 Mara Colombo1*
Mara Colombo1* Patrizia Mondini1Elisa Minenza2Claudia Foglia1Annamaria Mosconi2
Patrizia Mondini1Elisa Minenza2Claudia Foglia1Annamaria Mosconi2 Carmen Molica2Lorenza Pistola2
Carmen Molica2Lorenza Pistola2 Vienna Ludovini2
Vienna Ludovini2 Paolo Radice1
Paolo Radice1- 1Unit of Molecular Bases of Genetic Risk and Genetic Testing, Department of Experimental Oncology, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy
- 2Department of Medical Oncology, Santa Maria della Misericordia Hospital, Perugia, Italy
The widespread adoption of gene panel testing for cancer predisposition is leading to the identification of an increasing number of individuals with clinically relevant allelic variants in two or more genes. The potential combined effect of these variants on cancer risks is mostly unknown, posing a serious problem for genetic counseling in these individuals and their relatives, in whom the variants may segregate singly or in combination. We report a female patient who developed triple-negative high grade carcinoma in the right breast at the age of 36 years. The patient underwent bilateral mastectomy followed by combined immunotherapy and chemotherapy (IMpassion030 clinical trial). Two years later she developed a skin recurrence on the right anterior chest wall. Despite intensive treatment, the patient died at 40-year-old due to disease progression. Gene panel testing of patient’s DNA revealed the presence of a protein truncating variant in ATM [c.1672G>T; p.(Gly558Ter)] and of a not previously reported variant in the BRCA1 exon 22 donor splice site [c.5406+6T>G], whose clinical significance was unknown. The analysis of patient’s RNA revealed the up-regulation of two alternative BRCA1 mRNA isoforms derived from skipping of exon 22 and of exons 22-23. The corresponding predicted protein products, p.(Asp1778GlyfsTer27) and p.(Asp1778_His1822del) are both expected to affect the BRCA1 C Terminus (BRCT) domain. The two variants were observed to co-occur also in the proband’s brother who, in addition, was heterozygous for a common variant (c.4837A>G) mapped to BRCA1 exon 16. This allowed to ascertain, by transcript-specific amplification, the lack of functional mRNA isoforms expressed by the c.5406+6T>G allele and provided evidence to classify the BRCA1 variant as pathogenic, according to the guidelines of the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) consortium. To our knowledge, excluding two cases detected following the screening of population specific recurrent variants, only one ATM/BRCA1 double heterozygote has been reported in the literature, being the case here described the one with the youngest age at cancer onset. The systematic collection of cases with pathogenic variants in more than one cancer predisposition gene is needed to verify if they deserve ad hoc counseling and clinical management.
Introduction
Germline pathogenic variants in the BRCA1 (MIM# 113705) and BRCA2 (MIM# 600185) genes are the main risk factors for hereditary breast and ovarian cancer (HBOC). For several years, following their identification in the 1990s (1, 2), genetic tests in HBOC patients were limited to the screening of these two genes, using different mutation analysis techniques, including direct Sanger sequencing. However, it was soon apparent that only a fraction (approximately 15-25%) of HBOC families fulfilling the criteria for clinical testing carry pathogenic variants of BRCA genes (3, 4). Moreover, approximately 10-20% of tests detects the presence of variants, termed variants of uncertain significance (VUS), whose effect on cancer risk is unknown (5). More recently, other breast cancer (BC) and/or ovarian cancer (OC) predisposition genes have been identified (6). The recent advent of Next Generation Sequencing (NGS), in addition to minimizing the costs and time of genetic analyses, has enabled the simultaneous screening of multiple genes. Therefore, NGS has implemented the potential for the detection of pathogenic variants in HBOC genes other than BRCA1 and BRCA2 and for the identification of subjects with pathogenic variants in more than one cancer predisposition gene. In fact, although not frequently, HBOC patients have been described with double heterozygous pathogenic variants in ATM, BRCA1, BRCA2, CHEK2, MSH6, MUTYH, NBN/NBS1 and RAD50 following gene panel testing (7–13) with a frequency of approximately 0.3% (10, 13). Not surprisingly, double heterozygous carriers are more frequently detected in populations enriched with founder variants (14). To date, the contribute of a double heterozygosity condition for HBOC genes to the severity of the disease and its impact on the genetic counseling and clinical management of the carriers and their relatives is still debated. Further studies are therefore needed to clarify these issues.
In the present study, we report a female patient who developed triple-negative high grade carcinomas in the right breast at the ages of 36 and 38 years. Gene panel testing of patient’s constitutional DNA revealed, in addition to a protein truncating variant in ATM [c.1672G>T; p.(Gly558Ter)], the presence of a novel VUS in the consensus sequence of the donor splice site of BRCA1 exon 22 [c.5406+6T>G]. Gene transcript analysis revealed that the latter affected RNA splicing and allowed its classification as pathogenic. To the best of our knowledge, only three double heterozygotes for ATM and BRCA1 pathogenic variants had been previously reported, being the case here described the one with the youngest age at cancer onset.
