
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 30 January 2023
Sec. Cancer Genetics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1069467
This article is part of the Research TopicIdentification, Risk Stratification, and Optimized Management for Lynch SyndromeView all 14 articles
 Shujuan Pan*
Shujuan Pan* Hannah CoxJamie WillmottErin MundtHeidi GorringeMichelle LandonKarla R. BowlesBradford CoffeeBenjamin B. RoaDebora Mancini-DiNardo
Hannah CoxJamie WillmottErin MundtHeidi GorringeMichelle LandonKarla R. BowlesBradford CoffeeBenjamin B. RoaDebora Mancini-DiNardoBackground and Aims: Tumor immunohistochemical staining (IHC) of DNA mismatch repair (MMR) proteins is often used to guide germline genetic testing and variant classification for patients with suspected Lynch syndrome. This analysis examined the spectrum of germline findings in a cohort of individuals showing abnormal tumor IHC.
Methods: We assessed individuals with reported abnormal IHC findings and referred for testing with a six-gene syndrome-specific panel (n=703). Pathogenic variants (PVs) and variants of uncertain significance (VUS) in MMR genes were designated expected/unexpected relative to IHC results.
Results: The PV positive rate was 23.2% (163/703; 95% confidence interval [CI], 20.1%-26.5%); 8.0% (13/163; 95% CI, 4.3%-13.3%) of PV carriers had a PV in an unexpected MMR gene. Overall, 121 individuals carried VUS in MMR genes expected to be mutated based on IHC results. Based on independent evidence, in 47.1% (57/121; 95% CI, 38.0%-56.4%) of these individuals the VUSs were later reclassified as benign and in 14.0% (17/121; 95% CI, 8.4%-21.5%) of these individuals the VUSs were reclassified as pathogenic.
Conclusions: Among patients with abnormal IHC findings, IHC-guided single-gene genetic testing may miss 8% of individuals with Lynch syndrome. In addition, in patients with VUS identified in MMR genes predicted to be mutated by IHC, extreme caution must be taken when the IHC results are considered in variant classification.
Lynch syndrome is caused by an inherited germline pathogenic variant (PV) in one or more of the DNA mismatch repair (MMR) genes MLH1, MSH2, MSH6, and PMS2 (1, 2). Approximately 3% of colorectal cancer (CRC) cases result from Lynch syndrome, making it the most common heritable CRC syndrome. Lynch syndrome also is associated with endometrial, ovarian, gastric/small bowel, urothelial, central nervous system, pancreatic, and prostate cancers (3). Clinical management for patients with a Lynch syndrome-related cancer involves heightened secondary cancer surveillance and can include risk-reducing surgeries – measures that have been shown to reduce morbidity and mortality (4–6). Therefore, it is essential to distinguish between Lynch syndrome and sporadic disease in patients diagnosed with cancer.
One first-line approach to differentiate between Lynch syndrome and sporadic cancer is to use immunohistochemical (IHC) staining to assess MLH1, MSH2, MSH6, and PMS2 protein expression in tumor tissue from biopsy or surgical resection. The National Comprehensive Cancer Network (NCCN), the American Society for Clinical Oncology, and others recommend universal IHC screening of new CRC and endometrial cancer cases (3, 7). Abnormal tumor MMR protein expression by IHC suggests a deficiency in the corresponding gene(s) and compromised MMR. NCCN recommends referral for further genetic testing for patients with abnormal IHC findings (3, 8–11).
Reflex genetic testing after an abnormal tumor IHC result can follow numerous paths. For example, MLH1 protein expression can be disrupted either by a germline pathogenic variant (PV) in MLH1, DNA promoter hypermethylation that silences the gene, or by double somatic mutations. Historically, if MLH1 is absent on IHC for CRC tumors, subsequent testing can take several directions: (1) germline MLH1 testing; (2) tumor MLH1 methylation testing; (3) tumor testing for the BRAF p.V600E PV based on its association with MLH1 methylation status (3, 12). In recent years, tumor testing of the MMR genes to detect somatic mutations in MMR genes has also been recommended (13). Nevertheless, only when gene-specific testing fails to identify a mutation will germline testing of additional MMR genes and/or other genes associated with hereditary cancer syndromes typically be recommended (3). This stepwise approach has proven complex, confusing, and time-consuming. While NCCN guidelines recommend that an individual with expertise in genetics be involved in the diagnostic process (3), surveyed gastroenterologists reported that it is often unclear which specialist would be responsible for selecting and ordering the test (14). Each separate test adds time to the patient’s diagnostic journey and increases the risk of loss to follow-up, which can delay or prevent risk-reducing surgical procedures.
Another relevant concern is the sensitivity of MMR IHC, with a 5-10% false negative rate (3, 15). Staining quality can vary depending on the tumor microenvironment and tissue fixation conditions, leading to ambiguity, misinterpretation of results, and misinformed gene selection for testing (16). In addition, some individuals who have abnormal IHC are found to carry germline PVs in MMR genes not predicted by the IHC result (17–19) or in non-MMR genes associated with other cancer syndromes (20, 21), which would have been missed using gene-specific genetics testing guided by IHC results.
In addition to its use as a screening tool, IHC results may be employed as supportive evidence in determining the pathogenicity of variants identified in genes predicted by IHC to be mutated (22). For instance, the variant classification criteria used by the International Society for Gastrointestinal Hereditary Tumors (InSiGHT) indicate that: when the presence of a variant of uncertain significance (VUS) coincides with the absence of the corresponding MMR protein on IHC in two or more patients, it is deemed supporting evidence for class 5 (pathogenic) or class 4 (likely pathogenic); conversely, inconsistent IHC results observed in three or more tumors were considered as supportive evidence for class 2 (likely benign) or class 1 (benign) (23). This application is concerning given the low predictive value of IHC staining for Lynch syndrome (24, 25). Although IHC results generally are not used as stand-alone evidence for variant classification, there exists potential for an incorrect determination (22).
The frequency of discordance between IHC results and germline genetic findings across various tumor types has not been evaluated systematically in the clinical laboratory, meaning that it is unclear how many patients might be affected clinically by incomplete or misleading IHC results. The current analysis aimed to address this knowledge gap by evaluating germline genetic findings from multi-gene panel testing of individuals with abnormal IHC results in Lynch-associated tumor types. The objectives were to determine (1) the extent of PVs in genes not predicted by IHC, and (2) the possibility of misclassifying variants based on IHC findings.
The analysis included individuals who underwent clinical genetic testing that included the MMR genes (MLH1, MSH2, MSH6, PMS2) from May 2011 through April 2018. Individuals were included if they reported a personal history of cancer (e.g., colorectal, endometrial, ovarian) and/or colorectal polyps and an abnormal MMR IHC test result in a tumor sample type indicated for Lynch syndrome IHC testing (i.e., CRC or endometrial cancer). To eliminate pre-existing mutation bias and ensure the mutation status of all MMR genes were obtained, only patients whose genetic testing included all four genes were assessed. Therefore, the following criteria were not part of our data query: individuals who were tested for a subset of the MMR genes, for ancestry-specific founder mutations, or for a known familial mutation. Testing was performed by Myriad Genetic Laboratories, Inc. (Salt Lake City, UT), a national Clinical Laboratory Improvement Amendments- and College of American Pathology-certified facility. All individuals provided consent for clinical genetic testing, and test data were de-identified and aggregated for analysis. As a retrospective study performed on de-identified samples, this analytical validation was not subject to any additional review (HHS regulation 45 CFR 46 per section § 46.101). Clinical information, including personal history of cancer and the IHC tumor test result, was obtained from the test request form completed by the healthcare provider.
