
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 02 February 2023
Sec. Cancer Genetics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1112276
This article is part of the Research TopicNew Therapeutic Approaches in Microsatellite Stable Colorectal Cancer Patients.View all 7 articles
 Javier Ros1,2
Javier Ros1,2 Francesca Balconi3
Francesca Balconi3 Iosune Baraibar1Nadia Saoudi Gonzalez1Francesc Salva1Josep Tabernero1
Iosune Baraibar1Nadia Saoudi Gonzalez1Francesc Salva1Josep Tabernero1 Elena Elez1*
Elena Elez1*Immune checkpoint inhibitors have reshaped the prognostic of several tumor types, including metastatic colorectal tumors with microsatellite instability (MSI). However, 90-95% of metastatic colorectal tumors are microsatellite stable (MSS) in which immunotherapy has failed to demonstrate meaningful clinical results. MSS colorectal tumors are considered immune-cold tumors. Several factors have been proposed to account for this lack of response to immune checkpoint blockade including low levels of tumor infiltrating lymphocytes, low tumor mutational burden, a high rate of WNT/β-catenin pathway mutations, and liver metastases which have been associated with immunosuppression. However, studies with novel combinations based on immune checkpoint inhibitors are showing promising activity in MSS colorectal cancer. Here, we review the underlying biological facts that preclude immunotherapy activity, and detail the different immune checkpoint inhibitor combinations evaluated, along with novel immune-based therapies, to overcome innate mechanisms of resistance in MSS colorectal cancer.
Colorectal cancer (CRC) is the third most frequent cancer type diagnosed worldwide and the second cause of cancer-related deaths according to the Globocan data (1). Despite the improvement in early detection screening policies and the development of many novel treatments, in the USA, 5-year survival is only 12.5% (2), reflecting a clear unmet need for more effective treatments for patients with metastatic CRC (mCRC). Over the past decade, the development of immune checkpoint inhibitors has reshaped the prognostic of specific tumor types, notably melanoma and lung cancer, achieving deep, durable, and even complete responses. The identification of biomarkers of response is a much-needed paramount step forward in order to identify those patients who can benefit from immunotherapy strategies. In the case of mCRC, tumors with high microsatellite instability (MSI) or mismatch repair deficiency (MMRd), representing 5% of all mCRC, have achieved impressive and durable responses with immune checkpoint inhibitors. MSI tumors are highly enriched with neoantigens caused by the hypermutable status, typically presenting with high tumor mutational burden (TMB) and high levels of tumor infiltrating lymphocytes (TIL). Conversely, immune checkpoint inhibitors have demonstrated poor activity in most tumors that are mismatch-repair-proficient or microsatellite stable (MSS), which represent the vast majority (95%) of patients with mCRC.
MSI and hypermutated MSS tumors, also called “immune-hot tumors”, show high neoantigen loads and TMB, enriched activated CD4 and CD8 T cells, and conversely a low-frequency of myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) and increased expression of CTLA-4 and PD-1 and of its ligand PD-L1. In the majority of MSS tumors, on the other hand, immunotherapy alone has demonstrated no clinical activity, and they have been characterized as being enriched for MDSCs, poorly infiltrated by T cells, and have downregulated checkpoint inhibitors, HLA class-I and class-II, and CD4 and CD8 T cells (3). MSS tumors are considered “immune-cold tumors” characterized by low TMB and a lack of immune cell infiltration, which have been positioned as the main mechanisms of immune resistance (4, 5). The neoantigen enrichment of MSI mCRC is related to the hypermutable status in the context of MSI whereas MSS tumors harbor features of chromosomal instability, in which genomic aberrations of the genomic structure occur on a larger scale, leading to lower TMB and neoantigen generation (6). There is evidence suggesting that survival benefit among patients with CRC and high-TMB is limited to MSI and POL-E or POL-D1 tumors meaning that very few patients with MSS/high-TMB benefit from immune checkpoint blockade (7).
Overall, the lack of response to immune checkpoint inhibitors among patients with MSS mCRC may be driven by several molecular and histopathological factors. Mutations in the WNT/β-catenin signaling pathway genes, which is frequent in CRC, as well as liver metastases, may induce antitumor immunity inhibition and immune tolerance leading to T cell exclusion, explaining the upfront lack of response to immunotherapy. Furthermore, several clinical trials have demonstrated that patients with MSS mCRC without liver metastases are more prone to respond to immune checkpoint inhibitor combinations (8, 9). Several of the strategies attempting to increase immune checkpoint inhibitor activity in the MSS mCRC population have unfortunately given poor results, however, novel combinations appear promising. In this review, we describe the underlying biological features leading to MSS mCRC innate resistance to immune checkpoint inhibitors as well as the current clinical approaches and future directions to improve outcomes with immune checkpoint inhibitors in MSS mCRC patients.
The immunotherapy strategies tested to date have shown limited antitumor activity in MSS mCRC for multiple reasons including, among others, low TMB and low immune infiltration compared to MSI colorectal tumors or other hypermutated subtypes such as POL-E or POL-D mutated tumors. MSI tumors are inflamed and infiltrated with lymphocytes, T helper 1 cells, CD4+ cells, and macrophages and have a microenvironment rich in type I interferons (4, 10, 11), whereas their MSS counterparts are not inflamed and are usually PD-L1-negative (5, 12). Not only do MSS colorectal tumors have fewer TILs and lower TMB but their genomic alterations also seem to be poorly neo-antigenic (13). Thus, switching a tumor from an immune cold phenotype that abrogates T cell activity to a hot phenotype that leads to lymphocyte activation and tumor infiltration may improve immune checkpoint inhibitor efficacy. The complexity of MSS mCRC goes far beyond simple biomarkers such as PD-L1 expression, and TIL or TMB levels. Figure 1 presents the main differences between MSS and MSI mCRC.
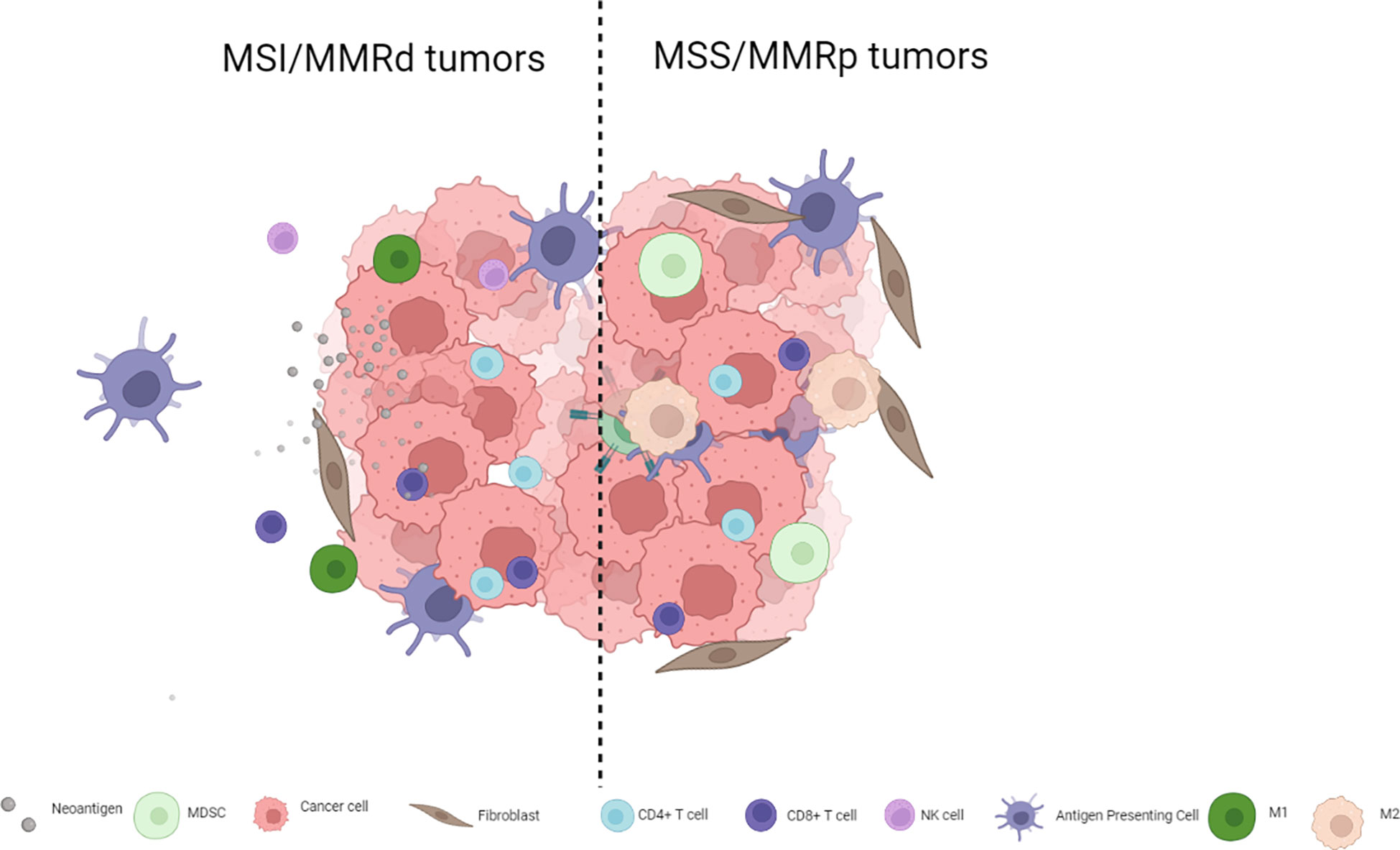
Figure 1 Key differences in immune cell infiltration for MSS and MSI mCRC.
