 Nouria Jantz-Naeem1
Nouria Jantz-Naeem1 Romy Böttcher-Loschinski2Katrin Borucki3
Romy Böttcher-Loschinski2Katrin Borucki3 Marisa Mitchell-Flack4
Marisa Mitchell-Flack4 Martin Böttcher2,5Burkhart Schraven1,5Dimitrios Mougiakakos2,5*
Martin Böttcher2,5Burkhart Schraven1,5Dimitrios Mougiakakos2,5* Sascha Kahlfuss1,5,6,7*
Sascha Kahlfuss1,5,6,7*- 1Institute of Molecular and Clinical Immunology, Medical Faculty, Otto-von-Guericke University Magdeburg, Magdeburg, Germany
- 2Department of Hematology and Oncology, University Hospital Magdeburg, Otto-von-Guericke University Magdeburg, Magdeburg, Germany
- 3Institute of Clinical Chemistry, Department of Pathobiochemistry, Medical Faculty, Otto-von-Guericke University Magdeburg, Magdeburg, Germany
- 4Department of Oncology, The Bloomberg~Kimmel Institute for Cancer Immunotherapy, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 5Health Campus Immunology, Infectiology and Inflammation (GCI), Medical Faculty, Otto-von-Guericke University Magdeburg, Magdeburg, Germany
- 6Institute of Medical Microbiology and Hospital Hygiene, Medical Faculty, Otto-von-Guericke University Magdeburg, Magdeburg, Germany
- 7Center for Health and Medical Prevention (CHaMP), Otto-von-Guericke-University, Magdeburg, Germany
One of the key challenges for successful cancer therapy is the capacity of tumors to evade immune surveillance. Tumor immune evasion can be accomplished through the induction of T cell exhaustion via the activation of various immune checkpoint molecules. The most prominent examples of immune checkpoints are PD-1 and CTLA-4. Meanwhile, several other immune checkpoint molecules have since been identified. One of these is the T cell immunoglobulin and ITIM domain (TIGIT), which was first described in 2009. Interestingly, many studies have established a synergistic reciprocity between TIGIT and PD-1. TIGIT has also been described to interfere with the energy metabolism of T cells and thereby affect adaptive anti-tumor immunity. In this context, recent studies have reported a link between TIGIT and the hypoxia-inducible factor 1-α (HIF1-α), a master transcription factor sensing hypoxia in several tissues including tumors that among others regulates the expression of metabolically relevant genes. Furthermore, distinct cancer types were shown to inhibit glucose uptake and effector function by inducing TIGIT expression in CD8+ T cells, resulting in an impaired anti-tumor immunity. In addition, TIGIT was associated with adenosine receptor signaling in T cells and the kynurenine pathway in tumor cells, both altering the tumor microenvironment and T cell-mediated immunity against tumors. Here, we review the most recent literature on the reciprocal interaction of TIGIT and T cell metabolism and specifically how TIGIT affects anti-tumor immunity. We believe understanding this interaction may pave the way for improved immunotherapy to treat cancer.
1 Introduction
1.1 Reciprocal metabolic interaction of tumor cells and T cells within the tumor microenvironment
Tumors are notorious for evading surveillance of the immune system via T cell hyporesponsiveness and dysfunction (1, 2). In particular, limited nutrient availability, in particular the scarcity of glucose (3) and tryptophan (4, 5) which are required for normal cell functionality, in the tumor microenvironment (TME) due to competition can impair CD8+ cytotoxic T cells (CTL) proliferation, survival, and effector function (4–6). In this context, tumor cells have been shown to express the enzyme indoleamine 2,3-dioxygenase (IDO), which on the one hand depletes tryptophan, a critical amino acid needed for T cell proliferation (4, 5), and on the other hand produces kynurenine, a T cell suppressive metabolic ‘waste’ product (7). It is noteworthy that the role of effector CD4+ T cells during anti-tumor immunity is not as well resolved as it is for CD8+ T cells (8). In addition, hypoxia within the TME can diminish anti-tumor activity directly by inhibiting NK cell-mediated killing (9), or by inducing T cell apoptosis through inhibition of CCR7 expression via the A2A receptor signaling pathway (10). Hypoxia has also been demonstrated to upregulate immune checkpoint proteins such as PD-L1 on tumor cells (11–13). Additionally, metabolites produced by tumor cells can promote tumor immune evasion. In this regard, adenosine, a byproduct of the enzymatic breakdown of adenosine 5’-triphosphate (ATP) via the ectonucleotidases CD39 and CD73, promotes tumor growth, survival, and metastasis and also impairs CD8+ T cell signaling and function (14–18). Furthermore, acidification of the TME through the generation of lactic acid by the tumor itself impairs respiration, chemotaxis, and cytokine production of CTLs (6, 19). Altogether, the TME is a unique metabolic niche that consists of several mechanisms to escape immune surveillance by impairing T cell metabolism and effector function.
1.2 The role of T cell and tumor cell metabolism for anti-tumor immunity
For T cells to be able to undergo essential processes such as proliferation, growth and differentiation, they need to metabolically adapt to their new requirements, a process also referred to as metabolic reprogramming (20, 21). Naïve T cells mainly make use of fatty acid oxidation, while activated T cells tend to shift from the energetically more favorable oxidative phosphorylation (OXPHOS) to the Warburg metabolism (22–24) to fulfill their need for various metabolic resources. In order to facilitate this kind of metabolic reprogramming during T cell activation, several different signaling cascades and transcription factors come into play. IL-2, a classical growth factor cytokine, and the ligation of costimulatory proteins will enable the metabolic transition to glycolysis by increasing the expression of nutrient transporters and activation of mTOR, a key metabolic regulator (25–27). Together with c-Myc, a protein that activates the transcription of metabolic genes essential for T cell activation (28), mTOR induces the increased expression of glucose transporter 1 (GLUT1) and CD98, a protein responsible for transporting amino acids into the cell (29). To summarize, it can be stated that the metabolic profile of T cells will determine their functional state.
There is increasing appreciation for the fact that a metabolic interplay between tumor and immune cells exists in the TME (30, 31). Further, there is evidence that immune checkpoint proteins themselves have an effect on T cell metabolism, reviewed comprehensively by Lim et al. (32). Kleffel et al. (33) have demonstrated that melanoma cell intrinsically expressed PD-1 upregulates the Akt/mTOR signaling pathway in cancer cells. In another study by Chang et al. (31), tumor PD-L1 expression promoted glycolysis and the activation of Akt/mTOR in tumor cells, while simultaneously suppressing the activity of mTOR in T cells by competing for glucose. The blocking of PD-L1, PD-1 and CTLA-4 resulted in altered concentrations of extracellular glucose (31). This is noteworthy as acidosis in the TME can limit the anti-tumor activity of CTL, as well as suppress their proliferation and cytokine production (34). It is plausible to assume that several immune checkpoint proteins can promote glycolysis in tumor cells, therefore creating a nutrient competitive scenario between tumor cells and immune cells within the TME.
1.3 Immune checkpoints in T cell immunity
To elicit a successful immune response against tumors, T cells need to become fully activated. This activation depends on two distinct signals. The first signal represents the engagement of the T cell receptor (TCR) by cognate peptide:MHC class I or II complexes (pMHC) presented by antigen presenting cells (APCs) (35). The second signal involves the co-stimulation via B7 proteins on APCs that interact with cluster of differentiation (CD)28 expressed on the surface of T cells (36, 37). Unchecked and/or persistent activation of T cells could lead to aberrant inflammation causing severe damage to host tissue. Because of this, it is necessary that T cell activation is closely regulated by co-stimulatory and co-inhibitory proteins, referred to as immune checkpoints (38).
In the past 10 years, the development of novel immunotherapies has been enormously successful especially within the areas of chimeric antigen receptor (CAR) T cells (39), bispecific antibodies capable of binding two targets simultaneously (40), and immune checkpoint inhibitors (ICI) (41). However, despite the enormous success of ICIs, many patients show or acquire resistance to treatment with ICIs (42). Consequently, the latter has resulted in a need for the identification of novel immune checkpoints such as lymphocyte activation gene-3 (LAG3) (43), V-domain Ig suppressor of T cell activation (VISTA) (44), B and T cell attenuator (BTLA) (45), B7 homolog 3 protein (B7-H3) (46), T cell immunoglobulin and mucin-domain containing-3 (TIM3) (47) and T cell immunoglobulin and ITIM domain (TIGIT) (48). These proteins each have distinct ligands and suppress T cell function through several mechanisms to ensure there is proper regulation of the T cell response. In the following paragraphs, we will briefly introduce several immune checkpoints by structure and function. Figure 1 details these structural differences and similarities between the different immune checkpoints.
