 Zhang Zhang
Zhang Zhang Yang Lu
Yang Lu Changle Shi
Changle Shi Min Chen
Min Chen Xin He
Xin He Hongying Zhang
Hongying Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 27 January 2023
Sec. Surgical Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1017154
This article is part of the Research TopicApplication of Molecular Genetics for the Diagnosis and Classification of Rare Cancers in Surgical PathologyView all 13 articles
Background: Dermatofibrosarcoma protuberans (DFSP) is an uncommon cutaneous tumor in children. Most published articles are sporadic or small series and lack systematically molecular analyses. The aim of our study is to better understand the clinicopathologic and genetic features of these rare lesions.
Methods: All patients diagnosed with DFSP aged ≤ 18 years were retrospectively reviewed from January 2006 to May 2022.
Results: A total of 66 cases (32 male and 34 female patients) were identified, with ages ranging from 0.3 to 18 years (median, 13 years). Tumor locations predominantly occurred on the trunk (38/66, 57.6%), followed by the extremities (20/66, 30.3%) and head/neck (8/66, 12.1%). Histological findings revealed classic (41/66, 62.1%), myxoid (4/66, 6.1%), pigmented (6/66, 9.1%), plaque-like (3/66, 4.5%), giant cell fibroblastoma (GCF; 6/66, 9.1%), and fibrosarcomatous (6/66, 9.1%) variants of DFSP. Immunochemistry revealed minority tumors (9/66, 13.6%) showing patchy or negative staining for CD34. Fluorescence in situ hybridization (FISH) indicated that 49 of 53 tested cases including all detected biopsy specimens (11/11) contained COL1A1-PDGFB fusion, in which the average copy number gain of COL1A1-PDGFB was 0.68. There were four cases negative for COL1A1-PDGFB rearrangement, one of which was found to harbor a novel COL3A1-PDGFB fusion by next-generation sequencing (NGS). Treatment for 63 patients comprised 40 marginal excisions and 23 wide local excisions (WLEs), including 1 with imatinib therapy. Follow-up information was available on 49 patients with a duration of 12–161 months (median, 60 months). Fourteen patients developed tumor recurrence, all with initial marginal excisions. The others survived with no evidence of disease.
Conclusions: This study of pediatric DFSP indicates certain discrepancies in clinicopathologic characteristics between children and adults. The majority of pediatric DFSPs contain COL1A1-PDGFB fusion, the same as their adult counterparts. The COL3A1-PDGFB chimerism might be associated with the special morphology of GCF, which needs further investigation. FISH is valuable in biopsy tissues and cases with atypical CD34 immunostaining, while supplementary NGS could be helpful to identify the cytogenetically cryptic DFSP. Overall, an urgent accurate diagnosis is needed to formulate an optimal therapeutic strategy in the pediatric population.
Dermatofibrosarcoma protuberans (DFSP), one of the most common dermal sarcomas, is a locally infiltrative dermal and subcutaneous fibroblastic tumor of intermediate malignancy (1). According to the 2020 World Health Organization (WHO) classification of soft tissue and bone tumors, the lesion divides into several histologic subtypes, including classic DFSP, pigmented DFSP, myxoid DFSP, DFSP with myoid differentiation, plaque-like DFSP, giant cell fibroblastoma (GCF), and fibrosarcomatous DFSP (FS-DFSP) (1). DFSP can locally recur with a rate of 20%–50%, of which FS-DFSP is the only subtype associated with more aggressive behavior. Approximately 10%–16% of FS-DFSP may develop distant metastases, resulting in a worse prognosis (1–4).
Cytogenetically, more than 90% of DFSP cases are characterized with the collagen type I alpha 1 and platelet-derived growth factor B (COL1A1-PDGFB) fusion deriving from supernumerary ring r (17;22) or translocation t (17;22) (q22; q13) (5). The chimeric formation could result in upregulating the PDGFB expression, associated with tumorigenesis and accordingly designed to be the target by tyrosine kinase inhibitors like imatinib mesylate (6). In clinical routine practice, the critical molecular abnormality is exploited to be detected by fluorescence in situ hybridization (FISH), aiding in diagnosis and validating targeted molecular therapy in certain circumstances (7, 8).
The incidence of DFSP is approximately 1 case per 1,000,000 person-years of adults predominantly within the second to fifth decades, while even rare in children (9–12). Notably, to the best of our knowledge, previous reports are mostly sporadic or small series and there are only six large analyses involving pediatric DFSP, most of which are absent of detailed histological analysis or systematic molecular studies (11, 13–17). Therefore, we retrospectively evaluated a large series of 66 pediatric DFSPs at the largest institution in Southwest China and aimed to further investigate the clinicopathological features and molecular characteristics.
This study was approved by the West China Hospital Institutional Review Board. A SNOMED search of the hospital surgical pathology and consultation files from January 2006 to May 2022 identified 926 DFSPs. All cases were independently reviewed by two pathologists (HZ and ZZ) who specialized in soft tissue tumors and two general surgical pathologists (XH and YL). The patients diagnosed with DFSP aged ≥ 19 years were considered adult DFSP and were excluded. Finally, 66 DFSP cases aged ≤ 18 years were confirmed as pediatric DFSP and included in this study.
Immunohistochemical analysis was performed using the EnVision Plus detection system (DAKO, Carpinteria, CA, USA) with positive and negative controls. Antibodies for the experiments included CD34 (EP88, ready-to-use; ZSGB-Bio), SMA (UMAB237, ready-to-use; ZSGB-Bio), Ki-67 (clone MIB-1, 1:100; Dako), desmin (D33, 1:100; Dako), S-100 protein (4C49, 1:100; Dako), myogenin (F5D, 1:50; ZSGB-Bio), Bcl-2 (EP36, 1:200; ZSGB-Bio), CD99 (EP8, ready-to-use; ZSGB-Bio), p16 (16P04/IC2, ready-to-use; Dako), p63 (4A4, 1:400; Dako), cytokeratin (AE1/AE3, 1:100; Dako), and EMA (E29, 1:100; Dako).
FISH analyses were performed using commercially available ZytoLight® SPEC PDGFB Dual Color Break Apart Probe and ZytoLight® SPEC COL1A1/PDGFB Dual Color Dual Fusion Probe (ZytoVision, Bremerhaven, Germany). The tests were performed on 4-μm sections according to an established laboratory protocol, as previously described (18). Two investigators independently counted at least 100 nuclei on each slide. A case was considered positive for PDGFB rearrangement when at least ≥10% of the cells exhibited a split signal pattern which showed that the distance between the green and red signals was greater than the diameter of two signals. The COL1A1-PDGFB fusion was considered positive when at least 10% of the cells showed one separate red signal, one separate green signal, and two red/green fusion signals. The COL1A1-PDGFB copy gain was calculated according to the description of Abbott et al. (19).
Genomic profiling was performed on FFPE tissues with capture-based panel targeting 481 soft tissue tumor-relevant genes. Genomic DNA was extracted from FFPE tissues using a QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The eligible DNA was hybridized with the capture probes, selected using magnetic beads, and polymerase chain reaction (PCR)-amplified. Then, library fragment size was determined by Bioanalyzer2100 (Agilent Technologies, Santa Clara, CA). The target-enriched library was sequenced on the Hiseq4000 platform (Illumina, Inc., San Diego, CA) following the manufacturer’s instructions. The average sequencing depth was 1000× for all targeted regions.
The COL3A1-PDGFB fusion was analyzed by PCR using primers (COL3A1-F: 5’-CTTCAGGGTGAGACAGCCAA-3’; PDGFB-R: 5’-CATAAGCCCCCGGATTTGGT-3’). Sanger sequencing was performed at Tsingke Biological Technology Co., Ltd. (Chengdu, China).
Statistical analysis was performed using the GraphPad Prism version 5 (GraphPad Software, San Diego, CA). Comparisons between different groups were evaluated using Student’s t-test and ANOVA for continuous variables. p < 0.05 indicates the statistical significance between different groups.
The relevant clinicopathologic data are summarized in Table 1. The study cohort comprised 32 male and 34 female patients (ratio, 1:1.1). The age of the patients ranged from 0.3 to 18 years (median, 13 years; mean, 11.38 years), including 2 infants (2/66, 3.0%; age ≤ 11 months) and 26 children aged less than 10 years (26/66, 39.4%). The tumor size ranged from 0.6 to 8 cm (median, 3 cm; mean, 2.9 cm). The majority of the tumors occurred on the trunk (38/66, 57.6%), followed by the extremities (20/66, 30.3%) and head/neck (8/66, 12.1%). The clinical manifestations commonly presented as nodular or multinodular masses (61/66, 92.4%) and much less in the plaque stage (5/66, 7.6%). The cut surfaces of DFSP were tan and yellow, with rubbery firm to soft gelatinous appearances. There were 15 cases (15/66, 22.7%) who underwent biopsies before treatment.
