational signature analysis was conducted, revealing that high-grade NSCLC exhibited a mutational signature similar to COSMIC SBS40, which is potentially related with age. To assess the influence of stages, the samples were divided into early stage (Stages 0, I, and III) and advanced stage (Stages III and IV) groups, and the mutational signatures of high-grade tumors were evaluated. Interestingly, both groups exhibited only one mutational signature, which was similar to COSMIC SBS40, with corresponding cosine similarities of 0.77 and 0.81 (Supplementary Figures 5A, B). The majority of the samples in this study (89%) were adenocarcinoma; thus, further analysis focused on adenocarcinoma samples. The only mutational signature identified in high-grade adenocarcinoma samples was also similar to COSMIC SBS40 (cosine similarity = 0.77) (Supplementary Figure 5C). Further analysis of the genes with mutations in the EBV infection pathway revealed that their mutational signature was also similar to COSMIC SBS40, with a cosine similarity of 0.69. To validate these findings, an independent cohort (MSK) was used, and the mutational signature of the genes in the EBV pathway was found to be similar to COSMIC SBS40, with a cosine similarity of 0.78. Between the GMU and MSK cohorts, the mutational signatures had a cosine similarity of 0.76, indicating mostly similar signatures to COSMIC SBS40. The etiology of COSMIC signature SBS40 is unknown, but it could potentially be related with age, as suggested by the COSMIC database. Our results suggest that SBS40 may also be related to EBV infection, possibly due to the continuous damage caused by the virus to lung tissue, leading to the formation of high-grade lung tumors with aging.
Targeted therapy and immunotherapy are commonly used in the treatment of lung cancer (42). Understanding the treatment efficacy of high-grade cancer could aid in the development of novel therapies. Our analysis of targeted genes revealed that high-grade NSCLC has a low percentage of actionable genes with LEVEL_1 evidence, and EGFR mutations are the most actionable with LEVEL_1 evidence. However, the lower percentage of EGFR mutations in high-grade NSCLC results in its low actionability. Despite the lower percentage of targeted therapy treatment, high-grade NSCLC may benefit from immunotherapy. TMB is a biomarker of immunotherapy, and high TMB is associated with a better prognosis (43, 44). High-grade NSCLC has a higher TMB value than low- and middle-grade NSCLC, which suggests that it may respond better to immunotherapy. Furthermore, in high-grade NSCLC, TMB is positively correlated with TP53 and FGFR1 mutations and negatively correlated with EGFR mutations, indicating that these genes could be used to predict the TMB value in high-grade NSCLC.
One limitation of this study is that it only analyzed 450 cancer-related genes, which may have precluded the identification of other related genes or functions. Additionally, the association between EBV infection status and high-grade NSCLC requires further investigation to validate the finding that the mutational signature of EBV infection is associated with high-grade NSCLC in this cohort and the MSK cohort. Future studies should analyze the association between EBV infection status and high-grade NSCLC.
In summary, our analysis revealed the defected pathway bias in different grades of NSCLC and demonstrated a possible association between EBV infection and high-grade NSCLC. We also found that the current targeted therapy needs more attention in treating high-grade NSCLC, while immunotherapy may be a promising approach for its treatment.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Affiliated Hospital of Guilin Medical University. The patients/participants provided their written informed consent to participate in this study.
Author contributions
MZ, YZ, and LL designed the research. YW, SQ, YL, LY, MZ, YZ, and LL performed data collection and analysis. YW, SQ, and YL wrote the manuscript draft. YW, SQ, YL, MZ, YZ, and LL prepared the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
YZ and LL are employees of Shanghai OrigiMed Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1119575/full#supplementary-material
Supplementary Figure 1 | Mutation profile of MSK cohort (A) Log-transformed P-value is shown for the functional enrichment of the top 30 mutated genes. (B) The gene mutation frequency is compared between the MSK and GMU cohorts.
Supplementary Figure 2 | Mutational frequency is compared among different grades of tumors (A–C) The frequency of mutated genes is compared within low-, middle-, and high-grade tumors.
Supplementary Figure 3 | The mutational signature of Epstein-Barr virus-related genes in the GMU cohort is compared to that in the MSK cohort. (A) The mutational signatures of Epstein-Barr virus-related genes in the GMU and MSK cohorts are compared to COMIC SBS40. (B) The age distribution shows no difference within low-, middle-, and high-grade lung tumors.
