 Jin-Young Min1
Jin-Young Min1 Do-Hee Kim
Do-Hee Kim- 1Department of Chemistry, Kyonggi University, Suwon, Gyeonggi-do, Republic of Korea
- 2College of Pharmacy, Keimyung University, Daegu, Republic of Korea
Owing to its unique nucleophilicity, cysteine is an attractive sulfhydryl-containing proteinogenic amino acid. It is also utilized in various metabolic pathways and redox homeostasis, as it is used for the component of major endogenous antioxidant glutathione and the generation of sulfur-containing biomolecules. In addition, cysteine is the most nucleophilic amino acid of proteins and can react with endogenous or exogenous electrophiles which can result in the formation of covalent bonds, which can alter the cellular states and functions. Moreover, post-translational modifications of cysteines trigger redox signaling and affect the three-dimensional protein structure. Protein phosphorylation mediated by kinases and phosphatases play a key role in cellular signaling that regulates many physiological and pathological processes, and consequently, the modification of cysteine regulates its activities. The modification of cysteine residues in proteins is critically important for the design of novel types of pharmacological agents. Therefore, in cancer metabolism and cancer cell survival, cysteine plays an essential role in redox regulation of cellular status and protein function. This review summarizes the diverse regulatory mechanisms of cysteine bound to or free from proteins in cancer. Furthermore, it can enhance the comprehension of the role of cysteine in tumor biology which can help in the development of novel effective cancer therapies.
Introduction
Cysteine, a sulfhydryl-containing proteinogenic amino acid essential for the human body, is employed in a variety of metabolic pathways, such as the regulation of reduced glutathione (GSH), a major endogenous antioxidant molecule and the generation of sulfur-containing biomolecules such as hydrogen sulfide (H2S), taurine, coenzyme A and biotin (1, 2). In addition, the reactive thiol (-SH) group of cysteine residues in proteins can undergo various post-translational modifications (PTMs) such as palmitoylation, glutathionylation, guanylation, cysteinylation, nitrosylation, and sulfhydration (Figure 1) (2). PTMs play a key role in various biological processes by affecting the structure, reactivity, stability, and function of the protein, thereby modulating a broad range of biological processes (3). Moreover, endogenous or exogenous electrophilic molecules can be covalently modified with the nucleophilic cysteine, which consequently alters the cellular states and functions. Some pharmacological agents that exploit the modification of cysteine residues present in many intracellular proteins affect the biological activity, thus exerting their drug effects (4). This review investigates the multifaceted role of cysteine bound to or free from proteins in cancer biology research.
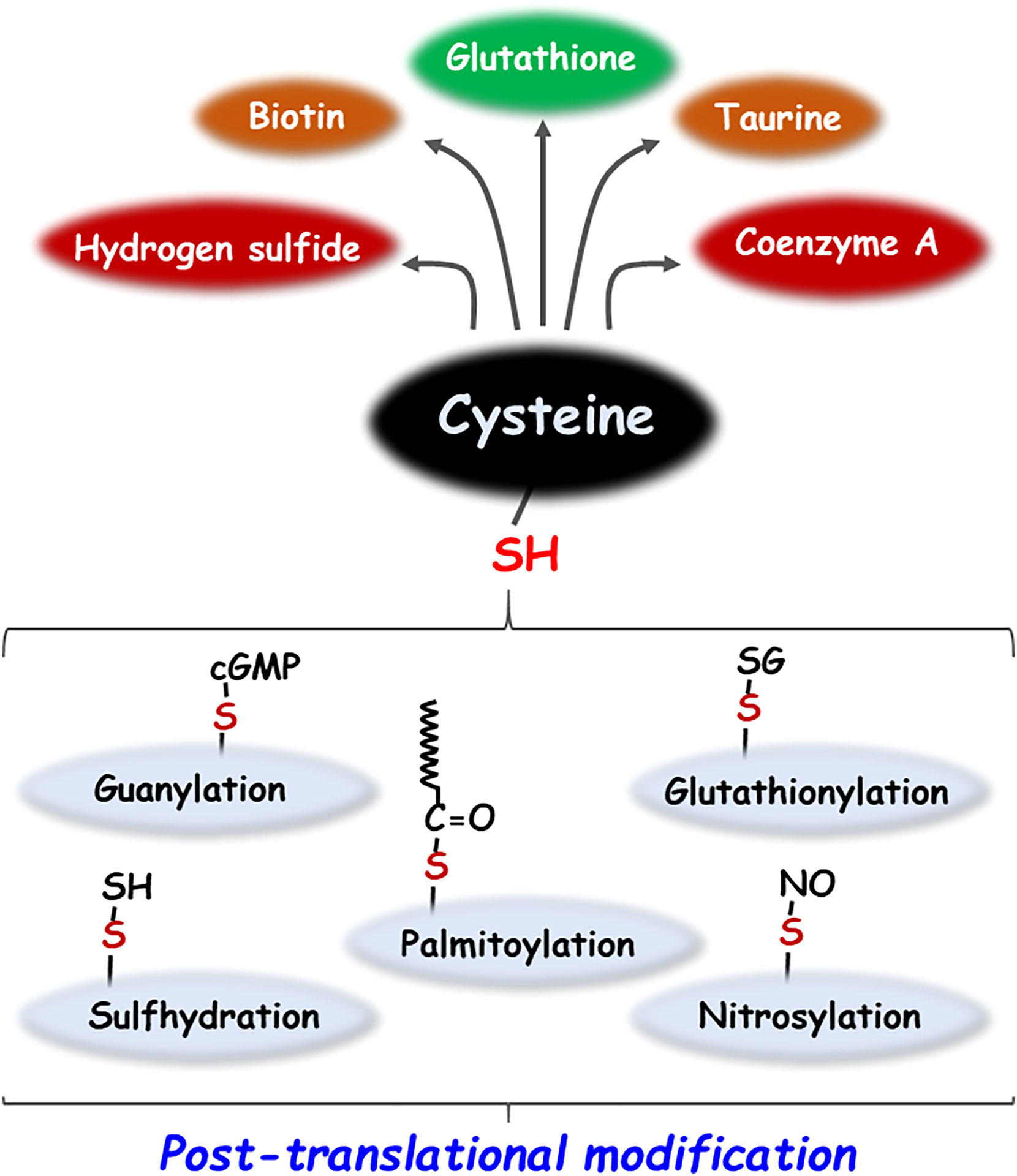
Figure 1 Metabolic products and post-translational modifications of cysteine. Cysteine generates sulfur-containing molecules such as glutathione, taurine, biotin, coenzyme A and hydrogen sulfide by various metabolic pathways. In addition, cysteine undergoes a variety of post-translational modifications including palmitoylation, glutathionylation, guanylation, nitrosylation, and sulfhydration.
