 Mario L. Marques-Piubelli
Mario L. Marques-Piubelli Catalina Amador
Catalina Amador Francisco Vega
Francisco Vega- 1Department of Translational Molecular Pathology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Division of Hematopathology, Department of Pathology and Laboratory Medicine, University of Miami, Miami, FL, United States
- 3Department of Hematopathology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
T-follicular helper (TFH) cells are one of the T-cell subsets with a critical role in the regulation of germinal center (GC) reactions. TFH cells contribute to the positive selection of GC B-cells and promote plasma cell differentiation and antibody production. TFH cells express a unique phenotype characterized by PD-1hi, ICOShi, CD40Lhi, CD95hi, CTLAhi, CCR7lo, and CXCR5hi. Three main subtypes of nodal TFH lymphomas have been described: 1) angioimmunoblastic-type, 2) follicular-type, and 3) not otherwise specified (NOS). The diagnosis of these neoplasms can be challenging, and it is rendered based on a combination of clinical, laboratory, histopathologic, immunophenotypic, and molecular findings. The markers most frequently used to identify a TFH immunophenotype in paraffin-embedded tissue sections include PD-1, CXCL13, CXCR5, ICOS, BCL6, and CD10. These neoplasms feature a characteristic and similar, but not identical, mutational landscape with mutations in epigenetic modifiers (TET2, DNMT3A, IDH2), RHOA, and T-cell receptor signaling genes. Here, we briefly review the biology of TFH cells and present a summary of the current pathologic, molecular, and genetic features of nodal lymphomas. We want to highlight the importance of performing a consistent panel of TFH immunostains and mutational studies in TCLs to identify TFH lymphomas.
Introduction
Nodal T-follicular helper (TFH) lymphomas represent a group of mature peripheral T-cell lymphomas (TCLs) with a gene expression profile and immunophenotype similar to that of nodal TFH cells (1, 2). This concept was initially described in angioimmunoblastic T-cell lymphoma (AITL) (3, 4) and more recently in a subset of peripheral T-cell lymphomas not otherwise specified (PTCL, NOS), which also has a gene expression profile, and immunophenotype suggestive of a TFH derivation (5, 6).
Here we present a brief summary of the biology of TFH cells and our current understanding of the clinicopathologic, molecular, and genetic features of nodal TFH lymphomas. Diagnostic considerations for this subgroup of lymphomas include distinguishing TFH lymphomas from other entities, particularly PTCL, NOS. For this, it is paramount to highlight the importance of performing a consistent set of at least 5 TFH immunomarkers and mutational studies in the work up of mature TCLs. Currently, we recommend using a five-marker panel that includes CD10, CXCL13, PD-1, ICOS and BCL6. This panel is used by other major academic centers in US and Europe. Per the WHO classification, the minimum criteria for assigning a TFH phenotype is 2 (but ideally 3 or more) markers in addition to CD4.
T-Follicular helper cells
The differentiation of naïve CD4+ T-cells through the stimulation of antigen-presenting cells (APC) is an essential step for the maintenance of adaptive immunity homeostasis. The CD4+ helper T (Th) subtype is essential for the effector functions of the T-cells, and it can be divided into four major subsets of effector cells that produce distinct cytokines to help in the recruitment and activation of different cell types. These subsets include Th1 and Th2 cells (for type 1 and type 2 helper T-cells, respectively), Th17 cells (due to their IL-17 signature), and follicular helper T (TFH) cells. The latter is a fundamental component of the germinal center (GC) reaction and B-cell specialization (7, 8). Other T-cell subsets include T regulatory cells (Treg), Th9, and Th22 (9).
Phenotypically, TFH cells are characterized by the expression of BCL6 and CXC chemokine receptors type 5 (CXCR5), which allow them to reach the GCs (10, 11). TFH cells also express the inducible T-cell costimulator (ICOS), the programmed cell death protein (PD-1), and the signaling lymphocytic activation molecule (SLAM)-associated protein (SAP) that contribute to the crosstalk between T- and B-cells. Inherited mutations in the ICOS gene cause some antibody deficiencies (12).
TFH cells also provide crucial signaling for the normal ontogeny of B-cells. Engagements of MHC class II, CD40, and ICOS-ligand on GC B-cells with the TCR, CD40L, and ICOS on TFH cells may generate the production of IL-21, CD40L, and IL-4 and support the formation and maintenance of GCs (13). These signals allow antigen-specific B-cells to survive, proliferate, undergo affinity maturation, and, ultimately, differentiate into memory B-cells or long-lived plasma cells.
BCL6 is the master transcriptional regulator in the differentiation of TFH cells. BLIMP1 is a transcription factor that antagonizes BCL6 function and prevents TFH differentiation (14). BCL-6 also inhibits transcription factors important to other T-cell subsets, such as T-bet, Gata 3, and RORyt needed for Th1, Th2, and Th17 differentiation, respectively (15–17). BCL-6 also prompts the upregulation of CXCR5, a required step for the relocation of these cells toward the GCs (18).
TET2-mediated demethylation of DNA at specific regulatory regions is required to balance the differentiation of CD4+ T-cells towards Th1 and TFH lineages. In the absence of TET2, CD4+ T-cell differentiation is skewed toward the generation of highly functional TFH cells (19). Note that TET2 loss-of-function mutations are frequently found in follicular helper T-cell lymphomas (see below).
Although TFH cells are needed for the GC reaction and therefore are located inside the GCs, they can also be found in other compartments outside the GCs. Circulating memory TFH cells appear to derive from GC TFH cells following downregulation of BCL6, ICOS, and PD-1 and upregulation of CCR7 (20–22). An activated subset of circulating TFH cells with expression of P-D1 and ICOS but with low expression of CCR7 has also been described (23). These circulating activated subsets of TFH cells are functional and associated with autoimmune processes (23, 24). T-follicular regulatory (Tfr) cells are another subset of TFH cells that share many characteristics of TFH cells, including the expression of BCL6, CXCR5, ICOS, and PD-1 but also express FOXP3, as conventional Tregs (25). Importantly, Tfr cells seem to have a suppressive function inside the GCs.
Nodal T-Follicular helper cell lymphomas
In the revised 4th edition of the Classification of Hematolymphoid Neoplasms of the World Health Organization (WHO), three lymphoma entities with a TFH gene expression signature were included and designated as AITL, nodal PTCL with a TFH phenotype and follicular T-cell lymphoma (FTCL) and grouped under the provisional umbrella category of nodal lymphomas of TFH origin (Table 1) (26). In the updated classification (5th Edition), these lymphomas are currently unified as nodal follicular helper T-cell lymphomas (TFH lymphoma) with three diseases, AITL-type, follicular-type, and not otherwise specified (NOS) (1). A similar approach and terminology have been adopted in the International Consensus Classification (ICC) (2). While the nomenclature is almost identical, there are minor differences, including the term nodal in the WHO, as implied in the ICC (Table 1) (1, 2). The immunomarkers most frequently used to establish a TFH immunophenotype in clinical practice include PD-1, CXCL13, CXCR5, ICOS, BCL6, and CD10.
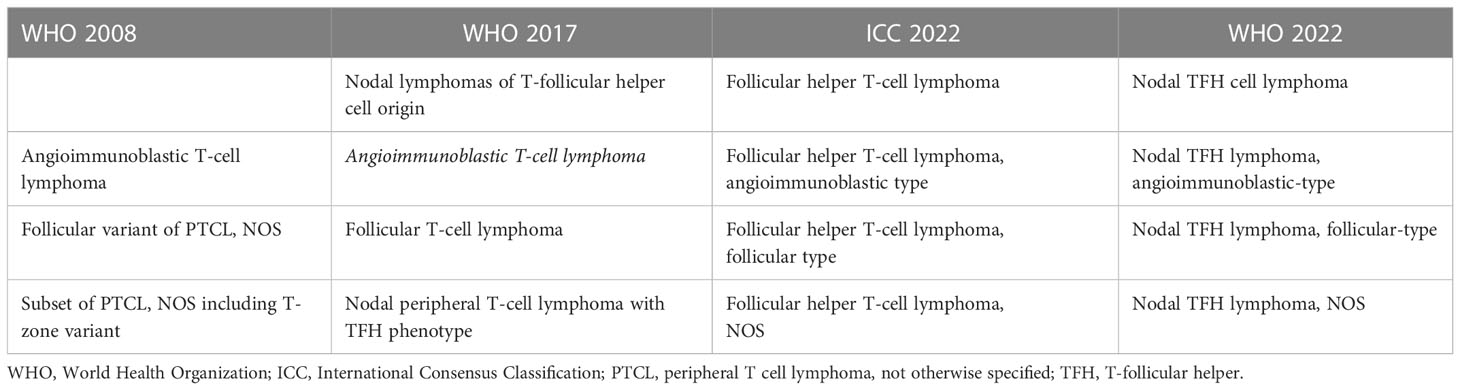
Table 1 Different designations for nodal T cell lymphomas with a TFH immunophenotype according to the different classifications schemas.