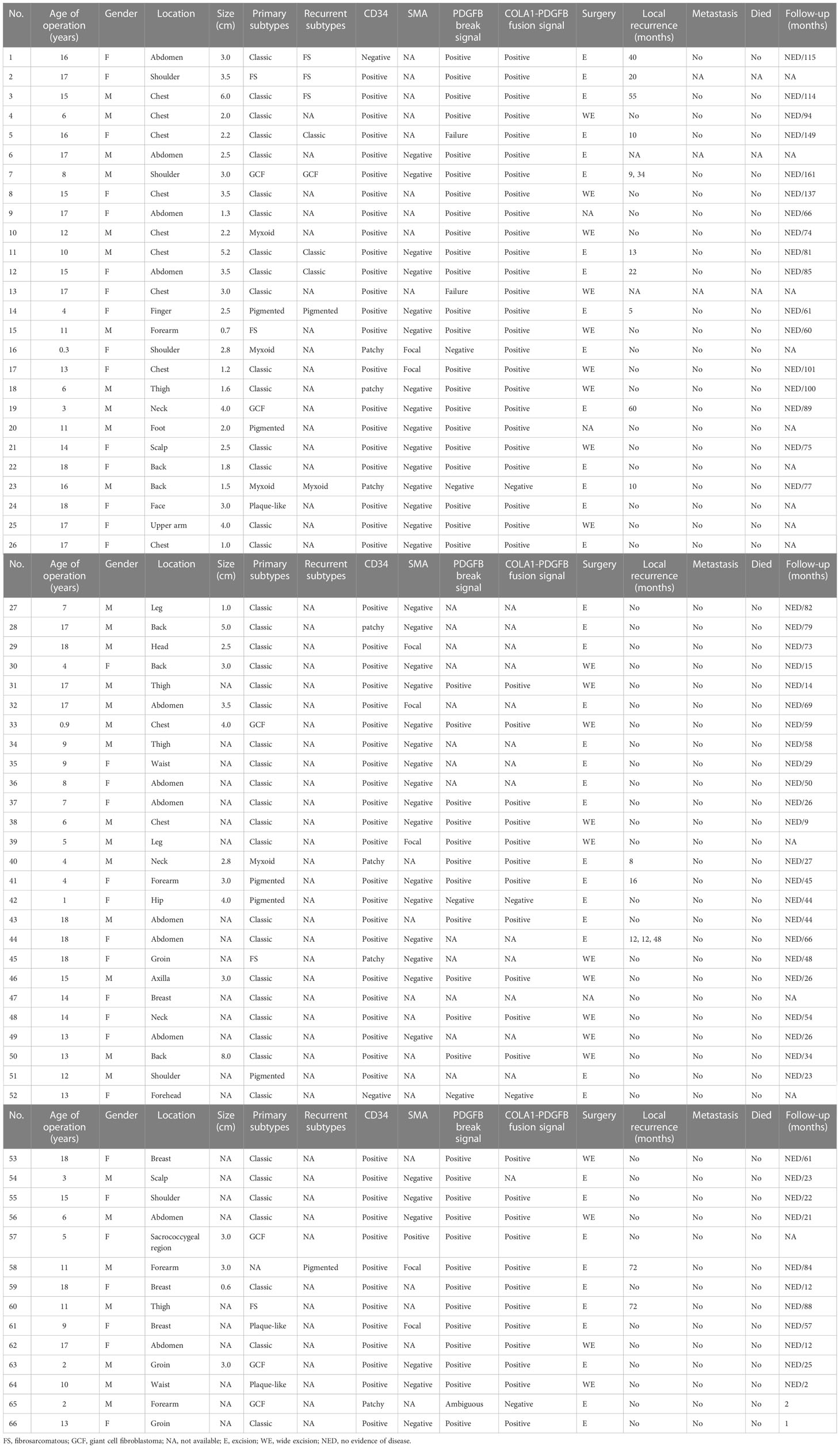
Table 1 Clinicopathological and cytogenetic features of 66 pediatric DFSPs.
The tumors were classified according to histologic subtypes into classic DFSP (41/66, 62.1%), myxoid DFSP (4/66, 6.1%), pigmented DFSP (6/66, 9.1%), plaque-like DFSP (3/66, 4.5%), giant cell fibroblastoma (GCF; 6/66, 9.1%), and fibrosarcomatous DFSP (6/66, 9.1%).
Microscopically, the DFSPs diffusely infiltrated into the dermis and subcutis with ill-defined borders, which, with infiltration into the subcutaneous fat, could result in a honeycomb-like appearance. Typically, uniform wavy or spindle tumor cells were proliferative and arranged in a storiform or cartwheel pattern (Figure 1A). In the myxoid subtype (cases 10, 16, 23, and 40), the abundant myxoid stroma with low cellularity and numerous vessels occupied more than 50% of the tumor (Figure 1B). There were six pigmented-subtype tumors (including cases 14, 20, 41, 42, 51, and 58; case 58 was a recurrent lesion), all of which could find that the pigmented dendritic cells were scattered over fibroblastic tumor cells (Figure 1C). The three cases of plaque-like DFSP (cases 24, 61, and 64) were dermal-based lesions, composed of regular plump tumor cells presenting a horizontally oriented arrangement and a focal storiform structure (Figure 1D). In six GCF tumors, a varying number of pleomorphic mononucleated or multinucleated giant cells admixed with spindle cells in the loose myxoid matrix or abundant collagenous stroma. Among them, three were pure GCF (cases 19, 33, and 65) and three were hybrid lesions (cases 7, 57, and 63) that consisted of conventional DFSP and GCF components. Most differently, case 65 uniquely consisted of a higher proportion of neoplasms that were with larger and more atypical giant nuclei compared to that of the other five typical GCFs, which predominantly contained slender wavy spindled cells and sporadic giant cells. The floret-like giant cell-lined pseudovascular spaces and infiltrated subcutaneous fat mimicking liposarcoma were easy to find (Figure 1E). Most of the above variants of DFSP presented low mitotic activity (0–5/10 high-power fields), while one conventional DFSP showed mitotic activity with 11/10 high-power fields. FS-DFSP presented in four primary cases (cases 2, 15, 45, and 60) and two recurrent lesions (cases 1 and 3), whose primary tumors were both confirmed as a conventional DFSP. The fibrosarcomatous component was composed of neoplastic cells with increased cellularity and mitotic activity, arranging in a fascicular pattern with a herringbone appearance (Figure 1F). Necrosis was not identified.
