 Charalampos G. Pontikoglou
Charalampos G. Pontikoglou Angelos Matheakakis1,2
Angelos Matheakakis1,2 Helen A. Papadaki
Helen A. Papadaki
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 25 January 2023
Sec. Hematologic Malignancies
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1102495
This article is part of the Research Topic Biological Aspects of Bone Marrow Failure View all 9 articles
Myelodysplastic syndromes include a broad spectrum of malignant myeloid disorders that are characterized by dysplastic ineffective hematopoiesis, reduced peripheral blood cells counts and a high risk of progression to acute myeloid leukemia. The disease arises primarily because of accumulating chromosomal, genetic and epigenetic changes as well as immune-mediated alterations of the hematopoietic stem cells (HSCs). However, mounting evidence suggests that aberrations within the bone marrow microenvironment critically contribute to myelodysplastic syndrome (MDS) initiation and evolution by providing permissive cues that enable the abnormal HSCs to grow and eventually establish and propagate the disease. Mesenchymal stromal cells (MSCs) are crucial elements of the bone marrow microenvironment that play a key role in the regulation of HSCs by providing appropriate signals via soluble factors and cell contact interactions. Given their hematopoiesis supporting capacity, it has been reasonable to investigate MSCs’ potential involvement in MDS. This review discusses this issue by summarizing existing findings obtained by in vitro studies and murine disease models of MDS. Furthermore, the theoretical background of targeting the BM-MSCs in MDS is outlined and available therapeutic modalities are described.
Within the adult Bone Marrow (BM) hematopoiesis take place in specialized microenvironments called niches that regulate the balance between quiescence, proliferation, differentiation and self-renewal of hematopoietic stem and progenitor cells (HSPCs) (1). The BM niche is composed of a network of cells of hematopoietic and non-hematopoietic origin as well as of extracellular matrix, which provide the structural support, the physical interactions and the molecular cues for hematopoietic stem cell (HSC) maintenance and function (1). Cells of hematopoietic lineage include lymphocytes, macrophages, osteoclasts, megakaryocytes as well as myeloid derived suppressor cells (MDSCs), while non-hematopoietic components comprise mesenchymal stromal cells (MSCs) and their progeny, vascular endothelial cells (ECs), fibroblasts, sympathetic neurons and non-myelinating Schwann cells collectively contributing to BM homeostasis (Figure 1). For a more detailed discussion of the niche the reader is referred to some excellent reviews (1–4).
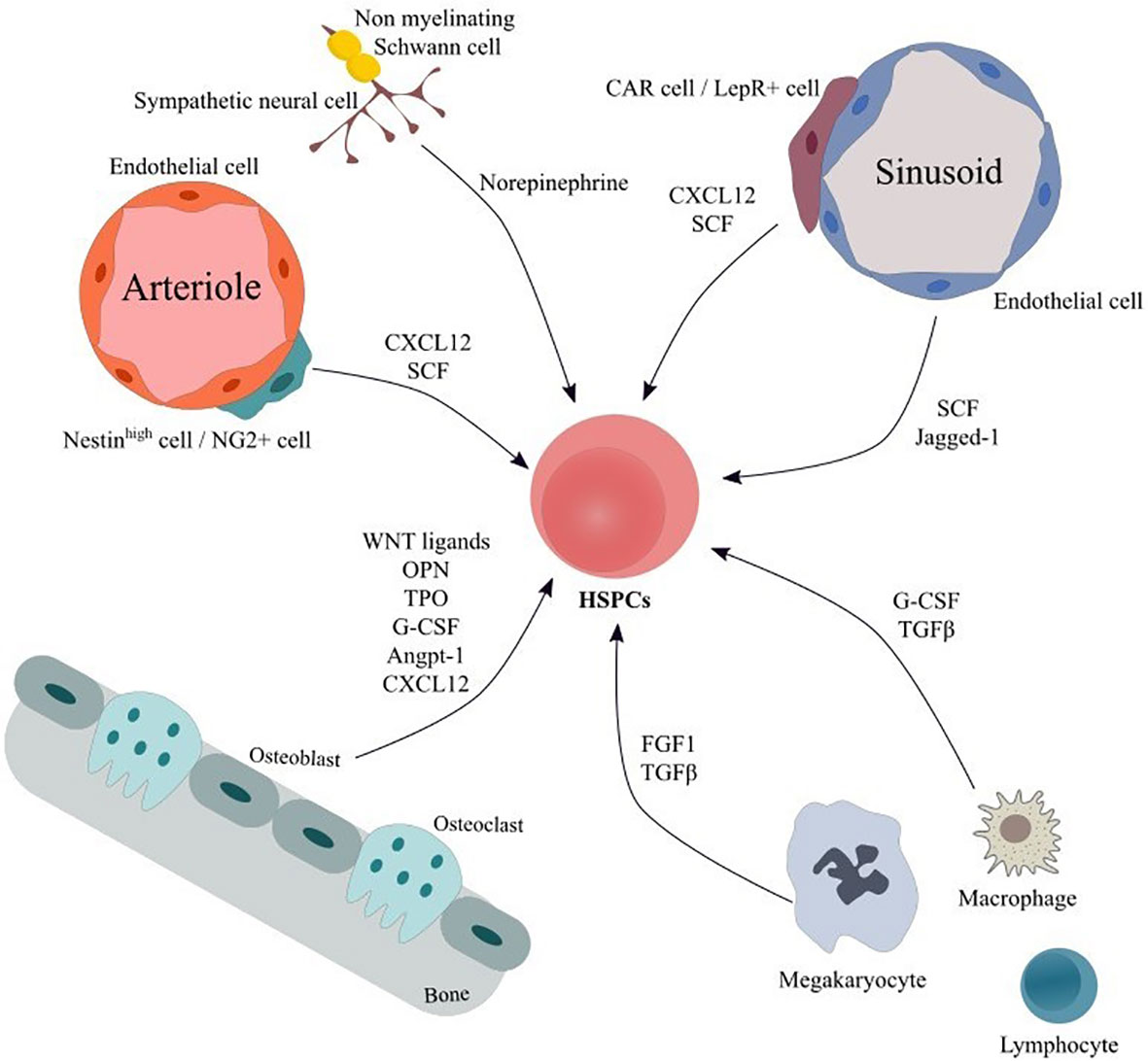
Figure 1 Main cellular components and soluble factors of hematopoietic HSC niche. The bone marrow niche comprises a variety of cellular populations embedded in the extracellular matrix. Cells of hematopoietic origin including macrophages, megakaryocytes and lymphocyte subgroups interact both via cell-to-cell connection and via secretion of soluble factor thus modulating HSPCs’ proliferation, differentiation and activation. ECs form a vast net of arterioles, sinusoids and capillaries and contribute in HSPC support by secreting factors, such as SCF and Notch ligands. MSC subpopulations modulate HSPCs’ maintenance, retention and proliferation and can differentiate to osteoblasts, adipocytes and chondrocytes. In addition, sympathetic neuronal cells act on HSPCs via adrenergic signaling, thereby inducing hematopoietic stem cells’ egress from the BM. Finally, Schwann cells may regulate hematopoietic stem cells’ quiescence. Angpt-1, Angiopoietin-1; CAR cells, CXCL12-abundant reticular cells; CXCL12, C-X-C motif chemokine ligand 12; ECs, endothelial cells; FGF1, fibroblast growth factor 1; G-CSF, granulocyte-colony stimulating factor; HSPCs, Hematopoietic stem and progenitor cells; LepR+ cell, leptin receptor+ cell; MSC, mesenchymal stromal cells; NG2+ cells, neural–glial antigen 2+ cells; OPN, Osteopontin; SCF, stem cell factor; TGFβ, transforming growth factor beta; TPO, Thrombopoietin.
As far as BM-MSCs are concerned, they consist of a minor and a heterogeneous population of perivascular cells with broad immunoregulatory properties that have the potential to differentiate into osteoblasts, chondrocytes and adipocytes and support hematopoiesis (1–3) (5). Using appropriate mouse models, various BM-MSC subsets with partially overlapping characteristics have been identified, such as CXCL12-abundant reticular (CAR) cells, leptin receptor (LepR)+ cells, nestin+ cells and neural–glial antigen 2 (NG2)+ cells (6–8) (Figure 1). Nestinbright and NG2+ cells are associated with arterioles, whereas CAR cells, LepR+ cells and nestindim cells are localized around sinusoids (9, 10). The aforementioned BM-MSC subpopulations are involved in the maintenance, proliferation, and retention of HSCs (1–3).
Due to the low frequency of MSCs within the BM, data regarding their impact in regulating the homeostasis of HSCs is mainly derived from studies using ex vivo-expanded cells. The latter are defined based on the three minimal criteria established by the International Society for Cellular Therapy (ISCT) (11) i.e. (a) adherence to plastic, (b) expression of the surface antigens CD73, CD90, CD105 while lacking the expression of the hematopoietic and endothelial molecules CD11b, CD14, CD19, CD34, CD45, CD79a, CD11b and HLA-DR, and (c) in vitro differentiation into three mesodermal lineages (osteoblasts, adipocytes, chondrocytes).