Cell infiltration plays a paramount role regulating immune checkpoint inhibitor activity. MDSCs are immature myeloid cells found in several tumor types. In CRC, tumor cells promote the induction of MDSCs that have an immunosuppressive role which facilitates tumor growth and spread by releasing factors such as reactive oxygen species, TGFβ, and nitric oxide (14). MSI and hypermutated tumors show a depletion of MDSCs and Tregs, unlike in MSS tumors (3). Furthermore, patients with a high level of MDSCs have shorter progression-free survival (PFS) with chemotherapy (15). MDSCs have been demonstrated to inhibit T cell proliferation, while blocking MDSC function was shown to restore INFγ secretion by T cells (16). In addition to MDSCs, tumor-associated macrophages (TAMs) also play a fundamental role in immune system modulation. TAMs can be divided into two major groups based in their phenotype. M1 macrophages are involved in tumor growth by secreting pro-inflammatory cytokines such as TNF-alfa, IL-1-B or IL-12, driving a Th1 response. On the other hand, M2 macrophages secrete immunosuppressive cytokines such as IL-10 and TGFβ which promote tumor progression and metastases. However, unlike other tumor types, the prognostic impact of TAMs in CRC remains unclear, likely in part because of high plasticity between macrophage subsets and a lack of well-defined markers to properly identify them (17–20). Likewise, the exact role of Tregs in CRC has not been fully elucidated. Overall, Tregs have a pro-inflammatory role, secreting TGFβ and IL-10, and suppressing CD4 and CD8 T cells and downregulating IFNγ and IL-2 secretion in tumors leading to an immunosuppressive microenvironment (21, 22).
Whereas MSI CRC tumors are enriched for hypermethylation, and BRAF and RNF43 mutations, MSS tumors are enriched in APC and KRAS mutations and chromosomal instability. These molecular features reflect the differences in the carcinogenesis pathway that may contribute to responses to immune checkpoint blockade strategies (23). β-catenin oncoprotein is a paramount activator of WNT signaling in CRC, particularly APC mutations (present in more than 70% of MSS CRC but in only 20% of MSI CRC) that regulate the function of β-catenin (<ξ>)(24). Studies in melanoma identified that increased β-catenin activation resulted in a decreased CD8 and CD103 dendritic cell population that hampered T cell recruitment into the tumor microenvironment, leading to a T cell exclusion status. Activated WNT/β-catenin signaling has been inversely correlated with the absence of T cell infiltration (CD8+ cells, CD45RO+ cells, and CD3+ cells but not with FOXP3+ cells) regardless of mutational load, and nuclear CTNNB1 protein expression (a marker of activation of the WNT pathway) was inversely associated with TIL levels in CRC samples. Non-inflamed CRC tumors were enriched in β-catenin signaling pathway (23, 25, 26), thus suggesting that WNT signaling inhibitors may be able to reverse immune exclusion (20). Furthermore, MSS tumors that harbor a lower mutational load exhibit less cancer immunoediting along with higher WNT pathway signaling (including RNF43 and Axin 1/2 mutations, and R-spondin gene fusions) and decreased T cell infiltration leading to “cold tumors” that are less responsive to immunotherapy strategies. Furthermore, APC biallelic mutations are associated with increased WNT signaling and decreased TILs in both MSS and MSI CRC. This finding is of particular interest considering that up to 70% of all CRC cases have APC biallelic loss. Finally, hypomethylation of AXIN2 super-enhancers has been associated with decreased TILs regardless of APC alterations in MSS tumors. Taken together, these data suggest there are multiple pathways that limit immune response in cancers with active WNT signaling (23, 27, 28).
Transforming growth factor-β (TGF-β) signaling is a well-known oncogenic pathway driving CRC; it acts as an oncogenic factor by creating an immune-suppressive tumor microenvironment, and inducing tumor cell proliferation, angiogenesis, and metastatic spread (29). TGF-β levels strongly correlate with consensus molecular subtype (CMS) 4, the mesenchymal subtype characterized by stromal infiltration and epithelial-mesenchymal transition based on genomic profiles. Increased TGF-β is associated with increased regulatory T cells and downregulation of natural killer cells, leading to an immunosuppressive microenvironment (29, 30). Whereas 76% of samples in CMS1 tumors -the inflamed subgroup- were MSI, among CMS4 tumors, 92% were MSS and 8% were MSI. Furthermore, TGF-β has been noted in liver metastases in which CD4 and CD8 T cells were downregulated and immune tolerance is abrogated by liver type 1 macrophage activation and a high number of Treg (31–33). This can partially explain what has been observed in some clinical trials in which patients with liver metastases show lower response rates with immune checkpoint inhibitors compared with patients without liver metastases (8, 9, 34–36). In addition, preclinical inhibition of TGF-β resulted in reduced immune-evasion process (37).
Finally, alterations in the mitogen-activated protein kinase pathway (MAPK) are common events in CRC, particularly RAS mutations (50%) and BRAF mutations (10%). Mutations in both RAS and BRAF perpetuates kinase activation, leading not only to cell growth, invasion, and metastases, but also to tumor microenvironment differentiation, and have been shown to reduce T cell CD8+ infiltration and diminish neoantigen presentation by impairing the interferon pathway. Nevertheless, treatment with either BRAF or KRAS G12C inhibitors led to reduced MDSC infiltration and increased TILs (38–41). Current studies combining a BRAF inhibitor with an anti-PD-1 have shown deep clinical impact, with prolonged PFS among patients with a BRAF-V600E mutation (42–44). Finally, while the predictive value of PD-L1 has been demonstrated in several tumor types, this has not been the case in CRC in either the CHECKMATE-142 trial with nivolumab plus ipilimumab, or the phase I trial with pembrolizumab (5, 12). The efficacy of combined pembrolizumab plus co-formulated favezelimab, in patients with PD-L1-positive refractory CRC is currently being evaluated in a phase III trial (NCT05064059).
The following section reviews the large body of research that has focused on strategies to identify and overcome the mechanisms of resistance to immune checkpoint inhibitors in MSS CRC. A summary of completed and ongoing clinical evaluations according to treatment strategy in MSS CRC is presented in Tables 1, 2 respectively, covering a range of settings with frontline, refractory, and maintenance therapies.
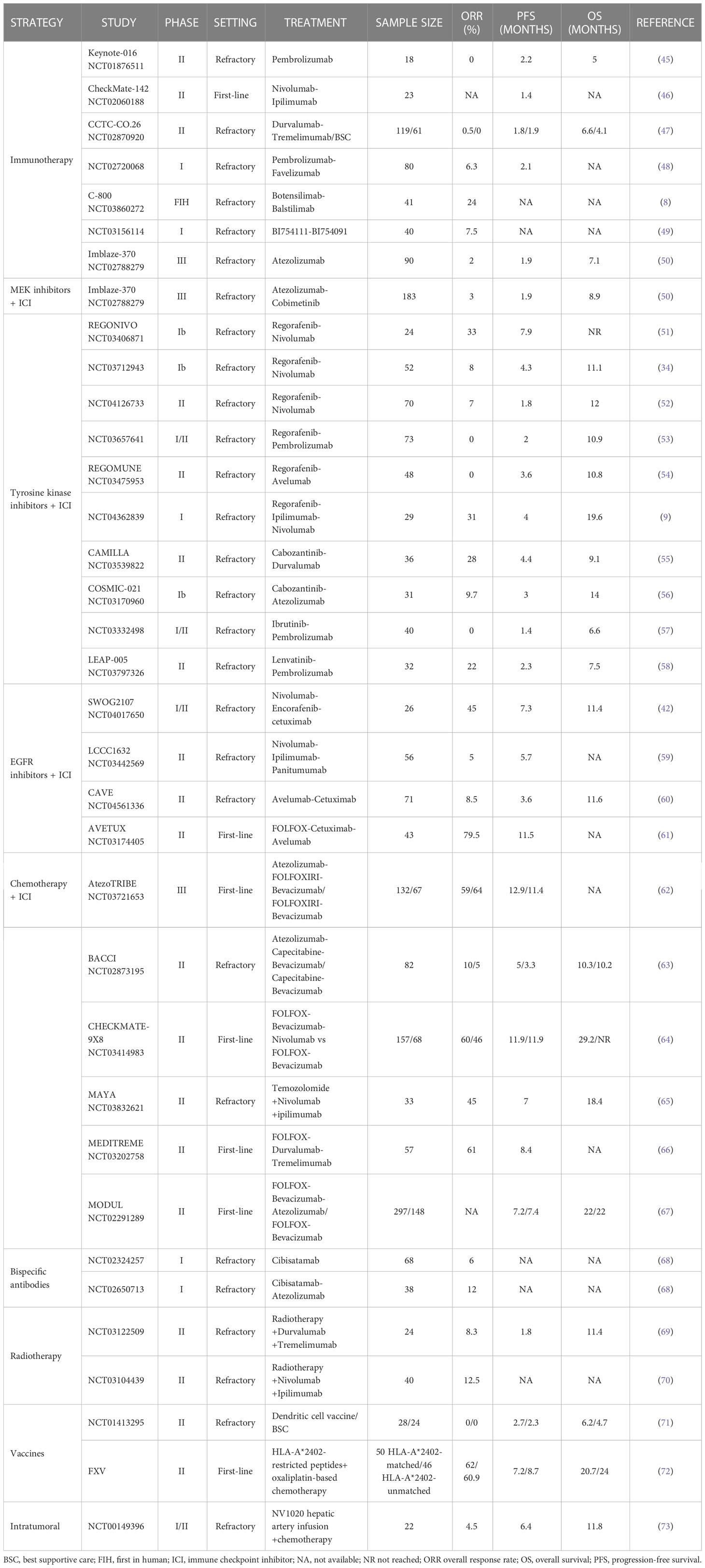
Table 1 Completed clinical trials evaluating therapeutic strategies with immune checkpoint inhibitors in MSS mCRC.