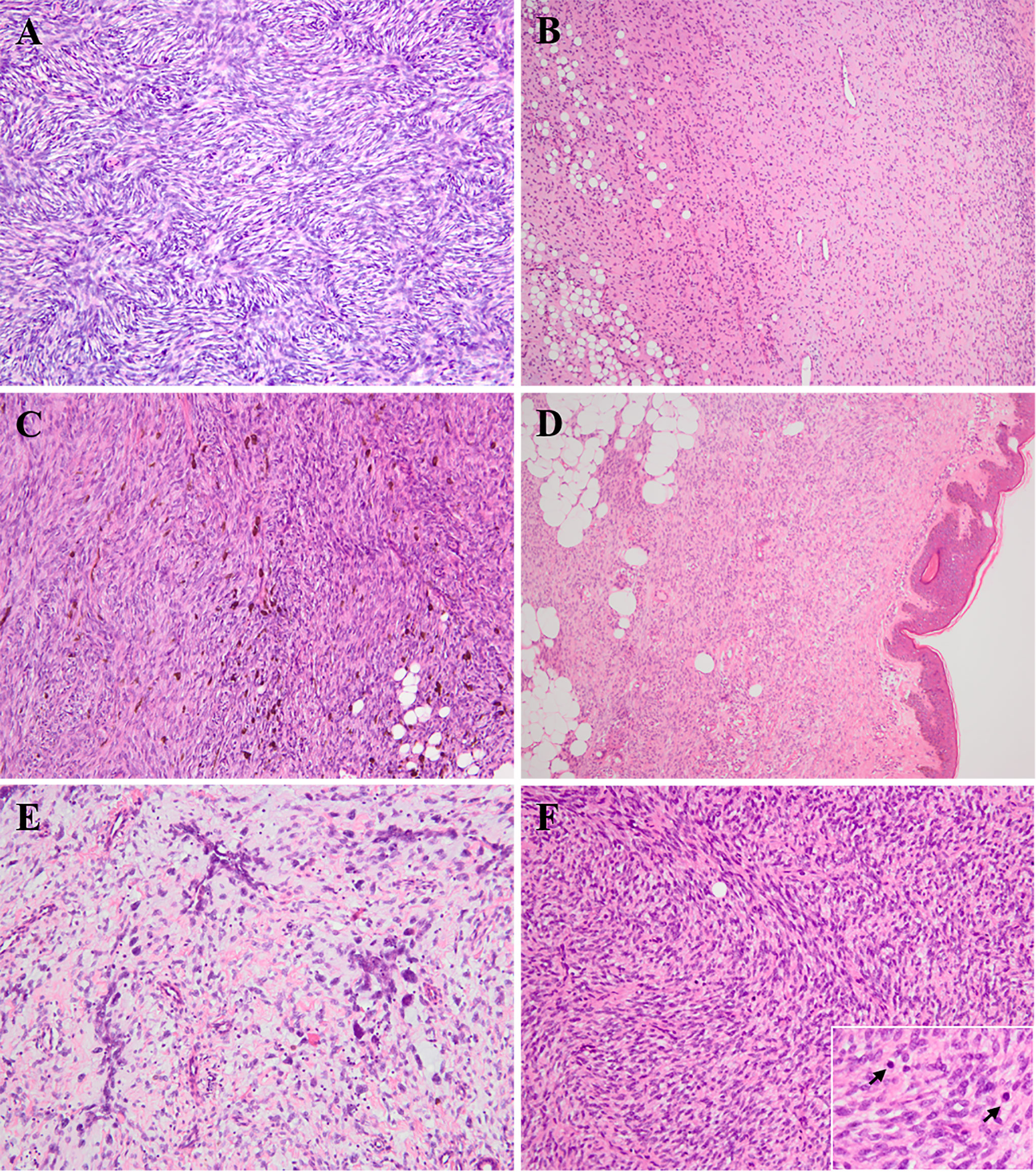
Figure 1 Histologic findings of various subtypes of pediatric DFSP. (A) Conventional DFSP. The lesion consisted of uniform bland spindled neoplastic cells arranged in a classic cartwheel pattern (H&E × 200). (B) Myxoid DFSP. The tumor had relatively low cellularity and consisted of pump spindle or stellated tumor cells diffusely distributed in prominent myxoid architecture with numerous vessels (H&E × 100). (C) Pigmented DFSP. The dendritic cells with melanin pigment punctuated within monotonous storiform area of conventional DFSP (H&E × 200). (D) Plaque-like DFSP. The dermal-based lesion composed of regular plump tumor cells presenting a horizontally oriented arrangement and a focal storiform structure (H&E × 100). (E) Giant cell fibroblastoma (case 65). High proportion of hyperchromatic multinucleated giant cells scattered in the loosely fibrous and myxoid stroma, and lined pseudovascular spaces (H&E × 200). (F) Fibrosarcomatous DFSP. The lesion was composed of high-grade fibrosarcoma-like component, showing a typical herringbone appearance with increasing mitoses (H&E × 200; insert × 400).
Immunohistochemical analysis revealed that most of the tumors (57/66, 86.3%) were diffusely positive for CD34, whereas the minority (9/66,13.6%) showed patchy or negative staining. The decreased or lost expression of CD34 was mainly observed in myxoid and fibrosarcomatous areas, originating from three conventional DFSPs, three myxoid DFSPs, two FS-DFSPs, and one GCF (Figure 2). Majority of tumors were negative for smooth muscle actin (SMA), while there were a few cases (9/46, 20%) exhibiting positivity or focal staining. All cases were negative for desmin, S-100 protein, myogenin, Bcl-2, CD99, p16, p63, cytokeratin, and EMA.
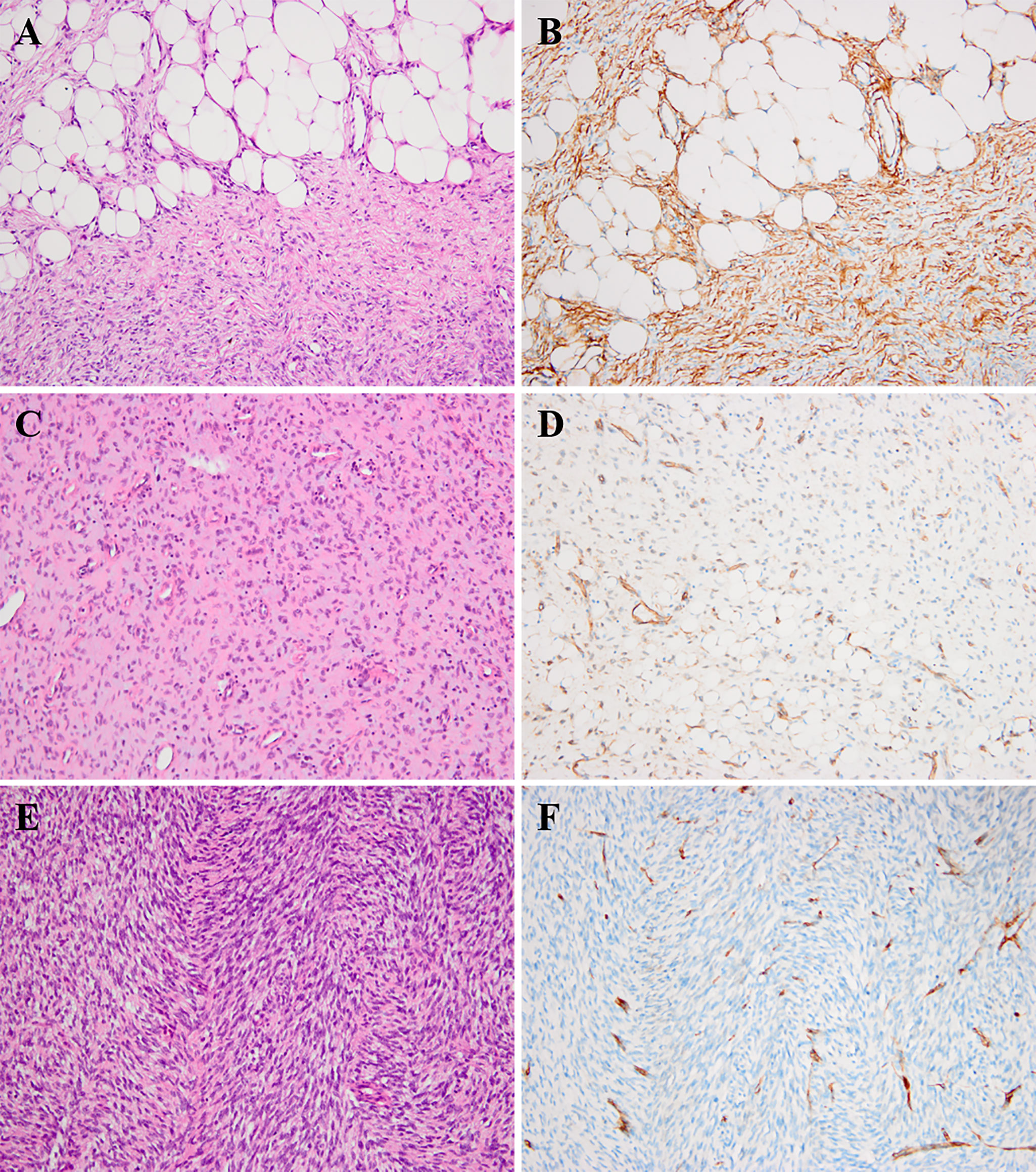
Figure 2 Histologic findings of DFSP and corresponding immunohistochemical images. (A, B) The neoplastic cells infiltrated into subcutaneous fat tissues forming a characteristic “honeycomb” pattern (A, H&E × 200). The corresponding component showed diffuse positivity for CD34 (B, × 200). (C, D) The myxoid component of DFSP (case 40) could show patchy or focal staining for CD34 (C, H&E × 200; D, × 200). (E, F) The fibrosarcomatous component of DFSP (case 1) could exhibit loss for CD34 staining (E, H&E × 200; F, × 200).
