 Hao Wang1
Hao Wang1 Kun Mi2*
Kun Mi2*- 1Breast Surgery, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, School of Medicine, University of Electronic Science and Technology of China, Chengdu, China
- 2Radiation Oncology Key Laboratory of Sichuan Province, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, School of Medicine, University of Electronic Science and Technology of China, Chengdu, China
Cellular plasticity is a well-known dynamic feature of tumor cells that endows tumors with heterogeneity and therapeutic resistance and alters their invasion–metastasis progression, stemness, and drug sensitivity, thereby posing a major challenge to cancer therapy. It is becoming increasingly clear that endoplasmic reticulum (ER) stress is a hallmark of cancer. The dysregulated expression of ER stress sensors and the activation of downstream signaling pathways play a role in the regulation of tumor progression and cellular response to various challenges. Moreover, mounting evidence implicates ER stress in the regulation of cancer cell plasticity, including epithelial–mesenchymal plasticity, drug resistance phenotype, cancer stem cell phenotype, and vasculogenic mimicry phenotype plasticity. ER stress influences several malignant characteristics of tumor cells, including epithelial-to-mesenchymal transition (EMT), stem cell maintenance, angiogenic function, and tumor cell sensitivity to targeted therapy. The emerging links between ER stress and cancer cell plasticity that are implicated in tumor progression and chemoresistance are discussed in this review, which may aid in formulating strategies to target ER stress and cancer cell plasticity in anticancer treatments.
1 Introduction
Cancer cell plasticity refers to the dynamic transition of cellular state that occurs during cancer initiation and progression (1, 2), which contributes to tumor heterogeneity and therapeutic resistance (3, 4). Epithelial-to-mesenchymal transition (EMT) and, the reversed process, mesenchymal-to-epithelial transition (MET) are the well-known forms of cellular plasticity, representing fundamental processes in the tumor invasion–metastasis cascade (5, 6). Epithelial–mesenchymal plasticity (EMP) encompasses EMT and MET, which are the key phenomena in tumor metastasis that are associated with cancer stem cell (CSC) generation and maintenance and therapeutic resistance (7–9), thereby posing a major challenge to effective therapy. Furthermore, CSCs exhibit epigenetic plasticity and therapeutic resistance, which contribute to cancer progression or relapse. Recent evidence also suggests that drug-resistant cells possess abnormal energetic and metabolic pathways that are involved in the induction, maintenance, and alteration of multidrug resistance (MDR) phenotype (10, 11).
The ER is a central organelle that facilitates protein synthesis, assembly, folding, and modification. The retention of unfolded or misfolded proteins within the ER lumen triggers the unfolded protein response (UPR), which leads to ER stress. Because of its roles in the regulation of multiple cancer cell functions, increasing evidence has linked ER stress to tumor progression (12, 13). ER stress has been shown to influence cancer cell proliferation, apoptosis, inflammatory response, and metastatic capacity (14). It has also been widely observed that when exposed to physiologic or pathologic stresses, cancer cells adopt various identities along a phenotypic spectrum, which results in cellular plasticity. However, the links between ER stress and cancer cell plasticity, such as EMP, MDR phenotype, CSC phenotype, and vasculogenic mimicry (VM) phenotype plasticity, have not been completely investigated, and new evidence is emerging. Here, we reviewed the roles of ER stress in cancer cell plasticity and the underlying molecular mechanisms.
2 Cellular plasticity in cancer
Cellular plasticity, which is observed during development, injury, and tumor progression, is a critical process that allows cells to assume distinct phenotypes to adapt to changing conditions (1, 15). Cellular plasticity is important in tumor proliferation, invasion, metastasis, and chemoresistance (16). Tumor cells can undergo phenotypic switch in response to cues from the surrounding microenvironment, such as EMP, CSC plasticity, drug resistance plasticity, and transdifferentiation, including VM (Figure 1).
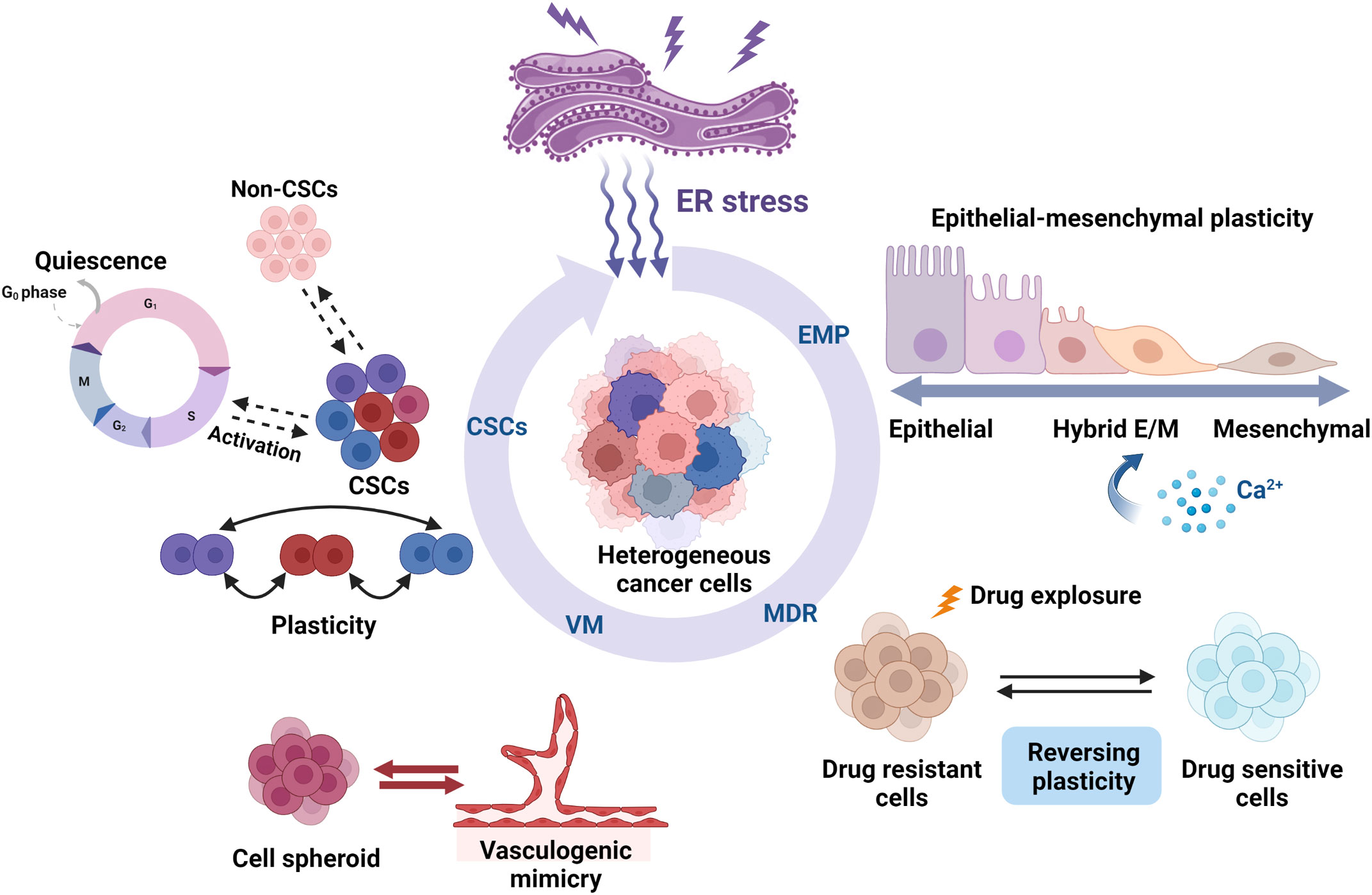
Figure 1 Roles of ER stress in the cellular plasticity of cancer cells. ER stress regulates cancer cell plasticity, including EMP, drug resistance phenotype, CSC phenotype, and VM phenotype plasticity. ER stress can regulate the EMP of cells that migrate along the EMT–MET axis, such as activation, inhibition, and pEMT induction. Several studies have found that ER stress influences the cell fate by promoting or suppressing the susceptibility to drug therapy. ER stress also plays an important role in regulating CSC differentiation and ratio and also regulates CSC plasticity, such as quiescence and activation. The interchange and coexistence of diverse phenotypes need to be studied further. VM is also another remarkable example of tumor cell plasticity. The activation of ER stress inhibits the formation of VM phenotypes in highly aggressive cells, thereby impeding tumor angiogenesis and progression. Created with BioRender.com.
2.1 EMP
Various human cancers exhibit plasticity between epithelial and mesenchymal states and the presence of EMT, MET, and hybrid epithelial/mesenchymal (E/M) or partial EMT (pEMT) phenotypes (17–20). EMT is defined as epithelial cells gradually losing epithelial characteristics while gaining motility and invasive characteristics of mesenchymal cells. MET is the reverse of EMT in that the cellular phenotype changes from mesenchymal cells to epithelial cells, thereby regaining apical–basal polarity. EMP refers to the ability of tumor cells to differentiate along the epithelial-mesenchymal spectrum and exhibit various intermediate hybrid E/M states (21, 22). As evidenced by pEMT, cells shift along the EMT-MET axis, which implies that EMT and MET are not binary fates (6). The underlying topographic map of EMT reveals a plethora of metastable hybrid phenotypic states, thus distinguishing stable epithelial and mesenchymal states (23). EMT, MET, and pEMT states can differ depending on tumor types, dissemination states, and the degrees of metastatic colonization or dormancy (7). In triple-negative breast cancer (TNBC), for example, EMP is a crucial mechanism that contributes to phenotype plasticity and heterogeneity, resulting in a heterogeneous clinical behavior associated with a poor prognosis (24). Primary breast CSCs of TNBC express epithelial and mesenchymal markers, thus indicating an EMP state (25). EMP is found in the majority of heterogeneous circulating tumor cell(CTC) phenotypes in the CTCs of patients with breast cancer (26).
In addition to the well-known transcriptional and post-transcriptional regulation that underpins EMP (27), plasticity is epigenetically regulated. Changing specific chromatin modifications demonstrates the roles of epigenetic regulators during EMP. In prostate cancer, for example, suppressing the epigenetic regulator chromatin remodeling proteinHMGA2 with a histone deacetylase (HDAC) inhibitor inhibits EMP and significantly reduces tumor growth and metastasis (28). HDAC, Methyl-CpG-binding domain 3 (Mbd3)/nucleosome remodeling and deacetylase (NuRD)complex, and Ten-eleven translocation 2 (Tet2) hydroxylase have all been identified as important regulators of EMP and metastasis in breast cancer (29).