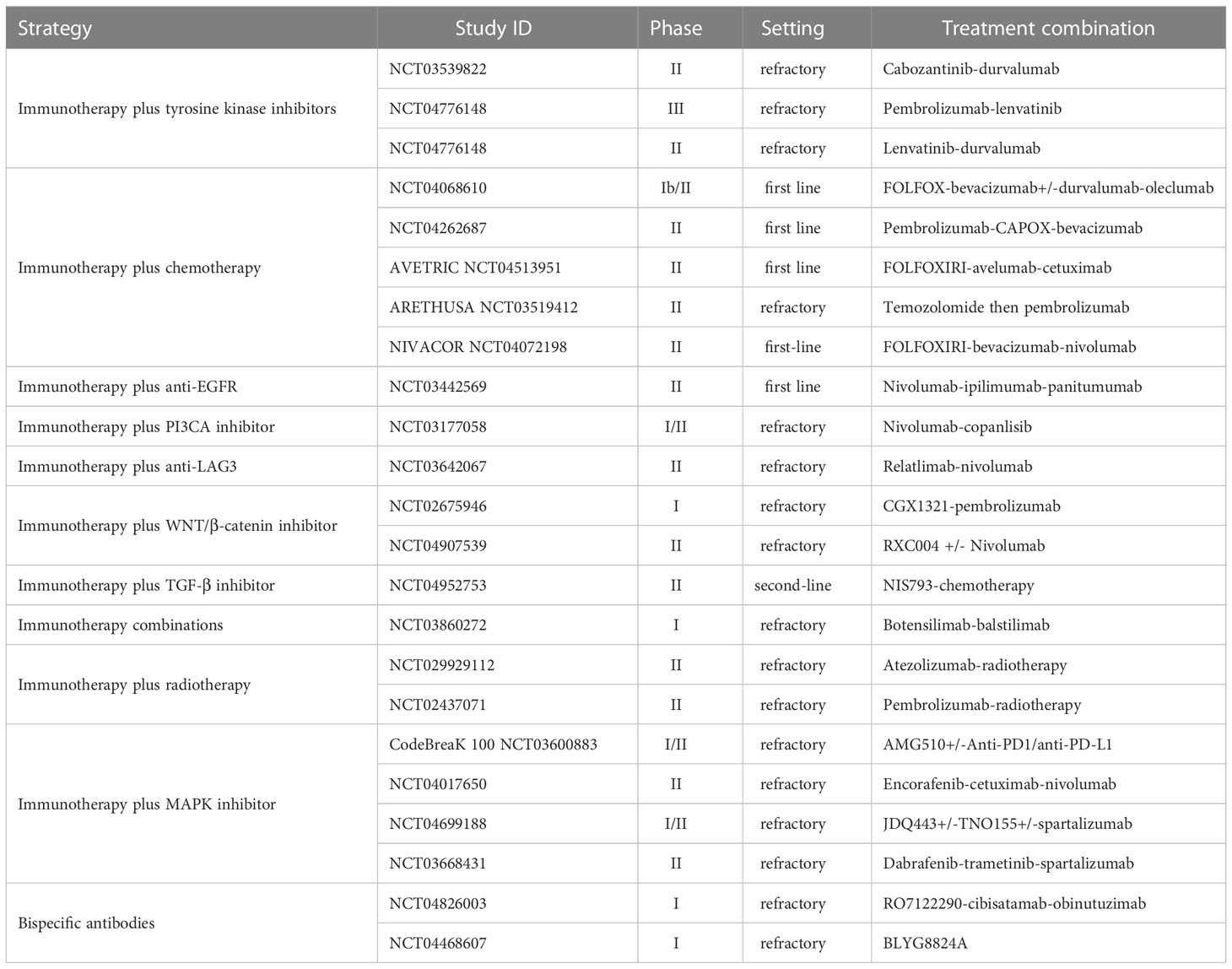
Table 2 Novel therapeutic strategies in ongoing clinical studies in MSS mCRC.
Immune checkpoint inhibitors lead to impressive, deep, and long-lasting responses in MSI mCRC in both the first-line and the refractory setting (12, 74). However, the results in the MSS population have not replicated those observed in the MSI setting. The anti-PD1-antibody pembrolizumab was evaluated in a phase II clinical trial which included 11 patients with MSI tumors and 21 MSS refractory tumors. The overall response rate (ORR) was 0% (95% confidence interval [CI], 0-20), and the PFS rate at 20 weeks was 11% (2 of 18 patients; 95% CI, 1-35). Somatic mutations per tumor, neoantigen load and CD8 infiltration were significantly lower in patients with MSS tumors suggesting that a hot-tumor microenvironment -not observed in MSS tumors- is needed to achieve immune response. Similar results were obtained in the phase I trial evaluating nivolumab in solid tumors, which included 19 patients with MSS tumors in the expansion cohort (75). Only one patient with CRC had a PD-L1 positive tumor and this patient did not respond. The combination of nivolumab and the anti-CTLA4 ipilimumab was also tested in the phase II CheckMate-142 trial. Overall, 23 patients with MSS tumors were enrolled; median PFS was 1.4 months, highlighting the lack of clinical activity of the double immune checkpoint combination (46). These results suggest there is no meaningful clinical activity with combined PD-1 and CTLA4 blockade in MSS mCRC, although a small number of patients achieved some clinical benefit (47).
Dual blockade of PD-1 and LAG-3 (an immune checkpoint inhibitor mainly expressed on T cells) has the potential to synergistically restore T-cell functionality, and therefore to boost the immune system antitumor activity (76, 77). The phase I trial (NCT03156114) evaluated the combination of BI754111 (an anti-LAG-3 monoclonal antibody) and BI754091 (an antiPD-1) in patients with advanced solid tumors. In the mCRC cohort, 40 patients with refractory tumors were included with an ORR of 7.5%, and a DCR of 35% (49). Other ongoing strategies include the combination of the LAG-3 inhibitor relatlimab with an anti-PD1 (NCT0306422067 and NCT05064059) (48) with the same results compared with immunotherapy alone.
More recently, promising activity has been seen in MSS tumors with novel immune checkpoint inhibitor combinations. Botensilimab, a novel innate/adaptive immune activator (Fc-enhanced CTLA4-inhibitor) in combination with balstilimab (anti-PD1) were tested in the first-in-human, phase I C-800 trial. The CRC MSS cohort included 41 heavily pretreated patients, and an impressive ORR of 24% and a disease control rate (DCR) of 73% was seen, with a well-tolerated safety profile (no grade 4 or 5 adverse events were reported). The exploratory analysis in terms of liver involvement showed enriched responses among the 24 patients without active liver metastases (ORR 42%, DCR 96%) suggesting that liver metastases may preclude immune system activation (8, 78, 79). Although the classical approach with immune-checkpoint inhibitors blockade has not been demonstrated to be as effective as in the MSI scenario, novel combinations nonetheless demonstrate promising activity, particularly among well-selected patients without liver metastases.
Activation of the MAPK pathway leads to proliferative tumor effects as well as decreased T cell infiltration and immunosuppression (38, 39). In preclinical models, MEK inhibition resulted in induced INFγ-dependent HLA and PDL1 upregulation, suggesting the potential synergic effect of MEK inhibitors with immunotherapy (80, 81). MEK inhibition also profoundly blocked naive CD8 T cell priming in tumor-bearing mice; however, it increased the number of effector-phenotype antigen-specific CD8 T cells within the tumor. In addition, MEK inhibition decrease tumor-infiltrating CD8 T cells from death driven by prolonged T cell receptor stimulation while sparing cytotoxic activity (81).