Genomic DNA was extracted from each patient’s blood sample (QIAsymphony; Qiagen, Venlo, The Netherlands), and subjected to genetic testing using a six-gene cancer panel designed for individuals with suspected Lynch syndrome or MUTYH-associated polyposis. The panel included MLH1, MSH2, MSH6, PMS2, EPCAM, and MUTYH. Testing included sequencing and large rearrangement analysis of all genes except EPCAM (large rearrangement analysis only).
For sequencing analysis, exonic regions and adjacent -20/+10 intron regions of each gene were amplified by Polymerase Chain Reaction (PCR) and sequenced in forward and reverse directions. For PMS2 exons 11-15 that have high homology to pseudogenes, a long-range PCR was first performed, and the regions of interest were amplified by nested PCR followed by sequencing.
For large rearrangement analysis of MLH2, MSH2, MSH6, EPCAM and MUTYH, a clinically validated high-density oligonucleotide microarray was used as the primary methodology (26) and multiplex ligation-dependent probe amplification (MLPA) (MRC-Holland, Amsterdam, The Netherlands) was used as confirmatory approach. For large rearrangements in PMS2, MLPA was used as the primary methodology. For any copy number changes revealed by MLPA in the pseudogene region of PMS2, long-range PCR was also performed to determine whether the large rearrangement was in PMS2 or the pseudogene.
Variant classification was consistent with guidelines from the American College of Medical Genetics and Genomics, as previously described (27, 28). Variants with a laboratory classification of pathogenic or likely pathogenic were considered PVs. Variants with a laboratory classification of benign or likely benign were considered benign (i.e., clinically insignificant). Variants for which clinical significance could not be determined were classified as VUS.
Genetic test results were considered “expected” if a germline PV was detected in an MMR gene consistent with the gene-specific testing strategy recommended by NCCN guidelines for the IHC test result (Supplemental Table 1) (3). For example, detection of a germline MLH1 or PMS2 mutation was considered “expected” in an individual who showed loss of MLH1 and/or PMS2 on IHC; however, a germline MSH2 mutation in this individual would be considered “unexpected”. The analysis also partitioned results based on “typical” and “atypical” MMR IHC patterns. In general, typical IHC patterns were those that involved only one of the two characteristic MMR heterodimer pairs, MSH2/MSH6 or MLH1/PMS2, and listed in the NCCN guidelines. Atypical patterns involved MMR proteins from both MMR heterodimer pairs. For proportions, an exact 95% confidence interval (CI) was calculated.
A total of 703 individuals were included in this analysis. Table 1 shows clinical and demographic characteristics according to the genetic test performed. Overall, CRC and endometrial were the most common cancers for these individuals, with CRC diagnosed in 76% and endometrial cancer in 23.5% of these individuals. Many individuals were diagnosed with two or more cancer types (e.g. CRC and endometrial cancer, endometrial cancer and ovarian cancer etc.) or one cancer type plus colorectal polyps. The majority of individuals were female (61.5%, N=432) and the median age at genetic testing was 59.2 years.
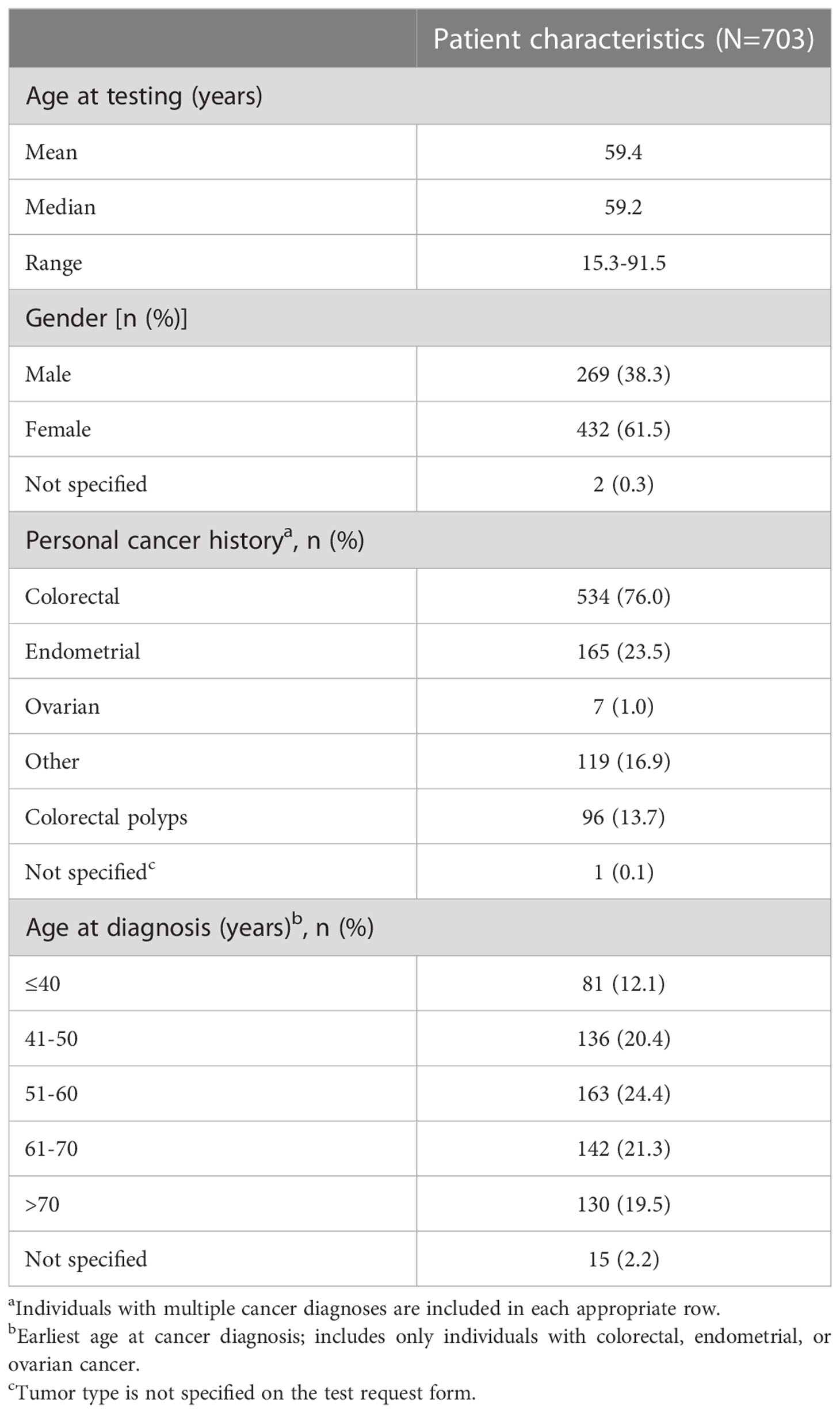
Table 1 Characteristics of the testing population.