Many functional proteins are involved in EMP regulation. The coxsackie and adenovirus receptor (CXADR), a tight junction protein, stabilizes AKT regulators and controls EMP in breast cancer (30). Syndecan-1, a cell surface proteoglycan, regulates EMP in tumor cells via nuclear translocation (31). Snail activation mediates EMP induction in prostate cancer (32). EMP regulators may also accommodate dynamic changes. The expression of the cell adhesion molecule CD44 is complex, with many isoforms, and the pattern of isoform expression changes during EMP transitions (33). Dynamic changes in the cohesin subunit RAD21 mediate chromatin architecture to control EMP for the regulation of cell fate in breast and gastric cancers (34). Furthermore, certain proteins can mediate the EMP process in a two-way manner. Secretary osteopontin (OPN) activates EMT to initiate cancer metastasis, whereas intracellular OPN (iOPN) induces MET to promote metastasis (35). Similarly, the kinesine-1 subunits kinesin family member 5B (KIF5B)/kinesin light chain 1 (KLC1) modulate the EMP process differently in breast cancer, with KIF5B being an inducer of EMT and KLC1 being its suppressor (36). Exosomes (37) and the classical transforming growth factor (TGF)β signaling pathway can also regulate tumor cell EMP (38, 39). Furthermore, the tumor microenvironment influences EMP regulation. Cancer-associated fibroblasts, for example, drive EMP and the formation of hybrid E/M states to induce invasive and metastatic tumor cell clusters (40). The EMP process also involves various phenotypic subtypes of macrophages in the tumor microenvironment (41).
EMP is a key mediator of metastatic dissemination and therapeutic resistance in several solid tumors. By restraining the EMP of tumor cells, researchers may be able to inhibit the progression of metastasis by facilitating an asymptomatic state of dormancy. A recent study has revealed that inhibition of spleen tyrosine kinase increases systemic tumor dormancy and, thus, reduces breast cancer metastasis (42).
2.2 Plasticity in drug resistance
According to emerging evidence, cancer therapies are hampered by reversible mechanisms that cause drug resistance. The plasticity of cancer cells drives their transformation to a phenotypic state that is not dependent on the original drug-responsive pathway. Because of intratumor heterogeneity and ongoing diversification in response to therapy, tumor cells survive the treatment and develop new resistant phenotypes (43). In melanoma, phenotype plasticity is a major cause for therapeutic resistance and is associated with increased levels of epidermal growth factor receptor (EGFR), receptor tyrosine kinaseAXL, or nerve growth factor receptor (NGFR), the expression of which is further upregulated by v-raf murine sarcoma viral oncogene homolog B1 (BRAF) inhibitors (44). Hence, therapeutic strategies could capitalize on this reversibility before relapse tumors develop genetic alterations that drive resistance. Furthermore, plasticity allows tumor cells to switch reversibly between drug resistance and drug sensitivity to escape and survive therapeutic challenges (45).
According to new research, a drug-tolerant population can switch between drug-sensitive and drug-tolerant states via non-genetic mechanisms, such as chromatin remodeling, and evolve into various resistant phenotypes (46). Plasticity in chemotherapy resistance is reflected in non-small cell lung cancer (NSCLC) by epigenetic alterations that allow tumor cells to adapt to new microenvironments after drug administration (47). Tamoxifen treatment causes acquired drug resistance in MCF7 breast cancer cells by altering the DNA methylation status (48). It has been demonstrated that epigenetic silencing of Spalt-like transcription factor 2 (SALL2) contributes to tamoxifen resistance in breast cancer by activating the AKT/mTOR pathway (49). Moreover, the ETS1/miR-23a-3p/ACSL4 axis may mediate sorafenib resistance via epigenetic regulation of ferroptosis in human hepatocellular carcinoma (50). Recent research provides more evidence demonstrating the crucial roles of epigenetic changes in regulating the resistant phenotype of tumor cells, which could serve as promising targets for overcoming clinical resistance. Furthermore, by activating cellular plasticity, tumor cells may be able to create a tumor-supportive microenvironment. The highly plastic cells in this microenvironment change dramatically to resist therapeutic drugs (51).
2.3 CSC plasticity
CSCs are a subpopulation of tumor cells capable of self-renewal and tumorigenesis. CSCs retain high stemness and plasticity (52), as evidenced by the processes of non-CSCs becoming CSCs, CSCs losing stemness, quiescent CSCs becoming active, and CSCs becoming quiescent. Non-CSCs in human basal breast cancers, for example, can acquire CSC phenotypes when zinc finger e-box binding homeobox 1 (ZEB1) is activated (53). Intravital lineage tracing in mammary tumors shows that existing CSCs disappear and new CSCs are formed during mammary tumor growth, thereby demonstrating the dynamic nature of plasticity in these cells (54). Cells expressing CSC-associated markers in glioblastoma do not represent a clonal entity but rather a plastic state that most cancer cells can adapt in response to microenvironmental signals (55). The dynamic coexistence of various phenotypes or states in CSCs is becoming common in various tumor contexts. Malignant cells in glioblastoma exist in four major cellular states that can interconvert and exhibit plasticity, which drives intratumoral heterogeneity (56). CSCs have been shown to exhibit three interchangeable phenotypes in breast cancer, namely, ALDH+, CD44+CD24−, and ALDH+CD44+CD24− CSCs, which indicates the plasticity and heterogeneity of CSCs (57).
CSCs can enter a dormant cellular state and exist in the G0 phase, which makes them resistant to conventional therapies that target actively dividing cells. Quiescence can be induced by altered microenvironmental cues or drug treatments. Breast cancer disseminated tumor cells (BC DTCs) may be instructed to enter dormancy by bone marrow NG2+/Nestin+ mesenchymal stem cells. When homeostasis of the bone marrow microenvironment changes, BC DTCs may emerge and cause a bone relapse (58). Laminin-332, as a component of the human hepatic CSC niche, plays a role in sustaining cell stemness and confers chemoresistance and quiescence (59). One understudied chemoresistance mechanism is the induction of quiescence. Nuclear factor of activated T cells cytoplasmic 4 (NFATC4) drives a quiescent phenotype in ovarian cancer and promotes chemotherapy resistance in vitro and in vivo (60). Apart from resistance to therapy, epigenetic determinants play an important role in CSC dormancy (61). SET domain-containing protein 4 (SETD4) alters heterochromatin formation to epigenetically regulate CSC quiescence in breast cancer (62). Alterations in cell states and switches to a dormant or quiescent state are major impediments to standard therapy (63).
Other studies indicate that quiescent CSCs can be reactivated under favorable conditions (64, 65). Transfer of mitochondrial DNA from extracellular vesicles acts as an oncogenic signal, potentially promoting the emergence of dormant cancer stem-like cells (66). Nuclear protein DEK is required for CSC activation in breast tumors as it upregulates cellular activation-related genes, including MYC targets (67). Understanding the mechanism underlying the activation of quiescent CSCs may lead to novel therapeutic strategies for overcoming quiescence-linked chemoradiotherapy resistance.
2.4 Plasticity in VM
VM is another remarkable example of tumor cell plasticity. VM is a functional microcirculation structure that is independent of endothelial vessels and describes the plasticity of highly aggressive tumor cells to develop vasculogenic-like, matrix-rich networks, thus mimicking endothelial cell activities and providing blood supply for tumor growth and metastasis (68, 69). EMT, which is based on EMP, plays a crucial role in the formation of VM during cancer progression. TGF-β1/ROCK signaling contributes to the formation of VM in hepatocellular carcinoma by inducing EMT (70). In ovarian carcinoma (71) and salivary adenoid cystic carcinoma, hypoxia may promote VM formation by inducing EMT (72). Dickkopf-1 promotes VM formation in NSCLC by increasing the expression of EMT-associated proteins (73). VM formation is influenced by several known EMP regulators, including ZEB1 (74), Twist1 (75), Snail (76), and Slug (77). The discovery of mechanisms underlying VM plasticity will shed light on the search for more precise targets in antiangiogenic treatment.
3 ER stress and cancer cell plasticity
According to emerging evidence, ER stress appears to play an important role in regulating cellular plasticity. Chronic ER stress promotes immunosuppressive phenotypes of immune cells in various diseases, such as cancer and inflammation (78) (79). inositol-requiring enzyme 1α (IRE1α)−X-Box Binding Protein 1 (XBP1) signaling, for example, promotes tumor immune evasion by enhancing the functions of tumor-associated myeloid cells (80, 81). Furthermore, thapsigargin-induced ER stress increases interleukin(Il)-10 transcription and promotes T cell phenotype plasticity (82). Recent studies in tumor cells show a comprehensive relationship between ER stress and cancer cell plasticity (Figure 1), including EMP, drug resistance phenotype, CSC phenotype, and VM phenotype plasticity. In the following sections, the emerging roles of ER stress in the regulation of tumor cell plasticity and the underlying mechanisms have been discussed.
3.1 ER stress and EMP
In recent years, the modulatory role of ER stress in EMP in various types of tumors has been studied. Increased ER stress and EMT, for example, have been linked to chemoresistance and poor survival in patients with lung cancer. ER stress caused by the activates of valosin-containing protein disrupt the EMT-like state and promote the migratory and invasive abilities of lung cancer (83). By inducing ER stress, IL-32 promotes EMT in human lung adenocarcinoma cells (84). The downstream signaling of the ER stress sensor IRE1α acts as an EMT regulator (Figure 2A). IRE1α promotes lung cancer progression and EMT via XBP1 mRNA splicing (85). Furthermore, IRE1α promotes miR-200 degradation in an IRE1-dependent decay (RIDD)-dependent manner (86), thereby leading to the depression of epithelial gene transcriptional repressors (Figure 2A) (87). Furthermore, sXBP1 is linked to the enhanced mesenchymal phenotypes of tumor cells. The IRE1–sXBP1 axis may be activated in response to stressful extracellular conditions that cause ER stress and regulate the expression of EMT transcription factor. The interaction of lysyl oxidase-like 2 (LOXL2) and Heat Shock Protein Family A (Hsp70) Member 5 (HSPA5) in the ER activates IRE1–XBP1 signaling and induces the expression of EMT markers in an XBP1-dependent manner (Figure 2A) (88).