Case presentation
The case here described (therein termed proband) is a woman who at 36 years old sough genetic counseling due to the cancer history of the mother, who was diagnosed with BC and OC (Figure 1). Furthermore, she reported additional cancer cases in second- and third-degree relatives, including lung, gastrointestinal, prostate and pancreatic cancers. Based on family history, the proband was considered eligible for mutation screening of the BRCA1, BRCA2 and PALB2 genes. An NGS analysis, performed using a small-size panel (Myriapod® NGS BRCA1-2 panel Kit CE-IVD, Diatech Pharmacogenetics), identified the c.5406+6T>G variant in BRCA1. At the time of gene testing (April 2018), no data on the clinical significance of this intronic variant were reported in the BRCA Exchange (15) and ClinVar (16) repositories, nor in the literature. A few months later the proband was diagnosed by ultrasound scan with a BC. She opted for a bilateral nipple-areola complex (NAC) sparing mastectomy. A right axillary lymph node dissection was performed for a stage IIB (pT2N1aM0) cancer. The histological examination revealed a triple-negative (ER, PgR, and HER2 negative) invasive ductal G3 carcinoma (ki67 = 50%) with in situ component (5%). Considering the diagnosis of BC, the family history and the absence of a definitely pathogenic variant in BRCA1, BRCA2 and PALB2, the genetic status of the proband was further investigated with a larger NGS-based panel (Hereditary Cancer Solution (HCS) kit, SOPHiA GENETICS) containing the following 26 genes, ATM, APC, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, EPCAM, FAM175A, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PIK3CA, PMS2, PTEN, RAD50, RAD51C, RAD51D, STK11, TP53, XRCC2. The analysis identified the ATM c.1672G>T variant, located in exon 11.
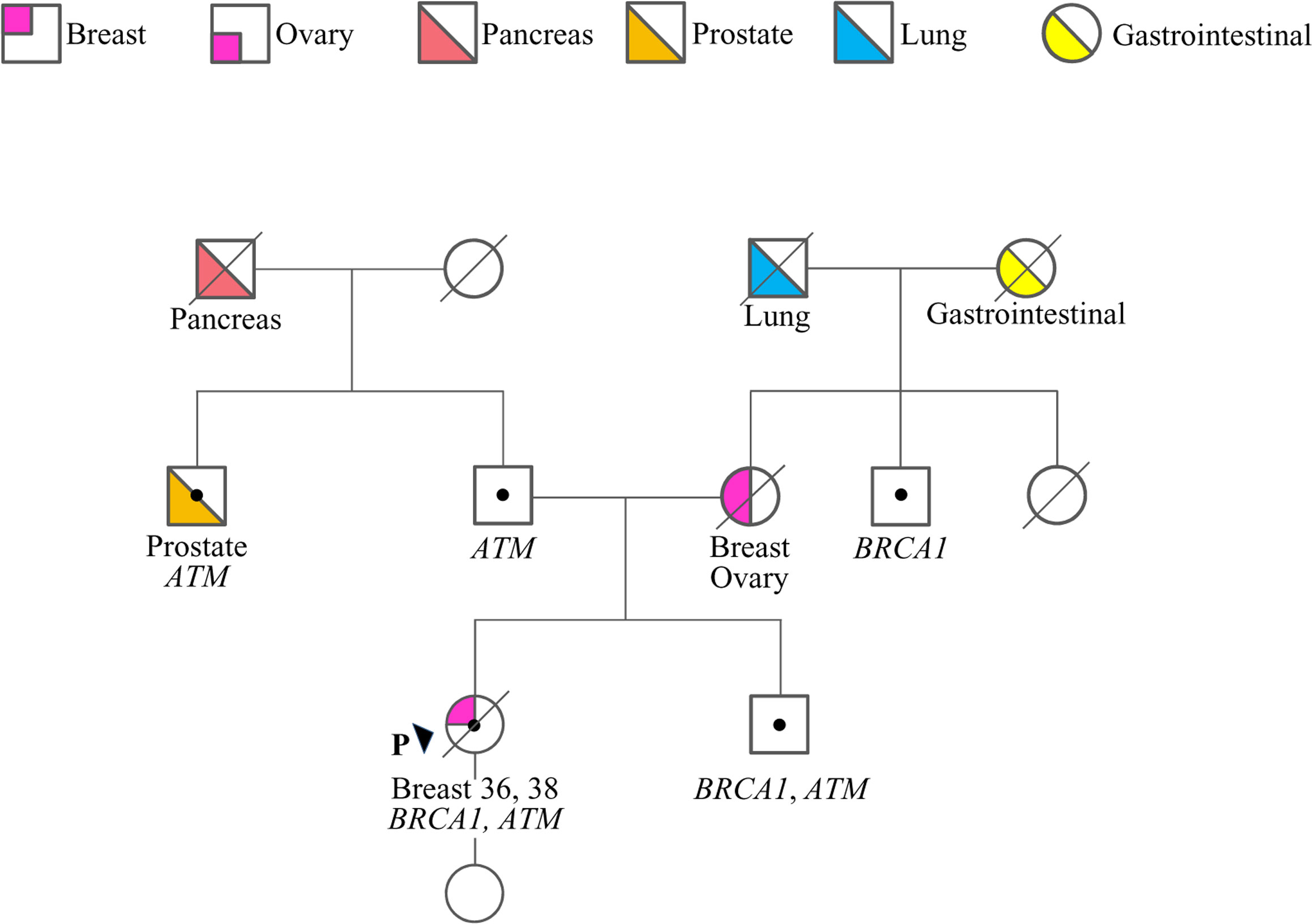
Figure 1 Family pedigree. Males and females are indicated by squares and circles, respectively. The proband (P) is indicated by the arrow. The ages of cancer diagnosis (proband only) and the phenotypes of affected family members are reported under the corresponding symbols, when known. Diagonal slashes indicate deceased individuals. Individuals who underwent genetic analysis are indicated by dots and the genes found mutated are reported.