The phase II randomized CO.26 trial evaluated the anti-PD1 durvalumab plus the MEK inhibitor tremelimumab compared with best supportive care. There were no significant differences in median PFS (1.9 and 1.8 months respectively). However, longer overall survival (OS) was observed among patients treated with durvalumab plus tremelimumab (HR, 0.66; 90% CI, 0.49-0.89; P = .02) with a particular benefit among patients with plasmatic TMB >28 variants per megabase. Atezolizumab plus the MEK inhibitor cobimetinib combination was evaluated in a phase Ib trail which included 84 patients with refractory CRC resulting in an ORR of 8% (82). Besides, a phase 3 trial randomized patients with refractory CRC to receive either atezolizumab plus cobimetinib, atezolizumab in monotherapy, or regorafenib. The trial did not meet its primary endpoint. Median OS was 8.9 months for the experimental combination therapy arm, 8.5 months for the control arm with regorafenib, and 7.1 months for patients receiving atezolizumab (50). These negative results can be partially explained by the enrollment of patients with refractory mCRC, without selecting by RAS/BRAF mutational status, and the fact that single MEK blockade fails to maintain MAPK inhibition due to adaptive feedback throughout EGFR receptor (83, 84).
Various combinations with immune checkpoint inhibitors and tyrosine kinase inhibitors have been investigated suggesting that tyrosine kinase inhibitors, particularly angiogenesis inhibitors, may decrease tumor-associated macrophages and Tregs, and enhance T cell infiltration and activation, as well as increase dendritic cell maturation increasing tumor antigenicity and tumor immunogenicity (85, 86).
The REGONIVO phase Ib study in Asia evaluated regorafenib in combination with nivolumab in patients with MSS and MSI-H CRC who had progress on previous systemic chemotherapy (34). Patients were treated with regorafenib and nivolumab. The ORR among patients with MSS disease was 33%, median PFS was 7.9 months, and median OS was not reached. The one-year PFS rate was 42%, suggesting durable disease control. However, among patients with liver metastasis, the ORR was 8%, whereas it was 64% for patients with lung metastasis, again suggesting that liver metastases may play an immunosuppressive role hampering immunotherapy activity. Other studies (NCT03712943 and NCT04126733) have investigated the same combination with median PFS of 4.3 and 1.8 months respectively (51, 52). Regorafenib was also combined with pembrolizumab in a phase I/II study in patients with refractory MSS CRC. No objective responses were observed. Median PFS was 2 months and median OS was 10.9 months (53). Another combination of regorafenib with another anti-PD-L1 antibody avelumab in a phase II trial examined the efficacy and safety of this combination in 48 patients with mismatch-repair-proficient CRC. The trial did not show any objective responses, however 23 patients presented stable disease (54%), and median PFS was 3.6 months (95% CI 1.8–5.4), and median OS was 10.8 months (95% CI 5.9–non-achieved-NA). Correlative analysis suggested an association between high tumor-associated macrophages and poor outcomes, whereas increased CD8 T cell infiltration suggested an improvement in clinical outcomes (54). Furthermore, a phase I trial combining regorafenib with ipilimumab and nivolumab in patients with MSS CRC (NCT04362839) gave an ORR of 31%, with a median PFS of 4 months. Cabozantinib was combined with durvalumab in a prospective, open-label, multicenter phase II trial giving a 28% ORR, median PFS of 4.4. months, and OS of 9.1 months (55). Of note, cabozantinib was also evaluated in combination with atezolizumab in the COSMIC-021 phase 1b trial which included 31 patients with MSS, refractory mCRC. Patients with wild-type RAS had numerically longer PFS and OS and higher ORR compared with patients with RAS mutated tumors (5.8 vs 2.7 months, 16.7 vs 8.7 months and 25% vs 0% respectively) (56). The pembrolizumab and lenvatinib combination in the non-randomized, phase II, LEAP-017 trial showed promising antitumor activity with an ORR of 22% and median PFS of 2.3 months among patients with refractory MSS CRC (58). Finally, the combination of ibrutinib, a Bruton’s tyrosine kinase (BTK) inhibitor, has also been tested in mCRC in combination with pembrolizumab showing poor clinical activity (ORR 0%, mPFS 1.4, and mOS 6.6 months) (57).
CRC harboring the BRAF-V600E mutation deserves specific attention. With its well-known poor prognostic, the BEACON trial was the first phase III trial demonstrating an advantage with the BRAF inhibitor encorafenib plus cetuximab with or without the MEK inhibitor binimetinib. Correlative analysis from paired biopsies demonstrated an increase in T cell infiltration and cytotoxic infiltration after the initiation of a BRAF inhibitor, suggesting potential cooperation between BRAF-targeting and the immune response (43). This is supported given that the combination of immune checkpoint inhibitors plus BRAF inhibitors has shown promising results: the phase I trial combining dabrafenib-trametinib and spartalizumab showed a 33% ORR with a 76% of DCR. In the same setting, the SWOG phase I/II trial demonstrated promising activity of the encorafenib-cetuximab-nivolumab triple combination, with an ORR of 50%, DCR of 95% and impressive 7.4 month median PFS and 15.1 month median OS in BRAF-V600E mutated MSS mCRC (42).
Targeting EGFR signaling—particularly the MAPK pathway—holds promise for creating synergy with immune checkpoint inhibitors, alone or in combination with chemotherapy. Anti-EGFR activity leads to NK cell activation and subsequent lytic activity on tumor cells by antibody-dependent cellular cytotoxicity, Treg immunosuppression and induces PD-L1 expression on tumor cells via INFγ (87–89). Panitumumab in combination with nivolumab and ipilimumab, has been investigated in a phase II study in patients with RAS/RAF wild-type mCRC. The ORR was 5%, with a median PFS of 5.7 months, favoring this triplet regimen over single-agent panitumumab (59). The combination of avelumab with cetuximab has been explored in the third-line chemorefractory setting in RAS wild-type mCRC with no selection regarding microsatellite status, in patients who had a complete or partial response to first-line chemotherapy plus anti-EGFR drugs. A promising median OS of 11.6 months and median PFS of 3.6 months were achieved (60). Together these data suggest potential synergism between immune checkpoint inhibitors and anti-EGFR drugs.
Combined chemotherapy and immunotherapy have been tested in several clinical trials. The rationale for these combinations relies on the fact that chemotherapy promotes immune cell infiltration, and dendritic cell maturation, enhances antigen presentation, as well as inhibits immunosuppressive cells (90–92). The addition of avelumab to FOLFOX and cetuximab in this setting was explored in the single-arm, phase II AVETUX trial which included 42 patients (2 MSI, 40 MSS). With RAS/BRAF wild-type tumors. Although the combined treatment gave an ORR of 79.5%, the PFS rate at 12 months was 40%, and the study did not reach its primary endpoint (12 months PFS 57%) (61). AtezoTRIBE, a randomized phase II trial, investigated the combination of FOLFOXIRI and bevacizumab, with or without atezolizumab, in patients with mCRC regardless of MSS/MSI status. Among the MSS subgroup, PFS improved from 11.4 to 12.9 months in patients treated with chemotherapy combined with atezolizumab (62). The BACCI phase II trial evaluated the addition of atezolizumab or placebo to capecitabine and bevacizumab in the chemo-refractory setting regardless of MSS status. The trial achieved its primary endpoint of median PFS (4.4 vs 3.6 months) in the overall population with a non-significant PFS improvement among the MSS subgroup (5.3 vs 3.3 months) (93). Another study, the phase III CheckMate9X8, evaluated the addition of nivolumab to standard first-line chemotherapy with FOLFOX-bevacizumab vs FOLFOX-bevacizumab in patients with previously untreated, unresectable, mCRC. However, the primary endpoint of PFS was not met, with the same median PFS of 11.9 months in both arms (64). Finally, the phase II MEDITREME trial evaluated the efficacy and safety of mFOLFOX6 (6 cycles) in combination with durvalumab and tremelimumab as induction therapy followed by maintenance therapy with durvalumab in patients with previously untreated RAS-mutated mCRC. ORR was 61% and mPFS was 8.4 months. Biomarker analysis showed that high baseline levels of Th2 and PDL1+ MDSC were associated with poor PFS (66).
After first-line induction therapy with FOLFOX-bevacizumab maintenance treatment with 5-FU plus atezolizumab has been also evaluated in the phase II MODUL trial which included patients with metastatic BRAF wild-type CRC. However, the trial did not show a significant difference in either PFS or OS (67). Results are awaited for the ongoing phase Ib/II COLUMBIA-1 trial is comparing FOLFOX and bevacizumab in combination with durvalumab and the anti CD37 oleclumab versus FOLFOX and bevacizumab (NCT04068610).
Also combining chemotherapy plus immunotherapy, the phase II MAYA trial (NCT03832621) evaluated the effect of temozolomide on TMB in terms of response to immune checkpoint inhibitors among MSS mCRC patients. Patients with chemo-refractory CRC were prescreened for MSS status and MGMT silencing. Eligible patients received temozolomide followed, in the absence of progression, by combination with ipilimumab and nivolumab. After a median follow-up of 23.1 months, the 8-month PFS rate was 36% and median PFS and OS were 7.0 and 18.4 months, respectively, with an ORR of 45% (65). Likewise, the ARETHUSA (NCT03519412) phase II trial is enrolling patients with MSS, RAS mutant CRC tumors that are MGMT negative and promoter methylation-positive and have TMB >20 mut/Mb after treatment with temozolomide. After this priming phase with temozolomide, patients with high TMB are treated with pembrolizumab (94) based on evidence that temozolomide can increase mutational burden in both tumor tissue and blood of CRC patients.
