
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 28 March 2023
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1063051
This article is part of the Research Topic Pharmaceutically Targeting Hypoxia in the Breast Cancer Microenvironment: Mechanistic and Translational Approaches View all 10 articles
 Juvin Ann Thomas1
Juvin Ann Thomas1 Athira Gireesh Gireesh Moly1
Athira Gireesh Gireesh Moly1 Hima Xavier1
Hima Xavier1 Priya Suboj2
Priya Suboj2 Amit Ladha3
Amit Ladha3 Gaurav Gupta4Santosh Kumar Singh5
Gaurav Gupta4Santosh Kumar Singh5 Partha Palit6*
Partha Palit6* Suboj Babykutty1*
Suboj Babykutty1*Breast cancer ranks second among the causes of cancer-related deaths in women. In spite of the recent advances achieved in the diagnosis and treatment of breast cancer, further study is required to overcome the risk of cancer resistance to treatment and thereby improve the prognosis of individuals with advanced-stage breast cancer. The existence of a hypoxic microenvironment is a well-known event in the development of mutagenesis and rapid proliferation of cancer cells. Tumor cells, purposefully cause local hypoxia in order to induce angiogenesis and growth factors that promote tumor growth and metastatic characteristics, while healthy tissue surrounding the tumor suffers damage or mutate. It has been found that these settings with low oxygen levels cause immunosuppression and a lack of immune surveillance by reducing the activation and recruitment of tumor infiltrating leukocytes (TILs). The immune system is further suppressed by hypoxic tumor endothelium through a variety of ways, which creates an immunosuppressive milieu in the tumor microenvironment. Non responsiveness of tumor endothelium to inflammatory signals or endothelial anergy exclude effector T cells from the tumor milieu. Expression of endothelial specific antigens and immunoinhibitory molecules like Programmed death ligand 1,2 (PDL–1, 2) and T cell immunoglobulin and mucin-domain containing-3 (TIM-3) by tumor endothelium adds fuel to the fire by inhibiting T lymphocytes while promoting regulatory T cells. The hypoxic microenvironment in turn recruits Myeloid Derived Suppressor Cells (MDSCs), Tumor Associated Macrophages (TAMs) and T regulatory cells (Treg). The structure and function of newly generated blood vessels within tumors, on the other hand, are aberrant, lacking the specific organization of normal tissue vasculature. Vascular normalisation may work for a variety of tumour types and show to be an advantageous complement to immunotherapy for improving tumour access. By enhancing immune response in the hypoxic tumor microenvironment, via immune-herbal therapeutic and immune-nutraceuticals based approaches that leverage immunological evasion of tumor, will be briefly reviewed in this article. Whether these tactics may be the game changer for emerging immunological switch point to attenuate the breast cancer growth and prevent metastatic cell division, is the key concern of the current study.
The most prevalent types of cancer diagnosed and the main reason for deaths due to cancer among women is breast cancer (BC) (1). The detection and treatment of BC have undergone a number of advancements in recent years. Since the early 1990s, there has been a 39% decrease in breast cancer mortality thanks to a combination of better screening, earlier detection/diagnosis, and anti-cancer medicines that have made substantial advances. The prognosis for patients with advanced-stage breast cancer can be improved, though further investigations are required to overcome the threat of cancer resistance to treatment.
Aggressive breast cancers with hypoxic cores account for 40% of cases; these tumours are highly metastatic, and resistant to the majority of treatments. Breast cancer stem cells (BCSCs) multiply in a hypoxic tumor microenvironment, which results in a variety of epigenetic changes that retain cancer stem cells in an undifferentiated state and aid in its development and recurrence (2).Unchecked tumor cell growth outgrows the surrounding vascular system, which leads to a reduction in oxygen delivery relative to demand. Chronic hypoxia refers to this restriction on oxygen diffusion, whereas acute hypoxia occurs when blood arteries abnormally close, leading to reduced perfusion (3–5). Chronic hypoxia causes DNA breakage, malfunctions in the mending systems and mutagenesis (6). On the other hand, short-term hypoxia boosts the generation of Reactive Oxygen Species (ROS), tumor survival, and spontaneous metastasis (7, 8). Cancer cells have demonstrated radio-resistance in vitro and in vivo under both chronic and acute hypoxia (9). Hypoxia-inducible factors (HIF), which is a transcription factor, serves as the catalyst for tumor growth (10, 11). HIFs are heterodimers made up of the oxygen-sensitive HIF–α and a subsequently expressed HIF–β (12). There are three isoforms; HIF1 and HIF2 are well known, while HIF 3 is also present but has not been well researched in relation to cancer (13). HIF-1 and HIF-2 under normoxic circumstances undergo hydroxylation at particular proline residues. This causes the tumor suppressor protein Von-Hippel Lindau (VHL) protein to bind to HIF 1 and 2, which then makes it easier for them to be degraded by the ubiquitin-proteaosome system (11, 14, 15). A variety of genes encoding proteins are used for anaerobic energy production, vascularization, extracellular matrix (ECM) remodelling, suppression of apoptosis, and metastasis progression. They are transcribed when hypoxia occurs due to which HIF–1 and HIF–2 get dimerized with the subunit. They (HIF–1 and HIF–2) are then translocated to the nucleus and where they activate hypoxia responsive elements (HRE) (11, 16). Patients with breast cancer who had higher levels of HIF expression in their primary tumor biopsies are more likely to develop metastases to the bone, lungs, liver, brain, and local lymph and causes major portion of breast cancer related mortalities (17).
Through a series of sequential multistep processes the original tumour transforms into a secondary tumour at a distant site (18). The EMT transcription factors (EMT-TFs), mainly belonging to the SNAIL, TWIST, and ZEB families, which play significant roles in all processes of tumour metastasis, cause the epithelial to mesenchymal transition (EMT), which is critical for cancer spread (19).
EMT type-1, type-2, and type-3 subtypes have been extensively investigated in a variety of physiological and pathological processes. Type -1 EMT is the exchange of Epithelial to Mesenchymal cells and associated with events in embryonic phase such as implantation, embryogenesis and organ development, while Type -2 is the transformation of epithelial to mesenchymal cells that occurs during wound healing and fibrosis driven by inflammation. Type-3 EMT is the transition of epithelial cells to mesenchymal cells and is active in different types of cancers including breast cancer. Therefore, Type-3 EMT is also known as “oncogenic epithelial-mesenchymal transition” (20). Different subtypes of breast cancer show distinct metastatic organ tropisms governed by different molecular mechanisms. Along with distant lymph nodes, common target organs for breast cancer metastasis include the bone, lung, liver, and brain (21). Though all breast cancer subtypes show bone metastasis, Luminal A and B have bone as their major metastatic site. Luminal B subtype is more probable than luminal A subtype to have bone as the first site of relapse when compared to other subtypes. Incidence of bone metastasis is much higher in luminal subtype tumors than in HER-2 positive and basal like subtypes. HER -2 positive subtype is more often positively tropic to liver and luminal B and basal-like subtypes present higher levels of lung -specific metastasis (22). Triple negative breast cancer (TNBC) is often associated with visceral metastases including lung, liver and brain (23). Additionally, it has been reported that hypoxia triggers EMT in different type of cancers including breast cancer, prostate cancer and oral cancer (24). Studies by Peng, Jianheng, et al. (2018). clearly show that TGF-1 and Suppressor of Mothers Against Decapentaplegic (SMAD3) expression levels were both dramatically raised by HIF-1 in breast cancer cells, however SMAD3 overexpression had no effect on either of these proteins’ expression levels (25). Moreover, HIF-1α upregulated the expression of EMT transcription factors, SWIFT and SNAIL. In case of SWIFT in breast cancer cell lines, HIF-1α could directly bind to proximal promoter of SWIFT and enhance transcription (26). Hypoxia triggers a significant up-regulation of angiogenic growth factors and their receptors, which causes endothelial cell migration with enhanced vascular permeability and promotes tumor angiogenesis. Due to the leaky vessels and haphazard arrangement, tumor and stromal cells have limited access to nutrients and oxygen during transformation and proliferation (27). However, tumors make up for this by producing metabolic intermediates that act as precursors for biosynthetic pathways, which allows cancer cells to adapt to these circumstances in the tumor microenvironment (TME) and continue to grow and multiply. Glucose metabolism is switched from the tricarboxylic acid pathway to the oxygen independent glycolysis through the activation of glycolytic pathway regulators such as glucose transport proteins (GLUTs), hexokinase 1 and 2 (HK1, 2), and pyruvate dehydrogenase kinase 1 (PDK1) (28–30). The role of HIF–1 in reshaping the topography of the ECM under hypoxia by collagen deposition and promoting the increased expression of remodelling enzymes such prolyl-4-hydroxylases and lysyl oxidases, which leads to ECM stiffness and metastasis, has been determined by studies. An increasing body of research indicates that hypoxia promotes the chemo-invasive and metastatic potential of breast cancer by activating metalloprotease 2 and 9 (MMP-2 and MMP-9), which also degrade ECM (31–33). According to multiple studies, hypoxic stress is expected to stimulate VEGF (Vascular Endothelial Growth Factor), a crucial regulator of angiogenesis (34). Immunosurveillance in breast cancer, as in many other tumor forms, is functionally represented by the presence of tumor infiltrating lymphocytes (TILs) into the neoplastic cellular mass (35). According to Ono et al., 2012, when compared to the HER–2-/HR+ subtype, TILs were significantly greater in TNBC (triple negative breast cancer) and HER–2+/HR- breast cancer subtypes. Furthermore, compared to TNBC with low TILs levels, the pathological complete response (pCR) rate was considerably higher in TNBC with high TILs scores (36). It has been found that low oxygen levels in the TME cause TILs to become less activated, which results in immunological suppression and less immune detection. Since hypoxia-associated transcription factors have long been suggested as a viable target for immunostimulatory therapy and immunological detection, numerous researchers have found therapeutic strategies for inhibiting these factors (37). In the TME, it has been demonstrated that overexpression of HIF-1 specifically affects many aspects of the immune cell’s capacity to fight tumors (38). The foregoing discussion has briefly outlined the role of hypoxia in the aggressiveness of breast cancer, specifically in the transformation of normal endothelium into tumorigenic endothelium, as well as the various therapeutic approaches by which immune modulators can specifically target breast cancer and how hypoxic tumor endothelial cells can be targeted within the TME.
Growing body of data indicates that, a hypoxic microenvironment may protect tumors from immunological treatments as well as naturally occurring anti-tumor immune responses by limiting anti-tumor immune effector cells and encouraging immune escape. It is clear that hypoxia affects immune cells either directly or indirectly, supporting the TME in a direction that is immunosuppressive (39–41). Understanding the disease biology of Breast Cancer (BC) requires an understanding of the immune surveillance in the tumor microenvironment since it has the potential to either facilitate the eradication of disease or encourage tumor growth. This dual role is sustained by the dynamic interactions between diverse immune effector cells, tumour cells, stromal cells, and soluble substances in the tumour microenvironment (42). Selection favours tumor variants with the genetic trait of escaping immunological detection, while variants lacking it are wiped off by immune surveillance. In addition to genetic variation, deficiencies in antigen presentation methods, T-cell receptor (TCR) signalling, interferon (IFN) signalling pathways, and expression of the MHC class I protein has been lost or mutated, along with tumour antigens (43, 44), which will impair the immune surveillance mechanism in the tumor microenvironment. Inhibition of tumor antigen-specific T cells by intratumoral myeloid-derived suppressor cells (MDSCs), T regulatory cells (Tregs), and the switch from an anti-tumorigenic T-helper type 1 (TH1) to a pro-tumorigenic T-helper type 2 (TH2) immune response are additional factors that significantly support tumor growth. Furthermore, a variety of soluble factors like TGF-β, IL–10 etc are released by tumor/stromal cells inhibit T-cell activation and dendritic function while promoting stromal remodelling and angiogenesis (45, 46). The hypoxic core of the tumors attracts more pro-tumorigenic leukocytes, including MDSCs, tumor-associated macrophages (TAMs), and regulatory T cells. This double down the body’s natural immune defensive measure to evacuate the aggressive tumor (47) (Figure 1). An in-depth study using Balb/c mice found that intra-tumoral hypoxia increases HIF-1 activation, which then sequentially activates the PDL-1, CD73, and CD47 genes to reduce the recruitment and activity of CD8+ T cells, NK cells, and macrophages, ultimately causing activity to evade both innate and adaptive immunity (48). Chemokines and their related receptors are another important hypoxic hotspot (49), which in turn affect tumor endothelial cells and increase the over expression of VEGF, CXCL12, and its receptor CXCR4, making all of them function on endothelial cells in an autocrine way. The expression of the pro-inflammatory chemokines CCL5 and CCL2 increased in HIF–1-deficient tumors, which in turn increased the infiltration of cytotoxic lymphocytes into the tumor (50). Additionally, the CXCR4-CXCR12 axis promotes metastasis to distant organs (51). Thus, hypoxic stress in TME along with activation of HIF transcription factors, is a principal cause for breast cancer angiogenesis, metastasis, immune suppression and overall poor survival rate. In the subsequent sections of this review, a detailed mechanism of hypoxia induced vascular abnormalities that activate endothelial cells (ECs) associated with tumor cells and immune suppression is furnished.
