 Jesus Gonzalez-Bosquet1S. John Weroha2Jamie N. Bakkum-Gamez3Amy L. Weaver4Michaela E. McGree4Sean C. Dowdy3Abimbola O. Famuyide3
Jesus Gonzalez-Bosquet1S. John Weroha2Jamie N. Bakkum-Gamez3Amy L. Weaver4Michaela E. McGree4Sean C. Dowdy3Abimbola O. Famuyide3 Benjamin R. Kipp5
Benjamin R. Kipp5 Kevin C. Halling5Siddhartha Yadav2
Kevin C. Halling5Siddhartha Yadav2 Fergus J. Couch4,5
Fergus J. Couch4,5 Karl C. Podratz3*
Karl C. Podratz3*- 1Department of Obstetrics and Gynecology, University of Iowa, Iowa City, IA, United States
- 2Division of Medical Oncology, Mayo Clinic, Rochester, MN, United States
- 3Department of Obstetrics and Gynecology, Mayo Clinic, Rochester, MN, United States
- 4Department of Quantitative Health Sciences, Mayo Clinic, Rochester, MN, United States
- 5Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, United States
Objective: To identify high-risk disease in clinicopathologic low-risk endometrial cancer (EC) with high microsatellite instability (MSI-H) or no specific molecular profile (NSMP) and therapeutic insensitivity in clinicopathologic high-risk MSI-H/NSMP EC.
Methods: We searched The Cancer Genome Atlas for DNA sequencing, RNA expression, and surveillance data regarding MSI-H/NSMP EC. We used a molecular classification system of E2F1 and CCNA2 expression and sequence variations in POLE, PPP2R1A, or FBXW7 (ECPPF) to prognostically stratify MSI-H/NSMP ECs. Clinical outcomes were annotated after integrating ECPPF and sequence variations in homologous recombination (HR) genes.
Results: Data were available for 239 patients with EC, which included 58 MSI-H and 89 NSMP cases. ECPPF effectively stratified MSI-H/NSMP EC into distinct molecular groups with prognostic implications: molecular low risk (MLR), with low CCNA2 and E2F1 expression, and molecular high risk (MHR), with high CCNA2 and E2F1 expression and/or PPP2R1A and/or FBXW7 variants. The 3-year disease-free survival (DFS) rate was 43.8% in the MHR group with clinicopathologic low-risk indicators and 93.9% in the MLR group (P<.001). In the MHR group, wild-type HR genes were present in 28% of cases but in 81% of documented recurrences. The 3-year DFS rate in patients with MSI-H/NSMP EC with clinicopathologic high-risk indicators was significantly higher in the MLR (94.1%) and MHR/HR variant gene (88.9%) groups than in the MHR/HR wild-type gene group (50.3%, P<.001).
Conclusion: ECPPF may resolve prognostic challenges for MSI-H/NSMP EC by identifying occult high-risk disease in EC with clinicopathologic low-risk indicators and therapeutic insensitivity in EC with clinicopathologic high-risk indicators.
Introduction
Over the past 4 decades, prognostic stratification of endometrial cancer (EC) has progressively incorporated serous histopathologic findings, lymphatic metastases, lymphovascular space involvement, TP53 variations, ERBB2 expression, mismatch repair and homologous recombination (HR) repair deficiencies, and other variables (1–5). More recently, The Cancer Genome Atlas (TCGA) generated a molecular-based prognostic classification system (6). Despite the evolution of EC risk stratification, corresponding risk-adjusted therapy has minimally affected EC mortality rates. The American Cancer Society 2020 Annual Report concluded that “cancer survival has improved since the mid-1970s for all of the most common cancers except uterine cervix and uterine corpus” and that “stagnant survival rates reflect a lack of major treatment advances” (7). Addressing this lack of major treatment advances requires an enhanced understanding of pivotal tumor-specific molecular drivers to facilitate the discovery of target-specific therapeutics.
TCGA classification of EC identified 4 unique molecular subgroups: 1) POLE ultramutated, characterized by POLE variations; 2) microsatellite instability hypermutated, classified by mismatch repair deficiency and high microsatellite instability (MSI-H); 3) copy-number low, denoted by no specific molecular profile (NSMP); and 4) copy-number high, primarily typified by TP53 variations and serouslike EC (6). Comparative prognoses associated with POLE ultramutated and copy-number high EC are contrasting, in which the prognosis is favorable for POLE ultramutated EC and substantially compromised for copy-number high EC. MSI-H and NSMP ECs have an intermediate prognosis but account for most EC tumors, thereby highly contributing to the annual EC mortality rate (6, 8–10). Therefore, determining the causes of treatment failures in MSI-H and NSMP ECs is paramount. Because MSI-H and NSMP tumors are predominantly low stage, with grade 1 and 2 histopathologic indicators (11), treatment failures may reflect our inability to identify clinicopathologic low-risk cases at high risk for recurrence (i.e., EC harboring occult high-risk disease). Radiotherapy (RT) and/or platinum-based chemotherapy (PbCT) are standard adjuvant treatment modalities for clinicopathologic high-risk MSI-H and NSMP EC. Treatment failures may also result from our inability to predict insensitivity to these standard adjuvant therapeutics (9, 12).
MSI-H EC generally harbors a high tumor mutation burden (TMB), and thus patients with MSI-H tumors are considered potential candidates for immunotherapy (6, 13–16). However, the high associated TMB presumably predicts a greater prevalence of HR gene variations, which portend sensitivity to DNA-damaging agents in cases with clinicopathologic high-risk indicators (17–20). Defining specific vulnerabilities in MSI-H tumors is necessary for decreasing the mortality rate in patients with this molecular profile. Furthermore, because of the absence of identifiable molecularly targetable pathways in NSMP, empiric therapy must be used for the largest EC cohort. Therefore, we used our previously described stratification system based on E2F1 and CCNA2 expression and sequence variations in POLE, PPP2R1A, and FBXW7 (termed ECPPF) to molecularly stratify MSI-H and NSMP ECs (21). The primary aim of our study was to use ECPPF to identify MSI-H and NSMP EC cases with clinicopathologic low-risk indicators that are at risk for occult extrauterine disease and recurrence and those with clinicopathologic high-risk indicators that are insensitive to standard therapeutic regimens.
Methods
Study population
The initial report of the ECs curated in the TCGA database consisted of 239 cases with documented DNA sequencing, RNA expression, and sufficient surveillance data (6). According to TCGA molecular classification of these cases, 30 tumors had POLE variations, 58 had MSI-H, 62 had TP53 variations (presumed copy-number high), and an estimated 89 had NSMP (i.e., copy-number low). We previously reported the integration of genomic, transcriptomic, and clinical outcomes data for these cases, which culminated in the generation of the ECPPF stratification system (21). However, the molecular-based stratification of clinical outcomes for these EC cases may have been biased by a relatively high percentage (26%) of tumors with high-risk TP53 variations; therefore, we limited the current study population to only the 147 cases of MSI-H and NSMP EC, which consisted predominantly of early-stage, low-grade tumors. As previously reported, the EC tumors included in TCGA were surgically staged, and adjuvant therapy consisted of chemotherapy and/or RT, with 98% of patients who received chemotherapy receiving PbCT (6).
Data acquisition
TCGA data relevant to EC (6) were downloaded as normalized, formatted, and organized data for integration and analysis as described in previous studies (21, 22). All data collection and processing, including the informed consent process, were performed after approval by the local institutional review board or ethics committee for each contributing institution and in accordance with TCGA Human Subjects Protection and Data Access Policies adopted by the National Cancer Institute and the National Human Genome Research Institute.
Sequence variation analysis
Only validated sequence variations (or TCGA level 3 variations) were used for analysis (6). Sequence variant information was abstracted from exome-sequencing data generated with the Illumina Genome Analyzer GAIIx or HiSeq 2000 sequencing platforms (Illumina, Inc). Silent variations were excluded from the analysis, and only frame-shift insertions and deletions, in-frame insertions or deletions, missense, nonsense, nonstop, and splice-site variations were included in the study. For our analysis, the number of variations for each selected gene and for each patient were recorded.
Gene expression
Normalized and log-transformed gene expression data were downloaded as level 3 RNA-sequencing data. These data were generated with Illumina HiSeq 2000 platforms and annotated with the hg19 version of the human genome. Statistical analyses of RNA expression data were performed with R software (The R Foundation) (23) and Bioconductor packages (Bioconductor) (24).