Fifteen distinct abnormal IHC patterns were reported. This included six IHC patterns categorized as typical, involving proteins from only one MMR heterodimer pair, and nine categorized as atypical, involving proteins from both pairs (Table 2). In total, 84.8% (N=596/703) of reported IHC patterns were typical, with the most common being a lack of MLH1/PMS2 expression. Among the 15.2% (N=107/703) atypical patterns, the most common involved disrupted expression of all four MMR proteins. Since atypical IHC patterns were rare, they were combined for subsequent analyses.
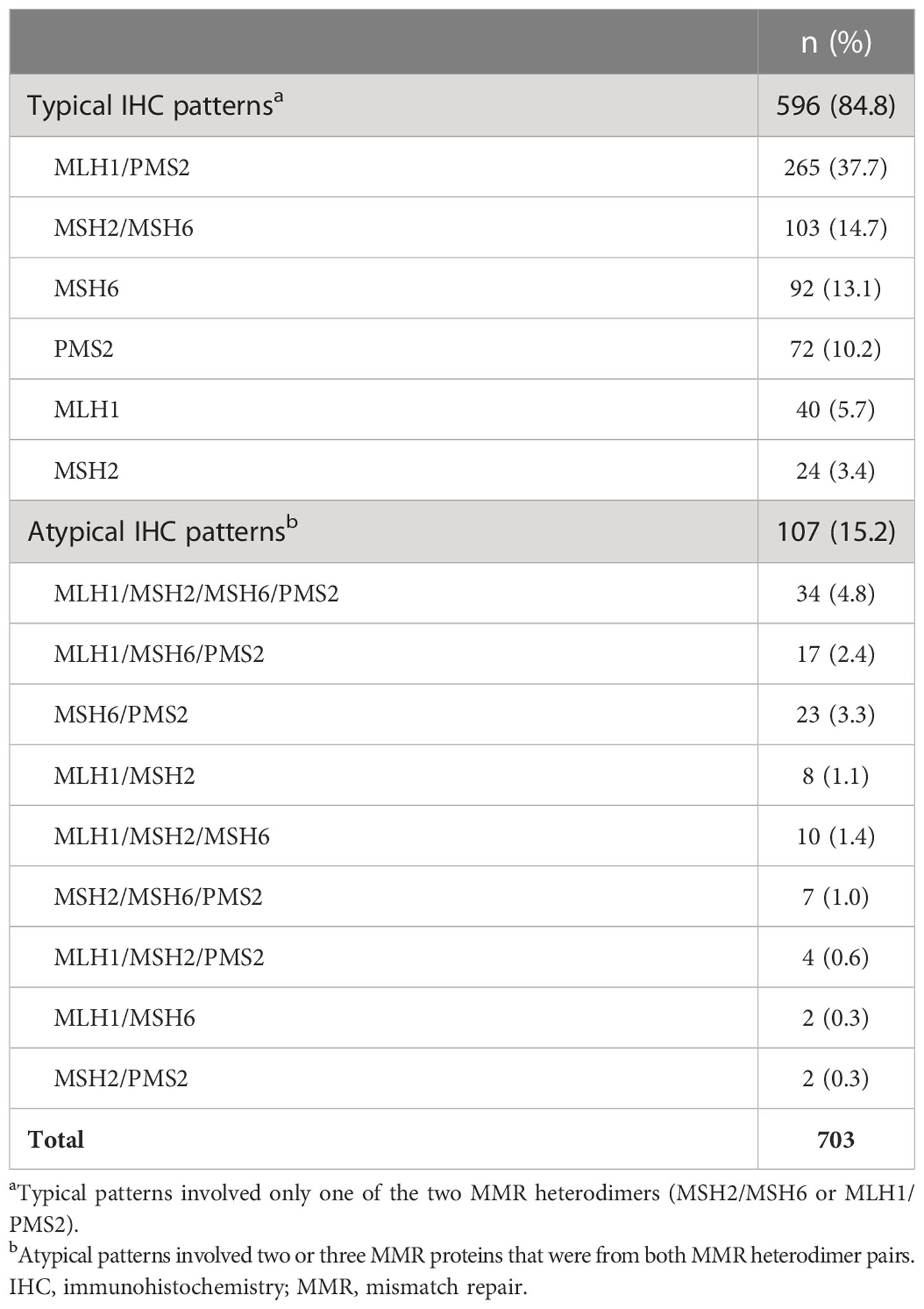
Table 2 Immunohistochemistry patterns for MMR proteins among tested individuals.
Among all the individuals included in this study, 23.2% (N=163/703; 95% CI, 20.1%-26.5%) carried germline PVs in MMR genes (Table 3). Expected PVs in MMR genes were seen in 21.3% individuals (N=150/703). Among the 163 PV carriers, 8.0% (N=13/163; 95% CI, 4.3%-13.3%) carried PVs in unexpected MMR genes. No individual carried more than one PV, and no EPCAM PVs were identified in this cohort. Monoallelic PVs in MUTYH were identified in 6 individuals (Supplemental Table 2). These were excluded from the analysis because only biallelic MUTYH PVs are considered as high risk for CRC (29, 30).
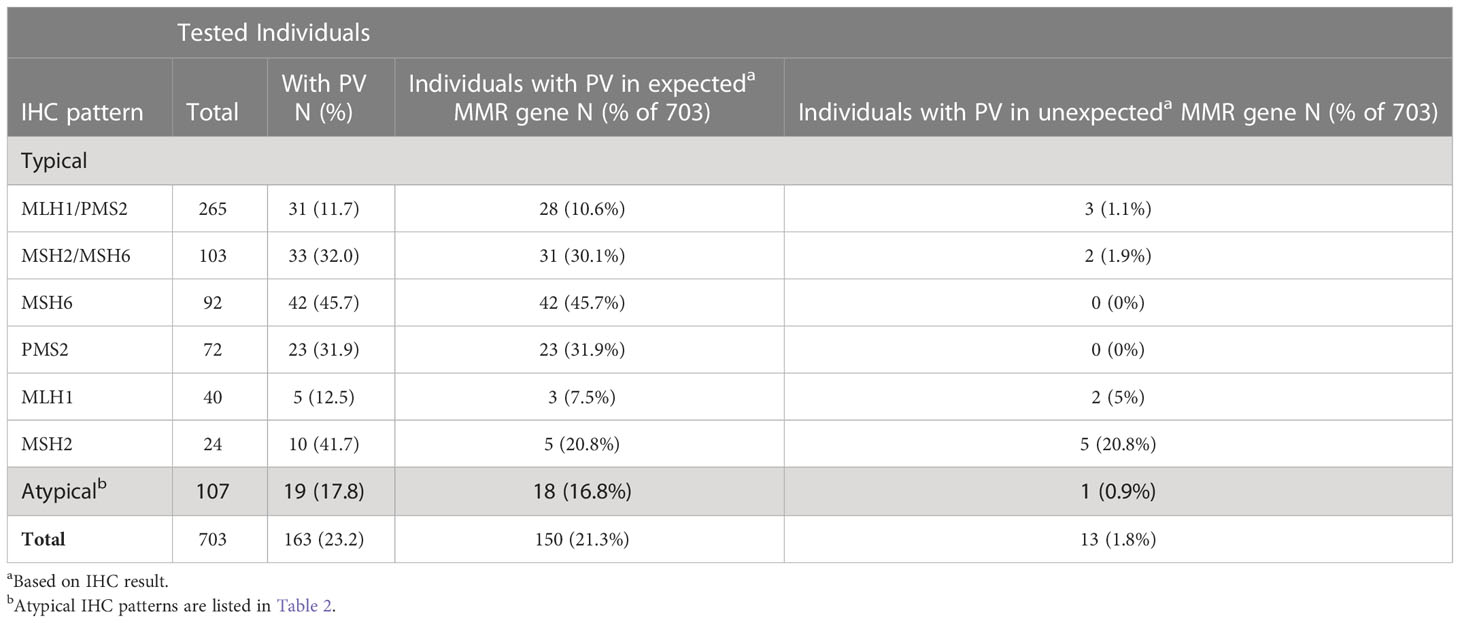
Table 3 Expected or unexpected germline pathogenic variants (PVs) identified among individuals tested with the six-gene panel, distributed by IHC pattern and type of variant.