As the crucial role of BM-MSCs and their progeny in the control of hematopoiesis is increasingly being acknowledged (12, 13), it may be reasonable to consider the possibility that defects of these cell populations are involved in the establishment and/or propagation of hematological malignancies. Myelodysplastic syndromes represent an attractive disease model to investigate this hypothesis.
Myelodysplastic syndromes consist of an heterogeneous group of clonal hematological disorders characterized by ineffective dysplastic hematopoiesis, peripheral blood cytopenias and an increased risk of transformation into acute myeloid leukemia (AML) (14). Patients with Myelodysplastic Syndrome (MDS) may be largely asymptomatic with mild cytopenias and long-life expectancy or they may exhibit profound symptoms, significantly reduced blood counts, and a very poor prognosis. The former are classified under the lower-risk (LR) group, whereas the latter under the higher-risk (HR) group, based on the International Prognostic Scoring System (IPSS) categorization (14). Notably, these groups differ in disease pathogenesis, risk of disease progression and survival and are managed with different therapeutic modalities (14). The pathogenesis of MDS development and disease progression to AML has long been recognized to involve accumulation of cytogenetic, genetic and epigenetic aberrations as well as immune-mediated alterations of hematopoietic cells (14). However, histologic studies in the 90s had already provided evidence for abnormalities in the components of the BM microenvironment and alterations in the localization of hematopoietic cells within the BM [reviewed in (15)]. These preliminary findings suggested that MDS should not merely be considered as a disorder of the hematopoietic compartment, but rather of the whole tissue.
In the present review, we will attempt to summarize existing knowledge provided by in vitro studies and mouse models supporting the notion of an impaired BM-MSC compartment in MDS and discuss its contribution in the pathogenesis of the disorder. Furthermore, the theoretical background of BM-MSCs’ therapeutic targeting in MDS will be outlined and an overview of relevant in vitro data will be provided (Figure 2).
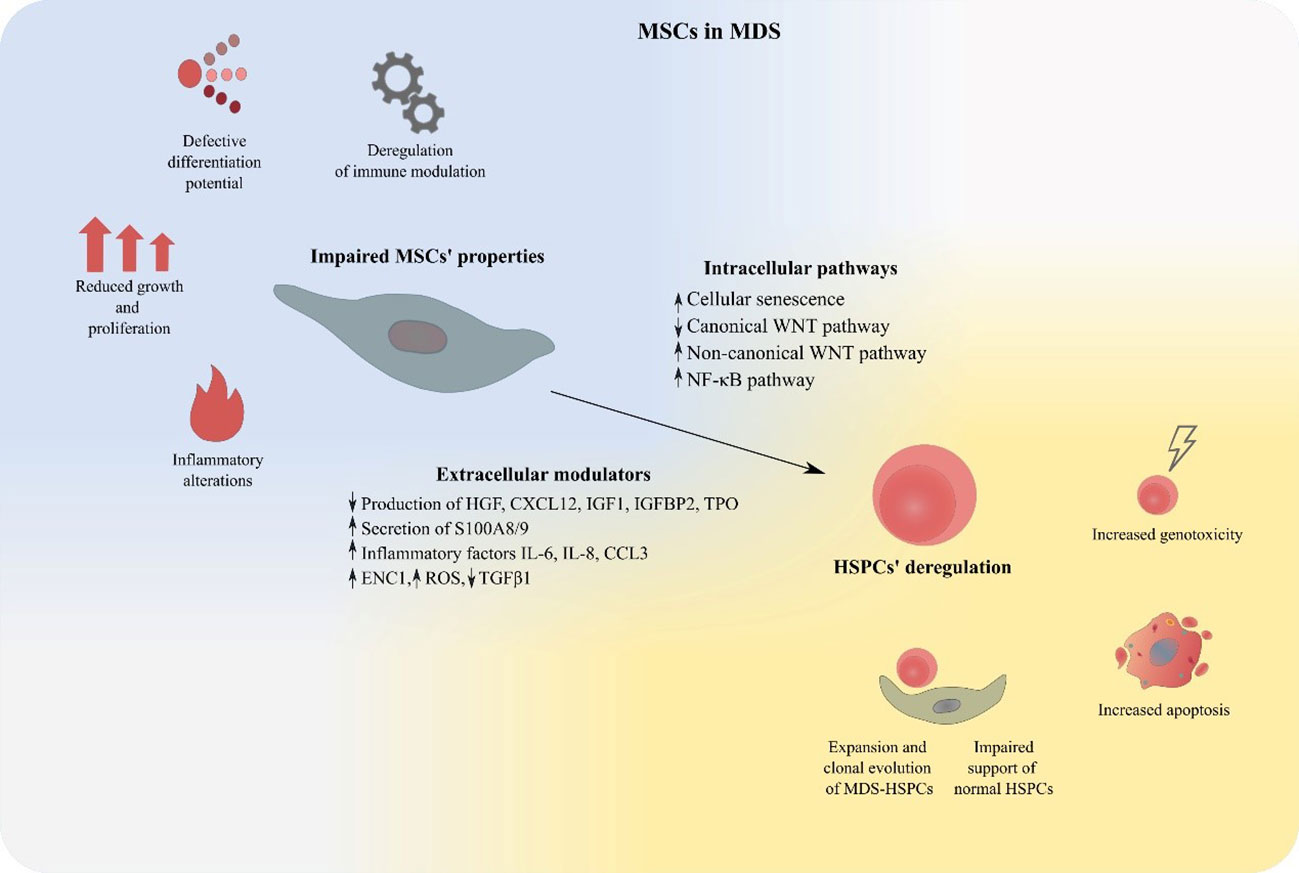
Figure 2 MSCs deregulation in MDS. MDS-derived BM-MSCs exhibit impaired properties regarding their proliferation, differentiation, modulation of the immune system and support of hematopoiesis. Expansion of malignant hematopoietic cells, suppression of normal HSPCs, increased apoptosis and genotoxicity are features that characterize MSC-mediated HSPC deregulation in MDS. BM-MSCs, Bone Marrow Mesenchymal Stromal Cells; HSPCs, Hematopoietic stem and progenitor cells; MDS, MDS Myelodysplastic Syndrome.
Early studies investigating the properties of MDS-derived BM-MSCs were conducted in ex vivo expanded cells, as their in situ counterparts represent only a minor fraction of BM nucleated cells (16). Published data are at times conflicting and this might be due to the variability of experimental approaches and to patient heterogeneity (16). Within this context, cultured MDS-derived BM-MSCs have been reported to exhibit an irregular morphology (Table 1) (17–22, 40), whereas other studies (Table 1) found no morphological abnormalities in patient BM-MSCs as compared to their normal counterparts (23–30). Moreover, most studies (Table 1) suggest that patient BM-MSCs do not differ in terms of immunophenotype from MSCs derived from healthy donors (25–30), although reduced expression of CD90, CD104 and CD105 has also been observed (17, 41).
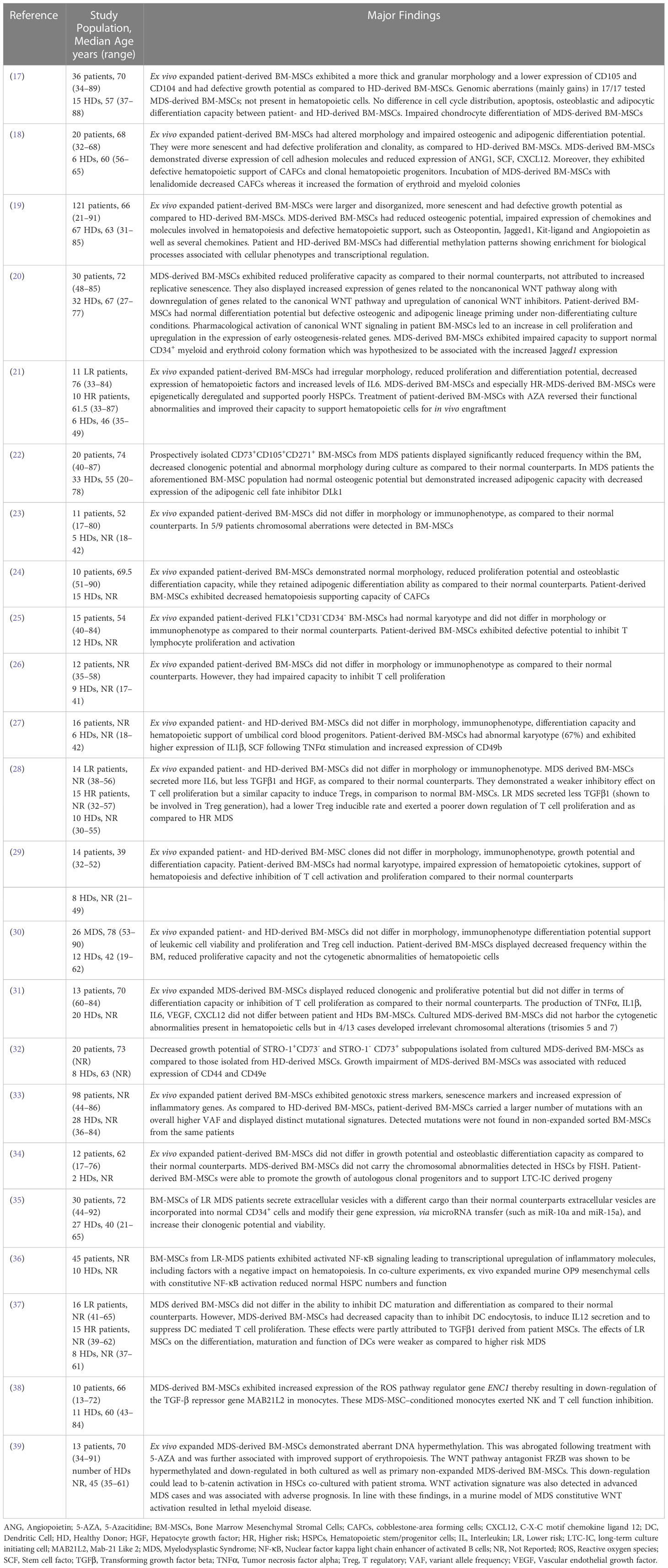
Table 1 In vitro studies of BM-MSCs in MDS.