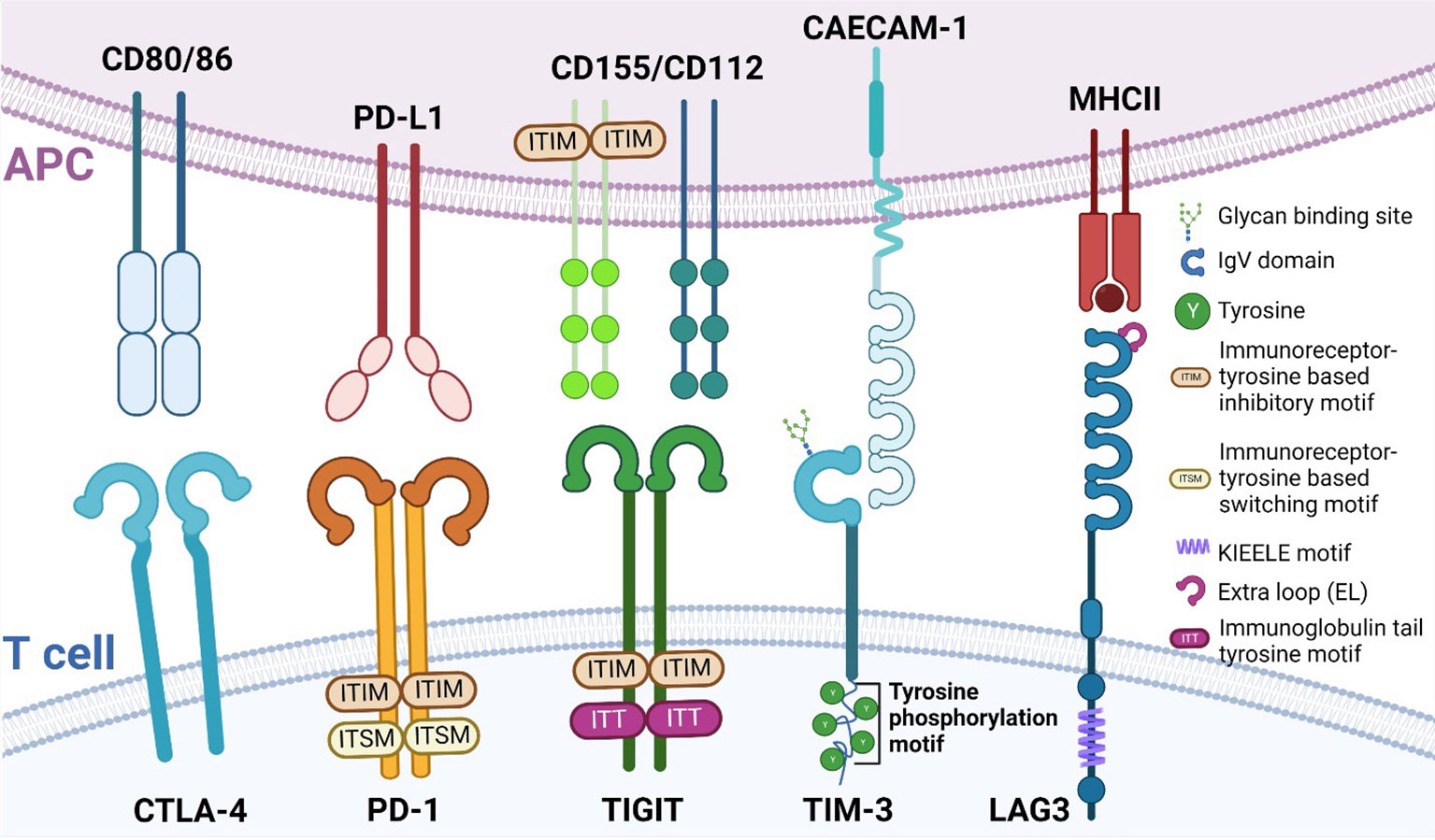
Figure 1 Structure of different immune checkpoints. Immune checkpoints and their structure expressed on T cells (bottom) and their respective ligands expressed on APCs (top). Depicted here are CTLA-4, PD-1, TIGIT, TIM-3 and LAG3. APC, antigen presenting cell; CTLA-4, Cytotoxic T lymphocyte antigen 4; PD-1, Programmed Death-1; TIGIT, T cell immunoglobulin and ITIM domain; TIM-3, T cell immunoglobulin domain and mucin domain 3; LAG3, lymphocyte activation gene-3.
1.3.1 PD-1
Programmed Death-1 (PD-1) is a type I transmembrane protein that is expressed in several immune cells, such as T, B, and NK cells. Structurally, it is composed of an extracellular immunoglobulin-like binding domain, a transmembrane region and a cytoplasmic domain containing an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-base switch motif (ITSM) (49). Engagement of PD-L1 with its receptor results in T cell dysfunction, exhaustion, and production of the immunosuppressive cytokine IL10 within the tumor (50). With the FDA approval for Nivolumab and Pembrolizumab, the potential of blocking PD-1 was realized and successfully applied to improve patient outcomes.
1.3.2 CTLA-4
Cytotoxic T lymphocyte antigen 4 (CTLA-4), also known as CD152, and CD28 are homologous receptors expressed on T cells. While structurally similar, they mediate opposing functions in T cell activation (51–54). Blockade of CTLA-4, such as with Ipilimumab (55), results in the amelioration of the immune response against tumors.
1.3.3 LAG3
LAG3 and CD4 share very similar structures in that they both have four extracellular Ig-like domains (56, 57). Interestingly, LAG3 has a 100-fold higher binding affinity with MHC class II (MHCII) compared to CD4, which is why MHCII is presumed to be the ligand for LAG3 (43) and why LAG3 may be a negative competitor of CD4 (58–62).
1.3.4 TIM3
Contrary to other immune checkpoint proteins, TIM3 does not consist of classical inhibitory signaling motifs such as ITIMS, but instead contains five conserved tyrosine residues (47). Two of these residues can be phosphorylated by Src kinases and are essential for downstream signal transduction (63, 64). Thus far, four distinct ligands, both soluble and surface-bound, have been found to interact with the IgV domain of TIM3 (phosphatidylserine (PtdSer), high-mobility group box-1 protein (HMGB1), carcinoembryonic antigen-related cell adhesion molecule 1 (CAECAM-1) and galectin-9 (Gal-9)) (65). It is noteworthy to mention that PD-1 and TIM-3 can share ligands, as is the case with Gal-9 (66). Tumor-infiltrating dendritic cells (DC) highly express TIM3, which can compete with nucleic acid binding to its ligand high-mobility group protein B1 (HMGB1), reducing anti-tumor immunity otherwise mediated by nucleic acids (67). TIM3 also works to inhibit T cells via interaction with the ligand Caecam1 (68).
1.3.5 TIGIT
T cell immunoglobulin and ITIM domain (TIGIT) was first identified in 2009 as an inhibitory immune checkpoint by Yu et al. (48). TIGIT has an extracellular immunoglobulin variable region, a transmembrane domain, as well as a cytoplasmic portion that contains an ITIM and an immunoglobulin tail tyrosine (ITT)-like phosphorylation motif (48), by which it delivers its inhibitory signals. TIGIT expression is restricted to lymphocyte and is found mainly on memory T cells and regulatory T cells (Tregs) as well as on NK cells (48, 69). Niebel et al. (70) have suggested that the expression of TIGIT mRNA is regulated via the methylation of the TIGIT gene. TIGIT binds to poliovirus receptor (PVR), also known as CD155 (71) with the highest binding affinity, as well as PVR ligand (PVRL) 2, also known as CD112 or Nectin-2 and PVRL3, also known as CD113 or Nectin-3 with lower affinity (71). Similar to CTLA-4/B7/CD28 pathway (72), TIGIT achieves its inhibitory effects by competing with other ligands such as CD266 or CD96 (73). The hypothesis that TIGIT inhibits T cell proliferation has been tested by several groups (74–76) and they reported a direct inhibitory effect.
Concerning the immunosuppressive effect of TIGIT, several mechanisms may explain its function. Among them, TIGIT signaling has been shown to inhibit NK cell degranulation and cytotoxicity (69, 77), where Stanietsky et al. (69) have demonstrated that this inhibitory effect is mediated directly via the ITIM of TIGIT. Additionally, TIGIT prevents CD226 signaling in T cells by preventing the homodimerization of the protein (78). CD226 transmits an activating signal and consequently induces the aggregation of LFA-1, an important integrin involved in T cell migration as well as cytotoxicity (79), where aggregation of integrins affects their conformation and the interaction with their ligand (80). The Treg response has also been reported to be modulated by TIGIT (78, 81). In these studies, TIGIT+ Tregs express higher levels of classical Treg genes, such as the transcription factor forkhead box P3 (FoxP3), and the surface molecules CD25 and CTLA-4. The engagement of TIGIT further leads to the secretion of IL10, a hallmark immunosuppressive cytokine, which selectively dampens T helper (Th)1 and Th17 immune responses (78). In certain types of cancer such as follicular lymphoma, TIGIT is strongly expressed by intratumoral Tregs as well as memory CD8+ T cells. Here, high numbers of TIGIT-expressing tumor infiltrating lymphocytes have been correlated with a poor survival rate (82). As such, TIGIT may in the future be used as a prognostic marker, since elevated expression in T and NK cells predicts negative clinical outcomes (83–90). Based on these findings, TIGIT has become the subject of increased research as a target for cancer therapy, especially in combination with other ICIs, such as PD-1 inhibitors (91).