Table 3 shows the distribution of IHC patterns among individuals found to have expected or unexpected MMR-gene PVs. It appears that PVs in expected MMR genes occurred most frequently in individuals showing isolated loss of MSH6 on IHC (45.7%; N=42/92) and least frequently among those showing isolated loss of MLH1 (7.5%; N=3/40). The most frequent unexpected MMR findings were observed in individuals who had isolated loss of MSH2 on IHC (20.8%; N=5/24). No unexpected MMR mutations were found in individuals with loss of MSH6 or PMS2 on IHC.
Table 4 lists the 13 individuals with unexpected germline PVs. The most common unexpected finding was germline PVs in MSH6 in individuals with isolated loss of MSH2 on IHC, which was seen in 4 out of 5 individuals.
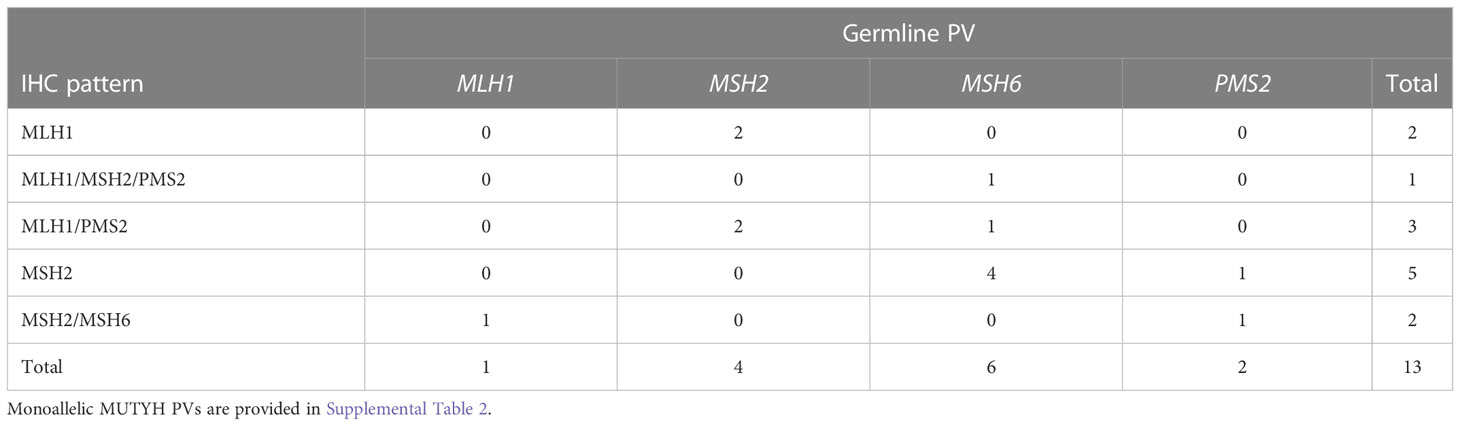
Table 4 Unexpected germline mismatch repair gene pathogenic variants found in individuals with different immunohistochemistry patterns.
199 MMR-gene VUSs were identified in nearly a quarter of the cohort (24.0%; N=169/703; 95% CI, 20.9%-27.4%) and some patients harbored more than one VUS. In 121 patients, 132 of these VUSs occurred in MMR genes that are expected to be mutated based on the IHC test result (17.2%; N=121/703; 95% CI,14.5%-20.2%).
For these 132 VUSs, there was a lack of sufficient evidence to support pathogenic or benign classifications at the time of variant identification (IHC was not considered as evidence for classification). During the timeline of this data query, May 2011 through April 2018, 44.7% (N=59/132; 95% CI, 36.0%-53.6%) of these observed VUSs were re-classified to likely benign or benign using evidence independent of IHC results, affecting 47.1% (N=57/121) individuals. By contrast, only 12.9% (N=17/132) of these VUSs were re-classified to PVs, affecting 14.0% (17/121) individuals (Table 5). The percentages of re-classified VUSs varied between different IHC patterns. For example, in individuals with isolated loss of PMS2 on IHC, 55.6% of PMS2 VUSs were downgraded to benign/likely benign variants. However, only 16.7% MLH1 VUSs were downgraded to benign/likely benign in patients with isolated loss of MLH1. On the other hand, only 3.6% of MSH6 VUSs were upgraded to PVs in patients with loss of MSH6 in IHC; however, 50% of MSH2 VUSs found in individuals with loss of MSH2 were determined to be pathogenic (Table 5).