Cysteine metabolism in cancer and tumor microenvironment
It is increasingly noted that altered levels of amino acids including cysteine in the tumor microenvironment, by uptake or processing of both intracellular and extracellular regions, have emerged as potential markers of tumor progression. Cysteine may interplay with the tumor microenvironment and cancer cell metabolism which affect the metastatic potential and drug resistance (5). The extracellular compartments are highly oxidizing, whereas cells generally maintain a reducing environment in the cytosol.Cysteine mainly exists as cystine, the oxidized dimer form of cysteine, in the extracellular space. Both forms of cysteine and cystine can be imported into cells through specific transporters, and amino acids exist as cysteine in a reducing environment. The cystine/cysteine redox cycle is characterized by a slight increase in the intracellular cysteine levels and exceedingly high extracellular cysteine concentrations, which efficiently protects the cells from oxidative stress-induced cell death (6). Tumors interact with the surrounding microenvironment and organs through the lymphatic network, composed of a complex mixture of cells, including tumor cells, fibroblasts and immune cells. Cystine is transported into stromal cells and converted to cysteine that supports glutathione (GSH) synthesis and secretion into the tumor microenvironment (7). Enhanced cysteine transport and its metabolism enable cancer cells to adapt to a challenging tumor microenvironment and acquire chemoresistance. This section discusses the role of cellular signaling molecules and proteins in regulating cysteine metabolism in cancer development and drug resistance.
xCT/SLC7A11
System xc- transporter is a heterodimer consisting of the disulfide-linked light (xCT/SLC7A11) and heavy (CD98/SLC3A2) chain subunits. Cystine/glutamate antiporter solute carrier family 7 member 11 (SLC7A11) is a major transporter regulating cysteine in tumor cells and is induced in response to a variety of stimuli, such as oxidative stress and electrophilic compounds (8). Lin et al. reported that SLC7A11 is widely expressed in multiple human cancers, and its up-regulation is correlated with poor survival outcomes in patients with breast cancer, prostate cancer, and papillary thyroid carcinoma (9). In prostate cancer, xCT protein expression is positively associated with invasion and metastasis by affecting the redox status of the tumor microenvironment. Zhong et al. reported that altering the extracellular cysteine/cystine ratio by xCT knockdown inhibits prostate cancer cell invasion (10). In addition, oncogenic KRAS-mutant cancer cells maintain the induction of xCT transcription, which enhance the GSH levels and protect tumor cells against oxidative stress (11). Moreover, cysteine and glutathione released from stromal fibroblasts in the tumor microenvironment confer resistance to cisplatin treatment in ovarian tumor cells by reducing the intracellular cisplatin accumulation. However, interferon (IFN)-γ derived from CD8+ T cells in the tumor stromal region attenuated platinum resistance through STAT1 phosphorylation and xCT downregulation in fibroblasts (Figure 2) (7). In contrast to the high expression of xCT in solid tumors, the xCT transporter is downregulated in hematologic malignancies such as chronic lymphocytic leukemia. Downregulation of xCT expression limits its ability to transport cystine for GSH synthesis (12). However, bone marrow stromal cells effectively import cystine, convert it to cysteine, and release it into the microenvironment for uptake by chronic lymphocytic leukemia cells to promote GSH synthesis (12). Interactions between stromal cells and leukemic cells are essential for the survival of chronic lymphocytic leukemia cells from drug-induced cytotoxicity (12). Furthermore, Cramer et al. reported that cysteinase, a glutathione inhibitor that degrades cysteine and cystine, suppressed the growth of prostate carcinoma allografts, reduced tumor growth in prostate and breast cancer xenografts due to depletion of intracellular GSH and consequent oxidative stress (13).