Bispecific antibodies are an large engineered family of molecules designed to recognize two different epitopes or antigens simultaneously, aiming to enhance the host’s immune activity against tumor cells by binding both tumor-enriched antigens (e.g., CEA, CEACAM, or CD276 antigen) and immune cells (mainly T cells using the CD3 receptor) (95). Cibisatamab (RO6958688, also known as CEA-TCB) is a T cell bispecific antibody targeting both CEA on tumor cells and CD3 on T cells, leading to crosslinking of tumor cells and lymphocyte T cell engagement and activation. In preclinical models, cibisatamab demonstrated deep antitumor activity, leading to increased intratumoral T cell infiltration and activation and PD-1/PD-L1 upregulation. In two ongoing dose escalation phase I studies, cibisatamab monotherapy or combined with atezolizumab, is administrated in patients with advanced CEA-positive solid tumors. The monotherapy group included 80 patients (70 patients with CRC), and the combination group included 45 patients (35 with CRC). CT scans revealed signs of tumor inflammation flare within 48 hours of the first dose. Additionally, tumor reduction was seen in MSS tumors in 13% and 36% of CRC patients in the cibisatamab monotherapy and combination groups respectively with an impressive DCR of 45% and 82%, respectively. Among patients with mCRC 68 patients treated with cibisatamab and 38 patients treated with cibisatamab in combination with atezolizumab, 10 (28%) and 6 (60%) respectively presented a metabolic partial response by PET scan 4-6 weeks after treatment initiation with evidence of antitumor activity during dose escalation (68).
Radiation therapy is an effective treatment for many cancers and a part of the multidisciplinary approach to cancer care, exploiting the mechanism of damage of DNA inside cancer cells. Distantly, radiotherapy can induce an abscopal effect (i.e., tumor shrinkage), which might results in tumor shrinkage outside the irradiated field due to an increase of immune cell tumor infiltrates and direct presentation of tumor antigens (96, 97). Durvalumab plus tremelimumab combined with radiotherapy in patients with mCRC was evaluated in a single arm phase II trial. ORR was 8.3%, and median PFS and median OS were 1.8 and 11.4 months respectively (69). The radiotherapy with ipilimumab and nivolumab combination has also been tested, leading to shrinkage of non-irradiated distant tumors with an ORR of 12.5% (70).
In a more recent approach, cancer vaccines may play a role in enhancing the immune system. stimulating tumor antigen-specific cytotoxic T lymphocytes that recognize and eliminate cancer cells (98). This strategy was evaluated in a randomized phase II trial with two groups of patients, one treated with the best supportive care and the other with autologous dendritic cells plus best supportive care (71). The study demonstrates that this antibody-drug conjugate generates a tumor-specific immune response but did not provide a survival benefit (median PFS was 2.7 vs 2.3 respectively and median OS was 6.2 vs 4.7, respectively). Another phase II trial evaluated the combination of a five peptides vaccine derived from tumor-associated antigens with oxaliplatin-based chemotherapy in the first-line setting, demonstrating the generation of a tumor-specific immune response but again without meaningful clinical benefit (72).
Finally, among novel cancer treatment strategies, oncolytic virotherapy has shown encouraging progress. Genetically modified herpes simplex viruses (HSVs) have demonstrated to induce cytolytic cell death and liberation of progeny virions, which infect adjacent tumor cells without adversely affecting untransformed parent cells. A phase I/II clinical trial genetically engineered oncolytic HSV NV1020 (73). Among the 22 patients treated, 50% showed stable disease after NV1020 administration with a 68% DCR and median OS of 11.8 months after treatment with chemotherapy.
Unlike MSI mCRC, the efficacy of immune checkpoint inhibitors alone or in combination with other therapeutics is limited to a narrow subgroup of patients. While the vast majority of MSS CRC tumors will not respond to immunotherapy, there appears to be a borderline immunogenic tumor subgroup that manifest with stable disease and partial responses. Classic predictive biomarkers such as PDL-1 expression or TMB do not appear to be valuable biomarkers for efficacy in MSS CRC. The main reasons for the lack of response to immunotherapy in MSS mCRC are the tumor cell infiltrate which has low levels of TIL and high infiltration of T cell suppression, Tregs, and macrophages. Other factors influencing poor outcomes are low TMB and low immunogenicity of MSS tumor-related neoantigens and genomic and clinical characteristics including a high rate of WNT/β-catenin pathway mutations, and the presence of liver metastases. These features highlight the non-inflamed reality of MSS tumors reflecting a low immunogenic environment. However, the tumor microenvironment is amenable to be modulated with other therapeutics that may increase immune checkpoint inhibitor responses. Furthermore, the genomic landscape of MSS CRC, mostly driven by the WNT/β-catenin pathway, has been identified as a critical factor of immunosuppression and T cell antitumor activity impairment, limiting immune checkpoint inhibitor activity. As a result, there are currently several clinical trials aiming to inhibit WNT/β-catenin pathway signaling using combination therapies with immune checkpoint inhibitors. Taking a similar approach, some studies have demonstrated that liver metastases promote an immunosuppressive microenvironment that limits immune checkpoint efficacy. Considering these upfront barriers that hamper immunotherapy activity, several combinations have been tested in combination with immune checkpoint inhibitors, including tyrosine kinase inhibitors, anti-EGFR, chemotherapy, and radiotherapy, while novel approached including bispecific antibodies, ADCs, and vaccines, with modest improvements in terms of clinical outcomes. Currently there are several ongoing trials assessing new drug combinations that aim to overcome the acquired mechanism of resistance in MSS CRC, as summarized in Table 2. These data form the basis of a better understanding of the biology underlying MSS colorectal tumors that will lead to improved clinical outcomes and the identification of clinical biomarkers relevant to this population. Recently several trials demonstrated that outcomes in patients with MSS CRC can be improved with either better designed drugs, such as botensilimab and balstilimab, or with bispecific antibodies, or with a smart trial design such as the MAYA or the ARETHUSA trials in which the TMB is successfully modulated using chemotherapy to boost the effect of subsequent immune checkpoint inhibitors. Similarly, liver metastases, mostly enriched in TGF-β, have been identified as a negative biomarker of response using immunotherapy combinations in several clinical trials. Besides, other treatment strategies are being developed for MSS CRC such as neoadjuvant immunotherapy. Indeed, the NICHE trial demonstrated that immune checkpoint inhibitors may have a role during early-stage disease in MSS CRC. The NICHE trial showed 27% pathological responses among patients with early-stage, low TMB, MSS, CRC treated with neoadjuvant nivolumab and ipilimumab combination. Of note, correlative analysis showed that CD8+ PD1+ cell infiltration was predictive of response (99). Despite these promising results, larger studies and longer follow-up are still required. Ultimately, a deep understanding of mechanisms of immunotherapy resistance and the heterogenous spectrum of mCRC is needed in order to improve therapeutical co-strategies to overcome primary resistance to immunotherapy. Furthermore, biomarker identification may help for an optimal patient selection.
JR, and FB wrote the first draft of the manuscript. All authors contributed to the manuscript revision. EE supervised the final version. All authors contributed to the article and approved the submitted version.
JR has received personal speaker honoraria from Sanofi, and AMGEN, and accommodation expenses from Pierre-Fabre, Servier, Amgen, and Merck. NS declares to have received honoraria from Amgen. IB declares to have received honoraria from Sanofi; and to have received travel and accommodations expenses from Amgen, Sanofi, Merck and Servier. FS reported Personal financial interests, honoraria for advisory role, travel grants, research grants past 5 years: Hoffman La-Roche, Sanofi Aventis, Amgen, Merck Serono, Servier, Bristol-Myers Squibb. Institutional financial interests, honoraria due to investigator contribution in clinical trials from: Hoffman La-Roche, Sanofi Aventis, Amgen, Merck Serono, MSD, Boehringer Ingelheim, AbbVie, Array Pharmaceuticals, Pierre-Fabre, Novartis, Bristol-Myers Squibb, GlaxoSmithKline, Medimmune. JT reports personal financial interest in form of scientific consultancy role for Array Biopharma, AstraZeneca, Bayer, Boehringer Ingelheim, Chugai, Daiichi Sankyo, F. Hoffmann-La Roche Ltd, Genentech Inc, HalioDX SAS, Hutchison MediPharma International, Ikena Oncology, Inspirna Inc, IQVIA, Lilly, Menarini, Merck Serono, Merus, MSD, Mirati, Neophore, Novartis, Ona Therapeutics, Orion Biotechnology, Peptomyc, Pfizer, Pierre Fabre, Samsung Bioepis, Sanofi, Scandion Oncology, Scorpion Therapeutics, Seattle Genetics, Servier, Sotio Biotech, Taiho, Tessa Therapeutics and TheraMyc. Stocks: Oniria Therapeutics and also educational collaboration with Imedex/HMP, Medscape Education, MJH Life Sciences, PeerView Institute for Medical Education and Physicians Education Resource PER EE has received personal speaker honoraria from Organon and Novartis; and personal advisory board honoraria from Amgen, Bayer, Hoffman-La Roche, Merck Serono, Sanofi, Pierre Fabre, MSD, and Servier. Her institution has received research funding from Amgen Inc, Array Biopharma Inc, AstraZeneca Pharmaceuticals LP, BeiGene, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Debiopharm International SA, F. Hoffmann-La Roche Ltd, Genentech Inc, HalioDX SAS, Hutchison MediPharma International, Janssen-Cilag SA, MedImmune, Menarini, Merck Health KGAA, Merck Sharp & Dohme, Merus NV, Mirati, Novartis Farmacéutica SA, Pfizer, Pharma Mar, Sanofi Aventis Recherche & Développement, Servier, Taiho Pharma USA Inc. She held/holds non-remunerated roles as Coordinator of the SEOM +MIR Section of Residents and Young Assistants of the Sociedad Española de Oncología Médica SEOM, Speaker of the ESMO Academy of the European Society for Medical Oncology ESMO, and Volunteer member of the ASCO Annual Meeting Scientific Program Committee: Developmental Therapeutics – Immunotherapy of the American Society of Clinical Oncology ASCO.