Molecular stratification of MSI-H and NSMP EC
We previously reported the applicability of ECPPF for stratifying the unabridged EC cohort in TCGA independent of histomorphologic variables (21). Restricting the study population to MSI-H and NSMP EC rendered analysis of POLE variations expendable from the ECPPF profiling panel. The log-transformed gene expression equal to or greater than 2.75 for either or both CCNA2 and E2F1 was converted to the quantitative expression sum of CCNA2+E2F1 and categorized as either low expression (sum<4.75) or high expression (sum≥4.75). Assessment of HR gene variations was restricted to the 10 most prevalent genes (ATR, ATM, BRCA2, BRCA1, CDK12, BARD1, NBN, PALB2, CHEK1, and RAD51) reportedly linked to EC and MSI-H (3, 25–28).
Statistical analysis
Data were descriptively summarized as frequency and percentage for categorical variables and median (IQR) for continuous variables. Clinicopathologic and molecular risk factors were compared between the MSI-H and NSMP molecular classification groups by using χ2 or Fisher exact tests for categorical variables. Follow-up duration was calculated from the date of surgical resection to the date of first documented recurrence or latest follow-up. Disease-free survival (DFS) rates were estimated by using the Kaplan-Meier method and compared between groups with log-rank tests. Cox proportional hazards models were fit to evaluate the association between molecular parameters and the risk of recurrence; associations were summarized with hazard ratios and 95% CIs estimated from the models. All calculated P values less than .05 were considered statistically significant. The data were analyzed with SAS, v9.4 (SAS Institute Inc).
Results
Clinicopathologic stratification of MSI-H and NSMP EC
MSI-H (n=58) and NSMP (n=89) tumors had prevalent early-stage, low-grade histopathologic characteristics and 50% or less myometrial invasion (MI) (Table 1). The median (IQR) age of the 147 patients with these tumors was 60 (54-67) years. Recurrences were documented in 26 (18%) cases, and the median (IQR) time to recurrence after surgical resection was 13.9 (10.6-22.9) months. Among the 121 cases without evidence of recurrence, the median (IQR) follow-up period was 28.3 (15.7-44.8) months.
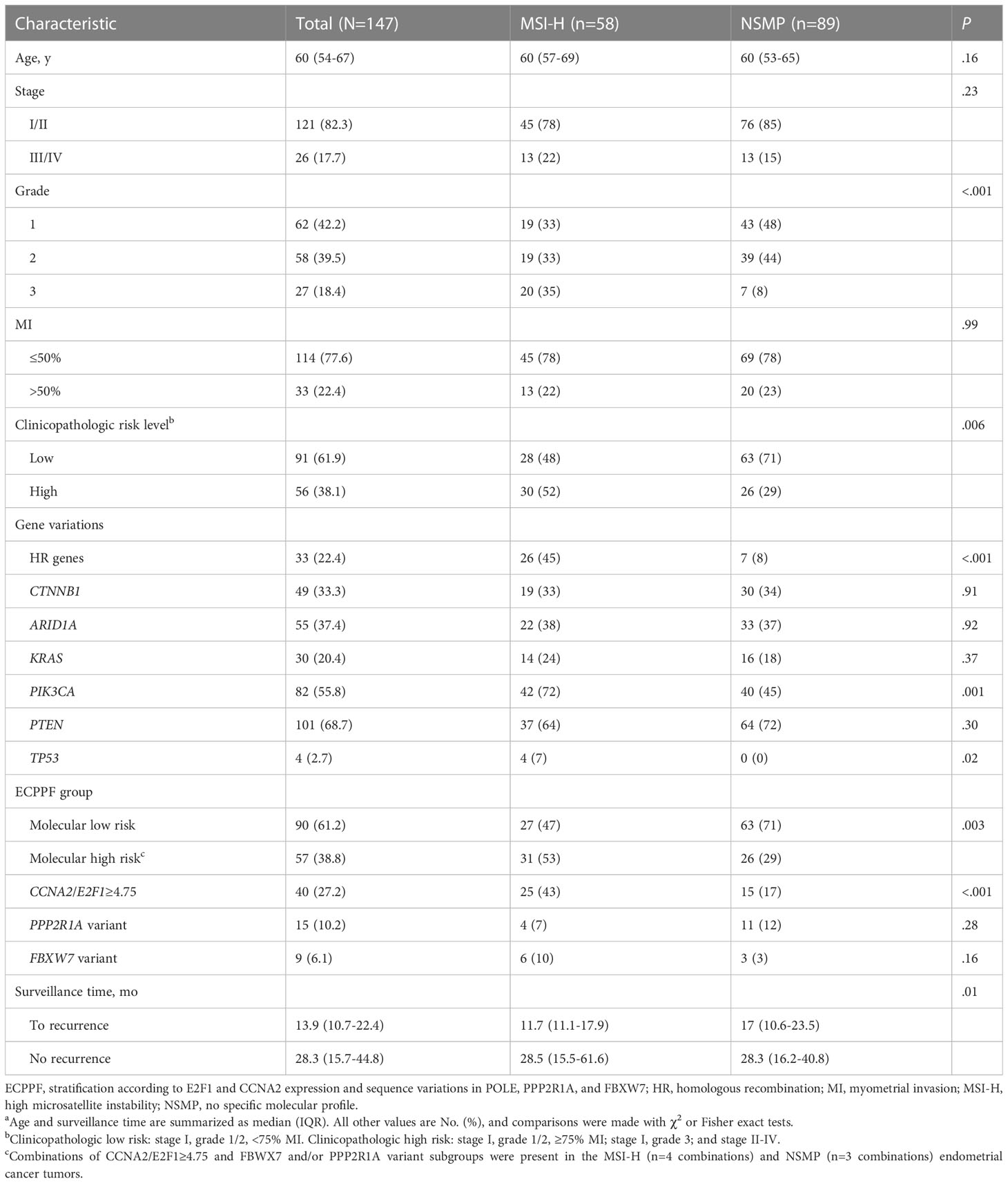
Table 1 Clinicopathologic and molecular characteristics of endometrial cancer with MSI-H and NSMPa.
We integrated tumor stage, grade, and MI data from the MSI-H and NSMP cases to analyze tumors with clinicopathologic low-risk indicators (i.e., stage I, grade 1/2, and <75% MI) and clinicopathologic high-risk indicators (i.e., stage I, grade 1/2, and ≥75% MI; stage I, grade 3; or stage II-IV). Although grade 3 histopathologic and clinicopathologic high-risk indicators were more prevalent in MSI-H tumors than in NSMP tumors, the 3-year DFS rates for patients with MSI-H and NSMP EC did not significantly differ (P=.34) (Figure 1A). The 3-year DFS rate (95% CI) was similar for patients with MSI-H tumors with clinicopathologic low-risk indicators and those with high-risk indicators (72.3% [53.9%-97.0%] vs 74.2% [59.4%-92.7%], P=.98) (Figure 1B). The 3-year DFS rate (95% CI) also did not significantly differ between patients with NSMP tumors with clinicopathologic low-risk indicators and those with high-risk indicators (79.0% [67.9%-92.0%] vs 87.3% [71.8%-100.0%], P=.42) (Figure 1C). Likewise, the 3-year DFS rate for the combined cohort of patients with MSI-H and NSMP ECs did not differ between those with clinicopathologic low-risk indicators (n=91) and those with high-risk indicators (n=56, P=.79) (Figure 1D).