We here set out to review the literature of the past 20 years on the reciprocal interaction of TIGIT and the T cell metabolism, how it affects anti-tumor immunity, and how a better understanding of this interaction can pave the way for improved immunotherapy to treat cancer.
2 Main review
2.1 Interaction of TIGIT and the metabolic TME
2.1.1 Inhibition of glucose metabolism in T cells
A recent study by Shao et al. (92) focused on the role of TIGIT in patients with colorectal cancer and revealed that upregulated TIGIT expression in CD3+ T cells correlated with poor survival. In this study the authors found that T cells expressing TIGIT had impaired proliferation, cytokine production, glucose uptake, and glycolytic function. Investigations by He et al. (86) demonstrated that TIGIT+ CD8+ T cells are impaired in their effector function, allowing for the hypothesis that immune escape in gastric cancer is at least in part mediated by the upregulation of TIGIT. These TIGIT+ CD8+ T cells had significantly reduced expression of glycolysis genes, including GLUT1 as well as (hexokinase) HK1 and HK2, which resulted in impaired glucose uptake and glycolysis (Figure 2). Aside from cancer, another study by Calvet-Mirabent et al. (93) has shown the relevance of the connection between glucose metabolism and TIGIT as an immune checkpoint in HIV infection. In their study, the authors utilized dual blockade of PD-1 and TIGIT, as well as employed the pro-glycolytic drug Metformin, and investigated the functional properties of CD8+ T cells from HIV-1 patients. Significant positive correlations were observed between the increase in maximum glycolytic activity after TCR activation and the percentages of single-positive TIGIT cells, while co-expression of PD-1 and TIGIT resulted in lower glycolysis rates. Further, treatment with Metformin together with dual blockade of the two checkpoints restored cytotoxic activity of CD8+ T cells (93). Thus, TIGIT seems to be capable to alter T cell function via the inhibition of glycolysis.
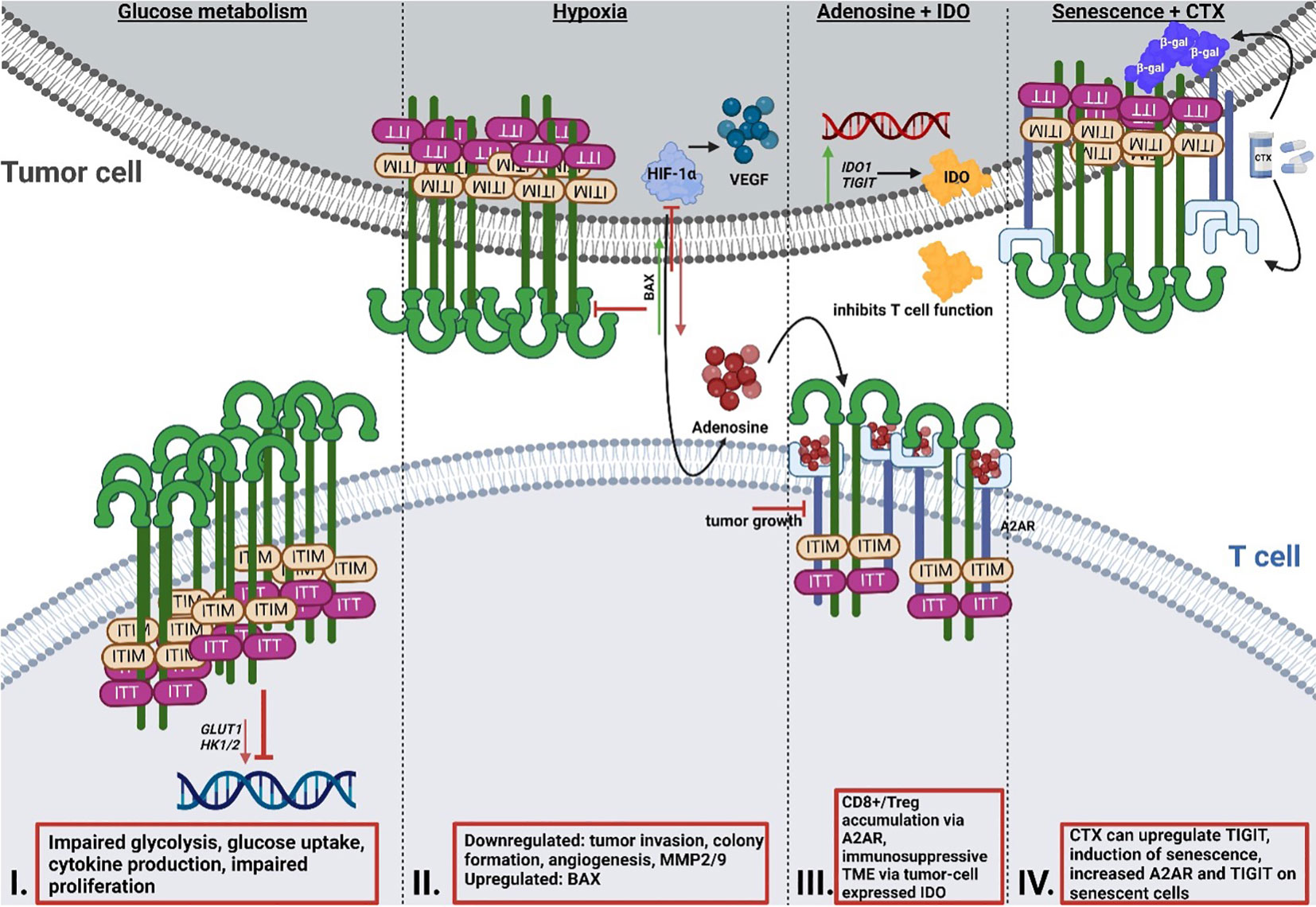
Figure 2 Reciprocal interaction of TIGIT signaling and T cell metabolism. I: Effect of TIGIT on glucose metabolism. Cancer cells inhibit T cell metabolism via enhancing the upregulation of TIGIT, resulting in impaired glycolysis gene expression of GLUT1 and HK1/2, glucose uptake and glycolysis, and reduced proliferation. II: Effect of TIGIT on hypoxia and hypoxia sensing. HIF1-α regulates the expression of immune checkpoints and the expression of VEGF, which mediates tumor neovascularization. Simultaneous blocking of HIF1-α and TIGIT results in reduced tumor invasion and colony formation, as well as impaired angiogenesis and reduced MMP2/9 expression. Dual blockade leads to induction of pro-apoptotic BAX. III: Interaction of TIGIT and adenosine signaling and IDO. A2AR regulates the accumulation of CD8+ T cells and Tregs. Altered metabolism and hypoxia result in increased adenosine in the TME. Deletion of A2AR leads to tumor rejection in mice. IDO is highly expressed by tumor cells and generates an immunosuppressive TME. Many cancer cells overexpress IDO1 and TIGIT simultaneously. IV: Interaction of chemotherapy and senescence and TIGIT. Chemotherapy regimens can result in the upregulation of TIGIT. A2AR is increased on the surface of senescent cells, with simultaneous upregulation of TIGIT. TIGIT, T cell immunoglobulin and ITIM domain; HK, hexokinase; GLUT1, glucose transporter 1; HIF1-α; hypoxia-inducible factor 1 alpha; MMP, matrix metalloproteinase; BAX, Bcl-2-associated X protein; IDO, indoleamine-pyrrole 2,3-dioxygenase; TME, tumor microenvironment.
2.1.2 Hypoxia
Hypoxia is widely accepted to be a critical mechanism responsible for the resistance of tumor cells to radio-, chemo-, and immunotherapy (94–97). As the volume of a tumor increases, increasing numbers of cells need to be supplied with blood and oxygen, which requires additional vascularization of the tumor tissue. Without this additional supply of blood and oxygen, a state of hypoxia sets in (98). It is well established that the transcription factor hypoxia-inducible factor 1α (HIF-1α) regulates the expression of immune checkpoint proteins such as PD-L1 and CD73 (99, 100). HIF-1α is a master regulator of the cell’s response to hypoxia (101). Under normoxic conditions, the activity of HIF-1α is repressed by proteasomal degradation via the oxygen-dependent prolyl hydroxylase domain (PHD) and the von Hippel-Lindau (VHL) protein (102). During tumor development, HIF-1α is pivotal to the cells’ metabolic adaptation to their surroundings, as growth success under metabolic duress strongly depends upon the cell’s ability to shift from oxidative phosphorylation (OXPHOS) to the more inefficient glycolytic metabolism for ATP generation. This is accomplished by HIF-1α-regulated genes encoding enzymes for glycolysis, such as the glucose transporters GLUT1 and GLUT3, HK1, and HK2 as well as phosphoglycerate kinase 1 (PGK1) (103). HIF-1α further regulates the expression of vascular endothelial growth factor (VEGF) (104), which enables neovascularization of tumors.