Nodal TFH cell lymphoma, angioimmunoblastic-type
For simplicity, we refer to this lymphoma as the historically used abbreviation AITL. AITL is the prototype of nodal TFH lymphomas, and it is one of the most common PTCL subtypes. There are some geographical variations in the frequency of AITL, with reported higher incidences in Europe than in the US; AITL in Europe represents about 35% of non-cutaneous T-cell lymphomas (TCLs) and ~16% in the US (27, 28). The diagnosis of AITL is based on a combination of clinical and laboratory findings, distinctive histopathologic features, and expression of TFH immunophenotype by the neoplastic cells. The neoplastic cells are characterized by the expression in variable rates of TFH markers such as CD10, Bcl-6, PD-1/CD279, ICOS, and CXCL13 (6, 26, 29). Other reported TFH markers that are not routinely used in clinical practice, include CXCR5, SAP, c-MAF, and CD200 (30–32).
Epidemiology and clinical features
AITL commonly affects middle-aged patients in the fifth to sixth decades, and there is a nearly equal incidence between genders and ethnicity. No racial predisposition is recognized. The most common clinical presentation is generalized lymphadenopathy (typically less than 3 cm), hepatosplenomegaly, constitutional symptoms, and skin rashes secondary to either neoplastic T-cell infiltration or as an autoimmune paraneoplastic manifestation (33). At diagnosis, most patients present with advanced stage and extranodal involvement. The most common sites of extranodal involvement are the spleen, bone marrow, skin, and lungs. Bone marrow involvement tends to occur early in the disease course, and its diagnosis can be extremely challenging (34). AITL is often associated with immune dysregulation, resulting in autoimmune complications and opportunistic infections (35, 36). The autoimmune and immunologic manifestations in TFH lymphomas are likely related to the functional and key role of TFH cells in B-cell activation, differentiation to plasma cells and B-cell recruitment. There are anecdotal reports of patients presenting with a smoldering course with waxing and waning lymphadenopathy (37, 38). While AITL is a systemic disease, rare cases of AITL and PTCL with TFH phenotype with localized disease and more indolent behavior have been reported, for example cases localized to the Waldeyer’s ring at presentation (39).
Morphologic features
Lymph nodes involved by AITL typically show partial or complete effacement with usually a diffuse or paracortical growth pattern and frequent perinodal extension but sparing of the subcortical sinuses (Figure 1). The neoplastic T-cells often constitute a minor part of a polymorphic inflammatory cellular infiltrate. The tumor cells are usually small- to medium-sized with mild nuclear atypia and clear cytoplasm. While they can be difficult to identify, they usually cluster around high endothelial venules (HEV) and are entrapped by the follicular dendritic cell (FDC) meshworks. Scattered or small groups of medium to large tumor cells with clear cytoplasm have been associated with IDH2R172 mutations (40). Additional distinctive pathologic features include the proliferation of arborizing high endothelial venules (HEV) surrounded by expanded networks of follicular dendritic cells (FDCs), and an inflammatory background with plasma cells, eosinophils, histiocytes, and scattered B-cell immunoblasts. Plasma cells may be abundant, in rare cases obscuring the neoplastic T-cells, and are usually polyclonal but may be monoclonal in a few cases (41).
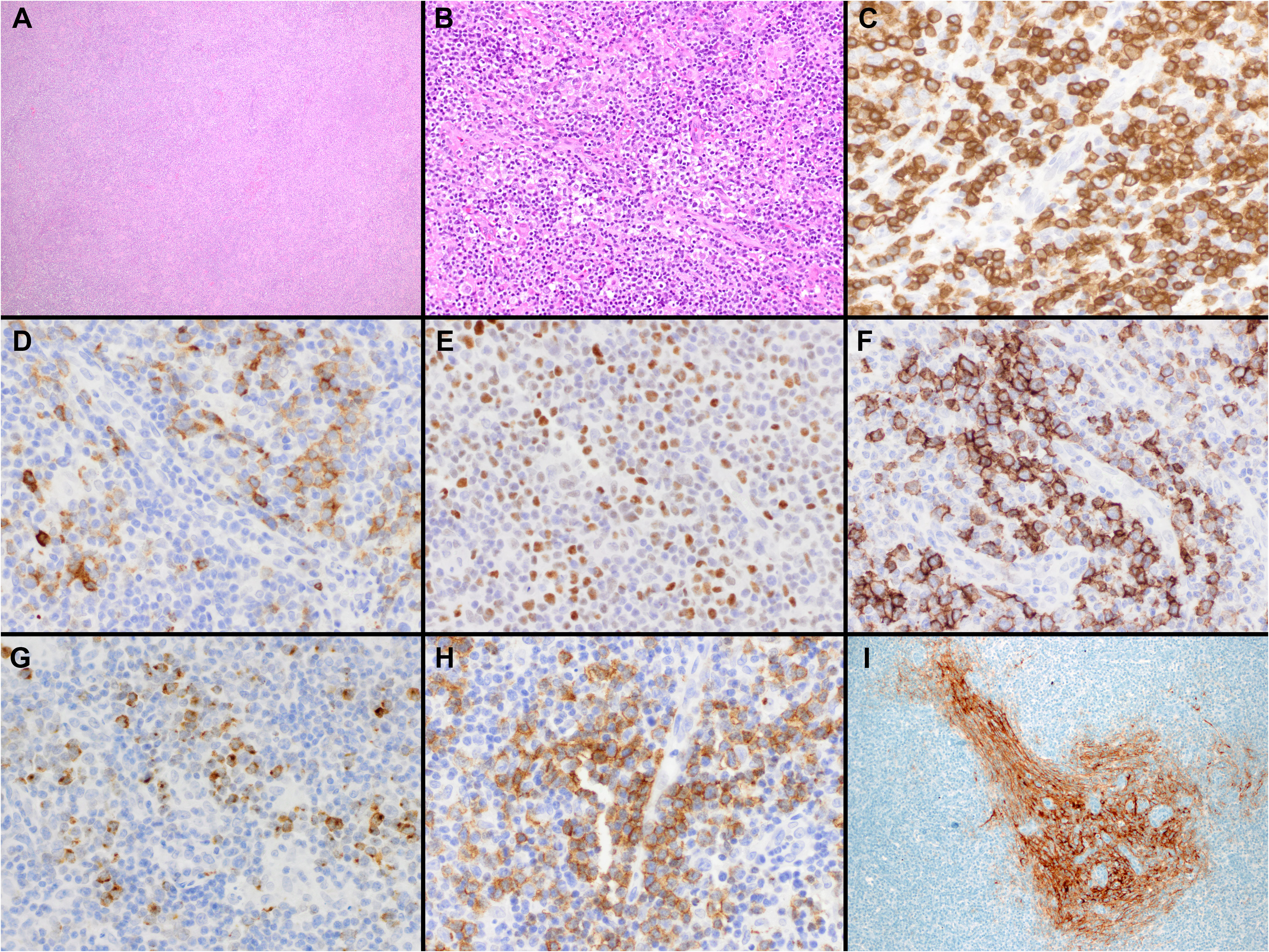
Figure 1 Histopathologic features of nodal T-follicular helper (TFH) cell lymphoma, angioimmunoblastic-type. (A, B) Hematoxylin & Eosin (H&E) shows that the neoplasm completely effaces the nodal architecture (A); the neoplasm is diffuse and composed of a heterogeneous cell infiltrate associated with numerous high endothelial venules (HEVs), inflammatory cells, and clusters of small lymphocytes with clear cytoplasm (B) (2x and 40x); (C) CD3 shows that most of the cells in the infiltrate are T-cells. Some of the T-cells are intermediate in size with irregular nuclear contours (40x); (D-H) The tumor cells are positive for CD10 (D), BCL6 (E), ICOS (F), CXCL13 (G) and PD-1 (H) supporting a TFH immunophenotype (all 40x). Note the clustering of the tumor cells around the HEVs. (I) CD21 highlights focally expanded follicular dendritic cell meshworks surrounding HEVs (20x).