A number of studies has demonstrated that MDS-derived BM-MSCs display defective growth potential (17–19, 21, 24, 30–32, 42, 43). This has been correlated with decreased expression of CD44 and CD49e (32) and at least in some cases it has been associated with increased cellular senescence (18, 19, 33) (Table 1). The impaired proliferative capacity of MDS-derived BM-MSCs was corroborated by our study as well (20) and it was suggested that this could be attributed to the decreased expression of the canonical WNT signaling pathway and the concomitant up-regulation of the non-canonical pathway. More recently, Falconi et al., confirmed the down-regulation of the canonical WNT signaling pathway in patient-derived BM-MSCs (40).
As far as the differentiation potential of MDS-derived BM-MSCs is concerned, data are contradictory and these discrepancies might be explained by diversities in patient categories and methodologies across different studies. On one hand, there have been studies demonstrating that these cells do no differ in their capacity to differentiate towards osteoblasts (20, 27, 30, 31, 34), adipocytes (19, 20, 27, 30, 31) and chondrocytes (19, 27, 31) as compared with BM-MSCs derived from healthy donors (Table 1). In contrast, reduced osteogenic (19, 21, 24), adipogenic (21) and chondrogenic differentiation (17) have also been reported (Table 1). As regards the deregylated osteogenic capacity of BM-MSCs derived from MDS patients, Geyh et al. have demonstrated that it could be partly triggered by TGFβ1 (44). Furthermore, in line with the impaired in vitro osteogenic differentiation potential of patients-derived BM-MSCs, an early study on transiliac bone biopsies obtained from MDS patients demonstrated abnormalities in bone remodeling consisting of decreased number of osteoclasts and osteoblasts and decreased bone formation as evidenced by the diminished mineral apposition rate (45). Bone loss in individuals suffering from MDS was also shown in a more recent report (46), which provided evidence that osteoporosis was more prevalent in patients as compared with age-matched controls.
The question whether MDS-derived BM-MSCs can effectively support hematopoiesis has been addressed in vitro by co-culturing them with HSPCs. Results are inconsistent and this may due to patient heterogeneity and differences in the experimental protocols in between studies. Within this context, some authors have suggested that patient BM-MSCs are able to sustain the growth of both autologous/leukemic HSPCs (30, 34) and HSPCs derived from healthy individuals (27) (Table 1). Muntion et al. (35), (Table 1) have shown that exosomes may be involved in the crosstalk between patient-derived BM-MSCs and hematopoietic cells. More precisely ex-vivo expanded BM-MSCs from lower risk MDS patients have been demonstrated to secrete exosomes with a different microRNA cargo than their normal counterparts. Patient BM-MSC-derived exosomes are incorporated into CD34+ cells from healthy donors and alter their gene expression via microRNA transfer (such as miR-10a and miR-15a) and increase their clonogenic potential and viability. Whether BM-MSC derived exosomes similarly support clonal hematopoiesis in the MDS setting remains to be seen. On the other hand, many studies have demonstrated that MDS-derived BM-MSCs have an impaired potential to support normal HSPCs (18–21, 24, 29, 31, 44)(Table 1). This could be attributed to TGFβ1 (44) as well as to the defective expression of niche-derived molecules known to be involved in hematopoiesis, such as osteopontin, angiopoietin, jagged-1, kit ligand, hepatocyte growth factor (HGF), C-X-C motif chemokine ligand 12 (CXCL12), insulin like growth factor-1 (IGF1), insulin growth factor binding protein 2 (IGFBP2), thrombopoietin (TPO) (18, 19, 29). In addition, in CD271+ BM-MSCs from LR-MDS the up-regulation of inflammatory factors and inhibitors of hematopoiesis such as interleukin 6 (IL-6), IL-8 and C-C motif chemokine ligand (CCL3) (21, 36, 47), the transcription of which is increased secondary to activated NF-kB signaling (36), has been associated with attenuation of HSPC numbers and function ex vivo. Finally, CD73+ MDS-derived BM-MSCs were shown to have a negative impact on the clonogenic potential of autologous hematopoietic cells, as compared to the effect of CD73+ BM-MSCs derived from healthy donors on normal hematopoietic cells (48). This was associated with the increased expression of focal adhesion kinase, a protein involved in various cellular processes including survival, proliferation, differentiation and adhesion (49), in patient-derived BM-MSCs (48).
It has been widely acknowledged that BM-MSCs possess broad immunoregulatory properties, involving cells associated with both innate and adaptive immunity (reviewed in (50)). A large body of evidence supports the role of immune abnormalities in MDS pathogenesis (reviewed in (51)) and accumulating data suggests that BM-MSCs may contribute herein (Table 1). More precisely, BM-MSCs from a specific MDS subtype (refractory anemia) deficiently inhibit in vitro T cell activation and proliferation (25). Another study has demonstrated that BM-MSCs derived from lower risk MDS patients differ in terms of immunoregulatory properties as compared to those from higher MDS patients (28). More specifically, the former were associated with a lower T cell apoptosis, a less potent inhibitory effect and a lower T-regulatory cell inducible rate. In addition, BM-MSCs from lower risk MDS patients exhibit decreased capacity to inhibit dendritic cell maturation and proliferation as compared to those derived from higher MDS patients (37). Moreover, MDS-derived BM-MSCs, but not those derived from healthy donors, have been reported to induce naive normal monocytes to acquire the properties of myeloid derived suppressor cells and eventually down-regulate NK and T cell function (38). Taken together these findings suggest that MDS-derived BM MSCs have impaired immuneregulatory functions. The fact that in our study (31) BM-MSCs from MDS patients effectively inhibited T cell proliferation might in part be attributed to differences in patient distribution within the MDS subtypes, along with dissimilar experimental protocols as compared to other studies.
Whereas clonal cytogenetic abnormalities in hematopoietic cells are detected in approximately 40-70% of patients with de novo MDS, data regarding the presence of genetic aberrations in MDS-derived BM MSCs have been contradictory. While several studies (17, 27, 52), including ours (53) (Table 1) have shown that patient ex-vivo expanded BM-MSCs harbor chromosomal aberrations, others report that these cells are normal in terms of cytogenetic analysis (25, 29, 34) (Table 1). Interestingly, clonal chromosomal abnormalities detected in MDS-derived BM-MSCs consistently differ from those in hematopoietic cells from the same individual (53), thereby suggesting that patient-derived BM-MSCs and hematopoietic cells do not derive from the same clone. In support of this notion, Fabiani et al. (54) reported that mutations of epigenetic and spliceosomal genes in the BM-mononuclear cells from MDS patients were not present in the mesenchymal compartment.
As some chromosomal aberrations in MDS-derived BM-MSCs were not detected in earlier passages, but only in later passages and as cytogenetic abnormalities were also found in cultured BM-MSCs derived from healthy donors (53), the possibility that these alterations occurred due to ex-vivo expansion and/or culture conditions could not be excluded. This issue was clarified in a recent study (33) (Table 1) investigating the occurrence of clonal mutations in patient and normal ex vivo expanded BM-MSCs by exome sequencing. MDS-derived BM-MSCs were shown to harbor increased mutational burden and distinct mutational signatures as compared to healthy BM-MSCs. However, highly recurrent mutations identified during culture could not be backtracked in primary -non expanded- stroma cells from the same patients (33). These findings suggest that there is no evidence for clonal mutations in the stroma compartment of MDS patients and that the mutations detected during ex vivo expansion of MDS-derived BM-MSCs are related to in vitro culture per se.