The proband was enrolled in the IMpassion030 clinical trial (NCT03498716) and received adjuvant immunotherapy (atezolizumab) combined with taxan- and anthracyclin-based chemotherapy and with adjuvant radiotherapy (40Gy) for 12 months after diagnosis. Two months after the end of the treatment, the patient underwent prophylactic bilateral hystero-adnexectomy surgery. At 38 years old a skin recurrence on the right anterior chest wall with lymphangitic carcinomatosis was diagnosed consistent with triple-negative BC (ki67 = 90%). She received a combination chemotherapy of carboplatin and gemcitabine for three months as first-line treatment followed by Olaparib 600 mg/day for two months due to disease progression. At the end of the treatment a revaluation computed tomography (CT) scan showed skin, lymph node, and pulmonary disease progression. The patient underwent a second line treatment with a combination of oral vinorelbine and capecitabine for four mouths and electrochemotherapy of skin metastases, followed by a further combination chemotherapy with cyclophosphamide-methotrexate-fluorouracil (CMF) for one month. An additional revaluation CT scan showed further disease progression. The patient died at the age of 40 years.
Following targeted genetic testing in the proband’s parents, the c.1672G>T in ATM and the c.5406+6T>G in BRCA1 variants were found to have been inherited from the father and the mother, respectively (Figure 1). While the ATM variant, which introduces a premature termination codon (PTC) resulting in a non-functional protein [p.(Gly558Ter)], could be considered pathogenic according to the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) (17), the clinical relevance of the BRCA1 variant remained uncertain.
Considering that the variant in BRCA1 is located in the consensus sequence of the donor splice site of exon 22 (according to the Breast Information Core (BIC) nomenclature) (18), a potential effect at mRNA splicing level was investigated by interrogating the Alamut Visual Plus software application (version v1.2.1 | © 2021 SOPHiA GENETICS). All tools integrated in the software predicted the variant to impact on the splicing by weakening the natural donor site of exon 22. The putative spliceogenic effect was then verified by characterizing the mRNA transcript profile as previously described (19, 20). Briefly, Epstein-Barr virus (EBV)-immortalized lymphoblastoid cell line (LCL) was established from the peripheral blood of the variant carrier. The degradation of transcripts containing premature termination codons (PTCs) via nonsense mediated mRNA decay (NMD) was prevented by growing the LCL in the presence of cycloheximide. Cytoplasmic RNA was isolated from the LCL and first strand cDNA was generated for RT-PCR analysis. The primers for the amplification were designed specifically for the variant under study. Furthermore, the reverse primer was labeled with 6-carboxyfluorescein (6-FAM) in order to obtain fluorescent amplification fragments detectable by capillary electrophoresis (CE). The CE profile obtained from the LCL carrying the BRCA1 variant was compared to that derived from BRCA1 wild type subjects (reference) and the aberrant products not present in the reference were characterized by Sanger sequencing. Experimental conditions are reported in Supplementary Table 1. The analysis revealed the up-regulation of two isoforms derived from the out-of-frame skipping of exon 22 (Δ22, major transcript) and the in-frame skipping of exons 22 and 23 (Δ22,23, minor transcript) (Figure 2), both previously described as naturally occurring isoforms (21). The corresponding predicted protein products, p.(Asp1778GlyfsTer27) and p.(Asp1778_His1822del), are both expected to affect the BRCA1 C Terminus (BRCT) of the protein, a clinically relevant functional domain. The BRCA1 and ATM variants were observed to co-occur also in the proband’s brother, unaffected at the time of genetic testing (at age 33 years), who in addition was heterozygous for the common variant c.4837A>G (rs1799966) mapped to BRCA1 exon 16. PCR fragments spanning the c.4837A>G variant were selectively amplified from the normal transcripts maintaining exon 22 using a reverse primer annealing to this exon and a forward primer annealing to the region upstream of the c.4837A>G variant. The sequence analysis of these amplification products, showing a mono-allelic expression, allowed us to ascertain the lack of functional mRNA isoforms expressed by the allele carrying the c.5406+6T>G variant (Figure 3) and to classify the variant as pathogenic (class 5) according to the BRCA1/2 Gene Variant Classification Criteria of the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) (22). Potential consequences on mRNA splicing were experimentally investigated following the above described approach also for the ATM c.1672G>T variant and excluded (data not shown).