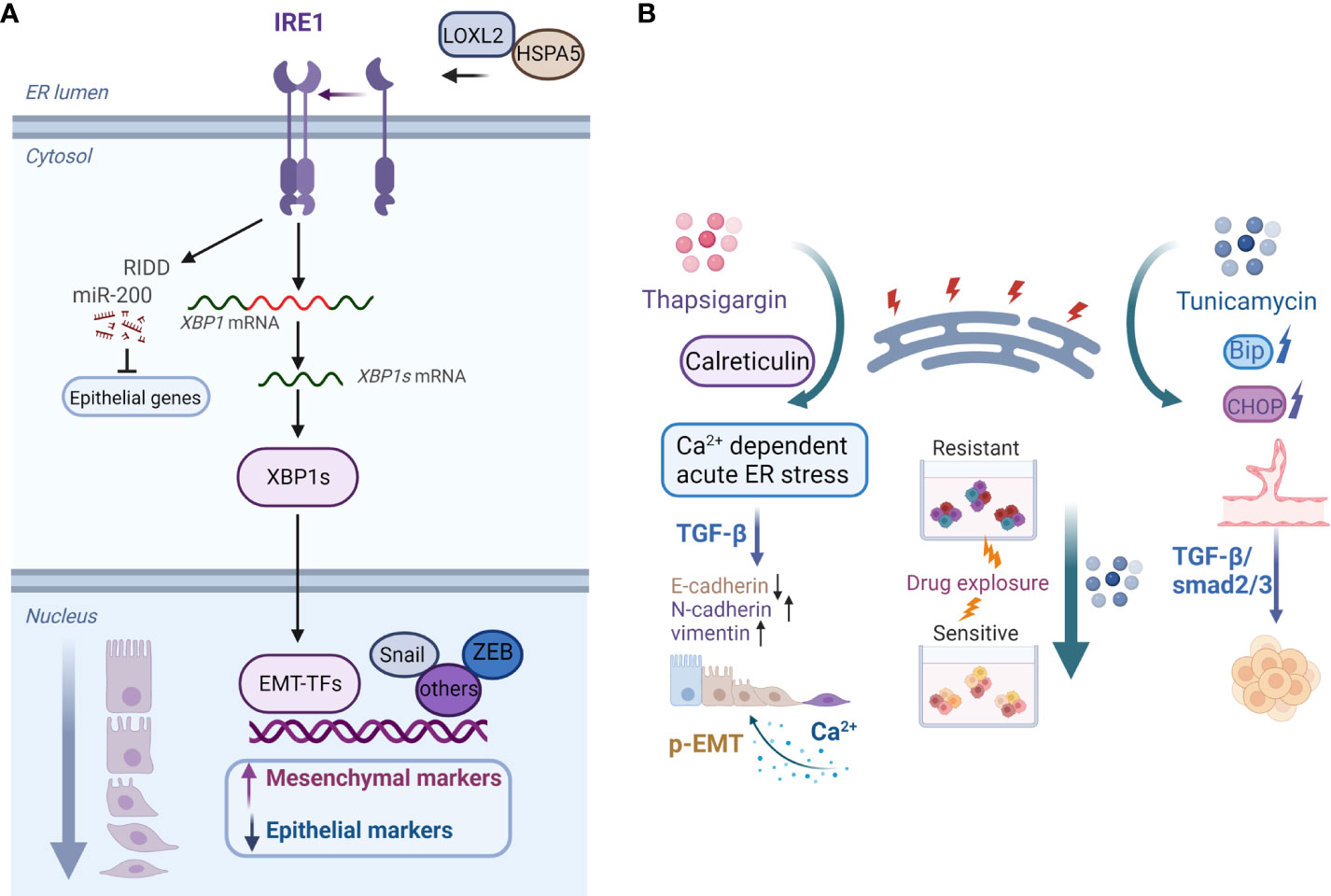
Figure 2 (A) Model of the IRE1–XBP1 axis that regulates EMP. Activation of the IRE1–XBP1 signaling pathway induces the expression of EMT transcription factors, which are direct transcriptional targets of XBP1. Additionally, IRE1α promotes miR-200 degradation via the RIDD process, which results in the derepression of epithelial gene transcriptional repressors. ER accumulation of LOXL2 interacts with HSPA5, activating the IRE1–XBP1 signaling pathway and inducing EMT. (B) Examples of roles of the ER stress inducers thapsigargin and tunicamycin in cancer cell plasticity regulation. Calreticulin promotes TGF-β-induced EMT by repressing E-cadherin and inducing N-cadherin and vimentin. Calreticulin induces EMT via Ca2+-dependent thapsigargin-induced acute ER stress. Moreover, prolonged calcium signaling induces pEMT in carcinoma cells. On the contrary, tunicamycin-induced ER stress inhibits the chemoresistance of hypopharyngeal carcinoma cells in 3D cultures. Furthermore, activation of ER stress inhibits the formation of VM phenotypes in TNBC cells via the TGF-β1/Smad2/3 signaling pathways.Created with BioRender.com.
ER stress induces the expression of cyclase-associated protein 2 (CAP2) and promotes EMT via the activation of Rac1 and ERK in liver cancer cells (89). In hepatocellular carcinoma cells, the expression of hepatitis B virus surface antigen induces ER stress, which increases the expression and secretion of fibroblast growth factor 19 (FGF19) to activate JAK2/STAT3 signaling and induce EMT (90). Additionally, as key players of UPR, activating transcription factor 6 (ATF6) upregulation and ATF4 downregulation activates PI3K/AKT/mTOR signaling but reduces Bone Morphogenetic Protein 2 (BMP2) signaling in colorectal cancer cells to enhance motility and invasion via EMT (91). ER stress also promotes the overexpression of T-synthase-specific molecular chaperone Cosmc in human colorectal cancer cells, which significantly enhances cell migration and invasion via activation of EMT (92). In squamous cell carcinomas, ER stress triggers the ectopic expression of Transmembrane and tetratricopeptide repeat containing protein 3 (TMTC3), which activates the GRP78/Protein kinase RNA-like ER kinase (PERK) signaling and increases the expression of EMT markers via an interleukin-like EMT inducer (93). ER stress suppression decreases the metastatic capacities of TNBC cells by inhibiting the Syntenin/SOX4/Wnt/β-catenin pathway, whereas heat shock protein A4 overexpression reverses these effects (94). Chemotherapeutic drugs commonly used to treat patients, such as cisplatin, gemcitabine, vinorelbine, and doxorubicin, also activate ER stress, which enhances EMT and proliferative phenotypes in cancer cells (95) (96).
In noncancerous cells, ER stress has been shown to exert a direct effect on EMP. ER stress, for example, induces EMT and, thus, increases the migration of lens epithelial cells (97). ER stress inhibits cell differentiation, downregulates the expression of cadherin-1 and cadherin-16, and upregulates the expression of vimentin and SNAI1, thereby indicating the loss of epithelial features and a shift toward a mesenchymal phenotype in thyroid cells (98). Alveolar epithelial cells undergo ER stress in a hypoxic microenvironment, which is accompanied by the increased expression of mesenchymal markers both in vivo and in vitro (99). In alveolar epithelial cells, ER stress causes a decrease in the expression of epithelial markers E-cadherin and Zonula occludens-1 and an increase in the expression of mesenchymal markers S100A4 and α-smooth muscle actin (100). EMT exerts a significant effect on pulmonary fibrosis. EMT is induced in human lung epithelial cells after treatment with the ER stress inducers tunicamycin and bleomycin via HDAC upregulation (101). Furthermore, advanced oxidation protein products activate ER stress in proximal tubular cells and induce EMT, as evidenced by p27 and α-SMA overexpression and E-cadherin downregulation in chronic kidney disease (102). Protein arginine methyltranferase-1 (PRMT1) causes ER stress and EMT in renal tubular epithelial cells as well (103).
ER stress and EMP may also regulate one another, according to research findings. Inducing EMT makes cells more vulnerable to ER stress by activating the PERK–eIF2α axis of the UPR. Moreover, PERK–eIF2α signaling is pivotal for maintaining ER homeostasis and is required for EMT cells to disseminate (104). ER stress in colorectal cancer cells is dependent on ZEB-1 induction. Colorectal cancer cells could not mount ER stress in response to microenvironmental stimuli in the absence of ZEB-1 (105).
However, apart from the usual role of ER stress in promoting EMT, some studies have reported the inhibition of EMT by ER stress. Chemical induction of ER stress inhibits EMT and migration in retinal pigment epithelial cells possibly by inhibiting TGF-β signaling (106). Honokiol-induced ER stress markedly inhibits histone deacetylase-3 expression and blocks EMP and metastatic dissemination in gastric cancer (107). Furthermore, melatonin induces ER stress and inhibits EMT via calpain-mediated C/EBP-β and NF-κB cleavage in gastric cancer (108). In human glioblastoma cells, sinomenine hydrochloride triggers ER stress, reverses endogenous and exogenous EMT, and inhibits migration and invasion. When ER stress is suppressed, the inhibition of mesenchymal markers (vimentin, Snail, and Slug) is abolished (109). In addition, a recent study shows that metabolism affects the ER stress and modulates EMT. In breast cancer cells, TGF-β-induced EMT could be suppressed by ER stress in response to cholesterol accumulation in the ER (110).
Additionally, ER stress is involved in the MET process. Hyperactivated ER stress is a significant reprogramming barrier that prevents the initial MET step to form induced pluripotent stem cells (iPSCs) from mesenchymal somatic cells (111). Adenosine Deaminase Acting on RNA (ADAR)1-dependent RNA editing could promote MET and induce iPSC reprogramming by alleviating ER stress (112). Overexpression of the ER secretion factor ER protein 29 in breast cancer cells results in enhanced MET phenotypes, including stress fiber loss, E-cadherin upregulation, and vimentin downregulation (113). The relationship between ER stress and EMP remains unknown, particularly in the process of cells shifting along the EMT-MET axis, as evidenced by intermediate hybrid E/M states.
3.2 ER stress and chemoresistance
The role of ER stress in promoting or counteracting cancer cell chemoresistance is debatable. There is no agreement on the relationship between ER stress and the development of drug-resistant phenotype in cancer cells. According to some studies, activating ER stress restores chemosensitivity, whereas contradictory results have been reported by other studies. Several investigations have highlighted the role of UPR in the determination of cell fate by either increasing or decreasing the susceptibility of cancer cells to chemotherapy drugs (114). Chemotherapeutic resistance caused by ER stress is common in aggressive tumors. One of the most important mechanisms promoting MDR development is ER stress. In cancer cells, ER stress adaptation results in an MDR phenotype with increased expression of the UPR sensor protein kinase PERK, which mediates Nuclear factor erythroid-derived 2-like 2 (Nrf2)-driven transcription of MDR related protein 1 (MRP1). Silencing PERK signaling inhibits tumor growth and enhances the susceptibility of tumor xenografts to chemotherapy (115). Adaptation to ER stress also improves DNA repair and damage tolerance, thereby increasing the resistance of stressed cancer cells to chemotherapeutics (96). ER stress increases chemoresistance in colon cancer cells by activating eukaryotic initiation factor 2 (eIF2)/ATF4 signaling (116). Moreover, by alleviating ER stress, astragaloside IV sensitizes NSCLC cells to cisplatin (117). Furthermore, ER stress upregulates the expression of the ZNF263–ARHGEF2 pathway, which contributes to ER stress-related treatment resistance (118). When exposed to ER stress, nasopharyngeal carcinoma cells secrete endoplasmic reticulum resident protein 44(ERp44)-containing exosomes, which boost the chemoresistance of neighboring cells (119). Induction of UPR promotes glioma cell metabolism and chemoresistance (120). Moreover, downregulation of ER stress response inhibits autophagy and overcomes temozolomide resistance in melanoma cells (121).