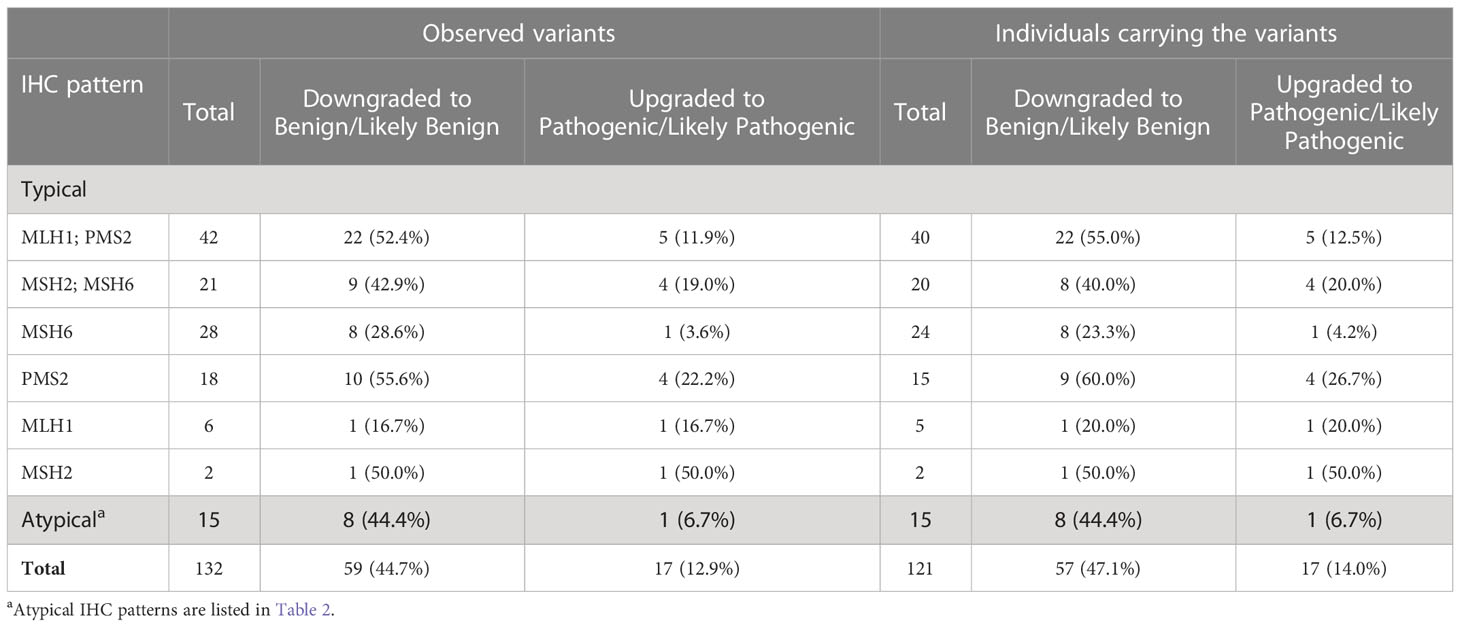
Table 5 Germline MMR gene VUSs that were observed in at least one individual with consistent IHC according to IHC pattern.
These 132 VUSs represented 103 unique variants, 35 of which were downgraded and 14 of which were upgraded (Table 6). InSiGHT guidelines indicate that IHC can be considered as evidence in variant classification if a VUS is observed in at least two patients when the gene is expected to be mutated based on IHC results (23). To assess the potential impact of IHC results in variant classification consistent with InSiGHT criteria, we evaluated the variants in genes expected to be mutated based on IHC in at least two patients. Of the 103 unique VUS, 17 were observed in at least 2 patients. Among these 17 variants that were classified as VUSs at the time of identification, 70.6% (N=12/17; 95% CI, 44.0%-89.7%) were downgraded to benign/likely benign and 17.6% (N=3/17; 95% CI, 3.8%-43.4%) were upgraded to pathogenic/likely pathogenic. The downgrade affected 77.8% (N=35/45; 95% CI, 62.9%-88.8%) of patients, and the upgrade affected 13.3% (N=6/45; 95% CI, 5.1%-26.8%) of patients (Table 6).
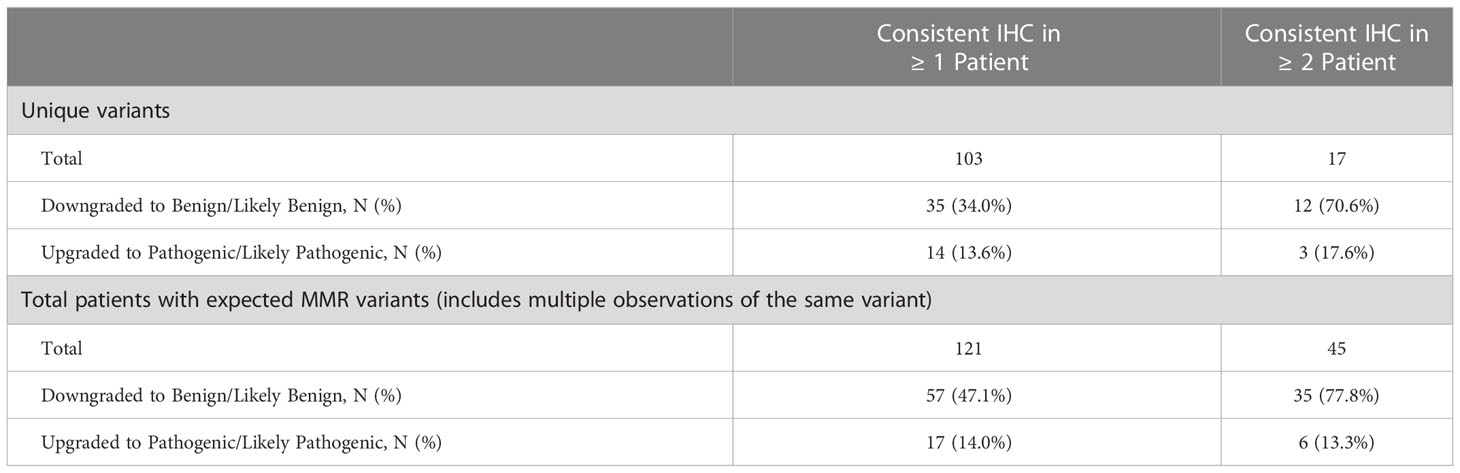
Table 6 Germline MMR gene VUS that were reclassified and patients affected by reclassification.
The evidence used for downgrading a VUS to benign/likely benign included an in-house cancer history weighting algorithm (Pheno) (31), in-trans observation with a PV in patients with no features of constitutional mismatch repair deficiency (CMMRD) syndrome (phase), functional RNA studies (splicing), updated population frequency estimate (population), and in-house algorithm for using multiple co-occurrence for evidence of pathogenicity called MCO (28, 31). Table 7 lists the basis for downgrade for these 35 variants. The most frequently used evidence was Pheno, accounting for downgrade of 17 variants. Downgrading based on phase (i.e., in-trans findings) in patients without clinical features of CMMRD was used for 13 variants. Seven variants were downgraded using population frequency.