Three overlapping architectural patterns (types I, II, and III) have been described by Attygale and colls (29). Pattern III is the most frequent (~80%) and is characterized by a total effacement of the nodal architecture without follicles. Pattern II shows multiple regressive “burnout”/atretic germinal centers, and pattern I (reactive hyperplasia-like) is characterized by reactive hyperplastic germinal centers with the lymphoma located in the interfollicular regions and associated with minimal expansion of FDC. A tumor cell-rich pattern has been recognized and likely represents a histological progression in patients with previous AITL (42). This pattern is enriched with tumor cells and is associated with minimal inflammatory background and limited or no HEV proliferation simulating PTCL-NOS. Other histologic variants of AITL include epithelioid-rich AITL, plasma cell-rich AITL, and AITL with Hodgkin Reed-Sternberg (HRS)-like cells. The epithelioid-rich AITL is characterized by a high content of epithelioid cells simulating lymphoepitheloid lymphoma/Lennert lymphoma or a granulomatous reaction (43, 44).
Frequently, AITL contains a variable number of B immunoblasts, which can be EBV-positive or negative. In some cases, B-immunoblasts can be monoclonal and form confluent sheets of large B-cells that morphologically meet the diagnostic criteria of large B cell lymphoma (27, 42, 45, 46). More rarely, the associated clonal B-cell proliferations are characterized by small lymphocytes and/or plasma cells, which may resemble nodal or extranodal marginal zone lymphoma (47).
HRS-like B-cells are not unusual in AITL, which are more frequently EBV-positive (48). These HRS-like cells can be rosetted by neoplastic T-cells, express B-cell markers such as CD20, or occasionally have overlapping immunophenotype with classical Hodgkin lymphoma (CHL), posing a diagnostic difficulty (42, 48).
Several reactive lymph node conditions with predominant paracortical and/or interfollicular patterns may morphologically resemble TCL. These include viral-induced lymphadenopathies, drug reactions in patients receiving anticonvulsant therapy (most commonly diphenylhydantoin), antibiotics or antivirals, and vaccination-induced reactions, as well as other non-specific etiologies. Drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome (also called Dilantin-associated lymphadenopathy) is a drug-induced severe adverse reaction that can be associated with lymphadenopathy, mimicking AITL (49).
Immunophenotypic features
The neoplastic T-cells are positive for alpha/beta TCR and CD4 and typically express pan-T cell markers, including CD2, CD3, and CD5. The aberrant loss or downregulation of one or more T-cell markers is frequently observed (50). The markers used to identify a TFH immunophenotype in paraffin-embedded tissue sections include PD-1, CXCL13, CXCR5, ICOS, BCL6, and CD10. PD-1 and ICOS are more sensitive than CXCL13 or CD10; conversely, CXCL13 and CD10 are more specific (51, 52). Partial expression of CD30 by the tumor cells is not unusual, and aberrant expression of CD20 by the lymphoma cells has also been reported (53). High expression of CD20 has been reported to be associated with a better overall survival (54–56). The use of follicular dendritic cell markers (e.g., CD21, CD23, or CD35) is helpful to assess for perivascular expansion of FDCs. CD23 has been recommended when staining for CD21 fails to show perivenular expansion of the FDCs (42). AITL with IDH2 mutations has stronger expression of CD10 and CXCL13. An antibody against IDH2R172K is available and can detect most cases with this mutation (57).
Flow cytometry frequently reveals decreased or absent expression of surface CD3 (58, 59). Also, the detection of a T-cell population by flow cytometry coexpressing CD4/CD10 or CD4/PD-1 (bright) on lymph nodes, bone marrow, or peripheral blood samples may help to support the diagnosis (60).
There have been occasional reports of AITL with cytotoxic phenotypes (61) and the possibility of a florid CD8+ cytotoxic cell proliferation obscuring a neoplastic TFH cell population has also been reported (62).
Molecular features
Mutational profile
AITL frequently shows a distinctive mutational profile with mutations involving RHOAG17V (50-70%), TET2 (40-80%), IDH2R172 (20-45%) and DNMT3A (20-30%) (Table 2) (5, 55, 56).
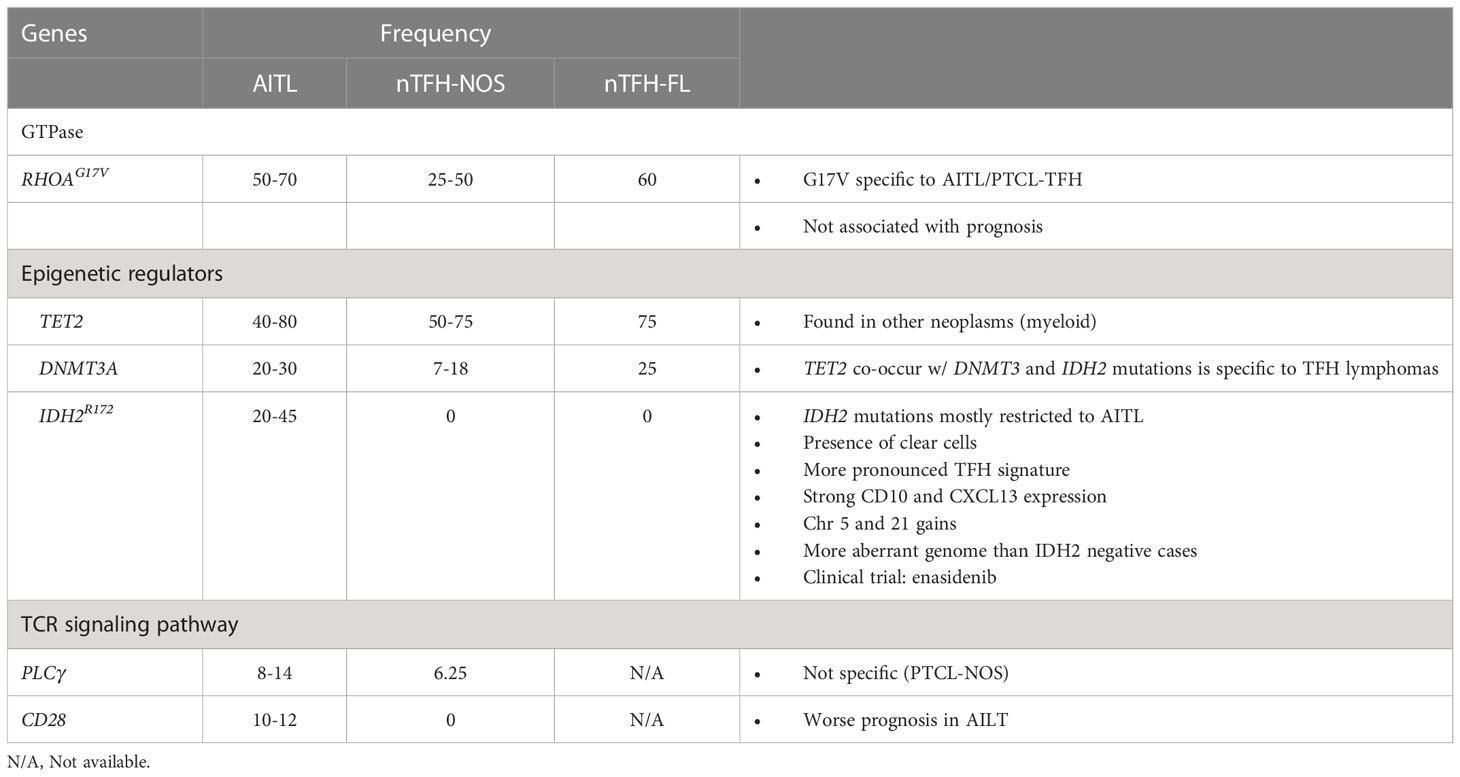
Table 2 Mutations associated with nodal follicular helper T cell lymphomas.
RHOA is a small GTPase involved in T-cell migration, polarization, and antigen recognition by cycling between GDP-bound (inactive) and GTP-bound (active states). The c.50G>T (p.Gly17Val) is the most frequent missense mutation of RHOA in AITL (56, 63). Mutations in RHOAG17V act as dominant negatives interfering with the signaling initiated by wild-type RHOA (64) and seem to be a secondary event contributing to the differentiation toward TFH phenotype (65). These mutations frequently co-occur with mutations in epigenetic regulators, especially TET2 mutations. Mutated RHOA acquires a novel function, binding and phosphorylating VAV1 protein (66). RHOAG17V mutation seems to increase TCR signaling through enhanced VAV1 resulting in stronger TCR signaling and preferential commitment to TFH rather than non-TFH lineage through enhancing ICOS signaling (67).