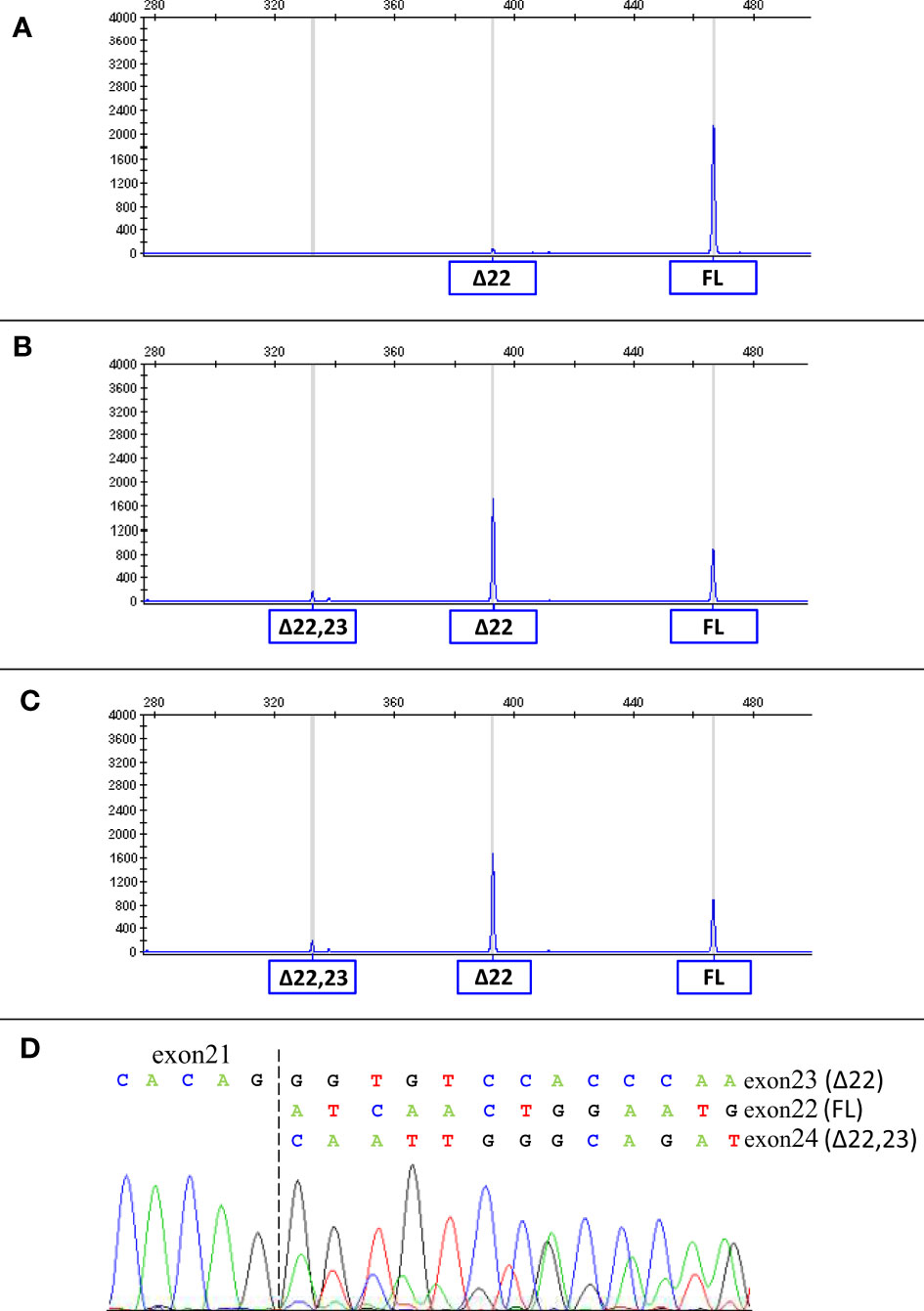
Figure 2 mRNA splicing analysis of the BRCA1 c.5406+6T>G variant. The capillary electrophoresis patterns of the cDNA fragments spanning BRCA1 exons 19 to 24 detected in LCLs from a BRCA1 wild type subject (A) and from c.5406+6T>G carriers (proband (B) and proband’s brother (C)) are shown. The peaks corresponding to normal transcripts maintaining exons 22 and 23 (full-length, FL) and aberrant transcripts (Δ22 and Δ22,23) are indicated. The sequencing of the PCR products (D) confirmed the presence of Δ22, Δ22,23 and normal transcripts in carriers of the BRCA1 c.5406+6T>G variant.