FISH analysis indicated that 46 cases (46/53, 86.8%) were positive for PDGFB in a split-signal pattern. There were four that showed negative results of PDGFB rearrangement (cases 16, 23, 42, and 52), in one of which (case 16) COL1A1-PDGFB fusion was detected. One case exhibited an ambiguous result of PDGFB rearrangement (case 65). The last two cases failed the experiment (cases 5 and 13). The median COL1A1-PDGFB copy gain of pediatric DFSP was 0.7 (range 0–1.8; mean ± SD, 0.68 ± 0.46). Not much different from the classic DFSP, the median COL1A1-PDGFB copy gain in the FS subtype was 0.6 (range 0–1.1; mean ± SD, 0.58 ± 0.40) (p = 0.64), while in the GCF subtype, the median COL1A1-PDGFB copy gain was 0.45 (range 0–1.25; mean ± SD, 0.49 ± 0.50) (p = 0.36) (Figures 3A–C).
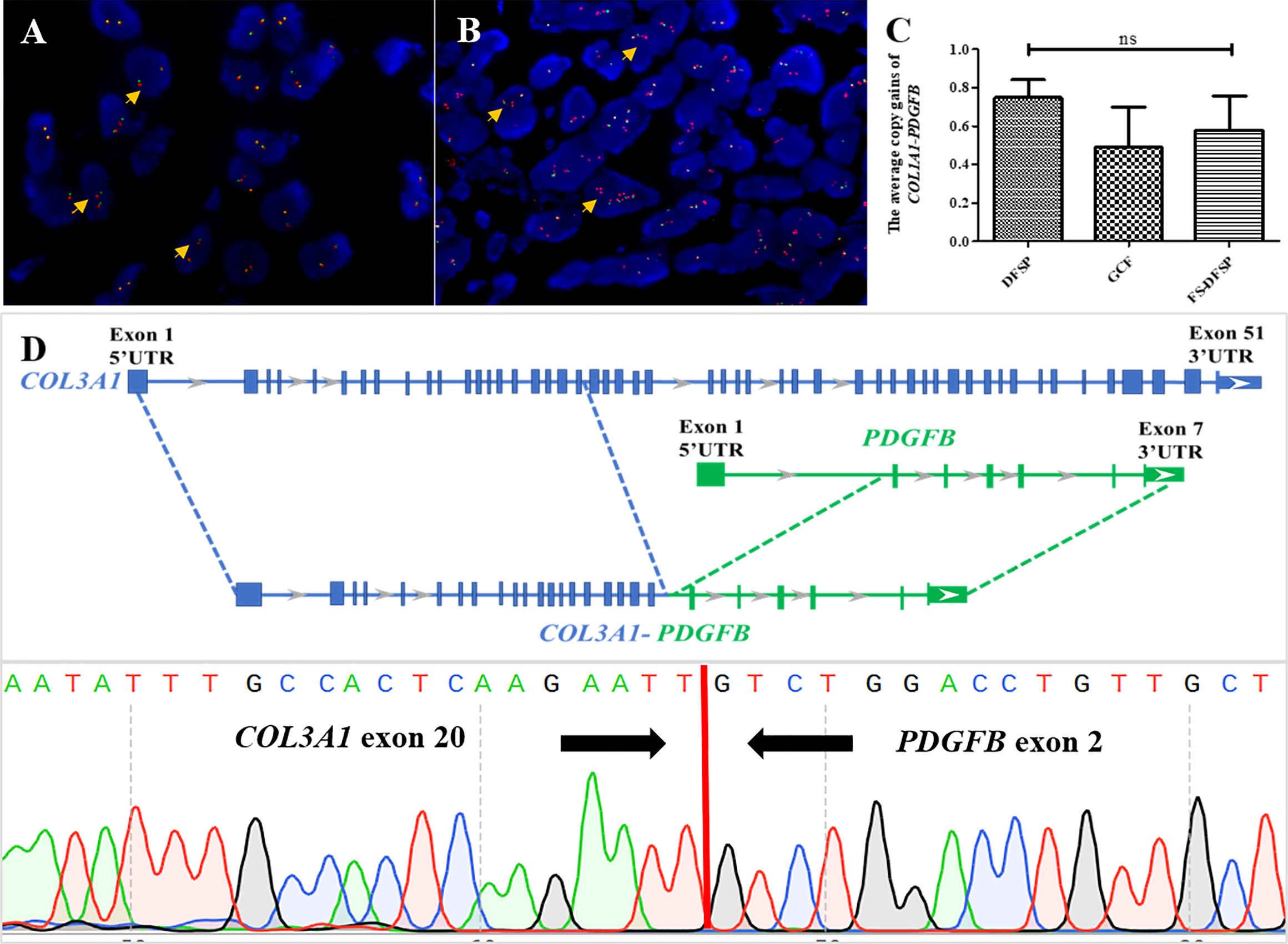
Figure 3 Molecular findings of pediatric DFSP. (A) FISH was performed on the DFSP component using a PDGFB break-apart probe to the locus on chromosome 22. The results showed several tumor cells with one red–green signal indicating a normal chromosome 22; one separate red and one separate green signal indicating a COL1A1-PDGFB fusion gene with no extra copies of red signal (orange arrows) (PDGFB, red signals and green signals). (B) FISH was performed on the DFSP component using a PDGFB break-apart probe to the locus on chromosome 22. The results showed several tumor cells with one red–green signal indicating a normal chromosome 22 and one to three extra copies of red signal (orange arrows) indicating a COL1A1-PDGFB fusion gene (PDGFB, red signals and green signals). (C) Comparison among DFSP (left column) and the GCF and the FS-DFSP (right two columns) indicated that there was not much difference in average COL1A1-PDGFB copy gains of pediatric DFSP. (D) Next-generation sequencing revealed case 65 with a novel COL3A1 (e20)-PDGFB (e2) fusion (upper part); Sanger sequencing results demonstrated the presence of the COL3A1-PDGFB fusion gene (lower part).
Moreover, 49 of 53 tested cases (92.5%) including all detected biopsy specimens (11/11, 100%) contained COL1A1-PDGFB fusion, while 4 cases were negative for COL1A1-PDGFB based on the routine FISH screening (cases 23, 42, 52, and 65). One case (case 65) with suspicious PDGFB split but COL1A1-PDGFB fusion negative was found to harbor a novel COL3A1-PDGFB fusion through NGS. The PCR and Sanger sequencing further confirmed the COL3A1-PDGFB fusion gene (Figure 3D).
The treatment and follow-up information is detailed in Table 1. Surgical strategies for 63 patients were composed of marginal excisions (40/63, 63.5%) and WLE (23/63, 36.5%). Eight cases (8/15, 53.3%) underwent WLE after biopsies. One (case 14) underwent finger amputation for recurrent tumor therapy. Targeted therapy with imatinib mesylate was administered to one patient (case 7) after tumor excision on the second relapse.
Follow-up information was available on 49 patients (49/66, 74.2%) with a duration from 12 to 161 months (median, 60 months). Fourteen patients (14/49, 28.5%) developed recurrence, all with marginal excisions with positive margins. The others survived with no evidence of disease.
The occurrence of DFSP could involve people of all ages, from neonates to old adults (13, 20–22). Pediatric DFSPs were uncommon, which accounted for only 7.1% (66/926) of all DFSPs in our medical center. Among the DFSPs, the GCF subtype occurred mainly within 10 years old, which was consistent with previous observations as a pediatric-predominance subtype. The cohort of DFSP displayed a slight female predilection (M:F = 1:1.1), which was similar to the US pediatric population (11). Consistently, GCF occurred significantly in the male sex (M:F = 5:1) as the tendency of literature presenting male predominance in children diagnosed GCF (13, 14). The tumors in our cases were widespread and were predominantly distributed on the trunk (57.6%), followed by the extremities (30.3%) and head/neck (12.1%). Moreover, functional vitals or cosmetic positions including breast (4/66, 6%), groin (3/66, 4.5%), scalp (2/66, 3%), and face (1/66, 1.5%) could be involved and were rarely mentioned in children according to previous findings (12, 23, 24).