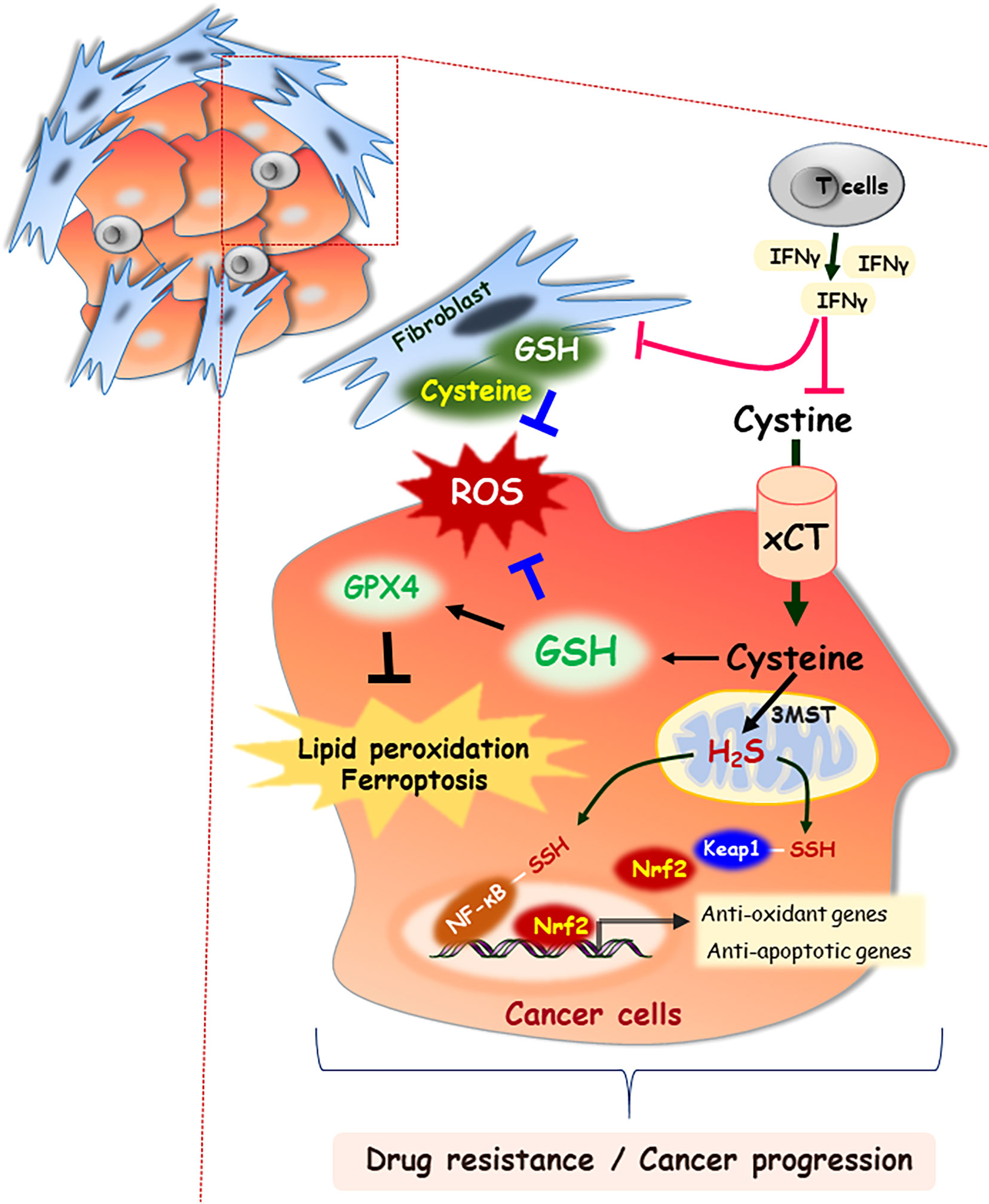
Figure 2 Cysteine as an important building block for cell-cell communication in tumor microenvironment. In the extracellular space, cysteine mainly exists as the oxidized dimer form, cystine. Both forms of cysteine and cystine can be imported into cells through specific transporters, xCT. GSH is synthesized from cysteine, which functions in concert to detoxify ROS. GSH acts as an essential cofactor for GPX4 enzymatic activity, which results in blocking lipid peroxidation and ferroptosis. Stromal fibroblast-derived cysteine and GSH also scavenge the ROS in tumor microenvironment, which contribute to cancer progression. However, IFN-γ derived from CD8+ T cells attenuates drug resistance through downregulation of xCT. In addition, 3MST resident in mitochondria generates H2S, and then it induces the NF-κB activation by persulfidation of its subunit or nuclear localization of Nrf2 via persulfidation of Keap1.