IDH2 is a metabolic mitochondrial enzyme involved in the generation of 2-oxoglutarate (2-HG). Mutant IDH2 forms have a neomorphic enzymatic activity leading instead to the generation of 2-hydroxyglutarate (2-HG), an oncometabolite that antagonizes the activity of α-KG-dependent dioxygenases (histone demethylases and the TET family of 5mC hydroxylases) (68, 69). They are described as a secondary event and might refine the differentiation of the premalignant clones towards a TFH signature (70). IDH2R172 mutations are associated with cases with a more pronounced TFH signature, strong CD10 and CXCL13 expression, gains of chromosomes 5 and 21, and more aberrant genome than the cases without IDH2R172 (40, 71). The more aberrant genome seen in cases with IDH2R172 mutations is likely due to the inhibitory effect of 2-HG oncometabolite on DNA repair enzymes (72). AITL with wild-type IDH2 show significant enrichment of PI3K-AKT activation pathways and have focal losses of negative regulators (phosphatases) of the PI3K-AKT pathway (71).
TET2, also known as ten-eleven translocation 2 (TET2), encodes a 2-oxoglutarate/Fe2+-dependent oxygease that participates in the epigenetic control of gene expression by catalyzing the oxidation of DNA 5-methylcytosine to 5-hydroxymethylcytosine (73, 74). Its loss-of function is an initial event in the neoplastic transformation and is associated with a worse outcome (75). DNMT3A encodes a DNA methyltransferase that controls cytosine methylation. Loss-of-function mutations in DNMT3A are also considered an initial event in the transformation process and frequently co-occur with TET2 mutations (69).
Other recurrent mutations frequently seen in AITL include TCR signaling genes, such as VAV1, PLCG1, CD28, and FYN (76). Those mutations, except for specific mutation and fusions of CD28, are not specific to AITL or TFH lymphomas. Mutations in the CD28 gene appear to show implications in outcomes since CD28-mutated AITL patients have inferior survival compared to patients with wild-type CD28 (77). Alterations in RHOA and VAV1 are mutually exclusive.
Relationship with clonal hematopoiesis
TET2 and DNMT3A mutations are not specific to AITL. However, different from other neoplasms these two mutations frequently co-occur in AITL. TET2 and DNMT3a mutations are the most frequent mutations in clonal hematopoiesis (CH). Many studies have now established that up to 80% of patients with TFH lymphomas carry the same TET2 and/or DNMT3A mutations identified in the T-cells and the myeloid cells (78–80). Furthermore, TET2 and DNMT3A mutations are not restricted to T-cells and myeloid cells but can also be identified in the admixed mature B-cells in the lymph nodes. On the contrary, RHOA and IDH2 mutations appear to be confined to the neoplastic T-cells and represent the “second” hit that contribute to the T-cell lymphomagenesis (65, 79, 81).
The background of clonal hematopoiesis of indetermined potential (CHIP) appears to be the source of myeloid neoplasm seen in TCL with a TFH phenotype, particularly after cytotoxic therapy (78, 79, 82). The occurrence of AML and other myeloid neoplasms after the diagnosis of AITL is significantly high (79). From a practical perspective, it is important to remember that in a patient being followed for AITL the presence of CHIP mutations does not necessary imply the presence of residual lymphoma.
Gene expression profile
In AITL, the expression profiling signatures are enriched in genes typically expressed by TFH cells (55, 56, 71). This demonstration of molecular similarities between AITL cells and TFH cells at a genome-wide level established the cellular “derivation” of AITL from TFH cells, initially suspected based on the expression of single TFH markers in AITL cells (3, 4, 6).The molecular profile of AITL is also dominated by a strong microenvironment imprint, including overexpression of B-cell and FDC-related genes, chemokines/chemokine receptors, and genes related to the extracellular matrix and vascular biology (52, 83, 84). Gene expression studies identified oncogenic pathways, including the NF-κB pathway, IL-6 signaling, and the TGFβ pathway enriched in AITL compared to other PTCLs (3, 4, 6, 55), but the genetic etiology of these activated pathways has not been completely elucidated.
Recently, Amador and colls have developed a digital gene expression classifier using specific signatures in nodal T-cell lymphomas and included a specific signature for nodal AITL (54). This assay can be used in paraffin-embedded tissue sections and thus can easily be translated to routine clinical practice to complement our conventional pathology approaches to better classify nodal TCLs.
Nodal TFH cell lymphoma, follicular type
For simplicity, we refer to this lymphoma as the historically used abbreviation FTCL. These are nodal TCLs with a nodular growth pattern that show significant histologic, immunophenotypic, transcriptomic, and genetic overlap with AITL (5). Isolated FTCL is very rare and usually occurs in association with AITL. Some patients at disease presentation have typical histologic and clinical features of AITL but, at relapse, show features of FTCL or vice versa (42, 85). Therefore it has been proposed that these two entities may constitute different morphologic representations of the same biological process (1, 2).
Epidemiology and clinical features
The median age of onset is 60-65 years with a slight male predominance (2). The clinical syndrome resembles AITL and other TFH lymphomas and is characterized by advanced-stage disease, generalized lymphadenopathy, splenomegaly, B symptoms, skin rash, and occasionally immune manifestations (85). Cases with localized disease have also been reported (86).
Morphologic features
Two patterns have been described. In the classic pathology description, FTCL has a follicular growth pattern mimicking follicular lymphoma (FL), where the follicles are populated by aberrant T-cells that express TFH markers (Figure 2) (85). Residual B-cells can be seen and are usually pushed to the periphery of the follicles by the neoplastic T-cells. Hodgkin and Reed-Sternberg (HRS)-like cells are also frequently noted. Alternatively, it can also mimic progressive transformation of germinal centers (PTGC). In this pattern, the nodules display a ‘moth-eaten’ appearance with aggregates of the neoplastic T-cells surrounded by small B-cells (85). Mixed FL-like and PTGC-like patterns can be seen. Focal paracortical hyperplasia is present with a polymorphic infiltrate of eosinophils and plasma cells and hyperplastic high endothelial venules.
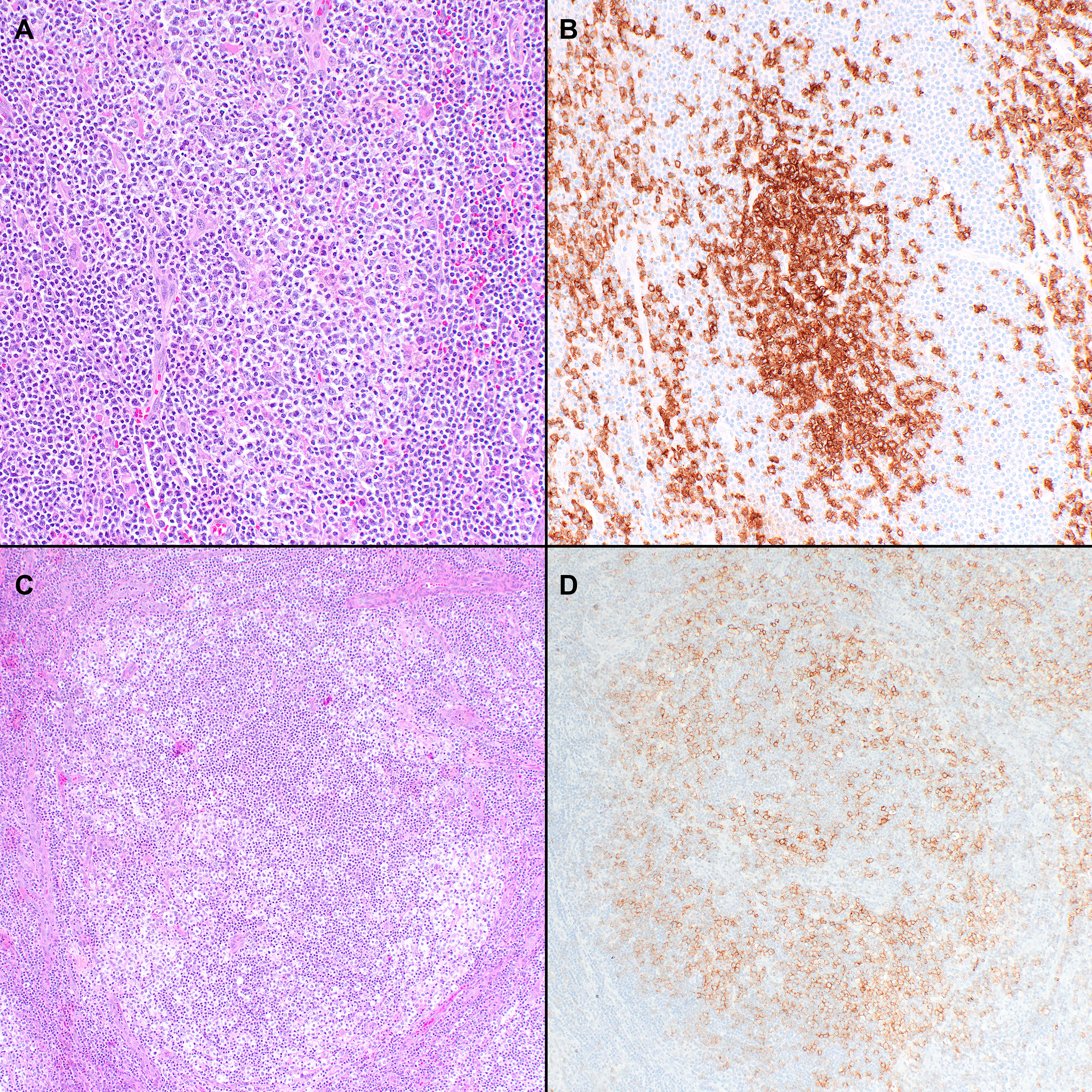
Figure 2 Histopathologic features of nodal T-follicular helper (TFH) cell lymphoma, follicular-type. (A) This case shows a predominantly follicular growth pattern, simulating follicular lymphoma, with the neoplastic follicles composed of small to medium-sized atypical lymphocytes admixed with scattered large cells (20x); (B) PD-1 shows that the tumor cells form solid clusters inside the nodules with residual B-cells pushed to the periphery of the follicles (not shown) (20x); (C) This other case shows that the neoplastic nodules have features of progressive transformation of germinal centers (PTGC). The nodules display a ‘moth-eaten’ appearance with aggregates of neoplastic T-cells surrounded by small B-cells (20x); (D) The neoplastic cells are positive for PD-1 in addition to other TFH markers (not shown) (20x).