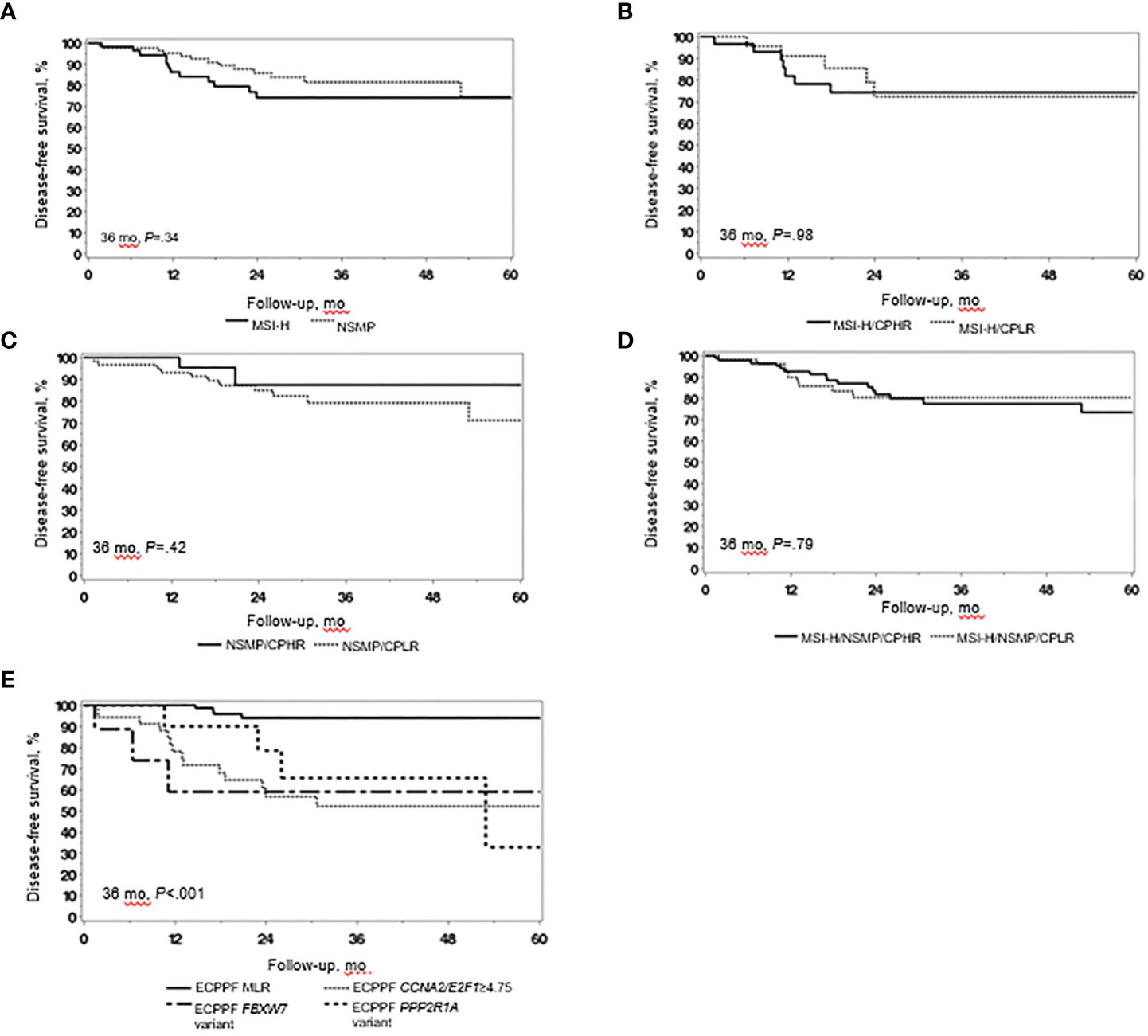
Figure 1 Clinical outcomes in endometrial cancer (EC) with high microsatellite instability (MSI-H) and no specific molecular profile (NSMP). (A) Disease-free survival (DFS) of patients with MSI-H EC (n=58) and NSMP EC (n=89). (B) DFS of patients with MSI-H EC with clinicopathologic low-risk (CPLR) indicators (n=28) and clinicopathologic high-risk (CPHR) indicators (n=30). (C) DFS of patients with NSMP EC with CPLR (n=63) and CPHR (n=26) indicators. (D) DFS of patients with combined MSI-H/NSMP EC with CPLR (n=91) and CPHR (n=56) indicators. (E) DFS of patients with combined MSI-H/NSMP EC (N=147) stratified by ECPPF into the molecular low-risk (MLR) group, characterized by CCNA2/E2F1<4.75 (n=90), and the following molecular high-risk subgroups: CCNA2/E2F1≥4.75 (n=34), FBXW7 variant (n=14), and PPP2R1A variant (n=9). Kaplan-Meier curves for DFS were compared with log-rank tests.
ECPPF stratification of MSI-H/NSMP EC
ECPPF stratified MSI-H/NSMP cases (N=147) into 2 distinct molecular groups with prognostic implications: 1) molecular low risk (MLR), characterized by low CCNA2 and/or E2F1 expression (i.e., CCNA2+E2F1 quantitative expression sum<4.75), and 2) molecular high risk (MHR). The MHR group was further stratified into 3 subgroups: 1) CCNA2/E2F1≥4.75, characterized by high CCNA2 and/or E2F1 expression (i.e., CCNA2+E2F1 quantitative expression sum≥4.75); 2) PPP2R1A variant; and 3) FBXW7 variant. The 3-year DFS rate of the MLR group (n=90) was significantly higher than that of the MHR subgroups (P<.001) (Figure 1E). The hazard ratio (95% CI) was 10.48 (3.46-31.77) for the CCNA2/E2F1≥4.75 subgroup, 16.59 (3.67-74.99) for the FBWX7 variant subgroup, and 7.74 (1.93-30.95) for the PPP2R1A variant subgroup (reference: MLR subgroup, P<.001).
ECPPF stratification of MSI-H EC
We assessed clinical outcomes according to the ECPPF-stratified groups for MSI-H cases (Figure 2A). The 3-year DFS rate (95% CI) was 95.0% (85.9%-100.0%) for the MLR group, 58.2% (39.4%-85.8%) for the CCNA2/E2F1≥4.75 subgroup, and 53.6% (25.7%-100.0%) for the combined PPP2R1A/FBXW7 variant subgroups (P=.01).
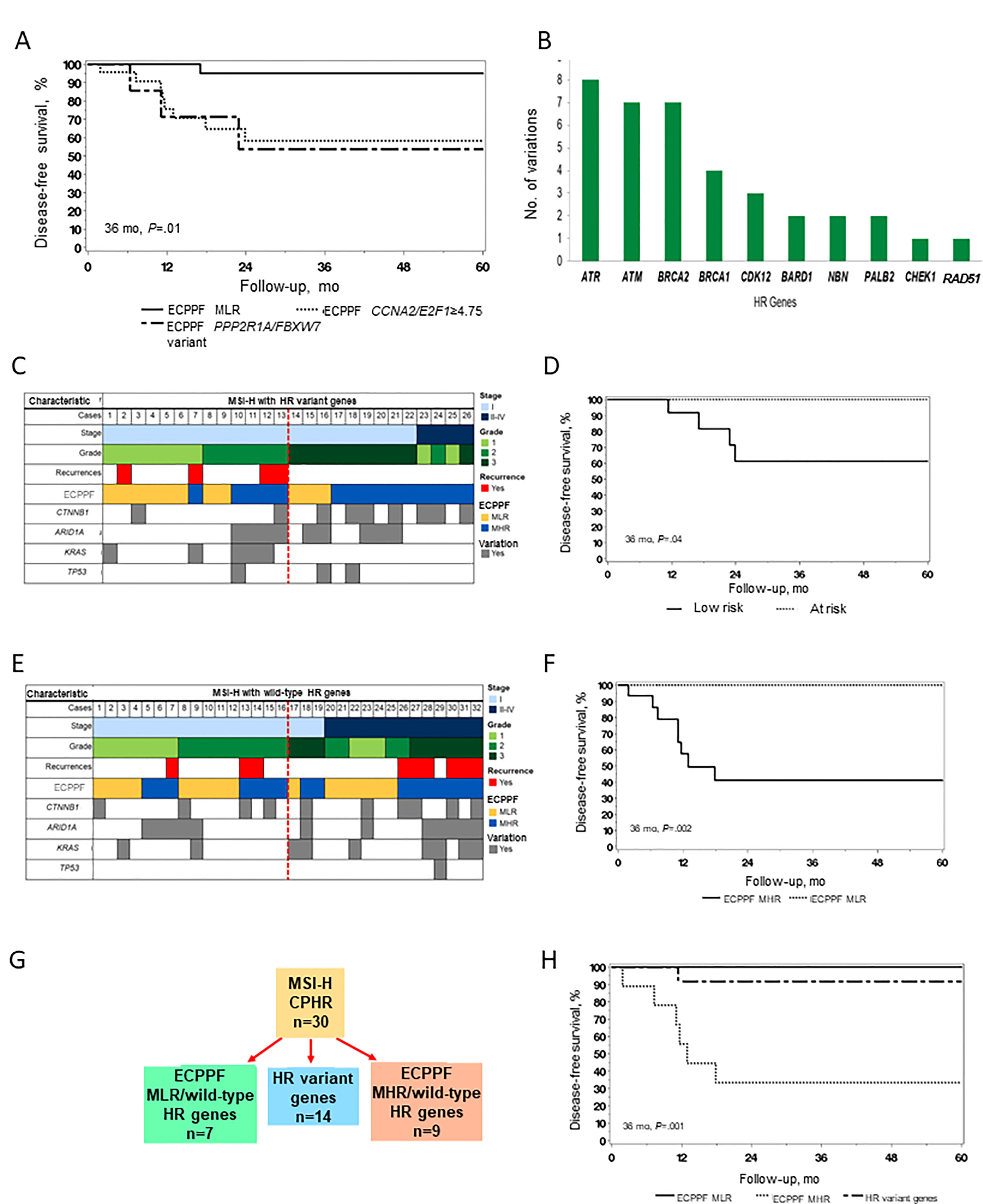
Figure 2 Molecular stratification and clinical outcomes of endometrial cancer (EC) with high microsatellite instability (MSI-H). (A) Disease-free survival (DFS) of patients with MSI-H EC (n=58) according to the ECPPF-stratified molecular low-risk (MLR) group, characterized by CCNA2/E2F1<4.75 (n=27), and the following molecular high-risk (MHR) subgroups: CCNA2/E2F1≥4.75 (n=22) and combined PPP2R1A/FBXW7 variant (n=9). (B) Number of documented sequence variations in select homologous recombination (HR) genes in the MSI-H cohort. (C) Pattern of stage, grade, recurrences, ECPPF-stratified MLR and MHR groups, and variations in CTNNB1, ARID1A, KRAS, and TP53 genes in cases of MSI-H EC with HR variant genes (red line delineates between stage I, grade 1/2 and other tumors). (D) DFS of patients with MSI-H EC with HR variant genes according to low-risk (n=13) and at-risk (n=13) tumors. (E) Pattern of stage, grade, recurrences, ECPPF-stratified MLR and MHR groups, and variations in CTNNB1, ARID1A, KRAS, and TP53 genes in cases of MSI-H EC with wild-type HR genes (red line delineates between stage I, grade 1/2 and other tumors). (F) DFS of patients with MSI-H EC with wild-type HR genes according to ECPPF-stratified MLR (n=16) and MHR (n=16) groups. (G) ECPPF stratification of MSI-H EC cases with clinicopathologic high-risk indicators (CPHR) according to MLR and MHR groups and HR variant gene status. (H) DFS of patients with MSI-H EC with clinicopathologic high-risk indicators according to ECPPF-stratified MLR group with wild-type HR genes (n=7), MLR group with HR variant genes (n=14), and MHR group with wild-type HR genes (n=9). Kaplan-Meier curves for DFS were compared with log-rank tests.
