
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 23 May 2023
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1090615
This article is part of the Research TopicCase Reports in Neuro-Oncology and Neurosurgical Oncology : 2022View all 32 articles
 Yingjie Wang1,2†Xuan Yang3†
Yingjie Wang1,2†Xuan Yang3† Qianquan Ma1,2†
Qianquan Ma1,2† Van Halm-Lutterodt Nicholas4Jianjun Sun1,2
Van Halm-Lutterodt Nicholas4Jianjun Sun1,2 Xiaofang Zhao1,2Weihai Liu1,2
Xiaofang Zhao1,2Weihai Liu1,2 Chenlong Yang1,2*
Chenlong Yang1,2*Background: Paraganglioma in the sellar region is an extremely rare entity, with a limited number of cases reported in the literature. Due to the paucity of clinical evidence, the diagnosis and treatment of paragangliomas in the sellar region remain challenging. Herein, we reported a case of sellar paraganglioma with parasellar and suprasellar extension. Particularly, the dynamic evolution of this benign tumor within a 7-year longitudinal observation was presented. Additionally, the relevant literature regarding sellar paraganglioma was comprehensively reviewed.
Case description: A 70-year-old woman presented with progressive visual deterioration and headache. Brain magnetic resonance imaging demonstrated a mass in the sellar region with parasellar and suprasellar extension. The patient refused surgical treatment. Seven years later, brain magnetic resonance imaging showed the lesion significantly progressed. Neurological examination revealed bilateral tubular contraction of visual fields. Laboratory examinations showed endocrine hormone levels were normal. Surgical decompression was performed via a subfrontal approach, and subtotal resection was achieved. Histopathological examination confirmed a diagnosis of paraganglioma. Postoperatively, she developed hydrocephalus, and ventriculoperitoneal shunting was performed. Eight months later, cranial CT showed no recurrence of the residual tumor, and the hydrocephalus had been relieved.
Conclusion: Paraganglioma occurring in the sellar region is rare, and the preoperative differential diagnosis is difficult. Owing to the infiltration to the cavernous sinus and internal carotid, complete surgical resection is usually impracticable. There has been no consensus regarding postoperative adjuvant radiochemotherapy for the tumor residue. In-situ recurrence and metastasis have been reported in the literature, and close follow-up is warranted.
Paragangliomas refer to benign neuroendocrine neoplasms originating from paraganglionic tissue, which are derived from neural crest progenitor cells. Except for the adrenal medulla, paragangliomas predominantly arise in the head and neck region, most commonly involving the carotid bodies, the jugular glomus, and the vagal bodies (1, 2). According to the literature, extra-adrenal paragangliomas account for only 10% to 15% of all paragangliomas (3); paragangliomas affecting the central nervous system are particularly rare, comprising approximately 0.6% of all head and neck neoplasms (4). Although paragangliomas are generally considered to be indolent entities corresponding histologically to World Health Organization (WHO) grade I (5), a high recurrence rate following surgical resection (10% for paragangliomas of carotid body and 50% to 60% for those occurring in other sites) and even metastasis (~10%) have been reported (6). Previously, we reported a consecutive surgical series of 19 patients with pathologically diagnosed spinal paragangliomas, in which we noted a