The remaining author declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin (2014) 64(2):104–17. doi: 10.3322/caac.21220
3. Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol (2015) 16(1):64. doi: 10.1186/s13059-015-0620-6
4. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagés C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi: 10.1126/science.1129139
5. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMoa1500596
6. Kim JE, Chun SM, Hong YS, Kim K-P, Kim SY, Kim J, et al. Mutation burden and I index for detection of microsatellite instability in colorectal cancer by targeted next-generation sequencing. J Mol Diagn (2019) 21(2):241–50. doi: 10.1016/j.jmoldx.2018.09.005
7. Rousseau B, Foote MB, Maron SB, Diplas BH, Lu S, Argilés G, et al. The spectrum of benefit from checkpoint blockade in hypermutated tumors. N Engl J Med (2021) 384(12):1168–70. doi: 10.1056/NEJMc2031965
8. Bullock AJ, Grossman JE, Fakih MG, Lenz HJ, Gordon MS, Margolin K, et al. (2022). Botensilimab, a novel innate/adaptive immune activator, plus balstilimab (anti-PD-1) for metastatic heavily pretreated microsatellite stable colorectal cancer, in: Present 2022 ESMO World Congr Gastrointest Cancers, Barcelona, Spain, June 29-July 2, 2022.
9. Fakih MG, Sandhu J, Lim D, Li SM, Wang C. 320MO a phase I clinical trial of regorafenib, ipilimumab, and nivolumab (RIN) in chemotherapy resistant MSS metastatic colorectal cancer (mCRC). Ann Oncol (2022) 33:S684. doi: 10.1016/j.annonc.2022.07.458
10. Young J, Simms LA, Biden KG, Wynter C, Whitehall V, Karamatic R, et al. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings. Am J Pathol (2001) 159(6):2107–16. doi: 10.1016/S0002-9440(10)63062-3
11. Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol (1994) 145(1):148–56.
12. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair–Deficient/Microsatellite instability–high metastatic colorectal cancer. J Clin Oncol (2018) 36(8):773–9. doi: 10.1200/JCO.2017.76.9901
13. Diaz LA. Barriers to efficacy of immunotherapy for microsatellite-stable metastatic colorectal cancer. J Clin Oncol (2022).
14. Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat Rev Gastroenterol Hepatol (2019) 16(6):361–75. doi: 10.1038/s41575-019-0126-x
15. Tada K, Kitano S, Shoji H, Nishimura T, Shimada Y, Nagashima K, et al. Pretreatment immune status correlates with progression-free survival in chemotherapy-treated metastatic colorectal cancer patients. Cancer Immunol Res (2016) 4(7):592–9. doi: 10.1158/2326-6066.CIR-15-0298
16. Kobayashi M, Chung JS, Beg M, Arriaga Y, Verma U, Courtney K, et al. Blocking monocytic myeloid-derived suppressor cell function via anti-DC-HIL/GPNMB antibody restores the In vitro integrity of T cells from cancer patients. Clin Cancer Res (2019) 25(2):828–38. doi: 10.1158/1078-0432.CCR-18-0330
17. Kang JC, Chen JS, Lee CH, Chang JJ, Shieh YS. Intratumoral macrophage counts correlate with tumor progression in colorectal cancer. J Surg Oncol (2010) 102(3):242–8. doi: 10.1002/jso.21617
18. Pinto ML, Rios E, Durães C, Ribeiro R, Machado JC, Mantovani A, et al. The two faces of tumor-associated macrophages and their clinical significance in colorectal cancer. Front Immunol (2019) 10:1875. doi: 10.3389/fimmu.2019.01875
19. Zhong X, Chen B, Yang Z. The role of tumor-associated macrophages in colorectal carcinoma progression. Cell Physiol Biochem (2018) 45(1):356–65. doi: 10.1159/000486816
20. Sahin IH, Ciombor KK, Diaz LA, Yu J, Kim R. Immunotherapy for microsatellite stable colorectal cancers: Challenges and novel therapeutic avenues. Am Soc Clin Oncol Educ B (2022) 42(42):242–53. doi: 10.1200/EDBK_349811
21. Horii M, Matsushita T. Regulatory b cells and T cell regulation in cancer. J Mol Biol (2021) 433(1):166685. doi: 10.1016/j.jmb.2020.10.019
22. Mougiakakos D. Regulatory T cells in colorectal cancer: from biology to prognostic relevance. Cancers (Basel) (2011) 3(2):1708–31. doi: 10.3390/cancers3021708
23. Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discovery (2018) 8(6):730–49. doi: 10.1158/2159-8290.CD-17-1327
24. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature (2012) 487(7407):330–7. doi: 10.1038/nature11252
25. Sun X, Liu S, Wang D, Zhang Y, Li W, Guo Y, et al. Colorectal cancer cells suppress CD4+ T cells immunity through canonical wnt signaling. Oncotarget (2017) 8(9):15168–81. doi: 10.18632/oncotarget.14834
26. Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res (2019) 25(10):3074–83. doi: 10.1158/1078-0432.CCR-18-1942
27. Bugter JM, Fenderico N, Maurice MM. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat Rev Cancer (2021) 21(1):5–21. doi: 10.1038/s41568-020-00307-z
28. Tu J, Park S, Yu W, Zhang S, Wu L, Carmon K, et al. The most common RNF43 mutant G659Vfs*41 is fully functional in inhibiting wnt signaling and unlikely to play a role in tumorigenesis. Sci Rep (2019) 9(1):18557. doi: 10.1038/s41598-019-54931-3
29. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med (2015) 21(11):1350–6. doi: 10.1038/nm.3967
30. Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PloS One (2018) 13(1):e0191358. doi: 10.1371/journal.pone.0197008
31. Tumeh PC, Hellmann MD, Hamid O, Tsai KK, Loo KL, Gubens MA, et al. Liver metastasis and treatment outcome with anti-PD-1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res (2017) 5(5):417–24. doi: 10.1158/2326-6066.CIR-16-0325
32. Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE, et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med (2021) 27(1):152–64. doi: 10.1038/s41591-020-1131-x
33. Katz SC, Bamboat ZM, Maker AV, Shia J, Pillarisetty VG, Yopp AC, et al. Regulatory T cell infiltration predicts outcome following resection of colorectal cancer liver metastases. Ann Surg Oncol (2013) 20(3):946–55. doi: 10.1245/s10434-012-2668-9
34. Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M, et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: An open-label, dose-escalation, and dose-expansion phase ib trial (REGONIVO, EPOC1603). J Clin Oncol (2020) 38(18):2053–61. doi: 10.1200/JCO.19.03296
35. Huang XM, Zhang NR, Lin XT, Zhu CY, Zou YF, Wu XJ, et al. Antitumor immunity of low-dose cyclophosphamide: Changes in T cells and cytokines TGF-beta and IL-10 in mice with colon-cancer liver metastasis. Gastroenterol Rep (2020) 8(1):56–65. doi: 10.1093/gastro/goz060
36. Kim R, Imanirad I, Strosberg J, Carballido E, Kim D. PD-2 final result of phase IB study of regorafenib and nivolumab in mismatch repair proficient advanced refractory colorectal cancer. Ann Oncol (2021) 32:S199. doi: 10.1016/j.annonc.2021.05.020
37. Ghahremanifard P, Chanda A, Bonni S, Bose P. TGF-β mediated immune evasion in cancer-spotlight on cancer-associated fibroblasts. Cancers (Basel) (2020) 12(12):3650. doi: 10.3390/cancers12123650
38. Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res an Off J Am Assoc Cancer Res (2012) 18(5):1386–94. doi: 10.1158/1078-0432.CCR-11-2479
39. Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, Rodriguez-Cruz T, et al. BRAFV600E Co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol Res (2015) 3(6):602–9. doi: 10.1158/2326-6066.CIR-15-0030
40. Ischenko I, D’Amico S, Rao M, Li J, Hayman MJ, Powers S, et al. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat Commun (2021) 12(1):1482. doi: 10.1038/s41467-021-21736-w
41. van Maldegem F, Downward J. Mutant KRAS at the heart of tumor immune evasion. Immunity (2020) 52(1):14–6. doi: 10.1016/j.immuni.2019.12.013