Supplementary Figure 4 | Actionability of mutations from tumors of different grades (A–C) The distribution of evidence levels in high-, middle-, and low-grade tumors is shown. (D–F) Tumor mutational burden (TMB) is compared between tumors with and without mutations in high-, middle-, and low-grade tumors.
Supplementary Figure 5 | Influence of histological subtypes and stages on high-grade tumors Mutational signatures are shown for lung adenocarcinoma (A), and early b and advanced-stage (C) tumors.
References
1. Cao M, Chen W. Epidemiology of lung cancer in China. Thorac Cancer (2019) 10(1):3–7. doi: 10.1111/1759-7714.12916
2. Chen P, Liu Y, Wen Y, Zhou C. Non-small cell lung cancer in China. Cancer Commun (Lond) (2022) 42(10):937–70. doi: 10.1002/cac2.12359
3. He S, Li H, Cao M, Sun D, Yang F, Yan X, et al. Survival of 7,311 lung cancer patients by pathological stage and histological classification: a multicenter hospital-based study in China. Transl Lung Cancer Res (2022) 11(8):1591–605. doi: 10.21037/tlcr-22-240
4. Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, et al. The 2015 world health organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol (2015) 10(9):1243–60. doi: 10.1097/JTO.0000000000000630
5. Barletta JA, Yeap BY, Chirieac LR. The prognostic significance of grading in lung adenocarcinoma. Cancer (2010) 116(3):659–69. doi: 10.1002/cncr.24831
6. Vanguri RS, Luo J, Aukerman AT, Egger JV, Fong CJ, Horvat N, et al. Multimodal integration of radiology, pathology and genomics for prediction of response to PD-(L)1 blockade in patients with non-small cell lung cancer. Nat Cancer (2022) 3(10):1151–64. doi: 10.1038/s43018-022-00416-8
7. Hao C, Wang C, Lu N, Zhao W, Li S, Zhang L, et al. Gene mutations associated with clinical characteristics in the tumors of patients with breast cancer. Front Oncol (2022) 12:778511. doi: 10.3389/fonc.2022.778511
8. Chen X, Bu Q, Yan X, Li Y, Yu Q, Zheng H, et al. Genomic mutations of primary and metastatic lung adenocarcinoma in Chinese patients. J Oncol (2020) 2020:e6615575. doi: 10.1155/2020/6615575
9. Xu S, Guo Y, Zeng Y, Song Z, Zhu X, Fan N, et al. Clinically significant genomic alterations in the Chinese and Western patients with intrahepatic cholangiocarcinoma. BMC Cancer (2021) 21(1):152. doi: 10.1186/s12885-021-07792-x
10. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol (2013) 31(3):213–9. doi: 10.1038/nbt.2514
11. Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics (2009) 25(17):2283–5. doi: 10.1093/bioinformatics/btp373
12. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol (2016) 12(4):e1004873. doi: 10.1371/journal.pcbi.1004873
13. Cao J, Chen L, Li H, Chen H, Yao J, Mu S, et al. An accurate and comprehensive clinical sequencing assay for cancer targeted and immunotherapies. Oncologist (2019) 24(12):e1294–302. doi: 10.1634/theoncologist.2019-0236
14. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature (2013) 499(7457):214–8. doi: 10.1038/nature12213
15. Meng H, Guo X, Sun D, Liang Y, Lang J, Han Y, et al. Genomic profiling of driver gene mutations in Chinese patients with non-small cell lung cancer. Front Genet (2019) 10:1008. doi: 10.3389/fgene.2019.01008
16. Motoaki Y, Sawabata N, Kawaguchi T, Kawai N, Nakai T, Ohbayashi C, et al. Histological grade: Analysis of prognosis of non-small cell lung cancer after complete resection. In Vivo (2018) 32(6):1505–12. doi: 10.21873/invivo.11407