GSH peroxidase 4 (GPX4)
The tripeptide GSH consists of glutamate, cysteine, and glycine and serves as a cysteine storage and transport form (14). It is synthesized by the consecutive action of two ATP-dependent enzymes, γ-glutamylcysteine synthetase, and GSH synthetase. The extracellular enzyme γ-glutamyltranspeptidase cleaves the γ-peptide linkage of GSH to release the product cysteinyl-glycine and glutamate. Cysteinyl-glycine can be hydrolyzed to cysteine and glycine, which can be imported into the cell (15, 16). Ferroptosis is an iron-dependent nonapoptotic cell death driven by lipid peroxidation and membrane damage, and is associated with a redox imbalance (17). GSH is an essential cofactor for GPX4 enzymatic activity (18). GPX4 converts GSH into oxidized glutathione (GSSG), which then reduces cytotoxic lipid peroxides to lipid alcohols. Inhibition of GPX4 activity leads to the accumulation of lipid peroxides, which triggers ferroptic cell death (19). When the cells encounter a decrease in cysteine levels, ferroptosis is triggered by the degradation of GPX4 via chaperone-mediated autophagy (20). Recently, Zhang et al. reported that cysteine starvation impairs GPX4 protein expression by inactivating mTORC1/4E-BP1-mediated protein translation. mTORC1 inhibitors also sensitize cancer cells to ferroptosis induced by GPX4 inhibitor in renal cancer UMRC6 cells (21). In addition, under dual depletion of cystine and GSH, hepatocellular carcinoma HepG2 cells undergo ferroptosis, characterized by a marked increase in lipid peroxidation, and this cell death program can be rescued by a ferroptosis inhibitor treatment (22). Moreover, Yang et al. reported that the administration of inhibitors targeting GPX4 directly or indirectly through GSH depletion suppresses tumor formation in mice bearing human fibrosarcoma HT-1080 xenografts (19). Fu et al. demonstrated that cisplatin-resistant gastric cancer cells had lower levels of ATF3 than their parental cells, which results in reduced ferroptosis due to low ROS, lipid peroxidation, and higher intracellular GSH levels (23). ATF3 sensitizes gastric carcinoma cells to cisplatin through ferroptosis induction by blocking the Nrf2/Keap1/xCT signaling pathway (23). In line with this notion, Fan et al. reported that the activation of the Nrf2/Keap1 pathway increased the xCT expression and diminished ferroptosis, which facilitating glioma cell growth (24). Also, Roh et al. showed that the inhibition of xCT sensitized cisplatin-resistant head and neck cancer (HNC) cell lines to cisplatin by inducing ferroptotic cell death via glutathione depletion and ROS accumulation (25).
Hydrogen sulfide
H2S is produced in mammalian cells by three enzymes, including cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtansferase (3MST) in mammalian cells. CBS and CSE are located in the cytosol of cells, whereas 3MST primarily resides in and generates H2S in mitochondria. Studies have reported that of H2S involved in both the inhibition and advancement of cancer. Knockdown of CBS decreases bioenergetics, actions such as oxygen consumption and ATP production in colon and ovarian cancer cells (26, 27). H2S might enhance glucose uptake by stimulating GLUT activity, thereby accelerating glycolysis. This supports the production of intracellular ATP required by cancer cell proliferation (28). In addition, vascular endothelial growth factor increases H2S level by upregulating CSE expression in endothelial cells, thereby promoting angiogenesis of endothelial cells obtained from breast carcinomas (B-TECs) (29). It also prompts to deliver nutrients and oxygen to cancer cells (29). CSE knockdown suppresses vascular endothelial growth factor--induced migration of B-TECs (29). H2S can mediate hypoxia-induced angiogenesis in cancer progression by inhibiting the catabolism of H2S and increases the expression of CSE (30, 31). Moreover, H2S exerts a protective effect against various apoptotic stimuli through the activation of NF-κB and Nrf2 mediated by H2S-linked persulfidation (32, 33). In the same context, H2S is able to accelerate the cell cycle in cancer cells by upregulating the expression of proliferating cell nuclear antigen and cyclin-dependent kinase 4, thereby promoting cell proliferation in oral squamous cell carcinoma (Figure 2) (34).
Taurine
Cysteine dioxygenases catalyzes the oxidation of cysteine to cysteine sulfinate. Cysteine sulfinic acid decarboxylase catalyzes the reaction, and carboxyl groups are removed to form hypotaurine, which subsequently generates taurine (35). Taurine, also known as 2-aminoethanesulfonic acid, is the most abundant free sulfur-containing amino acid in mammalian tissues, and possesses anti-oxidative, anti-inflammatory, and anti-apoptotic effects (36). Marcinkiewicz and Kontny reported that taurine has cytoprotection function and maintains the homeostasis of cells involved in acute and chronic inflammatory/oxidative stress (37). Several studies have confirmed that taurine displays a strong growth-inhibitory effect on various cancer types including colon cancer (38), lung cancer (39), hepatocarcinoma (40), melanoma (41), and breast cancer (42). El Agouza et al. reported that serum taurine levels decreased in patients with high breast cancer risk, which was linked to decreased angiogenesis (43). Moreover, taurine exerts a strong antitumor effect on rats harboring mammary carcinogenesis, which can be attributed to disturbances in the energy metabolism. Plasma concentrations of fumarate, malate, citrate, α-ketoglutarate, and pyruvate involved in glycolysis and the tricarboxylic acid (TCA) cycle are lower in the taurine-supplemented breast cancer mice group than their concentrations in a normally matched group of mice (44).