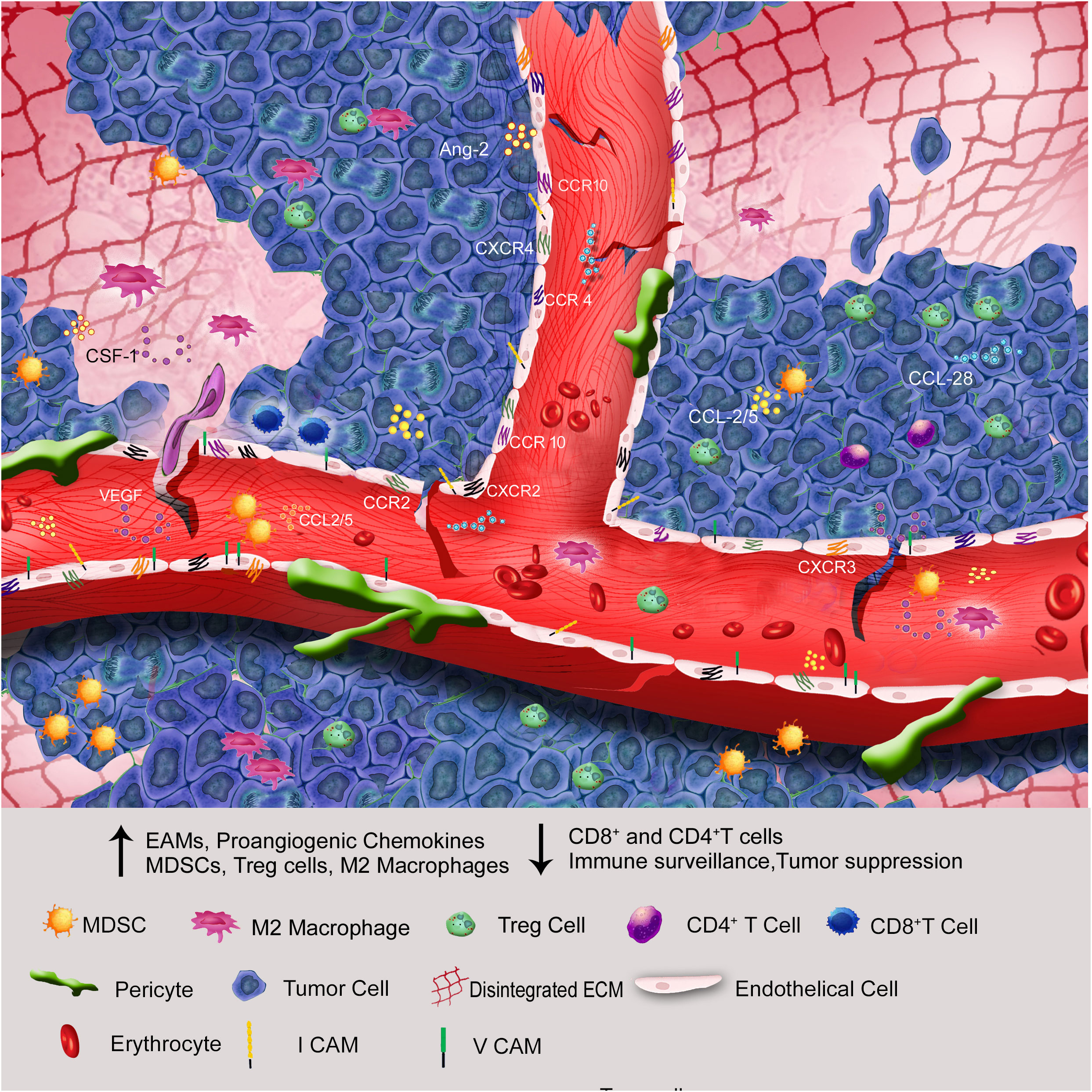
Figure 1 Through endothelial anergy, elevation of proangiogenic chemokines, cell adhesion molecules, and ECM disintegration, the hypoxic core of solid tumors attracts more immunosuppressive cells than immunostimulatory cells to the tumor microenvironment.
Tumor vasculature is different from healthy blood vessels in a number of aspects, including irregular structural dynamics, high permeability, and convoluted arteries, whereas healthy blood vessels are well-organized and provide for the best perfusion of nutrients and oxygen (52). Specialized mural cells called pericytes are seen in the normal vasculature, whereendothelium covers them uniformly (53). Patchy hypoperfusion and blood vessel leakage are caused by the loosening, unstable, and tumor-related pericyte phenotype (54).
Endothelial cells interact with tumor cells in numerous ways to promote angiogenesis (55) (Figure 2). They serve as the connecting link between cancer cells and immune cells (56). Proangiogenic substances like Vascular Endothelial Growth Factor (VEGF), basic fibroblast growth factor (bFGF), placental growth factor (PGF), and angiopoietin are released by tumor cells to initiate angiogenesis. According to studies, the hypoxic tumor microenvironment might boost VEGF synthesis, which in turn promotes the growth of new blood vessels (57). When the microenvironment experiences a lack of oxygen, endothelial cells and pericytes are stimulated to create VEGF, which functions in an autocrine and paracrine manner to increase the recruitment and activation of endothelial cells in the tumor site (58). Angiopoietin-2 (Ang–2) plays a critical function in destabilising vasculature for normal or pathological angiogenesis and is also up-regulated by hypoxia. It is only expressed at sites of vascular remodelling. Numerous studies have documented the essential function of Ang-2, a ligand for the endothelial cell-specific tyrosine kinase Tie2, in the vascular permeability and blood vessel instability that leads to tumor growth (59, 60). Thus, newly formed blood vessels have an uneven thickness of the basement membrane, a loose interaction between pericytes and endothelial cells, an increase in interstitial pressure, and ultimate vascular leakage (47). One of the main processes behind angiogenesis is the angiopoietin/Tie (tyrosine kinase) signalling pathway, which is composed of growth factors called angiopoietins. These include Ang-1, a strong angiogenic growth factor that communicates with Tie2, and Ang-2, a vascular disruptor with a negative effect that also uses Tie2 as a conduit (61). To cause pericyte separation from the basement membrane and migration, Ang-2 and Tie2 bind in the hypoxic tumor microenvironment. Mice lacking in pericytes had higher Ang–2 levels, suggesting that pericytes may control Ang–2 levels and limit vascular permeability. This finding highlights the importance of Ang–2 in reducing vessel leakiness (62, 63). In a different study, it was reported that reduced pericyte coverage increased IL-6 expression in the hypoxic tumor microenvironment and MDSC transmigration and circulating malignant cell phenotype (64). Rgs5 overexpression in pericytes and endothelium seen in the hypoxic tumor microenvironment causes the vasculature to become unstable. Better pericyte coverage, less vascular leakage, and adequate oxygen perfusion was all observed in the Rgs5-deficient mice (65). In-depth research found that the development of a receptor complex made up of PDGF-R and VEGF-R2 during PDGF-induced angiogenesis is what causes VEGF to activate VEGF-R2 and suppress PDGF-R signalling in vascular smooth muscle cells (66), which then decouples neo-vasculature from pericyte covering. Researchers have shown that inhibiting VEGF and Ang–2 jointly results in tumor necrosis, vascular regression, intra-tumoral phagocyte antigen presentation, and a reduction in breast cancer-brain metastases (67, 68).
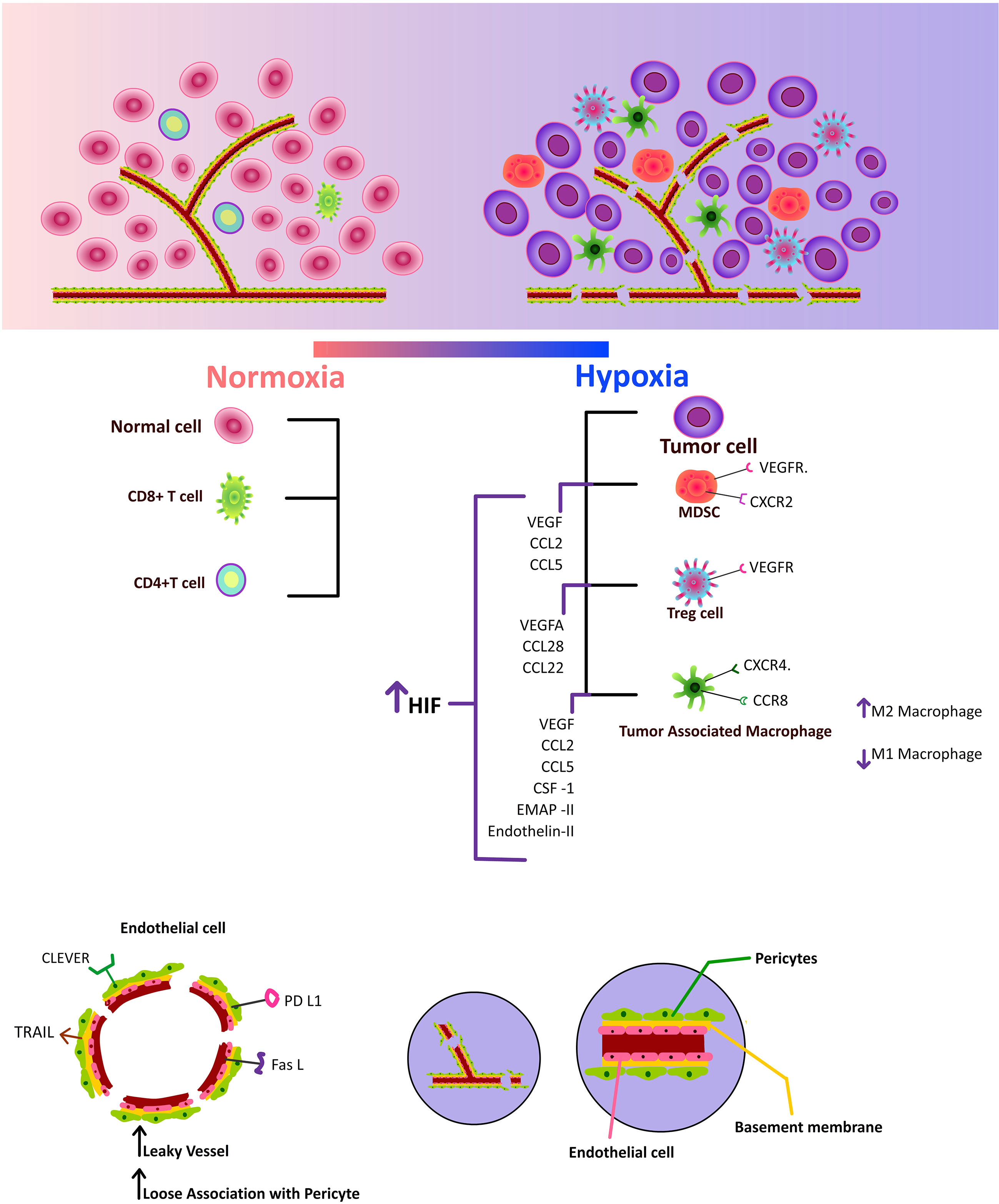
Figure 2 Increased hypoxia inducible factor, which upregulates pro-angiogenic factors and chemokines, draws immunosuppressive cells to the tumor microenvironment. Additionally, increased CLEVER, TRAIL, PD-L1, and FAS-L expression on tumor endothelium impairs effector T cells.