Immunophenotypic features
The neoplastic T-cells have a TFH phenotype and typically express pan-T cell markers, including CD2, CD3, and CD5, although aberrant loss or downregulation of CD7 can be seen. By flow cytometry, dim expression of surface CD3 is frequently observed. CD4 is positive in most cases, with few instances double negative for CD4 and CD8 (85). The HRS-like cells are of B-cell lineage, positive for CD30 and positive for CD15 and EBER in some cases. This phenomenon raises concern for classic Hodgkin lymphoma. Neoplastic rosetting by T-cells around the HRS-like cells is seen in virtually all cases of FTCL, and a retained network of FDCs is seen underlying the neoplastic follicles. FTCL usually lacks the other typical features of AITL, including the characteristic expanded FDC networks surrounding HEVs (85).
Molecular features
The few cases included in gene expression profiling studies have shown that FTCL clusters closer to AITL than PTCL-NOS (5). Similarly, the mutational profile of FTCL seems to be similar to other nodal TFH lymphomas with mutations in TET2, DNMT3A, and RHOAG17V, but not in IDH2 which are usually restricted to AITL (5). FTCL can harbor a characteristic t(5;9)(q33;q22) resulting in an ITK-SYK fusion in approximately 40% of cases (87). This fusion acts as a constitutively active SYK tyrosine kinase and drives lymphomagenesis by triggering antigen-independent activation of TCR signaling. Translocations involving FER and FES have been recently described, including ITK-FER and RLTPR-FES, that result in the activation of the STAT3 signaling (88).
Nodal TFH cell lymphoma, not otherwise specified
Nodal TFH lymphoma, NOS includes those TCLs with TFH phenotype, confirmed by the expression of CD4 and at least 2 TFH markers that lack the morphologic features of AITL and FTCL. This group includes cases previously categorized as PTCL-NOS that have shown significant molecular and genetic overlap with other TFH lymphomas (5, 56, 71). It is possible that TFH lymphoma, NOS, represents more than a single entity as currently defined.
Epidemiology and clinical features
The frequency of this neoplasm is unknown. However, it is our personal experience that up to 30% of the PTCL-NOS are reclassified as nodal TFH, NOS when a panel of TFH immunomarkers is analyzed. The patients usually present with disseminated lymphadenopathy associated, which can be associated with autoimmune manifestations (5).
Morphologic features
The overall morphologic spectrum of these tumors has not been completely elucidated. Most cases are characterized by a diffuse tumor-cell-rich infiltrate of variably-sized lymphoid cells without the typical HEC and FDC proliferations (Figure 3) (1, 2). Additionally, cases previously described as lymphoepithelioid lymphomas with TFH marker expression are currently included in this group (89). A systematic evaluation of these cases shows that they can frequently have one or two AITL-like features not commonly seen in PTCL-NOS cases (51). There are some overlapping features between nodal TFH lymphoma, NOS and the tumor cell-rich pattern of AITL. However, it has been mentioned that the tumor cell-rich pattern of AITL still maintains the focal perivenular FDC expansions (1).
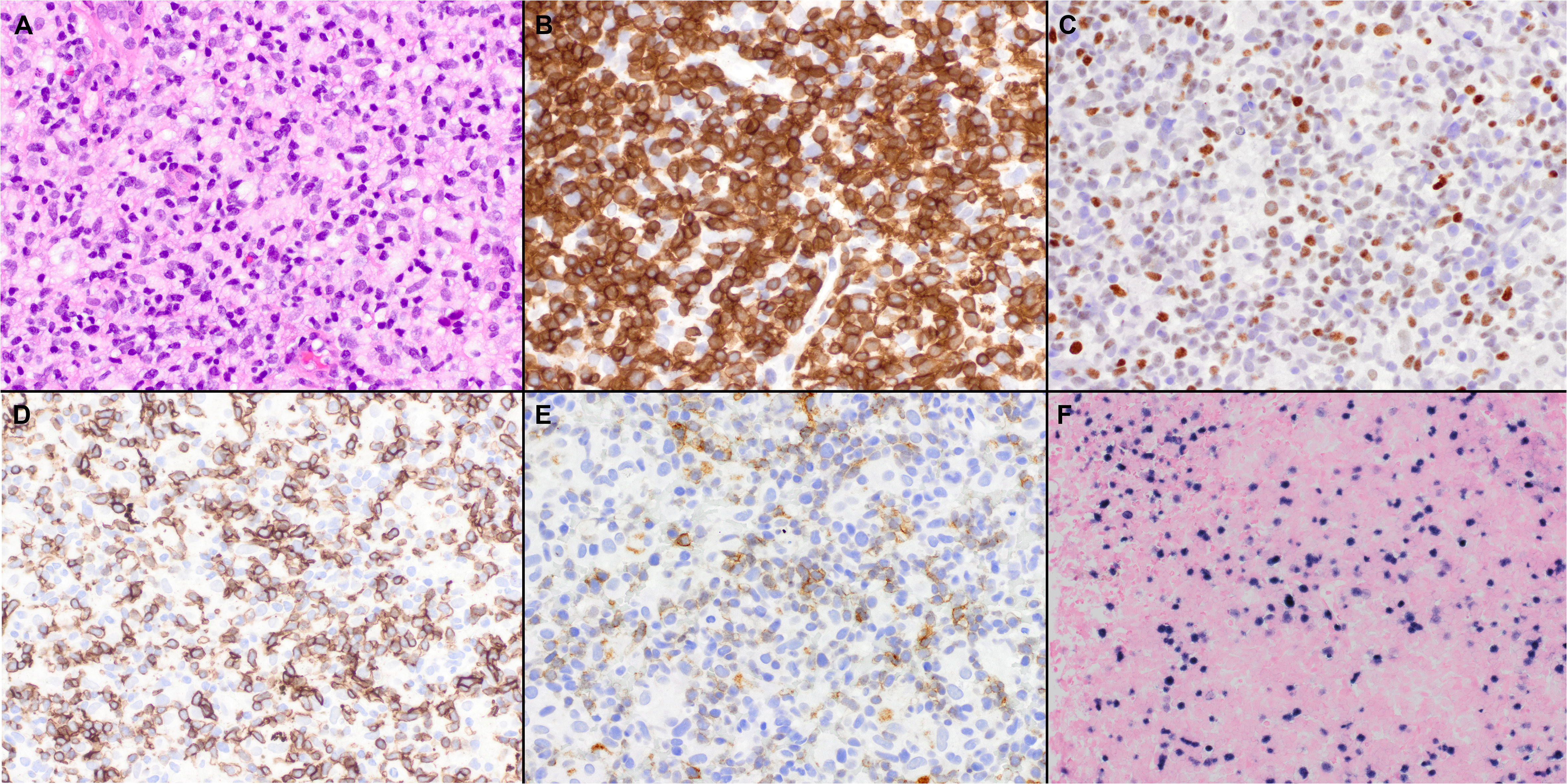
Figure 3 Histopathologic features of nodal TFH cell lymphoma, not otherwise specified (NOS). (A) In the case shown, the neoplasm is diffuse and composed predominantly of intermediate atypical lymphoid cells (40x); Features of AITL were not seen. (B) The tumor cells are positive for CD3 (40x); (C-E) BCL6 (C), ICOS (D), and PD-1 (E) are also variably positive in the neoplastic cells (40x); (F) Epstein-Barr virus-encoded small RNAs (EBER) shows positivity in scattered cells (20x).