considerable risk of in-situ recurrence Sellar paragangliomas are extremely unusual as paraganglia do not normally exist in this region, which has been only reported in very limited cases (7–36). This may be secondary to the persistence of paraganglionic tissue caused by deficient involution during early life. In this study, we presented a case of sellar paraganglioma with parasellar and suprasellar extension. Particularly, the dynamic evolution of this benign tumor within a 7-year longitudinal observation was presented. Additionally, the relevant literature regarding sellar paraganglioma was comprehensively reviewed.
A 70-year-old woman presented with a 3-year history of visual deterioration and headache. Physical examination revealed binocular severe visual disturbance. Brain magnetic resonance imaging (MRI) was requested, yielding a space-occupying lesion in the sellar region with right parasellar and suprasellar extension (Figure 1). The mass was irregular in shape displaying isointensity on T1-weighted imaging and heterogenous signals on T2-weighted imaging, and contrast enhancement was notable after the administration of gadolinium diethylenetriamine pentaacetic acid (Gd-DTPA). Surgical resection of the lesion via a subfrontal approach was recommended, but the patient refused it considering the advanced age and mild symptoms.
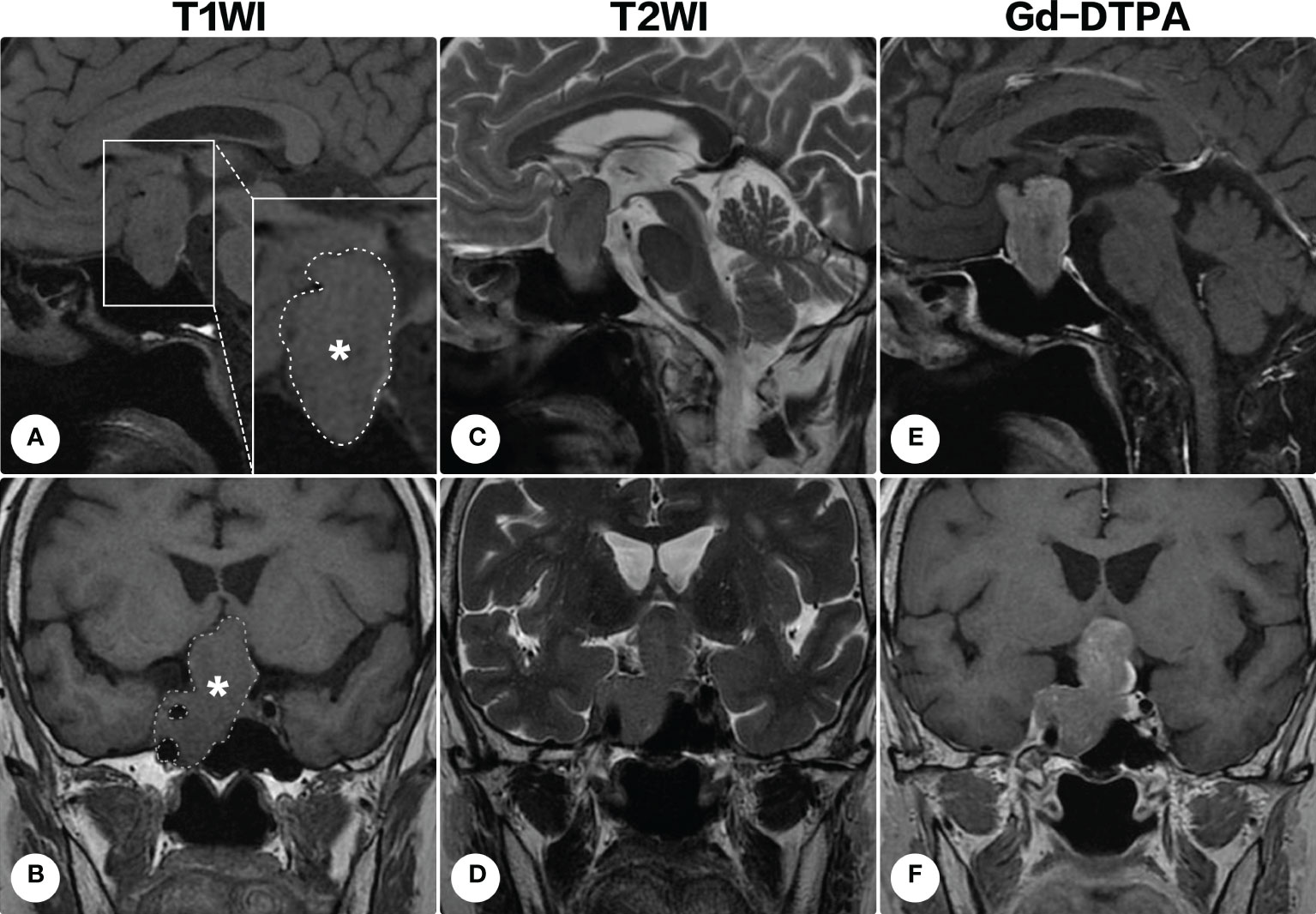
Figure 1 Brain magnetic resonance imaging on the first admission. Brain magnetic resonance imaging revealed a space-occupying lesion in the sellar region with right parasellar and suprasellar extension. The lesion (indicated by asterisks and outlined by dashed lines) appeared isointense on T1-weighted imaging [(A), sagittal; (B) coronal] and heterogenous signals on T2-weighted imaging [(C), sagittal; (D) coronal]. Contrast-enhanced T1-weighted images demonstrated a remarkable homogeneous enhancement [(E) sagittal; (F), coronal].
During the following years, the visual field defect was progressively aggravated. The latest brain MRI after a 7-year conservative observation demonstrated the lesion significantly progressed (Figure 2), and the patient was readmitted. Ophthalmologic examination showed bilateral visual field defects and pathologic myopia with posterior staphyloma and tigroid fundus (Figure 3). Other cranial nerves were normal, and there were no sensorimotor dysfunctions. Laboratory examinations showed endocrine hormone levels (including prolactin [PRL], growth hormone [GH], follicle stimulating hormone [FSH], luteinizing hormone [LH], adrenocorticotropic hormone [ACTH], and thyroid hormones) were all within normal limits. Cranial computed tomography (CT) and MRI demonstrated a giant lobulated sellar mass with parasellar and suprasellar extension, and the optic chiasm was remarkably displaced; the lesion was isointense on T1-weighted imaging and heterogeneously hyperintense on T2-weighted imaging, with remarkable enhancement after administration of contrast medium (Figures 2A-G). CT angiography did not identify any significant vascular aberrance (Figure 2H).
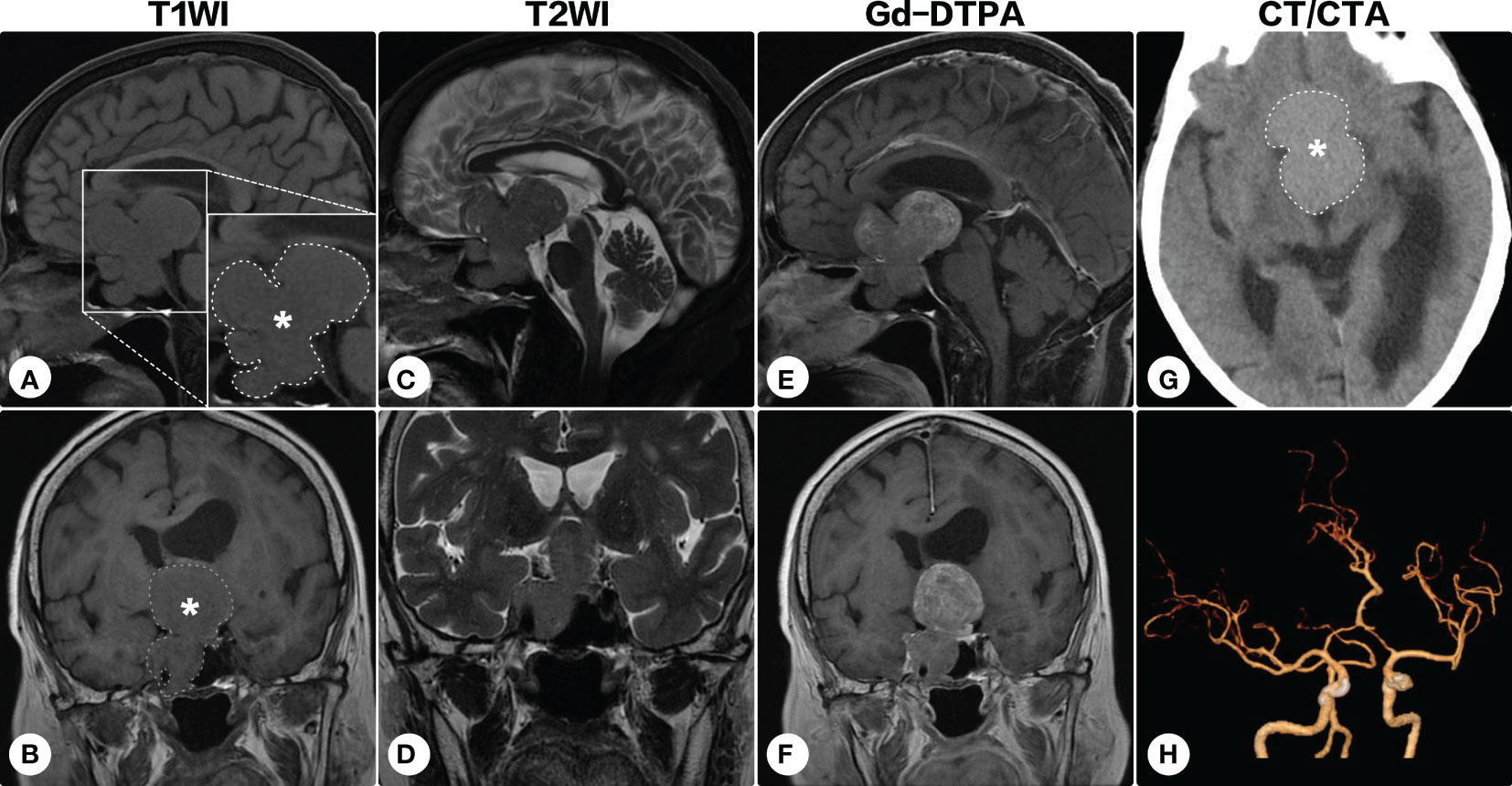
Figure 2 Brain magnetic resonance imaging after a 7-year conservative observation. Brain magnetic resonance imaging showed the lesion (indicated by asterisks and outlined by dashed lines) significantly progressed [(A, C, E) sagittal; (B, D, F) coronal]. On cranial computed tomography, the mass was hyperdense (G). Computed tomographic angiography identified no significant vascular aberrance (H).