Endothelial Cells (ECs) ECs have specific functions based on their locations and exhibit distinct heterogeneity across vascular beds. They have a critical role in the regulation of immune responses, inflammation, angiogenesis and actively control the degree of vascular relaxation and constriction (69). ECs that line tumor blood vessels are initially derived from the surrounding tissue, and in due course of the tumor progression, they reprogram to a tumoral phenotype (70). Normal physiologic conditions do not often need the activation of ECs, but in tumors, hypoxia and other inflammatory signals cause ECs in the tumor microenvironment to become active, causing aberrant angiogenesis and impeding normal immune surveillance (71). Numerous inflammatory reactions in the tumor microenvironment, such as inflammatory cytokines, chemokines, reactive oxygen species, etc., directly or indirectly stimulate the tumor endothelium. Tumor endothelium has a very different genetic profile than healthy endothelium (Table 1), with the main variations influencing a number of cell adhesion molecules (such as ICAM1, VCAM1, E-selectin), antigen presentation, and chemokines (such as CCL2, CCL18, CXCL10, and CXCL11) and cytokines involved (such as TNFα, IFNγ, and IL-1) in immune cell recruitment. All of these elements have a deleterious impact on the immune surveillance of tumor cells in the tumor microenvironment. This unique character of TEC that enables them to avoid immune cell extravasation and, unresponsiveness of TECs to pro-inflammatory stimulation is Endothelial cell anergy, also called vascular immune checkpoint (72). ECs lining tumour blood arteries have a very different metabolic profile from ECs in normal tissue. A stronger dependence of tumour ECs on glucose metabolism is supported by the transcriptional elevation of the glycolytic pathway gene, PFKFB3, in comparison to other normal EC. According to a report, tumor ECs and tumor associated macrophages (TAMs) engage in fierce competition for glucose, and the tumor ECs’ intake of the metabolite boosts the angiogenic response in the tumor microenvironment (73). TECs produce energy through aerobic glycolysis and fatty acid oxidation, rather than oxidative phosphorylation. This metabolic reprogramming enables the tumor ECsto check the production of reactive oxygen species (ROS), and also allows the production of ATP more rapidly than through oxidative metabolism (74). Alam et al. (2014) has identified suprabasin as a new marker for TECs. Suprabasin, an upstream component of the AKT pathway, was substantially expressed in TECs compared to normal ECs and linked favourably with TECs’ capacity for migration and tube formation (75). On the other hand, microarray and immunohistochemical studies revealed that biglycan is a specific marker of TEC and an autocrine angiogenic factor of TECs (76). Because of the aggressive behaviour of tumor endothelial cells and their particular molecular, cytogenetic, and metabolic characteristics, the tumor microenvironment can therefore selectively draw in immune suppressive cells. The role of TECs in increasing immunological suppression is discussed in the following sections of this review.

Table 1 Factors converting normal endothelium to tumor endothelium.
The success of immunotherapy depends on adequate immune cell infiltration, and low immune cell infiltration reduces the effectiveness of immunotherapy. Despite the fact that angiogenesis triggered by tumors is crucial for the growth of solid tumors, mounting evidences indicate that it also aids in immune evasion by fostering a highly immunosuppressive TME by increasing the proportion of T reg cells and MDSCs. Furthermore, by preventing dendritic cell (DC) maturation and T cell growth, VEGF impacts immunological responses in TME (77). Leukocytes are extravasated to tissues from blood arteries during typical inflammation. These leukocytes in circulation were halted and firmly adhered to the endothelium cells by a multi-step procedure. This mechanism calls for the endothelial adhesion molecules (EAM), E-selectin (rolling), ICAM1 and VCAM1 (firm rest), VE-cadherin and CD31 (trans-endothelial migration). In addition to EAMs, surface antigens (such as HLA molecules) must be upregulated, pro-thrombotic endothelial cell changes (such as the loss of the surface anticoagulant molecules thrombomodulin and heparan sulphate), cytokines (such as IL–6, IL–8, and MCP–1) production, and changes in vascular tone (such as the loss of vascular integrity and expression of vasodilators) are associated with inflammation (78). When inflammatory cytokines like TNF, IFN, and IL–1 are released, normal ECs are activated. This causes an up-regulation of adhesion molecules, which in turn triggers the extravasation of leukocytes. However, the continual secretion of pro-angiogenic molecules VEGF and bFGF in hypoxic microenvironments adversely affects this process. Even when TNF is present, they prevent ICAM1, VCAM1, and E-selectin from being overstimulated (79–81). The hypoxia inducing factors favour the heterogeneity as well as inflammation of tumor endothelial cell (82). In vitro and in vivo studies by Tellier et al. (2015) showed that ECs exposed to hypoxia expressed tumor-promoting pro-inflammatory cytokines and chemokine such as IL-6, IL-8, and CXCL1 (83). In addition, hypoxic tumor niche affects protein glycosylation and favours recruitment of immune suppressive cells to TME, thereby impacting tumor progression (84). Research shows that the tumor endothelium downregulates the expression of endothelial adhesion molecules (EAM) such as ICAM1/2, VCAM1, E-selectin, and CD34. This lessens immune cell infiltration or leucocyte extravasation in the TME, enabling the TME to adopt an immune evasion strategy and accelerate tumor growth (72, 79, 85). Hypoxic tumor microenvironment alters the tumor endothelium leading to its diversity and also induces its “stemness”. This induction of stemness is favourable for cancer stem cells, which can drive tumor initiation and progression (86). Moreover, breast cancer stem cells enhance its tumorigenic phenotype (87). Cancer stem cells and their role in tumor progression is a vast horizon and beyond the scope of this review. Stemness genes such stem cell antigen-1 (Sca-1), MDR-1, and aldehyde dehydrogenase (ALDH) have been found to have elevated expression in TECs. This tumour stromal stem-like cell population affects the TEC population in the TME (57). With higher VEGF expression, ovarian, oesophageal, and colorectal tumors showed worse survival rates and a higher chance of relapse. VEGF synthesis in tumors affects tumor behaviour by reducing T cell numbers in addition to promoting angiogenesis. In pre-clinical tumor models, it was found that using anti-VEGF antibodies increased the recruitment of T lymphocytes into the tumor microenvironment (88, 89). Increased nitric oxide (NO) in the TME has been found to upregulate VEGF in solid tumors and further regulate the expression of several adhesion molecules involved in the interaction between EC and leukocytes. FoxP3+ T regulatory (Treg) cells predominated and there was little CD8+ infiltration in solid tumors of both humans and mice that express specific Fas ligands in the tumor vasculature. Similarly, blockade of Fas-Fas L signalling increased intra-tumoral CD8+ T cells and subsequent reduction in tumor size (90). It is now understood that the tumor endothelium plays a crucial dual role in the development of the tumor by favouring a niche where inflammation is increased by upregulating EAMs and by negatively regulating the influx of leucocytes through endothelial anergy (Figure 3). The role of hypoxia in attracting immune suppressive cells to the TME and its relationship with cancer endothelium are discussed in the forthcoming sections of this review.
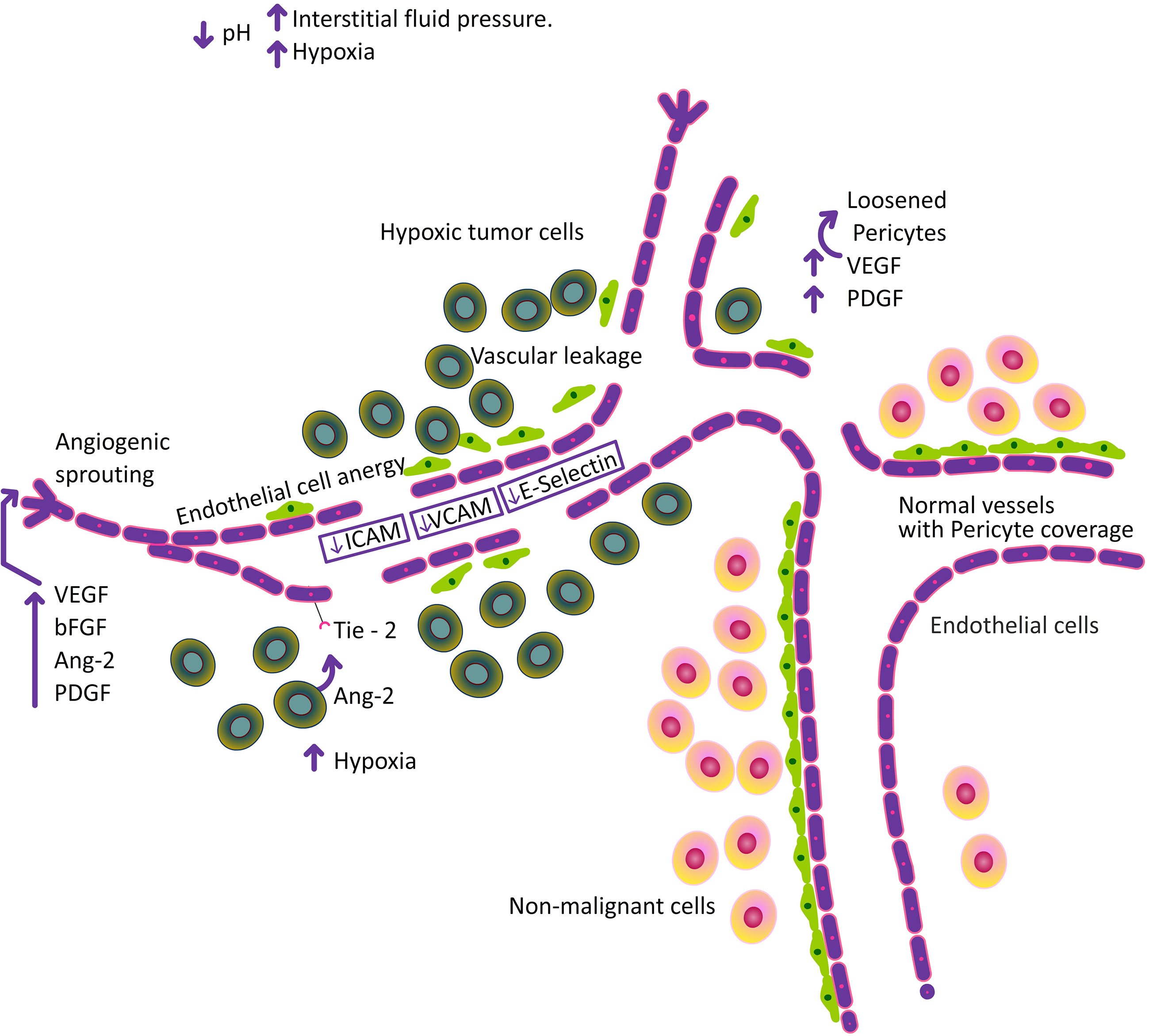
Figure 3 Increase in hypoxia, alter the pH and increase in interstitial fluid pressure followed by structural abnormalities in the tumor vasculature.
MDSCs are one of the main immunosuppressive elements in TME. Their activation and expansion coincide with metastasis and progression in many types of cancer (91). Those patients with higher levels of MDSCs in the tumor microenvironment have exhibited higher metastatic burden and poor survival (92).Various inflammatory cytokines such as IL-13, IL-4, and transforming growth factor (TGF–β)- are linked to MDSC proliferation, whereas granulocyte-macrophage colony-stimulating factor (GM-CSF), Prostaglandin 2 (PGE2), IL-6, stem cell factor, and vascular endothelial growth factor (VEGF) are linked to MDSC activation (93).The expression of VEGF receptor on MDSCs, explains the correlation between the up-regulation of VEGF in hypoxia and the accumulation of MDSCs in the TME (94). The presence of hypoxia in the tumor microenvironment boosts the production of CCL 5, which in turn stimulates the HIF 1α and VEGF signalling pathways. Hypoxia in the tumor microenvironment increases the production of CCL 5, and which in turn activates VEGF signalling mechanism. The expression of PD L-1 in MDSCs is increased by VEGF production in the tumor microenvironment, which limits the recruitment of cytotoxic lymphocytes (85). In line with this, a related study showed that numerous MDSC chemoattractants were upregulated in the VEGF overexpression group, suggesting that the immunosuppressive effects of VEGF are partially mediated by MDSC recruitment into the tumor microenvironment (95). Similarly, lymphatic endothelial cells (LECs) have shown to recruit MDSCs to the tumor microenvironment and increase tumor progression in TNBC cells via pro-angiogenic receptor CXCR2 on MDSCs. According to the study, tumor-derived vascular endothelial growth factor-C (VEGF-C) stimulated LECs to produce chemokines, which in turn helped MDSCs find their way to lymph nodes. Additionally, LEC-released chemokines increased lymphatic invasion by upregulating VE-cadherin phosphorylation and junction disruption, which in turn increased serum amyloid A1 (SAA1) expression in breast cancer cells (96). In a recent study by Roberts et al. (2022) reported that co-culturing of murine TNBC-4T1 cells with MDSC and murine LECs (iLECs) in culture inserts showed an increase in MDSC invasion in the presence of iLEC (97). Monocyte/M-MDSC recruitment to malignancies is mostly facilitated by CCL2 and CCL5 (C-C motif ligand 2/5) chemokines. There is evidence that CCL2 is crucial for recruiting PMN-MDSC as well (98). Apart from endothelial cells, the absence or presence of immature pericytes, a component of vasculature also acts as a signal for MDSC recruitment in breast cancer patients (64). Taken together, MDSC play a critical role in tumor immune suppression where its activity is tightly regulated by hypoxia via endothelial receptors and growth factors, which overall aids in tumor progression. The trafficking of MDSCs by TECs has not been elicited clearly this far. Thus, establishing this link may offer novel targets of anti-tumor therapies.