Conclusive evidence for the key role of the mesenchymal components of the BM microenvironment in the emergence of MDS-like disease in mice was firstly reported by Raaijmakers et al. (55). In this pivotal study, the authors demonstrated that targeted deletion of Dicer1 -an RNAse III endonuclease involved in miRNA biogenesis- from murine osterix-expressing osteoprogenitors, but not terminally differentiated osteoblasts, resulted in defective osteoblast differentiation and in the initiation and propagation of a form of myelodysplasia associated with leukopenia and lymphocytopenia. Osteolineage cells from mutant mice expressed significantly lower levels of the Schwachman-Bodian-Diamond syndrome (Sbds) gene (55). Mutations of this gene are found in Schwachman-Diamond syndrome (SDS) which is characterized by exocrine pancreatic dysfunction, cytopenias- especially neutropenia and bone abnormalities (56). Of note, patients with SDS are at risk for developing MDS and AML (56). Sbds deletion in murine osteoprogenitors resulted in leukopenia, lymphopenia, and myelodysplasia, thus recapitulating the phenotype of mice harboring Dicer1 deletion within the same cells (55). Consistent with these findings, reduced expression of Dicer1 mRNA and protein levels as well down-regulated Sbds gene expression were also reported in BM-MSCs from MDS patients (57). On the other hand, a recent study showed that none out of 121 individuals with germline pathogenic Dicer1 variants developed MDS or leukemia (58). Thus, the clinical significance of Dicer1 in the MDS setting remains elusive. In addition Sbds deletion in osterix+ mesenchymal stem/progenitor cells induces genotoxic stress in HSPCs via the inflammatory p53-S100A8/9-TLR signaling (59). Activation of this axis seems to have clinical relevance as it was also observed in a subset of lower risk MDS patients who were characterized by a significantly shorter progression free survival and leukemic evolution (59).
The NUP98 gene encodes a protein that is involved in RNA and protein transport across the nuclear membrane (60). NUP98 fusion genes have been identified in various hematologic malignancies, including MDS. The NUP98-HOXD13 fusion gene has been detected in patients with MDS (60) and mice expressing the hematopoietic compartment specific vav-driven NUP98-HOXD13 fusion transgene eventually develop MDS-like features (46, 60). Interestingly, these mice exhibit also a disrupted microenvironment, which has been reported to contribute significantly in disease progression (46, 61). Alterations in the BM microenvironment include increased number of osteoblasts, reduced numbers of osteoclasts and increased amount of non-mineralized bone (46). Notably, neutralization of the increased serum levels of fibroblast growth factor 23 (FGF23), a regulator of phosphate homeostasis and bone mineralization and an inhibitor of erythropoiesis, restored bone microarchitecture and improved osteoid mineralization and anemia (61). Elevated FGF23 levels and increased amount of non mineralized bone were also observed in samples from MDS patients, thereby corroborating the findings in NHD13 mice (61)
Impaired bone metabolism was also reported recently in the Abcg2 MDS/AML mouse model (62). In this model, expression of a mutant enhancer of zeste homolog 2 (EZH2-the catalytic subunit of polycomp repressive complex 2, one of the 10 most frequently mutated genes in MDS (63)) lacking the catalytic SET domain (EZH2-dSET) induces Abcg2 (a drug efflux transporter) up-regulation. Mice transplanted with syngeneic bone marrow cells retrovirally tranduced with EZH2-dSET developed an MDS-like disease (63). These mice were also shown to have reduced bone volume, due to decreased bone formation. Similar findings were observed in NHD13 mice and in MDS patients as well. Furthermore, the authors demonstrated that murine MDS/AML cells inhibited the commitment of BM-MSCs towards osteoprogenitors, thereby deregulating the capacity of MSCs to support normal hematopoiesis. This suppression was mediated via extracellular vesicles derived from MDS/AML cells (62). Collectively, this data add to the established notion that malignant hematopoietic cells are able to modify MSCs, so that the latter promote disease propagation and evolution.
The Apcdel/+ MDS mouse model is a conditional knock-out mouse based on the Mx1-Cre system the most commonly used system to delete the gene of interest in experimental hematology (64–66). Apc is a crucial negative regulator of the canonical b-catenin (Ctnnb1)/WNT-pathway. In the Apcdel/+ MDS mouse model the deletion of 1 Apc allele occurs not only in hematopoietic cells but also in BM stromal cells (65, 66). Apc haploinsufficiency in the mouse BM niche induces the development of myelodysplasia, which is characterized by severe macrocytic anemia (65). Interestingly, MDS is mediated via aberrant WNT signaling in the BM microenvironment (66). In support of this notion drug inhibition of the WNT/Ctnnb1 pathway by the anthelminthic agent pyrvinium delays and/or inhibits MDS development in Apcdel/+ mice (66).
As far as human MDS is concerned, in vivo evidence for the facilitating role of BM-MSCs was provided by a study using a xenograft model (67). More specifically, co-transplantation of CD34+ cells derived from lower risk MDS patients along with ex vivo expanded BM-MSCs from the same patients in NOD/LtSzcid-IL2rg-/- (NSG) mice significantly increased engraftment of hematopoietic cells as compared to transplantation in the absence of BM-MSCs or with BM-MSCs derived from healthy donors. Furthermore, ex vivo expanded MDS-derived BM-MSCs were shown to differ from their normal counterparts regarding expression of genes related with osteogenesis, adipogenesis, fibrosis, inflammation, cell adhesion, extracellular matrix remodeling and cytokine signaling. Of note, normal MSCs acquired similar properties upon co-culture with patient-derived bone marrow cells, substantiating the issue of hematopoietic MDS cells inducing niche alterations, favoring malignant cell expansion and disease evolution (67). However, in contrast to the aforementioned xenograft model, femoral co-transplantation of BM mononuclear cells derived from MDS patients in combination with autologous or allogeneic patient-derived BM-MSCs or BM-MSCs derived from healthy individuals in NSG mice or in NSG mice with knock-in for human stem cell factor (SCF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-3, did not enhance MDS engraftment (68). The discrepancy between the two studies is not clear. Yet, the fact that in the latter study injected MSCs did not exhibit long-term engraftment suggests that human microenvironment was only transiently established, which might explain -at least in part- the lack of influence of MSCs on MDS engraftment
The contribution of human MDS-derived BM-MSCs for the engraftment and maintenance of MDS HSCs in mice was also corroborated in a recent study by Mian et al. (69). The authors injected patient derived HSCs into gelatin-based scaffolds that were previously seeded with autologous or allogeneic MDS-derived BM-MSCs and the construct was subsequently transplanted in mice. Engraftment of MDS HSCs was achieved in 94% of cases whereas persistent long-term engraftment within scaffolds was observed in 0.2-86%. Interestingly, patient-derived HSCs were shown to move out of the scaffold and home to an adjacent scaffold previously seeded with human BM-MSCs but not with murine BM- MSCs (69), thereby highlighting the critical interplay between bone marrow microenvironment and malignant cells in the MDS setting.
The aforementioned preclinical studies support the critical contribution of the BM-MSC compartment in the initiation and/or propagation of MDS and provide the rationale for its therapeutic targeting as a strategy to delay and/or halt disease evolution. In line with this notion, the beneficial effects of some of the therapeutic modalities currently applied in MDS may be exerted via amelioration of the BM-MSCs’ impairment, as will be outlined below (Table 2).
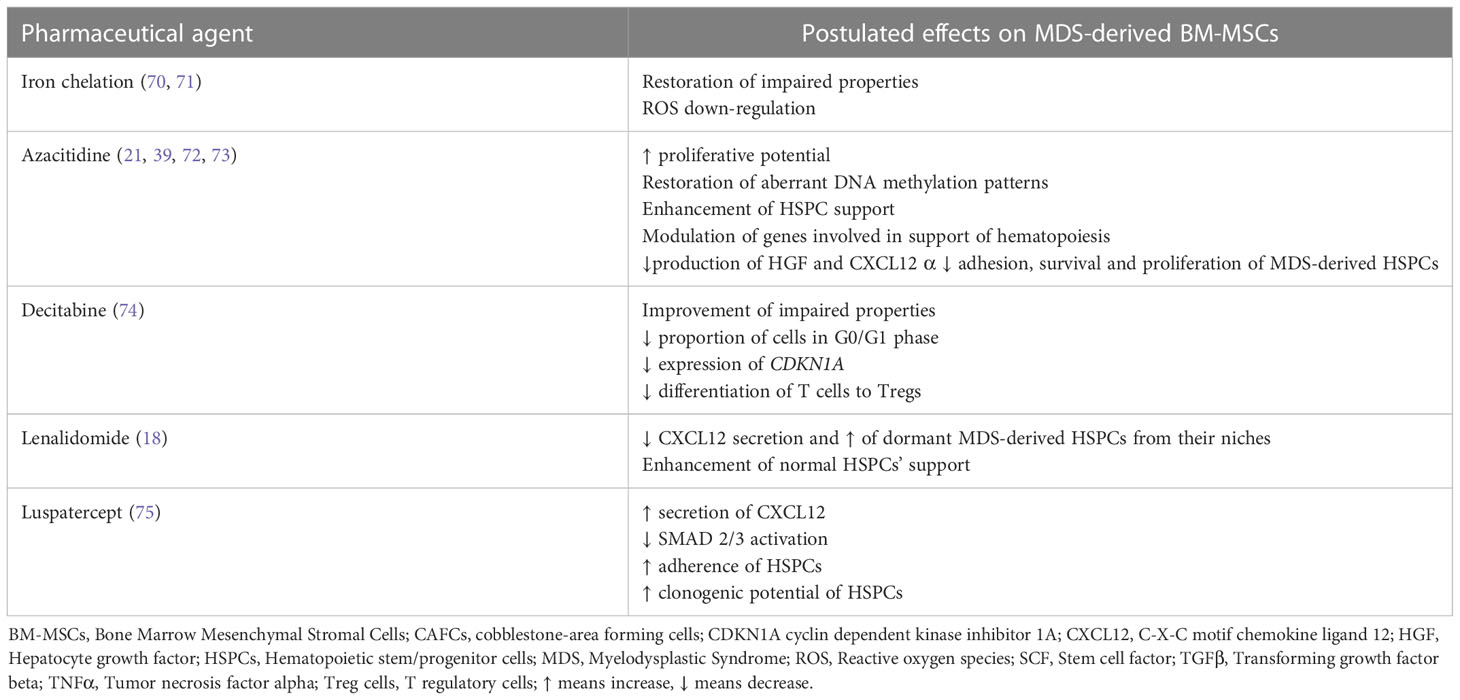
Table 2 Treatment modalities in MDS and their potential effects on patient-derived BM-MSCs.