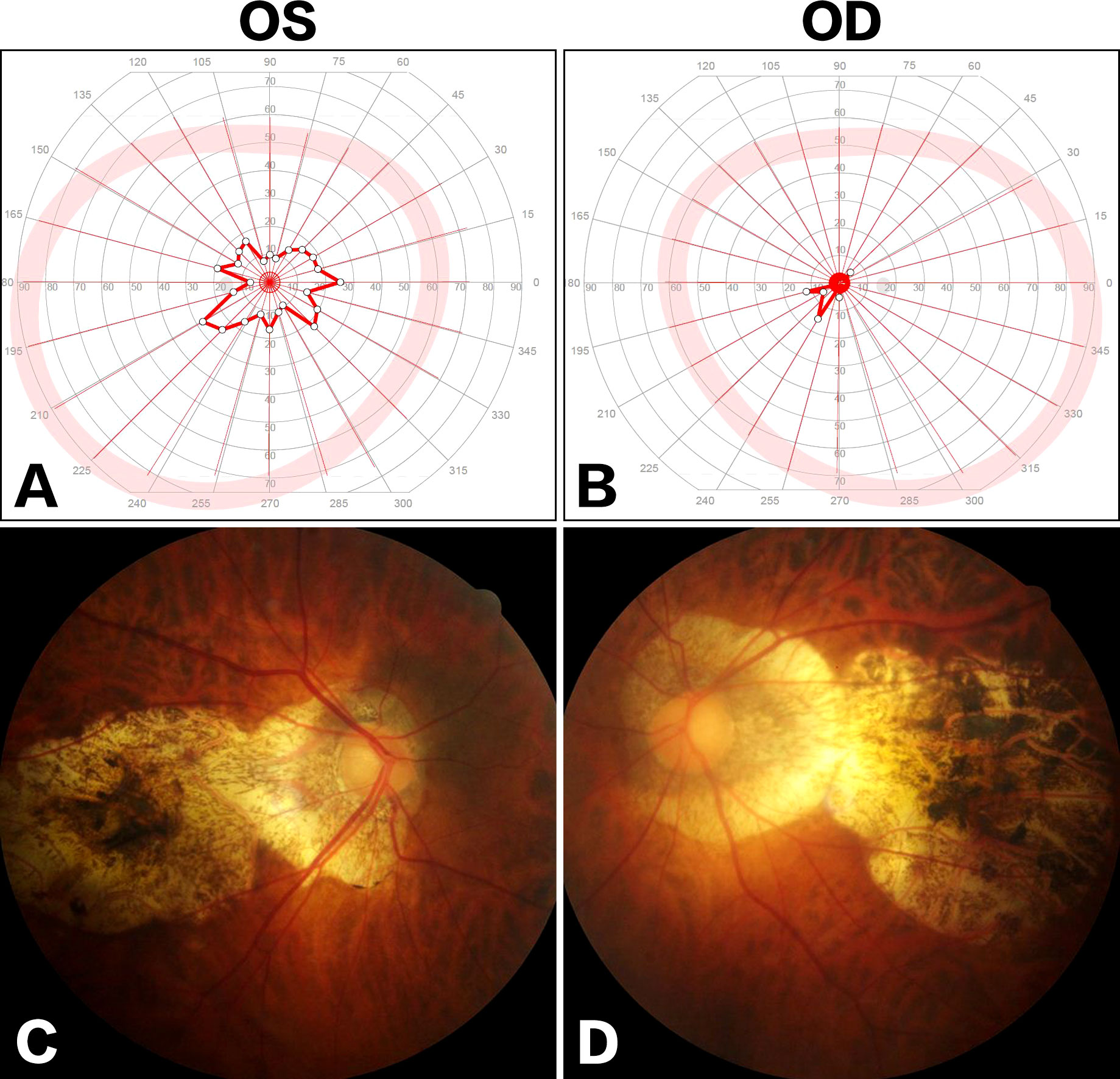
Figure 3 Ophthalmologic examinations. Visual field examination showed bilateral tubular contraction of visual fields [(A) OS; (B) OD]. Fundus examination demonstrated posterior staphyloma and tigroid fundus [(C), OS; (D) OD].
Preoperatively, a diagnosis of nonfunctional pituitary adenoma was suspected. As the main body of the lesion extended to the suprasellar and parasellar areas, the transnasal-sphenoidal approach may be difficult to achieve complete tumor resection. The patient underwent a surgical decompression via a subfrontal approach. Intraoperatively, the tumor was found to be rubbery with an abundant blood supply. The tumor tissue was closely attached to the internal carotid artery and cavernous sinus, and subtotal resection was eventually achieved.
Histopathological sections revealed a tumor with a lobulated pattern and cellular nests surrounded by vascular fibrous septa (Figure 4). The irregular and lobulated clusters of cuboidal cells were consistent with the morphological characteristics of paraganglioma (Zellballen pattern). Immunohistochemical stains showed the tumor cells were strongly positive for synaptophysin (SYN), chromogranin A (CgA), and microtubule-associated protein 2 (MAP-2), but negative for glial fibrillary acidic protein (GFAP), S100 protein, epithelial membrane antigen (EMA), cytokeratin (CK), CD68, and neuronal nuclear antigen (NeuN). Additionally, the tumor cells showed no immunoreactivity against pituitary cell-lineage transcription factors (T-PIT, PIT-1, SF-1), thyroid transcription factor 1 (TTF-1), or endocrine markers (PRL, GH, thyroid stimulating hormone [TSH], ACTH, FSH, LH). The Ki-67 proliferation index was <5%. A diagnosis of paraganglioma was made.