So far, one study has recently addressed the synergy between TIGIT and HIF-1α (105). In this study, Fathi et al. demonstrated that simultaneous blocking of both TIGIT and HIF-1α results in a significant reduction of tumor cell invasion, decreased colony formation, and inhibited angiogenesis (105). Both matrix metalloproteinases (MMP) 2 and MMP9 as well as VEGF mRNA expression levels were decreased under the dual blockade. Additionally, expression of the anti-apoptotic protein B-cell lymphoma (BCL)2 was downregulated, whereas mRNA expression of the pro-apoptotic protein Bcl-2-associated X protein (BAX) was upregulated. What remains unclear is if and how, precisely, these two proteins interact with one another. Since a correlation between TIGIT and HIF1a was demonstrated by Fathi et al., further research is required to unravel the precise mechanisms of relation of the two proteins in T cells, especially when considering that HIF1a increases the expression of other immune checkpoints such as PD-L1 (11–13).
2.1.3 Adenosine
Originally, adenosine receptors (ARs) were categorized into A1 or A2 ARs, depending on whether they have an inhibitory or stimulatory effect on cyclic adenosine monophosphate (cAMP) in the brain (106). Currently, ARs are categorized into four subtypes, A1, A2A, A2B and A3 (107). The majority of A2ARs are distributed in organs of the respiratory system, heart and lung, as well as in the central nervous system (CNS), and the immune system (108, 109). The adenosine receptor A2A (ADORA2) plays an important role in protecting tissues from immune-mediated damage following noninfectious inflammation, as well as in regulating the accumulation of CD8+ T cells and NK cells (110, 111). An altered metabolism, increased expression of CD73 as well as hypoxia (112) in the tumor can lead to higher adenosine levels in the TME (111, 113) via signaling through the A2A adenosine receptors (114). In this context, Ohta et al. (111) investigated the effect of A2A receptor deficiency on anti-tumor immunity mediated by CD8+ T cells and observed that genetic deletion of the A2A receptor results in tumor rejection in mice. Additionally, A2A receptor antagonists considerably delayed tumor growth via anti-tumor CD8+ T cells. Ohta et al. (115) have shown that immunosuppressive Tregs were induced by increased levels of extracellular adenosine, as mediated via A2AR stimulation. As of yet, only very few studies (116, 117) have investigated in detail the correlation between the A2A receptor and TIGIT so far. Brauneck et al. (116) investigated the correlation between the A2A receptor and TIGIT on NK cells and showed that NK-cell mediated killing of acute myeloid leukemia (AML) cells could be ameliorated by co-blockade of TIGIT and A2AR, or of TIGIT and CD39, indicating a link between the two proteins. Another study by Muhammad et al. (117) revealed that the stimulation of the A2A receptor is necessary for the emergence of TIGIT-positive Tregs in mice and that this axis is impaired in uveitis patients. This study appears to have identified a subset of TIGIT+ Tregs that are functionally dependent on the expression of the A2A receptor.
2.1.4 IDO
IDO1 plays a pivotal role in the conversion of tryptophan to kynurenine (118). IDO1 is highly expressed in tumor cells and contributes to the establishment of a local immunosuppressive TME by enabling immune tolerance (119). It has been demonstrated that IDO1 inhibition induced a robust anti-tumor immune response in a mouse model when employed both as a single agent (120–127), or in combination with chemotherapeutic drugs (121, 128), highlighting the potential of IDO1 as a therapeutic target.
A recent study by Robertson et al. (129) has shown that CD8+ T cell tumor infiltrates from uveal melanoma (UM) overexpress the genes encoding for both IDO1 and TIGIT. As previously mentioned, IDO is known to limit T cell function and induce mechanisms of tolerance (130, 131). Stålhammer et al. (83) have demonstrated that not only the number of IDO+ cells in tumor tissues of UM appear higher than in normal choroid tissues, but that the same is true for TIGIT+ cells. Importantly, the number of IDO+ cells correlated with the number of TIGIT+ cells in tumor cores and full tumor sections (83). The association of TIGIT expression with IDO and PD-L1 has also been observed in the tumor core of glioblastoma (GBM) (132), underlining the necessity to further study the correlation between these proteins.
2.1.5 Chemotherapy and senescence
TIGIT has recently been described as a marker for senescence due to its higher expression in aged T cells (133). The blocking of TIGIT results in improved functional capacity of senescent T cells as demonstrated by Song et al. (133), Chew et al. (134) and Kong et al. (84). The latter study also demonstrated that TIGIT expression on CD8+ T cells is not only elevated in acute myeloid leukemia (AML) patients, but that high TIGIT levels also correlate with primary refractory disease, as well as leukemia relapse following allogenic stem cell transplantation. TIGIT-high CD8+ T cells presented as functionally impaired and exhausted, whereas TIGIT blockade rescued functionality and anti-tumor response, highlighting TIGIT blockade as a potential therapeutic approach for leukemia.
Cancer treatment options in terms of chemotherapy are varied and often rely on combinatorial therapies. Some common agents used for different types of cancer are 5-Fluorouracil, an antimetabolite, DNA intercalators such as oxaliplatin and taxanes that target microtubules (135). A recent study by Davern et al. (136) revealed certain chemotherapy regimens give rise to an immune-resistant phenotype via the upregulation of inhibitory immune checkpoint ligands, among them TIGIT, in oesophageal adenocarcinoma (OAC). The study aimed to elucidate the effect of OAC chemotherapy approaches on the induction of a senescent-like state in cancer cells as senescent cancer cells are involved in conferring treatment resistance and promoting a microenvironment conducive to tumor growth via secretion of several pro-inflammatory markers, referred to as senescence-associated phenotype (SASP) (136).Using ß-galactosidase (ß-gal), an enzyme involved in the process of producing galactosylated proteins, as a marker for senescence, the authors demonstrated that the number of senescent-like cells increased significantly following chemotherapy, prompting the question whether immune checkpoints were expressed on these senescent cells or even upregulated following the treatment. The immune checkpoint TIM-3 was significantly upregulated in OE33 cells, whereas TIGIT was significantly upregulated in the SK-GT-4 cells. We know that immune checkpoints are essential for immune evasion, and if these immune checkpoints are present on senescent OAC cells, this may represent a drugable target for future therapies. Returning to another protein already addressed in this review, the adenosine receptor A2A was significantly increased on the surface of senescent-like SK-GT-4 cells, which were also shown to have increased TIGIT expression following a chemotherapy regimen. While senescent cells do have an activated glucose metabolism, they at the same time display an unbalanced lipid metabolism, which results in an altered expression of lipid metabolic enzymes, ultimately culminating in senescence induction and thereby limited functionality (137). Senescent T cells also demonstrate loss of cell surface CD28 (138–140), a protein required for lipid raft formation, IL-2 gene transcription and T cell activation. Since CD28 has also been linked to metabolic fitness of a T cell (141), the loss of this protein due to senescence can dramatically affect T cell functionality (142). Liu et al. (137) have demonstrated that the prevention of T cell senescence resulted in enhanced anti-tumor immunity, therefore maybe providing another point of potential therapeutic application.
Interestingly, TIGIT has also been shown to be intrinsically expressed in murine colorectal cell lines (143). To elucidate the functional effect of this intrinsic TIGIT, Zhou et al. (143) deleted the protein using CRISPR/Cas9 and observed that knockout resulted in significantly impaired tumor growth, together with increased IFNy secretion and cytotoxicity by NK cells, indicating that tumor cell-intrinsic TIGIT has a considerable effect on tumor growth and may present a potential therapeutic target.
2.2 Current status of anti-TIGIT therapeutics in clinical studies
As of August 2021, several anti-TIGIT antibodies were registered in preclinical and active clinical trials (clinicaltrials.org, anti-TIGIT). For example, two antibodies had progressed to the Phase III status (Tiragolumab (144), Ociperlimab (145)) and two were active in Phase II trials Vibostolimab, Domvanalimab) (146, 147), all of which also in combination with Atelizumab (anti-PD-L1), Pembrolizumab (anti-PD-1) and other agents. Additionally, a bispecific antibody targeting both PD-1 and TIGIT (HLX301, NCT05102214) simultaneously is under current clinical review. As discussed, TIGIT expression has been observed, among others, with PD-L1 in the tumor core (132), hinting at some kind of link between these two proteins. Currently, an anti-TIGIT candidate in combination with an anti-PD-1 antibody is being evaluated for the application for recurrent glioblastoma (148). Furthermore, increased levels of extracellular adenosine, as mediated by A2AR stimulation (114), have been shown to have a detrimental effect on anti-tumor activity (111, 115–117). Etrumadenant, an A2AR antagonist, is currently being investigated in a clinical trial in combination with Domvanalimab and Zimbrelimab (anti-PD-1) (149). It is noteworthy that the majority of the anti-TIGIT antibodies in clinical trials currently are fully human and demonstrate good tolerance by patients, also in combination with anti-PD-1 and anti-PD-L1 antibodies (150). As previously discussed in this review, TIGIT monotherapy does not result in significantly altered disease outcomes, underlining this as a potential caveat of TIGIT as a therapeutic target and highlighting the necessity for a combinatorial approach with other agents. Immune checkpoint therapy using Ipilimumab and Nivolumab as the most prominent agents has proved successful, and, taken together with the low efficacy of anti-TIGIT monotherapy, prompts the question which cohort of patients could additionally benefit from either a monotherapy or a combinatorial treatment.