Iron overload (IO) is a common finding in MDS patients that occurs as a result of ineffective hematopoiesis as well as red blood cell transfusions (76, 77). Aside from its deleterious effect on hematopoiesis (76, 77) IO has also been demonstrated to impair BM-MSCs. More precisely, Huang et al. (78) have demonstrated that BM-MSCs from iron overloaded higher-risk MDS patients exhibit decreased quantity, defective proliferation capacity and reduced osteogenic differentiation potential. Furthermore, they express lower levels of hematopoiesis-associated genes such as VEGFA, CXCL12, and TGFβ1 (78). In addition IO induces apoptosis in MDS-derived BM-MSCs via increased level of reactive oxygen species (ROS) and the ROS-associated Wnt/b-catenin pathway (78). IO has also been reported to induce mitochondrial fragmentation via ROS and the activation of the AMPK/MFF/Drp1 pathway in MDS-derived BM-MSCs. Of note, all these effects of IO in patient BM-MSCs are reversed, at least in part, by antioxidants and iron chelation. Moreover, in an IO mouse model, antioxidant and iron chelation have been shown to partially restore the defective BM-MSCs’ hematopoietic support. Iron chelation is commonly administered in MDS patients, especially in those with lower-risk MDS and its treatment is associated with improvement of cytopenias in a significant proportion of patients (70). The aforementioned data imply that in the MDS setting the beneficial role of iron chelation in hematopoiesis may also result from the restoration, to a certain extent, of the MSCs deficits and further support the application of this treatment in myelodysplasia.
On the other hand Hu et al. (71) have reported that IO upregulates the expression of IL-6, IL-8, TGFβ and VEGF in MDS-derived BM-MSCs through ROS upregulation and subsequent HIF-1a overexpression (71). Antioxidants and iron chelation down-regulated the levels of the aforementioned cytokines. These data provide the theoretical background for probing more deeply into the role of HIF-1a in the BM microenvironment in MDS and provide further justification for the use of HIF-1 inhibitors in this disorder, an issue that is currently under investigation (79). Of note, the discrepancies in terms of the effect of IO in the expression of VEGF and TGFβ between the study Huang et al. (56) and that of Hu et al. (71) may reflect differences in patient cohorts regarding disease categorization and IPSS risk classification.
Previous studies have demonstrated that ex vivo expanded MDS-derived BM-MSCs have significantly different DNA methylation patterns from their normal counterparts (19, 21)and are characterized by aberrant hypermethylation (19, 39). These findings set the stage for the investigation of the potential effect on patient MSCs of the hypomethylating agent azacitidine (AZA), which is widely used in higher risk MDS and may also be effective in lower risk MDS refractory to first line treatments (80, 81). AZA treatment has thus been shown to restore the aberrant hypermethylation pattern of MDS-derived BM-MSCs (39) to increase their proliferation potential and osteogenic capacity, and improve their ability to support HSPCs for in vivo engraftment (21). Another study (72) has reported that in patient BM-MSCs AZA regulates various genes involved in the support of hematopoiesis, essentially genes associated with IFN-γ and extracellular matrix receptor interaction pathways. This transcriptional modulation could account for the improved capacity of AZA-treated MDS-derived BM-MSCs to preferentially support healthy-derived HSPCs, over patient-derived HSPCs (72). AZA has also been shown to upregulate the expression of the serine protease inhibitor kunitz-type2 (SPINT2/HAI-2), an inhibitor of hepatocyte growth factor (HGF) activation, in patient-derived BM-MSCs (73). Based on data derived from the HS-5 stromal line, it has been hypothesized that AZA may down-regulate HGF and CXL12 production by MDS-MSCs and consequently increase cell-adhesion, proliferation and survival of MDS-derived HSPCs (73). The aforementioned findings provide evidence for the potential of AZA in targeting the defective BM-MSCs, thereby extending the effects of this therapeutic modality in the MDS-setting.
Decitabine is another hypomethylating agent which has been approved by the Food and Drug Administration (FDA) for the treatment of patients with MDS (82). Incubation of culture expanded BM-MSCs from MDS patients in the presence of Decitabine resulted in a significant decrease in the proportion of cells in the G0/G1 phases as compared to MDS-derived BM-MSCs incubated in the absence of the drug (control group). This effect was associated with a reduced gene expression of cyclin dependent kinase inhibitor 1A (CDKN1A) (74). Moreover the ability of BM-MSCs from MDS patients incubated in the presence of decitabine to induce the differentiation of T cells into Tregs was significantly reduced compared with control BM-MSCs and this was linked to decreased gene expression of programmed death-ligand 1 (PDL1) (74). Taken together these findings suggest that decitabine may improve, at least to a certain extent, the impaired properties of MDS-derived BM-MSCs
Lenalidomide is an immunomodulatory agent showing efficacy in lower risk MDS (80, 81). Of note, apart from acting directly on MDS cells, lenalidomide has been demonstrated to target the BM microenvironment as well (83), including BM-MSCs (18). More precisely lenalidomide reduces CXCL12 secretion by MDS-derived BM-MSCs and this has been hypothesized to induce the egress of dormant MDS cells from their niches and render them more sensitive to the drug. In addition, treatment of ex vivo expanded lower risk MDS-derived BM-MSCs with lenalidomide has been reported to improve their capacity to support normal clonogenic HSPCs (18).
The downstream key mediators of TGF-β superfamily signaling, SMAD2/3 are constitutively activated in MDS CD34+ cells and this has been associated with impaired late-stage erythroid maturation and subsequent anemia (84, 85). Luspatercept is a therapeutic agent that sequesters TGF-β superfamily ligands and consequently decreases SMAD2/3 activation thereby restoring erythroid maturation and improving anemia. It has been approved for the treatment of selected patient with lower risk MDS (84, 85). Interestingly, a recent study has provided evidence that luspatercept affects also the BM-MSCs (75). More precisely, it decreases SMAD2/3 activation in ex-vivo expanded MDS-derived BM-MSCs and increases CXCL12 secretion. Furthermore, pre-treatment of MDS-derived BM-MSCs with luspatercept improved the clonogenic potential of co-cultured HSPCs, their adherence and expression of CXCR4, the CXCL12 receptor, as well as their homing in zebrafish embryos. Finally, BM-MSCs derived from patients treated with luspatercept exhibited a greater capacity to sustain the clonogenic potential of normal, but not MDS-derived HSPCs (75).
As previously mentioned, MDS-derived hematopoietic cells have been demonstrated to impair BM-MSCs (62, 67). Therefore, it is tempting to speculate that the therapeutic effect of allogeneic HSPC transplantation, which remains the only curative option in MDS (80), may also be exerted via favorable modulations of the functions and properties of the mesenchymal compartment. This issue requires further investigation by comparative analyses on the characteristics of MDS-derived BM-MSCs prior and post allogeneic BM transplantation. To the best of our knowledge, such studies have not been reported thus far.