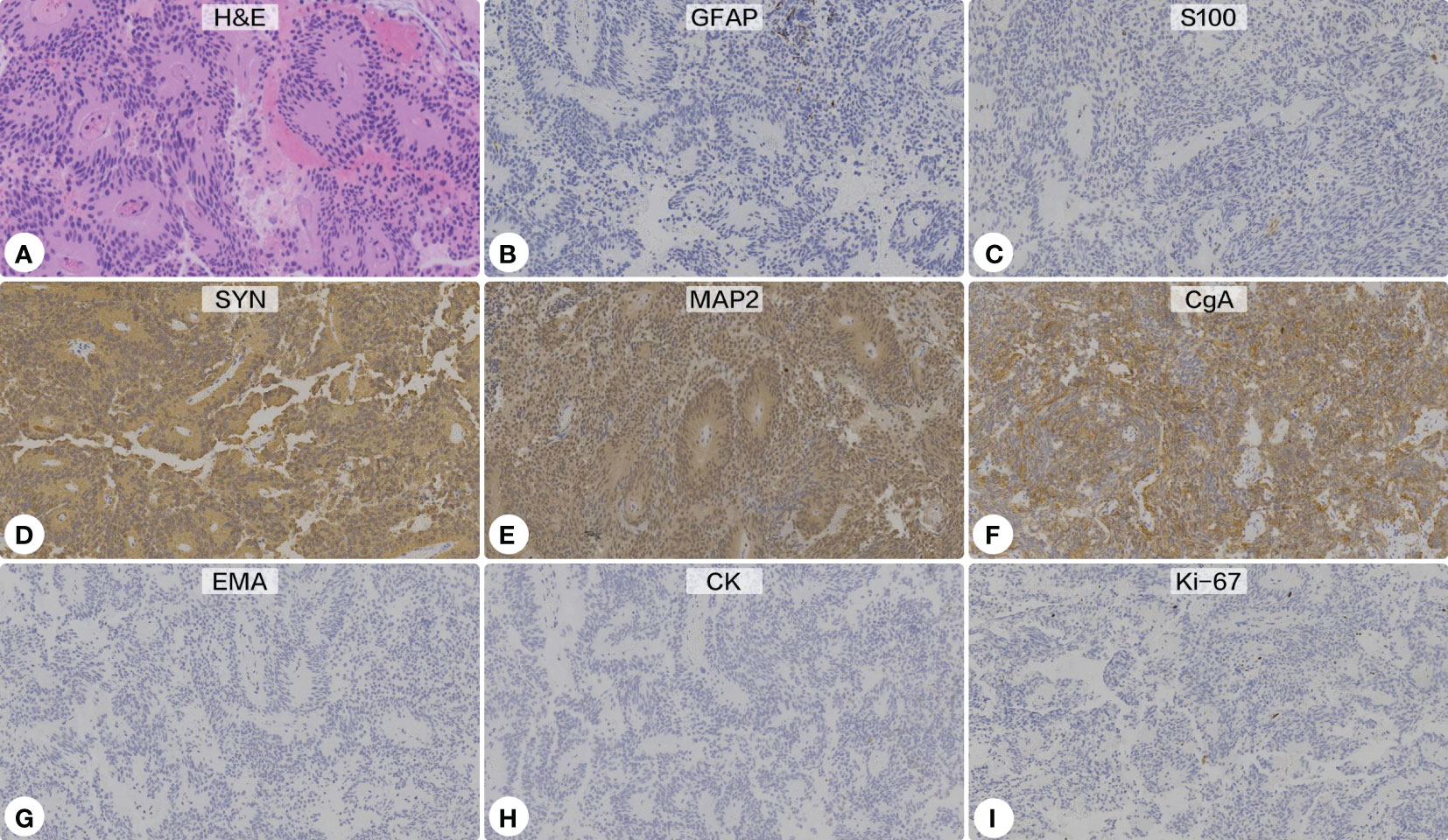
Figure 4 Histopathological and immunohistochemical examinations. (A) Hematoxylin-eosin staining showed a lobulated pattern and cellular nests surrounded by vascular fibrous septa, which were consistent with the morphological characteristics of paraganglioma (Zellballen pattern) (Original magnification ×200). Immunohistochemical stains showed the tumor cells were negative for glial fibrillary acidic protein (B) and S100 protein (C), but strongly positive for synaptophysin (D), microtubule-associated protein 2 (E), and chromogranin A (F). Additionally, the tumor showed no immunoreactivity against epithelial membrane antigen (G) or cytokeratin (H). The Ki-67 proliferation index was <5% (I).
Postoperative CT confirmed the tumor was subtotally removed, and the tumor cavity was filled with hemostatic material (Figure 5A). The patient’s visual function showed no significant improvement. One month after the operation, she developed gait disturbance and urinary incontinence. Repeated cranial CT showed ventricle dilation with interstitial edema (Figure 5B). A secondary hydrocephalus was diagnosed and a ventriculoperitoneal shunting was performed (Figure 5C). A week postoperatively, the neurological deficiencies were partially improved, and CT showed the ventricles shrank (Figure 5D). Eight months postoperatively, cranial CT showed no recurrence of the residual tumor, and the hydrocephalus had been relieved (Figures 5E, F). However, she got pneumonia two months later and succumbed to respiratory failure.