2.3 The potential of PD-1, CTLA-4 and other negative regulators as biomarkers
Predictive biomarkers are essential to evaluate the outcome of therapeutic approaches, or at least, to provide an indication before commencement of the therapy regimen. Especially in the case of highly multifactorial diseases such as cancer and autoimmunity, such biomarkers should ideally indicate whether a monotherapy or a combinatorial approach is necessary. Here, the induction of negative regulators results in the suppression of, among other mediators, cell death mechanisms (151). Specifically these negative regulators of cell death signaling, such as heat shock proteins (HSP) (152), the Bcl-2 family (153), the PI3K/Akt/mTOR pathway (154) and others, as reviewed in detail by Razaghi et al. in (155), have found clinical application as prognostic biomarkers. In summary, negative regulators of cell death signaling appear to have great potential and present clinical application as prognostic biomarkers, raising the question whether this is also the case for the immune checkpoint proteins. When considering anti-PD-1 or anti-PD-L1 therapy, using (over-)expression of PD-L1 as biomarker appears plausible. In this context, Teng et al. (156) came up with a classification that describes PD-L1 positive tumors with infiltrating lymphocytes as a type 1 TME, proposing it to be the most likely to respond to immune checkpoint blockade. However, also PD-L1 negative tumors have been shown to be able to respond positively to antibodies targeting the PD-1/PD-L1 axis (157, 158). This consequently raises the concern that the predictive value of PD-1 and PD-L1 as biomarkers may not be optimal and universally valid across all patients, as intrapatient and even intratumor heterogeneity has been observed (159).
Other studies have hinted at the possible prognostic power of CLTA-4 expression. Here, Liu et al. (160) have demonstrated that, in some cancers, patients with higher CTLA-4 expression had a shorter overall survival than those with lower expression. However, an association between the expression levels of PD-1 and CTLA-4 and tumor-infiltrating cells exists (160). Liu et al. point out that the expression of these two immune checkpoint proteins varies across different cancers and that many cancer types demonstrate PD-1 and CTLA-4 mutations, leading to their abnormal expression, which may be used as a prognostic biomarker.
Whether TIGIT can be used in a similar manner remains to be investigated and demonstrated. Since TIGIT in its effects appears to be functionally and mechanistically tethered to other negative immune regulators such as PD-1, TIGIT alone may not prove a reliable and unambiguous prognostic biomarker. To assess the protein’s capacity of serving as a prognostic factor, large amounts of correlation data from different kinds of cancers, across different genders, ages and perhaps even ethnicities are necessary, providing information on its function and mechanistics on its own and together with other proteins that TIGIT is known to interact with. It may well be possible that a combination of factors, such as presence of PD-1, TIGIT and senescence markers will be able to form a prognostic unit of response to and success of immunotherapy in different cancers.
3 Discussion
While the exact role of TIGIT within the TME is still not fully elucidated, the apparent synergy between TIGIT and HIF-1α as well as PD-1 (161) does allow for the assumption that this protein does not simply have a redundant role. Based upon the literature reviewed here, blockade or targeting of TIGIT alone does not appear to have a major effect on either the progression or even curative approaches in different oncologic diseases. It is rather the combination of TIGIT blockade together with blocking of another checkpoint, such as PD-1. The fact that a synergy exists between the two is well documented and accepted to the point that several clinical trials aiming to block both proteins simultaneously are currently ongoing (162). The challenge of such a therapy, even if successful, lies in the fact that not all cancers are PD-L1 positive, thereby restricting the potential applications from the beginning. Another potential caveat is that the precise mechanism of the synergistic effects observed between the two checkpoint proteins is not fully understood, and as such it may prove difficult to design effective and individualized therapies without fully understanding the mechanistic foundations of the observed effects.
In terms of metabolism, it can be hypothesized that presence or overexpression of TIGIT poses a metabolic barrier to T cell function. Data by Gilmour et al. (163) suggest that the co-expression of TIGIT with VISTA may lead to an altered metabolic phenotype of CTL. It was been detailed in the introductory section of this review that several other immune checkpoint proteins, such as
PD-1 and CTLA-4 appear to have an effect on glycolysis of tumor cells, and thereby on the ability of immune cells to perform glycolysis due to nutrient competition within the TME. Limited nutrient ability, such as the scarcity of glucose, will lead to impaired T cell function and therefore an impaired anti-tumor response of those T cells. It is therefore crucial to further investigate the potential direct and indirect effects of TIGIT on the metabolism of T cells and other immune cells in the context of anti-tumor immunity.
It is well-known that hypoxia plays a major role in creating hostile microenvironments that are toxic to immune cells yet conducive to tumor growth. So far, only one study has investigated the direct interaction between HIF-1α and TIGIT. It remains an open question whether a potential three-way synergy might exist between blocking not only TIGIT and PD-1, but also HIF-1α. Along this line, it would be important to assess whether a co-blockade of TIGIT and HIF-1α is as effective as the blockade of TIGIT and PD-1 as a therapeutic possibility for those cancers which are not PD-L1 positive.
The interplay between TIGIT and adenosine as well as the A2A receptor makes for another interesting point of further investigation. The genetic deletion of the A2A receptor in mice resulted in tumor rejection (162), allowing for the hypothesis that some connection may also exist between these proteins. Additionally, it is known that hypoxia leads to higher adenosine levels in the TME, prompting the question whether the TIGIT-A2AR-HIF-1α axis could provide another possible three-way blockade for therapeutic purposes. The A2A receptor was additionally observed to be upregulated on the surface senescent cancer cells, which at the same time showed increased TIGIT expression following some chemotherapy regimens.
The potential of TIGIT expression as a biomarker has been suggested, although for this, larger association studies are needed. Future experiments should aim to elucidate the connection between TIGIT and other immune checkpoints, particularly those involved in the immune response against cancers which do not express PD-L1, as well as the interplay with HIF-1α and the A2A receptor. Perhaps this will lead to a better understanding of the exact mechanisms governing the synergistic inhibitory effects of combination treatments. Taken together, TIGIT appears to have a therapeutic potential, especially in the context of combinatorial therapies and alleviating the metabolic barrier that immune checkpoint proteins are able to pose, that should not be overlooked and disregarded for further research, both of basic and translational nature.
Author contributions
NJ-N and SK designed the study. NJ-N performed literature search. NJ-N and SK drafted the manuscript. NJ-N designed Figures 1 and 2. RB-L, KB, MM-F, MB, BS and DM provided critical input throughout the work and corrected the manuscript. SK and BS together with DM supervised the work. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants of the state of Saxony-Anhalt (SI2 and SI3) to BS and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 361210922/GRK2408 – (Project 12) to SK. For the latter, a monthly academic individual plan (toc2FzY2hhLmthaGxmdXNzQG1lZC5vdmd1LmRl) was purchased.