Integrating ECPPF and HR gene variations in MSI-H EC
Among 58 MSI-H cases, 26 (45%) had at least 1 HR gene variation (Table 1). ATR, ATM, BRCA2, and BRCA1 had the most sequence variations (Figure 2B). We used ECPPF to compare clinical outcomes of patients with stage I, grade 1/2 (i.e., low-risk) MSI-H EC tumors with HR variant genes and those with stage I, grade 3 or stage II-IV (i.e., at-risk) MSI-H EC tumors with HR variant genes (generally adjuvant therapy candidates) (Figure 2C). Most of the at-risk MSI-H cases with HR variant genes were stratified by ECPPF as MHR; however, no recurrences were documented in these cases. In contrast, several recurrences were documented in the low-risk MSI-H cases with HR variant genes, and most of these were stratified by ECPPF as MHR (Figure 2C). The 3-year DFS rate was also significantly lower for patients with low-risk MSI-H tumors with HR variant genes than those with at-risk tumors (P=.04) (Figure 2D).
MSI-H tumors with wild-type HR genes were equally distributed among low-risk and at-risk cases. ECPPF stratification of these cases showed differential outcomes for the MLR and MHR groups (Figure 2E). No recurrences were documented in the MSI-H cases with wild-type HR genes that were ECPPF-stratified as MLR, but recurrences were documented in most of the ECPPF-stratified MHR cases. The 3-year DFS rate (95% CI) for patients with MSI-H tumors with wild-type HR genes that were ECPPF-stratified as MHR was 41.0% (21.4%-78.6%), which was significantly lower than that for patients with ECPPF-stratified MLR EC (P=.002) (Figure 2F).
Clinical outcomes were assessed after segregating MSI-H tumors with clinicopathologic high-risk indicators and HR variant genes (n=30). Generally, patients with these tumors are candidates for adjuvant therapy. Clinicopathologic high-risk MSI-H cases were ECPPF-stratified as MLR, HR variant gene, and MHR groups (Figure 2G). The 3-year DFS rate (95% CI) was 100% (100.0%-100.0%) for the MLR group, 91.7% (77.3%-100.0%) for the HR variant gene group, and 33.3% (13.2%-84.0%) for the MHR group (P=.001) (Figure 2H).
ECPPF stratification of NSMP EC
Clinicopathologic low-risk indicators were present more often in NSMP (n=63, 71%) than in MSI-H (n=28, 48%) tumors (P=.006) (Table 1) but failed to predict superior DFS (Figure 1C). ECPPF stratification into the MLR group and MHR CCNA2/E2F1≥4.75 and combined PPP2R1/FBXW7 variant subgroups resulted in significantly different 3-year DFS rates for patients with NSMP EC (P<.001) (Figure 3A). The 3-year DFS rate (95% CI) was 93.6% (86.7%-100.0%) for the MLR group, 36.7% (14.0%-96.0%) for the CCNA2/E2F1≥4.75 subgroup, and 61.1% (32.4%-100.0%) for the combined PPP2R1A/FBXW7 variant subgroups. Furthermore, the hazard ratio (95% CI) was 14.29 (3.39-60.19) for the CCNA2/E2F1≥4.75 subgroup and 9.72 (2.16-43.74) for the combined PPP2R1A/FBXW7 variant subgroups (reference: MLR subgroup, P<.001) (Figure 3B).
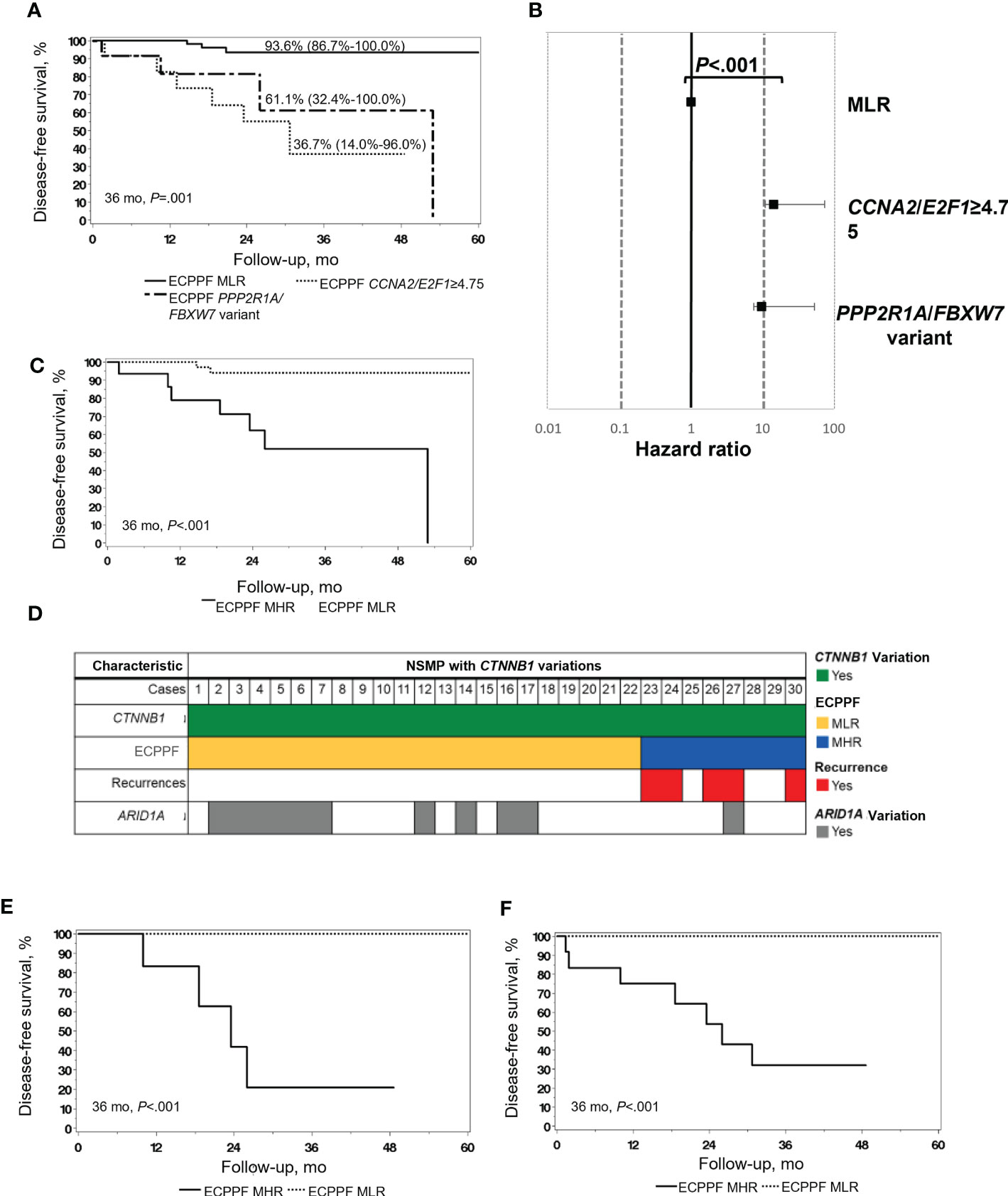
Figure 3 Molecular stratification and clinical outcomes of endometrial cancer (EC) with no specific molecular profile (NSMP). (A) Disease-free survival (DFS) of patients with NSMP EC (n=89) according to the ECPPF-stratified molecular low-risk (MLR) group, characterized by CCNA2/E2F1<4.75 (n=63), and the following molecular high-risk (MHR) subgroups: CCNA2/E2F1≥4.75 (n=12) and combined PPP2R1A/FBXW7 variant (n=14). DFS (95% CI) at 36 months is indicated for each group. (B) Forest plot of Cox proportional hazard ratios for the 3 ECPPF-stratified groups (reference: CCNA2/E2F1<4.75). (C) DFS of patients with stage I, grade 1/2, ≤50% myometrial invasion (MI) NSMP EC (n=55) according to ECPPF-stratified MLR (n=39) and MHR (n=16) groups. (D) Pattern of recurrences, ECPPF-stratified MLR and MHR groups, and variations in CTNNB1 and ARID1A in cases of NSMP with CTNNB1 variations (n=30). (E) DFS of patients with stage I, grade 1/2, ≤50% MI NSMP EC with CTNNB1 variations (n=21) according to ECPPF-stratified MLR (n=15) and MHR (n=6) groups. (F) DFS of patients with NSMP EC with CTNNB1 and ARID1A variations (n=52) according to ECPPF-stratified MLR (n=39) and MHR (n=13) groups. Kaplan-Meier curves for DFS were compared with log-rank tests.