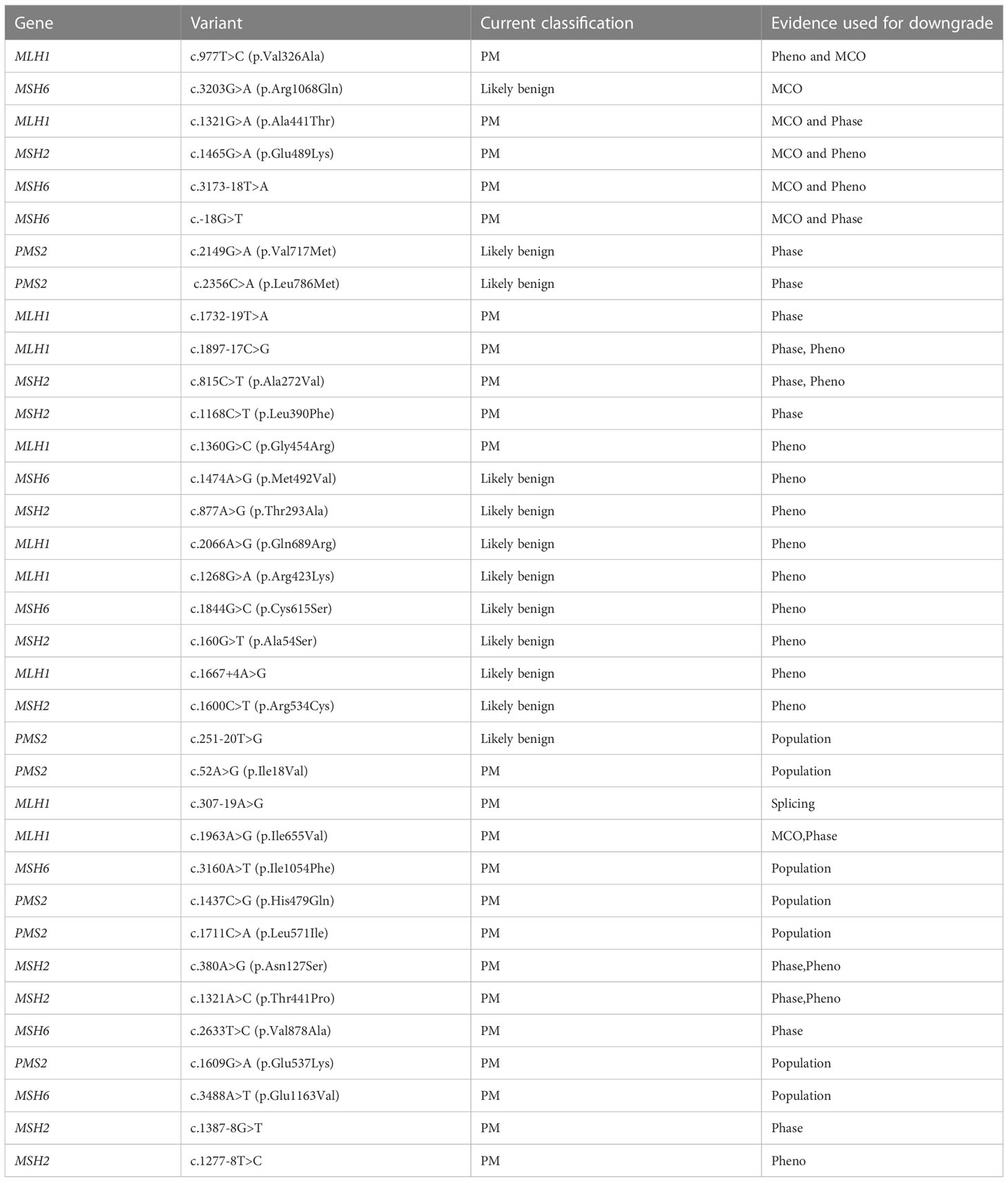
Table 7 35 variants downgraded from VUS to Benign/Likely benign.
In this study, we analyzed germline findings in 703 individuals with Lynch syndrome-associated cancer types and abnormal IHC findings to evaluate the concordance of IHC with the germline findings. To our knowledge, this is the first study to show the prevalence and spectrum of germline MMR gene mutations detected in a heterogeneous population with abnormal IHC that are referred to a commercial molecular diagnostic laboratory.
Within the entire cohort, only 21.3% of the individuals carried PVs in MMR genes predicted by IHC. This is lower than the previously reported germline PV positive rates in CRC patients with combined IHC and somatic BRAF testing (13). Somatic BRAF mutation p.V600E is present in 69% of methylation cases (32), which can contribute to the absence of MLH1/PMS2 on IHC. This study is solely based on the IHC results provided by the health care provider on the test requisition forms without any information on the somatic BRAF mutation status, therefore many of the cases with MLH1/PMS2 missing on IHC may be resulting from a somatic BRAF mutation. This might contribute to the lower PV positive rate in our cohort as we did not have information on somatic BRAF mutation status.
Overall, 8% of PV carriers identified by our panel testing carry a PV in an MMR gene not predicted by IHC results. This is particularly prevalent in individuals with isolated loss of MSH2 by IHC, where a single gene testing strategy would lead to MSH2 sequencing. Of the 10 individuals with loss of MSH2 by IHC and PV positive in this cohort, 5 harbored PVs in genes other than MSH2. These findings suggests that IHC-guided single gene testing can extend the patient’s diagnostic journey, potentially miss a Lynch syndrome diagnosis, and delay appropriate medical management. Therefore, MMR gene panel tests should be offered to all patients with abnormal IHC to prevent missing a Lynch syndrome diagnosis.
Our data showed that over 17% of patients with abnormal IHC had a VUS initially identified in the MMR gene predicted to be mutated by IHC. More importantly, nearly 1/3 of these variants were observed in more than one individual with concordant IHC results, which would be considered supporting evidence for a class 5 (pathogenic) or a class 4 (likely pathogenic) classification according to InSiGHT guidelines (23). However, in nearly half of these patients, the VUSs identified were downgraded to benign variants. If IHC results had been used as evidence for pathogenicity, these variants might have been classified in error as likely pathogenic, potentially leading to unnecessary overtreatment in the form of intensified screening and risk-reducing surgeries. These findings warrant great caution for the use of IHC results as evidence of pathogenicity for variants in MMR genes.
We observed 15 different IHC patterns in our patient cohort, 9 of which we considered “atypical” because proteins from both MLH1/PMS2 and MSH2/MSH6 heterodimers were affected. It has been reported that the IHC-null phenotype, in which all four MMR proteins are absent, can be caused by a combination of MLH1 promoter methylation and double somatic mutation in MSH2(33). Double somatic mutations have been demonstrated as an important mechanism affecting MMR protein expression (13, 34, 35). We suspect that many of these atypical IHC patterns in our cohort are caused by this mechanism, affecting both heterodimers; or in certain cases by a combination of a germline mutation affecting one heterodimer and double somatic mutation affecting the other heterodimer. Some of these IHC patterns, such as isolated loss of MLH1, may represent certain artifacts due to antibody reactivity (36). Nevertheless, the findings of PVs in these cases underscores the necessity of testing all MMR genes for patients suspected for Lynch syndrome, irrespective of the IHC results.
One limitation of the analysis was the assumption that the IHC results reported on the test request form were accurate and that results were based on a staining method that included all four MMR proteins. In practice, to reduce costs, laboratories often begin with two-protein staining for MSH6 and PMS2, with reflex to MSH2 and/or MLH1 if a defect is detected. The rationale for two-protein staining stems from the fact that the stabilities of MSH6 and PMS2 depend largely on their dimerization with MSH2 and MLH1, respectively. However, it has been shown that the two-staining method can miss a small number of Lynch syndrome patients who have solitary loss of MSH2 (37). Based on this observation, patients with intact PMS2 or MSH6, but isolated loss of MLH1 or MSH2 may have been inadvertently excluded from this patient cohort. However, these findings are considered rare, and since they are not included in the concordance calculation, we do not anticipate these patients to greatly impact our conclusion. Another limitation of our study is the lack of information of the MLH1 promoter methylation status. Promoter methylation assays often are conducted for individuals showing loss of MLH1 and/or PMS2 patterns, and only upon testing negative for promoter methylation would these individuals be referred for germline genetic testing (17, 38, 39). However, MLH1 promoter methylation status was unknown for individuals in this study since methylation assay information was not captured on the laboratory’s test request form. Therefore, the low PV-positive rates correlating with loss of MLH1 or concurrent loss of MLH1/PMS2 on IHC might reflect MLH1 promoter methylation in some individuals.