Immunophenotypic features
By definition, the tumor cells are positive for CD4 and express at least 2 TFH makers. Some cases show focal positivity for TFH markers and positivity in a small subset of the tumor cells, being difficult to classify. Currently, there is no standard percentage of TFH positivity and the intensity of expression in tumor cells needed to establish a reproducible diagnosis of TFH lymphoma, NOS over PTCL, NOS.
Molecular features
Similarly to FTCL, gene expression studies show that TFH lymphoma, NOS clusters closer to AITL than PTCL-NOS (5, 76). The mutational profile of TFH lymphoma, NOS, seems similar to AITL mutations in TET2, DNMT3A, and RHOAG17V (5, 56, 76). IDH2R172 mutation is characteristic of AITL, although it can rarely be seen in TFH lymphoma, NOS (90). RHOAG17V mutations were identified in 60% of the cases (76). TET2 mutations seem slightly more frequent in TFH lymphoma, NOS (5). Mutations in genes of the TCR signaling pathway (including CD28) can be seen in a subset of cases (76, 90). It has been reported that the presence of TCR-signaling-related mutations correlated with early disease progression (76).
Conclusions
TFH lymphomas are a group of mature peripheral T-cell lymphomas with distinctive clinicopathologic and molecular features. Although AITL is well-characterized and has unique morphologic features that facilitate its diagnosis, the other subtypes, mainly the TFH lymphoma, NOS, is one of exclusion. Additional studies are required to better understand and delineate this category of nodal TFH lymphomas.
Since the diagnosis of nodal TFH lymphomas is very challenging, especially in small core needle biopsies, the diagnostic evaluation of TCLs requires the integration of clinicopathologic features together with a complete panel of TFH markers. We envision that incorporating mutational studies and gene expression profiling in the near future into the routine diagnostic armamentarium will facilitate the diagnosis of TFH lymphomas.
Author contributions
All the authors performed writing, reviewing, and revisions of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
FV receives research funding from CRISPR Therapeutics, Allogene Therapeutics, and Geron Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: Lymphoid neoplasms. Leukemia (2022) 36(7):1720–48. doi: 10.1038/s41375-022-01620-2
2. Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH, Anderson KC, et al. The international consensus classification of mature lymphoid neoplasms: A report from the clinical advisory committee. Blood (2022) 140(11):1229–53. doi: 10.1182/blood.2022015851
3. de Leval L, Rickman DS, Thielen C, Reynies A, Huang YL, Delsol G, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood (2007) 109(11):4952–63. doi: 10.1182/blood-2006-10-055145
4. Piccaluga PP, Agostinelli C, Califano A, Carbone A, Fantoni L, Ferrari S, et al. Gene expression analysis of angioimmunoblastic lymphoma indicates derivation from T follicular helper cells and vascular endothelial growth factor deregulation. Cancer Res (2007) 67(22):10703–10. doi: 10.1158/0008-5472.CAN-07-1708
5. Dobay MP, Lemonnier F, Missiaglia E, Bastard C, Vallois D, Jais JP, et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica (2017) 102(4):e148–51. doi: 10.3324/haematol.2016.158428
6. Grogg KL, Attygalle AD, Macon WR, Remstein ED, Kurtin PJ, Dogan A. Angioimmunoblastic T-cell lymphoma: A neoplasm of germinal-center T-helper cells? Blood (2005) 106(4):1501–2. doi: 10.1182/blood-2005-03-1083
7. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
8. Ribeiro F, Perucha E, Graca L. T Follicular cells: The regulators of germinal center homeostasis. Immunol Lett (2022) 244:1–11. doi: 10.1016/j.imlet.2022.02.008
9. Liu Z, Fan H, Jiang S. CD4(+) T-cell subsets in transplantation. Immunol Rev (2013) 252(1):183–91. doi: 10.1111/imr.12038
10. Crotty S. T Follicular helper cell biology: A decade of discovery and diseases. Immunity (2019) 50(5):1132–48. doi: 10.1016/j.immuni.2019.04.011
11. Ioannidou K, Ndiaye DR, Noto A, Fenwick C, Fortis SP, Pantaleo G, et al. In situ characterization of follicular helper CD4 T cells using multiplexed imaging. Front Immunol (2020) 11:607626. doi: 10.3389/fimmu.2020.607626
12. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Drager R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol (2003) 4(3):261–8. doi: 10.1038/ni902
13. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center b cell and T follicular helper cell development initiates in the interfollicular zone. Immunity (2011) 34(6):947–60. doi: 10.1016/j.immuni.2011.03.024
14. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325(5943):1006–10. doi: 10.1126/science.1175870
15. Nakayamada S, Kanno Y, Takahashi H, Jankovic D, Lu KT, Johnson TA, et al. Early Th1 cell differentiation is marked by a tfh cell-like transition. Immunity (2011) 35(6):919–31. doi: 10.1016/j.immuni.2011.11.012
16. Hercor M, Anciaux M, Denanglaire S, Debuisson D, Leo O, Andris F. Antigen-presenting cell-derived IL-6 restricts the expression of GATA3 and IL-4 by follicular helper T cells. J Leukoc Biol (2017) 101(1):5–14. doi: 10.1189/jlb.1HI1115-511R
17. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325(5943):1001–5. doi: 10.1126/science.1176676
18. Chen X, Ma W, Zhang T, Wu L, Qi H. Phenotypic tfh development promoted by CXCR5-controlled re-localization and IL-6 from radiation-resistant cells. Protein Cell (2015) 6(11):825–32. doi: 10.1007/s13238-015-0210-0
19. Baessler A, Novis CL, Shen Z, Perovanovic J, Wadsworth M, Thiede KA, et al. Tet2 coordinates with Foxo1 and Runx1 to balance T follicular helper cell and T helper 1 cell differentiation. Sci Adv (2022) 8(24):eabm4982. doi: 10.1126/sciadv.abm4982
20. Sage PT, Alvarez D, Godec J, von Andrian UH, Sharpe AH. Circulating T follicular regulatory and helper cells have memory-like properties. J Clin Invest (2014) 124(12):5191–204. doi: 10.1172/JCI76861
21. Weber JP, Fuhrmann F, Hutloff A. T-Follicular helper cells survive as long-term memory cells. Eur J Immunol (2012) 42(8):1981–8. doi: 10.1002/eji.201242540
22. Herati RS, Muselman A, Vella L, Bengsch B, Parkhouse K, Del Alcazar D, et al. Successive annual influenza vaccination induces a recurrent oligoclonotypic memory response in circulating T follicular helper cells. Sci Immunol (2017) 2(8). doi: 10.1126/sciimmunol.aag2152
23. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3-CXCR5+ memory tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity (2013) 39(4):758–69. doi: 10.1016/j.immuni.2013.08.031
24. Wang J, Shan Y, Jiang Z, Feng J, Li C, Ma L, et al. High frequencies of activated b cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin Exp Immunol (2013) 174(2):212–20. doi: 10.1111/cei.12162
25. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17(8):975–82. doi: 10.1038/nm.2425
26. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of tuours of haematopoietic and lymphoid tissues. 4th edition. Lyon: IARC (2017).