Acknowledgments
Figures 1 and 2 were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T Cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol (2013) 25:214–21. doi: 10.1016/j.coi.2012.12.003
3. Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol (2015) 6:1. doi: 10.3389/fimmu.2015.00001
4. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol (2013) 34:137–43. doi: 10.1016/j.it.2012.10.001
5. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189:1363–72. doi: 10.1084/jem.189.9.1363
6. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
7. Siska PJ, Jiao J, Matos C, Singer K., Berger RS, Dettmer K, et al. Kynurenine induces T cell fat catabolism and has limited suppressive effects in vivo. EBioMedicine (2021) 74:103734. doi: 10.1016/j.ebiom.2021.103734
8. Schreiber S, Hammers CM, Kaasch AJ, Schraven B, Dudeck A, Kahlfuss S. Metabolic interdependency of Th2 cell-mediated type 2 immunity and the tumor microenvironment. Front Immunol (2021) 12:632581. doi: 10.3389/fimmu.2021.632581
9. Sarkar S, Germeraad WT, Rouschop KM, Steeghs EM, van Gelder M, Bos GM, et al. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PloS One (2013) 8:e64835. doi: 10.1371/journal.pone.0064835
10. Sun J, Zhang Y, Yang M, Zhang Y, Xie Q, Li Z, et al. Hypoxia induces T-cell apoptosis by inhibiting chemokine c receptor 7 expression: the role of adenosine receptor A(2). Cell Mol Immunol (2010) 7:77–82. doi: 10.1038/cmi.2009.105
11. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res (2014) 74:665–74. doi: 10.1158/0008-5472.CAN-13-0992
12. Palsson-McDermott EM, Dyck L, Zaslona Z, Menon D, McGettrick AF, Mills KHG, et al. Pyruvate kinase M2 is required for the expression of the immune checkpoint PD-L1 in immune cells and tumors. Front Immunol (2017) 8:1300. doi: 10.3389/fimmu.2017.01300
13. Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest. (2018) 128:5137–49. doi: 10.1172/JCI96268
14. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev (2017) 276:121–44. doi: 10.1111/imr.12528
15. d'Almeida SM, Kauffenstein G, Roy C, Basset L, Papargyris L, Henrion D, et al. The ecto-ATPDase CD39 is involved in the acquisition of the immunoregulatory phenotype by m-CSF-macrophages and ovarian cancer tumor-associated macrophages: Regulatory role of IL-27. Oncoimmunology (2016) 5:e1178025. doi: 10.1080/2162402X.2016.1178025
16. Gourdin N, Bossennec M, Rodriguez C, Vigano S, Machon C, Jandus C, et al. Autocrine adenosine regulates tumor polyfunctional CD73(+)CD4(+) effector T cells devoid of immune checkpoints. Cancer Res (2018) 78:3604–18. doi: 10.1158/0008-5472.CAN-17-2405
17. Li L, Wang L, Li J, Fan Z, Yang L, Zhang Z, et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res (2018) 78:1779–91. doi: 10.1158/0008-5472.CAN-17-2460
18. Linnemann C, Schildberg FA, Schurich A, Diehl L, Hegenbarth SI, Endl E, et al. Adenosine regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology (2009) 128(1 Suppl):e728–37. doi: 10.1111/j.1365-2567.2009.03075.x
19. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res (2012) 72:2746–56. doi: 10.1158/0008-5472.CAN-11-1272
20. Peng HY, Lucavs J, Ballard D, Das JK, Kumar A, Wang L, et al. Metabolic reprogramming and reactive oxygen species in T cell immunity. Front Immunol (2021) 12:652687. doi: 10.3389/fimmu.2021.652687
21. Balyan R, Gautam N, Gascoigne NRJ. The ups and downs of metabolism during the lifespan of a T cell. Int J Mol Sci (2020) 21(21):7972. doi: 10.3390/ijms21217972
22. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol (2013) 14:1064–72. doi: 10.1038/ni.2687
23. Cham CM, Gajewski TF. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol (2005) 174:4670–7. doi: 10.4049/jimmunol.174.8.4670
24. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
25. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi: 10.1016/s1074-7613(02)00323-0
26. Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity (2007) 27(2):173–8. doi: 10.1016/j.immuni.2007.07.008
27. Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell (2007) 18(4):1437–46. doi: 10.1091/mbc.e06-07-0593
28. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35(6):871–82. doi: 10.1016/j.immuni.2011.09.021
29. Geltink RIK, Kyle RL, Pearce EL. Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol (2018) 36:461–88. doi: 10.1146/annurev-immunol-042617-053019
30. Ghesquière B, Wong BW, Kuchnio A, Carmeliet P. Metabolism of stromal and immune cells in health and disease. Nature (2014) 511(7508):167–76. doi: 10.1038/nature13312
31. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
32. Lim S, Phillips JB, Madeira da Silva L, Zhou M, Fodstad O, Owen LB, et al. Interplay between immune checkpoint proteins and cellular metabolism. Cancer Res (2017) 77(6):1245–9. doi: 10.1158/0008-5472.CAN-16-1647
33. Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell (2015) 162(6):1242–56. doi: 10.1016/j.cell.2015.08.052
34. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
35. Sharpe AH. Mechanisms of costimulation. Immunol Rev (2009) 229:5–11. doi: 10.1111/j.1600-065X.2009.00784.x
36. Bour-Jordan H, Bluestone JA. CD28 function: A balance of costimulatory and regulatory signals. J Clin Immunol (2002) 22(1):1–7. doi: 10.1023/a:1014256417651
37. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: From mechanism to therapy. Immunity (2016) 44(5):973–88. doi: 10.1016/j.immuni.2016.04.020
38. Chen L. Co-Inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol (2004) 4:336–47. doi: 10.1038/nri1349
39. Yu S, Li A, Liu Q, Li T, Yuan X, Han X, et al. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J Hematol Oncol (2017) 10:78. doi: 10.1186/s13045-017-0444-9
40. Yu S, Li A, Liu Q, Yuan X, Xu H, Jiao D, et al. Recent advances of bispecific antibodies in solid tumors. J Hematol Oncol (2017) 10:155. doi: 10.1186/s13045-017-0522-z
41. Ballas ZK. The 2018 Nobel prize in physiology or medicine: an exemplar of bench to bedside in immunology. J Allergy Clin Immunol (2018) 142:1752–3. doi: 10.1016/j.jaci.2018.10.021
42. Yi M, Yu S, Qin S, Liu Q, Xu H, Zhao W, et al. Gut microbiome modulates efficacy of immune checkpoint inhibitors. J Hematol Oncol (2018) 11:47. doi: 10.1186/s13045-018-0592-6
43. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev (2017) 276:80–96. doi: 10.1111/imr.12519
44. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208:577–92. doi: 10.1084/jem.20100619
45. Yu X, Zheng Y, Mao R, Su Z, Zhang J. BTLA/HVEM signaling: milestones in research and role in chronic hepatitis b virus infection. Front Immunol (2019) 10:617. doi: 10.3389/fimmu.2019.00617
46. Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: A costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol (2001) 2:269–74. doi: 10.1038/85339
47. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415:536–41. doi: 10.1038/415536a
48. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10:48–57. doi: 10.1038/ni.1674
49. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol (2007) 8:239–45. doi: 10.1038/ni1443
50. Sun Z, Fourcade J, Pagliano O, Chauvin JM, Sander C, Kirkwood JM. IL10 and PD-1 cooperate to limit the activity of tumor-specific CD8+ T cells. Cancer Res (2015) 75:1635–44. doi: 10.1158/0008-5472.CAN-14-3016
51. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: A moving target in immunotherapy. Blood (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
52. Schwartz JC, Zhang X, Fedorov AA, Nathenson SG, Almo SC. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature (2001) 410(6828):604–8. doi: 10.1038/35069112
53. Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, et al. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature (2001) 410(6828):608–11. doi: 10.1038/35069118
54. Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, et al. The interaction properties of costimulatory molecules revisited. Immunity (2002) 17(2):201–10. doi: 10.1016/S1074-7613(02)00362-X
55. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. (2015) 112(19):6140–5. doi: 10.1073/pnas.1417320112
56. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med (1990) 171:1393–405. doi: 10.1084/jem.171.5.1393
57. Mastrangeli R, Micangeli E, Donini S. Cloning of murine LAG-3 by magnetic bead-bound homologous probes and PCR (gene-capture PCR). Anal Biochem (1996) 241:93–102. doi: 10.1006/abio.1996.0382
58. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell (2019) 176:334–347.e12. doi: 10.1016/j.cell.2018.11.010
59. Huard B, Gaulard P, Faure F, Hercend T, Triebel F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics (1994) 39:213–7. doi: 10.1007/BF00241263
60. Workman CJ, Rice DS, Dugger KJ, Kurschner C, Vignali DAA. Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur J Immunol (2002) 32:2255–63. doi: 10.1002/1521-4141(200208)32:8<2255::AID-IMMU2255>3.0.CO;2-A
61. Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol (1995) 25:2718–21. doi: 10.1002/eji.1830250949
62. Workman C, Vignali D. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol (2003) 33:970–9. doi: 10.1002/eji.200323382
63. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev (2017) 276:97–111. doi: 10.1111/imr.12520
64. van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophys Res Commun (2006) 351:571–6. doi: 10.1016/j.bbrc.2006.10.079