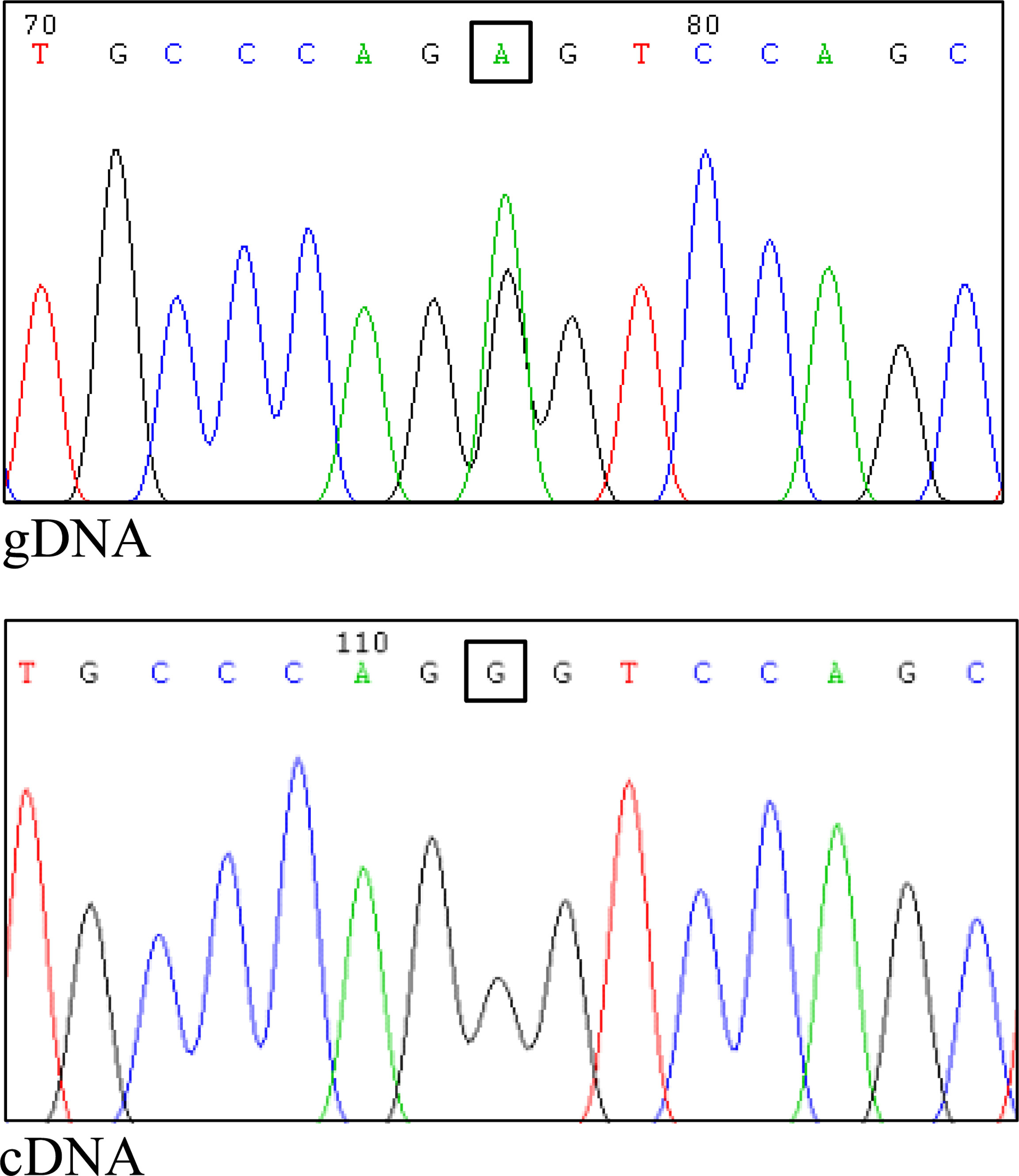
Figure 3 Assessment of allele-specific expression of the normal BRCA1 transcript. The common variant c.4837A>G, mapped to BRCA1 exon 16, was proven to be heterozygous in the genomic DNA (gDNA) of the proband’s brother (upper panel). The specific amplification of cDNA fragment maintaining exon 22 (normal transcript) and including the c.4837A>G variant was performed using a forward primer in exon 16 and a reverse primer in exon 22. The sequencing of the RT-PCR product showed a mono-allelic expression of the normal transcript (lower panel), indicating a complete spliceogenic effect of the c.5406+6T>G variant.