Cysteine in carbon metabolism reprogramming
Metabolic reprogramming, an important cancer hallmark, refers to the ability of cancer cells to modify their metabolism, to support the increased energy demand due to the continuous growth and rapid proliferation of cancerous cells. In fact, the metabolic changes in glucose, lipids, and amino acids provide the cancer cells with the energy and substances needed for biosynthesis and the maintenance of biological functions (45). Amino acids participate in important processes such as oncogenesis and progression and are important raw materials for cell anabolism. Recently, Nunes et al. reported that cysteine promotes sulfur and carbon metabolic reprogramming, the underlying the adaptation of ovarian cancer cells to hypoxic microenvironment. (46). In fact, the xCT transporter localizes in mitochondria of ovarian cancer cells, and then an increase in intracellular cysteine facilitates ATP production under hypoxia conditions (46). Nunes et al. reported that cysteine allows ovarian cancer cells to adapt to hypoxic environments and to escape from carboplatin cytotoxicity (47, 48). Although it plays a minor role, transsulfuration contributes to de novo cysteine synthesis from methionine which is recognized as an additional mechanism for maintaining cysteine pools in the tumor microenvironment (49). Enhanced transsulfuration activity driven by the glycine N-methyltransferase may contribute to cysteine biosynthesis and promote cancer cell survival in cysteine-limited microenvironment, which has been demonstrated to support tumor growth in vivo (50). Liu et al. reported that upregulated transsulfuration pathway for cysteine synthesis in erastin-resistant ovarian cancer cells compensates for cysteine deprivation by xCT blockage, which is mediated by Nrf2-mediated CBS activation (51). Knockdown of CBS promotes cellular oxidative stress and lipid peroxidation, thus enhancing ferroptosis susceptibility (51). Moreover, patient-derived basal-like breast cancer tumors exhibited elevated expression of CBS. Anti-proliferative effect and diminished malignant transformation are observed after CBS silencing in basal-like breast cancer cells. Disruption of CBS inhibits both hypoxic response and tumor angiogenesis in basal-like breast cancer cell-derived xenograft tumors, which have larger intratumoral necrotic areas. It is due to the increased vulnerability to oxidative stress and ferroptosis induced by cysteine deprivation (52). Floros et al. reported that MYCN stimulates the transsulfuration pathway through the induction of the key enzymes CBS and methylthioadenosine phosphorylase, and further protects neuroblastomas from ferroptotic cell death (53). Cysteine is a valuable carbon source, since its catabolism produces organic compounds such as pyruvate, α-ketobutyrate, glutamate, serine, propionyl-CoA, succinate, and acetyl-CoA which supply the TCA cycle, and are intermediates for fatty acid synthesis (54, 55).
Post-translational modifications targeting cysteine of protein in cancer
PTMs is a biochemical modification occurring to one or more amino acids on a protein during or after protein translation. PTMs are critical molecular events in a series of biological processes such as cell growth, proliferation, differentiation, metabolism, and apoptosis (56). In most proteomes, cysteine residues are frequently low but with high chemical reactivity. The cysteine thiols could be the nucleophilic residue attacking the substrate. Cysteine residues also react with each other to form disulfide bonds which stabilize the three-dimensional structure and alter the redox state (57). Protein oxidation, lipidation, and metabolites-mediated protein modification occur in cysteine (58). These PTMs are involve in various pathological events or diseases such as inflammation, carcinogenesis, aging, and neurodegenerative disorders (59–61).
Protein oxidation
Cysteine residues in proteins are easily oxidized by ROS, reactive nitrogen species, reactive sulfur species, or GSH. Protein S-nitrosylation, the covalent attachment of nitric oxide (NO) moiety to the reactive thiol group of a cysteine residue to form S-nitrosothiol is an important PTM for most classes of proteins (62). Numerous S-nitrosylated proteins such as Bcl-2, p53, HIF-1α, PTEN, and Src are involved in cell survival, angiogenesis, tumorigenesis, and response to cancer treatment (63–68). NO impairs the apoptotic function of cells and increases resistance to cisplatin-induced cell death in human lung carcinoma cells. NO production induces S-nitrosylation of Bcl-2, which inhibits its ubiquitination and subsequent proteasomal degradation (64). In addition, S-nitrosylation on Cys498 residue of Src kinase induces autophosphorylation, which promotes nitric oxide-mediated cell invasion and resistance to anoikis in cancer cells (68, 69). NO donors such as sodium nitroprusside and S-nitrosoglutathione promote S-nitrosylation at the Cys183 residue of extracellular signal-regulated kinase 1/2 (ERK1/2), which leads to inducing apoptosis in U251 glioma cells (70). Therefore, S-nitrosylation affects a variety of proteins that play important roles in the cellular dysfunctions and contribute to cancer progression and response to chemotherapy.
Protein lipidation
Protein lipidation is an important PTM that can reversibly or irreversibly attach lipid types to proteins, and the three major lipidation processes are palmitoylation, myristoylation, and farnesylation. Palmitoylated proteins are modified by the attachment of fatty acid to cysteine residues via thioester linkage. Palmitoylation of protein regulates its binding at the plasma membrane, lipid raft localization, and protein stability (71). In prostate and breast cancer cells, palmitoylation occurs at the cysteine residue 797 of the epidermal growth factor receptor (EGFR) residing in mitochondria, which stimulates the activation of EGFR. It promotes mitochondrial fusion and cell survival by upregulating mitochondrial prohibitin 2 and optic atrophy 1 protein levels (72). In addition, the isoprenyl group can react with cysteine thiol and bind to proteins, thus forming irreversible S-prenylation reactions such as farnesylation. Farnesyltransferase is an enzyme for farnesylation on the cysteine residue of the CAAX motif region of cytosolic RAS protein, resulting in RAS protein association with cellular membranes. Fatty acid synthase is a metabolic enzyme involved in liponeogenesis and its overexpression has been associated with poor prognosis and shorter disease-free survival in patients with prostate cancer, lung cancer, and sarcoma (73–75). Scribble (Scrib) organizes cell polarity gradients and suppresses aberrant growth signals in various human cancers. Scrib Cys4 and Cys10 residues are required for palmitoylation of Scrib by ZDHHC protein acyl transferases (76). The expression of the epithelial-mesenchymal transition transcription factor Snail leads to Scrib displacement from the plasma membrane to the cytosol, which is associated with disrupting S-palmitoylation of Scrib in epithelial cancer cells (77).