42. Morris VK, Parseghian CM, Escano M, Johnson B, Raghav KPS, Dasari A, et al. Phase I/II trial of encorafenib, cetuximab, and nivolumab in patients with microsatellite stable, BRAF V600E metastatic colorectal cancer. J Clin Oncol (2022) 40(4 suppl):12. doi: 10.1200/JCO.2022.40.4_suppl.012
43. Corcoran R, Giannakis M, Allen J, Chen J, Pelka K, Chao S, et al. SO-26 clinical efficacy of combined BRAF, MEK, and PD-1 inhibition in BRAFV600E colorectal cancer patients. Ann Oncol (2020) 31(226–227):S226–7. doi: 10.1016/j.annonc.2020.04.041
44. Tabernero J, Ros J, Élez E. The evolving treatment landscape in BRAF-V600E –mutated metastatic colorectal cancer. Am Soc Clin Oncol Educ B (2022) 42(42):254–63. doi: 10.1200/EDBK_349561
45. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
46. Overman MJ, Kopetz S, McDermott RS, Leach J, Lonardi S, Lenz HJ, et al. Nivolumab ± ipilimumab in treatment (tx) of patients (pts) with metastatic colorectal cancer (mCRC) with and without high microsatellite instability (MSI-h): CheckMate-142 interim results. J Clin Oncol (2016) 34(15 suppl):3501. doi: 10.1200/JCO.2016.34.15_suppl.3501
47. Chen EX, Jonker DJ, Loree JM, Kennecke HF, Berry SR, Couture F, et al. Effect of combined immune checkpoint inhibition vs best supportive care alone in patients with advanced colorectal cancer. JAMA Oncol (2020) 6(6):831. doi: 10.1001/jamaoncol.2020.0910
48. Garralda E, Sukari A, Lakhani NJ, Patnaik A, Lou Y, Im SA, et al. A phase 1 first-in-human study of the anti-LAG-3 antibody MK4280 (favezelimab) plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. J Clin Oncol (2021) 39(15 suppl):3584. doi: 10.1200/JCO.2021.39.15_suppl.3584
49. Bendell J, Ulahannan SV, Chu Q, Patel M, George B, Auguste A, et al. Abstract 779: A phase I study of BI 754111, an anti-LAG-3 monoclonal antibody (mAb), in combination with BI 754091, an anti-PD-1 mAb: Biomarker analyses from the microsatellite stable metastatic colorectal cancer (MSS mCRC) cohort. Cancer Res (2020) 80(16 Supplement):779. doi: 10.1158/1538-7445.AM2020-779
50. Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol (2019) 20(6):849–61. doi: 10.1016/S1470-2045(19)30027-0
51. Kim RD, Kovari BP, Martinez M, Xie H, Sahin IH, Mehta R, et al. A phase I/Ib study of regorafenib and nivolumab in mismatch repair proficient advanced refractory colorectal cancer. Eur J Cancer (2022) 169:93–102. doi: 10.1016/j.ejca.2022.03.026
52. Fakih M, Raghav KPS, Chang DZ, Bendell JC, Larson T, Cohn AL, et al. Single-arm, phase 2 study of regorafenib plus nivolumab in patients with mismatch repair-proficient (pMMR)/microsatellite stable (MSS) colorectal cancer (CRC). J Clin Oncol (2021) 39(15 suppl):3560–0. doi: 10.1200/JCO.2021.39.15_suppl.3560
53. Barzi A, Azad NS, Yang Y, Tsao-Wei D, Rehman R, Fakih M, et al. Phase I/II study of regorafenib (rego) and pembrolizumab (pembro) in refractory microsatellite stable colorectal cancer (MSSCRC). J Clin Oncol (2022) 40(4 suppl):15. doi: 10.1200/JCO.2022.40.4_suppl.015
54. Cousin S, Cantarel C, Guegan JP, Gomez-Roca C, Metges JP, Adenis A, et al. Regorafenib-avelumab combination in patients with microsatellite stable colorectal cancer (REGOMUNE): A single-arm, open-label, phase II trial. Clin Cancer Res (2021) 27(8):2139–47. doi: 10.1158/1078-0432.CCR-20-3416
55. Saeed A, Park R, Dai J, Al-Rajabi RMT, Kasi A, Saeed A, et al. Phase II trial of cabozantinib (Cabo) plus durvalumab (Durva) in chemotherapy refractory patients with advanced mismatch repair proficient/microsatellite stable (pMMR/MSS) colorectal cancer (CRC): CAMILLA CRC cohort results. J Clin Oncol (2022) 40(4 suppl):135. doi: 10.1200/JCO.2022.40.4_suppl.135
56. Abrams TA, Kazmi SMA, Winer IS, Subbiah V, Falchook GS, Reilley M, et al. A phase 1b multitumor cohort study of cabozantinib plus atezolizumab in advanced solid tumors (COSMIC-021): Results of the colorectal cancer cohort. J Clin Oncol (2022) 40(4 suppl):121. doi: 10.1200/JCO.2022.40.4_suppl.121
57. Kim DW, Tan E, Zhou JM, Schell MJ, Martinez M, Yu J, et al. A phase 1/2 trial of ibrutinib in combination with pembrolizumab in patients with mismatch repair proficient metastatic colorectal cancer. Br J Cancer (2021) 124(11):1803–8. doi: 10.1038/s41416-021-01368-z
58. Gomez-Roca C, Yanez E, Im SA, Castanon Alvarez E, Senellart H, Doherty M, et al. LEAP-005: A phase II multicohort study of lenvatinib plus pembrolizumab in patients with previously treated selected solid tumors–results from the colorectal cancer cohort. J Clin Oncol (2021) 39(3 suppl):94. doi: 10.1200/JCO.2021.39.3_suppl.94
59. Lee MS, Loehrer PJ, Imanirad I, Cohen S, Ciombor KK, Moore DT, et al. Phase II study of ipilimumab, nivolumab, and panitumumab in patients with KRAS / NRAS / BRAF wild-type (WT) microsatellite stable (MSS) metastatic colorectal cancer (mCRC). J Clin Oncol (2021) 39(3 suppl):7. doi: 10.1200/JCO.2021.39.3_suppl.7
60. Martinelli E, Martini G, Famiglietti V, Troiani T, Napolitano S, Pietrantonio F, et al. Cetuximab rechallenge plus avelumab in pretreated patients with RAS wild-type metastatic colorectal cancer. JAMA Oncol (2021) 7(10):1529. doi: 10.1001/jamaoncol.2021.2915
61. Stein A, Binder M, Goekkurt E, Lorenzen S, Riera-Knorrenschild J, Depenbusch R, et al. Avelumab and cetuximab in combination with FOLFOX in patients with previously untreated metastatic colorectal cancer (MCRC): Final results of the phase II AVETUX trial (AIO-KRK-0216). J Clin Oncol (2020) 38(4 suppl):96. doi: 10.1200/JCO.2020.38.4_suppl.96
62. Antoniotti C, Borelli B, Rossini D, Pietrantonio F, Morano F, Salvatore L, et al. AtezoTRIBE: A randomised phase II study of FOLFOXIRI plus bevacizumab alone or in combination with atezolizumab as initial therapy for patients with unresectable metastatic colorectal cancer. BMC Cancer (2020) 20(1):683. doi: 10.1186/s12885-020-07169-6
63. Mettu NB, Ou FS, Zemla TJ, Halfdanarson TR, Lenz HJ, Breakstone RA, et al. Assessment of capecitabine and bevacizumab with or without atezolizumab for the treatment of refractory metastatic colorectal cancer. JAMA Netw Open (2022) 5(2):e2149040. doi: 10.1001/jamanetworkopen.2021.49040
64. Lenz HJ, Parikh AR, Spigel DR, Cohn AL, Yoshino T, Kochenderfer MD, et al. Nivolumab (NIVO) + 5-fluorouracil/leucovorin/oxaliplatin (mFOLFOX6)/bevacizumab (BEV) versus mFOLFOX6/BEV for first-line (1L) treatment of metastatic colorectal cancer (mCRC): Phase 2 results from CheckMate 9X8. J Clin Oncol (2022) 40(4 suppl):8. doi: 10.1200/JCO.2022.40.4_suppl.008
65. Morano F, Raimondi A, Pagani F, Lonardi S, Salvatore L, Cremolini C, et al. Temozolomide followed by combination with low-dose ipilimumab and nivolumab in patients with microsatellite-stable, O 6 -Methylguanine–DNA methyltransferase–silenced metastatic colorectal cancer: The MAYA trial. J Clin Oncol (2022) 40(14):1562–73. doi: 10.1200/JCO.21.02583
66. Fumet JD, Chibaudel B, Bennouna J, Borg C, Martin-Babau J, Cohen R, et al. 433P durvalumab and tremelimumab in combination with FOLFOX in patients with previously untreated RAS-mutated metastatic colorectal cancer: First results of efficacy at one year for phase II MEDITREME trial. Ann Oncol (2021), 32:S551. doi: 10.1016/j.annonc.2021.08.954
67. Grothey A, Tabernero J, Arnold D, De Gramont A, Ducreux MP, O’Dwyer PJ, et al. Fluoropyrimidine (FP) + bevacizumab (BEV) + atezolizumab vs FP/BEV in BRAFwt metastatic colorectal cancer (mCRC): Findings from cohort 2 of MODUL – a multicentre, randomized trial of biomarker-driven maintenance treatment following first-line induction th. Ann Oncol (2018) 29:viii714–5. doi: 10.1093/annonc/mdy424.020