We have summarized herein the current views on the implication of BM-MSCs in the MDS pathogenesis. Collectively, there is large number of studies that support the notion that BM-MSCs play a major part in the hematopoietic failure that characterizes MDS and demonstrate their involvement in the initiation and progression of the disease. The interactions between BM-MSCs and HSCs have been historically assessed in vitro. Although discrepancies exist, due to different experimental approaches and patient heterogeneity in between studies, most reports have shown aberrancies in patient-derived BM-MSCs and defective hematopoiesis-supporting capacity. Nevertheless, in vitro findings may not adequately recapitulate the complexity of the BM and thus may not allow for the accurate dissection of the interactions between BM-MSCs and HSPCs within the niche. More recently, mouse models of MDS and xenotransplantation studies have shed some light into the crosstalk between BM-MSCs and their progeny on one hand and HSPCs on the other.Evidence has been provided for two –non mutually exclusive – aspects of the role of BM-MSCs in MDS pathophysiology: a) aberrant BM-MSCs promote the development of the myeloid malignancy and b) malignant HSPCs alter BM-MSCs causing them to facilitate disease propagation and/or evolution. Based on these notions a matter of utmost importance that has not been investigated thus far is the delineation of the potential impact of BM-MSCs in the setting of clonal hematopoiesis detected in individuals with otherwise normal complete blood cell counts and without any overt underlying hematological malignancy (86). Such clonal hematopoiesis, called clonal hematopoiesis of indeterminate potential (CHIP) (86) may be linked to aging (87) and is characterized by mutations in genes associated with MDS and AML (86, 87). Notably, CHIP has been associated with an increased risk of hematologic malignancies (87, 88), including MDS. Whether perturbed BM-MSCs can create a permissive soil herein and thus contribute to malignant transformation of CHIP cases remains to be seen.
At present, we have only gained a first insight on the dependence of MDS cells on the BM-microenvironment and lots of issues still remain obscure. The emergence of studies using humanized bone marrow like structures (89, 90) that employ the seeding of AML/MDS-derived BM-MSCs on appropriate scaffolds, the subsequent implantation of AML/MDS-HSPCs into the scaffolds and finally the introduction of the constructs subcutaneously into a mouse is anticipated to provide a robust and accurate mode that will pave the way for the decoding the complex interplay between AML/MDS cells and their microenvironment. In addition, as the critical contribution of BM-MSCs in MDS pathophysiology is being acknowledged, there is an increasing interest in their potential therapeutic targeting. Of note, some of the already available MDS treatments have indeed been recognized to affect the mesenchymal compartment as well. However, as the interactions between HSPCs and BM-MSCs in the MDS setting is progressively being unraveled, opportunities for the development of novel therapies to regulate this crosstalk is expected to emerge and possibly change the treatment landscape in this myeloid malignancy.
Conceptualization, Writing-Review and Editing, CP. Writing-Original Draft, AM. Writing-Review and Editing, HP. All authors contributed to the article and approved the submitted version.
The authors wish to thank Mrs. Maria-Ioanna Tsertou for her contribution in creating Figures 1 and 2.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Comazzetto S, Shen B, Morrison SJ. Niches that regulate stem cells and hematopoiesis in adult bone marrow. Dev Cell (2021) 56(13):1848–60. doi: 10.1016/j.devcel.2021.05.018
2. Hoggatt J, Kfoury Y, Scadden DT. Hematopoietic stem cell niche in health and disease. Annu Rev Pathol (2016) 11:555–81. doi: 10.1146/annurev-pathol-012615-044414
3. Pinho S, Frenette PS. Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol (2019) 20(5):303–20. doi: 10.1038/s41580-019-0103-9
4. Xiao Y, McGuinness CS, Doherty-Boyd WS, Salmeron-Sanchez M, Donnelly H, Dalby MJ. Current insights into the bone marrow niche: From biology in vivo to bioengineering ex vivo. Biomaterials (2022) 286:121568. doi: 10.1016/j.biomaterials.2022.121568
5. Pontikoglou C, Deschaseaux F, Sensebe L, Papadaki HA. Bone marrow mesenchymal stem cells: Biological properties and their role in hematopoiesis and hematopoietic stem cell transplantation. Stem Cell Rev (2011) 7(3):569–89. doi: 10.1007/s12015-011-9228-8
6. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature (2012) 481(7382):457–62. doi: 10.1038/nature10783
7. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by Cxcl12-Cxcr4 chemokine signaling in bone marrow stromal cell niches. Immunity (2006) 25(6):977–88. doi: 10.1016/j.immuni.2006.10.016
8. Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature (2003) 425(6960):836–41. doi: 10.1038/nature02041
9. Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature (2013) 502(7473):637–43. doi: 10.1038/nature12612
10. Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature (2015) 526(7571):126–30. doi: 10.1038/nature15250
11. Dominici M, Le BK, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. the international society for cellular therapy position statement. Cytotherapy (2006) 8(4):315–7. doi: 10.1080/14653240600855905
12. Crippa S, Bernardo ME. Mesenchymal stromal cells: Role in the bm niche and in the support of hematopoietic stem cell transplantation. HemaSphere (2018) 2(6):e151. doi: 10.1097/hs9.0000000000000151
13. Crippa S, Santi L, Berti M, De Ponti G, Bernardo ME. Role of ex vivo expanded mesenchymal stromal cells in determining hematopoietic stem cell transplantation outcome. Front Cell Dev Biol (2021) 9:663316. doi: 10.3389/fcell.2021.663316
14. Adès L, Itzykson R, Fenaux P. Myelodysplastic syndromes. Lancet (London England) (2014) 383(9936):2239–52. doi: 10.1016/s0140-6736(13)61901-7
15. Raaijmakers MH. Myelodysplastic syndromes: Revisiting the role of the bone marrow microenvironment in disease pathogenesis. IntJHematol (2012) 95(1):17–25. doi: 10.1007/s12185-011-1001-x
16. Kastrinaki MC, Pontikoglou C, Klaus M, Stavroulaki E, Pavlaki K, Papadaki HA. Biologic characteristics of bone marrow mesenchymal stem cells in myelodysplastic syndromes. Curr Stem Cell Res Ther (2011) 6(2):122–30. doi: 10.2174/157488811795495422
17. Lopez-Villar O, Garcia JL, Sanchez-Guijo FM, Robledo C, Villaron EM, Hernández-Campo P, et al. Both expanded and uncultured mesenchymal stem cells from mds patients are genomically abnormal, showing a specific genetic profile for the 5q- syndrome. Leukemia (2009) 23(4):664–72. doi: 10.1038/leu.2008.361
18. Ferrer RA, Wobus M, List C, Wehner R, Schönefeldt C, Brocard B, et al. Mesenchymal stromal cells from patients with myelodyplastic syndrome display distinct functional alterations that are modulated by lenalidomide. Haematologica (2013) 98(11):1677–85. doi: 10.3324/haematol.2013.083972
19. Geyh S, Oz S, Cadeddu RP, Frobel J, Bruckner B, Kundgen A, et al. Insufficient stromal support in mds results from molecular and functional deficits of mesenchymal stromal cells. Leukemia (2013) 27(9):1841–51. doi: 10.1038/leu.2013.193
20. Pavlaki K, Pontikoglou CG, Demetriadou A, Batsali AK, Damianaki A, Simantirakis E, et al. Impaired proliferative potential of bone marrow mesenchymal stromal cells in patients with myelodysplastic syndromes is associated with abnormal wnt signaling pathway. Stem Cells Dev (2014) 23(14):1568–81. doi: 10.1089/scd.2013.0283
21. Poon Z, Dighe N, Venkatesan SS, Cheung AMS, Fan X, Bari S, et al. Bone marrow mscs in mds: Contribution towards dysfunctional hematopoiesis and potential targets for disease response to hypomethylating therapy. Leukemia (2019) 33(6):1487–500. doi: 10.1038/s41375-018-0310-y
22. Weickert MT, Hecker JS, Buck MC, Schreck C, Rivière J, Schiemann M, et al. Bone marrow stromal cells from mds and aml patients show increased adipogenic potential with reduced delta-Like-1 expression. Sci Rep (2021) 11(1):5944. doi: 10.1038/s41598-021-85122-8