Metabolite-mediated PTMs
Metabolites have been shown to be involved in critical biological changes and regulations. Itaconate, derived from citrate produced in the TCA cycle, contains α,β-unsaturated carboxylic acid, and covalently modifies at the Cys151 residue of Keap1 (78). Itaconate promotes tumor growth via oxidative phosphorylation-driven ROS generation in peritoneal tissue-resident macrophages and concomitant ROS-mediated mitogen-activated protein kinases (MAPKs) activation in tumor cells (79). Interestingly, Nrf2 expression is significantly downregulated in peritoneal tissue-resident macrophages isolated from Immune-responsive gene 1 (Irg1) shRNA-injected tumor-bearing mouse (79). Irg1 is a mitochondrial enzyme that produces itaconates. Cys151 residue is required to inhibit Keap1-mediated Nrf2 degradation, thus itaconate upregulates Nrf2 levels via Keap1 alkylation (Figure 3). Moreover, 4-hydroxy-2-nonenal (HNE), a major α,β-unsaturated aldehyde product of n-6 fatty acid oxidation, is involved in metabolic and neurodegenerative diseases, inflammatory diseases, and cancer (80). 4-HNE can form adducts with Fas, a death receptor protein with a cysteine-rich extracellular domain (81). Also, 4-HNE induces Daxx protein level and promotes the export of Daxx from the nucleus to the cytosol in Jurkat T lymphocyte cells. Daxx is then bound to Fas, which leads to suppression of apoptosis (81). Moreover, cytoplasmic translocation of Daxx induces up-regulation of heat shock factor 1 associated stress-responsive genes, which may contribute to resistance to apoptosis.
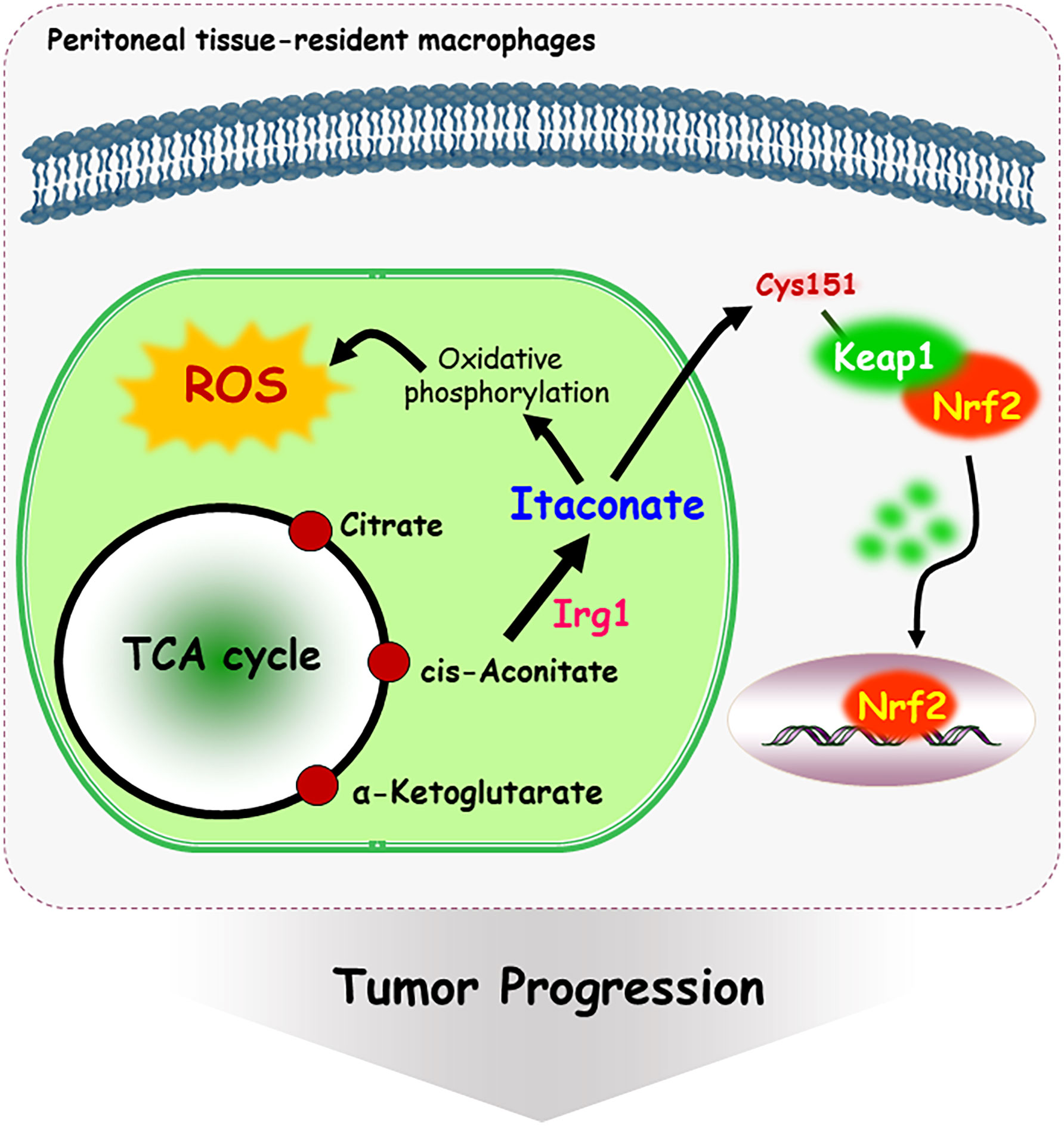
Figure 3 Mechanism of itaconate-mediated cysteine modification. Immune-responsive gene 1 (Irg1) is a mitochondrial enzyme-producing itaconate. Itaconate, covalently modifies at the Cys 151 residue of Keap1, following Nrf2 nuclear localization and activation. In addition, itaconate promotes tumor growth via oxidative phosphorylation-driven ROS generation in peritoneal tissue-resident macrophages.