ECPPF stratification of early-stage, low-grade NSMP EC
Because 55 of 89 (62%) NSMP cases were stage I, grade 1/2 with 50% or less MI, which generally confers excellent outcomes without adjuvant chemotherapeutic treatment, we next used ECPPF to identify cases of occult high-risk disease in conventionally low-risk EC. ECPPF stratification of these NSMP early-stage, low-grade cases into MLR and MHR groups resulted in significantly different 3-year DFS rates (P<.001) (Figure 3C). The 3-year DFS rate (95% CI) was 93.9% (86.1%-100.0%) for the MLR group and 51.8% (29.7%-90.5%) for the MHR group. The hazard ratio (95% CI) for the MHR group was 12.10 (2.46-59.42) (reference: MLR group, P=.002).
ECPPF stratification of CTNNB1 variations in NSMP EC
Among 30 CTNNB1-variant NSMP cases, all were grade 1/2, and 22 were ECPPF-stratified as MLR (15 stage I with ≤50% MI, 4 with >50% MI, and 3 stage II/III), and 8 were ECPPF-stratified as MHR (all stage I, and 2 with >50% MI). All documented CTNNB1-variant–associated recurrences occurred in the MHR group (Figure 3D). The 3-year DFS rate for patients with early-stage, low-grade NSMP tumors with CTNNB1 variations significantly differed between the MLR and MHR groups (P<.001) (Figure 3E). ECPPF stratification of NSMP tumors with ARID1A and/or CTNNB1 variations into MLR (n=39) and MHR (n=13) groups showed that all documented recurrences occurred in the MHR group. Consequently, the 3-year DFS rate (95% CI) for the MHR group was 32.1% (12.9%-80.3%), which was significantly lower than that for the MLR group (P<.001) (Figure 3F).
ECPPF stratification predicts recurrence in MSI-H/NSMP EC with clinicopathologic low-risk indicators
ECPPF stratification of MSI-H/NSMP EC with clinicopathologic low-risk indicators (n=91) into the MLR and MHR groups (Figure 4A) yielded distinctly diverse clinical outcomes. The 3-year DFS rate (95% CI) was 93.9% (87.4%-100.0%) for the MLR group (n=61) and 43.8% (26.1%-73.6%) for the MHR group (n=30) (P<.001, Figure 4B). The hazard ratio (95% CI) for the MHR group was 12.37 (3.54-43.30) (reference: MLR group, P<.001).
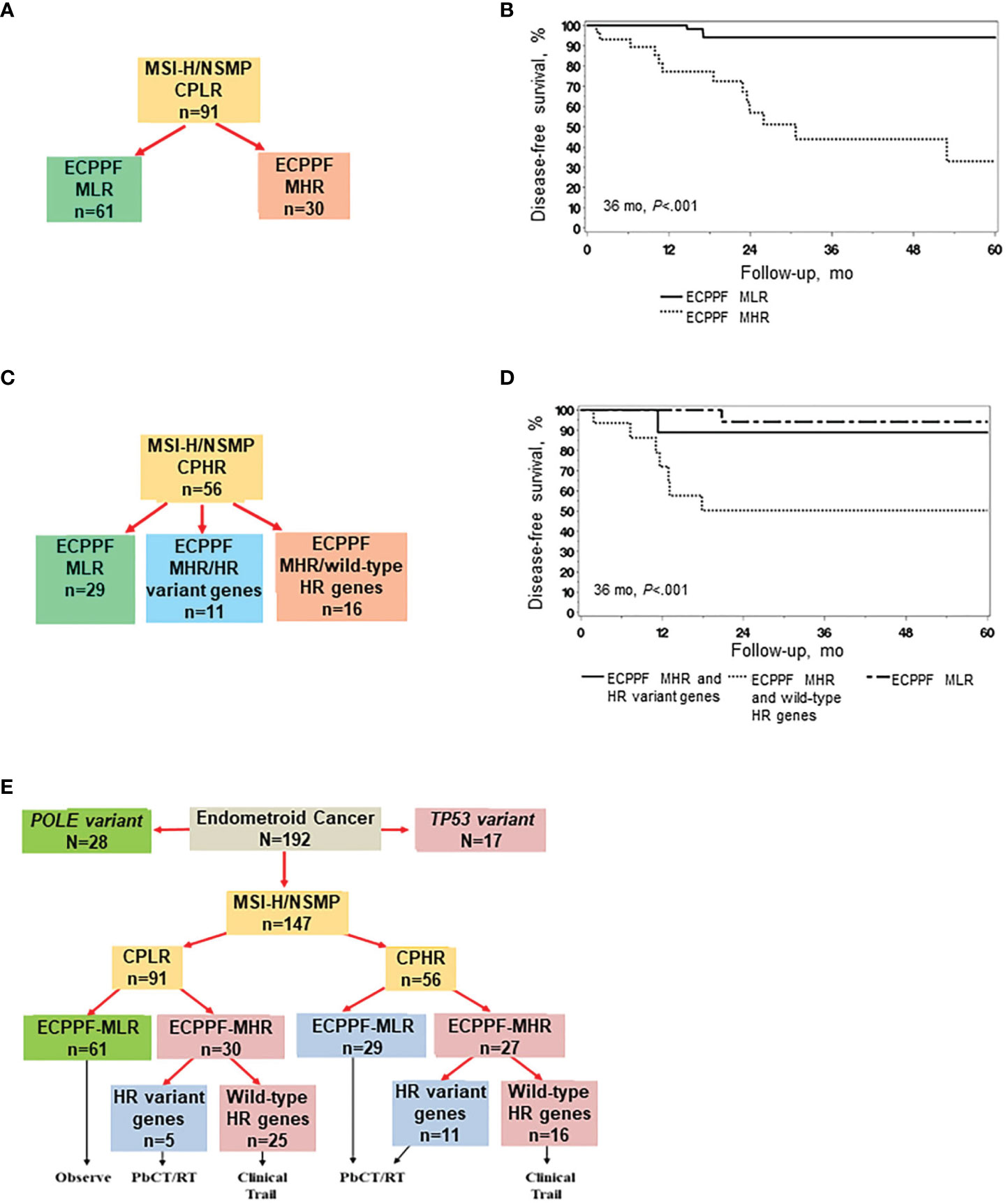
Figure 4 ECPPF stratification of endometrial cancer (EC) with high microsatellite instability (MSI-H) or no specific molecular profile (NSMP) and clinicopathologic low-risk (CPLR) or high-risk (CPHR) indicators. CPLR indicators comprise stage I, grade 1/2, <75% myometrial invasion (MI) tumors, and CPHR indicators comprise stage I, grade 1/2, ≥75% MI; stage I, grade 3; and stage II-IV tumors. (A) Stratification of MSI-H/NSMP cases with CPLR indicators according to the ECPPF-stratified molecular low-risk (MLR) and molecular high-risk (MHR) groups. (B) Disease-free survival (DFS) of patients with MSI-H/NSMP EC with CPLR indicators according to the ECPPF-stratified MLR (n=61) and MHR (n=30) groups. (C) Stratification of MSI-H/NSMP EC cases with CPHR indicators according to the ECPPF-stratified MLR group, MHR group with homologous recombination (HR) variant genes, and MHR group with wild-type HR genes. (D) DFS of patients with MSI-H/NSMP EC with CPHR indicators according to the ECPPF-stratified MLR group (n=29), MHR group with HR variant genes (n=11), and MHR group with wild-type HR genes (n=16). (E) ECPPF and HR variant gene stratification of MSI-H/NSMP cases with CPLR and CPHR indicators. Linkage to molecular-based treatment options is shown for each stratified group. Kaplan-Meier curves for DFS were compared with log-rank tests. PbCT indicates platinum-based chemotherapy; RT, radiotherapy.