17. Sun Z, Aubry MC, Deschamps C, Marks RS, Okuno SH, Williams BA, et al. Histologic grade is an independent prognostic factor for survival in non–small cell lung cancer: An analysis of 5018 hospital- and 712 population-based cases. J Thorac Cardiovasc Surg (2006) 131(5):1014–20. doi: 10.1016/j.jtcvs.2005.12.057
18. Zhou Y, Ge F, Du Y, Li Q, Cai J, Liu X, et al. Unique profile of driver gene mutations in patients with non-Small-Cell lung cancer in qujing city, yunnan province, southwest China. Front Oncol (2021) 11:644895. doi: 10.3389/fonc.2021.644895
19. Wang Y, Wang Y, Li J, Li J, Che G. Clinical significance of PIK3CA gene in non-Small-Cell lung cancer: A systematic review and meta-analysis. BioMed Res Int (2020) 2020:1–9. doi: 10.1155/2020/3608241
20. Boyarskikh UA, Gulyaeva LF, Avdalyan AM, Kechin AA, Khrapov EA, Lazareva DG, et al. Spectrum of TP53 mutations in BRCA1/2 associated high-grade serous ovarian cancer. Front Oncol (2020) 10:1103. doi: 10.3389/fonc.2020.01103
21. Adamczyk-Gruszka O, Horecka-Lewitowicz A, Strzelecka A, Wawszczak-Kasza M, Gruszka J, Lewitowicz P. The roles of TP53 and FGFR2 in progress made treating endometrial cancer. Diagnostics (2022) 12(7):1737. doi: 10.3390/diagnostics12071737
22. Ungerleider NA, Rao SG, Shahbandi A, Yee D, Niu T, Frey WD, et al. Breast cancer survival predicted by TP53 mutation status differs markedly depending on treatment. Breast Cancer Res (2018) 20(1):115. doi: 10.1186/s13058-018-1044-5
23. Zhang D, Tian J, Xia Q, Yang Z, Gu B. Significance and mechanisms analyses of RB1 mutation in bladder cancer disease progression and drug selection by bioinformatics analysis. BLC (2021) 7(2):133–42. doi: 10.3233/BLC-200368
24. Brat DJ, Aldape K, Colman H, Figrarella-Branger D, Fuller GN, Giannini C, et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol (2020) 139(3):603–8. doi: 10.1007/s00401-020-02127-9
25. Muto Y, Ryo E, Namikawa K, Takahashi A, Ogata D, Fujimura T, et al. RB1 gene mutations are a distinct predictive factor in merkel cell carcinoma. Pathol Int (2021) 71(5):337–47. doi: 10.1111/pin.13090
26. Gao H, Kang N, Hu C, Zhang Z, Xu Q, Liu Y, et al. Ginsenoside Rb1 exerts anti-inflammatory effects in vitro and in vivo by modulating toll-like receptor 4 dimerization and NF-kB/MAPKs signaling pathways. Phytomedicine (2020) 69:153197. doi: 10.1016/j.phymed.2020.153197
27. Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun (2015) 6(1):1–10. doi: 10.1038/ncomms7377
28. Li J, Wei B, Feng J, Wu X, Chang Y, Wang Y, et al. Case report: TP53 and RB1 loss may facilitate the transformation from lung adenocarcinoma to small cell lung cancer by expressing neuroendocrine markers. Front Endocrinol (2022) 13:1006480. doi: 10.3389/fendo.2022.1006480
29. Han AJ, Xiong M, Zong YS. Association of Epstein-Barr virus with lymphoepithelioma-like carcinoma of the lung in southern China. Am J Clin Pathol (2000) 114(2):220–6. doi: 10.1309/148K-ND54-6NJX-NA61
30. Chau SL, Tong JHM, Chow C, Kwan JSH, Lung RWM, Chung LY, et al. Distinct molecular landscape of Epstein–Barr virus associated pulmonary lymphoepithelioma-like carcinoma revealed by genomic sequencing. Cancers (2020) 12(8):2065. doi: 10.3390/cancers12082065
31. Lawson JS, Glenn WK. Catching viral breast cancer. Infect Agents Cancer (2021) 16(1):37. doi: 10.1186/s13027-021-00366-3
32. Nakano H, Saito M, Nakajima S, Saito K, Nakayama Y, Kase K, et al. PD-L1 overexpression in EBV-positive gastric cancer is caused by unique genomic or epigenomic mechanisms. Sci Rep (2021) 11(1):1982. doi: 10.1038/s41598-021-81667-w