23. Flores-Figueroa E, Arana-Trejo RM, Gutiérrez-Espíndola G, Pérez-Cabrera A, Mayani H. Mesenchymal stem cells in myelodysplastic syndromes: Phenotypic and cytogenetic characterization. Leuk Res (2005) 29(2):215–24. doi: 10.1016/j.leukres.2004.06.011
24. Varga G, Kiss J, Várkonyi J, Vas V, Farkas P, Pálóczi K, et al. Inappropriate notch activity and limited mesenchymal stem cell plasticity in the bone marrow of patients with myelodysplastic syndromes. Pathol Oncol Res POR (2007) 13(4):311–9. doi: 10.1007/bf02940310
25. Han Q, Sun Z, Liu L, Chen B, Cao Y, Li K, et al. Impairment in immuno-modulatory function of Flk1(+)Cd31(-)Cd34(-) mscs from mds-Ra patients. Leuk Res (2007) 31(11):1469–78. doi: 10.1016/j.leukres.2006.12.016
26. Zhi-Gang Z, Wei-Ming L, Zhi-Chao C, Yong Y, Ping Z. Immunosuppressive properties of mesenchymal stem cells derived from bone marrow of patient with hematological malignant diseases. Leuk Lymphoma (2008) 49(11):2187–95. doi: 10.1080/10428190802455875
27. Flores-Figueroa E, Montesinos JJ, Flores-Guzman P, Gutierrez-Espindola G, Arana-Trejo RM, Castillo-Medina S, et al. Functional analysis of myelodysplastic syndromes-derived mesenchymal stem cells. Leuk Res (2008) 32(9):1407–16. doi: 10.1016/j.leukres.2008.02.013
28. Zhao Z, Wang Z, Li Q, Li W, You Y, Zou P. The different immunoregulatory functions of mesenchymal stem cells in patients with low-risk or high-risk myelodysplastic syndromes. PloS One (2012) 7(9):e45675. doi: 10.1371/journal.pone.0045675
29. Zhao ZG, Xu W, Yu HP, Fang BL, Wu SH, Li F, et al. Functional characteristics of mesenchymal stem cells derived from bone marrow of patients with myelodysplastic syndromes. Cancer Lett (2012) 317(2):136–43. doi: 10.1016/j.canlet.2011.08.030
30. Corradi G, Baldazzi C, Očadlíková D, Marconi G, Parisi S, Testoni N, et al. Mesenchymal stromal cells from myelodysplastic and acute myeloid leukemia patients display in vitro reduced proliferative potential and similar capacity to support leukemia cell survival. Stem Cell Res Ther (2018) 9(1):271. doi: 10.1186/s13287-018-1013-z
31. Klaus M, Stavroulaki E, Kastrinaki MC, Fragioudaki P, Giannikou K, Psyllaki M, et al. Reserves, functional, immunoregulatory, and cytogenetic properties of bone marrow mesenchymal stem cells in patients with myelodysplastic syndromes. Stem Cells Dev (2010) 19(7):1043–54. doi: 10.1089/scd.2009.0286
32. Aanei CM, Flandrin P, Eloae FZ, Carasevici E, Guyotat D, Wattel E, et al. Intrinsic growth deficiencies of mesenchymal stromal cells in myelodysplastic syndromes. Stem Cells Dev (2012) 21(10):1604–15. doi: 10.1089/scd.2011.0390
33. Jann J-C, Mossner M, Riabov V, Altrock E, Schmitt N, Flach J, et al. Bone marrow derived stromal cells from myelodysplastic syndromes are altered but not clonally mutated in vivo. Nat Commun (2021) 12(1):6170. doi: 10.1038/s41467-021-26424-3
34. Soenen-Cornu V, Tourino C, Bonnet ML, Guillier M, Flamant S, Kotb R, et al. Mesenchymal cells generated from patients with myelodysplastic syndromes are devoid of chromosomal clonal markers and support short- and long-term hematopoiesis in vitro. Oncogene (2005) 24(15):2441–8. doi: 10.1038/sj.onc.1208405
35. Muntión S, Ramos TL, Diez-Campelo M, Rosón B, Sánchez-Abarca LI, Misiewicz-Krzeminska I, et al. Microvesicles from mesenchymal stromal cells are involved in hpc-microenvironment crosstalk in myelodysplastic patients. PloS One (2016) 11(2):e0146722. doi: 10.1371/journal.pone.0146722
36. Ping Z, Chen S, Hermans SJF, Kenswil KJG, Feyen J, van Dijk C, et al. Activation of nf-Kb driven inflammatory programs in mesenchymal elements attenuates hematopoiesis in low-risk myelodysplastic syndromes. Leukemia (2019) 33(2):536–41. doi: 10.1038/s41375-018-0267-x
37. Wang Z, Tang X, Xu W, Cao Z, Sun L, Li W, et al. The different immunoregulatory functions on dendritic cells between mesenchymal stem cells derived from bone marrow of patients with low-risk or high-risk myelodysplastic syndromes. PloS One (2013) 8(3):e57470. doi: 10.1371/journal.pone.0057470
38. Sarhan D, Wang J, Sunil Arvindam U, Hallstrom C, Verneris MR, Grzywacz B, et al. Mesenchymal stromal cells shape the mds microenvironment by inducing suppressive monocytes that dampen nk cell function. JCI Insight (2020) 5(5):e130155. doi: 10.1172/jci.insight.130155
39. Bhagat TD, Chen S, Bartenstein M, Barlowe AT, Von Ahrens D, Choudhary GS, et al. Epigenetically aberrant stroma in mds propagates disease Via Wnt/Beta-catenin activation. Cancer Res (2017) 77(18):4846–57. doi: 10.1158/0008-5472.CAN-17-0282
40. Falconi G, Fabiani E, Fianchi L, Criscuolo M, Raffaelli CS, Bellesi S, et al. Impairment of Pi3k/Akt and Wnt/B-catenin pathways in bone marrow mesenchymal stem cells isolated from patients with myelodysplastic syndromes. Exp Hematol (2016) 44(1):75–83.e1-4. doi: 10.1016/j.exphem.2015.10.005
41. Campioni D, Rizzo R, Stignani M, Melchiorri L, Ferrari L, Moretti S, et al. A decreased positivity for Cd90 on human mesenchymal stromal cells (Mscs) is associated with a loss of immunosuppressive activity by mscs. Cytom Part B Clin Cytom (2009) 76(3):225–30. doi: 10.1002/cyto.b.20461
42. Wu Y, Aanei CM, Kesr S, Picot T, Guyotat D, Campos Catafal L. Impaired expression of focal adhesion kinase in mesenchymal stromal cells from low-risk myelodysplastic syndrome patients. Front Oncol (2017) 7:164. doi: 10.3389/fonc.2017.00164
43. Aanei CM, Catafal LC. Evaluation of bone marrow microenvironment could change how myelodysplastic syndromes are diagnosed and treated. Cytom Part A (2018) 93(9):916–28. doi: 10.1002/cyto.a.23506
44. Geyh S, Rodríguez-Paredes M, Jäger P, Koch A, Bormann F, Gutekunst J, et al. Transforming growth factor B1-mediated functional inhibition of mesenchymal stromal cells in myelodysplastic syndromes and acute myeloid leukemia. Haematologica (2018) 103(9):1462–71. doi: 10.3324/haematol.2017.186734
45. Mellibovsky L, Diez A, Serrano S, Aubia J, Pérez-Vila E, Mariñoso ML, et al. Bone remodeling alterations in myelodysplastic syndrome. Bone (1996) 19(4):401–5. doi: 10.1016/s8756-3282(96)00210-4
46. Weidner H, Rauner M, Trautmann F, Schmitt J, Balaian E, Mies A, et al. Myelodysplastic syndromes and bone loss in mice and men. Leukemia (2017) 31(4):1003–7. doi: 10.1038/leu.2017.7
47. Chen S, Zambetti NA, Bindels EM, Kenswill K, Mylona AM, Adisty NM, et al. Massive parallel rna sequencing of highly purified mesenchymal elements in low-risk mds reveals tissue-Context-Dependent activation of inflammatory programs. Leukemia (2016) 30(9):1938–42. doi: 10.1038/leu.2016.91
48. Aanei CM, Eloae FZ, Flandrin-Gresta P, Tavernier E, Carasevici E, Guyotat D, et al. Focal adhesion protein abnormalities in myelodysplastic mesenchymal stromal cells. Exp Cell Res (2011) 317(18):2616–29. doi: 10.1016/j.yexcr.2011.08.007
49. Schaller MD. Cellular functions of fak kinases: Insight into molecular mechanisms and novel functions. J Cell Sci (2010) 123(Pt 7):1007–13. doi: 10.1242/jcs.045112
50. Matheakakis A, Batsali A, Papadaki HA, Pontikoglou CG. Therapeutic implications of mesenchymal stromal cells and their extracellular vesicles in autoimmune diseases: From biology to clinical applications. Int J Mol Sci (2021) 22(18):10132. doi: 10.3390/ijms221810132
51. Peng X, Zhu X, Di T, Tang F, Guo X, Liu Y, et al. The yin-yang of immunity: Immune dysregulation in myelodysplastic syndrome with different risk stratification. Front Immunol (2022) 13:994053. doi: 10.3389/fimmu.2022.994053
52. Blau O, Baldus CD, Hofmann WK, Thiel G, Nolte F, Burmeister T, et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood (2011) 118(20):5583–92. doi: 10.1182/blood-2011-03-343467
53. Kouvidi E, Stratigi A, Batsali A, Mavroudi I, Mastrodemou S, Ximeri M, et al. Cytogenetic evaluation of mesenchymal Stem/Stromal cells from patients with myelodysplastic syndromes at different time-points during ex vivo expansion. Leuk Res (2016) 43:24–32. doi: 10.1016/j.leukres.2016.02.007
54. Fabiani E, Falconi G, Fianchi L, Guidi F, Bellesi S, Voso MT, et al. Mutational analysis of bone marrow mesenchymal stromal cells in myeloid malignancies. Exp Hematol (2014) 42(9):731–3. doi: 10.1016/j.exphem.2014.04.011
55. Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature (2010) 464(7290):852–7. doi: 10.1038/nature08851
56. Huang JN, Shimamura A. Clinical spectrum and molecular pathophysiology of shwachman-diamond syndrome. Curr Opin Hematol (2011) 18(1):30–5. doi: 10.1097/MOH.0b013e32834114a5
57. Santamaria C, Muntion S, Roson B, Blanco B, Lopez-Villar O, Carrancio S, et al. Impaired expression of dicer, drosha, sbds and some micrornas in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica (2012) 97(8):1218–24. doi: 10.3324/haematol.2011.054437
58. Vasta LM, Khan NE, Higgs CP, Harney LA, Carr AG, Harris AK, et al. Hematologic indices in individuals with pathogenic germline Dicer1 variants. Blood Adv (2021) 5(1):216–23. doi: 10.1182/bloodadvances.2020002651
59. Zambetti NA, Ping Z, Chen S, Kenswil KJG, Mylona MA, Sanders MA, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell (2016) 19(5):613–27. doi: 10.1016/j.stem.2016.08.021
60. Lin YW, Slape C, Zhang Z, Aplan PD. Nup98-Hoxd13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood (2005) 106(1):287–95. doi: 10.1182/blood-2004-12-4794
61. Weidner H, Baschant U, Lademann F, Ledesma Colunga MG, Balaian E, Hofbauer C, et al. Increased fgf-23 levels are linked to ineffective erythropoiesis and impaired bone mineralization in myelodysplastic syndromes. JCI Insight (2020) 5(15):e137062. doi: 10.1172/jci.insight.137062
62. Hayashi Y, Kawabata KC, Tanaka Y, Uehara Y, Mabuchi Y, Murakami K, et al. Mds cells impair osteolineage differentiation of mscs Via extracellular vesicles to suppress normal hematopoiesis. Cell Rep (2022) 39(6):110805. doi: 10.1016/j.celrep.2022.110805
63. Kawabata KC, Hayashi Y, Inoue D, Meguro H, Sakurai H, Fukuyama T, et al. High expression of Abcg2 induced by Ezh2 disruption has pivotal roles in mds pathogenesis. Leukemia (2018) 32(2):419–28. doi: 10.1038/leu.2017.227
64. Velasco-Hernandez T, Säwén P, Bryder D, Cammenga J. Potential pitfalls of the Mx1-cre system: Implications for experimental modeling of normal and malignant hematopoiesis. Stem Cell Rep (2016) 7(1):11–8. doi: 10.1016/j.stemcr.2016.06.002
65. Stoddart A, Wang J, Fernald AA, Karrison T, Anastasi J, Le Beau MM. Cell intrinsic and extrinsic factors synergize in mice with haploinsufficiency for Tp53, and two human Del(5q) genes, Egr1 and apc. Blood (2014) 123(2):228–38. doi: 10.1182/blood-2013-05-506568
66. Stoddart A, Wang J, Hu C, Fernald AA, Davis EM, Cheng JX, et al. Inhibition of wnt signaling in the bone marrow niche prevents the development of mds in the Apc(Del/+) mds mouse model. Blood (2017) 129(22):2959–70. doi: 10.1182/blood-2016-08-736454
67. Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell (2014) 14(6):824–37. doi: 10.1016/j.stem.2014.02.014
68. Krevvata M, Shan X, Zhou C, Dos Santos C, Habineza Ndikuyeze G, Secreto A, et al. Cytokines increase engraftment of human acute myeloid leukemia cells in immunocompromised mice but not engraftment of human myelodysplastic syndrome cells. Haematologica (2018) 103(6):959–71. doi: 10.3324/haematol.2017.183202
69. Mian SA, Bonnet D. Nature or nurture? role of the bone marrow microenvironment in the genesis and maintenance of myelodysplastic syndromes. Cancers (Basel) (2021) 13(16):4116. doi: 10.3390/cancers13164116
70. Parisi S, Finelli C. Prognostic factors and clinical considerations for iron chelation therapy in myelodysplastic syndrome patients. J Blood Med (2021) 12:1019–30. doi: 10.2147/jbm.s287876
71. Hu J, Meng F, Hu X, Huang L, Liu H, Liu Z, et al. Iron overload regulate the cytokine of mesenchymal stromal cells through Ros/Hif-1α pathway in myelodysplastic syndromes. Leuk Res (2020) 93:106354. doi: 10.1016/j.leukres.2020.106354
72. Wenk C, Garz AK, Grath S, Huberle C, Witham D, Weickert M, et al. Direct modulation of the bone marrow mesenchymal stromal cell compartment by azacitidine enhances healthy hematopoiesis. Blood Adv (2018) 2(23):3447–61. doi: 10.1182/bloodadvances.2018022053
73. Roversi FM, Cury NM, Lopes MR, Ferro KP, Machado-Neto JA, Alvarez MC, et al. Up-regulation of Spint2/Hai-2 by azacytidine in bone marrow mesenchymal stromal cells affects leukemic stem cell survival and adhesion. J Cell Mol Med (2019) 23(2):1562–71. doi: 10.1111/jcmm.14066
74. Pang Y, Geng S, Zhang H, Lai P, Liao P, Zeng L, et al. Phenotype of mesenchymal stem cells from patients with myelodyplastic syndrome maybe partly modulated by decitabine. Oncol Lett (2019) 18(5):4457–66. doi: 10.3892/ol.2019.10788
75. Wobus M, Mies A, Asokan N, Oelschlägel U, Möbus K, Winter S, et al. Luspatercept restores sdf-1-Mediated hematopoietic support by mds-derived mesenchymal stromal cells. Leukemia (2021) 35(10):2936–2947. doi: 10.1038/s41375-021-01275-5
76. Gattermann N. Iron overload in myelodysplastic syndromes (Mds). Int J Hematol (2018) 107(1):55–63. doi: 10.1007/s12185-017-2367-1
77. Brissot E, Bernard DG, Loréal O, Brissot P, Troadec M-B. Too much iron: A masked foe for leukemias. Blood Rev (2020) 39:100617. doi: 10.1016/j.blre.2019.100617
78. Huang L, Liu Z. Iron overload impairs bone marrow mesenchymal stromal cells from higher-risk mds patients by regulating the ros-related Wnt/B-catenin pathway. Stem Cells Int (2020) 2020:8855038. doi: 10.1155/2020/8855038
79. Henry DH, Glaspy J. Roxadustat for the treatment of anemia in patients with lower-risk myelodysplastic syndrome: Open-label, dose-selection, lead-in stage of a phase 3 study. Am J Hematol (2022) 97(2):174–84. doi: 10.1002/ajh.26397
80. Fenaux P, Haase D, Santini V, Sanz GF, Platzbecker U, Mey U. Myelodysplastic syndromes: Esmo clinical practice guidelines for diagnosis, treatment and follow-up(†☆). Ann Oncol (2021) 32(2):142–56. doi: 10.1016/j.annonc.2020.11.002
81. Carraway HE, Saygin C. Therapy for lower-risk mds. Hematol Am Soc Hematol Educ Program (2020) 2020(1):426–33. doi: 10.1182/hematology.2020000127
82. Short NJ, Kantarjian H. Hypomethylating agents for the treatment of myelodysplastic syndromes and acute myeloid leukemia: Past discoveries and future directions. Am J Hematol (2022) 97(12):1616–1626. doi: 10.1002/ajh.26667
83. Stahl M, Zeidan AM. Lenalidomide use in myelodysplastic syndromes: Insights into the biologic mechanisms and clinical applications. Cancer (2017) 123(10):1703–13. doi: 10.1002/cncr.30585
84. Verma A, Suragani RN, Aluri S, Shah N, Bhagat TD, Alexander MJ, et al. Biological basis for efficacy of activin receptor ligand traps in myelodysplastic syndromes. J Clin Invest (2020) 130(2):582–9. doi: 10.1172/jci133678
85. Kubasch AS, Fenaux P, Platzbecker U. Development of luspatercept to treat ineffective erythropoiesis. Blood Adv (2021) 5(5):1565–75. doi: 10.1182/bloodadvances.2020002177
86. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood (2015) 126(1):9–16. doi: 10.1182/blood-2015-03-631747
87. Gondek LP. Chip: Is clonal hematopoiesis a surrogate for aging and other disease? Hematol Am Soc Hematol Educ Program (2021) 2021(1):384–9. doi: 10.1182/hematology.2021000270
88. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. New Engl J Med (2014) 371(26):2488–98. doi: 10.1056/NEJMoa1408617
89. Antonelli A, Noort WA, Jaques J, de Boer B, de Jong-Korlaar R, Brouwers-Vos AZ, et al. Establishing human leukemia xenograft mouse models by implanting human bone marrow-like scaffold-based niches. Blood (2016) 128(25):2949–59. doi: 10.1182/blood-2016-05-719021
Keywords: hematopoiesis, bone marrow, mesenchymal stromal cells, myelodysplastic syndromes, iron overload, hypomethylating agents, lenalidomide, luspatercept
Citation: Pontikoglou CG, Matheakakis A and Papadaki HA (2023) The mesenchymal compartment in myelodysplastic syndrome: Its role in the pathogenesis of the disorder and its therapeutic targeting. Front. Oncol. 13:1102495. doi: 10.3389/fonc.2023.1102495
Received: 18 November 2022; Accepted: 06 January 2023;
Published: 25 January 2023.
Edited by:
Elena Solomou, University of Patras, GreeceReviewed by:
Eleni Gavriilaki, G. Papanikolaou General Hospital, GreeceCopyright © 2023 Pontikoglou, Matheakakis and Papadaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charalampos G. Pontikoglou, cG9udGlrb2dsb3VAeWFob28uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.