Discussion
In this study, we describe a female patient diagnosed with triple-negative high grade BC, with two heterozygous variants in genes associated with increased risk of BC, namely the pathogenic (protein truncating) variant c.1672G>T in ATM and the suspected spliceogenic variant c.5406+6T>G in BRCA1. Neither of the two variants had been previously published or reported in publicly available databases. Due to the uncertainty on the clinical relevance of the BRCA1 variant, we analyzed the LCL RNAs of the proband and her brother, who also carried the same variant. These investigations allowed us to classify the BRCA1 variant as pathogenic, according to the ENIGMA guidelines. Notably, our observations are consistent with the outcomes of a saturation genome editing (SGE) assay, which were published after the identification of the variant in the case here described, that suggested a loss-of-function effect of the BRCA1 c.5406+6T>G variant (23).
To the best of our knowledge, only three double heterozygotes for germline pathogenic variants in the BRCA1 and ATM genes had been previously reported. Notably, two cases, both presenting the c.181T>G [p.(Cys61Gly)] variant in BRCA1 and the c.5932G>T [p.(Glu1978Ter)] variant in ATM, had been detected following targeted screening of population-specific pathogenic variants (24). Conversely, the third case, carrying the c.5123C>A [p.(Ala1708Glu)] variant in BRCA1 and c.2413C>T [p.(Arg805Ter)] variant in ATM was identified following a gene panel NGS analysis (7). The latter case reported multiple cancer diagnoses in addition to breast cancer, including an infiltrating carcinoma of the ampulla and a clear-cell endometrial adenocarcinoma.
Both the ATM and BRCA1 genes code for key proteins involved in DNA double-strand breaks (DSBs) repair by homologous recombination (HR), a crucial mechanism for maintaining genomic integrity and preventing cancer development (25). It is debated whether, given the involvement of the two proteins in the same pathway, the risk of cancer and the severity of the disease can be increased in carriers of pathogenic variants in both genes, compared to single pathogenic variant carriers. Notably, it has been observed that in Brca1-null murine mammary tissue the heterozygous loss of Atm affects both mammary development, reducing ductal branching during puberty, and tumor severity, increasing invasiveness and causing undifferentiated tumor types, a phenotype not observed in the presence of Atm wild-type alleles (26). A later study demonstrated that in murine thymocytes the hemizygosity for both Atm and Brca1 decreases radiation-induced apoptosis compared to single Atm or Brca1 hemizygous cells (27). Finally, it has been observed that, compared to single Atm and Brca1 heterozygous cells, mouse embryo fibroblasts double mutated in Atm and Brca1 show an increased frequency of neoplastic transformation, a higher genomic instability, a delayed recognition of DNA damage induced by photon irradiation and an incomplete DNA repair (28).
Loss of function (LOF) BRCA1 variants are associated with a high chance of cancer, with estimated cumulative risks by age 80 years of 72% and 44% for breast and ovarian cancer, respectively (29). Conversely, LOF ATM variants are considered moderate risk factors for breast cancer (cumulative risk by age 80 years of 27%) (30), with some limited evidence of association with ovarian cancer risk (31). To date, several common genetic variants, conferring a modest increase of breast/ovarian cancer risk in the general population, have been ascertained to modulate the penetrance of BRCA1 pathogenic variants (32). It is conceivable that the same modifier effect could be elicited by rare pathogenic variants in genes associated with moderate cancer risk. Consistent with this hypothesis, it has been recently reported that the occurrence of additional truncating variants in DNA repair genes might lead to an earlier onset of breast cancer in carriers of pathogenic BRCA1 variants (33). However, in the four BRCA1/ATM double heterozygotes reported to date (7, 24 and present study), the age of first breast cancer diagnosis ranged from 36 to 55 years (the patient here described being the one with the earliest age of onset) with a median age of 43,2 years, similar to that (42 years) observed in carriers of single pathogenic variants in BRCA1 (34). Analogously, when considering the tumor pathological characteristics, no specific phenotypes were observed in double heterozygous carriers compared to BRCA1 pathogenic variant carriers. In all four cases the diagnosed breast cancers were estrogen receptor negative and in two of them (the patient described by Andres et al. (7) and our patient) they were classified as high grade triple-negative carcinomas, which are usually associated with an unfavorable prognosis. However, they are also the more represented breast cancer subtype associated with BRCA1 pathogenic variants, being detected in approximately 65% of carriers with a breast cancer diagnosis overall (35). Conversely, the observation that both our proband and the case described by Andres et al. (7) developed multiple malignancies is in favor of a particularly aggressive clinical phenotype possible associated with the simultaneous occurrence of BRCA1 and ATM pathogenic variants in the same individual. Based on genetic status and family history, the proband’s healthy brother, carrying both BRCA1 and ATM pathogenic variants, was recommended to undergo intensive surveillance, including endoscopic examination of the lower gastrointestinal tract every five years, yearly screening including mammography with breast magnetic resonance imaging (MRI), abdominal ultrasound scan and prostate specific antigen (PSA) test, starting from the age of 40 years.