33. Tsao SW, Tsang CM, Lo KW. Epstein–Barr Virus infection and nasopharyngeal carcinoma. Philos Trans R Soc B: Biol Sci (2017) 372(1732):20160270. doi: 10.1098/rstb.2016.0270
34. Gómez-Román JJ, Martínez MN, Fernández SL, Val-Bernal JF. Epstein–Barr Virus-associated adenocarcinomas and squamous-cell lung carcinomas. Mod Pathol (2009) 22(4):530–7. doi: 10.1038/modpathol.2009.7
35. Wang S, Xiong H, Yan S, Wu N, Lu Z. Identification and characterization of Epstein-Barr virus genomes in lung carcinoma biopsy samples by next-generation sequencing technology. Sci Rep (2016) 6(1):26156. doi: 10.1038/srep26156
36. Osorio JC, Blanco R, Corvalán AH, Muñoz JP, Calaf GM, Aguayo F. Epstein–Barr Virus infection in lung cancer: Insights and perspectives. Pathogens (2022) 11(2):132. doi: 10.3390/pathogens11020132
37. Rae W, Sowerby JM, Verhoeven D, Youssef M, Kotagiri P, Savinykh N, et al. Immunodeficiency, autoimmunity, and increased risk of b cell malignancy in humans with TRAF3 mutations. Sci Immunol (2022) 7(74):eabn3800. doi: 10.1126/sciimmunol.abn3800
38. Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell (2003) 4(3):181–9. doi: 10.1016/S1535-6108(03)00220-4
39. Cai L, Chen Y, Tong X, Wu X, Bao H, Shao Y, et al. The genomic landscape of young and old lung cancer patients highlights age-dependent mutation frequencies and clinical actionability in young patients. Int J Cancer (2021) 149(4):883–92. doi: 10.1002/ijc.33583
40. Tao J, Kahn L. Epstein-Barr Virus–associated high-grade b-cell lymphoma of mucosal-associated lymphoid tissue in a 9-Year-Old boy. Arch Pathol Lab Med (2000) 124(10):1520–4. doi: 10.5858/2000-124-1520-EBVAHG
41. Glaser SL, Hsu JL, Gulley ML. Epstein-Barr Virus and breast cancer: state of the evidence for viral carcinogenesis. Cancer Epidemiol Biomarkers Prev (2004) 13(5):688–97. doi: 10.1158/1055-9965.688.13.5
42. Zulfiqar B, Farooq A, Kanwal S, Asghar K. Immunotherapy and targeted therapy for lung cancer: Current status and future perspectives. Front Pharmacol (2022) 13:1035171. doi: 10.3389/fphar.2022.1035171
43. Subbiah V, Solit DB, Chan TA, Kurzrock R. The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB)≥ 10: a decision centered on empowering patients and their physicians. Ann Oncol (2020) 31(9):1115–8. doi: 10.1016/j.annonc.2020.07.002
44. Herbst RS, Lopes G, Kowalski DM, Nishio M, Wu YL, de Castro Junior G, et al. Association between tissue TMB (tTMB) and clinical outcomes with pembrolizumab monotherapy (pembro) in PD-L1-positive advanced NSCLC in the KEYNOTE-010 and-042 trials. Ann Oncol (2019) 30:v916–7. doi: 10.1093/annonc/mdz394.077
Keywords: lung cancer, tumor grade, genomic mutations, Epstein-Barr virus, pathway analysis
Citation: Wang Y, Qin S, Liang Y, Yan L, Zheng M, Zeng Y and Lu L (2023) Tumor grade-associated genomic mutations in Chinese patients with non-small cell lung cancer. Front. Oncol. 13:1119575. doi: 10.3389/fonc.2023.1119575
Received: 09 December 2022; Accepted: 06 March 2023;
Published: 20 March 2023.
Edited by:
Francesco Pepe, University of Naples Federico II, ItalyReviewed by:
Andrea Moreno Manuel, Fundación de Investigación del Hospital General Universitario de Valencia, SpainZhixi Su, Singlera Genomic Ltd, China
Copyright © 2023 Wang, Qin, Liang, Yan, Zheng, Zeng and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Zheng, emhlbmdtNjYzQDEyNi5jb20=; Yanwu Zeng, emVuZ3l3QG9yaWdpbWVkLmNvbQ==; Leilei Lu, bHVsbEBvcmlnaW1lZC5jb20=
†These authors have contributed equally to this work