On the contrary, ER stress can counteract cancer cell chemoresistance and mediate cell apoptosis. Tunicamycin, for example, significantly increases chemotherapy-induced apoptosis by inducing ER stress in multidrug-resistant gastric cancer cells (122). Accordingly, our previous study found that tunicamycin-induced ER stress reduces the chemoresistance of hypopharyngeal carcinoma cells both in vitro and in vivo (Figure 2B) (123). WW domain containing oxidoreductase (WWOX) makes epithelial ovarian cancer cells more sensitive to paclitaxel via ER stress-induced apoptosis (124). Betulinic acid treatment increases GRP78-dependent ER stress and exerts chemosensitizing effects in breast cancer (125). transmembrane 9 superfamily 4 (TM9SF4) knockdown increases ER stress, reduces cell growth, and induces cell death in chemoresistant breast cancer cells (126). Furthermore, PERK activation induces ER stress and improves the chemosensitivity to taxol treatment in colorectal cancer cells (127), and a combination of 5-FU and withaferin-A upregulates the expression of ER stress sensors and induces PERK axis-mediated apoptosis (128). Hence, a thorough understanding of the opposing roles of ER stress in regulating the drug resistance and sensitivity of cancer cells may have significant implications for the selection of different treatment strategies.
3.3 ER stress and CSCs
ER stress plays a crucial role in regulating the functions of stem-like cells. Endodermal differentiation of mouse embryonic stem cells could be induced by the ER stress-inducing agents thapsigargin and tunicamycin (129). ER stress response promotes BMP9-induced bone formation and matrix mineralization in mesenchymal stem cells (130). According to research findings, UPR activates ER stress, which causes rapid loss of stemness in intestinal epithelial cells (IECs) (131). A recent study found that excessive ER stress causes apoptosis in intestinal epithelial stem cells, thereby resulting in aggravated colitis (132). Another study made a similar observation in IECs. ER stress is induced during the transition from stem cells to transit-amplifying cells and mediates stem cell loss in a PERK–eIF2α–dependent manner. Similarly, ER stress disrupts Wnt signaling downstream of nuclear β-catenin, which causes the death of Apc-mutated intestinal epithelial stem cells (133). Furthermore, XBP1 has been shown to decrease the stemness of IECs (134).
In tumors, ER proteostasis is important for maintaining CSC integrity. In breast cancer, stem-like cells express high levels of ER-associated p97, the loss of which activates UPR and alters the expressions of multiple stemness-associated genes, thus leading to the demise of CSCs (135). ER stress also activates UPR, which promotes the differentiation of colon CSCs, thus leading to enhanced chemosensitivity (136). For instance, overexpression of ATF6 and XBP1 reduces the proliferation and stemness of colorectal cancer cells by activating PERK signaling (137). Moreover, brefeldin A, an inducer of ER stress in eukaryotic cells, inhibits CSC-like properties in colorectal (138) and breast cancer cells (139). Interestingly, CSCs are sensitive to the mitochondrial targeting antibiotic doxycycline, which induces ATF4-mediated ER stress and leads to apoptosis selectively in the cancer stem-like cells (140). Furthermore, the adaptation to ER stress drives the malignancy and drug resistance of tumor cells. Study of the relationship between CSCs and adaptation to ER stress has revealed that the proportion of apoptosis-resistant CSCs is elevated in ER stress-resistant melanoma. Similarly, Homeobox B9 (HOXB9) regulates the self-renewal of CSCs and antagonizes ER stress-induced apoptosis by modulating the miR–765–FOXA2 axis in melanoma cells (141). Therefore, apart from the role of ER stress in CSC differentiation and apoptosis, mechanisms involving ER stress regulation on CSC plasticity, such as quiescence and activation, and the interchange of coexistence of diverse phenotypes need to be further investigated.
3.4 ER stress and angiogenesis and VM
Evidence suggests that ER stress alters the expression and activity of vascular growth factors, thereby modulating the functions of vascular endothelial cells and tumor angiogenesis (142–145). Hepatitis B virus small envelope protein-induced ER stress activates UPR signaling, thereby increasing the expression and secretion of vascular endothelial growth factor (VEGF) A and, consequently, the angiogenic capacity of hepatocellular carcinoma cells (146). In malignant glioma, IRE1 is a critical regulator of tumor angiogenesis and metastasis. Inhibiting IRE1α is associated with a decrease in proangiogenic cytokines, such as VEGFA, IL-1beta, IL-6, and IL-8, and an increase in antiangiogenic gene transcripts (147). In collaboration with hypoxia-inducing factor 1α (HIF1α), XBP1s drive the angiogenesis and progression of TNBC as the downstream transcription factor of IRE1α (148). However, ER stress has been shown to drive antiangiogenic responses. ER stress-induced miR-153 expression in breast cancer cells activates IRE1α and XBP1, which inhibits HIF1α expression and tumor angiogenesis by decreasing VEGFA production (149). Neuronal ER stress inhibits myeloid cell-induced vascular regeneration by promoting the degradation of IRE1α-dependent netrin-1 (150). Moreover, C/EBP homologous protein-10 (CHOP-10) can activate an antiangiogenic response in postnatal neovascularization under ER stress (151). By activating p38-mediated ER stress, low-intensity pulsed ultrasound increases apoptosis and inhibits angiogenesis in endothelial cells (152).
The majority of studies linking ER stress to tumor angiogenesis have focused on VEGF and other factors with vasomodulatory properties in angiogenic cascades modulated by the UPR. However, the influences of ER stress on the VM phenotypes of cancer cells remain poorly understood. TNBC cells with mesenchymal phenotypes form tubular VM networks in three-dimensional (3D) matrigel cultures. For the first time, Liu et al. reported that the activation of ER stress reduces VM phenotypes in TNBC cells via regulation of TGF-β1/Smads and β-catenin signaling pathways (Figure 2B) (153). As a potential transdifferentiation event indicative of the unique capability of certain aggressive tumor cells associated with EMT and stemness, VM plasticity has profound implications in tumor progression. Therefore, the potential regulatory mechanisms involved in ER stress and VM should be clarified to facilitate the development of targeted therapies that prevent tumor angiogenesis and consequently impede tumor progression.
3.5 ER stress, calcium signaling, and EMP
The ER must maintain a tightly controlled oxidizing and Ca2+-rich folding environment for protein synthesis, folding, and modification. ER-resident chaperones, such as immunoglobulin binding protein, calreticulin, calnexin, and protein disulfide isomerases, play critical roles in ER protein folding and Ca2+ buffering. ER-Ca2+ depletion may disrupt ER homeostasis and the balance between protein folding load and capacity (154). Many aspects of tumor activity, including proliferation, angiogenesis, invasion, EMT, and drug resistance, involve calcium signaling pathways (155). TGF-β-induced EMT is associated with alterations in ER calcium homeostasis in human breast cancer cells (156). Calreticulin is an ER-resident multifunctional protein that promotes TGF-β-induced EMT (Figure 2B) (157). Moreover, the effect of cyclophilin B (CypB) regulation on Slug expression in renal tubular epithelial cells is dependent on its interaction with calreticulin and calreticulin-dependent calcium signaling in the ER lumen (158). Another study found that the ER transmembrane protein transmembrane and coiled-coil domains 1 (TMCO1) is important for maintaining calcium homeostasis, promoting EMT in human gliomas, and inducing cell migration and invasion (159). Furthermore, EMT remodels Ca2+ influx in breast cancer cells, possibly by changing the functions of the store-operated Ca2+ channel poreforming subunit ORAI1 and transient receptor potential canonical type 1 (TRPC1) channels (160). In terms of the regulation of specific mesenchymal markers, ER ATPase inhibitor thapsigargin is an inducer of vimentin in breast cancer cells, which involves store-operated Ca2+ entry (161). Calreticulin induces EMT in pancreatic cancer via intracellular free Ca2+-dependent, thapsigargin-induced acute stress and IRE1α-mediated chronic ER stress (Figure 2B) (162).
EMP-associated hybrid E/M or pEMT states are distinct from classical EMT and could confer unique malignant properties to tumor cells (17, 22, 163). Recent studies have revealed the links between calcium signaling and pEMT (Figure 2B). Prolonged calcium signaling induces pEMT in carcinoma cells, which is accompanied by the internalization of membrane-associated E-cadherin and other epithelial proteins and an increase in cellular migration and invasion (164). Tumor cells within a mesenchymal state have more intracellular calcium, and ER, as one of the Ca2+ stores in cells, might play a role to facilitate intracellular Ca2+ reaching levels sufficient for P-EMT. However, the point of convergence of the P-EMT and complete EMT transitions remains unclear. It was reported that ER stressor thapsigargin increased cytosolic Ca2+ concentration, while it was also shown to increase levels of active TGF-β1 (165). It is possible that Ca2+ dysregulation induced the mesenchymal transformation of cells and then TGF-β1 activation leads to a complete EMT response. Thus, extracellular signals prompt an increase in Ca2+ flux, and the release of Ca2+ from ER stores mediates EMT in multiple ways. Therefore, exploring the potential therapeutic benefits of targeting Ca2+ signaling to block EMP in cancer cells could provide a novel complement to standard therapies.
4 Discussion
Cellular plasticity has emerged as a well-recognized mode of therapeutic resistance in various cancers in recent years. Tumor progression, metastasis, and drug resistance are driven by cellular plasticity. The mechanisms governing this cell state switch have also been elucidated. Persistent ER stress, an emerging cancer hallmark, is caused by various factors that disrupt ER homeostasis in malignant cells. It is well known that unresolved ER stress promotes tumor cell malignancy and drug resistance, thus contributing to the acquisition of EMP, stemness, and drug resistance plasticity to promote tumor progression.
However, our understanding of the interactions between ER stress and cancer cell plasticity and the levels and specificities of regulation for a specific type of plasticity is still limited and warrants further investigation. Decoding how the ER stress pathway regulates cell plasticity is a major challenge for researchers and necessitates defining the rationale for drug design and application. Deciphering the molecular connections between ER stress and cancer cell plasticity will definitely contribute to the development of new therapeutic strategies that, when combined with existing anticancer treatments, will provide better clinical responses in patients.
Author contributions
HW prepared figures and wrote the first draft of the manuscript. KM conceptualized and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by The National Natural Science Foundation of China (grant no. 81872402) and grants from Sichuan Science and Technology Department Key Research and Development Project Fund (grant no. 2022YFS0215), Sichuan Natural Science Foundation (grant no.2022NSFSC0707) and Medical Science and Technology Project of Sichuan Health Commission (grant no. 21PJ114).