ECPPF stratification and HR variant gene status predict treatment sensitivity in MSI-H/NSMP EC with clinicopathologic high-risk indicators
We next assessed treatment sensitivity according to ECPPF stratification and HR variant gene status (Figure 4C). The 3-year DFS rate (95% CI) was significantly higher for the ECPPF-stratified MLR group (94.1% [83.6%-100.0%]) and the MHR group with HR variant genes (88.9% [70.6%-100.0%]) than that for the ECPPF-stratified MHR group with wild-type HR genes (50.3% [29.8%-84.6%]; P<.001) (Figure 4D).
ECPPF stratification, clinicopathologic indicators, and wild-type HR gene status predict treatment sensitivity in MSI-H/NSMP EC
Segregating MSI-H and NSMP cases according to clinicopathologic low-risk and high-risk indicators first and then according to ECPPF-stratified MLR, MHR with HR variant gene, and MHR with wild-type HR gene groups may provide a molecular-based strategy for managing EC. Of 26 total documented recurrences, 21 (81%) occurred in the 41 (51%) ECPPF-stratified MHR group tumors with wild-type HR genes, including 14 of 25 (56%) of these tumors with clinicopathologic low-risk indicators.
Integrating ECPPF molecular stratification and treatment algorithms for MSI-H/NSMP EC in a hypothetical but similar population would most likely entail the following molecular-based therapy recommendations: observation for 41% (67% with clinicopathologic low-risk indicators) of patients, RT and/or PbCT for 31% of patients (including 5% with clinicopathologic low-risk indicators), and innovative clinical trials for 28% of patients (including 27% with clinicopathologic low-risk indicators) (Figure 4E).
Discussion
Endometrioid histopathologic characteristics are present in more than 80% of EC tumors, with most classified as MSI-H or NSMP, low stage, and low grade (6, 11, 29). The observation that MSI-H/NSMP EC with clinicopathologic low-risk indicators accounted for most of the MSI-H/NSMP recurrences denotes major limitations in conventional risk assignment strategies. Improving clinical outcomes requires the identification of molecular vulnerabilities to permit target-specific risk designation and choice of therapeutics. Consequently, we assessed our previously described ECPPF molecular stratification system for its ability to resolve MSI-H/NSMP EC prognoses (21).
Because sensitivity to RT and chemotherapeutics are in part predicated on attenuated DNA-damage repair pathway elements (18, 30, 31), we integrated HR variant gene status into our stratification system. Our findings show that ECPPF prognostically stratifies MSI-H and NSMP ECs and that integration of HR variant gene status may facilitate development of new molecular-based therapeutic strategies. The ECPPF-stratified MHR group identified cases at high risk for recurrence in those conventionally classified as clinicopathologic low risk. ECPPF stratification also identified NSMP EC with variant CTNNB1 as a subgroup with a high risk of recurrence and the MHR group with wild-type HR genes as insensitive to standard adjuvant PbCT and/or RT. In contrast, the ECPPF-stratified MLR group predicted sensitivity to standard therapies (PbCT and/or RT) in EC with clinicopathologic high-risk indicators and/or HR variant genes.
The high TMB associated with MSI-H EC has understandably led to embracing immunotherapy in persistent or recurrent disease (13–16). High TMB also predicts an increased prevalence of HR variant genes and most likely sensitivity to RT and DNA-damaging agents (17). Consistent with this precept, recent studies reported improved clinical outcomes with RT in patients with MSI-H EC (32, 33). In the current study, ECPPF effectively stratified MSI-H cases, in which patients in the MHR group with early-stage, low-grade tumors, who are rarely candidates for adjuvant therapy, had a poor DFS rate. Moreover, the ECPPF-stratified MLR and MHR groups with HR variant genes and clinicopathologic high-risk indicators had higher DFS rates than did the MHR group with wild-type HR genes and clinicopathologic high-risk indicators.
These observations advocate for strategizing treatment options for MSI-H and NSMP EC tumors with clinicopathologic low-risk indicators according to ECPPF-stratified MLR and MHR groups. Thus, pending HR gene status, conservative surveillance is recommended for the MLR group, and early therapeutic intervention (innovative clinical trials) is recommended for the MHR group. Furthermore, a therapeutic strategy for MSI-H and NSMP EC with clinicopathologic high-risk indicators would consist of treatment with contemporary PbCT and/or RT for patients with ECPPF-stratified MLR tumors and MHR tumors with HR variant genes but innovative clinical trials for those with ECPPF-stratified MHR tumors with wild-type HR genes.
Although NSMP EC is primarily early stage and low grade, it is associated with an intermediate prognosis (6, 8–10). This paradox suggests that its high recurrence rate is linked to unrecognized high-risk disease in conventionally classified low-risk EC. ECPPF appeared to effectively stratify NSMP EC cases, particularly for early-stage, low-risk tumors. The MLR group had a distinctly favorable DFS rate over that of the MHR group with occult high-risk disease. CTNNB1 variations in low-grade NSMP EC are reportedly associated with poor patient outcomes (18, 34, 35). ECPPF efficiently stratified NSMP tumors with variant CTNNB1, and all documented recurrences occurred in the MHR group. ECPPF appears to provide a molecular profile for TCGA-designated copy-number low tumors by identifying occult high-risk disease within conventional low-risk cases and high-risk disease that is insensitive to standard adjuvant PbCT and/or RT.
The recently published version of the National Comprehensive Cancer Network guidelines encourages molecular assessment of EC and states that probing for POLE hotspot variations and performing immunohistochemical analysis of mismatch repair deficiency and wild-type TP53 expression may identify MSI-H and NSMP, respectively (36). However, without specific profiling of adverse molecular drivers, targetable vulnerabilities remain unrecognized. Therefore, ECPPF classification provides new profiling insights to refine prognoses and improve therapeutic efficacy.