Role of cysteine residues for inhibitors targeting protein kinases and phosphatases
PTMs can control the biological function of numerous phosphatases, kinases, and transcription factors, which can alter the intracellular localization of target proteins or their interaction with binding partner proteins. Several studies have been conducted on cysteine modification by various endogenous and exogenous molecules/chemicals in various transcription factors (82). Recently, the ability of oxidative stress and small molecule inhibitor-induced cysteine modifications have received considerable attention for regulating the function of both protein phosphatases and kinases (83).
Protein phosphatases belong to two major families of phosphatases: serine/threonine protein phosphatases and protein tyrosine phosphatases (PTPs). PTPs regulate signal transduction pathways involving tyrosine phosphorylation and have been implicated in the development of cancer (84, 85). The active site of most classical PTPs is formed by the P-loop, which contains the conserved PTP signature motif (H/V)C(X)5R(S/T). The cellular redox state is involved in regulating tyrosine phosphatase activity through the reversible oxidation of catalytic cysteine to sulfenic/sulfinic acid (86). The catalytic cysteine is highly susceptible to oxidation and nitrosylation, leading to reversible or irreversible modifications that abolish its nucleophilic function and inactivate its enzyme activity (87, 88). Oxidation of the Cys residue in the active site of PTPs by ROS abrogates its nucleophilic properties, thereby inhibiting PTP activity (87). PTP1B acts as a negative regulator of multiple receptor tyrosine kinases, including the EGFR. NADPH oxidase 4 (NOX4)-mediated oxidation of PTP1B in the endoplasmic reticulum enhances EGFR phosphorylation (89). Bile acids can trigger mitochondrial ROS generation in hepatocytes that in turn act to mediate the inactivation of PTPs, resulting in the activation of EGFR (90). In addition, S-nitrosylation of the Cys215 residue by NO donors shields PTP1B from subsequent hydrogen peroxide-induced irreversible oxidation (88). PTPN22 (short for protein tyrosine phosphatase non-receptor type 22), known as a risk factor in multiple autoimmune disorders, reduces T-cell activity by removing phosphate groups from phosphorylated proteins such as LCK, Fyn, and Zap70, which are associated with T-cell receptor signaling pathway. Interaction between the non-catalytic cysteine at position 129 and the catalytic cysteine 227 shows the formation of a disulfide bond, which maintains its catalytic activity (91, 92). However, when Cys 129 residue of PTPN22 is mutated, the disulfide bond cannot form and the enzyme is exposed by oxidation, resulting in inactivation (93).
Recently, the discovery of reversible/irreversible covalent inhibitors targeting cysteine residues in and around the ATP-binding pocket of kinases has been gaining considerable attention (94). Hypothemycin, one of the resorcylic acid lactones, is a representative inhibitor targeting cysteine residues located in the ATP site of Ser/Thr/Tyr protein kinases. The α,β-unsaturated enone moiety of resorcylic acid lactones is susceptible to Michael addition reaction with a conserved cysteine residue (Cys166 in human ERK2) (95). In addition, hypothemycin inhibits the phosphorylation of the mitogen-activated protein kinase kinase (MEK)3/6 substrate p38, the MEK4/7 substrate c-Jun N-terminal kinase (JNK), and the TGF-β-activated kinase 1 (TAK1) substrate IκB kinase β. MEK and TAK1 contain the conserved cysteine residue corresponding to ERK2 (95). Afatinib, the first covalent inhibitor of EGFR approved by the FDA, binds to Cys797 residue on the kinase domain of EGFR in the “DFG-in” conformation (94). Tan et al., reported that FIIN-2, an irreversible covalent FGFR inhibitor, formed the covalent binding mode at the Cys477 residue of FGFR4 in the “DGF-out” conformation (96). Furthermore, a newly synthesized compound targeting Cys174 at the DFG-1 position in TAK1 is considered a type II inhibitor (97). There is a growing interest in the discovery of kinase inhibitors that can be reversibly or irreversibly modulated by targeting cysteine. Many researchers have focused on understanding the mode of action of inhibitors that can act differently depending on the DGF-in or DFG-out conformation.
Conclusion and future perspectives
As shown in the Figure 4, cysteine has different fates including the synthesis of cysteine-derived molecule, sulfur/carbon metabolic reprogramming, and venue for post-translational protein modification of various proteins or discovery of drug inhibitors. Intracellular and extracellular alteration of amino acid metabolism in the tumor microenvironment can influence cancer growth, progression, and metastasis. Cysteine, as a multifaceted precursor, plays a central role in cellular metabolism and contributes to the survival and proliferation of cancer cells. Changes in the redox state of cells by cystine/cysteine circuitry control the ROS levels, which modulates cellular signal transduction pathways involved in cell survival and resistance to chemotherapy. Several proteins, involving covalent or non-covalent cysteine modifications have been also identified. Naturally or synthetic chemotherapeutic agents exert their effects through oxidation or modification of cysteine thiol groups present in the cellular signal molecules mediated by phosphatases and kinases. Covalent or non-covalent inhibitors are again attracting attention, and they target site-specifically a cysteine residue near the active pocket of druggable proteins. Further studies are needed to validate the reversible/irreversible modes of action of covalent inhibitors.