In conclusion, the overall germline PV positive rate of abnormal IHC in a population of patients with Lynch-associated cancer types who were referred to a clinical molecular diagnostic lab is approximately 20%, and nearly 2% of these individuals carried a germline PV in an MMR gene that was not consistent with the IHC result. In addition, VUS findings in genes that appear consistent with IHC findings were often downgraded to benign based on independent evidence. These findings raise two clinical issues. First, in currently recommended testing procedures there exists true risk for missing germline PVs in Lynch syndrome, as well as other conditions that may predispose patients to hereditary cancers and have characteristics overlapping the hallmarks of Lynch syndrome. The outcome can be misdiagnosis and undertreatment of patients. Second, IHC results are not reliable as supportive evidence for variant classification. Our findings support revisiting guideline recommendations for diagnostic testing of individuals diagnosed with CRC or other Lynch syndrome-related cancers with consideration given to first-line use of comprehensive germline panel testing that combines analytical accuracy with robust variant classification. This recommendation is aligned with the recent update of the NCCN guideline to consider germline multigene panel testing for all individuals with CRC (3).
The data analyzed in this study is subject to the following licenses/restrictions: Data are not publicly available due to patient privacy concerns, but can be provided upon reasonable request from the corresponding author. Requests to access these datasets should be directed to Shujuan Pan, c3BhbkBteXJpYWQuY29t.
Ethical review and approval was not required for the study of human participants in accordance with the local legislation and institutional requirements. Written informed consent from the patients/participants OR patients/participants legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
SP, JW, EM, DM-D, and BC contributed to conceptualization. HC contributed to data curation. HC contributed to formal analysis. SP contributed to methodology and project administration. HG, ML, KB, BR and DMD contributed to supervision. KB contributed to validation. SP wrote the original draft, and JW, KB and DM-D contributed additional writing, review, and editing. All authors contributed to the article and approved the submitted version.
This work was supported by Myriad Genetic Laboratories, Inc.
We would like to thank Sarah Ratzel, PhD, Elizabeth Cogan, PhD, Krystal Brown, PhD and Jennifer Logan, PhD for editing and reviewing the manuscript.
All authors were employees of Myriad Genetic Laboratories, Inc., at the time of this work and received salary and stock options as compensation.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1069467/full#supplementary-material
CRC, colorectal cancer; IHC, immunohistochemistry; LR, large rearrangement; MMR, mismatch repair; PV, pathogenic variant; VUS, variant of uncertain significance.
1. Kastrinos F, Samadder NJ, Burt RW. Use of family history and genetic testing to determine risk of colorectal cancer. Gastroenterology (2020) 158(2):389–403. doi: 10.1053/j.gastro.2019.11.029
2. Nguyen LH, Goel A, Chung DC. Pathways of colorectal carcinogenesis. Gastroenterology (2020) 158(2):291–302. doi: 10.1053/j.gastro.2019.08.059
3. Gupta S, Weiss JM, Axell L, et al. NCCN clinical practice guidelines in oncology: Genetic/Familial high-RIsk assessment: Colorectal (Version 1.2022) (2022). National Comprehensive Cancer Network. Available at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf (Accessed August 19, 2022).
4. Engel C, Rahner N, Schulmann K, Holinski-Feder E, Goecke TO, Schackert HK, et al. Efficacy of annual colonoscopic surveillance in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol hepatol: Off Clin Pract J Am Gastroenterol Assoc (2010) 8(2):174–82. doi: 10.1016/j.cgh.2009.10.003
5. Järvinen HJ, Mecklin JP, Sistonen P. Screening reduces colorectal cancer rate in families with hereditary nonpolyposis colorectal cancer. Gastroenterology (1995) 108(5):1405–11. doi: 10.1016/0016-5085(95)90688-6
6. Järvinen HJ, Renkonen-Sinisalo L, Aktán-Collán K, Peltomäki P, Aaltonen LA, Mecklin JP. Ten years after mutation testing for lynch syndrome: Cancer incidence and outcome in mutation-positive and mutation-negative family members. J Clin Oncol (2009) 27(28):4793–7. doi: 10.1200/JCO.2009.23.7784
7. Sepulveda AR, Hamilton SR, Allegra CJ, Grody W, Cushman-Vokoun AM, Funkhouser WK, et al. Molecular biomarkers for the evaluation of colorectal cancer: Guideline from the American society for clinical pathology, college of American pathologists, association for molecular pathology, and the American society of clinical oncology. J Clin Oncol (2017) 35(13):1453–86. doi: 10.1200/JCO.2016.71.9807
8. Stoffel EM, Mangu PB, Gruber SB, Hamilton SR, Kalady MF, Lau MW, et al. Hereditary colorectal cancer syndromes: American society of clinical oncology clinical practice guideline endorsement of the familial risk-colorectal cancer: European society for medical oncology clinical practice guidelines. J Clin Oncol (2015) 33(2):209–17. doi: 10.1200/JCO.2014.58.1322
9. Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Screening for the lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med (2005) 352(18):1851–60. doi: 10.1056/NEJMoa043146
10. Vasen HF, Moslein G, Alonso A, Bernstein I, Bertario L, Blanco I, et al. Guidelines for the clinical management of lynch syndrome (hereditary non-polyposis cancer). J Med Genet (2007) 44(6):353–62. doi: 10.1136/jmg.2007.048991
11. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst (2004) 96(4):261–8. doi: 10.1093/jnci/djh034
12. Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: A literature review assessing utility of tumour features for MMR variant classification. J Med Genet (2012) 49(3):151–7. doi: 10.1136/jmedgenet-2011-100714
13. Hampel H, Pearlman R, Beightol M, Zhao W, Jones D, Frankel WL, et al. Assessment of tumor sequencing as a replacement for lynch syndrome screening and current molecular tests for patients with colorectal cancer. JAMA Oncol (2018) 4(6):806–13. doi: 10.1001/jamaoncol.2018.0104
14. Noll A, JP P, Zhou M, Weber TK, Ahnen D, Wu XC, et al. Barriers to lynch syndrome testing and preoperative result availability in early-onset colorectal cancer: A national physician survey study. Clin Transl Gastroenterol (2018) 9(9):185. doi: 10.1038/s41424-018-0047-y
15. Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. part i. the utility of immunohistochemistry. J Mol Diagn. (2008) 10(4):293–300. doi: 10.2353/jmoldx.2008.080031
16. Bartley AN, Luthra R, Saraiya DS, Urbauer DL, Broaddus RR. Identification of cancer patients with lynch syndrome: Clinically significant discordances and problems in tissue-based mismatch repair testing. Cancer Prev Res (Phila). (2012) 5(2):320–7. doi: 10.1158/1940-6207.CAPR-11-0288
17. Buchanan DD, Clendenning M, Rosty C, Eriksen SV, Walsh MD, Walters RJ, et al. Tumor testing to identify lynch syndrome in two Australian colorectal cancer cohorts. J Gastroenterol Hepatol (2017) 32(2):427–38. doi: 10.1111/jgh.13468
18. Roth RM, Haraldsdottir S, Hampel H, Arnold CA, Frankel WL. Discordant mismatch repair protein immunoreactivity in lynch syndrome-associated neoplasms: A recommendation for screening Synchronous/Metachronous neoplasms. Am J Clin Pathol (2016) 146(1):50–6. doi: 10.1093/ajcp/aqw067
19. Espenschied CR, LaDuca H, Li S, McFarland R, Gau CL, Hampel H. Multigene panel testing provides a new perspective on lynch syndrome. J Clin Oncol (2017) 35(22):2568–75. doi: 10.1200/JCO.2016.71.9260
20. Yurgelun MB, Allen B, Kaldate RR, Bowles KR, Judkins T, Kaushik P, et al. Identification of a variety of mutations in cancer predisposition genes in patients with suspected lynch syndrome. Gastroenterology (2015) 149(3):604–13. doi: 10.1053/j.gastro.2015.05.006
21. Lorans M, Dow E, Macrae FA, Winship IM, Buchanan DD. Update on hereditary colorectal cancer: Improving the clinical utility of multigene panel testing. Clin Colorectal Cancer. (2018) 17(2):e293–305. doi: 10.1016/j.clcc.2018.01.001
22. Tricarico R, Kasela M, Mareni C, Thompson BA, Drouet A, Staderini L, et al. Assessment of the InSiGHT interpretation criteria for the clinical classification of 24 MLH1 and MSH2 gene variants. Hum Mutat (2017) 38(1):64–77. doi: 10.1002/humu.23117
23. Tumours ISfGH. the InSiGHT variant interpretation committee: Mismatch repair gene variant classification criteria. Available at: https://www.insight-group.org/content/uploads/2018/08/2018-06_InSiGHT_VIC_v2.4.pdf (Accessed 10/21/2020, 2020).