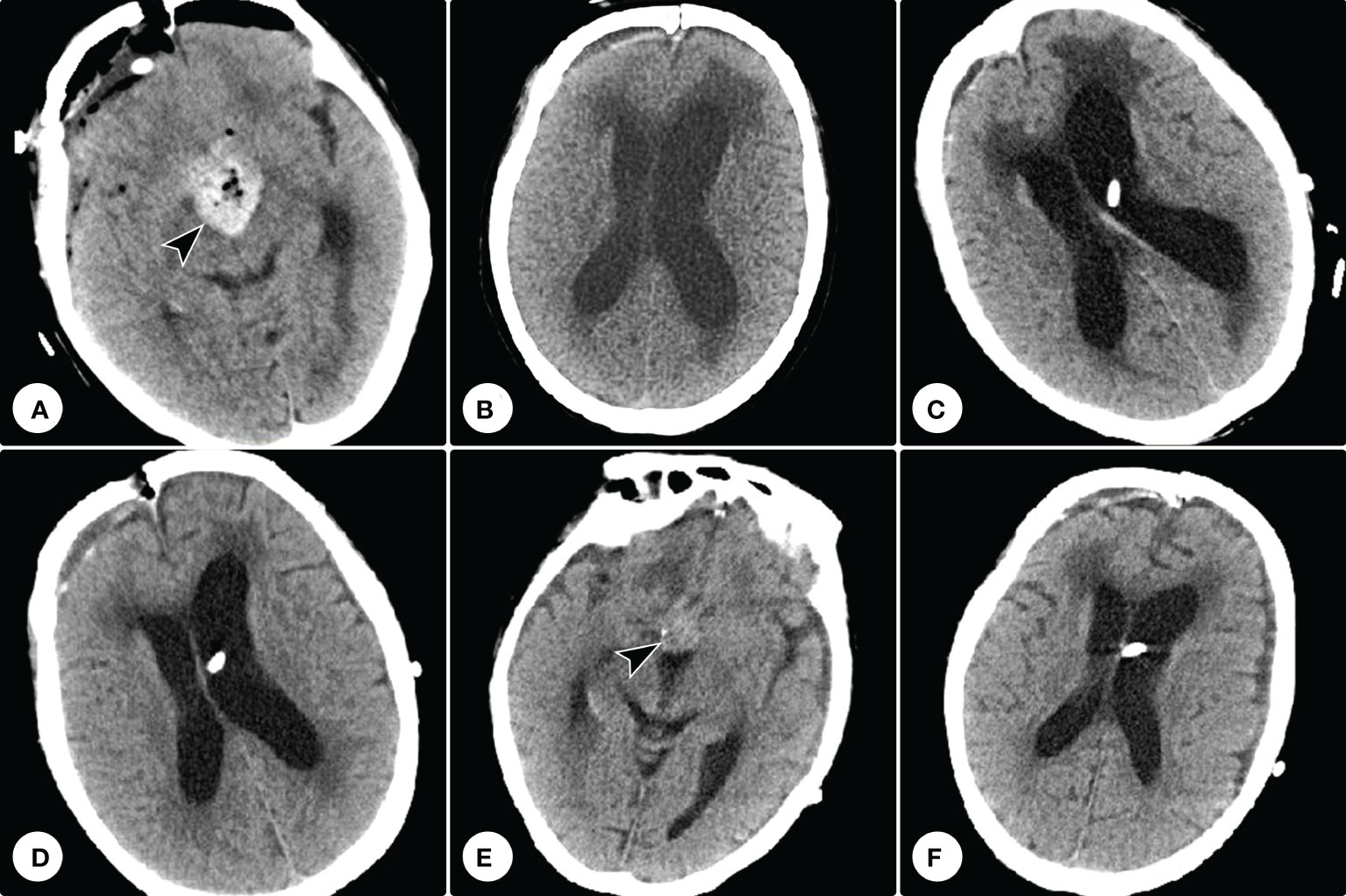
Figure 5 Postoperative and follow-up computed tomography examinations. (A) Postoperative computed tomography confirmed the tumor was subtotally removed, and the tumor cavity was filled with hemostatic material (arrowhead). (B) Repeated computed tomography showed ventricle dilation with interstitial edema. (C) Computed tomography showed a ventriculoperitoneal shunting was performed. (D) A week postoperatively, computed tomography showed the ventricles shrank. (E, F) Eight months postoperatively, cranial CT showed no recurrence of the tumor residue and the hemostatic material had been absorbed (E), and the hydrocephalus had been relieved (F).
We conducted a literature review to identify articles on sellar paragangliomas published up to November 1, 2022. We performed a systematic search of the PubMed and Embase databases using the terms “paraganglioma” and “sellar”. A total of 30 publications were retrieved, and all of them are case reports.
There were 13 females and 22 males (including our current case), with a female-to-male ratio of 1:1.69. The average age was 46.9 ± 22.5 (range, 13~84 years). The most common onset symptoms were visual deficits (68.6%) and headache (65.7%). All the patients underwent surgical treatment, and no complete resection was described. Postoperative adjuvant radiation was administered for controlling the tumor residue in 45.7% of all cases (16 of 35 cases), with a dosage of 40~50 Gy (20~28 fractions). Follow-up data were available in 23 cases; after an average follow-up period of 21.5 ± 28.8 months, two patients experienced in-situ recurrence, and metastasis was noted in two other cases. Demographic and clinical profiles were summarized in Table 1. Radiological characteristics were presented in Table 2.
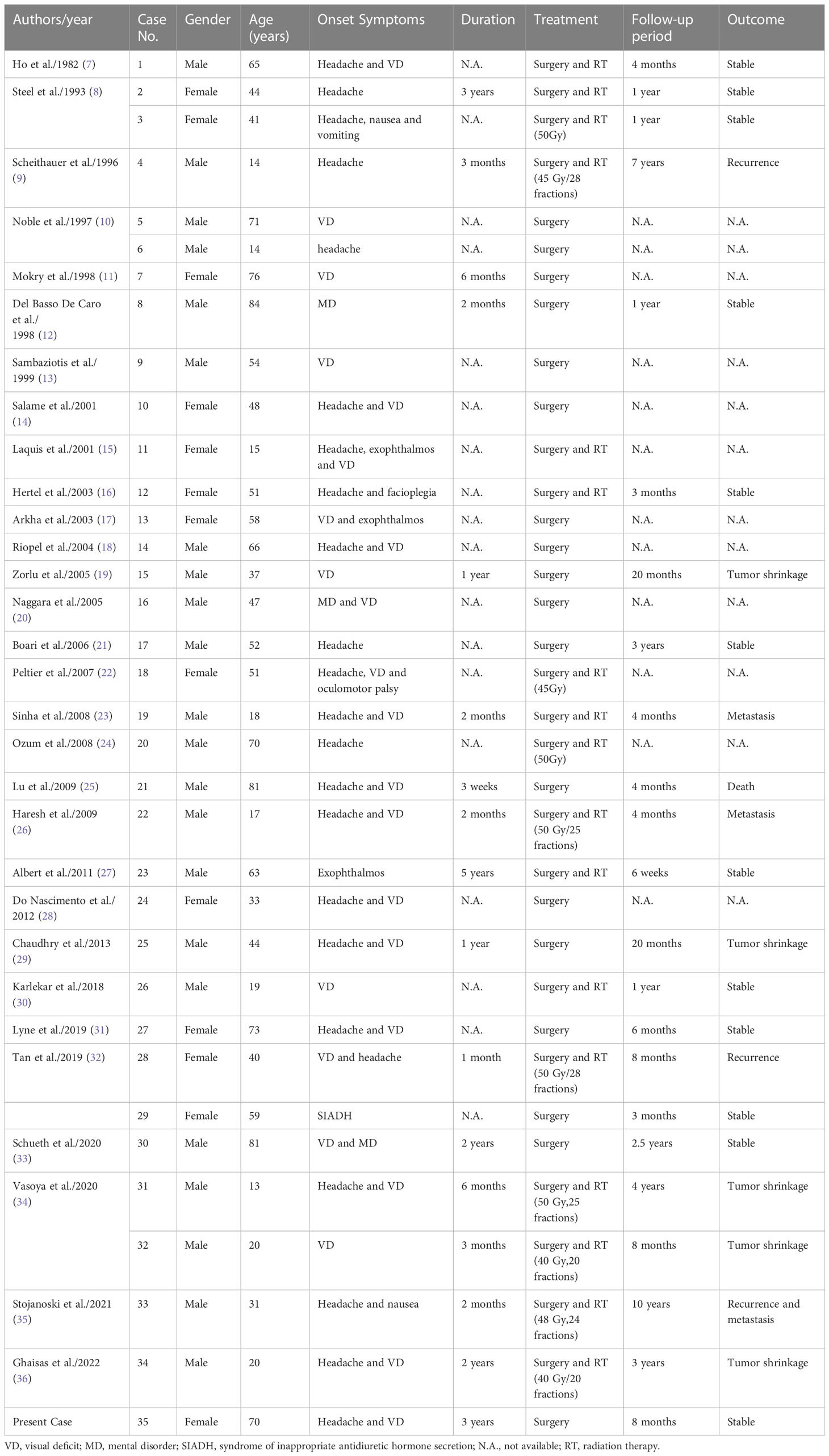
Table 1 Demographic and clinical features of paragangliomas in the sellar region.