The distribution spectrum of the subtype exhibited some differences between pediatric and adult DFSPs. In the adult population, conventional DFSP was the most predominant subtype, which constituted approximately 61.3%–91% of DFSPs in previous studies, followed by FS-DFSP (10%–16%), myxoid DFSP (7.6%), pigmented DFSP (2.7%–5%), GCF (2.7%), and plaque-like DFSP (1.3%–1.7%) (3, 25–29). According to the results with detailed constitution of pediatric DFSP variants in this large cohort, the incidence of conventional DFSP (62.1%), FS-DFSP (9.1%), and myxoid DFSP (6.1%) was close to the lower limit of the adult counterparts, while pigmented DFSP (9.1%), GCF (9.1%), and plaque-like DFSP (4.5%) were higher than that of the adult.
CD34 is the most frequently used immunohistochemical marker for the diagnosis of DFSP. Typically, DFSP stains positive for CD34 in about 90% of tumors and mostly shows negativity for SMA (30, 31). In our series, 86.3% of cases were diffusely positive for CD34 expression, whereas diminished or absent staining presented in approximately 13.6% of pediatric DFSP cases. The decreased or lost expression of CD34 was mainly observed in myxoid and fibrosarcomatous areas, as published (3, 14, 32, 33). One case of classic DFSP showed a lack of CD34 expression (case 52), which two pathologists independently reviewed, arriving at a diagnosis of classic DFSP based on the typical histology. FISH utilizing the PDGFB break-apart probe revealed unbalanced translocation presenting additional 3’-red signals in 5% of these tumor cells, while the COL1A1-PDGFB fusion probe showed yellow signal denoting a fusion pattern in 2% of tumor cells. This case might show cryptic rearrangement associated with DFSP. Regretfully, the specimen could not be further investigated using NGS because of poor quality.
Evidence of COL1A1-PDGFB rearrangement is the key to the differential diagnosis of difficult cases and is inevitable for the effective application of targeted treatment. FISH has shown the validity for confirmation of the COL1A1-PDGFB fusion and it was widely applied to clinical detection. In our children’s series, 92.5% of children’s DFSPs were confirmed positive results by using the PDGFB break-apart probe and the COL1A1-PDGFB fusion probe as routine screening methods, similar to the previous FISH studies with detectable rate ranging from 86% to 96% (7, 8, 34). In addition, the fusion product COL1A1-PDGFB was amplified with low levels of the17q and 22q sequences (usually one to three copies), which could be detectable by either FISH or comparative genomic hybridization (GCH) (19, 35). In our study, we found that the average gains of the COL1A1-PDGFB fusion gene showed no statistical difference in ordinary DFSP, FS-DFSP, and GCF groups of children, among which the GCF cases presented the lowest average genomic gains. While correlated studies were scarce in children, more samples and deeper investigations are needed to further reveal the meaning of the molecular characters in pediatric patients.
Nevertheless, based on previous studies, a minority of DFSP cases with uncertain or negative results based on routine FISH assays are considered to be molecular unconfirmed DFSP and might result in inaccurate diagnosis (36). Application of supplementary NGS approaches would be of value to fusion detection (37). In our cohort, 7.5% (4/53) of pediatric DFSPs exhibited molecular unconfirmed characteristics. After carefully reviewing the published English articles, we included two studies and collected a total of seven pediatric DFSPs (including the four pediatric cases in our cohort) considered to be molecular unconfirmed based on routine FISH detection, the clinicopathological and molecular characteristics of which are summarized in Table 2. The clinicopathological features of most cases seem to not differ from the corresponding subtypes except one GCF (present case 65) with atypical morphology. Genetically important, NGS revealed the GCF containing a novel COL3A1-PDGFB fusion that was first presented in DFSP. COL3A1, located in 2q32.2, belongs to the collagen genes together with COL1A1 and COL1A2, and encodes a structural protein of type III collagen, which is found in abundance in extensible connective tissues, such as skin, blood vessels, gastrointestinal tract, and the developing brain (38–41). The translocation of COL3A1 had been reported as a rare partner fusing to PLAG1-rearranged neoplasms (including lipoblastoma and unclassified spindle cell neoplasm) and USP6-rearranged neoplasms (including cranial fasciitis, cellular fibroma of the tendon sheath, and unclassified benign myofibroblastic tumor). The breakpoints seem to constantly occur at exon 1 of COL3A1 in COL3A1-PLAG1 and COL3A1-USP6 cases, whereas they occurred at exon 20 of COL3A1 in the present GCF case (40, 42–46). In addition, atypical variants of COL3A1 were associated with Ehlers–Danlos syndrome (EDS) involving connective tissue disorders (39, 47). Furthermore, though without unique clinical presentation, some distinctive pathological morphology was observed that could be associated with the COL3A1-rearranged GCF, which contained a higher proportion of multinucleated giant cells with larger and more atypical nuclei than the COL1A1-rearranged GCFs. However, more studies are needed to confirm the relationship between the specific chimerism and morphology. In addition, there was one classic DFSP with COL6A3-PDGFD and one FS-DFSP with EMILIN2-PDGFD in children from the work of Lee et al. (48). Dadone-Montaudié et al. described one pediatric pigmented DFSP, which was not found to have suspicious transcript even after using RNA sequencing, indicating that a more complicated mechanism might exist (36). There were three cases (present cases 23, 42, and 52) of genetic aberrations in the current study that could not be further identified using NGS because of the poor quality of the specimens, which could be associated with alternative rearrangement (including PDGFD rearrangement and PDGFB rearrangement with novel partner genes), cryptic COL1A1-PDGFB fusion, and even other sophisticated chromosomal aberrations (36, 49–52). We still needed more specimens to reveal the genetic characteristics of cryptic pediatric DFSP. Altogether, NGS could be a helpful strategy to identify the molecular unconfirmed DFSP and provide detailed information about abnormal genes for further investigation.
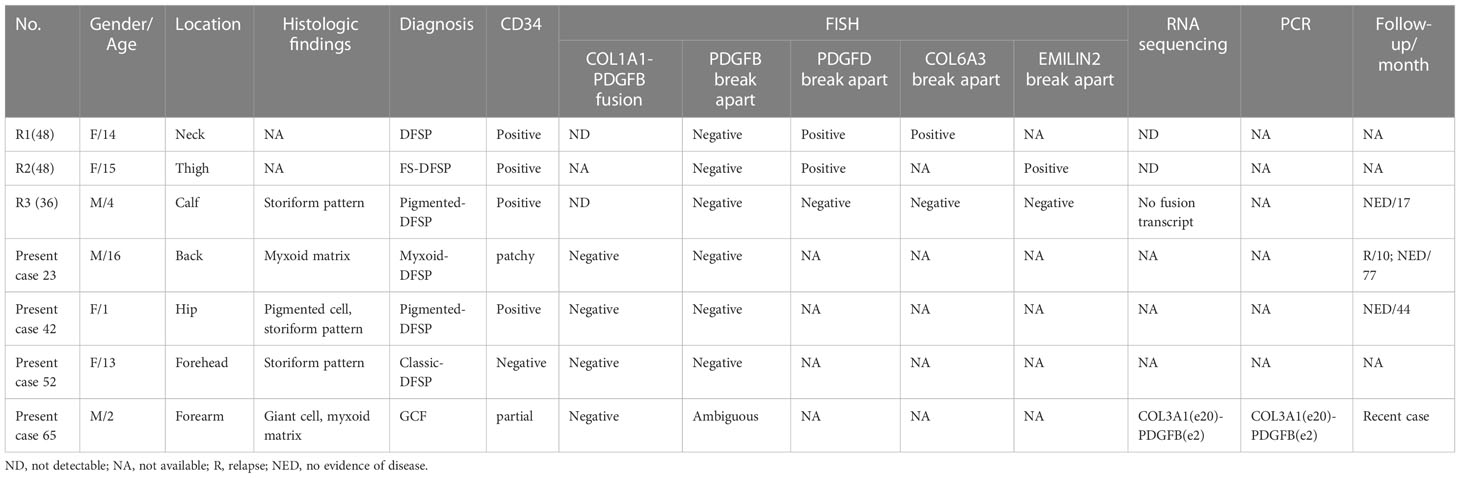
Table 2 Clinicopathologic and molecular characteristics of 7 cytogenetically cryptic DFSPs in children.