24. Ferguson SE, Aronson M, Pollett A, Eiriksson LR, Oza AM, Gallinger S, et al. Performance characteristics of screening strategies for lynch syndrome in unselected women with newly diagnosed endometrial cancer who have undergone universal germline mutation testing. Cancer (2014) 120(24):3932–9. doi: 10.1002/cncr.28933
25. Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Feasibility of screening for lynch syndrome among patients with colorectal cancer. J Clin Oncol (2008) 26(35):5783–8. doi: 10.1200/JCO.2008.17.5950
26. Mancini-DiNardo D, Judkins T, Woolstenhulme N, Burton C, Schoenberger J, Ryder M, et al. Design and validation of an oligonucleotide microarray for the detection of genomic rearrangements associated with common hereditary cancer syndromes. J Exp Clin Cancer Res (2014) 33(1):74. doi: 10.1186/s13046-014-0074-9
27. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet med: Off J Am Coll Med Genet (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
28. Esterling L, Wijayatunge R, Brown K, Morris B, Hughes E, Pruss D, et al. Impact of a cancer gene variant reclassification program over a 20-year period. JCO Precis Oncol (2020) 4:944–54. doi: 10.1200/PO.20.00020
29. Win AK, Dowty JG, Cleary SP, Kim H, Buchanan DD, Young JP, et al. Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology (2014) 146(5):1208–1211.e1201-1205. doi: 10.1053/j.gastro.2014.01.022
30. Lubbe SJ, Di Bernardo MC, Chandler IP, Houlston RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol (2009) 27(24):3975–80. doi: 10.1200/JCO.2008.21.6853
31. Morris B, Hughes E, Rosenthal E, Gutin A, Bowles KR. Classification of genetic variants in genes associated with lynch syndrome using a clinical history weighting algorithm. BMC Genet (2016) 17(1):99. doi: 10.1186/s12863-016-0407-0
32. Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from lynch syndrome. Genet med: Off J Am Coll Med Genet (2009) 11(1):42–65. doi: 10.1097/GIM.0b013e31818fa2db
33. Wang T, Stadler ZK, Zhang L, Weiser MR, Basturk O, Hechtman JF, et al. Immunohistochemical null-phenotype for mismatch repair proteins in colonic carcinoma associated with concurrent MLH1 hypermethylation and MSH2 somatic mutations. Fam Cancer. (2018) 17(2):225–8. doi: 10.1007/s10689-017-0031-9
34. Mensenkamp AR, Vogelaar IP, van Zelst-Stams WA, Goossens M, Ouchene H, Hendriks-Cornelissen SJ, et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in lynch syndrome-like tumors. Gastroenterology (2014) 146(3):643–646.e648. doi: 10.1053/j.gastro.2013.12.002
35. Sourrouille I, Coulet F, Lefevre JH, Colas C, Eyries M, Svrcek M, et al. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam Cancer. (2013) 12(1):27–33. doi: 10.1007/s10689-012-9568-9
36. Bosch DE, Yeh MM, Salipante SJ, Jacobson A, Cohen SA, Konnick EQ, et al. Isolated MLH1 loss by immunohistochemistry because of benign germline MLH1 polymorphisms. JCO Precis Oncol (2022) 6:e2200227. doi: 10.1200/PO.22.00227
37. Pearlman R, Markow M, Knight D, Pritchard CC, Hampel H, Frankel WL, et al. Two-stain immunohistochemical screening for lynch syndrome in colorectal cancer may fail to detect mismatch repair deficiency. Mod Pathol (2018) 31(12):1891–900. doi: 10.1038/s41379-018-0058-y
38. Buchanan DD, Tan YY, Walsh MD, Clendednning M, Metcalf AM, Ferguson K, et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J Clin Oncol (2014) 32(2):90–100. doi: 10.1200/JCO.2013.51.2129
39. Goodfellow PJ, Billingsley CC, Lankes HA, Ali S, Cohn DE, Broaddus RJ, et al. Combined microsatellite instability, MLH1 methylation analysis, and immunohistochemistry for lynch syndrome screening in endometrial cancers from GOG210: An NRG oncology and gynecologic oncology group study. J Clin Oncol (2015) 33(36):4301–8. doi: 10.1200/JCO.2015.63.9518
Keywords: hereditary cancer syndrome, clinical genetic testing, cancer diagnosis, universal tumor screening, IHC – immunohistochemistry
Citation: Pan S, Cox H, Willmott J, Mundt E, Gorringe H, Landon M, Bowles KR, Coffee B, Roa BB and Mancini-DiNardo D (2023) Discordance between germline genetic findings and abnormal tumor immunohistochemistry staining of mismatch repair proteins in individuals with suspected Lynch syndrome. Front. Oncol. 13:1069467. doi: 10.3389/fonc.2023.1069467
Received: 13 October 2022; Accepted: 11 January 2023;
Published: 30 January 2023.
Edited by:
Inge Bernstein, Aalborg University Hospital, DenmarkReviewed by:
Serena Bonin, University of Trieste, ItalyCopyright © 2023 Pan, Cox, Willmott, Mundt, Gorringe, Landon, Bowles, Coffee, Roa and Mancini-DiNardo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shujuan Pan, c3BhbkBteXJpYWQuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.