Macrophages make up a significant portion of the leukocyte infiltrate, which is found in all cancers to variable degrees (99). Macrophages, which are derived from the blood compartment, are renowned for their flexible and variable genomes (100). Overall, macrophage matrix metalloproteinase-12 dampens inflammation and neutrophil influx in arthritis (101). Depending upon their nature of activation and the TME, macrophages can either increase or inhibit the immune responses that fight cancer. According to reports, VEGF stimulates the growth of endothelial cells in breast cancer and attracts macrophages through the VEGF receptor. The synthesis of several pro-angiogenic substances by recruited macrophages, such as VEGF, tumor necrosis factor (TNF), and thymidine phosphorylase (TP), is able to accelerate angiogenesis in the TME. This is a form of mutual activation/dependence between TAM and tumor endothelial cells in the TME (102). Tumor cells and stroma, which make up the hypoxic core of the tumor microenvironment, promote the production of VEGF, CCL2, CCL5, CSF-1, EMAP-II, endothelin-2, SEMA3A, oncostatin M, and eotaxin. The overproduction of migratory molecules promotes macrophage infiltration of the tumor (103).Macrophages can be classified as pro inflammatory-immune stimulatory (M1) or alternatively activated anti-inflammatory-immunosuppressive (M2) macrophages depending on the environment in which they are recruited.It has been shown that the recruitment of TAM into the TME is a poor prognostic indicator for overall survival and treatment effectiveness (104). In addition, non-responsive tumor EC-derived IL-6 is a cytokine that encourages macrophage M2-like polarisation in the tumor microenvironment. The EC biomarker ESM1, which is linked to a poor prognosis in human gastrointestinal and hepatocellular carcinomas, is also highly expressed in tumor ECs in a number of mouse tumor models. Furthermore, studies have shown that ESM1 induces ECs to express ICAM1, which draws and polarises M2 macrophages toward the tumor microenvironment (105–107). As tumors grows, increased levels of hypoxia cause M1-polarized macrophages to secrete less pro-inflammatory mediators including IL-1, TNFα, and CCL17 and accelerate macrophage differentiation toward the M2-like phenotype. Despite the fact that hypoxia does not affect the proportion of different macrophage subsets, it does cause the M2-like macrophage subset to activate the transcription of pro-tumor genes, including growth factors like FGF2, PDGF, and VEGF (108, 109). Therefore, the dichotomy in macrophage differentiation into M1 and M2 type can have varied outcome on tumor progression via hypoxia and angiogenesis machineries (110). The polarisation of macrophages from the anti-tumor M1 phenotype to the pro-tumor M2 phenotype is clearly influenced by hypoxic stress. Focusing on TAMs that are produced by hypoxia and their trafficking across tumor endothelium may be beneficial given that it has been suggested that TAMs and cancer endothelium interact. The elucidation of this mechanism might produce brand-new indicators and therapeutic outcomes that are promising.
Hypoxic TME can regulate T cell response in two major ways depending upon the T cell subset. Effector T cells, in the form of CD4+ and CD8+ T cells, play a critical role in resolving tumor growth by releasing inflammatory cytokines or by direct lysis of tumor cells. However, these effector T cell responses undergo immune suppression in the tumor microenvironment which is typically hypoxic (111). Multiple factors such as tumor growth factor (TGF-β), IL-10, VEGF, indoleamine 2, 3-dioxygenase (IDO) and arginase contribute to their immune suppression (112). Another mechanism by which hypoxia mediated immune suppression in effector T cells is through the loss of expression of co-stimulatory molecules (CD80, CD86 and CD40) on dendritic cells that interact with T cells and promote immune tolerance in them (113). Cytotoxic CD8+ T cells are capable of infiltrating tumors making them critical for tumor clearance. However, under hypoxic conditions, these CD8+ T cells also undergo immune suppression via defective antigen presentation of tumor cells through low MHC expression, down regulation of transporter associated with antigen processing protein (TAP) and tumor antigen (114–116). This tumor hypoxia specifically promotes the immunosuppressive function of T regulatory cells including its migration and activation at the tumor site (117). Treg cells are characterized by the expression of specific cell surface molecules (such as CD25, GITR, CTLA4) and nuclear transcription factor (FOXP3) (118), mediate immune suppression by downregulating activated T cell function through increased production of immunosuppressive cytokines such as IL-10 and TGF-β or via interaction between CTLA-4 on Tregs and CD80/86 on antigen presenting cells or by sequestering IL-2 from naïve T-cells by its IL-2 receptor (CD25) on Tregs (119). Hypoxia induced HIF-1α results in increased expression of FOXP3 in Treg cells (120) and at the same time, also promotes CCL28 in the tumor microenvironment. This CCL28 binds to its cognate receptor CCR10 on Treg cells and thereby promotes the migration of Treg cells (117). HIF-2α is also involved in Treg stability as HIF-2α-deficient Tregs are functionally defective at suppressing effector T-cell function (121). Tumor infiltration by T lymphocytes has shown to increase overall survival in different types of cancer such as colorectal, ovarian, breast and melanoma. Tumor endothelial cells act as a major barrier for the extravasation of effector T lymphocytes into the tumor niche through the downregulation of ICAM1 and VCAM1. Furthermore, TECs can increase the expression of molecules such as common lymphatic and vasculature endothelial receptor 1 (CLEVER1) on their surface to recruit immunosuppressive Treg cells (80). (Figure 2) Interestingly, the expression of TRAIL and FasL on TECs selectively kill effector T cells while not hampering Treg cells. The pre-clinical studies by actively immunizing tumor endothelial expressing antigens via DNA vaccines and protein pulsed DCs successfully inhibited tumor growth and increased the infiltration of CD8+ T cells in the TME. Also, DNA vaccines targeting tumor endothelial marker 1 (TEM1), specific TEC expressing antigen could escalate intratumoral infiltration of endogenous CD3+ T cells (122). Thus, tumor induced hypoxia and tumor associated endothelial cells mediate immune suppression by directly or indirectly regulating T cell functions, which promotes tumor growth and invasiveness.
Immunosuppressive T cells known as regulatory T (T reg) cells control homeostasis and self-tolerance by limiting erroneous immune responses (123). One of the essential immune cells favouring tumor growth and regulating the immunesurveillance is the CD4+CD25+Foxp3+ Treg cell (124). FoxP3, a sign of Treg activity, has been proposed as a marker of tumor progression and metastasis in breast carcinoma. In order to determine the progression and prognosis of BC, measuring Tregs recruitment and activity has been proven to be a useful approach (125). Hypoxia has been proven to associate with the infiltration of regulatory T cells in breast tumor microenvironment by the upregulation of CXCR4 receptor on Tregs. Hypoxic stress induced expression of CXCR4 by activating HIF pathway has been reported in different stromal cells including endothelial cells in TME. By increasing the expression of FOXP3, a lineage transcriptional regulator of Tregs, HIF-1 may also indirectly stimulate CXCR4 expression.
By increasing the expression of FOXP3, a lineage transcriptional regulator of Tregs, HIF-1 may also indirectly stimulate CXCR4 expression. Thus, there are opportunities for clinically targeting Tregs by blocking CXCR4 to stratify patients for anti-HIF therapies (126). Mounting evidences suggests the prime role of hypoxia in stimulating the secretion of cytokines and chemo- attractants from cancer cells and tumor associated macrophages, including CCL28, CCL22 and IL-10, that recruit Treg cells from the circulation. Hypoxia induces the expression of CD 73 on various cell types including T regs, and actively involves in the generation of immunosuppressive metabolite adenosine which negatively affects T cell function (127, 128). Treg infiltration in tumor locations can be correlated with increased microvessel density and upregulation of angiogenesis indicators like VEGF in breast and endometrial malignancies, illustrating the relation between Tregs and tumor angiogenesis (127). According to Andrea Facciabene et al., hypoxic intraperitoneal tumors recruit Treg cells, which impair effector T cell activity and promote tumor angiogenesis via VEGF-A. Furthermore, CCL28 secreted by hypoxic tumor cells attract Treg cells in to the tumor niche. Treg cells can directly contribute to the overproduction of VEGF-A and can promote the proliferation and recruitment of endothelial cells (129). In fact, Tregs contribute to angiogenesis indirectly by inhibiting Th1 effector T cells and secreting interferon-induced chemokines like CXCL9, 10 and 11 as well as angiostatic cytokines like TNF and IFN. Another work by Andrea Facciabene et al. shown that depletion of Treg cells reduce VEGF upregulation and angiogenesis (52). Increased expression of CCR8 was noticed in tumor infiltrating Tregs compared to circulating T regs. A promising immunotherapeutic strategy for the treatment of breast cancer would involve targeting CCR8 to prevent the migration of tumor-resident Tregs (130). Targeting T reg cells in tumors using selective immunotoxin against CD 25 (Treg marker) increased CD8+ T cell-dependent antitumor immune response in experimental tumor models (131). T reg cell elimination and subsequent anti-VEGF therapy, restored IFN- production in CD8+ T cells and enhanced the antitumor response from anti-VEGF therapy in tumors (132). Although there are lot of studies depicting hypoxia mediated T reg infiltration, there are no confirmatory studies on the role of hypoxic cancer endothelium, especially breast cancer endothelium in recruiting regulatory T cells to the TME.
Recently, immune checkpoint blockade therapy has gained attention since it allows patients’ natural immune systems to combat cancer. Immune checkpoint molecules like CTLA4, PD1, PDL–1, LAG3, and TIM–3 inhibit the immune response in different tumor types at different phases of tumor development (133). A significant development in the field of cancer immunotherapy is the discovery ofproteins like programmed cell death protein 1 (PD1), programmed cell death ligand 1 (PDL–1), and cytotoxic T lymphocyte-associated antigen 4 (CTLA4). These molecules block the signals that result from the activation of the T cell receptor (TCR), which eliminates cytotoxic T cells (CTLs) and blocks anti-tumor immunity.
The FDA recently approved the use of two mouse antibodies (immune checkpoint inhibitors) known as anti-CTLA-4 and anti-PD-1 for the treatment of humans (134). Immune checkpoint (IC) molecules are found in TME cells including cancer cells, immune cells, and stromal cells like TEC. It has been discovered that TECs express PD-L1 and PD-L2 along with other well-known immunoinhibitory molecules such TIM-3, which raises the possibility that they may be able to directly suppress T cell activation when the vasculature is present. It has been shown that pro-inflammatory cytokines like IFN and TNF encourage PDL–1 up-regulation on ECs. Although the most often used biomarker in immune-oncology to determine treatment options and patient stratification is now PDL–1 expression by cancer cells, its therapeutic value has not yet been determined (82). Studies conducted in vitro and in vivo by Barsoum I et al. gave the information for the upregulation of PDL-1 in an HIF dependent way. By utilising glyceryl trinitrate (GTN), an agonist of nitric oxide (NO) for signalling, they were also able to prevent the HIF-1 accumulation and the hypoxia-dependent PDL-1 production and Cytotoxic T Lymphocyte resistance (135). The expression of PD-L1/2 may begin to make T lymphocytes lethargic and exhausted before they even enter the cancer microenvironment. In the pancreatic neuroendocrine tumour model and the polyoma middle T oncoprotein (PyMT) breast cancer model (RT2-PNET), the concomitant administration of anti-VEGFR2 and PDL–1 antibodies promoted the development of specialised vessels known as High Endothelial Venules (HEVs), which support lymphocyte trafficking and enhance T-cell infiltration. Even before T cells penetrate the tumour microenvironment, the expression of PDL–1/2 may start to cause them to become anergic and worn out. In the pancreatic neuroendocrine tumour model and the polyoma middle T oncoprotein (PyMT) breast cancer model (RT2-PNET), the combination of both anti-VEGFR2 antibodies and PDL–1 antibodies stimulated the formation of specialised vessels called High Endothelial Venules (HEVs), which contribute to lymphocyte trafficking and improved T-cell infiltration (129).
It has proven that Immune checkpoint inhibitors or drugs could increase immune surveillance. Thus, combination therapies targeting Immune checkpoint and metabolism of cancer endothelium could be a promising strategy to reduce tumor progression.