The diagnosis of pediatric DFSP could be challenging, and it should be differentiated from not only benign mimics but also malignant neoplasms, similar to its subtypes in terms of the diversity of histological morphology in the pediatric tumor. Basically, the histologically and immunophenotypically (CD34 positive) overlapping pediatric lesions are often considered in the differential diagnoses, such as plaque-like CD34-positive dermal fibroma (lack of COL1A1-PDGFB), fibroblastic connective tissue nevus, fibrous hamartoma of infancy (EGFR exon 20 insertion/duplication mutations), pediatric NTRK-rearranged spindle cell neoplasm (co-expression of CD34 and S100, and NTRK-positive expression or rearrangement), lipofibromatosis, and plexiform myofibroblastoma. Although DFSPs are generally centered within the dermis or subcutis and characterized by spindle cells with a storiform to the whorled pattern, when these tumors have similar morphological features, the cytogenetic method can help with diagnosis, especially for small or superficial biopsy samples. In addition, there are a minority of DFSPs (especially myxoid DFSP and FS-DFSP) with patchy or negative staining for CD34 that are more likely to result in diagnostic pitfalls. The myxoid DFSP could be easily confused with other myxoid lesions such as superficial acral fibromyxoma, solitary fibrous tumor, and low-grade fibromyxoid sarcoma (LGFMS). Despite developing typically in old adults, pediatric liposarcoma should rarely be excluded, especially myxoid liposarcoma (ML) (53). FS-DFSP with atypical staining for CD34 could be confused with high-grade sarcomas, such as high-grade infantile fibrosarcoma (IFS) with ETV6-NTRK3 fusion, especially dedifferentiated liposarcoma (DDL) without a well-differentiated liposarcoma (WDL) component. Notably, the situation we had reported could be extremely challenging due to the presence of MDM2/FRS2 amplification and the lack of evidence for COL1A1-PDGFB fusion by routine FISH screening (54). Importantly, carefully looking for conventional components and searching for characteristics of fusion genes could be helpful for pathologists to confirm complicated cases.
Tissue biopsy, as one of the gold standard diagnostic examinations, could effectively assist in identifying uncertain lesions at an early stage. There were 15 (15/66, 22.7%) children who underwent biopsy, and 11 biopsy specimens detected by FISH all identified with COL1A1-PDGFB fusion, which reconfirmed the valuable function of FISH in biopsy samples as shown in our previous report (55). Therefore, it encouraged us to increase biopsy in children and boosted FISH application in pediatric specimens.
Accurate and early diagnosis is critical to guide an appropriate therapeutic scheme in children. Nowadays, the recommended treatment for DFSP is either WLE (2–4 cm) with tumor-negative margins or Mohs micrographic surgery (MMS) (56). In our group, 40 patients (63.5%) underwent marginal excisions, and the other 23 patients (36.5%) received WLE. The overall prognosis of pediatric DFSP was favorable, with all patients surviving without metastasis. However, 15 patients (15/49, 30.6%) developed tumor recurrence. It should be noted that there were 2 of 15 (13.3%) recurrent tumors transforming into fibrosarcomatous DFSP, which was more aggressive than the primary conventional types. Importantly, the recurrence of DFSP was closely related to surgical margin. A wide local excision (2–3 cm) with tumor-negative margins reduces the local recurrence of DFSP from 50%–75% to 0%–30% (24, 57). Moreover, MMS could further decrease the recurrence rate to 0.6%–6% (58). Noteworthy, a higher proportion of patients (8/15, 53.3%) were subjected to WLE, which was a benefit from early diagnosis through biopsy. It highlighted the pivotal role of biopsy in providing definitive evidence for patients to select an optimal therapeutic strategy. Moreover, 11 patients suffered defects from surgery involving anatomic critical regions including the breast, groin, finger, scalp, and face in this cohort. Actually, there are numerous issues arising from surgery for the special population: poor compliance of young patients, prolonged anesthesia duration, increasing surgical difficulty and risk, and unacceptable cosmetic and functional mutilation.
Another treatment opportunity that is typically reserved for adults in surgically unresectable, recurrent, or metastatic cases is targeted therapy (7). However, there is no standard clinical guidance about targeted therapy for pediatric patients. Hitherto, imatinib was applied on six child patients postoperatively for continuous remission and preoperatively for tumor reduction, all of whom achieved the desired effects (23, 59–62). In our study, we reported another patient (case 7) diagnosed with the GCF subtype who benefited from targeted therapy with imatinib after marginal excision in the second relapse, with no evidence of disease after 10 years. Therefore, using imatinib mesylate could act as a neoadjuvant or adjuvant therapy to help control tumor progression, in tandem with other treatments in children, but it still warrants further evaluation and large-scale investigation.
In conclusion, we present the largest series study of 66 cases of pediatric DFSP with genetic investigation. There are certain differences in clinicopathology between children and adults. Most pediatric DFSPs contain classical COL1A1-PDGFB fusion as compared with adults. The COL3A1-PDGFB chimerism might be associated with the special morphology of GCF, which needs further investigation. Furthermore, FISH screening, and even supplementary NGS detection, should be used in identifying pediatric lesions with typical or uncertain morphology involving dermis and subcutis. The overall prognosis would be favorable with appropriate treatment, while more attention should be paid to recurrence prevention and mutilation reduction in DFSP management in the special population.
The data presented in the study are deposited in the NCBI repository (https://www.ncbi.nlm.nih.gov/), accession number OQ262947.
ZZ and YL analyzed the data and prepared the manuscript. CS collected the clinicopathological data of the patients. MC carried out the molecular studies. HZ, ZZ, XH and YL were responsible for diagnosis and review. HZ supervised and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (No. 81972520), Sichuan Science and Technology Program (No. 2022YFS0376), and the 1·3·5 project for disciplines of excellence–Clinical Research Incubation Project, West China Hospital, Sichuan University (NO.2018HXFH011).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mentzel T, Pedeutour F. Dermatofibrosarcoma protuberans. In: Who classification of tumours of soft tissue and bone, 5th Edition. Lyon (France: International Agency for Research on Cancer (2020).
2. Abbott JJ, Oliveira AM, Nascimento AG. The prognostic significance of fibrosarcomatous transformation in dermatofibrosarcoma protuberans. Am J Surg Pathol (2006) 30(4):436–43. doi: 10.1097/00000478-200604000-00002
3. Llombart B, Monteagudo C, Sanmartín O, López-Guerrero JA, Serra-Guillén C, Poveda A, et al. Dermatofibrosarcoma protuberans: A clinicopathological, immunohistochemical, genetic (Col1a1-pdgfb), and therapeutic study of low-grade versus high-grade (Fibrosarcomatous) tumors. J Am Acad Dermatol (2011) 65(3):564–75. doi: 10.1016/j.jaad.2010.06.020