Acknowledgments
Authors thank other members of KM’s laboratory for their critical reading of the manuscript. We apologize to those investigators whose work has not been cited in this review due to space constraints.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yuan S, Norgard RJ, Stanger BZ. Cellular plasticity in cancer. Cancer Discov (2019) 9:837–51. doi: 10.1158/2159-8290.CD-19-0015
2. Marjanovic ND, Hofree M, Chan JE, Canner D, Wu K, Trakala M, et al. Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell (2020) 38:229–246.e13. doi: 10.1016/j.ccell.2020.06.012
3. Jung E, Osswald M, Ratliff M, Dogan H, Xie R, Weil S, et al. Tumor cell plasticity, heterogeneity, and resistance in crucial microenvironmental niches in glioma. Nat Commun (2021) 12:1014. doi: 10.1038/s41467-021-21117-3
4. Boumahdi S, de Sauvage FJ. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov (2020) 19:39–56. doi: 10.1038/s41573-019-0044-1
5. Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol (2019) 29:212–26. doi: 10.1016/j.tcb.2018.12.001
6. Bakir B, Chiarella AM, Pitarresi JR, Rustgi AK. EMT. MET, plasticity, and tumor metastasis. Trends Cell Biol (2020) 30:764–76. doi: 10.1016/j.tcb.2020.07.003
7. Lambert AW, Weinberg RA. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer (2021) 21:325–38. doi: 10.1038/s41568-021-00332-6
8. Williams ED, Gao D, Redfern A, Thompson EW. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat Rev Cancer (2019) 19:716–32. doi: 10.1038/s41568-019-0213-x
9. Garg M. Emerging roles of epithelial-mesenchymal plasticity in invasion-metastasis cascade and therapy resistance. Cancer Metastasis Rev (2022) 41:131–45. doi: 10.1007/s10555-021-10003-5
10. Lettnin AP, Wagner EF, Carrett-Dias M, Dos Santos Machado K, Werhli A, Cañedo AD, et al. Silencing the OCT4-PG1 pseudogene reduces OCT-4 protein levels and changes characteristics of the multidrug resistance phenotype in chronic myeloid leukemia. Mol Biol Rep (2019) 46:1873–84. doi: 10.1007/s11033-019-04639-4
11. Alexa-Stratulat T, Pešić M, Gašparović AČ, Trougakos IP, Riganti C. What sustains the multidrug resistance phenotype beyond ABC efflux transporters? looking beyond the tip of the iceberg. Drug Resist Update (2019) 46:100643. doi: 10.1016/j.drup.2019.100643
12. Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer (2021) 21:71–88. doi: 10.1038/s41568-020-00312-2
13. Limia CM, Sauzay C, Urra H, Hetz C, Chevet E, Avril T. Emerging roles of the endoplasmic reticulum associated unfolded protein response in cancer cell migration and invasion. Cancers (2019) 11:631. doi: 10.3390/cancers11050631
14. Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. BioMed Pharmacother (2019) 118:109249. doi: 10.1016/j.biopha.2019.109249
15. Tata PR, Rajagopal J. Cellular plasticity: 1712 to the present day. Curr Opin Cell Biol (2016) 43:46–54. doi: 10.1016/j.ceb.2016.07.005
16. Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic plasticity: Driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell (2019) 24:65–78. doi: 10.1016/j.stem.2018.11.011
17. Kröger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL, et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci U.S.A. (2019) 116:7353–62. doi: 10.1073/pnas.1812876116
18. Ungefroren H, Thürling I, Färber B, Kowalke T, Fischer T, De Assis LVM, et al. The quasimesenchymal pancreatic ductal epithelial cell line PANC-1-A useful model to study clonal heterogeneity and EMT subtype shifting. Cancers (2022) 14:2057. doi: 10.3390/cancers14092057
19. Zhu N, Xu X, Wang Y, Zeng M-S, Yuan Y. EBV latent membrane proteins promote hybrid epithelial-mesenchymal and extreme mesenchymal states of nasopharyngeal carcinoma cells for tumorigenicity. PloS Pathog (2021) 17:e1009873. doi: 10.1371/journal.ppat.1009873
20. Vilchez Mercedes SA, Bocci F, Ahmed M, Eder I, Zhu N, Levine H, et al. Nrf2 modulates the hybrid Epithelial/Mesenchymal phenotype and notch signaling during collective cancer migration. Front Mol Biosci (2022) 9:807324. doi: 10.3389/fmolb.2022.807324
21. Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2020) 21:341–52. doi: 10.1038/s41580-020-0237-9
22. Bornes L, Belthier G, van Rheenen J. Epithelial-to-Mesenchymal transition in the light of plasticity and hybrid E/M states. J Clin Med (2021) 10:2403. doi: 10.3390/jcm10112403
23. Font-Clos F, Zapperi S, La Porta CAM. Topography of epithelial-mesenchymal plasticity. Proc Natl Acad Sci U.S.A. (2018) 115:5902–7. doi: 10.1073/pnas.1722609115
24. Kvokačková B, Remšík J, Jolly MK, Souček K. Phenotypic heterogeneity of triple-negative breast cancer mediated by epithelial-mesenchymal plasticity. Cancers (2021) 13:2188. doi: 10.3390/cancers13092188
25. Strietz J, Stepputtis SS, Follo M, Bronsert P, Stickeler E, Maurer J. Human primary breast cancer stem cells are characterized by epithelial-mesenchymal plasticity. Int J Mol Sci (2021) 22:1808. doi: 10.3390/ijms22041808
26. Tashireva LA, Savelieva OE, Grigoryeva ES, Nikitin YV, Denisov EV, Vtorushin SV, et al. Heterogeneous manifestations of epithelial-mesenchymal plasticity of circulating tumor cells in breast cancer patients. Int J Mol Sci (2021) 22:2504. doi: 10.3390/ijms22052504
27. Migault M, Sapkota S, Bracken CP. Transcriptional and post-transcriptional control of epithelial-mesenchymal plasticity: why so many regulators? Cell Mol Life Sci (2022) 79:182. doi: 10.1007/s00018-022-04199-0
28. Ruscetti M, Dadashian EL, Guo W, Quach B, Mulholland DJ, Park JW, et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene (2016) 35:3781–95. doi: 10.1038/onc.2015.444
29. Nihan Kilinc A, Sugiyama N, Reddy Kalathur RK, Antoniadis H, Birogul H, Ishay-Ronen D, et al. Histone deacetylases, Mbd3/NuRD, and Tet2 hydroxylase are crucial regulators of epithelial-mesenchymal plasticity and tumor metastasis. Oncogene (2020) 39:1498–513. doi: 10.1038/s41388-019-1081-2
30. Nilchian A, Johansson J, Ghalali A, Asanin ST, Santiago A, Rosencrantz O, et al. CXADR-mediated formation of an AKT inhibitory signalosome at tight junctions controls epithelial-mesenchymal plasticity in breast cancer. Cancer Res (2019) 79:47–60. doi: 10.1158/0008-5472.CAN-18-1742
31. Kumar-Singh A, Parniewska MM, Giotopoulou N, Javadi J, Sun W, Szatmári T, et al. Nuclear syndecan-1 regulates epithelial-mesenchymal plasticity in tumor cells. Biology (2021) 10:521. doi: 10.3390/biology10060521
32. Miao L, Yang L, Li R, Rodrigues DN, Crespo M, Hsieh J-T, et al. Disrupting androgen receptor signaling induces snail-mediated epithelial-mesenchymal plasticity in prostate cancer. Cancer Res (2017) 77:3101–12. doi: 10.1158/0008-5472.CAN-16-2169
33. Primeaux M, Gowrikumar S, Dhawan P. Role of CD44 isoforms in epithelial-mesenchymal plasticity and metastasis. Clin Exp Metastasis (2022) 39:391–406. doi: 10.1007/s10585-022-10146-x
34. Yun J, Song S-H, Kim H-P, Han S-W, Yi EC, Kim T-Y. Dynamic cohesin-mediated chromatin architecture controls epithelial-mesenchymal plasticity in cancer. EMBO Rep (2016) 17:1343–59. doi: 10.15252/embr.201541852
35. Jia R, Liang Y, Chen R, Liu G, Wang H, Tang M, et al. Osteopontin facilitates tumor metastasis by regulating epithelial-mesenchymal plasticity. Cell Death Dis (2016) 7:e2564. doi: 10.1038/cddis.2016.422
36. Moamer A, Hachim IY, Binothman N, Wang N, Lebrun J-J, Ali S. A role for kinesin-1 subunits KIF5B/KLC1 in regulating epithelial mesenchymal plasticity in breast tumorigenesis. EBioMedicine (2019) 45:92–107. doi: 10.1016/j.ebiom.2019.06.009
37. Gao L, Nie X, Gou R, Hu Y, Dong H, Li X, et al. Exosomal ANXA2 derived from ovarian cancer cells regulates epithelial-mesenchymal plasticity of human peritoneal mesothelial cells. J Cell Mol Med (2021) 25:10916–29. doi: 10.1111/jcmm.16983
38. Rafehi S, Ramos Valdes Y, Bertrand M, McGee J, Préfontaine M, Sugimoto A, et al. TGFβ signaling regulates epithelial-mesenchymal plasticity in ovarian cancer ascites-derived spheroids. Endocr Relat Cancer (2016) 23:147–59. doi: 10.1530/ERC-15-0383
39. Santos M, Ferreira M, Oliveira P, Mendes N, André A, Vieira AF, et al. Epithelial-mesenchymal plasticity induced by discontinuous exposure to TGFβ1 promotes tumour growth. Biology (2022) 11:1046. doi: 10.3390/biology11071046
40. Matsumura Y, Ito Y, Mezawa Y, Sulidan K, Daigo Y, Hiraga T, et al. Stromal fibroblasts induce metastatic tumor cell clusters via epithelial-mesenchymal plasticity. Life Sci alliance (2019) 2:e201900425. doi: 10.26508/lsa.201900425
41. Yang M, Ma B, Shao H, Clark AM, Wells A. Macrophage phenotypic subtypes diametrically regulate epithelial-mesenchymal plasticity in breast cancer cells. BMC Cancer (2016) 16:419. doi: 10.1186/s12885-016-2411-1
42. Shinde A, Hardy SD, Kim D, Akhand SS, Jolly MK, Wang W-H, et al. Spleen tyrosine kinase-mediated autophagy is required for epithelial-mesenchymal plasticity and metastasis in breast cancer. Cancer Res (2019) 79:1831–43. doi: 10.1158/0008-5472.CAN-18-2636
43. Marusyk A, Janiszewska M, Polyak K. Intratumor heterogeneity: The Rosetta stone of therapy resistance. Cancer Cell (2020) 37:471–84. doi: 10.1016/j.ccell.2020.03.007
44. Arozarena I, Wellbrock C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat Rev Cancer (2019) 19:377–91. doi: 10.1038/s41568-019-0154-4
45. Li J, Li X, Guo Q. Drug resistance in cancers: A free pass for bullying. Cells (2022) 11:3383. doi: 10.3390/cells11213383