Due to metastasis and disease recurrence, breast cancer is the cancer that claims the lives of more women than any other (136). Journey of tumor cells across the endothelial membrane is a key step in the process of tumor invasion and metastasis. Tumor cells cross the vascular membrane stimulated by several cytokines and growth factors such as Transforming Growth Factor-Beta (TGF-β) superfamily of proteins,Bone Morphogenetic Proteins (BMPs). This is called Endothelial-mesenchymal transition (EndMT). EndMT induced by TGF-β shows a decrease in the expression of endothelial markers such as VE-cadherin, claudin and zona-occludens 1 (ZO-1) and an elevation in EMT transcription factors such as Snail, Slug and ZEB-1 which marks metastasis. TGF-β promotes ECs to change into CAF-like cells, which results in the loss of endothelial adhesion molecules and remodelling of the endothelium cytoskeleton via the Rho and Rac-1 signalling pathway, which is the primary characteristic of EndMT (76, 137, 138). It has been found that hypoxia associated with inflammation or tissue damage can also cause EndEMT (139). Rokana, et al. (2021) have recently studied the role of ICAM in trans-endothelial migration in breast cancer and also assessed the therapeutic efficacy of anti-ICAM1 neutralizing antibody on breast tumor cell aggregation and trans-endothelial migration. This anti-ICAM treatment inhibited the cluster formation of TNBC cells in suspension. Clinical data also showed high levels of ICAM1 mRNA expression in breast tumors which might mediate distant metastasis. This makes ICAM as a potential therapeutic target in TNBC metastasis (140). S100P and Ezrin, two members of the S100 family of short calcium binding proteins, also encourage the trans-endothelial migration of triple negative breast cancer cells. Furthermore, S100P activity has been linked to a variety of malignancies, a poor prognosis, metastasis and recurrence, and a low rate of survival in TNBC patients (141). An investigation on the SDF-1/CXCR4 axis in a breast carcinoma model revealed their role of hypoxia. It was confirmed that in a hypoxic tumor niche activation of HIF leads to the transcription of an array of HIF target genes including SDF-1 and CXCR4 which contributes to tumor cell migration and adhesion to endothelial cells in breast cancer cells. Moreover, the study also illustrated that SDF-1 binding to CXCR4 stimulated tube formation in endothelial cells, which point towards its role in angiogenesis and trans-endothelial migration (142). Since metastasis and related poor survival is a hallmark of breast cancer, pathways targeting trans-endothelial migration could have a significant impact in clinical trials.
The hypoxic microenvironment and impaired immune response to cancer cells by innate immune cells continue to be major obstacles for immunotherapy. Recent advancements in nano-immunotherapy, however, might lessen immunosuppression brought on by hypoxia and enhance systemic antitumor immune responses to eradicate metastatic breast cancer cells. Similar to this, in situ O2 generation, O2 delivery, normalisation of tumor vasculature, and mitochondrial-respiration inhibition could be the alluring therapy to overcome hypoxia-driven immune suppression for preventing the growth and progression of metastatic breast cancer cell lines. Monoclonal antibodies, immune modulators, and biodegradable bio-nanomaterials could address the problems in a very satisfactory way when used in conjunction with such methodologies. Nowadays, there is a great deal of interest in using nanomaterials as treatments to target the downstream pathways that lead to hypoxia-driven immune suppression in breast cancer (143). In this sense, PD1 receptor antagonist delivered in nanoform has the potential to effectively address the problem of hypoxic endothelium-driven immunosuppression during the treatment of breast cancer. The specific ligand antagonist of the upstream hyper-expressive biomarkers, which is responsible for the immunosuppressive state caused by hypoxic endothelium around the breast cancer microenvironment, can be used to develop new strategies. This may be a new strategy to improve immune monitoring in the treatment of breast cancer, especially for managing disease progression. Additionally, by preventing hypoxia in TME, such an approach could activate the immune cells, strengthen immune surveillance, and destroy breast cancer cells. By disrupting the hypoxia-mediated cancer signaling pathway, they may specifically target the hypoxic TME (144). Through the generation of oxygen-derived free radicals, radiation destroys tumor cells by causing DNA fragmentation. Due to the absence of oxygen and a reduction in DNA repair processes, hypoxia confers resistance to radiation therapy and reduces the effectiveness of radiation. The main cause of chemotherapy resistance in hypoxic tumors is the fact that many of the commonly used medications need oxygen to release the deadly free radicals that kills the tumor cells (145). In addition, another study that looked at the connection between HIF-1α/CAIX and the response to epirubicin found a strong correlation between high HIF-1α expression and a subpar response to treatment (146). This gives a clear insight that hypoxic pathway is involved in significant events that have a direct impact on the efficacy of numerous treatment methods. In the earlier sections of the review, we addressed the role of pro-angiogenic factor stimulation that causes tumor-associated endothelial cells to become anergic, losing the capacity to react to inflammatory signals and rendering them unable to activate EAMs (79, 147). It was discovered that this anti-infiltration barrier helped tumors avoid being destroyed by the immune system. Consequently, it is thought that using angiogenesis inhibitors to encourage leukocyte infiltration into the tumor is a useful way to increase the effectiveness of ICIs (148, 149). Bevacizumab, a VEGFA neutralizing antibody, has been shown to increase the number and activation of DC (149–151) as well as the number of cytotoxic T cells (152, 153) and to reverse VEGF-induced T cell exhaustion (154). Whereas, Sutinib, a TKI of VEGFR and other kinases, was shown to decrease the number of MDSCs and T reg cell (155, 156). Xiaodong et al. (2020). identified a tumor endothelial specific marker CLEC14A, which specifically recruits T reg recruitment and subsequently enhance immune suppression in the TME. CLEC 14 A specific CART cells, exhibited substantial decrease in tumor growth through IFN-γ indicating their antitumor potential (157).
TNBCs exhibit a markedly increased HIF transcriptional activity and a subpar response to the existing therapeutic strategies (158). Therefore, it makes sense to speculate that a novel therapeutic approach to treat TNBCs might involve targeting hypoxia in the TME. Preclinical investigations indicate that the combination of cytotoxic chemotherapy with molecules that block HIFs is particularly promising. Therapeutic studies with Digoxin and acriflavine, two HIF-1α inhibitors, demonstrated reduced initial tumor development, vascularization, invasion, and metastasis in breast cancer animal models. Furthermore, digoxin prevents HIF–1α dependent transcriptional responses that encourage cancer stem cell (CSC) resistance to chemotherapy, which causes tumor regression in TNBC when combined with paclitaxel or gemcitabine (159). Numerous synthetic and natural substances have been shown to block the regulation of HIF-1 on downstream target genes by lowering HIF-1 mRNA levels, accelerating the protein’s breakdown, and preventing HIF-1 and HIF-1 dimerization (160, 161) (Table 2). An effective anticancer therapy may include normalizing the tumor vasculature rather than destroying it. Anti-angiogenic drugs must be dosed carefully during vascular normalization in order to reverse the aberrant phenotype of the tumor vasculature and increase blood flow and oxygenation. Through vessel maturation and the alleviation of immunosuppression brought on by hypoxia and/or VEGF, it has been demonstrated that vascular normalization enhances immunological responses (16, 162). It indicates that restoring the structural and functional integrity of the tumour vasculature is a viable method for polarising TAMs to an anti-tumor phenotype (163). The hypoxic tumor endothelium is hyperglycolytic, thus, targeting tumor endothelial metabolism might offer a novel therapeutic strategy. Glycolysis is the energy source for endothelial sprouting in angiogenesis rather than oxidative phosphorylation for ATP production. Thus, blocking PFKFB3, a key molecule involved in endothelial glycolysis pathway, reduced vessel sprouting and angiogenesis (164). VEGF and PFKFB3, which are both implicated in the TEC’s glycolytic pathway, were downregulated when tumor-cell-specific cyclooxygenase (COX-2) was pharmacologically inhibited. The restoration of glucose metabolism in TECs and the reduction of tip cells, filopodia, and branching are affected by COX-2 inhibitor therapy-induced inhibition of PFKFB3-mediated endothelial cell motility (165). The role of anti-angiogenic drugs in regulating the tumor vasculature and reducing hypoxia-induced immune suppression has been shown in a number of preclinical investigations. These anti-angiogenic therapies have also been successful in overcoming endothelial anergy, which results in normalized production of endothelial adhesion molecules, which are necessary for leukocyte trans-endothelial migration into the TMC. Treatment that targets the VEGF pathway may result in increased immune infiltration and ICAM1 overexpression in renal cell carcinoma (166). Since TECs have higher glycolytic rates than typical proliferating ECs, reducing glycolysis by blocking PFKFB3 with 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) may be able to suppress tumor growth. According to studies, 3PO therapy increased the effectiveness of chemotherapy, aided in the normalisation of tumor vessels, tightened the EC barrier to prevent cancer metastasis, and encouraged tumor vessel normalisation. In mouse tumor models, proliferating ECs were treated with the weak mitochondrial uncoupler Embelin, which resulted in reduced mitochondrial oxidative phosphorylation, inhibited tumor growth, and reduced microvessel density (164). The endothelium of tumors significantly differs from endothelium of healthy tissues in a number of aspects, including metabolic reprogramming, alternative morphology, cytogenetics, and molecular genetics (82). So, a viable strategy to restore normal tumor vasculature involves attacking tumor endothelium. The research carried out by our group has also shown a rise in the infiltration of effector T cells, which in mouse TNBC inhibit the pro-angiogenic CXCR2 receptor. Targeting CXCR2 or other pro-angiogenic expressed on tumor endothelium may produce breakthrough outcomes in accordance with the promising results. (Unpublished data). Similar results were seen in vitro when a novel synthetic quinoline derivative was administered to block the pro-angiogenic chemokine (Unpublished data). Targeting the molecules involved in the regulation of tumors would open up the potential of a wide range of therapeutic options, normalising vascular function, and improving immune surveillance. Future therapeutic implications may benefit from knowledge of the variations in chemokines or pro-angiogenic substances produced by NECs and TECs in a hypoxic environment, as well as the expression of these receptors in normal and malignant cells.
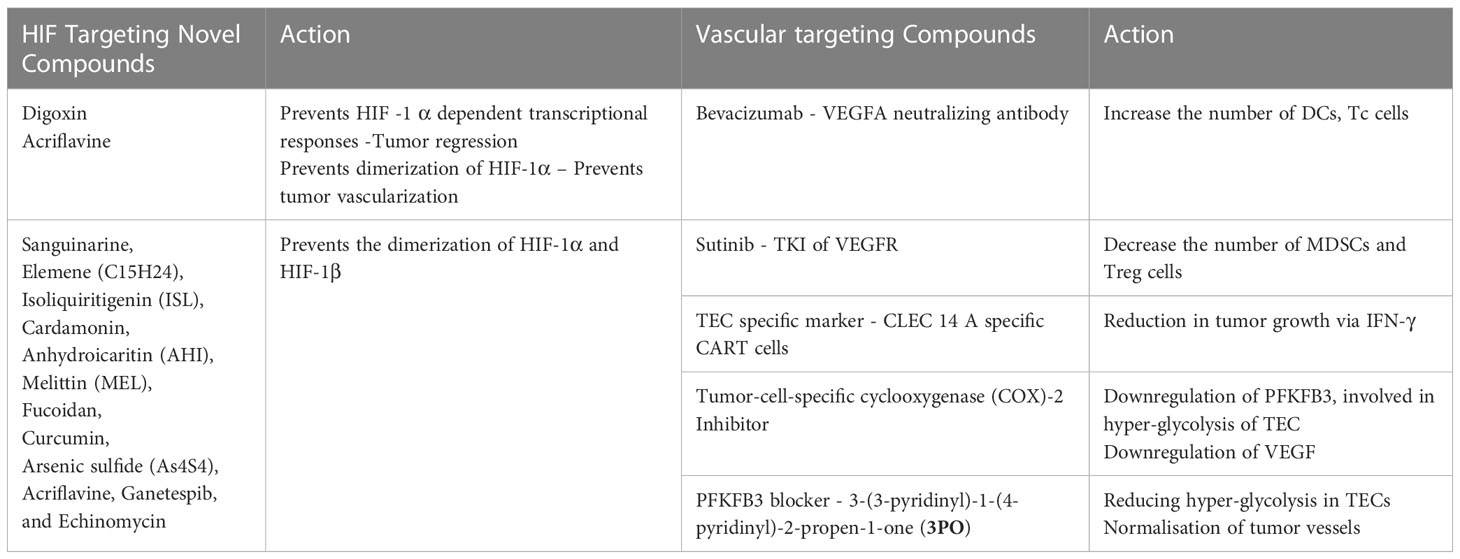
Table 2 Hypoxia and vascular targeting compounds.