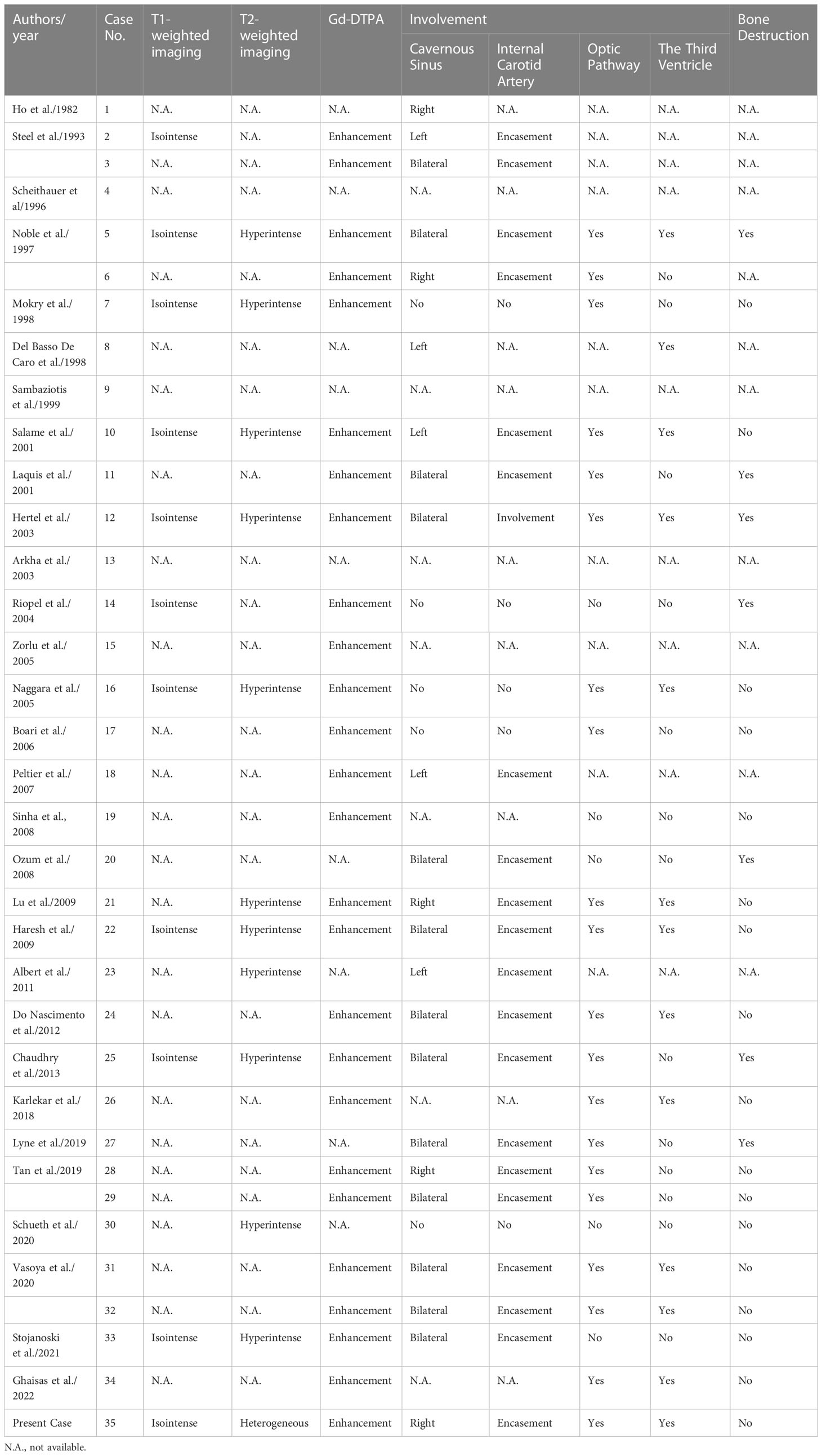
Table 2 Radiological characteristics of paragangliomas in the sellar region.
Paragangliomas are relatively rare tumors that develop from the paraganglia of the autonomic nervous system. Because the sellar region lacks autonomic nervous tissue and there are no paraganglia, the derivation and etiology of sellar paragangliomas remain a mystery. Regarding its origin, there are two hypotheses: 1) the ‘embryonic remnants’ theory, proposing that paraganglioma may arise from the embryonic paraganglionic cells trapped around the pituitary; or 2) the ‘ectopic migration’ theory, holding that paraganglioma may originate from paraganglionic cells abnormally migrated across the tympanic or ciliary branches of the glossopharyngeal nerve to the cavernous sinus (8, 20). In this study, we found the majority of reported cases showed involvement of the cavernous sinus, which seems to support the latter view. Additionally, the most common onset symptom was visual decline rather than abnormal pituitary endocrine function, highly implying paraganglioma is an extra-pituitary lesion. In our current case, a longitudinal conservative observation demonstrated remarkable tumor growth. At the early stage, the tumor invaded the right cavernous sinus and encased the right internal carotid artery, while no osseous destruction or pituitary dysfunctions were noted. These clinicoradiological features prompted the cavernous sinus origin of sellar paragangliomas.
Preoperative diagnosis of sellar paragangliomas is extremely challenging, and differential diagnoses mainly include pituitary adenomas, meningiomas, craniopharyngiomas, germinomas, and Rathke cleft cysts. In most cases, sellar paragangliomas tend to be misdiagnosed as nonfunctional pituitary adenomas. The following potential characteristics may assist the diagnosis: 1) despite the giant size of the tumor, the patient’s endocrinal functions are usually normal; 2) just like the “salt and pepper” appearance on T2-weighted imaging that is frequently seen in peripheral paragangliomas, the high vascularization leads to heterogeneous intensity on T2-weighted imaging in sellar paragangliomas. Some scholars also proposed that functional imaging, such as 18F-DOPA positron emission tomography, may help confirm the neuroendocrine nature of tumors with high specificity (37, 38). Moreover, the imaging also shows additional advantages in diagnosing multifocality (co-existing paragangliomas in the rest of the body) and/or distant metastases (38, 39).