65. Liu F, Liu Y, Chen Z. Tim-3 expression and its role in hepatocellular carcinoma. J Hematol Oncol (2018) 11:126. doi: 10.1186/s13045-018-0667-4
66. Yang R, Sun L, Li CF, Wang YH, Yao J, Li H, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun (2021) 12(1):832. doi: 10.1038/s41467-021-21099-2
67. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13:832–42. doi: 10.1038/ni.2376
68. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517:386–90. doi: 10.1038/nature13848
69. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A. (2009) 106:17858–63. doi: 10.1073/pnas.0903474106
70. Niebel D, Fröhlich A, Zarbl R, Fietz S, de Vos L, Vogt TJ, et al. DNA Methylation regulates TIGIT expression within the melanoma microenvironment, is prognostic for overall survival, and predicts progression-free survival in patients treated with anti-PD-1 immunotherapy. Clin Epigenetics. (2022) 14(1):50. doi: 10.1186/s13148-022-01270-2
71. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT Co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 17:989–1004. doi: 10.1016/j.immuni.2016.05.001
72. Le Mercier I, Lines JL, Noelle RJ. Beyond CTLA-4 and PD-1, the generation z of negative checkpoint regulators. Front Immunol (2015) 6:418. doi: 10.3389/fimmu.2015.00418
73. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol Rev (2017) 276:112–20. doi: 10.1111/imr.12518
74. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol (2011) 41:902–15. doi: 10.1002/eji.201041136
75. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol (2012) 188:3869–75. doi: 10.4049/jimmunol.1103627
76. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol (2011) 186:1338–42. doi: 10.4049/jimmunol.1003081
77. Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R, et al. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur J Immunol (2013) 43:2138–50. doi: 10.1002/eji.201243072
78. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
79. Shibuya K, Lanier LL, Phillips JH, Ochs HD, Shimizu K, Nakayama E, et al. Physical and functional association of LFA-1 with DNAM-1 adhesion molecule. Immunity (1999) 11:615–23. doi: 10.1016/S1074-7613(00)80136-3
80. Reyes R, Monjas A, Yánez-Mó M, Cardeñes B, Morlino G, Gilsanz A, et al. Different states of integrin LFA-1 aggregation are controlled through its association with tetraspanin CD9. Biochim Biophys Acta (2015) 1853(10 Pt A):2464–80. doi: 10.1016/j.bbamcr.2015.05.018
81. Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, et al. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol (2015) 195:145–55. doi: 10.4049/jimmunol.1402381
82. Yang ZZ, Kim HJ, Wu H, Jalali S, Tang X, Krull J, et al. TIGIT expression is associated with T-cell suppression and exhaustion and predicts clinical outcome and anti-PD-1 response in follicular lymphoma. Clin Cancer Res (2020) 26(19):5217–31. doi: 10.1158/1078-0432.CCR-20-0558
83. Stålhammar G, Seregard S, Grossniklaus HE. Expression of immune checkpoint receptors indoleamine 2,3-dioxygenase and T cell ig and ITIM domain in metastatic versus nonmetastatic choroidal melanoma. Cancer Med (2019) 8(6):2784–92. doi: 10.1002/cam4.2167
84. Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC, et al. T-Cell immunoglobulin and ITIM domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin Cancer Res (2016) 22:3057–66. doi: 10.1158/1078-0432.CCR-15-2626
85. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Investig (2015) 125:2046–58. doi: 10.1172/JCI80445
86. He W, Zhang H, Han F, Chen X, Lin R, Wang W, et al. CD155T/TIGIT signaling regulates CD8(+) T-cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res (2017) 77:6375–88. doi: 10.1158/0008-5472.CAN-17-0381
87. Lucca LE, Lerner BA, Park C, DeBartolo D, Harnett B, Kumar VP, et al. Differential expression of the T-cell inhibitor TIGIT in glioblastoma and MS. Neurol Neuroimmunol. Neuroinflamm. (2020) 7. doi: 10.1212/NXI.0000000000000712
88. Ostroumov D, Duong S, Wingerath J, Woller N, Manns MP, Timrott K, et al. Transcriptome profiling identifies TIGIT as a marker of T cell exhaustion in liver cancer. Hepatology (2021) 73(4):1399–418. doi: 10.1002/hep.31466
89. Xu D, Zhao E, Zhu C, Zhao W, Wang C, Zhang Z, et al. TIGIT and PD-1 may serve as potential prognostic biomarkers for gastric cancer. Immunobiology (2020) 225:151915. doi: 10.1016/j.imbio.2020.151915
90. Duan X, Liu J, Cui J, Ma B, Zhou Q, Yang X, et al. Expression of TIGIT/CD155 and correlations with clinical pathological features in human hepatocellular carcinoma. Mol Med Rep (2019) 20:3773–81. doi: 10.3892/mmr.2019.10641
91. Solomon BL, Garrido-Laguna I. TIGIT: A novel immunotherapy target moving from bench to bedside. Cancer Immunol Immunother. (2018) 67:1659–67. doi: 10.1007/s00262-018-2246-5
92. Shao Q, Wang L, Yuan M, Jin X, Chen Z, Wu C. TIGIT induces (CD3+) T cell dysfunction in colorectal cancer by inhibiting glucose metabolism. Front Immunol (2021) 12:688961. doi: 10.3389/fimmu.2021.688961
93. Calvet-Mirabent M, Sánchez-Cerrillo I, Martín-Cófreces N, Martínez-Fleta P, de la Fuente H, Tsukalov I, et al. Antiretroviral therapy duration and immunometabolic state determine efficacy of ex vivo dendritic cell-based treatment restoring functional HIV-specific CD8+ T cells in people living with HIV. EBioMedicine (2022) 81:104090. doi: 10.1016/j.ebiom.2022.104090
94. Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer (2002) 2:38–47. doi: 10.1038/nrc704
95. Shannon AM, Bouchier-Hayes DJ, Condron CM, Toomey D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat Rev (2003) 29:297–307. doi: 10.1016/S0305-7372(03)00003-3
96. Daniel SK, Sullivan KM, Labadie KP, Pillarisetty VG. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma, clin. Transl Med (2019) 8:10. doi: 10.1186/s40169-019-0226-9
97. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. (2013) 123(9):3664–71. doi: 10.1172/JCI67230
98. Li Y, Zhao L, Li XF. Hypoxia and the tumor microenvironment. Technol Cancer Res Treat (2021) 20. doi: 10.1177/15330338211036304
99. Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, et al. Blocking triggering receptor expressed on myeloid cells-1-positive tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-programmed cell death ligand 1 resistance in liver cancer. Hepatology (2019) 70(1):198–214. doi: 10.1002/hep.30593
100. Lequeux A, Noman MZ, Xiao M, Sauvage D, Van Moer K, Viry E, et al. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett (2019) 458:13–20. doi: 10.1016/j.bbcan.2018.07.002
101. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. (1995) 92(12):5510–4. doi: 10.1073/pnas.92.12.5510
102. Samanta D, Semenza GL. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim Biophys Acta Rev Cancer. (2018) 1870(1):15–22. doi: 10.1016/j.bbcan.2018.07.002
103. Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol (2014) 49:1–15. doi: 10.3109/10409238.2013.838205
104. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol (1996) 16:4604–13. doi: 10.1111/j.1471-4159.1979.tb05236.x
105. Fathi M, Bahmanpour S, Barshidi A, Rasouli H, Karoon Kiani F, Mahmoud Salehi Khesht A, et al. Simultaneous blockade of TIGIT and HIF-1α induces synergistic anti-tumor effect and decreases the growth and development of cancer cells. Int Immunopharmacol. (2021) 101(Pt A):108288. doi: 10.1016/j.intimp.2021.108288
106. Van Calker D, Muller M, Hamprecht B. Adenosine regulates via two different types of receptors, the accumulation of cyclic AMP in cultured brain cells. J Neurochem (1979) 33:999–1005. doi: 10.1111/j.1471-4159.1979.tb05236.x
107. Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International union of pharmacology. XXV. nomenclature and classification of adenosine receptors. Pharmacol Rev (2001) 53(4):527–52. doi: 10.1016/j.pneurobio.2007.07.005
108. Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W. Structure and function of adenosine receptors and their genes. Naunyn. Schmiedebergs. Arch Pharmacol (2000) 362:364–74. doi: 10.1007/s002100000313
109. Fredholm BB, Chern Y, Franco R, Sitkovsky M. Aspects of the general biology of adenosine A2A signaling. Prog Neurobiol (2007) 83:263–76. doi: 10.1016/j.pneurobio.2007.07.005
110. Sitkovsky MV, Hatfield S, Abbott R, Belikoff B, Lukashev D, Ohta A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol Res (2014) 2:598–605. doi: 10.1161/01.RES.81.2.154
111. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U.S.A. (2006) 103:13132–7. doi: 10.1016/j.ccell.2016.06.025
112. Decking UK, Schlieper G, Kroll K, Schrader J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res (1997) 81(2):154–64. doi: 10.1161/01.res.81.2.154
113. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-Inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell (2016) 30:391–403. doi: 10.3389/fimmu.2012.00190
114. Hatfield SM, Kjaergaard J, Lukashev D, Belikoff B, Schreiber TH, Sethumadhavan S, et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J Mol Med (Berl). (2014) 92(12):1283–92. doi: 10.1007/s00109-014-1189-3
115. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol (2012) 3:190. doi: 10.3389/fimmu.2012.00190