In the light of the foregoing discussion, it will be wise to more effectively employ therapeutic alternatives in BCs with treatment resistance. Understanding the control of selective immune cell trafficking via hypoxic tumour endothelium may also be necessary. Clinical studies have found that the early preventative methods for the development of new blood vessels have only limited effects. The establishment of resistant mechanisms and enhanced tumor hypoxia are the causes of this insufficient efficacy. The development of effective treatment combinations can be facilitated by an understanding of how tumors use hypoxic endothelium cells to evade the immune system. Hypoxia regulating molecules and Immune Checkpoint Inhibitors (ICIs) should be combined to improve the prognosis of patients with hypoxic breast cancer in the light of the role played by the hypoxic TME in immune evasion. Therefore, molecules linked to selective trafficking may offer brand-new prognostic criteria to justify the use of particular immunotherapy regimens in conjunction with vascular targeting therapeutics. Though it will take time to investigate and comprehend such issues, promising research on anti-angiogenic adjuvant immunotherapy techniques offers hope for the improvement of treatment ways to improve the outcomes of breast cancer patients. A concerted effort from all investigators is imperative to usher in an era free from the woes of life-threatening diseases such as the BCs.
SB and PP conceived the concept and contributed writing and editing of the review. All authors contributed to the article and approved the submitted version.
This study was supported by Teachers Associateship for Research Excellence, SERB, Govt. of. India. (TAR/2021/000147) and Kairali Research Award (Biological Science), KSCSTE, Govt. of Kerala (KSHECA3/ 352/Kairali Research Award/109/2021).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA: Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
2. Yun Z, Lin Q. Hypoxia and regulation of cancer cell stemness. Tumor Microenviron Cell Stress (2014) 772:41–53. doi: 10.1007/978-1-4614-5915-6_2
3. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer (2019) 18(1):1–15. doi: 10.1186/s12943-019-1089-9
4. Chen A, Sceneay J, Gödde N, Kinwel T, Ham S, Thompson EW, et al. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene (2018) 37(31):4214–25. doi: 10.1038/s41388-018-0259-3
5. Saxena K, Jolly MK. Acute vs. chronic vs. cyclic hypoxia: Their differential dynamics, molecular mechanisms, and effects on tumor progression. Biomolecules (2019) 9(8):339. doi: 10.3390/biom9080339
6. Chan N, Koritzinsky M, Zhao H, Bindra R, Glazer PM, Powell S, et al. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res (2008) 68(2):605–14. doi: 10.1158/0008-5472.CAN-07-5472
7. Rofstad EK, Gaustad JV, Egeland TA, Mathiesen B, Galappathi K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int J Cancer (2010) 127(7):1535–46. doi: 10.1002/ijc.25176
8. Bayer C, Vaupel P. Acute versus chronic hypoxia in tumors. Strahlenther Onkol (2012) 188(7):616–27. doi: 10.1007/s00066-012-0085-4
9. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (2015) 3:83. doi: 10.2147/HP.S93413
10. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The hypoxic tumor microenvironment. Oncogenesis (2018) 7(1):1–13. doi: 10.1038/s41389-017-0011-9
11. Emami Nejad A, Najafgholian S, Rostami A, Sistani A, Shojaeifar S, Esparvarinha M, et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int (2021) 21(1):1–26. doi: 10.1186/s12935-020-01719-5
12. Chao Y, Zhong ZF, Sheng-Peng W, Chi-Teng V, Bin Y, Yi-Tao W. HIF-1: Structure, biology and natural modulators. Chin J Nat Med (2021) 19(7):521–7. doi: 10.1016/S1875-5364(21)60051-1
13. Tolonen JP, Heikkilä M, Malinen M, Lee HM, Palvimo JJ, Wei GH, et al. A long hypoxia-inducible factor 3 isoform 2 is a transcription activator that regulates erythropoietin. Cell Mol Life Sci (2020) 77(18):3627–42. doi: 10.1007/s00018-019-03387-9
14. Gilreath C, Boerma M, Qin Z, Hudson MK, Wang S. The hypoxic microenvironment of breast cancer cells promotes resistance in radiation therapy. Front Oncol (2021) 10:629422. doi: 10.3389/fonc.2020.629422
15. Vettori A, Greenald D, Wilson GK, Peron M, Facchinello N, Markham E, et al. Glucocorticoids promote Von hippel lindau degradation and hif-1α stabilization. Proc Natl Acad Sci (2017) 114(37):9948–53. doi: 10.1073/pnas.1705338114
16. Kim J w, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3(3):177–85. doi: 10.1016/j.cmet.2006.02.002
17. Gilkes DM, Semenza GL. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol Lond Engl (2013) 9(11):1623–36. doi: 10.2217/fon.13.92
18. Park M, Kim D, Ko S, Kim A, Mo K, Yoon H. Breast cancer metastasis: Mechanisms and therapeutic implications. Int J Mol Sci (2022) 23(12):6806. doi: 10.3390/ijms23126806
19. Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer (2018) 18(2):128–34. doi: 10.1038/nrc.2017.118
20. Eskiizmir G, Özgür E. Epithelial-mesenchymal transition in tumor microenvironment induced by hypoxia. Cancer Metastasis (2018). doi: 10.5772/intechopen.78717
21. Cao J, Zhang M, Wang B, Zhang L, Zhou F, Fang M. Chemoresistance and metastasis in breast cancer molecular mechanisms and novel clinical strategies. Front Oncol (2021) 11:658552. doi: 10.3389/fonc.2021.658552
22. Wang L, Zhang S, Wang X. The metabolic mechanisms of breast cancer metastasis. Front Oncol (2021) 10:602416. doi: 10.3389/fonc.2020.602416
23. O’Reilly D, Al Sendi M, Kelly CM. Overview of recent advances in metastatic triple negative breast cancer. World J Clin Oncol (2021) 12(3):164. doi: 10.5306/wjco.v12.i3.164
24. Joseph JP, Harishankar MK, Pillai AA, Devi A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol (2018) 80:23–32. doi: 10.1016/j.oraloncology.2018.03.004
25. Peng J, Wang X, Ran L, Song J, Luo R, Wang Y. Hypoxia-inducible factor 1α regulates the transforming growth factor β1/SMAD family member 3 pathway to promote breast cancer progression. J Breast Cancer (2018) 21(3):259–66. doi: 10.4048/jbc.2018.21.e42
26. Tam SY, Wu VW, Law HK. Hypoxia-induced epithelial-mesenchymal transition in cancers: HIF-1α and beyond. Front Oncol (2020) 10:486. doi: 10.3389/fonc.2020.00486
27. Ezdakova MI, Andreeva ER, Gurieva TS, Dadasheva OA, Orlova VS, Buravkova LB. Effects of hypoxia and growth factors on the angiogenic activity of multipotent mesenchymal stromal cells. Aviakosmicheskaia Ekol Meditsina Aerosp Environ Med (2015) 49(5):29–35.
28. Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol (2012) 24(6):650. doi: 10.1097/CCO.0b013e328356da72
29. Courtnay R, Ngo DC, Malik N, Ververis K, Tortorella SM, Karagiannis TC. Cancer metabolism and the warburg effect: The role of HIF-1 and PI3K. Mol Biol Rep (2015) 42(4):841–51. doi: 10.1007/s11033-015-3858-x
30. Shin E, Koo JS. Glucose metabolism and glucose transporters in breast cancer. Front Cell Dev Biol (2021) 2404. doi: 10.3389/fcell.2021.728759
31. Dekker Y, Le Dévédec SE, Danen EH, Liu Q. Crosstalk between hypoxia and extracellular matrix in the tumor microenvironment in breast cancer. Genes (2022) 13(9):1585. doi: 10.3390/genes13091585
32. Munoz-Najar UM, Neurath KM, Vumbaca F, Claffey KP. Hypoxia stimulates breast carcinoma cell invasion through MT1-MMP and MMP-2 activation. Oncogene (2006) 25(16):2379–92. doi: 10.1038/sj.onc.1209273
33. Quintero-Fabián S, Arreola R, Becerril-Villanueva E, Torres-Romero JC, Arana-Argáez V, Lara-Riegos J, et al. Role of matrix metalloproteinases in angiogenesis and cancer. Front Oncol (2019) 9:1370. doi: 10.3389/fonc.2019.01370
34. Zhu H, Zhang S. Hypoxia inducible factor-1α/vascular endothelial growth factor signaling activation correlates with response to radiotherapy and its inhibition reduces hypoxia-induced angiogenesis in lung cancer. J Cell Biochem (2018) 119(9):7707–18. doi: 10.1002/jcb.27120
35. El Bairi K, Haynes HR, Blackley E, Fineberg S, Shear J, Turner S, et al. The tale of TILs in breast cancer: A report from the international immuno-oncology biomarker working group. NPJ Breast Cancer (2021) 7(1):1–17. doi: 10.1038/s41523-021-00346-1
36. Ono M, Tsuda H, Shimizu C, Yamamoto S, Shibata T, Yamamoto H, et al. Tumor-infiltrating lymphocytes are correlated with response to neoadjuvant chemotherapy in triple-negative breast cancer. Breast Cancer Res Treat (2012) 132(3):793–805. doi: 10.1007/s10549-011-1554-7
37. Wigerup C, Påhlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther (2016) 164:152–69. doi: 10.1016/j.pharmthera.2016.04.009
38. Pezzuto A, Carico E. Role of HIF-1 in cancer progression: Novel insights. A review. Curr Mol Med (2018) 18(6):343–51. doi: 10.2174/1566524018666181109121849
39. Duechler M, Peczek L, Zuk K, Zalesna I, Jeziorski A, Czyz M. The heterogeneous immune microenvironment in breast cancer is affected by hypoxia-related genes. Immunobiology (2014) 219(2):158–65. doi: 10.1016/j.imbio.2013.09.003
40. Chouaib S, Messai Y, Couve S, Escudier B, Hasmim M, Noman MZ. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front Immunol (2012) 3:21. doi: 10.3389/fimmu.2012.00021
41. Wei J, Wu A, Kong LY, Wang Y, Fuller G, Fokt I, et al. Hypoxia potentiates glioma-mediated immunosuppression. PloS One (2011) 6(1):e16195. doi: 10.1371/journal.pone.0016195
42. Cimino-Mathews A, Foote JB, Emens LA. Immune targeting in breast cancer. Oncology (2015) 29(5):375–5.
43. DeNardo DG, Coussens LM. Inflammation and breast cancer. balancing immune response: Crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res (2007) 9(4):1–10. doi: 10.1186/bcr1746
44. Singh R, Paterson Y. Immunoediting sculpts tumor epitopes during immunotherapy. Cancer Res (2007) 67(5):1887–92. doi: 10.1158/0008-5472.CAN-06-3960
45. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: From immunosurveillance to tumor escape. Nat Immunol (2002) 3(11):991–8. doi: 10.1038/ni1102-991
46. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest (2007) 117(5):1137–46. doi: 10.1172/JCI31405
47. Hida K, Maishi N, Ryo Takeda D, Hida Y. The roles of tumor endothelial cells in cancer metastasis. Exon Publ (2022), 137–48. doi: 10.36255/exon-publications.metastasis.endothelial-cells
48. Semenza GL. Intratumoral hypoxia and mechanisms of immune evasion mediated by hypoxia-inducible factors. Physiology (2021) 36(2):73–83. doi: 10.1152/physiol.00034.2020
49. Korbecki J, Kojder K, Kapczuk P, Kupnicka P, Gawrońska-Szklarz B, Gutowska I, et al. The effect of hypoxia on the expression of CXC chemokines and CXC chemokine receptors–a review of literature. Int J Mol Sci (2021) 22(2):843. doi: 10.3390/ijms22020843
50. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res (2009) 69(7):3077–85. doi: 10.1158/0008-5472.CAN-08-2281
51. Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Lett (2007) 256(2):137–65. doi: 10.1016/j.canlet.2007.05.013
52. Rajani C, Borisa P, Karanwad T, Borade Y, Patel V, Rajpoot K, et al. Cancer-targeted chemotherapy: Emerging role of the folate anchored dendrimer as drug delivery nanocarrier. Pharm Appl Dendrimers (2020) p:151–98. doi: 10.1016/B978-0-12-814527-2.00007-X
53. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov (2011) 10(6):417–27. doi: 10.1038/nrd3455
54. Berthiaume AA, Grant RI, McDowell KP, Underly RG, Hartmann DA, Levy M, et al. Dynamic remodeling of pericytes in vivo maintains capillary coverage in the adult mouse brain. Cell Rep (2018) 22(1):8–16. doi: 10.1016/j.celrep.2017.12.016
55. Lopes-Bastos BM, Jiang WG, Cai J. Tumor–endothelial cell communications: Important and indispensable mediators of tumor angiogenesis. Anticancer Res (2016) 36(3):1119–26.