27. de Leval L, Parrens M, Le Bras F, Jais JP, Fataccioli V, Martin A, et al. Angioimmunoblastic T-cell lymphoma is the most common T-cell lymphoma in two distinct French information data sets. Haematologica (2015) 100(9):e361–4. doi: 10.3324/haematol.2015.126300
28. Vose J, Armitage J, Weisenburger D, International TCLP. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J Clin Oncol (2008) 26(25):4124–30. doi: 10.1200/JCO.2008.16.4558
29. Attygalle A, Al-Jehani R, Diss TC, Munson P, Liu H, Du MQ, et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood (2002) 99(2):627–33. doi: 10.1182/blood.V99.2.627
30. Murakami YI, Yatabe Y, Sakaguchi T, Sasaki E, Yamashita Y, Morito N, et al. C-maf expression in angioimmunoblastic T-cell lymphoma. Am J Surg Pathol (2007) 31(11):1695–702. doi: 10.1097/PAS.0b013e318054dbcf
31. Dorfman DM, Shahsafaei A. CD200 (OX-2 membrane glycoprotein) is expressed by follicular T helper cells and in angioimmunoblastic T-cell lymphoma. Am J Surg Pathol (2011) 35(1):76–83. doi: 10.1097/PAS.0b013e31820065c9
32. Roncador G, Garcia Verdes-Montenegro JF, Tedoldi S, Paterson JC, Klapper W, Ballabio E, et al. Expression of two markers of germinal center T cells (SAP and PD-1) in angioimmunoblastic T-cell lymphoma. Haematologica (2007) 92(8):1059–66. doi: 10.3324/haematol.10864
33. Lachenal F, Berger F, Ghesquieres H, Biron P, Hot A, Callet-Bauchu E, et al. Angioimmunoblastic T-cell lymphoma: Clinical and laboratory features at diagnosis in 77 patients. Med (Baltimore) (2007) 86(5):282–92. doi: 10.1097/MD.0b013e3181573059
34. Khokhar FA, Payne WD, Talwalkar SS, Jorgensen JL, Bueso-Ramos CE, Medeiros LJ, et al. Angioimmunoblastic T-cell lymphoma in bone marrow: A morphologic and immunophenotypic study. Hum Pathol (2010) 41(1):79–87. doi: 10.1016/j.humpath.2009.06.016
35. Dunleavy K, Wilson WH, Jaffe ES. Angioimmunoblastic T cell lymphoma: Pathobiological insights and clinical implications. Curr Opin Hematol (2007) 14(4):348–53. doi: 10.1097/MOH.0b013e328186ffbf
36. Mourad N, Mounier N, Briere J, Raffoux E, Delmer A, Feller A, et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the groupe d'Etude des lymphomes de l'Adulte (GELA) trials. Blood (2008) 111(9):4463–70. doi: 10.1182/blood-2007-08-105759
37. Federico M, Rudiger T, Bellei M, Nathwani BN, Luminari S, Coiffier B, et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: Analysis of the international peripheral T-cell lymphoma project. J Clin Oncol (2013) 31(2):240–6. doi: 10.1200/JCO.2011.37.3647
38. Tan LH, Tan SY, Tang T, Lim ST, Tan D, Lim LC, et al. Angioimmunoblastic T-cell lymphoma with hyperplastic germinal centres (pattern 1) shows superior survival to patterns 2 and 3: A meta-analysis of 56 cases. Histopathology (2012) 60(4):570–85. doi: 10.1111/j.1365-2559.2011.04097.x
39. Wang J, Tang W, Zhang W, Wang J, Chen F, Zhong L, et al. Clinicopathological characterization of follicular helper T-cell-derived peripheral T-cell lymphoma involving waldeyer's ring. Virchows Arch (2021) 479(2):355–63. doi: 10.1007/s00428-021-03071-z
40. Steinhilber J, Mederake M, Bonzheim I, Serinsoz-Linke E, Muller I, Fallier-Becker P, et al. The pathological features of angioimmunoblastic T-cell lymphomas with IDH2(R172) mutations. Mod Pathol (2019) 32(8):1123–34. doi: 10.1038/s41379-019-0254-4
41. Huppmann AR, Roullet MR, Raffeld M, Jaffe ES. Angioimmunoblastic T-cell lymphoma partially obscured by an Epstein-Barr virus-negative clonal plasma cell proliferation. J Clin Oncol (2013) 31(2):e28–30. doi: 10.1200/JCO.2012.43.3797
42. Attygalle AD, Cabecadas J, Gaulard P, Jaffe ES, de Jong D, Ko YH, et al. Peripheral T-cell and NK-cell lymphomas and their mimics; taking a step forward - report on the lymphoma workshop of the XVIth meeting of the European association for haematopathology and the society for hematopathology. Histopathology (2014) 64(2):171–99. doi: 10.1111/his.12251
43. Hartmann S, Agostinelli C, Klapper W, Korkolopoulou P, Koch K, Marafioti T, et al. Revising the historical collection of epithelioid cell-rich lymphomas of the Kiel lymph node registry: What is lennert's lymphoma nowadays? Histopathology (2011) 59(6):1173–82. doi: 10.1111/j.1365-2559.2011.04069.x
44. Agostinelli C, Hartmann S, Klapper W, Korkolopoulou P, Righi S, Marafioti T, et al. Peripheral T cell lymphomas with follicular T helper phenotype: A new basket or a distinct entity? revising Karl lennert's personal archive. Histopathology (2011) 59(4):679–91. doi: 10.1111/j.1365-2559.2011.03981.x
45. Tokunaga T, Shimada K, Yamamoto K, Chihara D, Ichihashi T, Oshima R, et al. Retrospective analysis of prognostic factors for angioimmunoblastic T-cell lymphoma: A multicenter cooperative study in Japan. Blood (2012) 119(12):2837–43. doi: 10.1182/blood-2011-08-374371
46. Zettl A, Lee SS, Rudiger T, Starostik P, Marino M, Kirchner T, et al. Epstein-Barr Virus-associated b-cell lymphoproliferative disorders in angloimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, unspecified. Am J Clin Pathol (2002) 117(3):368–79. doi: 10.1309/6UTX-GVC0-12ND-JJEU
47. Kaffenberger B, Haverkos B, Tyler K, Wong HK, Porcu P, Gru AA. Extranodal marginal zone lymphoma-like presentations of angioimmunoblastic T-cell lymphoma: A T-cell lymphoma masquerading as a b-cell lymphoproliferative disorder. Am J Dermatopathol (2015) 37(8):604–13. doi: 10.1097/DAD.0000000000000266
48. Nicolae A, Pittaluga S, Venkataraman G, Vijnovich-Baron A, Xi L, Raffeld M, et al. Peripheral T-cell lymphomas of follicular T-helper cell derivation with Hodgkin/Reed-sternberg cells of b-cell lineage: Both EBV-positive and EBV-negative variants exist. Am J Surg Pathol (2013) 37(6):816–26. doi: 10.1097/PAS.0b013e3182785610
49. Chen HC, Wang RC, Tsai HP, Medeiros LJ, Chang KC. Morphologic spectrum of lymphadenopathy in drug reaction with eosinophilia and systemic symptoms syndrome. Arch Pathol Lab Med (2022) 146(9):1084–93. doi: 10.5858/arpa.2021-0087-OA
50. Lee SS, Rudiger T, Odenwald T, Roth S, Starostik P, Muller-Hermelink HK. Angioimmunoblastic T cell lymphoma is derived from mature T-helper cells with varying expression and loss of detectable CD4. Int J Cancer (2003) 103(1):12–20. doi: 10.1002/ijc.10758
51. Basha BM, Bryant SC, Rech KL, Feldman AL, Vrana JA, Shi M, et al. Application of a 5 marker panel to the routine diagnosis of peripheral T-cell lymphoma with T-follicular helper phenotype. Am J Surg Pathol (2019) 43(9):1282–90. doi: 10.1097/PAS.0000000000001315
52. Gaulard P, de Leval L. Follicular helper T cells: Implications in neoplastic hematopathology. Semin Diagn Pathol (2011) 28(3):202–13. doi: 10.1053/j.semdp.2011.03.003
53. Onaindia A, Martinez N, Montes-Moreno S, Almaraz C, Rodriguez-Pinilla SM, Cereceda L, et al. CD30 expression by b and T cells: A frequent finding in angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma-not otherwise specified. Am J Surg Pathol (2016) 40(3):378–85. doi: 10.1097/PAS.0000000000000571
54. Amador C, Bouska A, Wright G, Weisenburger DD, Feldman AL, Greiner TC, et al. Gene expression signatures for the accurate diagnosis of peripheral T-cell lymphoma entities in the routine clinical practice. J Clin Oncol (2022) 40(36):JCO2102707. doi: 10.1200/JCO.21.02707
55. Iqbal J, Weisenburger DD, Greiner TC, Vose JM, McKeithan T, Kucuk C, et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood (2010) 115(5):1026–36. doi: 10.1182/blood-2009-06-227579
56. Rodríguez M, Alonso-Alonso R, Tomás-Roca L, Rodríguez-Pinilla SM, Manso-Alonso R, Cereceda L, et al. Peripheral T-cell lymphoma: Molecular profiling recognizes subclasses and identifies prognostic markers. Blood Adv (2021) 5(24):5588–98. doi: 10.1182/bloodadvances.2021005171