68. Tabernero J, Melero I, Ros W, Argiles G, Marabelle A, Rodriguez-Ruiz ME, et al. Phase ia and ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J Clin Oncol (2017) 35(15 suppl):3002. doi: 10.1200/JCO.2017.35.15_suppl.3002
69. Segal NH, Cercek A, Ku G, Wu AJ, Rimner A, Khalil DN, et al. Phase II single-arm study of durvalumab and tremelimumab with concurrent radiotherapy in patients with mismatch repair–proficient metastatic colorectal cancer. Clin Cancer Res (2021) 27(8):2200–8. doi: 10.1158/1078-0432.CCR-20-2474
70. Parikh AR, Clark JW, Wo JYL, Yeap BY, Allen JN, Blaszkowsky LS, et al. A phase II study of ipilimumab and nivolumab with radiation in microsatellite stable (MSS) metastatic colorectal adenocarcinoma (mCRC). J Clin Oncol (2019) 37(15 suppl):3514. doi: 10.1200/JCO.2019.37.15_suppl.3514
71. Caballero-Baños M, Benitez-Ribas D, Tabera J, Varea S, Vilana R, Bianchi L, et al. Phase II randomised trial of autologous tumour lysate dendritic cell plus best supportive care compared with best supportive care in pre-treated advanced colorectal cancer patients. Eur J Cancer (2016), 64:167–74. doi: 10.1016/j.ejca.2016.06.008
72. Hazama S, Nakamura Y, Tanaka H, Hirakawa K, Tahara K, Shimizu R, et al. A phase II study of five peptides combination with oxaliplatin-based chemotherapy as a first-line therapy for advanced colorectal cancer (FXV study). J Transl Med (2014) 12(1):108. doi: 10.1186/1479-5876-12-108
73. Geevarghese SK, Geller DA, de Haan HA, Hörer M, Knoll AE, Mescheder A, et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther (2010) 21(9):1119–28. doi: 10.1089/hum.2010.020
74. André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-Instability-High advanced colorectal cancer. N Engl J Med (2020) 383(23):2207–18. doi: 10.1056/NEJMoa2017699
75. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
76. Turnis ME, Andrews LP, Vignali DAA. Inhibitory receptors as targets for cancer immunotherapy. Eur J Immunol (2015) 45(7):1892–905. doi: 10.1002/eji.201344413
77. Andrews LP, Marciscano AE, Drake CG, Vignali DAA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev (2017) 276(1):80–96. doi: 10.1111/imr.12519
78. Waight JD, Chand D, Dietrich S, Gombos R, Horn T, Gonzalez AM, et al. Selective FcγR Co-engagement on APCs modulates the activity of therapeutic antibodies targeting T cell antigens. Cancer Cell (2018) 33(6):1033–1047.e5. doi: 10.1016/j.ccell.2018.05.005
79. El-Khoueiry A, Bullock A, Tsimberidou A, Mahadevan D, Wilky B, Twardowski P, et al. 479 AGEN1181, an fc-enhanced anti-CTLA-4 antibody, alone and in combination with balstilimab (anti-PD-1) in patients with advanced solid tumors: Initial phase I results. J Immunother Cancer (2021) 9(Suppl 2):A509–9. doi: 10.1136/jitc-2021-SITC2021.479
80. Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: Effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res (2015) 21(7):1639–51. doi: 10.1158/1078-0432.CCR-14-2339
81. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity (2016) 44(3):609–21. doi: 10.1016/j.immuni.2016.01.024
82. Hellmann MD, Kim TW, Lee CB, Goh BC, Miller WH, Oh DY, et al. Phase ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann Oncol (2019) 30(7):1134–42. doi: 10.1093/annonc/mdz113
83. Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated reactivation of MAPK signaling contributes to insensitivity of BRAF-mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discovery (2012) 2(3):227–35. doi: 10.1158/2159-8290.CD-11-0341
84. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature (2012) 483(7387):100–3. doi: 10.1038/nature10868
85. Saeed A, Park R, Sun W. The integration of immune checkpoint inhibitors with VEGF targeted agents in advanced gastric and gastroesophageal adenocarcinoma: A review on the rationale and results of early phase trials. J Hematol Oncol (2021) 14(1):13. doi: 10.1186/s13045-021-01034-0
86. Ou DL, Chen CW, Hsu CL, Chung CH, Feng ZR, Lee BS, et al. Regorafenib enhances antitumor immunity via inhibition of p38 kinase/Creb1/Klf4 axis in tumor-associated macrophages. J Immunother Cancer (2021) 9(3):e001657. doi: 10.1136/jitc-2020-001657
87. Bourhis J, Stein A, Paul de Boer J, Van Den Eynde M, Gold KA, Stintzing S, et al. Avelumab and cetuximab as a therapeutic combination: An overview of scientific rationale and current clinical trials in cancer. Cancer Treat Rev (2021) 97:102172. doi: 10.1016/j.ctrv.2021.102172
88. Ferris RL, Lenz HJ, Trotta AM, García-Foncillas J, Schulten J, Audhuy F, et al. Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: Harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat Rev (2018) 63:48–60. doi: 10.1016/j.ctrv.2017.11.008
89. Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol (2013) 6(1):1. doi: 10.1186/1756-8722-6-1
90. Heinhuis KM, Ros W, Kok M, Steeghs N, Beijnen JH, Schellens JHM. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol (2019) 30(2):219–35. doi: 10.1093/annonc/mdy551
91. Apetoh L, Ladoire S, Coukos G, Ghiringhelli F. Combining immunotherapy and anticancer agents: the right path to achieve cancer cure? Ann Oncol (2015) 26(9):1813–23. doi: 10.1093/annonc/mdv209
92. Duffy AG, Greten TF. Immunological off-target effects of standard treatments in gastrointestinal cancers. Ann Oncol (2014) 25(1):24–32. doi: 10.1093/annonc/mdt349
93. Mettu NB, Niedzwiecki D, Boland PM, Fakih M, Arrowood C, Bolch E, et al. BACCI: A phase II randomized, double-blind, placebo-controlled study of capecitabine bevacizumab plus atezolizumab versus capecitabine bevacizumab plus placebo in patients with refractory metastatic colorectal cancer. J Clin Oncol (2018) 36(4 suppl):TPS873. doi: 10.1200/JCO.2018.36.4_suppl.TPS873
94. Crisafulli G, Sartore-Bianchi A, Lazzari L, Pietrantonio F, Amatu A, Macagno M, et al. Temozolomide treatment alters mismatch repair and boosts mutational burden in tumor and blood of colorectal cancer patients. Cancer Discovery (2022) 12(7):1656–75. doi: 10.1158/2159-8290.CD-21-1434
95. Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discovery (2019) 18(8):585–608. doi: 10.1038/s41573-019-0028-1
96. Abuodeh Y, Venkat P, Kim S. Systematic review of case reports on the abscopal effect. Curr Probl Cancer (2016) 40(1):25–37. doi: 10.1016/j.currproblcancer.2015.10.001
97. Rodriguez-Ruiz ME, Rodriguez I, Barbes B, Mayorga L, Sanchez-Paulete AR, Ponz-Sarvise M, et al. Brachytherapy attains abscopal effects when combined with immunostimulatory monoclonal antibodies. Brachytherapy (2017) 16(6):1246–51. doi: 10.1016/j.brachy.2017.06.012
98. Liu J, Fu M, Wang M, Wan D, Wei Y, Wei X. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J Hematol Oncol (2022) 15(1):28. doi: 10.1186/s13045-022-01247-x
Keywords: colorectal cancer, immunotherapy, tyrosine kinase inhibitors, liver metastases, microsatellite stable (MSS)
Citation: Ros J, Balconi F, Baraibar I, Saoudi Gonzalez N, Salva F, Tabernero J and Elez E (2023) Advances in immune checkpoint inhibitor combination strategies for microsatellite stable colorectal cancer. Front. Oncol. 13:1112276. doi: 10.3389/fonc.2023.1112276
Received: 30 November 2022; Accepted: 18 January 2023;
Published: 02 February 2023.
Edited by:
Andreas Seeber, Innsbruck Medical University, AustriaReviewed by:
Yoshiaki Nakamura, National Cancer Center Hospital East, JapanCopyright © 2023 Ros, Balconi, Baraibar, Saoudi Gonzalez, Salva, Tabernero and Elez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Elez, bWVlbGV6QHZoaW8ubmV0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.