4. King L, López-Terrada D, Jakacky J, McCarville MB, Spunt SL, Bridge JA, et al. Primary intrathoracic dermatofibrosarcoma protuberans. Am J Surg Pathol (2012) 36(12):1897–902. doi: 10.1097/PAS.0b013e31826b7919
5. Sirvent N, Maire G, Pedeutour F. Genetics of dermatofibrosarcoma protuberans family of tumors: From ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosomes Cancer (2003) 37(1):1–19. doi: 10.1002/gcc.10202
6. Malhotra B, Schuetze SM. Dermatofibrosarcoma protruberans treatment with platelet-derived growth factor receptor inhibitor: A review of clinical trial results. Curr Opin Oncol (2012) 24(4):419–24. doi: 10.1097/CCO.0b013e328353d78d
7. Patel KU, Szabo SS, Hernandez VS, Prieto VG, Abruzzo LV, Lazar AJ, et al. Dermatofibrosarcoma protuberans Col1a1-pdgfb fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol (2008) 39(2):184–93. doi: 10.1016/j.humpath.2007.06.009
8. Karanian M, Pérot G, Coindre JM, Chibon F, Pedeutour F, Neuville A. Fluorescence in situ hybridization analysis is a helpful test for the diagnosis of dermatofibrosarcoma protuberans. Mod Pathol (2015) 28(2):230–7. doi: 10.1038/modpathol.2014.97
9. Jing C, Zhang H, Zhang X, Yu S. Dermatofibrosarcoma protuberans: A clinicopathologic and therapeutic analysis of 254 cases at a single institution. Dermatol Surg (2021) 47(2):e26–30. doi: 10.1097/dss.0000000000002578
10. Valdivielso-Ramos M, Torrelo A, Campos M, Feito M, Gamo R, Rodriguez-Peralto JL. Pediatric dermatofibrosarcoma protuberans in Madrid, Spain: Multi-institutional outcomes. Pediatr Dermatol (2014) 31(6):676–82. doi: 10.1111/pde.12371
11. Rubio GA, Alvarado A, Gerth DJ, Tashiro J, Thaller SR. Incidence and outcomes of dermatofibrosarcoma protuberans in the us pediatric population. J Craniofac Surg (2017) 28(1):182–4. doi: 10.1097/scs.0000000000003203
12. Sleiwah A, Wright TC, Chapman T, Dangoor A, Maggiani F, Clancy R. Dermatofibrosarcoma protuberans in children. Curr Treat Options Oncol (2022) 23(6):843–54. doi: 10.1007/s11864-022-00979-9
13. Terrier-Lacombe MJ, Guillou L, Maire G, Terrier P, Vince DR, de Saint Aubain Somerhausen N, et al. Dermatofibrosarcoma protuberans, giant cell fibroblastoma, and hybrid lesions in children: Clinicopathologic comparative analysis of 28 cases with molecular data–a study from the French federation of cancer centers sarcoma group. Am J Surg Pathol (2003) 27(1):27–39. doi: 10.1097/00000478-200301000-00004
14. Jha P, Moosavi C, Fanburg-Smith JC. Giant cell fibroblastoma: An update and addition of 86 new cases from the armed forces institute of pathology, in honor of dr. Franz m. enzinger. Ann Diagn Pathol (2007) 11(2):81–8. doi: 10.1016/j.anndiagpath.2006.12.010
15. Janz TA, Nagasubramanian R, Wei JL. Pediatric head and neck fibrosarcomas: A demographical, treatment, and survival analysis and review of a rare case. Int J Pediatr Otorhinolaryngol (2019) 116:92–6. doi: 10.1016/j.ijporl.2018.09.031
16. van Lee CB, Kan WC, Gran S, Mooyaart A, Mureau MAM, Williams HC, et al. Dermatofibrosarcoma protuberans re-excision and recurrence rates in the Netherlands between 1989 and 2016. Acta Derm Venereol (2019) 99(12):1160–5. doi: 10.2340/00015555-3287
17. Shmookler BM, Enzinger FM, Weiss SWJC. Giant cell fibroblastoma. A Juvenile Form Dermatofibrosarcoma Protuberans (1989) 64(10):2154. doi: 10.1002/1097-0142(19891115)64:10<2154::aidcncr2820641030>3.0.co;2-n
18. Zhang H, Erickson-Johnson M, Wang X, Oliveira JL, Nascimento AG, Sim FH, et al. Molecular testing for lipomatous tumors: Critical analysis and test recommendations based on the analysis of 405 extremity-based tumors. Am J Surg Pathol (2010) 34(9):1304–11. doi: 10.1097/PAS.0b013e3181e92d0b
19. Abbott JJ, Erickson-Johnson M, Wang X, Nascimento AG, Oliveira AM. Gains of Col1a1-pdgfb genomic copies occur in fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Mod Pathol (2006) 19(11):1512–8. doi: 10.1038/modpathol.3800695
20. Weinstein JM, Drolet BA, Esterly NB, Rogers M, Bauer BS, Wagner AM, et al. Congenital dermatofibrosarcoma protuberans: Variability in presentation. Arch Dermatol (2003) 139(2):207–11. doi: 10.1001/archderm.139.2.207
21. Maire G, Fraitag S, Galmiche L, Keslair F, Ebran N, Terrier-Lacombe MJ, et al. A clinical, histologic, and molecular study of 9 cases of congenital dermatofibrosarcoma protuberans. Arch Dermatol (2007) 143(2):203–10. doi: 10.1001/archderm.143.2.203
22. Thway K, Noujaim J, Jones RL, Fisher C. Dermatofibrosarcoma protuberans: Pathology, genetics, and potential therapeutic strategies. Ann Diagn Pathol (2016) 25:64–71. doi: 10.1016/j.anndiagpath.2016.09.013
23. Posso-De Los Rios CJ, Lara-Corrales I, Ho N. Dermatofibrosarcoma protuberans in pediatric patients: A report of 17 cases. J Cutan Med Surg (2014) 18(3):180–5. doi: 10.2310/7750.2013.13099
24. Fezza R, Shahzeidi P, Downing C, Cognetta A. A rare presentation of dermatofibrosarcoma protuberans of the scalp treated with "Slow-mohs" micrographic surgery. J Drugs Dermatol (2022) 21(5):534–5. doi: 10.36849/jdd.6719
25. Zhou X, Sun D, Liu Y, Sun Q, Yuan Z, Luo X, et al. Dermatofibrosarcoma protuberans: Our 10-year experience on 80 patients. J Dermatolog Treat (2020) 31(6):554–8. doi: 10.1080/09546634.2019.1622629
26. Fletcher CD, Theaker JM, Flanagan A, Krausz T. Pigmented dermatofibrosarcoma protuberans (Bednar tumour): Melanocytic colonization or neuroectodermal differentiation? a clinicopathological and immunohistochemical study. Histopathology (1988) 13(6):631–43. doi: 10.1111/j.1365-2559.1988.tb02094.x
27. Hoesly PM, Lowe GC, Lohse CM, Brewer JD, Lehman JS. Prognostic impact of fibrosarcomatous transformation in dermatofibrosarcoma protuberans: A cohort study. J Am Acad Dermatol (2015) 72(3):419–25. doi: 10.1016/j.jaad.2014.11.020
28. Xu S, Zhao L, Wang J. Atrophic dermatofibrosarcoma protuberans: A clinicopathological study of 16 cases. Pathology (2019) 51(6):615–20. doi: 10.1016/j.pathol.2019.06.002
29. Nishio J, Iwasaki H, Nabeshima K, Naito M. Cytogenetics and molecular genetics of myxoid soft-tissue sarcomas. Genet Res Int (2011) 2011:497148. doi: 10.4061/2011/497148
30. Llombart B, Sanmartín O, López-Guerrero JA, Monteagudo C, Serra C, Requena C, et al. Dermatofibrosarcoma protuberans: Clinical, pathological, and genetic (Col1a1-pdgfb ) study with therapeutic implications. Histopathology (2009) 54(7):860–72. doi: 10.1111/j.1365-2559.2009.03310.x
31. Abenoza P, Lillemoe T. Cd34 and factor xiiia in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans. Am J Dermatopathol (1993) 15(5):429–34. doi: 10.1097/00000372-199310000-00003
32. Lo CH, Tsang PM, Cheng SY. Myoid differentiation in dermatofibrosarcoma protuberans and its fibrosarcomatous variant: 10 years' experience in a tertiary hospital. Autops Case Rep (2022) 12:e2021368. doi: 10.4322/acr.2021.368
33. Reilly DJ, Loo YL, Alexander WM, Wilks DJ, MacGregor D, Coombs CJ. Diagnostic and management considerations in pediatric dermatofibrosarcoma protuberans. Ann Plast Surg (2022) 88(3):319–22. doi: 10.1097/sap.0000000000002970
34. Segura S, Salgado R, Toll A, Martín-Ezquerra G, Yébenes M, Sáez A, et al. Identification of T(17;22)(Q22;Q13) (Col1a1/Pdgfb) in dermatofibrosarcoma protuberans by fluorescence in situ hybridization in paraffin-embedded tissue microarrays. Hum Pathol (2011) 42(2):176–84. doi: 10.1016/j.humpath.2010.07.015
35. Kiuru-Kuhlefelt S, El-Rifai W, Fanburg-Smith J, Kere J, Miettinen M, Knuutila S. Concomitant DNA copy number amplification at 17q and 22q in dermatofibrosarcoma protuberans. Cytogenet Cell Genet (2001) 92(3-4):192–5. doi: 10.1159/000056901