The most common symptom, visual deterioration, is caused by the tumor compression to the optic chiasma, and thus surgical resection of the tumor is the most effective treatment. The optional surgical strategies include the subfrontal approach, the transpterional approach, and transnasal-sphenoidal operations. In some cases, sellar paragangliomas are highly vascularized, and preoperative angiography may provide essential information for the identification of tumor blood supply. When a definitive supplying artery can be identified, preoperative endovascular embolization may significantly reduce the risk of intraoperative bleeding. In our case, the tumor was moderately enhanced after the administration of contrast medium and computed tomographic angiography showed no vascular aberrance, and thus endovascular embolization was not performed. Sellar paragangliomas are usually lobulated, and the cavernous sinus and internal carotid artery are always involved; therefore, intraoperative complete resection of the tumor may be extraordinarily difficult. For controlling the tumor residue, postoperative radiotherapy showed potential efficacies. Chemotherapy has not yet been reported for sellar paragangliomas.
The surgical outcomes of sellar paragangliomas varied distinctly. In most cases, surgery with or without radiation therapy leads to residual stability or even tumor shrinkage. Metastasis can also occur in a few cases, and close follow-up is indispensable.
Paragangliomas occurring in the sellar region are rare, and preoperative differential diagnosis is difficult. Complete surgical resection of the tumor is usually impracticable, and postoperative adjuvant radiotherapy can be considered for the tumor residue. In-situ recurrence and metastasis have been reported in the literature, and close follow-up should be highlighted.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual legal guardian for the publication of any potentially identifiable images or data included in this article.
Conception and design: CY. Acquisition of data: YW, XZ, WL, and QM. Analysis and interpretation of data: XY, QM, JS, and VHN. Drafting and critically revising the article: YW, XY, and CY. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81901202 to CY; 82002883 to XY), Beijing Natural Science Foundation (7222217 to CY), the Capital Health Research and Development of Special (2022-4-40918 to CY), AO Spine Research Start-up Grant (AOS-Startup-21-016 to CY), Clinical Medicine Plus X-Young Scholars Project, Peking University, the Fundamental Research Funds for the Central Universities (PKU2021LCXQ007 to CY), and the priming scientific research foundation for the junior researcher in Beijing Tongren Hospital, Capital Medical University (2020-YJJ-ZZL-017 to XY).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer (2011) 18(6):R253–76. doi: 10.1530/ERC-11-0170
2. Kliewer KE, Cochran AJ. A review of the histology, ultrastructure, immunohistology, and molecular biology of extra-adrenal paragangliomas. Arch Pathol Lab Med (1989) 113(11):1209–18.
3. Brahmbhatt P, Patel P, Saleem A, Narayan R, Young M. Retroperitoneal paraganglioma presenting as a chest pain: a case report. Case Rep Oncol Med (2013) 2013:329472. doi: 10.1155/2013/329472
4. Chang H, Silva MA, Torres AA, Weng J, de Lima Guido LP, Velez-Torres J, et al. A carcinoid tumor of the middle ear masquerading as a glomus tympanicum presenting with temporal lobe hemorrhage in a 70-year-old woman: case report and review of the literature. Neurochirurgie (2022) 68(6):654–60. doi: 10.1016/j.neuchi.2022.07.008
5. Yang C, Li G, Fang J, Wu L, Yang T, Deng X, et al. Clinical characteristics and surgical outcomes of primary spinal paragangliomas. J Neurooncol (2015) 122(3):539–47. doi: 10.1007/s11060-015-1742-0
6. Reithmeier T, Gumprecht H, Stolzle A, Lumenta CB. Intracerebral paraganglioma. Acta Neurochir (Wien) (2000) 142(9):1063–6. doi: 10.1007/s007010070064
7. Ho KC, Meyer G, Garancis J, Hanna J. Chemodectoma involving the cavernous sinus and semilunar ganglion. Hum Pathol (1982) 13(10):942–3. doi: 10.1016/S0046-8177(82)80058-0
8. Steel TR, Dailey AT, Born D, Berger MS, Mayberg MR. Paragangliomas of the sellar region: report of two cases. Neurosurgery (1993) 32(5):844–7. doi: 10.1227/00006123-199305000-00021
9. Scheithauer BW, Parameswaran A, Burdick B. Intrasellar paraganglioma: report of a case in a sibship of von hippel-lindau disease. Neurosurgery (1996) 38(2):395–9. doi: 10.1097/00006123-199602000-00034
10. Noble ER, Smoker WR, Ghatak NR. Atypical skull base paragangliomas. AJNR Am J Neuroradiol (1997) 18(5):986–90.
11. Mokry M, Kleinert R, Clarici G, Obermayer-Pietsch B. Primary paraganglioma simulating pituitary macroadenoma: a case report and review of the literature. Neuroradiology (1998) 40(4):233–7. doi: 10.1007/s002340050573
12. Del Basso De Caro ML, Siciliano A, Cappabianca P, Alfieri A, de Divitiis E. Intrasellar paraganglioma with suprasellar extension: case report. Tumori (1998) 84(3):408–11. doi: 10.1177/030089169808400319
13. Sambaziotis D, Kontogeorgos G, Kovacs K, Horvath E, Levedis A. Intrasellar paraganglioma presenting as nonfunctioning pituitary adenoma. Arch Pathol Lab Med (1999) 123(5):429–32. doi: 10.5858/1999-123-0429-IPPANP
14. Salame K, Ouaknine GE, Yossipov J, Rochkind S. Paraganglioma of the pituitary fossa: diagnosis and management. J Neurooncol (2001) 54(1):49–52. doi: 10.1023/A:1012535230135
15. Laquis SJ, Vick V, Haik BG, Fleming JC, Wilson MW. Intracranial paraganglioma (glomus tumor) with orbital extension. Ophthalmic Plast Reconstr Surg (2001) 17(6):458–61. doi: 10.1097/00002341-200111000-00015
16. Hertel F, Bettag M, Morsdorf M, Feiden W. Paragangliomas of the parasellar region. Neurosurg Rev (2003) 26(3):210–4. doi: 10.1007/s10143-003-0266-9
17. Arkha Y, Boutarbouch M, Lamalmi N, Halefadl S, Laamarti A, Derraz S, et al. Paraganglioma of the latero-sellar area. case report. Neurochirurgie (2003) 49(5):540–4.