56. Solimando AG, Summa SD, Vacca A, Ribatti D. Cancer-associated angiogenesis: The endothelial cell as a checkpoint for immunological patrolling. Cancers (2020) 12(11):3380. doi: 10.3390/cancers12113380
57. Maishi N, Annan DA, Kikuchi H, Hida Y, Hida K. Tumor endothelial heterogeneity in cancer progression. Cancers (2019) 11(10):1511. doi: 10.3390/cancers11101511
58. Nomura M, Yamagishi S i, Harada S i, Hayashi Y, Yamashima T, Yamashita J, et al. Possible participation of autocrine and paracrine vascular endothelial growth factors in hypoxia-induced proliferation of endothelial cells and pericytes. J Biol Chem (1995) 270(47):28316–24. doi: 10.1074/jbc.270.47.28316
59. Reiss Y, Knedla A, Tal AO, Schmidt MH, Jugold M, Kiessling F, et al. Switching of vascular phenotypes within a murine breast cancer model induced by angiopoietin-2. J Pathol J Pathol Soc G B Irel (2009) 217(4):571–80. doi: 10.1002/path.2484
60. Imanishi Y, Hu B, Jarzynka MJ, Guo P, Elishaev E, Bar-Joseph I, et al. Angiopoietin-2 stimulates breast cancer metastasis through the α5β1 integrin-mediated pathway. Cancer Res (2007) 67(9):4254–63. doi: 10.1158/0008-5472.CAN-06-4100
61. Zhang Y, Kontos CD, Annex BH, Popel AS. Angiopoietin-tie signaling pathway in endothelial cells: A computational model. Iscience (2019) 20:497–511. doi: 10.1016/j.isci.2019.10.006
62. Geranmayeh MH, Rahbarghazi R, Farhoudi M. Targeting pericytes for neurovascular regeneration. Cell Communication Signaling (2019) 17(1):1–3. doi: 10.1186/s12964-019-0340-8
63. Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature (2010) 468(7323):562–6. doi: 10.1038/nature09513
64. Hong J, Tobin NP, Rundqvist H, Li T, Lavergne M, García-Ibáñez Y, et al. Role of tumor pericytes in the recruitment of myeloid-derived suppressor cells. J Natl Cancer Inst (2015) 107(10):djv209. doi: 10.1093/jnci/djv209
65. Enström A, Carlsson R, Özen I, Paul G. RGS5: A novel role as a hypoxia-responsive protein that suppresses chemokinetic and chemotactic migration in brain pericytes. Biol Open (2022) 11(10):bio059371. doi: 10.1242/bio.059371
66. Mamer SB, Chen S, Weddell JC, Palasz A, Wittenkeller A, Kumar M, et al. Discovery of high-affinity PDGF-VEGFR interactions: redefining RTK dynamics. Sci Rep (2017) 7(1):1–14. doi: 10.1038/s41598-017-16610-z
67. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassará A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med (2017) 9(385):eaak9670. doi: 10.1126/scitranslmed.aak9670
68. Bohn KA, Sechrest ER, Adkins CE, Mittapalli RK, Nounou MI, Terrell-Hall TB, et al. Inhibition of VEGF and angiopoietin-2 to reduce brain metastases of breast cancer burden. Cancer Res (2015) 75:1388–8. doi: 10.1158/1538-7445.AM2015-1388
69. Krüger-Genge A, Blocki A, Franke RP, Jung F. Vascular endothelial cell biology: An update. Int J Mol Sci (2019) 20(18):4411. doi: 10.3390/ijms20184411
70. Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF. Heterogeneity of the tumor vasculature. InSeminars Thromb Hemostasis (2010) 36, No. 03:321–31. doi: 10.1055/s-0030-1253454
71. Griffioen AW, Damen CA, Mayo KH, Barendsz-Janson AF, Martinotti S, Blijham GH, et al. Angiogenesis inhibitors overcome tumor induced endothelial cell anergy. Int J Cancer (1999) 80(2):315–9. doi: 10.1002/(SICI)1097-0215(19990118)80:2<315::AID-IJC23>3.0.CO;2-L
72. Klein D. The tumor vascular endothelium as decision maker in cancer therapy. Front Oncol (2018) 8:367. doi: 10.3389/fonc.2018.00367
73. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumor angiogenesis. Nat Rev Cancer (2017) 17(8):457–74. doi: 10.1038/nrc.2017.51
74. Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, et al. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell (2016) 30(6):968–85. doi: 10.1016/j.ccell.2016.10.006
75. Alam MT, Nagao-Kitamoto H, Ohga N, Akiyama K, Maishi N, Kawamoto T, et al. Suprabasin as a novel tumor endothelial cell marker. Cancer Sci (2014) 105(12):1533–40. doi: 10.1111/cas.12549
76. Liang J, Wang S, Zhang G, He B, Bie Q, Zhang B. A new antitumor direction: Tumor-specific endothelial cells. Front Oncol (2021) 11. doi: 10.3389/fonc.2021.756334
77. Huinen ZR, Huijbers EJ, van Beijnum JR, Nowak-Sliwinska P, Griffioen AW. Anti-angiogenic agents–overcoming tumor endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol (2021) 18(8):527–40. doi: 10.1038/s41571-021-00496-y
78. Kwaifa IK, Bahari H, Yong YK, Noor SM. Endothelial dysfunction in obesity-induced inflammation: Molecular mechanisms and clinical implications. Biomolecules (2020) 10(2):291. doi: 10.3390/biom10020291
79. Griffioen AW, Damen CA, Blijham GH, Groenewegen G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood (1996) 667–73. doi: 10.1182/blood.V88.2.667.bloodjournal882667
80. De Sanctis F, Ugel S, Facciponte J, Facciabene A. The dark side of tumor-associated endothelial cells. Semin Immunol (2018) 35:35–47. doi: 10.1016/j.smim.2018.02.002
81. Flati V, Pastore LI, Griffioen AW, Satijn S, Tomato E, D’alimonte I, et al. Endothelial cell anergy is mediated by bFGF through the sustained activation of p38-MAPK and NF-κB inhibition. Int J Immunopathol Pharmacol (2006) 19(4):761–73. doi: 10.1177/039463200601900406
82. Nagl L, Horvath L, Pircher A, Wolf D. Tumor endothelial cells (TECs) as potential immune directors of the tumor microenvironment–new findings and future perspectives. Front Cell Dev Biol (2020) 766. doi: 10.3389/fcell.2020.00766
83. Tellier C, Desmet D, Petit L, Finet L, Graux C, Raes M, et al. Cycling hypoxia induces a specific amplified inflammatory phenotype in endothelial cells and enhances tumor-promoting inflammation in vivo. Neoplasia (2015) 17(1):66–78. doi: 10.1016/j.neo.2014.11.003
84. Brown Chandler K E, Costello C, Rahimi N. Glycosylation in the tumor microenvironment: implications for tumor angiogenesis and metastasis. Cells (2019) 8(6):544. doi: 10.3390/cells8060544
85. Dirkx AE, oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Steege JCB t, et al. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res (2003) 63(9):2322–9.
86. Zeng H, Ji J, Song X, Huang Y, Li H, Huang J, et al. Stemness related genes revealed by network analysis associated with tumor immune microenvironment and the clinical outcome in lung adenocarcinoma. Front Genet (2020) 1095. doi: 10.3389/fgene.2020.549213
87. Pei J, Wang Y, Li Y. Identification of key genes controlling breast cancer stem cell characteristics via stemness indices analysis. J Transl Med (2020) 18(1):1–15. doi: 10.1186/s12967-020-02260-9
88. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci (2012) 109(43):17561–6. doi: 10.1073/pnas.1215397109
89. Dings RP, Vang KB, Castermans K, Popescu F, Zhang Y, Mescher MF, et al. Enhancement of t-cell–mediated antitumor response: Angiostatic adjuvant to immunotherapy against CancerEnhancement of t-cell–mediated antitumor response. Clin Cancer Res (2011) 17(10):3134–45. doi: 10.1158/1078-0432.CCR-10-2443
90. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med (2014) 20(6):607–15. doi: 10.1038/nm.3541
91. Dysthe M, Parihar R. Myeloid-derived suppressor cells in the tumor microenvironment. Tumor Microenviron (2020) 1224:117–40. doi: 10.1007/978-3-030-35723-8_8
92. Safarzadeh E, Hashemzadeh S, Duijf PH, Mansoori B, Khaze V, Mohammadi A, et al. Circulating myeloid-derived suppressor cells: An independent prognostic factor in patients with breast cancer. J Cell Physiol (2019) 234(4):3515–25. doi: 10.1002/jcp.26896
93. Li Y, He H, Jihu R, Zhou J, Zeng R, Yan H. Novel characterization of myeloid-derived suppressor cells in tumor microenvironment. Front Cell Dev Biol (2021) 9:698532. doi: 10.3389/fcell.2021.698532
94. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell–mediated antitumor immunity. Nat Med (2003) 9(5):562–7. doi: 10.1038/nm863
95. Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor CellsVEGF induces MDSCs and blocks tumor immunity in HGSOC. Clin Cancer Res (2017) 23(2):587–99. doi: 10.1158/1078-0432.CCR-16-0387
96. Chen JY, Lai YS, Chu PY, Chan SH, Wang LH, Hung WC. Cancer-derived VEGF-c increases chemokine production in lymphatic endothelial cells to promote CXCR2-dependent cancer invasion and MDSC recruitment. Cancers (2019) 11(8):1120. doi: 10.3390/cancers11081120
97. Roberts LM, Perez MJ, Balogh KN, Mingledorff G, Cross JV, Munson JM. Myeloid derived suppressor cells migrate in response to flow and lymphatic endothelial cell interaction in the breast tumor microenvironment. Cancers (2022) 14(12):3008. doi: 10.3390/cancers14123008
98. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37(3):208–20. doi: 10.1016/j.it.2016.01.004
99. Noy R, Pollard JW. Tumor-associated macrophages: From mechanisms to therapy. Immunity (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
100. Poh AR, Ernst M. Targeting macrophages in cancer: From bench to bedside. Front Oncol (2018) 8:49. doi: 10.3389/fonc.2018.00049
101. Bellac CL, Dufour A, Krisinger MJ, Loonchanta A, Starr AE, auf dem Keller U, et al. Macrophage matrix metalloproteinase-12 dampens inflammation and neutrophil influx in arthritis. Cell Rep (2014) 9(2):618–32. doi: 10.1016/j.celrep.2014.09.006
102. Leek RD, Hunt NC, Landers RJ, Lewis CE, Royds JA, Harris AL. Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. J Pathol (2000) 190(4):430–6. doi: 10.1002/(SICI)1096-9896(200003)190:4<430::AID-PATH538>3.0.CO;2-6
103. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumor-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14(7):399–416. doi: 10.1038/nrclinonc.2016.217
104. Jayasingam SD, Citartan M, Thang TH, Mat Zin AA, Ang KC, Ch’ng ES. Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: Technicalities and challenges in routine clinical practice. Front Oncol (2020) 9:1512. doi: 10.3389/fonc.2019.01512
105. Wang Q, He Z, Huang M, Liu T, Wang Y, Xu H, et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2α. Nat Commun (2018) 9(1):559. doi: 10.1038/s41467-018-03050-0
106. Abid MR, Yi X, Yano K, Shih SC, Aird WC. Vascular endocan is preferentially expressed in tumor endothelium. Microvascular Res (2006) 72(3):136–45. doi: 10.1016/j.mvr.2006.05.010
107. Huang X, Chen C, Wang X, Zhang JY, Ren BH, Ma DW, et al. Prognostic value of endocan expression in cancers: Evidence from meta-analysis. OncoTargets Ther (2016) 9:6297–304. doi: 10.2147/OTT.S110295
108. White JR, Harris RA, Lee SR, Craigon MH, Binley K, Price T, et al. Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics (2004) 83(1):1–8. doi: 10.1016/S0888-7543(03)00215-5
109. Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, et al. Hypoxia-induced gene expression in human macrophages: Implications for ischemic tissues and hypoxia-regulated gene therapy. Am J Pathol (2003) 163:1233–43. doi: 10.1016/S0002-9440(10)63483-9
110. Li X, Liu R, Su X, Pan Y, Han X, Shao C, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer (2019) 18(1):1–6. doi: 10.1186/s12943-019-1102-3
111. Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1α and adenosine receptors. Nat Rev Immunol (2005) 5(9):712–21. doi: 10.1038/nri1685
112. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stag0g J, Elkord E, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. In: Semin Cancer Biol Elsevier; (2015) p:S185–98. doi: 10.1016/j.semcancer.2015.03.004
113. Winning S, Fandrey J. Dendritic cells under hypoxia: How oxygen shortage affects the linkage between innate and adaptive immunity. J Immunol Res (2016) 2016. doi: 10.1155/2016/5134329
114. Garrido F, Ruiz-Cabello F, Cabrera T, Perez-Villar JJ, López-Botet M, Duggan-Keen M, et al. Implications for immunosurveillance of altered HLA class I phenotypes in human tumors. Immunol Today (1997) 18(2):89–95. doi: 10.1016/S0167-5699(96)10075-X
115. Johnsen AK, Templeton DJ, Sy MS, Harding CV. Deficiency of transporter for antigen presentation (TAP) in tumor cells allows evasion of immune surveillance and increases tumorigenesis. J Immunol (1999) 163(8):4224–31. doi: 10.4049/jimmunol.163.8.4224
116. Maeurer MJ, Gollin SM, Martin D, Swaney W, Bryant J, Castelli C, et al. Tumor escape from immune recognition:Llethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/Melan-A antigen. J Clin Invest (1996) 98(7):1633–41. doi: 10.1172/JCI118958
117. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumor hypoxia promotes tolerance and angiogenesis via CCL28 and treg cells. Nature (2011) 475(7355):226–30. doi: 10.1038/nature10169
118. Corthay A. How do regulatory T cells work? Scand J Immunol (2009) 70(4):326–36. doi: 10.1111/j.1365-3083.2009.02308.x
119. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
120. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha–dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci (2012) 109(41):E2784–93. doi: 10.1073/pnas.1202366109
121. Hsu TS, Lin YL, Wang YA, Mo ST, Chi PY, Lai ACY, et al. HIF-2α is indispensable for regulatory T cell function. Nat Commun (2020) 11(1):1–16.
122. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
123. Niederlova V, Tsyklauri O, Chadimova T, Stepanek O. CD8+ tregs revisited: A heterogeneous population with different phenotypes and properties. Eur J Immunol (2021) 51(3):512–30. doi: 10.1002/eji.202048614
124. Verma A, Mathur R, Farooque A, Kaul V, Gupta S, Dwarakanath BS. T-Regulatory cells in tumor progression and therapy. Cancer Manag Res (2019) 11:10731. doi: 10.2147/CMAR.S228887
125. Stenström J, Hedenfalk I, Hagerling C. Regulatory T lymphocyte infiltration in metastatic breast cancer–an independent prognostic factor that changes with tumor progression. Breast Cancer Res (2021) 23(1):1–12. doi: 10.1186/s13058-021-01403-0
126. Yan M, Jene N, Byrne D, Millar EK, O’Toole SA, McNeil CM, et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Res (2011) 13(2):1–10. doi: 10.1186/bcr2869
127. Lužnik Z, Anchouche S, Dana R, Yin J. Regulatory T cells in angiogenesis. J Immunol (2020) 205(10):2557–65. doi: 10.4049/jimmunol.2000574
128. Chambers AM, Lupo KB, Matosevic S. Tumor microenvironment-induced immunometabolic reprogramming of natural killer cells. Front Immunol (2018) 9:2517. doi: 10.3389/fimmu.2018.02517
129. Georganaki M, van Hooren L, Dimberg A. Vascular targeting to increase the efficiency of immune checkpoint blockade in cancer. Front Immunol (2018) 9:3081. doi: 10.3389/fimmu.2018.03081
130. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity (2016) 45(5):1122–34. doi: 10.1016/j.immuni.2016.10.032
131. Onda M, Kobayashi K, Pastan I. Depletion of regulatory T cells in tumors with an anti-CD25 immunotoxin induces CD8 T cell-mediated systemic antitumor immunity. Proc Natl Acad Sci (2019) 116(10):4575–82. doi: 10.1073/pnas.1820388116
132. Long Y, Tao H, Karachi A, Grippin AJ, Jin L, Chang YE, et al. Dysregulation of glutamate transport enhances treg function that promotes VEGF blockade resistance in GlioblastomaAnti-VEGF therapy promotes treg function. Cancer Res (2020) 80(3):499–509. doi: 10.1158/0008-5472.CAN-19-1577
133. Hu M, Li Y, Lu Y, Wang M, Li Y, Wang C, et al. The regulation of immune checkpoints by the hypoxic tumor microenvironment. PeerJ (2021) 9:e11306. doi: 10.7717/peerj.11306
134. Rupp T, Genest L, Babin D, Legrand C, Hunault M, Froget G, et al. Anti-CTLA-4 and anti-PD-1 immunotherapies repress tumor progression in preclinical breast and colon model with independent regulatory T cells response. Transl Oncol (2022) 20:101405. doi: 10.1016/j.tranon.2022.101405
135. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res (2014) 74(3):665–74. doi: 10.1158/0008-5472.CAN-13-0992
136. DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding Sauer A, et al. Breast cancer statistics, 2019. CA Cancer J Clin (2019) 69(6):438–51. doi: 10.3322/caac.21583
137. Maishi N, Kikuchi H, Sato M, Nagao-Kitamoto H, Annan DA, Baba S, et al. Development of immortalized human tumor endothelial cells from renal cancer. Int J Mol Sci (2019) 20(18):4595. doi: 10.3390/ijms20184595
138. Ciszewski WM, Sobierajska K, Wawro ME, Klopocka W, Chefczyńska N, Muzyczuk A, et al. The ILK-MMP9-MRTF axis is crucial for EndMT differentiation of endothelial cells in a tumor microenvironment. Biochim Biophys Acta BBA-Mol Cell Res (2017) 1864(12):2283–96. doi: 10.1016/j.bbamcr.2017.09.004
139. Xu X, Tan X, Tampe B, Sanchez E, Zeisberg M, Zeisberg EM. Snail is a direct target of hypoxia-inducible factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J Biol Chem (2015) 290(27):16653–64. doi: 10.1074/jbc.M115.636944
140. Taftaf R, Liu X, Singh S, Jia Y, Dashzeveg NK, Hoffmann AD, et al. ICAM1 initiates CTC cluster formation and trans-endothelial migration in lung metastasis of breast cancer. Nat Commun (2021) 12(1):1–15. doi: 10.1038/s41467-021-25189-z
141. Kikuchi K, McNamara KM, Miki Y, Iwabuchi E, Kanai A, Miyashita M, et al. S100P and ezrin promote trans-endothelial migration of triple negative breast cancer cells. Cell Oncol (2019) 42(1):67–80. doi: 10.1007/s13402-018-0408-2
142. Jin F, Brockmeier U, Otterbach F, Metzen E. New insight into the SDF-1/CXCR4 axis in a breast carcinoma model: Hypoxia-induced endothelial SDF-1 and tumor cell CXCR4 are required for tumor cell intravasation. Mol Cancer Res (2012) 10(8):1021–31. doi: 10.1158/1541-7786.MCR-11-0498
143. Wang Y, Shang W, Niu M, Tian J, Xu K. Hypoxia-active nanoparticles used in tumor theranostic. Int J Nanomedicine (2019) 14:3705. doi: 10.2147/IJN.S196959
144. Bosco MC, D’Orazi G, Del Bufalo D. Targeting hypoxia in tumor: a new promising therapeutic strategy. J Exp Clin Cancer Res (2020) 39(1):1–3. doi: 10.1186/s13046-019-1517-0
145. Minassian LM, Cotechini T, Huitema E, Graham CH. Hypoxia-induced resistance to chemotherapy in cancer. Hypoxia Cancer Metastasis (2019) 1136:123–39. doi: 10.1007/978-3-030-12734-3_9
146. Generali D, Berruti A, Brizzi MP, Campo L, Bonardi S, Wigfield S, et al. Hypoxia-inducible factor-1α expression predicts a poor response to primary chemoendocrine therapy and disease-free survival in primary human breast cancer. Clin Cancer Res (2006) 12(15):4562–8. doi: 10.1158/1078-0432.CCR-05-2690
147. Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: the role of angiogenic factors. Cancer Res (1996) 56(5):1111–7.
148. Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol (2018) 15(5):310–24. doi: 10.1038/nrclinonc.2018.9
149. Rahma OE, Hodi FS. The intersection between tumor angiogenesis and immune SuppressionAntiangiogenesis and immunotherapy. Clin Cancer Res (2019) 25(18):5449–57. doi: 10.1158/1078-0432.CCR-18-1543
150. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumors. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
151. Boucher Y, Kumar AS, Posada JM, Gjini E, Pfaff K, Lipschitz M, et al. Bevacizumab improves tumor infiltration of mature dendritic cells and effector T-cells in triple-negative breast cancer patients. NPJ Precis Oncol (2021) 5(1):1–6. doi: 10.1038/s41698-021-00197-w
152. Liu Y, Zhang T, Zhang L, Zhao C, Zhang Z, Wang Z, et al. Combined application of bevacizumab and PD-1 blockade displays durable treatment effects by increasing the infiltration and cytotoxic function of CD8+ T cells in lung cancer. Immunotherapy (2022) 14(9):695–708. doi: 10.2217/imt-2021-0196
153. Martino EC, Misso G, Pastina P, Costantini S, Vanni F, Gandolfo C, et al. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discov (2016) 2(1):1–8. doi: 10.1038/cddiscovery.2016.25
154. Bourhis M, Palle J, Galy-Fauroux I, Terme M. Direct and indirect modulation of T cells by VEGF-a counteracted by anti-angiogenic treatment. Front Immunol (2021) 12:616837. doi: 10.3389/fimmu.2021.616837
155. Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res (2009) 69(6):2514–22. doi: 10.1158/0008-5472.CAN-08-4709
156. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res (2009) 15(6):2148–57. doi: 10.1158/1078-0432.CCR-08-1332
157. Zhuang X, Maione F, Robinson J, Bentley M, Kaul B, Whitworth K, et al. CAR T cells targeting tumor endothelial marker CLEC14A inhibit tumor growth. JCI Insight (2020) 5(19). doi: 10.1172/jci.insight.138808
158. Xu R. P4HA1 is a new regulator of the HIF-1 pathway in breast cancer. Cell Stress (2019) 3(1):27. doi: 10.15698/cst2019.01.173
159. Zhang Y, Zhang H, Wang M, Schmid T, Xin Z, Kozhuharova L, et al. Hypoxia in breast cancer–scientific translation to therapeutic and diagnostic clinical applications. Front Oncol (2021) 11:652266. doi: 10.3389/fonc.2021.652266
160. Liu Q, Guan C, Liu C, Li H, Wu J, Sun C. Targeting hypoxia-inducible factor-1alpha: A new strategy for triple-negative breast cancer therapy. BioMed Pharmacother (2022) 156:113861. doi: 10.1016/j.biopha.2022.113861
161. Bailey CM, Liu Y, Peng G, Zhang H, He M, Sun D, et al. Liposomal formulation of HIF-1α inhibitor echinomycin eliminates established metastases of triple-negative breast cancer. Nanomedicine Nanotechnol Biol Med (2020) 29:102278. doi: 10.1016/j.nano.2020.102278
162. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat Rev Clin Oncol (2018) 15(5):325–40. doi: 10.1038/nrclinonc.2018.29
163. He Z, Zhang S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.741305
164. Li J, Eu JQ, Kong LR, Wang L, Lim YC, Goh BC, et al. Targeting metabolism in cancer cells and the tumor microenvironment for cancer therapy. Molecules (2020) 25(20):4831. doi: 10.3390/molecules25204831
165. Zhang L, Li S, Li L, Chen Z, Yang Y. COX-2 inhibition in the endothelium induces glucose metabolism normalization and impairs tumor progression. Mol Med Rep (2018) 17(2):2937–44. doi: 10.3892/mmr.2017.8270
Keywords: tumor endothelial cells (TECs), hypoxia inducible factors (HIFs), myeloid derived suppressor cells (MDSCs), T regulatory cells (Treg cells), angiogenesis, hypoxic tumor microenvironment, immunological switch point
Citation: Thomas JA, Gireesh AGM, Xavier H, Suboj P, Ladha A, Gupta G, Singh SK, Palit P and Babykutty S (2023) Enhancement of immune surveillance in breast cancer by targeting hypoxic tumor endothelium: Can it be an immunological switch point? Front. Oncol. 13:1063051. doi: 10.3389/fonc.2023.1063051
Received: 06 October 2022; Accepted: 17 February 2023;
Published: 28 March 2023.
Edited by:
Subhadeep Roy, Indian Institute of Technology Delhi, IndiaCopyright © 2023 Thomas, Gireesh, Xavier, Suboj, Ladha, Gupta, Singh, Palit and Babykutty. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suboj Babykutty, c3Vib2ouYmFieWt1dHR5QG1pYy5hYy5pbg==; Partha Palit, aXRzcGFydGhhX3BAeWFob28uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.