In conclusion, the number of carriers of constitutional pathogenic variants in both BRCA1 and ATM reported to date are too limited to assess if their cancer risks differ from those of individuals carrying a single BRCA1 pathogenic variant. Therefore, the systematic collection of these cases is needed to address this issue and to define if, due to their particular genetic condition, they deserve ad hoc counseling and clinical management, including specific risk reduction measures.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Santa Maria della Misericordia Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
MC conceived the study, designed and performed the in silico and in vitro splicing analyses, and drafted the manuscript. VL contributed to conception of the study. EM, AM, and CM treated and followed the patient and her family. LP and VL performed the NGS analysis. PM provided technical assistance to the in vitro splicing experiments. CF established the LCLs. VL and EM contributed to the draft. PR made substantial contributions to conception of the study and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was partially supported by a grant from the Italian Association for Cancer Research to PR (AIRC IG 2018 Id.22093).
Acknowledgments
The authors wish to thank the patient and her family for participating in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1102184/full#supplementary-material
References
1. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science (1994) 266:66–71. doi: 10.1126/science.7545954
2. Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature (1995) 378:789–92. doi: 10.1038/378789a0
3. Thomas SF, Amie MD, Julia ER, Mark H, Brian EW, Beth L, et al. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: Analysis of 10,000 individuals. J Clin Oncol (2002) 20:1480–90. doi: 10.1200/JCO.2002.20.6.1480
4. Karin K, Kerstin R, Barbara W, Eric H, Jan H, Britta B, et al. Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J Med Genet (2016) 53:465–71. doi: 10.1136/jmedgenet-2015-103672
5. Eccles BK, Copson E, Maishman T, Abraham JE, Eccles DM. Understanding of BRCA VUS genetic results by breast cancer specialists. BMC Cancer (2015) 15:936. doi: 10.1186/s12885-015-1934-1
6. Bellcross CA. Hereditary breast and ovarian cancer: An updated primer for OB/GYNs. Obstet Gynecol Clin North Am (2022) 49:117–47. doi: 10.1016/j.ogc.2021.11.005
7. Andrés R, Menao S, Arruebo M, Quílez E, Cardiel MJ. Double heterozygous mutation in the BRCA1 and ATM genes involved in development of primary metachronous tumours: A case report. Breast Cancer Res Treat (2019) 177:767–70. doi: 10.1007/s10549-019-05343-4
8. Agaoglu NB, Doganay L. Concurrent pathogenic variations in patients with hereditary cancer syndromes. Eur J Med Genet (2021) 64:104366. doi: 10.1016/j.ejmg.2021.104366
9. Duzkale Teker N, Eyerci N. Double heterozygous mutations in the BRCA2 and ATM genes: A case report and review of the literature. Breast Care (Basel) (2021) 16:412–7. doi: 10.1159/000511430
10. Macquere P, Orazio S, Bonnet F, Jones N, Bubien V, Chiron J, et al. Increased incidence of pathogenic variants in ATM in the context of testing for breast and ovarian cancer predisposition. J Hum Genet (2022) 67:339–45. doi: 10.1038/s10038-022-01014-3
11. Mampel A, Sottile ML, Denita-Juárez SP, Vargas AL, Vargas-Roig LM. Double heterozygous pathogenic variants in the BRCA1 and BRCA2 genes in a patient with bilateral metachronous breast cancer. Cancer Genet (2022) 260-261:14–7. doi: 10.1016/j.cancergen.2021.11.003
12. Sánchez Castro EE, Ziegler-Rodriguez G, Castro Mujica MDC. Double heterozygous mutation in RAD50 and ATM genes in a Peruvian family with five cancer types: A case report. Rev Fac Cien Med Univ Nac Cordoba (2022) 79:53–6. doi: 10.31053/1853.0605.v79.n1.32795
13. Vietri MT, Caliendo G, D'Elia G, Resse M, Casamassimi A, Minucci PB, et al. Five Italian families with two mutations in BRCA genes. Genes (Basel) (2020) 11:1451. doi: 10.3390/genes11121451
14. Nurmi A, Muranen TA, Pelttari LM, Kiiski JI, Heikkinen T, Lehto S, et al. Recurrent moderate-risk mutations in Finnish breast and ovarian cancer patients. Int J Cancer (2019) 145:2692–700. doi: 10.1002/ijc.32309
15. BRCA Exchange, in: November 18, 2022. Available at: https://brcaexchange.org/variants.
16. ClinVar, in: November 18, 2022. Available at: https://www.ncbi.nlm.nih.gov/clinvar/.
17. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
18. Breast Information Core (BIC), in: November 18, 2022. Available at: https://research.nhgri.nih.gov/bic/.
19. Caleca L, Putignano AL, Colombo M, Congregati C, Sarkar M, Magliery TJ, et al. Characterization of an Italian founder mutation in the RING-finger domain of BRCA1. PloS One (2014) 9:e86924. doi: 10.1371/journal.pone.0086924
20. Colombo M, Lopez-Perolio I, Meeks HD, Caleca L, Parsons MT, Li HY, et al. The BRCA2 c.68-7T > a variant is not pathogenic: A model for clinical calibration of spliceogenicity. Hum Mutat (2018) 39:729–41. doi: 10.1002/humu.23411
21. Colombo M, Blok MJ, Whiley P, Santamariña M, Gutiérrez-Enríquez S, Romero A, et al. Comprehensive annotation of splice junctions supports pervasive alternative splicing at the BRCA1 locus: A report from the ENIGMA consortium. Hum Mol Genet (2014) 23:3666–80. doi: 10.1093/hmg/ddu075
22. Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) consortium, in: November 18, 2022. Available at: https://enigmaconsortium.org/.
23. Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature (2018) 562:217–22. doi: 10.1038/s41586-018-0461-z
24. Sokolenko AP, Bogdanova N, Kluzniak W, Preobrazhenskaya EV, Kuligina ES, Iyevleva AG, et al. Double heterozygotes among breast cancer patients analyzed for BRCA1, CHEK2, ATM, NBN/NBS1, and BLM germ-line mutations. Breast Cancer Res Treat (2014) 145:553–62. doi: 10.1007/s10549-014-2971-1
25. Yamamoto H, Hirasawa A. Homologous recombination deficiencies and hereditary tumors. Int J Mol Sci (2021) 23:348. doi: 10.3390/ijms23010348
26. Bowen TJ, Yakushiji H, Montagna C, Jain S, Ried T, Wynshaw-Boris A. Atm heterozygosity cooperates with loss of Brca1 to increase the severity of mammary gland cancer and reduce ductal branching. Cancer Res (2005) 65:8736–46. doi: 10.1158/0008-5472.CAN-05-1598
27. Su F, Smilenov LB, Ludwig T, Zhou L, Zhu J, Zhou G, et al. Hemizygosity for atm and Brca1 influence the balance between cell transformation and apoptosis. Radiat Oncol (2010) 5:15. doi: 10.1186/1748-717X-5-15
28. Wang J, Su F, Smilenov LB, Zhou L, Hu W, Ding N, et al. Mechanisms of increased risk of tumorigenesis in atm and Brca1 double heterozygosity. Radiat Oncol (2011) 6:96. doi: 10.1186/1748-717X-6-96
29. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA (2017) 317:2402–16. doi: 10.1001/jama.2017.7112
30. Easton DF, Pharoah PD, Antoniou AC, Tischkowitz M, Tavtigian SV, Nathanson KL, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med (2015) 372:2243–57. doi: 10.1056/NEJMsr1501341
31. Lilyquist J, LaDuca H, Polley E, Davis BT, Shimelis H, Hu C, et al. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol Oncol (2017) 147:375–80. doi: 10.1016/j.ygyno.2017.08.030
32. Barnes DR, Rookus MA, McGuffog L, Leslie G, Mooij TM, Dennis J, et al. Polygenic risk scores and breast and epithelial ovarian cancer risks for carriers of BRCA1 and BRCA2 pathogenic variants. Genet Med (2020) 22:1653–66. doi: 10.1038/s41436-020-0862-x
33. Sepahi I, Faust U, Sturm M, Bosse K, Kehrer M, Heinrich T, et al. Investigating the effects of additional truncating variants in DNA-repair genes on breast cancer risk in BRCA1-positive women. BMC Cancer (2019) 19:787. doi: 10.1186/s12885-019-5946-0
34. Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst (2002) 94:1365–72. doi: 10.1093/jnci/94.18.1365
35. Spurdle AB, Couch FJ, Parsons MT, McGuffog L, Barrowdale D, Bolla MK, et al. Refined histopathological predictors of BRCA1 and BRCA2 mutation status: A large-scale analysis of breast cancer characteristics from the BCAC, CIMBA, and ENIGMA consortia. Breast Cancer Res (2014) 16:3419. doi: 10.1186/s13058-014-0474-y
Keywords: BRCA1, ATM, double heterozygote, spliceogenic variant, case report
Citation: Colombo M, Mondini P, Minenza E, Foglia C, Mosconi A, Molica C, Pistola L, Ludovini V and Radice P (2023) A novel BRCA1 splicing variant detected in an early onset triple-negative breast cancer patient additionally carrying a pathogenic variant in ATM: A case report. Front. Oncol. 13:1102184. doi: 10.3389/fonc.2023.1102184
Received: 18 November 2022; Accepted: 27 February 2023;
Published: 21 March 2023.
Edited by:
Athina Markou, National and Kapodistrian University of Athens, GreeceReviewed by:
Marcus Vetter, University Hospital of Basel, SwitzerlandBo Hu, Zhejiang Chinese Medical University, China
Laura Ancuta Pop, University of Medicine and Pharmacy Iuliu Hatieganu, Romania
Copyright © 2023 Colombo, Mondini, Minenza, Foglia, Mosconi, Molica, Pistola, Ludovini and Radice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mara Colombo, bWFyYS5jb2xvbWJvQGlzdGl0dXRvdHVtb3JpLm1pLml0