The predominant challenges for treating MSI-H and NSMP ECs are distinguishing occult high-risk disease in conventionally characterized histomorphologic low-risk EC and predicting insensitivity of EC with clinicopathologic high-risk indicators to contemporary therapies (12). The 43.8% 3-year DFS rate of the ECPPF-stratified MHR group with clinicopathologic low-risk indicators is the first evidence that molecular stratification can distinguish high-risk disease in conventionally classified low-risk MSI-H/NSMP EC. Importantly, 81% of documented recurrences occurred in the ECPPF-stratified MHR group with wild-type HR genes which accounted for only 28% of all TCGA MSI-H/NSMP EC cases. Furthermore, our analysis of MSI-H/NSMP EC with clinicopathologic high-risk indicators showed that the 3-year DFS rate of the MLR group and MHR group with HR variant genes was considerably disparate from that of the MHR group with wild-type HR genes. This finding is consistent with treatment sensitivity to standard therapeutic regimens in the MLR group and MHR group with HR variant genes and treatment insensitivity in the MHR group with wild-type HR genes. These observations introduce a potential molecular-based paradigm shift for precision therapeutics in MSI-H and NSMP ECs. On the basis of our previously reported molecular schematic of disrupted EC signaling pathways (21), reasonable therapeutic options may include modulators of the CCNA2-E2F1-CIP2A axis or PP2A activity and FBXW7 substrates alone or in combination with PbCT or PARP inhibitors (27, 36–40).
Strengths of our analyses include the size of the patient population, centralized pathology review, and availability of robust genomic and transcriptomic databases. Assessing the primary clinical outcome of DFS rather than overall survival is an additional strength of the study that afforded analysis of limitations in contemporary staging, prognostication, and therapeutic efficacy. Limitations of this study included the variability and length of follow-up periods. Because the upper quartile of time to recurrence exceeded the lower quartile of follow-up time, the extent of recurrences in our study population may be underestimated. Recent meta-analyses of prognostic indicators independent of the TCGA signature suggest that lymphovascular space involvement (LVSI) has prognostic value for EC and that deep MI may be a predictor of recurrence (41, 42). LVSI information was not accessible in the TCGA study population and warrants inclusion in subsequent ECPPF stratification studies. In previously reported multivariate analyses incorporating the ECPPF elements with numerous traditional risk factors, deep MI was not an independently significant risk factor (21, 43). Moreover, definitive information in TCGA regarding treatment strategies for the patients was not available. However, because specimens were submitted chiefly from comprehensive cancer centers, it is reasonable to assume that the therapies were consistent with contemporary established guidelines. Such therapies include surgical staging and adjuvant RT and/or chemotherapy in patients with clinically identified at-risk disease; as such, 98% of patients receiving chemotherapy received PbCT (6). Knowledge of the sites of recurrence (e.g., local, regional, or distant) would have also been preferable for this study, particularly for tumors with predominantly clinicopathologic low-risk indicators.
In summary, ECPPF identifies occult high-risk disease in MSI-H/NSMP EC with clinicopathologic low-risk indicators and appears to efficiently triage treatment sensitivity of MSI-H/NSMP EC with clinicopathologic high-risk indicators to contemporary adjuvant therapies. Consequently, ECPPF stratification confers a new therapeutic paradigm for MSI-H/NSMP EC: 1) observation only for MLR cases with clinicopathologic low-risk indicators; 2) PbCT and/or RT for MHR cases with HR variant genes and clinicopathologic low-risk indicators, MLR cases with clinicopathologic high-risk indicators, and MHR cases with HR variant genes; and 3) innovative clinical trials for MHR cases with wild-type HR genes and clinicopathologic low-risk or high-risk indicators.
Expanded gene symbols
CCNA2, cyclin A2; CTNNB1, catenin beta 1; E2F1, E2F transcription factor 1; FBXW7, F-box and WD repeat domain-containing protein 7; POLE, DNA polymerase epsilon, catalytic subunit; PPP2R1A, protein phosphatase 2 scaffold subunit alpha.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article.
Author contributions
Conceptualization: JG-B and KP; Data curation: JG-B and KP; Formal analysis: JG-B, JB-G, BK, KH, and KP; Funding acquisition: SD, AF, and KP; Investigation: JG-B, SW, JB-G, AW, MM, SD, AF, BK, KH, SY, FC, and KP; Methodology: JG-B, BK, KH, and KP; Project administration: KP; Resources: JG-B and KP; Software: JG-B, AW, and MM; Supervision: KP; Pathology: BK, KH, and FC; Writing – original draft: JG-B, JB-G, AW, MM, BK, KH, SY, and KP; Writing – review and editing: JG-B, SW, JB-G, AW, MM, SD, AF, BK, KH, SY, FC, and KP. All authors contributed to the article and approved the submitted version.
Funding
The study was internally funded by Mayo Clinic, which had no involvement in the study design; collection, analysis, and interpretation of data; writing of the report; or decision to submit the article for publication.
Acknowledgments
Nisha Badders, PhD, ELS, Mayo Clinic, substantively edited the manuscript. The Scientific Publications staff at Mayo Clinic provided proofreading and administrative and clerical support. Portions of the Methods section were previously published in PLoS One. 2021 Jan;16(1):e0245664 and Gynecol Oncol. 2021 Jul;162(1):182-9.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
DFS, disease-free survival; EC, endometrial cancer; ECPPF, stratification with E2F1 and CCNA2 expression and sequence variations in POLE, PPP2R1A, and FBXW7; HR, homologous recombination; LVSI, lymphovascular space involvement; MHR, molecular high risk; MI, myometrial invasion; MLR, molecular low risk; MSI-H, high microsatellite instability; NSMP, no specific molecular profile; PbCT, platinum-based chemotherapy; RT, radiotherapy; TCGA, The Cancer Genome Atlas; TMB, tumor mutation burden.
References
1. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol (1983) 15:10–7. doi: 10.1016/0090-8258(83)90111-7
2. Hamilton CA, Pothuri B, Arend RC, Backes FJ, Gehrig PA, Soliman PT, et al. Endometrial cancer: a society of gynecologic oncology evidence-based review and recommendations. Gynecol Oncol (2021) 160:817–26. doi: 10.1016/j.ygyno.2020.12.021
3. Deshpande M, Romanski PA, Rosenwaks Z, Gerhardt J. Gynecological cancers caused by deficient mismatch repair and microsatellite instability. Cancers (Basel). (2020) 12. doi: 10.3390/cancers12113319
4. Ashley CW, Da Cruz Paula A, Kumar R, Mandelker D, Pei X, Riaz N, et al. Analysis of mutational signatures in primary and metastatic endometrial cancer reveals distinct patterns of DNA repair defects and shifts during tumor progression. Gynecol Oncol (2019) 152:11–9. doi: 10.1016/j.ygyno.2018.10.032
5. Wallbillich JJ, Morris RT, Ali-Fehmi R. Comparing mutation frequencies for homologous recombination genes in uterine serous and high-grade serous ovarian carcinomas: a case for homologous recombination deficiency testing in uterine serous carcinoma. Gynecol Oncol (2020) 159:381–6. doi: 10.1016/j.ygyno.2020.08.012
6. Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. (2013) 497:67–73. doi: 10.1038/nature12113
7. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
8. Stelloo E, Nout RA, Osse EM, Jurgenliemk-Schulz IJ, Jobsen JJ, Lutgens LC, et al. Improved risk assessment by integrating molecular and clinicopathological factors in early-stage endometrial cancer-combined analysis of the PORTEC cohorts. Clin Cancer Res (2016) 22:4215–24. doi: 10.1158/1078-0432.CCR-15-2878
9. Leon-Castillo A, de Boer SM, Powell ME, Mileshkin LR, Mackay HJ, Leary A, et al. Molecular classification of the PORTEC-3 trial for high-risk endometrial cancer: impact on prognosis and benefit from adjuvant therapy. J Clin Oncol (2020) 38:3388–97. doi: 10.1200/JCO.20.00549
10. Pasanen A, Loukovaara M, Ahvenainen T, Vahteristo P, Butzow R. Differential impact of clinicopathological risk factors within the 2 largest ProMisE molecular subgroups of endometrial carcinoma. PloS One (2021) 16:e0253472. doi: 10.1371/journal.pone.0253472
11. Kolehmainen A, Pasanen A, Tuomi T, Koivisto-Korander R, Butzow R, Loukovaara M. Clinical factors as prognostic variables among molecular subgroups of endometrial cancer. PloS One (2020) 15:e0242733. doi: 10.1371/journal.pone.0242733
12. Jamieson A, Bosse T, McAlpine JN. The emerging role of molecular pathology in directing the systemic treatment of endometrial cancer. Ther Adv Med Oncol (2021) 13. 17588359211035959. doi: 10.1177/17588359211035959
13. Eggink FA, Van Gool IC, Leary A, Pollock PM, Crosbie EJ, Mileshkin L, et al. Immunological profiling of molecularly classified high-risk endometrial cancers identifies POLE-mutant and microsatellite unstable carcinomas as candidates for checkpoint inhibition. Oncoimmunology. (2017) 6:e1264565. doi: 10.1080/2162402X.2016.1264565
14. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite Instability/Mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol (2020) 38:1–10. doi: 10.1200/JCO.19.02105
15. Makker V, Taylor MH, Aghajanian C, Oaknin A, Mier J, Cohn AL, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer. J Clin Oncol (2020) 38:2981–92. doi: 10.1200/JCO.19.02627
16. Makker V, Colombo N, Herráez AC, Santin A, Colomba E, Miller D, et al. A multicenter, open-label, randomized, phase III study to compare the efficacy and safety of lenvatinib in combination with pembrolizumab versus treatment of physician's choice in patients with advanced endometrial cancer. Gynecologic Oncol (2021) 162:S4. doi: 10.1016/S0090-8258(21)00657-0
17. Zhao H, Thienpont B, Yesilyurt BT, Moisse M, Reumers J, Coenegrachts L, et al. Mismatch repair deficiency endows tumors with a unique mutation signature and sensitivity to DNA double-strand breaks. Elife. (2014) 3:e02725. doi: 10.7554/eLife.02725.041
18. Damia G, Broggini M. Platinum resistance in ovarian cancer: role of DNA repair. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11010119
19. Garutti M, Pelizzari G, Bartoletti M, Malfatti MC, Gerratana L, Tell G, et al. Platinum salts in patients with breast cancer: a focus on predictive factors. Int J Mol Sci (2019) 20:3390. doi: 10.3390/ijms20143390
20. da Costa A, Baiocchi G. Genomic profiling of platinum-resistant ovarian cancer: the road into druggable targets. Semin Cancer Biol (2021) 77:29–41. doi: 10.1016/j.semcancer.2020.10.016
21. Bosquet JG, Zhang Q, Cliby WA, Bakkum-Gamez JN, Cen L, Dowdy SC, et al. Association of a novel endometrial cancer biomarker panel with prognostic risk, platinum insensitivity, and targetable therapeutic options. PloS One (2021) 16:e0245664. doi: 10.1371/journal.pone.0245664
22. Gonzalez-Bosquet J, Bakkum-Gamez JN, Weaver AL, McGree ME, Dowdy SC, Famuyide AO, et al. PP2A and E3 ubiquitin ligase deficiencies: seminal biological drivers in endometrial cancer. Gynecol Oncol (2021) 162:182–9. doi: 10.1016/j.ygyno.2021.04.008
23. Core Team R. R: a language and environment for statistical computing (2013). Available at: https://www.R-project.org.
24. Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. Orchestrating high-throughput genomic analysis with bioconductor. Nat Methods (2015) 12:115–21. doi: 10.1038/nmeth.3252
25. Bilbao C, Ramirez R, Rodriguez G, Falcon O, Leon L, Diaz-Chico N, et al. Double strand break repair components are frequent targets of microsatellite instability in endometrial cancer. Eur J Cancer. (2010) 46:2821–7. doi: 10.1016/j.ejca.2010.06.116
26. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. (2016) 16:110–20. doi: 10.1038/nrc.2015.21
27. Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen WJ, et al. Prevalence of homologous recombination-related gene mutations across multiple cancer types. JCO Precis Oncol (2018) 2018. doi: 10.1200/PO.17.00286
28. de Jonge MM, Auguste A, van Wijk LM, Schouten PC, Meijers M, Ter Haar NT, et al. Frequent homologous recombination deficiency in high-grade endometrial carcinomas. Clin Cancer Res (2019) 25:1087–97. doi: 10.1158/1078-0432.CCR-18-1443
29. Bell DW, Ellenson LH. Molecular genetics of endometrial carcinoma. Annu Rev Pathol (2019) 14:339–67. doi: 10.1146/annurev-pathol-020117-043609
30. Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med (2015) 66:129–43. doi: 10.1146/annurev-med-081313-121208
31. Brasseur K, Gevry N, Asselin E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget. (2017) 8:4008–42. doi: 10.18632/oncotarget.14021
32. Reijnen C, Kusters-Vandevelde HVN, Prinsen CF, Massuger L, Snijders M, Kommoss S, et al. Mismatch repair deficiency as a predictive marker for response to adjuvant radiotherapy in endometrial cancer. Gynecol Oncol (2019) 154:124–30. doi: 10.1016/j.ygyno.2019.03.097
33. McEachron J, Zhou N, Spencer C, Chatterton C, Shanahan L, Katz J, et al. Adjuvant chemoradiation associated with improved outcomes in patients with microsatellite instability-high advanced endometrial carcinoma. Int J Gynecol Cancer. (2021) 31:203–8. doi: 10.1136/ijgc-2020-001709
34. Kurnit KC, Kim GN, Fellman BM, Urbauer DL, Mills GB, Zhang W, et al. CTNNB1 (beta-catenin) mutation identifies low grade, early stage endometrial cancer patients at increased risk of recurrence. Mod Pathol (2017) 30:1032–41. doi: 10.1038/modpathol.2017.15
35. De Leo A, de Biase D, Lenzi J, Barbero G, Turchetti D, Grillini M, et al. ARID1A and CTNNB1/beta-catenin molecular status affects the clinicopathologic features and prognosis of endometrial carcinoma: implications for an improved surrogate molecular classification. Cancers (Basel) (2021) 13:950. doi: 10.3390/cancers13050950
36. Abu-Rustum N, Yashar C, Arend R, Barber E, Bradley K, Brooks R, et al. Uterine neoplasms, version 1.2023, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw (2023) 21:181–209. doi: 10.6004/jnccn.2023.0006
37. Westermarck J. Targeted therapies don't work for a reason; the neglected tumor suppressor phosphatase PP2A strikes back. FEBS J (2018) 285:4139–45. doi: 10.1111/febs.14617
38. Remmerie M, Janssens V. PP2A: a promising biomarker and therapeutic target in endometrial cancer. Front Oncol (2019) 9:462. doi: 10.3389/fonc.2019.00462
39. Kim H, Ronai ZA. PRMT5 function and targeting in cancer. Cell Stress. (2020) 4:199–215. doi: 10.15698/cst2020.08.228
40. Lodovichi S, Cervelli T, Pellicioli A, Galli A. Inhibition of DNA repair in cancer therapy: toward a multi-target approach. Int J Mol Sci (2020) 21:6684. doi: 10.3390/ijms21186684
41. Raffone A, Travaglino A, Raimondo D, Neola D, Renzulli F, Santoro A, et al. Prognostic value of myometrial invasion and TCGA groups of endometrial carcinoma. Gynecol Oncol (2021) 162:401–6. doi: 10.1016/j.ygyno.2021.05.029
42. Raffone A, Travaglino A, Raimondo D, Neola D, Maletta M, Santoro A, et al. Lymphovascular space invasion in endometrial carcinoma: a prognostic factor independent from molecular signature. Gynecol Oncol (2022) 165:192–7. doi: 10.1016/j.ygyno.2022.01.013
43. Gonzalez-Bosquet J, Weroha SJ, Bakkum-Gamez JN, Weaver AL, McGree ME, Dowdy SC, et al. ECPPF (E2F1, CCNA2, POLE, PPP2R1A, FBXW7) stratification: profiling high-risk subtypes of histomorphologically low-risk and treatment-insensitive endometrioid endometrial cancer. PloS One (2022) 17:e0278408. doi: 10.1371/journal.pone.0278408
Keywords: CCNA2, ECPPF, E2F1, endometrial cancer, FBXW7, MSI, NSMP, PPP2R1A
Citation: Gonzalez-Bosquet J, Weroha SJ, Bakkum-Gamez JN, Weaver AL, McGree ME, Dowdy SC, Famuyide AO, Kipp BR, Halling KC, Yadav S, Couch FJ and Podratz KC (2023) Prognostic stratification of endometrial cancers with high microsatellite instability or no specific molecular profile. Front. Oncol. 13:1105504. doi: 10.3389/fonc.2023.1105504
Received: 22 November 2022; Accepted: 09 May 2023;
Published: 23 May 2023.
Edited by:
Sarah M. Temkin, National Institutes of Health (NIH), United StatesReviewed by:
Diego Raimondo, University of Bologna, ItalyKomsun Suwannarurk, Thammasat University, Thailand
Copyright © 2023 Gonzalez-Bosquet, Weroha, Bakkum-Gamez, Weaver, McGree, Dowdy, Famuyide, Kipp, Halling, Yadav, Couch and Podratz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karl C. Podratz, cG9kcmF0ei5rYXJsQG1heW8uZWR1