116. Brauneck F, Seubert E, Wellbrock J, Schulze Zur Wiesch J, Duan Y, Magnus T, et al. Combined blockade of TIGIT and CD39 or A2AR enhances NK-92 cell-mediated cytotoxicity in AML. Int J Mol Sci (2021) 22(23):12919. doi: 10.3390/ijms222312919
117. Muhammad F, Wang D, McDonald T, Walsh M, Drenen K, Montieth A, et al. TIGIT+ A2Ar-dependent anti-uveitic treg cells are a novel subset of tregs associated with resolution of autoimmune uveitis. J Autoimmun (2020) 111:102441. doi: 10.1016/j.jaut.2020.102441
118. Löb S, Königsrainer A, Rammensee HG, Opelz G, Terness P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer (2009) 9:445–52. doi: 10.1038/nm934
119. Vacchelli E, Aranda F, Eggermont A, Sautès-Fridman C, Tartour E, Kennedy EP, et al. Trial watch: IDO inhibitors in cancer therapy. OncoImmunology (2014) 3:e957994. doi: 10.1038/nm1196
120. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9:1269–74. doi: 10.1002/ijc.10645
121. Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med (2005) 11:312–19. doi: 10.1038/nm1196
122. Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer (2002) 101:151–55. doi: 10.1002/ijc.10645
123. Hoffman RM. Tumor growth control with IDO-silencing salmonella-letter. Cancer Res (2013) 73:4591. doi: 10.1158/0008-5472.CAN-12-4719
124. Manuel ER, Diamond DJ. A road less traveled paved by IDO silencing: harnessing the antitumor activity of neutrophils. Oncoimmunology (2013) 2:e23322. doi: 10.4161/onci.23322
125. Zheng X, Koropatnick J, Chen D, Velenosi T, Ling H, Zhang X, et al. Silencing IDO in dendritic cells: a novel approach to enhance cancer immunotherapy in a murine breast cancer model. Int J Cancer (2013) 132:967–77. doi: 10.1002/ijc.27710
126. Blache CA, Manuel ER, Kaltcheva TI, Wong AN, Ellenhorn JD, Blazar BR, et al. Systemic delivery of salmonella typhimurium transformed with IDO shRNA enhances intratumoral vector colonization and suppresses tumor growth. Cancer Res (2012) 72:6447–56. doi: 10.1158/0008-5472.CAN-12-0193
127. Wang D, Saga Y, Mizukami H, Sato N, Nonaka H, Fujiwara H, et al. Indoleamine-2,3-dioxygenase, an immunosuppressive enzyme that inhibits natural killer cell function, as a useful target for ovarian cancer therapy. Int J Oncol (2012) 40:929–34. doi: 10.1016/j.ccell.2017.07.003
128. Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res (2007) 67:792–801. doi: 10.1016/j.it.2012.10.001
129. Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell (2017) 32(204–220):e215. doi: 10.1097/PPO.0b013e3181eb3343
130. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol (2012) 34:137–43. doi: 10.3390/jpm10030112
131. Soliman H, Mediavilla-Varela M, Antonia S. Indoleamine 2,3-dioxygenase: is it an immune suppressor? Cancer J (2010) 16:354–9. doi: 10.1111/acel.12716
132. Rahimi Koshkaki H, Minasi S, Ugolini A, Trevisi G, Napoletano C, Zizzari IG, et al. Immunohistochemical characterization of immune infiltrate in tumor microenvironment of glioblastoma. J Pers Med (2020) 10(3):112. doi: 10.3390/jpm10030112
133. Song Y, Wang B, Song R, Hao Y, Wang D, Li Y, et al. T-Cell immunoglobulin and ITIM domain contributes to CD8+ T-cell immunosenescence. Aging Cell (2018) 17(2):e12716. doi: 10.1111/acel.12716
134. Chew GM, Fujita T, Webb GM, Burwitz BJ, Wu HL, Reed JS, et al. TIGIT marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV infection. PloS Pathog (2016) 12(1):e1005349. doi: 10.1371/journal.ppat.1005349
135. Holzheimer R, Mannick JA, Zuckschwerdt. Surgical treatment: Evidence-based and problem-oriented. (2001). doi: 10.1126/scitranslmed.aaz6314
136. Davern M, Donlon NE, Sheppard A, Connell FO, Hayes C, Bhardwaj A, et al. Chemotherapy regimens induce inhibitory immune checkpoint protein expression on stem-like and senescent-like oesophageal adenocarcinoma cells. Transl Oncol (2021) 14(6):101062. doi: 10.1016/j.tranon.2021.101062
137. Liu X, Hartman CL, Li L, Albert CJ, Si F, Gao A, et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci Transl Med (2021) 13:eaaz6314. doi: 10.1046/j.1365-2567.1996.d01-689.x
138. Effros RB, Boucher N, Porter V, Zhu X, Spaulding C, Walford RL, et al. Decline in CD28+ T cells in centenarians and in long-term T cell cultures: A possible cause for both in vivo and in vitro immunosenescence. Exp Gerontol. (1994) 29(6):601–9. doi: 10.1016/0531-5565(94)90073-6
139. Fagnoni FF, Vescovini R, Mazzola M, Bologna G, Nigro E, Lavagetto G, et al. Expansion of cytotoxic CD8+ CD28- T cells in healthy ageing people, including centenarians. Immunology (1996) 88(4):501–7. doi: 10.1046/j.1365-2567.1996.d01-689.x
140. Weng NP, Akbar AN, Goronzy J. CD28(-) T cells: Their role in the age-associated decline of immune function. Trends Immunol (2009) 30(7):306–12. doi: 10.1016/j.it.2009.03.013
141. Thompson CB, Lindsten T, Ledbetter JA, Kunkel SL, Young HA, Emerson SG, et al. CD28 activation pathway regulates the production of multiple T-cell-derived lymphokines/cytokines. Proc Natl Acad Sci U S A. (1989) 86(4):1333–7. doi: 10.1073/pnas.86.4.1333
142. Klein Geltink RI, O'Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, et al. Mitochondrial priming by CD28. Cell (2017) 171(2):385–397.e11. doi: 10.1016/j.cell.2017.08.018
143. Zhou XM, Li WQ, Wu YH, Han L, Cao XG, Yang XM, et al. Intrinsic expression of immune checkpoint molecule TIGIT could help tumor growth in vivo by suppressing the function of NK and CD8+ T cells. Front Immunol (2018) 9:2821. doi: 10.3389/fimmu.2018.02821
149. Safety and Efficacy of Zimberelimab (AB122) in Combination With Domvanalimab (AB154) and Etrumadenant (AB928) in Patients With Previously Treated Non-Small Cell Lung Cancer - Full Text View - ClinicalTrials.gov.
150. Rotte A, Sahasranaman S, Budha N. Targeting TIGIT for immunotherapy of cancer: Update on clinical development. Biomedicines (2021) 9(9):1277. doi: 10.3390/biomedicines9091277
151. Hata AN, Engelman JA, Faber AC. The BCL2 family: Key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discovery (2015) 5(5):475–87. doi: 10.1158/2159-8290.CD-15-0011
152. Khalil AA, Kabapy NF, Deraz SF, Smith C. Heat shock proteins in oncology: diagnostic biomarkers or therapeutic targets? Biochim Biophys Acta (2011) 1816(2):89–104. doi: 10.1016/j.bbcan.2011.05.001
153. Del Gaizo Moore V, Letai A. BH3 profiling–measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett (2013) 332(2):202–5. doi: 10.1016/j.canlet.2011.12.021
154. Owonikoko TK, Khuri FR. Targeting the PI3K/AKT/mTOR pathway: Biomarkers of success and tribulation. Am Soc Clin Oncol Educ Book (2013) 10.1200/EdBook_AM.2013.33.e395. doi: 10.14694/EdBook_AM.2013.33.e395
155. Razaghi A, Heimann K, Schaeffer PM, Gibson SB. Negative regulators of cell death pathways in cancer: perspective on biomarkers and targeted therapies. Apoptosis (2018) 23(2):93–112. doi: 10.1007/s10495-018-1440-4
156. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res (2015) 75(11):2139–45. doi: 10.1158/0008-5472.CAN-15-0255
157. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
158. Mahoney KM, Atkins MB. Prognostic and predictive markers for the new immunotherapies. Oncol (Williston Park). (2014) 28 Suppl 3:39–48. doi: 10.3389/fimmu.2020.02048
159. Mansfield AS, Murphy SJ, Peikert T, Yi ES, Vasmatzis G, Wigle DA, et al. Heterogeneity of programmed cell death ligand 1 expression in multifocal lung cancer. Clin Cancer Res (2016) 22(9):2177–82. doi: 10.1158/1078-0432.CCR-15-2246
160. Liu JN, Kong XS, Huang T, Wang R, Li W, Chen QF. Clinical implications of aberrant PD-1 and CTLA4 expression for cancer immunity and prognosis: A pan-cancer study. Front Immunol (2020) 11:2048. doi: 10.3389/fimmu.2020.02048
161. Ma L, Gai J, Qiao P, Li Y, Li X, Zhu M, et al. A novel bispecific nanobody with PD-L1/TIGIT dual immune checkpoint blockade. Biochem Biophys Res Commun (2020) 531(2):144–51. doi: 10.1016/j.bbrc.2020.07.072
162. Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. (2020) 8(2):e000957. doi: 10.1136/jitc-2020-000957
163. Gilmour C, Yoo S-K, Chan T, Wang L. The co-expression of VISTA and TIGIT on cytotoxic T cells defines subpopulation with altered immunometabolism [abstract]. in: Abstracts: AACR virtual special conference: Tumor immunology and immunotherapy; 2021 Oct 5-6. Philadelphia (PA): AACR. Cancer Immunol Res (2022) 10(1 Suppl). doi: 10.1002/hep.30593
Keywords: T cells, metabolism, cancer, therapy, microenviroment
Citation: Jantz-Naeem N, Böttcher-Loschinski R, Borucki K, Mitchell-Flack M, Böttcher M, Schraven B, Mougiakakos D and Kahlfuss S (2023) TIGIT signaling and its influence on T cell metabolism and immune cell function in the tumor microenvironment. Front. Oncol. 13:1060112. doi: 10.3389/fonc.2023.1060112
Received: 02 October 2022; Accepted: 11 January 2023;
Published: 17 February 2023.
Edited by:
Carlos Pérez-Plasencia, National Autonomous University of Mexico, MexicoReviewed by:
Bernd Heinrich, National Institutes of Health (NIH), United StatesMohamed S. Abdel-Hakeem, Emory University, United States
Copyright © 2023 Jantz-Naeem, Böttcher-Loschinski, Borucki, Mitchell-Flack, Böttcher, Schraven, Mougiakakos and Kahlfuss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sascha Kahlfuss, c2FzY2hhLmthaGxmdXNzQG1lZC5vdmd1LmRl; Dimitrios Mougiakakos, ZGltaXRyaW9zLm1vdWdpYWtha29zQG1lZC5vdmd1LmRl