57. Dupuy A, Lemonnier F, Fataccioli V, Martin-Garcia N, Robe C, Pelletier R, et al. Multiple ways to detect IDH2 mutations in angioimmunoblastic T-cell lymphoma from immunohistochemistry to next-generation sequencing. J Mol Diagn (2018) 20(5):677–85. doi: 10.1016/j.jmoldx.2018.05.012
58. Singh A, Schabath R, Ratei R, Stroux A, Klemke CD, Nebe T, et al. Peripheral blood sCD3(-) CD4(+) T cells: A useful diagnostic tool in angioimmunoblastic T cell lymphoma. Hematol Oncol (2014) 32(1):16–21. doi: 10.1002/hon.2080
59. Yabe M, Gao Q, Ozkaya N, Huet S, Lewis N, Pichardo JD, et al. Bright PD-1 expression by flow cytometry is a powerful tool for diagnosis and monitoring of angioimmunoblastic T-cell lymphoma. Blood Cancer J (2020) 10(3):32. doi: 10.1038/s41408-020-0301-x
60. Loghavi S, Wang SA, Medeiros LJ, Jorgensen JL, Li X, Xu-Monette ZY, et al. Immunophenotypic and diagnostic characterization of angioimmunoblastic T-cell lymphoma by advanced flow cytometric technology. Leuk Lymphoma (2016) 57(12):2804–12. doi: 10.3109/10428194.2016.1170827
61. Liao YL, Chang ST, Kuo SY, Lin SH, Chen CK, Chang KM, et al. Angioimmunoblastic T-cell lymphoma of cytotoxic T-cell phenotype containing a large b-cell proliferation with an undersized b-cell clonal product. Appl Immunohistochem Mol Morphol (2010) 18(2):185–9. doi: 10.1097/PAI.0b013e3181c0692b
62. Attygalle AD, Dobson R, Chak PK, Vroobel KM, Wren D, Mugalaasi H, et al. Parallel evolution of two distinct lymphoid proliferations in clonal haematopoiesis. Histopathology (2022) 80(5):847–58. doi: 10.1111/his.14619
63. Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet (2014) 46(2):171–5. doi: 10.1038/ng.2872
64. Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet (2014) 46(2):166–70. doi: 10.1038/ng.2873
65. Nguyen TB, Sakata-Yanagimoto M, Asabe Y, Matsubara D, Kano J, Yoshida K, et al. Identification of cell-type-specific mutations in nodal T-cell lymphomas. Blood Cancer J (2017) 7(1):e516. doi: 10.1038/bcj.2016.122
66. Fujisawa M, Sakata-Yanagimoto M, Nishizawa S, Komori D, Gershon P, Kiryu M, et al. Activation of RHOA-VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia (2018) 32(3):694–702. doi: 10.1038/leu.2017.273
67. Cortes JR, Ambesi-Impiombato A, Couronne L, Quinn SA, Kim CS, da Silva Almeida AC, et al. RHOA G17V induces T follicular helper cell specification and promotes lymphomagenesis. Cancer Cell (2018) 33(2):259–273.e7. doi: 10.1016/j.ccell.2018.01.001
68. Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood (2011) 118(17):4509–18. doi: 10.1182/blood-2010-12-325241
69. Cortes JR, Palomero T. The curious origins of angioimmunoblastic T-cell lymphoma. Curr Opin Hematol (2016) 23(4):434–43. doi: 10.1097/MOH.0000000000000261
70. Wang C, McKeithan TW, Gong Q, Zhang W, Bouska A, Rosenwald A, et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood (2015) 126(15):1741–52. doi: 10.1182/blood-2015-05-644591
71. Heavican TB, Bouska A, Yu J, Lone W, Amador C, Gong Q, et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood (2019) 133(15):1664–76. doi: 10.1182/blood-2018-09-872549
72. Wang P, Wu J, Ma S, Zhang L, Yao J, Hoadley KA, et al. Oncometabolite d-2-Hydroxyglutarate inhibits ALKBH DNA repair enzymes and sensitizes IDH mutant cells to alkylating agents. Cell Rep (2015) 13(11):2353–61. doi: 10.1016/j.celrep.2015.11.029
73. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (2009) 324(5929):930–5. doi: 10.1126/science.1170116
74. Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature (2010) 466(7310):1129–33. doi: 10.1038/nature09303
75. Lemonnier F, Couronne L, Parrens M, Jais JP, Travert M, Lamant L, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood (2012) 120(7):1466–9. doi: 10.1182/blood-2012-02-408542
76. Vallois D, Dobay MP, Morin RD, Lemonnier F, Missiaglia E, Juilland M, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood (2016) 128(11):1490–502. doi: 10.1182/blood-2016-02-698977
77. Rohr J, Guo S, Huo J, Bouska A, Lachel C, Li Y, et al. Recurrent activating mutations of CD28 in peripheral T-cell lymphomas. Leukemia (2016) 30(5):1062–70. doi: 10.1038/leu.2015.357
78. Tiacci E, Venanzi A, Ascani S, Marra A, Cardinali V, Martino G, et al. High-risk clonal hematopoiesis as the origin of AITL and NPM1-mutated AML. N Engl J Med (2018) 379(10):981–4. doi: 10.1056/NEJMc1806413
79. Lewis NE, Petrova-Drus K, Huet S, Epstein-Peterson ZD, Gao Q, Sigler AE, et al. Clonal hematopoiesis in angioimmunoblastic T-cell lymphoma with divergent evolution to myeloid neoplasms. Blood Adv (2020) 4(10):2261–71. doi: 10.1182/bloodadvances.2020001636
80. Cheng S, Zhang W, Inghirami G, Tam W. Mutation analysis links angioimmunoblastic T-cell lymphoma to clonal hematopoiesis and smoking. Elife (2021) 10. doi: 10.7554/eLife.66395
81. Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell (2011) 20(1):25–38. doi: 10.1016/j.ccr.2011.06.003
82. Yao WQ, Wu F, Zhang W, Chuang SS, Thompson JS, Chen Z, et al. Angioimmunoblastic T-cell lymphoma contains multiple clonal T-cell populations derived from a common TET2 mutant progenitor cell. J Pathol (2020) 250(3):346–57. doi: 10.1002/path.5376
83. Iqbal J, Wright G, Wang C, Rosenwald A, Gascoyne RD, Weisenburger DD, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood (2014) 123(19):2915–23. doi: 10.1182/blood-2013-11-536359
84. Iqbal J, Wilcox R, Naushad H, Rohr J, Heavican TB, Wang C, et al. Genomic signatures in T-cell lymphoma: How can these improve precision in diagnosis and inform prognosis? Blood Rev (2016) 30(2):89–100. doi: 10.1016/j.blre.2015.08.003
85. Huang Y, Moreau A, Dupuis J, Streubel B, Petit B, Le Gouill S, et al. Peripheral T-cell lymphomas with a follicular growth pattern are derived from follicular helper T cells (TFH) and may show overlapping features with angioimmunoblastic T-cell lymphomas. Am J Surg Pathol (2009) 33(5):682–90. doi: 10.1097/PAS.0b013e3181971591
86. Miyoshi H, Sato K, Niino D, Arakawa F, Kimura Y, Kiyasu J, et al. Clinicopathologic analysis of peripheral T-cell lymphoma, follicular variant, and comparison with angioimmunoblastic T-cell lymphoma: Bcl-6 expression might affect progression between these disorders. Am J Clin Pathol (2012) 137(6):879–89. doi: 10.1309/AJCPBPNV86VZENGV
87. Streubel B, Vinatzer U, Willheim M, Raderer M, Chott A. Novel t(5;9)(q33;q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma. Leukemia (2006) 20(2):313–8. doi: 10.1038/sj.leu.2404045
88. Debackere K, van der Krogt JA, Tousseyn T, Ferreiro JAF, Van Roosbroeck K, Marcelis L, et al. FER and FES tyrosine kinase fusions in follicular T-cell lymphoma. Blood (2020) 135(8):584–8. doi: 10.1182/blood.2019002401
89. Kurita D, Miyoshi H, Yoshida N, Sasaki Y, Kato S, Niino D, et al. A clinicopathologic study of lennert lymphoma and possible prognostic factors: The importance of follicular helper T-cell markers and the association with angioimmunoblastic T-cell lymphoma. Am J Surg Pathol (2016) 40(9):1249–60. doi: 10.1097/PAS.0000000000000694
Keywords: peripheral T cell lymphomas, angioimmunoblastic T cell lymphoma, follicular T helper, next-generation sequencing, molecular and genetic profiling
Citation: Marques-Piubelli ML, Amador C and Vega F (2023) Pathologic and molecular insights in nodal T-follicular helper cell lymphomas. Front. Oncol. 13:1105651. doi: 10.3389/fonc.2023.1105651
Received: 22 November 2022; Accepted: 13 January 2023;
Published: 30 January 2023.
Edited by:
Ryan Wilcox, University of Michigan, United StatesReviewed by:
Carlos Murga-Zamalloa, University of Illinois at Chicago, United StatesShannon Carty, University of Michigan, United States
Copyright © 2023 Marques-Piubelli, Amador and Vega. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario L. Marques-Piubelli, TUxQaXViZWxsaUBtZGFuZGVyc29uLm9yZw==
†These authors have contributed equally to this work