18. Riopel C, Courville P, Fabre B, Callonnec F, Bolognini B, Marie JP, et al. Parasellar paraganglioma: a case report. Ann Pathol (2004) 24(1):62–7. doi: 10.1016/S0242-6498(04)93903-X
19. Zorlu F, Selek U, Ulger S, Donmez T, Erden E. Paraganglioma in sella. J Neurooncol (2005) 73(3):265–7. doi: 10.1007/s11060-004-5673-4
20. Naggara O, Varlet P, Page P, Oppenheim C, Meder JF. Suprasellar paraganglioma: a case report and review of the literature. Neuroradiology (2005) 47(10):753–7. doi: 10.1007/s00234-005-1422-4
21. Boari N, Losa M, Mortini P, Snider S, Terreni MR, Giovanelli M. Intrasellar paraganglioma: a case report and review of the literature. Acta Neurochir (Wien) (2006) 148(12):1311–4. doi: 10.1007/s00701-006-0895-1
22. Peltier J, Fichten A, Lefranc M, Grunewald P, Thelu F, Toussaint P, et al. Paraganglioma of the cavernous sinus. case report. Neurochirurgie (2007) 53(5):391–4. doi: 10.1016/j.neuchi.2007.06.006
23. Sinha S, Sharma MC, Sharma BS. Malignant paraganglioma of the sellar region mimicking a pituitary macroadenoma. J Clin Neurosci (2008) 15(8):937–9. doi: 10.1016/j.jocn.2007.03.029
24. Ozum U, Egilmez R, Yildirim A. Paraganglioma in pituitary fossa. Neuropathology (2008) 28(5):547–50. doi: 10.1111/j.1440-1789.2008.00885.x
25. Lu JQ, Khalil M, Hu W, Sutherland GR, Clark AW. Tumor-to-tumor metastasis: esophageal carcinoma metastatic to an intracranial paraganglioma. J Neurosurg (2009) 110(4):744–8. doi: 10.3171/2008.9.JNS08397
26. Haresh KP, Prabhakar R, Anand Rajan KD, Sharma DN, Julka PK, Rath GK. A rare case of paraganglioma of the sella with bone metastases. Pituitary (2009) 12(3):276–9. doi: 10.1007/s11102-008-0099-1
27. Albert A, Ramirez JA, Codere F, Petrecca K. Sellar paraganglioma: a unique route to a rare destination case report and literature review. Clin Neurol Neurosurg (2011) 113(8):675–7. doi: 10.1016/j.clineuro.2011.04.002
28. do Nascimento A, Maranha LA, Corredato RA, Araujo JC, Bleggi-Torres LF. 33 year-old woman with a large sellar tumor. Brain Pathol (2012) 22(6):869–70. doi: 10.1111/j.1750-3639.2012.00640.x
29. Chaudhry NS, Ahmad F, Blieden C, Morcos JJ. Suprasellar and sellar paraganglioma presenting as a nonfunctioning pituitary macroadenoma. J Clin Neurosci (2013) 20(11):1615–8. doi: 10.1016/j.jocn.2013.02.004
30. Karlekar M, Kumar S, Jadhav S, Lila A, Bandgar T, Shah N. Sellar paraganglioma. Clin Nucl Med (2018) 43(8):591–2. doi: 10.1097/RLU.0000000000002157
31. Lyne SB, Polster SP, Fidai S, Pytel P, Yamini B. Primary sellar paraganglioma: case report with literature review and immunohistochemistry resource. World Neurosurg (2019) 125:32–6. doi: 10.1016/j.wneu.2019.01.094
32. Tan CL, Pang YH, Lim KHC, Sein L, Codd PJ, McLendon RE. Two extraordinary sellar neuronal tumors: literature review and comparison of clinicopathologic features. Am J Clin Pathol (2019) 151(3):241–54. doi: 10.1093/ajcp/aqy155
33. Schueth EA, Martinez DC, Kulwin CG, Bonnin JM, Payner TD, Ting JY. Recurrent primary intrasellar paraganglioma. Case Rep Otolaryngol (2020) 2020:2580160. doi: 10.1155/2020/2580160
34. Vasoya P, Aryan S, Thakar S, Sivaraju L, Ghosal N, Hegde AS. Sellar-suprasellar paraganglioma: report of 2 cases and review of literature. World Neurosurg (2020) 140:293–300. doi: 10.1016/j.wneu.2020.04.157
35. Stojanoski S, Boldt HB, Kozic D, Patocs A, Korbonits M, Medic-Stojanoska M, et al. Case report: malignant primary sellar paraganglioma with unusual genetic and imaging features. Front Oncol (2021) 11:739255. doi: 10.3389/fonc.2021.739255
36. Ghaisas S, Rao KS, Preethi A, Rani PK. Suprasellar paraganglioma in a clinical setting of von hippel-lindau syndrome. BMJ Case Rep (2022) 15(3):e245907. doi: 10.1136/bcr-2021-245907
37. Taieb D, Kaliski A, Boedeker CC, Martucci V, Fojo T, Adler JR Jr., et al. Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev (2014) 35(5):795–819. doi: 10.1210/er.2014-1026
38. Timmers HJ, Taieb D, Pacak K. Current and future anatomical and functional imaging approaches to pheochromocytoma and paraganglioma. Horm Metab Res (2012) 44(5):367–72. doi: 10.1055/s-0031-1299712
39. Archier A, Varoquaux A, Garrigue P, Montava M, Guerin C, Gabriel S, et al. Prospective comparison of (68)Ga-DOTATATE and (18)F-FDOPA PET/CT in patients with various pheochromocytomas and paragangliomas with emphasis on sporadic cases. Eur J Nucl Med Mol Imaging (2016) 43(7):1248–57. doi: 10.1007/s00259-015-3268-2
Keywords: paraganglioma, sellar, extra-adrenal, surgical treatment, case report
Citation: Wang Y, Yang X, Ma Q, Nicholas VH-L, Sun J, Zhao X, Liu W and Yang C (2023) Case Report: Paraganglioma in the sellar region: longitudinal observation and surgical outcome. Front. Oncol. 13:1090615. doi: 10.3389/fonc.2023.1090615
Received: 05 November 2022; Accepted: 05 May 2023;
Published: 23 May 2023.
Edited by:
Marcos Vinicius Calfat Maldaun, Hospital Sirio Libanes, BrazilReviewed by:
Li Cai, CHI St. Vincent, United StatesCopyright © 2023 Wang, Yang, Ma, Nicholas, Sun, Zhao, Liu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chenlong Yang, dmlrLnlhbmdAcGt1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.