36. Dadone-Montaudié B, Alberti L, Duc A, Delespaul L, Lesluyes T, Pérot G, et al. Alternative pdgfd rearrangements in dermatofibrosarcomas protuberans without pdgfb fusions. Mod Pathol (2018) 31(11):1683–93. doi: 10.1038/s41379-018-0089-4
37. Zhu R, Yan J, Li B, Tan F, Yan W, Shen J, et al. Determination of Col1a1-pdgfb breakpoints by next-generation sequencing in the molecular diagnosis of dermatofibrosarcoma protuberans. Exp Mol Pathol (2021) 122:104672. doi: 10.1016/j.yexmp.2021.104672
38. Ucaryilmaz Metin C, Ozcan G. Comprehensive bioinformatic analysis reveals a cancer-associated fibroblast gene signature as a poor prognostic factor and potential therapeutic target in gastric cancer. BMC Cancer (2022) 22(1):692. doi: 10.1186/s12885-022-09736-5
39. Sølyst S, Oksjoki R, Farholt S, Nielsen DG, Christensen AH, Fagerberg CR, et al. Carriers of Col3a1 pathogenic variants in Denmark: Interfamilial variability in severity and outcome of elective surgical procedures. Clin Genet (2022) 102(3):191–200. doi: 10.1111/cge.14176
40. Santisukwongchote S, Thorner PS, Desudchit T, Techavichit P, Jittapiromsak N, Amornfa J, et al. Pediatric fibromyxoid tumor with Plag1 fusion: An emerging entity with a novel intracranial location. Neuropathology (2022) 42(4):315–22. doi: 10.1111/neup.12837
41. Thiele BJ, Doller A, Kähne T, Pregla R, Hetzer R, Regitz-Zagrosek V. Rna-binding proteins heterogeneous nuclear ribonucleoprotein A1, E1, and K are involved in post-transcriptional control of collagen I and iii synthesis. Circ Res (2004) 95(11):1058–66. doi: 10.1161/01.Res.0000149166.33833.08
42. Fritchie K, Wang L, Yin Z, Nakitandwe J, Hedges D, Horvai A, et al. Lipoblastomas presenting in older children and adults: Analysis of 22 cases with identification of novel Plag1 fusion partners. Mod Pathol (2021) 34(3):584–91. doi: 10.1038/s41379-020-00696-4
43. Mantilla JG, Gross JM, Liu YJ, Hoch BL, Ricciotti RW. Characterization of novel Usp6 gene rearrangements in a subset of so-called cellular fibroma of tendon sheath. Mod Pathol (2021) 34(1):13–9. doi: 10.1038/s41379-020-0621-1
44. Malik F, Wang L, Yu Z, Edelman MC, Miles L, Clay MR, et al. Benign infiltrative myofibroblastic neoplasms of childhood with Usp6 gene rearrangement. Histopathology (2020) 77(5):760–8. doi: 10.1111/his.14182
45. Paulson VA, Stojanov IA, Wasman JK, Restrepo T, Cano S, Plunkitt J, et al. Recurrent and novel Usp6 fusions in cranial fasciitis identified by targeted rna sequencing. Mod Pathol (2020) 33(5):775–80. doi: 10.1038/s41379-019-0422-6
46. Logan SJ, Schieffer KM, Conces MR, Stonerock E, Miller AR, Fitch J, et al. Novel morphologic findings in Plag1-rearranged soft tissue tumors. Genes Chromosomes Cancer (2021) 60(8):577–85. doi: 10.1002/gcc.22953
47. Colman M, Castori M, Micale L, Ritelli M, Colombi M, Ghali N, et al. Atypical variants in Col1a1 and Col3a1 associated with classical and vascular ehlers-danlos syndrome overlap phenotypes: Expanding the clinical phenotype based on additional case reports. Clin Exp Rheumatol (2022) 40 Suppl 134(5):46–62. doi: 10.55563/clinexprheumatol/kzkq6y
48. Lee PH, Huang SC, Wu PS, Tai HC, Lee CH, Lee JC, et al. Molecular characterization of dermatofibrosarcoma protuberans: The clinicopathologic significance of uncommon fusion gene rearrangements and their diagnostic importance in the exclusively subcutaneous and circumscribed lesions. Am J Surg Pathol (2022) 46(7):942–55. doi: 10.1097/pas.0000000000001866
49. Nakamura I, Kariya Y, Okada E, Yasuda M, Matori S, Ishikawa O, et al. A novel chromosomal translocation associated with Col1a2-pdgfb gene fusion in dermatofibrosarcoma protuberans: Pdgf expression as a new diagnostic tool. JAMA Dermatol (2015) 151(12):1330–7. doi: 10.1001/jamadermatol.2015.2389
50. Dickson BC, Hornick JL, Fletcher CDM, Demicco EG, Howarth DJ, Swanson D, et al. Dermatofibrosarcoma protuberans with a novel Col6a3-pdgfd fusion gene and apparent predilection for breast. Genes Chromosomes Cancer (2018) 57(9):437–45. doi: 10.1002/gcc.22663
51. Chen Y, Shi YZ, Feng XH, Wang XT, He XL, Zhao M. Novel tnc-pdgfd fusion in fibrosarcomatous dermatofibrosarcoma protuberans: A case report. Diagn Pathol (2021) 16(1):63. doi: 10.1186/s13000-021-01123-1
52. Bianchini L, Maire G, Guillot B, Joujoux JM, Follana P, Simon MP, et al. Complex T(5;8) involving the Cspg2 and Ptk2b genes in a case of dermatofibrosarcoma protuberans without the Col1a1-pdgfb fusion. Virchows Arch (2008) 452(6):689–96. doi: 10.1007/s00428-008-0580-2
53. Peng R, Li N, Lan T, Chen H, Du T, He X, et al. Liposarcoma in children and young adults: A clinicopathologic and molecular study of 23 cases in one of the largest institutions of China. Virchows Arch (2021) 479(3):537–49. doi: 10.1007/s00428-021-03076-8
54. Lu Y, Li T, Chen M, Peng H, Du T, Qiu Y, et al. Coamplification of 12q15 and 12p13 and homozygous Cdkn2a/2b deletion: Synergistic role of fibrosarcomatous transformation in dermatofibrosarcoma protuberans with a cryptic Col1a1-pdgfb fusion. Virchows Arch (2022) 481(2):313–9. doi: 10.1007/s00428-022-03297-5
55. Zhang Z, Chen H, Chen M, He X, Wang Y, Zhang H. Application of Col1a1-pdgfb fusion gene detection by fluorescence In situ hybridization in biopsy tissue of dermatofibrosarcoma protuberans. J Dermatol (2017) 44(7):798–802. doi: 10.1111/1346-8138.13767
56. Miller SJ, Alam M, Andersen JS, Berg D, Bichakjian CK, Bowen GM, et al. Dermatofibrosarcoma protuberans. J Natl Compr Canc Netw (2012) 10(3):312–8. doi: 10.6004/jnccn.2012.0032
57. Lemm D, Mügge LO, Mentzel T, Höffken K. Current treatment options in dermatofibrosarcoma protuberans. J Cancer Res Clin Oncol (2009) 135(5):653–65. doi: 10.1007/s00432-009-0550-3
58. Foroozan M, Sei JF, Amini M, Beauchet A, Saiag P. Efficacy of mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: Systematic review. Arch Dermatol (2012) 148(9):1055–63. doi: 10.1001/archdermatol.2012.1440
59. Yen H, Pan SC, Huang CH, Wong TW. Complete remission of a periorbital dermatofibrosarcoma protuberans with adjuvant imatinib mesylate in a child. JAAD Case Rep (2015) 1(4):172–4. doi: 10.1016/j.jdcr.2015.04.003
60. Gooskens SL, Oranje AP, van Adrichem LN, de Waard-van der Spek FB, den Hollander JC, van de Ven CP, et al. Imatinib mesylate for children with dermatofibrosarcoma protuberans (Dfsp). Pediatr Blood Cancer (2010) 55(2):369–73. doi: 10.1002/pbc.22494
61. Suzuki D, Kobayashi R, Yasuda K, Yamamoto H, Morioka K, Mikawa M, et al. Congenital dermatofibrosarcoma protuberans in a newborn infant with a massive back tumor: Favorable effects of oral imatinib on the control of residual tumor growth. J Pediatr Hematol Oncol (2011) 33(7):e304–6. doi: 10.1097/MPH.0b013e31822d4d21
Keywords: dermatofibrosarcoma protuberans, giant cell fibroblastoma, pediatric sarcoma, COL1A1-PDGFB fusion, COL3A1-PDGFB fusion, fluorescence in situ hybridization, next generation sequencing
Citation: Zhang Z, Lu Y, Shi C, Chen M, He X and Zhang H (2023) Pediatric dermatofibrosarcoma protuberans: A clinicopathologic and genetic analysis of 66 cases in the largest institution in Southwest China. Front. Oncol. 13:1017154. doi: 10.3389/fonc.2023.1017154
Received: 11 August 2022; Accepted: 02 January 2023;
Published: 27 January 2023.
Edited by:
Ming Zhao, Zhejiang Provincial People’s Hospital, ChinaReviewed by:
Thomas G. Salopek, University of Alberta, CanadaCopyright © 2023 Zhang, Lu, Shi, Chen, He and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongying Zhang, aHlfemhhbmdAc2N1LmVkdS5jbg==; aHlfemhAMjYzLm5ldA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.