46. Ramirez M, Rajaram S, Steininger RJ, Osipchuk D, Roth MA, Morinishi LS, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun (2016) 7:10690. doi: 10.1038/ncomms10690
47. Xue Y, Hou S, Ji H, Han X. Evolution from genetics to phenotype: reinterpretation of NSCLC plasticity, heterogeneity, and drug resistance. Protein Cell (2017) 8:178–90. doi: 10.1007/s13238-016-0330-1
48. Zhao H, Lin LF, Hahn J, Xie J, Holman HF, Yuan C. Single-cell image-based analysis reveals chromatin changes during the acquisition of tamoxifen drug resistance. Life (2022) 12:438. doi: 10.3390/life12030438
49. Ye L, Lin C, Wang X, Li Q, Li Y, Wang M, et al. Epigenetic silencing of SALL2 confers tamoxifen resistance in breast cancer. EMBO Mol Med (2019) 11:e10638. doi: 10.15252/emmm.201910638
50. Lu Y, Chan Y-T, Tan H-Y, Zhang C, Guo W, Xu Y, et al. Epigenetic regulation of ferroptosis via ETS1/miR-23a-3p/ACSL4 axis mediates sorafenib resistance in human hepatocellular carcinoma. J Exp Clin Cancer Res (2022) 41:3. doi: 10.1186/s13046-021-02208-x
51. da Silva-Diz V, Lorenzo-Sanz L, Bernat-Peguera A, Lopez-Cerda M, Muñoz P. Cancer cell plasticity: Impact on tumor progression and therapy response. Semin Cancer Biol (2018) 53:48–58. doi: 10.1016/j.semcancer.2018.08.009
52. Paul R, Dorsey JF, Fan Y. Cell plasticity, senescence, and quiescence in cancer stem cells: Biological and therapeutic implications. Pharmacol Ther (2022) 231:107985. doi: 10.1016/j.pharmthera.2021.107985
53. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell (2013) 154:61–74. doi: 10.1016/j.cell.2013.06.005
54. Zomer A, Ellenbroek SIJ, Ritsma L, Beerling E, Vrisekoop N, Van Rheenen J. Intravital imaging of cancer stem cell plasticity in mammary tumors. Stem Cells (2013) 31:602–6. doi: 10.1002/stem.1296
55. Dirkse A, Golebiewska A, Buder T, Nazarov PV, Muller A, Poovathingal S, et al. Stem cell-associated heterogeneity in glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat Commun (2019) 10:1787. doi: 10.1038/s41467-019-09853-z
56. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell (2019) 178:835–849.e21. doi: 10.1016/j.cell.2019.06.024
57. Zhang R, Tu J, Liu S. Novel molecular regulators of breast cancer stem cell plasticity and heterogeneity. Semin Cancer Biol (2022) 82:11–25. doi: 10.1016/j.semcancer.2021.03.008
58. Nobre AR, Risson E, Singh DK, Di Martino JS, Cheung JF, Wang J, et al. Bone marrow NG2(+)/Nestin(+) mesenchymal stem cells drive DTC dormancy via TGFβ2. Nat Cancer (2021) 2:327–39. doi: 10.1038/s43018-021-00179-8
59. Govaere O, Wouters J, Petz M, Vandewynckel Y-P, Van den Eynde K, Van den Broeck A, et al. Laminin-332 sustains chemoresistance and quiescence as part of the human hepatic cancer stem cell niche. J Hepatol (2016) 64:609–17. doi: 10.1016/j.jhep.2015.11.011
60. Cole AJ, Iyengar M, Panesso-Gómez S, O’Hayer P, Chan D, Delgoffe GM, et al. NFATC4 promotes quiescence and chemotherapy resistance in ovarian cancer. JCI Insight (2020) 5:e131486. doi: 10.1172/jci.insight.131486
61. Ferrer AI, Trinidad JR, Sandiford O, Etchegaray J-P, Rameshwar P. Epigenetic dynamics in cancer stem cell dormancy. Cancer Metastasis Rev (2020) 39:721–38. doi: 10.1007/s10555-020-09882-x
62. Ye S, Ding Y-F, Jia W-H, Liu X-L, Feng J-Y, Zhu Q, et al. SET domain-containing protein 4 epigenetically controls breast cancer stem cell quiescence. Cancer Res (2019) 79:4729–43. doi: 10.1158/0008-5472.CAN-19-1084
63. Chen W, Dong J, Haiech J, Kilhoffer M-C, Zeniou M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int (2016) 2016:1740936. doi: 10.1155/2016/1740936
64. Xie XP, Laks DR, Sun D, Ganbold M, Wang Z, Pedraza AM, et al. Quiescent human glioblastoma cancer stem cells drive tumor initiation, expansion, and recurrence following chemotherapy. Dev Cell (2022) 57:32–46.e8. doi: 10.1016/j.devcel.2021.12.007
65. Sistigu A, Musella M, Galassi C, Vitale I, De Maria R. Tuning cancer fate: Tumor microenvironment’s role in cancer stem cell quiescence and reawakening. Front Immunol (2020) 11:2166. doi: 10.3389/fimmu.2020.02166
66. Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci U.S.A. (2017) 114:E9066–75. doi: 10.1073/pnas.1704862114
67. Yang Y-S, Jia X-Z, Lu Q-Y, Cai S-L, Huang X-T, Yang S-H, et al. Exosomal DEK removes chemoradiotherapy resistance by triggering quiescence exit of breast cancer stem cells. Oncogene (2022) 41:2624–37. doi: 10.1038/s41388-022-02278-x
68. Hendrix MJC, Seftor EA, Hess AR, Seftor REB. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer (2003) 3:411–21. doi: 10.1038/nrc1092
69. Zheng N, Zhang S, Wu W, Zhang N, Wang J. Regulatory mechanisms and therapeutic targeting of vasculogenic mimicry in hepatocellular carcinoma. Pharmacol Res (2021) 166:105507. doi: 10.1016/j.phrs.2021.105507
70. Zhang X, Zhang J, Zhou H, Liu G, Li Q. Rho kinase mediates transforming growth factor-β1-induced vasculogenic mimicry formation: involvement of the epithelial-mesenchymal transition and cancer stemness activity. Acta Biochim Biophys Sin (2020) 52:411–20. doi: 10.1093/abbs/gmaa014
71. Du J, Sun B, Zhao X, Gu Q, Dong X, Mo J, et al. Hypoxia promotes vasculogenic mimicry formation by inducing epithelial-mesenchymal transition in ovarian carcinoma. Gynecol Oncol (2014) 133:575–83. doi: 10.1016/j.ygyno.2014.02.034
72. Wang H-F, Wang S-S, Zheng M, Dai L-L, Wang K, Gao X-L, et al. Hypoxia promotes vasculogenic mimicry formation by vascular endothelial growth factor a mediating epithelial-mesenchymal transition in salivary adenoid cystic carcinoma. Cell Prolif (2019) 52:e12600. doi: 10.1111/cpr.12600
73. Yao L, Zhang D, Zhao X, Sun B, Liu Y, Gu Q, et al. Dickkopf-1-promoted vasculogenic mimicry in non-small cell lung cancer is associated with EMT and development of a cancer stem-like cell phenotype. J Cell Mol Med (2016) 20:1673–85. doi: 10.1111/jcmm.12862
74. Wang H, Huang B, Li BM, Cao KY, Mo CQ, Jiang SJ, et al. ZEB1-mediated vasculogenic mimicry formation associates with epithelial-mesenchymal transition and cancer stem cell phenotypes in prostate cancer. J Cell Mol Med (2018) 22:3768–81. doi: 10.1111/jcmm.13637
75. Zhao N, Sun H, Sun B, Zhu D, Zhao X, Wang Y, et al. miR-27a-3p suppresses tumor metastasis and VM by down-regulating VE-cadherin expression and inhibiting EMT: an essential role for twist-1 in HCC. Sci Rep (2016) 6:23091. doi: 10.1038/srep23091
76. Lin X, Sun R, Zhao X, Zhu D, Zhao X, Gu Q, et al. C-myc overexpression drives melanoma metastasis by promoting vasculogenic mimicry via c-myc/snail/Bax signaling. J Mol Med (2017) 95:53–67. doi: 10.1007/s00109-016-1452-x
77. Zhao J, Wu J, Qin Y, Zhang W, Huang G, Qin L. LncRNA PVT1 induces aggressive vasculogenic mimicry formation through activating the STAT3/Slug axis and epithelial-to-mesenchymal transition in gastric cancer. Cell Oncol (Dordr) (2020) 43:863–76. doi: 10.1007/s13402-020-00532-6
78. Salminen A, Kaarniranta K, Kauppinen A. ER stress activates immunosuppressive network: implications for aging and alzheimer’s disease. J Mol Med (Berl) (2020) 98:633–50. doi: 10.1007/s00109-020-01904-z
79. Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, et al. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity (2014) 41:389–401. doi: 10.1016/j.immuni.2014.08.015
80. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell (2015) 161:1527–38. doi: 10.1016/j.cell.2015.05.025
81. Condamine T, Dominguez GA, Youn J-I, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol (2016) 1:aaf8943. doi: 10.1126/sciimmunol.aaf8943
82. Franco A, Almanza G, Burns JC, Wheeler M, Zanetti M. Endoplasmic reticulum stress drives a regulatory phenotype in human T-cell clones. Cell Immunol (2010) 266:1–6. doi: 10.1016/j.cellimm.2010.09.006
83. Shah PP, Beverly LJ. Regulation of VCP/p97 demonstrates the critical balance between cell death and epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget (2015) 6:17725–37. doi: 10.18632/oncotarget.3918
84. Gong L, Liu G, Zhu H, Li C, Li P, Liu C, et al. IL-32 induces epithelial-mesenchymal transition by triggering endoplasmic reticulum stress in A549 cells. BMC Pulm Med (2020) 20:278. doi: 10.1186/s12890-020-01319-z
85. Tavernier Q, Legras A, Didelot A, Normand C, Gibault L, Badoual C, et al. High expression of spliced X-box binding protein 1 in lung tumors is associated with cancer aggressiveness and epithelial-to-mesenchymal transition. Sci Rep (2020) 10:10188. doi: 10.1038/s41598-020-67243-8
86. Wang J-M, Qiu Y, Yang Z, Kim H, Qian Q, Sun Q, et al. IRE1α prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci Signal (2018) 11:eaao4617. doi: 10.1126/scisignal.aao4617
87. Perdigão-Henriques R, Petrocca F, Altschuler G, Thomas MP, Le MTN, Tan SM, et al. miR-200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the Zeb2 and Snail1 transcriptional repressor complexes. Oncogene (2016) 35:158–72. doi: 10.1038/onc.2015.69
88. Cuevas EP, Eraso P, Mazón MJ, Santos V, Moreno-Bueno G, Cano A, et al. LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signalling pathway. Sci Rep (2017) 7:44988. doi: 10.1038/srep44988
89. Yoon S, Shin B, Woo HG. Endoplasmic reticulum stress induces CAP2 expression promoting epithelial-mesenchymal transition in liver cancer cells. Mol Cells (2021) 44:569–79. doi: 10.14348/molcells.2021.0031
90. Wu S, Ye S, Lin X, Chen Y, Zhang Y, Jing Z, et al. Small hepatitis b virus surface antigen promotes malignant progression of hepatocellular carcinoma via endoplasmic reticulum stress-induced FGF19/JAK2/STAT3 signaling. Cancer Lett (2021) 499:175–87. doi: 10.1016/j.canlet.2020.11.032
91. Li R, Zhou H, Li M, Mai Q, Fu Z, Jiang Y, et al. Gremlin-1 promotes colorectal cancer cell metastasis by activating ATF6 and inhibiting ATF4 pathways. Cells (2022) 11:2136. doi: 10.3390/cells11142136
92. Gao T, Du T, Hu X, Dong X, Li L, Wang Y, et al. Cosmc overexpression enhances malignancies in human colon cancer. J Cell Mol Med (2020) 24:362–70. doi: 10.1111/jcmm.14740
93. Yuan H, Zhao Z, Guo Z, Ma L, Han J, Song Y. A novel ER stress mediator TMTC3 promotes squamous cell carcinoma progression by activating GRP78/PERK signaling pathway. Int J Biol Sci (2022) 18:4853–68. doi: 10.7150/ijbs.72838
94. Nan J, Hu X, Guo B, Xu M, Yao Y. Inhibition of endoplasmic reticulum stress alleviates triple-negative breast cancer cell viability, migration, and invasion by Syntenin/SOX4/Wnt/β-catenin pathway via regulation of heat shock protein A4. Bioengineered (2022) 13:10564–77. doi: 10.1080/21655979.2022.2062990
95. Shah PP, Dupre TV, Siskind LJ, Beverly LJ. Common cytotoxic chemotherapeutics induce epithelialmesenchymal transition (EMT) downstream of ER stress. Oncotarget (2017) 8:22625–39. doi: 10.18632/oncotarget.15150
96. Chen C-Y, Kawasumi M, Lan T-Y, Poon C-L, Lin Y-S, Wu P-J, et al. Adaptation to endoplasmic reticulum stress enhances resistance of oral cancer cells to cisplatin by up-regulating polymerase η and increasing DNA repair efficiency. Int J Mol Sci (2020) 22:355. doi: 10.3390/ijms22010355
97. Zhou S, Yang J, Wang M, Zheng D, Liu Y. Endoplasmic reticulum stress regulates epithelial−mesenchymal transition in human lens epithelial cells. Mol Med Rep (2020) 21:173–80. doi: 10.3892/mmr.2019.10814
98. Ulianich L, Mirra P, Garbi C, Calì G, Conza D, Treglia AS, et al. The pervasive effects of ER stress on a typical endocrine cell: Dedifferentiation, mesenchymal shift and antioxidant response in the thyrocyte. Front Endocrinol (Lausanne) (2020) 11:588685. doi: 10.3389/fendo.2020.588685
99. Delbrel E, Uzunhan Y, Soumare A, Gille T, Marchant D, Planès C, et al. ER stress is involved in epithelial-to-mesenchymal transition of alveolar epithelial cells exposed to a hypoxic microenvironment. Int J Mol Sci (2019) 20:1299. doi: 10.3390/ijms20061299
100. Tanjore H, Cheng D-S, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem (2011) 286:30972–80. doi: 10.1074/jbc.M110.181164
101. Liu D, Zhu H, Gong L, Pu S, Wu Y, Zhang W, et al. Histone deacetylases promote ER stress induced epithelial mesenchymal transition in human lung epithelial cells. Cell Physiol Biochem (2018) 46:1821–34. doi: 10.1159/000489367
102. Tang X, Rong G, Bu Y, Zhang S, Zhang M, Zhang J, et al. Advanced oxidation protein products induce hypertrophy and epithelial-to-mesenchymal transition in human proximal tubular cells through induction of endoplasmic reticulum stress. Cell Physiol Biochem (2015) 35:816–28. doi: 10.1159/000369740
103. Chen Y-Y, Peng X-F, Liu G-Y, Liu J-S, Sun L, Liu H, et al. Protein arginine methyltranferase-1 induces ER stress and epithelial-mesenchymal transition in renal tubular epithelial cells and contributes to diabetic nephropathy. Biochim Biophys Acta Mol basis Dis (2019) 1865:2563–75. doi: 10.1016/j.bbadis.2019.06.001
104. Feng Y-X, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JHL, Proia TA, et al. Epithelial-to-mesenchymal transition activates PERK-eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discovery (2014) 4:702–15. doi: 10.1158/2159-8290.CD-13-0945
105. Zeindl-Eberhart E, Brandl L, Liebmann S, Ormanns S, Scheel SK, Brabletz T, et al. Epithelial-mesenchymal transition induces endoplasmic-reticulum-stress response in human colorectal tumor cells. PloS One (2014) 9:e87386. doi: 10.1371/journal.pone.0087386
106. Ouyang S, Ji D, He S, Xia X. Endoplasmic reticulum stress as a novel target to inhibit transdifferentiation of human retinal pigment epithelial cells. Front Biosci (Landmark Ed (2022) 27:38. doi: 10.31083/j.fbl2702038
107. Wu S-M, Jan Y-J, Tsai S-C, Pan H-C, Shen C-C, Yang C-N, et al. Targeting histone deacetylase-3 blocked epithelial-mesenchymal plasticity and metastatic dissemination in gastric cancer. Cell Biol Toxicol (2022). doi: 10.1007/s10565-021-09673-2
108. Wu SM, Lin WY, Shen CC, Pan HC, Wang KB, Chen YC, et al. Melatonin set out to ER stress signaling thwarts epithelial mesenchymal transition and peritoneal dissemination via calpain-mediated C/EBPβ and NFκB cleavage. J Pineal Res (2016) 60:142–54. doi: 10.1111/jpi.12295
109. Jiang Y, Jiao Y, Liu Y, Zhang M, Wang Z, Li Y, et al. Sinomenine hydrochloride inhibits the metastasis of human glioblastoma cells by suppressing the expression of matrix metalloproteinase-2/-9 and reversing the endogenous and exogenous epithelial-mesenchymal transition. Int J Mol Sci (2018) 19:844. doi: 10.3390/ijms19030844
110. Zhao Y, He L, Wang T, Zhu L, Yan N. 2-hydroxypropyl-β-cyclodextrin regulates the epithelial to mesenchymal transition in breast cancer cells by modulating cholesterol homeostasis and endoplasmic reticulum stress. Metabolites (2021) 11:562. doi: 10.3390/metabo11080562
111. Fuentes-Iglesias A, Ameneiro C, Guallar D, Fidalgo M. Detecting and modulating ER stress to improve generation of induced pluripotent stem cells. Methods Mol Biol (2022) 2454:743–54. doi: 10.1007/7651_2021_354
112. Guallar D, Fuentes-Iglesias A, Souto Y, Ameneiro C, Freire-Agulleiro O, Pardavila JA, et al. ADAR1-dependent RNA editing promotes MET and iPSC reprogramming by alleviating ER stress. Cell Stem Cell (2020) 27:300–314.e11. doi: 10.1016/j.stem.2020.04.016
113. Bambang IF, Xu S, Zhou J, Salto-Tellez M, Sethi SK, Zhang D. Overexpression of endoplasmic reticulum protein 29 regulates mesenchymal-epithelial transition and suppresses xenograft tumor growth of invasive breast cancer cells. Lab Invest (2009) 89:1229–42. doi: 10.1038/labinvest.2009.87
114. Mehdizadehtapeh L, Obakan Yerlİkaya P. Endoplasmic reticulum stress and oncomir-associated chemotherapeutic drug resistance mechanisms in breast cancer tumors. Turkish J Biol (2021) 45:1–16. doi: 10.3906/biy-2010-62
115. Salaroglio IC, Panada E, Moiso E, Buondonno I, Provero P, Rubinstein M, et al. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol Cancer (2017) 16:91. doi: 10.1186/s12943-017-0657-0
116. Guo B, Xiong X, Hasani S, Wen Y-A, Li AT, Martinez R, et al. Downregulation of PHLPP induced by endoplasmic reticulum stress promotes eIF2α phosphorylation and chemoresistance in colon cancer. Cell Death Dis (2021) 12:960. doi: 10.1038/s41419-021-04251-0
117. Lai S-T, Wang Y, Peng F. Astragaloside IV sensitizes non-small cell lung cancer cells to cisplatin by suppressing endoplasmic reticulum stress and autophagy. J Thorac Dis (2020) 12:3715–24. doi: 10.21037/jtd-20-2098
118. Zhu Y, Liu W, Wang Z, Wang Y, Tan C, Pan Z, et al. ARHGEF2/EDN1 pathway participates in ER stress-related drug resistance of hepatocellular carcinoma by promoting angiogenesis and malignant proliferation. Cell Death Dis (2022) 13:652. doi: 10.1038/s41419-022-05099-8
119. Xia T, Tian H, Zhang K, Zhang S, Chen W, Shi S, et al. Exosomal ERp44 derived from ER-stressed cells strengthens cisplatin resistance of nasopharyngeal carcinoma. BMC Cancer (2021) 21:1003. doi: 10.1186/s12885-021-08712-9
120. Epple LM, Dodd RD, Merz AL, Dechkovskaia AM, Herring M, Winston BA, et al. Induction of the unfolded protein response drives enhanced metabolism and chemoresistance in glioma cells. PloS One (2013) 8:e73267. doi: 10.1371/journal.pone.0073267
121. Ryabaya O, Prokofieva A, Khochenkov D, Abramov I, Zasedatelev A, Stepanova E. Inhibition of endoplasmic reticulum stress-induced autophagy sensitizes melanoma cells to temozolomide treatment. Oncol Rep (2018) 40:385–94. doi: 10.3892/or.2018.6430
122. Wu J, Chen S, Liu H, Zhang Z, Ni Z, Chen J, et al. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting n-glycosylation. J Exp Clin Cancer Res (2018) 37:272. doi: 10.1186/s13046-018-0935-8
123. Gu C, Zhang Y, Chen D, Liu H, Mi K. Tunicamycin-induced endoplasmic reticulum stress inhibits chemoresistance of FaDu hypopharyngeal carcinoma cells in 3D collagen I cultures and in vivo. Exp Cell Res (2021) 405:112725. doi: 10.1016/j.yexcr.2021.112725
124. Janczar S, Nautiyal J, Xiao Y, Curry E, Sun M, Zanini E, et al. WWOX sensitises ovarian cancer cells to paclitaxel via modulation of the ER stress response. Cell Death Dis (2017) 8:e2955. doi: 10.1038/cddis.2017.346
125. Cai Y, Zheng Y, Gu J, Wang S, Wang N, Yang B, et al. Betulinic acid chemosensitizes breast cancer by triggering ER stress-mediated apoptosis by directly targeting GRP78. Cell Death Dis (2018) 9:636. doi: 10.1038/s41419-018-0669-8
126. Zhu Y, Xie M, Meng Z, Leung L-K, Chan FL, Hu X, et al. Knockdown of TM9SF4 boosts ER stress to trigger cell death of chemoresistant breast cancer cells. Oncogene (2019) 38:5778–91. doi: 10.1038/s41388-019-0846-y
127. Lei Y, He L, Yan C, Wang Y, Lv G. PERK activation by CCT020312 chemosensitizes colorectal cancer through inducing apoptosis regulated by ER stress. Biochem Biophys Res Commun (2021) 557:316–22. doi: 10.1016/j.bbrc.2021.03.041
128. Alnuqaydan AM, Rah B, Almutary AG, Chauhan SS. Synergistic antitumor effect of 5-fluorouracil and withaferin-a induces endoplasmic reticulum stress-mediated autophagy and apoptosis in colorectal cancer cells. Am J Cancer Res (2020) 10:799–815.
129. Xu H, Tsang KS, Wang Y, Chan JC, Xu G, Gao W-Q. Unfolded protein response is required for the definitive endodermal specification of mouse embryonic stem cells via Smad2 and β-catenin signaling. J Biol Chem (2014) 289:26290–301. doi: 10.1074/jbc.M114.572560
130. Zhang J, Weng Y, Liu X, Wang J, Zhang W, Kim SH, et al. Endoplasmic reticulum (ER) stress inducible factor cysteine-rich with EGF-like domains 2 (Creld2) is an important mediator of BMP9-regulated osteogenic differentiation of mesenchymal stem cells. PloS One (2013) 8:e73086. doi: 10.1371/journal.pone.0073086
131. Heijmans J, van Lidth de Jeude JF, Koo B-K, Rosekrans SL, Wielenga MCB, van de Wetering M, et al. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep (2013) 3:1128–39. doi: 10.1016/j.celrep.2013.02.031
132. Xiao P, Hu Z, Lang J, Pan T, Mertens RT, Zhang H, et al. Mannose metabolism normalizes gut homeostasis by blocking the TNF-α-mediated proinflammatory circuit. Cell Mol Immunol (2022) 20:119–30. doi: 10.1038/s41423-022-00955-1
133. van Lidth de Jeude JF, Meijer BJ, Wielenga MCB, Spaan CN, Baan B, Rosekrans SL, et al. Induction of endoplasmic reticulum stress by deletion of Grp78 depletes apc mutant intestinal epithelial stem cells. Oncogene (2017) 36:3397–405. doi: 10.1038/onc.2016.326
134. Niederreiter L, Fritz TMJ, Adolph TE, Krismer A-M, Offner FA, Tschurtschenthaler M, et al. ER stress transcription factor Xbp1 suppresses intestinal tumorigenesis and directs intestinal stem cells. J Exp Med (2013) 210:2041–56. doi: 10.1084/jem.20122341
135. Li C, Huang Y, Fan Q, Quan H, Dong Y, Nie M, et al. p97/VCP is highly expressed in the stem-like cells of breast cancer and controls cancer stemness partly through the unfolded protein response. Cell Death Dis (2021) 12:286. doi: 10.1038/s41419-021-03555-5
136. Wielenga MCB, Colak S, Heijmans J, van Lidth de Jeude JF, Rodermond HM, Paton JC, et al. ER-Stress-Induced differentiation sensitizes colon cancer stem cells to chemotherapy. Cell Rep (2015) 13:489–94. doi: 10.1016/j.celrep.2015.09.016
137. Spaan CN, Smit WL, van Lidth de Jeude JF, Meijer BJ, Muncan V, van den Brink GR, et al. Expression of UPR effector proteins ATF6 and XBP1 reduce colorectal cancer cell proliferation and stemness by activating PERK signaling. Cell Death Dis (2019) 10:490. doi: 10.1038/s41419-019-1729-4
138. Tseng C-N, Huang C-F, Cho C-L, Chang H-W, Huang C-W, Chiu C-C, et al. Brefeldin a effectively inhibits cancer stem cell-like properties and MMP-9 activity in human colorectal cancer colo 205 cells. Molecules (2013) 18:10242–53. doi: 10.3390/molecules180910242
139. Tseng C-N, Hong Y-R, Chang H-W, Yu T-J, Hung T-W, Hou M-F, et al. Brefeldin a reduces anchorage-independent survival, cancer stem cell potential and migration of MDA-MB-231 human breast cancer cells. Molecules (2014) 19:17464–77. doi: 10.3390/molecules191117464
140. Matsumoto T, Uchiumi T, Monji K, Yagi M, Setoyama D, Amamoto R, et al. Doxycycline induces apoptosis via ER stress selectively to cells with a cancer stem cell-like properties: importance of stem cell plasticity. Oncogenesis (2017) 6:397. doi: 10.1038/s41389-017-0009-3
141. Lin J, Zhang D, Fan Y, Chao Y, Chang J, Li N, et al. Regulation of cancer stem cell self-renewal by HOXB9 antagonizes endoplasmic reticulum stress-induced melanoma cell apoptosis via the miR-765-FOXA2 axis. J Invest Dermatol (2018) 138:1609–19. doi: 10.1016/j.jid.2018.01.023
142. Takayanagi T, Kawai T, Forrester SJ, Obama T, Tsuji T, Fukuda Y, et al. Role of epidermal growth factor receptor and endoplasmic reticulum stress in vascular remodeling induced by angiotensin II. Hypertension (2015) 65:1349–55. doi: 10.1161/HYPERTENSIONAHA.115.05344
143. Binet F, Sapieha P. ER stress and angiogenesis. Cell Metab (2015) 22:560–75. doi: 10.1016/j.cmet.2015.07.010
144. Bouvier N, Fougeray S, Beaune P, Thervet E, Pallet N. The unfolded protein response regulates an angiogenic response by the kidney epithelium during ischemic stress. J Biol Chem (2012) 287:14557–68. doi: 10.1074/jbc.M112.340570
145. Pereira ER, Liao N, Neale GA, Hendershot LM. Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PloS One (2010) 5:e12521. doi: 10.1371/journal.pone.0012521
146. Wu S-X, Ye S-S, Hong Y-X, Chen Y, Wang B, Lin X-J, et al. Hepatitis b virus small envelope protein promotes hepatocellular carcinoma angiogenesis via endoplasmic reticulum stress signaling to upregulate the expression of vascular endothelial growth factor a. J Virol (2022) 96:e0197521. doi: 10.1128/JVI.01975-21
147. Auf G, Jabouille A, Guérit S, Pineau R, Delugin M, Bouchecareilh M, et al. Inositol-requiring enzyme 1alpha is a key regulator of angiogenesis and invasion in malignant glioma. Proc Natl Acad Sci U.S.A. (2010) 107:15553–8. doi: 10.1073/pnas.0914072107
148. Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature (2014) 508:103–7. doi: 10.1038/nature13119
149. Liang H, Xiao J, Zhou Z, Wu J, Ge F, Li Z, et al. Hypoxia induces miR-153 through the IRE1α-XBP1 pathway to fine tune the HIF1α/VEGFA axis in breast cancer angiogenesis. Oncogene (2018) 37:1961–75. doi: 10.1038/s41388-017-0089-8
150. Binet F, Mawambo G, Sitaras N, Tetreault N, Lapalme E, Favret S, et al. Neuronal ER stress impedes myeloid-cell-induced vascular regeneration through IRE1αdegradation of netrin-1. Cell Metab (2013) 17:353–71. doi: 10.1016/j.cmet.2013.02.003
151. Loinard C, Zouggari Y, Rueda P, Ramkhelawon B, Cochain C, Vilar J, et al. C/EBP homologous protein-10 (CHOP-10) limits postnatal neovascularization through control of endothelial nitric oxide synthase gene expression. Circulation (2012) 125:1014–26. doi: 10.1161/CIRCULATIONAHA.111.041830
152. Su Z, Xu T, Wang Y, Guo X, Tu J, Zhang D, et al. Low−intensity pulsed ultrasound promotes apoptosis and inhibits angiogenesis via p38 signaling−mediated endoplasmic reticulum stress in human endothelial cells. Mol Med Rep (2019) 19:4645–54. doi: 10.3892/mmr.2019.10136
153. Liu H, Wang H, Chen D, Gu C, Huang J, Mi K. Endoplasmic reticulum stress inhibits 3D matrigel-induced vasculogenic mimicry of breast cancer cells via TGF-β1/Smad2/3 and β-catenin signaling. FEBS Open Bio (2021) 11:2607–18. doi: 10.1002/2211-5463.13259
154. Verfaillie T, Garg AD, Agostinis P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett (2013) 332:249–64. doi: 10.1016/j.canlet.2010.07.016
155. Santoni G, Morelli MB, Marinelli O, Nabissi M, Santoni M, Amantini C. Calcium signaling and the regulation of chemosensitivity in cancer cells: Role of the transient receptor potential channels. Adv Exp Med Biol (2020) 1131:505–17. doi: 10.1007/978-3-030-12457-1_20
156. Mahdi SHA, Cheng H, Li J, Feng R. The effect of TGF-beta-induced epithelial-mesenchymal transition on the expression of intracellular calcium-handling proteins in T47D and MCF-7 human breast cancer cells. Arch Biochem Biophys (2015) 583:18–26. doi: 10.1016/j.abb.2015.07.008
157. Karimzadeh F, Opas M. Calreticulin is required for TGF-β-Induced epithelial-to-Mesenchymal transition during cardiogenesis in mouse embryonic stem cells. Stem Cell Rep (2017) 8:1299–311. doi: 10.1016/j.stemcr.2017.03.018
158. Sarró E, Durán M, Rico A, Bou-Teen D, Fernández-Majada V, Croatt AJ, et al. Cyclophilins a and b oppositely regulate renal tubular epithelial cell phenotype. J Mol Cell Biol (2020) 12:499–514. doi: 10.1093/jmcb/mjaa005
159. Gao L, Ye Z, Liu J-H, Yang J-A, Li Y, Cai J-Y, et al. TMCO1 expression promotes cell proliferation and induces epithelial-mesenchymal transformation in human gliomas. Med Oncol (2022) 39:90. doi: 10.1007/s12032-022-01687-y
160. Davis FM, Peters AA, Grice DM, Cabot PJ, Parat M-O, Roberts-Thomson SJ, et al. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PloS One (2012) 7:e36923. doi: 10.1371/journal.pone.0036923
161. Stewart TA, Azimi I, Marcial D, Peters AA, Chalmers SB, Yapa KTDS, et al. Differential engagement of ORAI1 and TRPC1 in the induction of vimentin expression by different stimuli. Lab Invest (2020) 100:224–33. doi: 10.1038/s41374-019-0280-3
162. Sheng W, Wang G, Tang J, Shi X, Cao R, Sun J, et al. Calreticulin promotes EMT in pancreatic cancer via mediating Ca(2+) dependent acute and chronic endoplasmic reticulum stress. J Exp Clin Cancer Res (2020) 39:209. doi: 10.1186/s13046-020-01702-y
163. Liao C, Wang Q, An J, Long Q, Wang H, Xiang M, et al. Partial EMT in squamous cell carcinoma: A snapshot. Int J Biol Sci (2021) 17:3036–47. doi: 10.7150/ijbs.61566
164. Norgard RJ, Pitarresi JR, Maddipati R, Aiello-Couzo NM, Balli D, Li J, et al. Calcium signaling induces a partial EMT. EMBO Rep (2021) 22:e51872. doi: 10.15252/embr.202051872
Keywords: cellular plasticity, ER stress, epithelial-mesenchymal plasticity, resistance, cancer stem cell, vasculogenic mimicry
Citation: Wang H and Mi K (2023) Emerging roles of endoplasmic reticulum stress in the cellular plasticity of cancer cells. Front. Oncol. 13:1110881. doi: 10.3389/fonc.2023.1110881
Received: 29 November 2022; Accepted: 31 January 2023;
Published: 20 February 2023.
Edited by:
Majid Momeny, University of California, San Francisco, United StatesReviewed by:
Hira Lal Goel, UMass Chan Medical School, United StatesSolmaz AghaAmiri, University of Texas Health Science Center at Houston, United States
Copyright © 2023 Wang and Mi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun Mi, bWlrdW5AdWVzdGMuZWR1LmNu