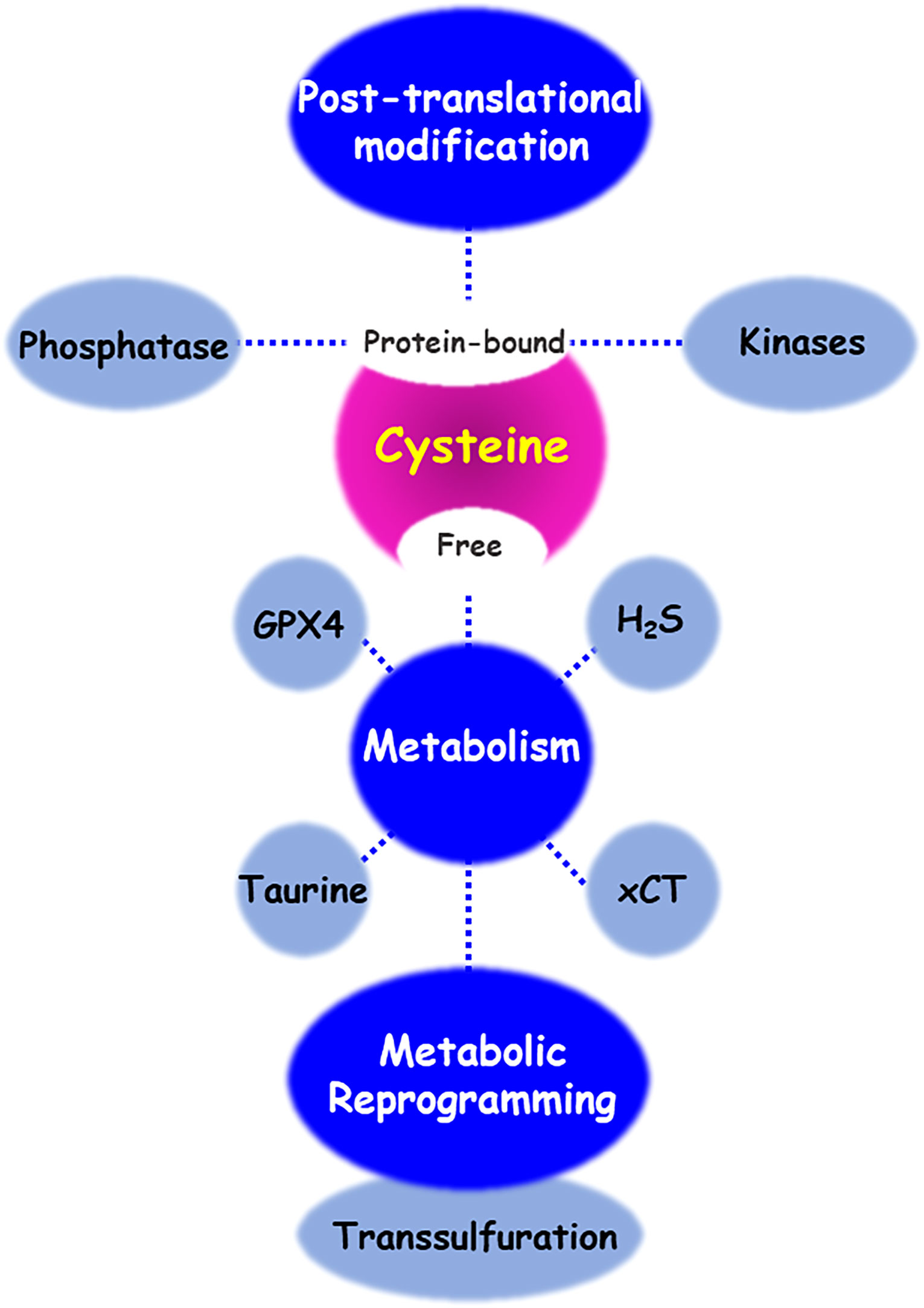
Figure 4 Cysteine metabolic fate. There are diverse regulatory mechanisms of cysteine bound to or free from proteins. Enzymes and metabolites of cysteine transport and metabolism enables cancer cells to contributing cancer progression and acquiring chemoresistance. In addition, cysteine residue of proteins undergoes various post-translational modifications, to maintain its redox homeostasis. Oxidative stress and small molecule inhibitors-induced cysteine modifications regulate the function of both protein phosphatases and kinases.
Author contributions
J-YM documented paper, drew figures, and summarized data. K-SC and D-HK designed paper and supervised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Kyonggi University’s Graduate Research Assistantship 2021, and the Basic Science Research Program grant (No. 2020R1A2C1103139) from the National Research Foundation (NRF) of the Republic of Korea.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chevallier V, Zoller M, Kochanowski N, Andersen MR, Workman CT, Malphettes L. Use of novel cystine analogs to decrease oxidative stress and control product quality. J Biotechnol (2021) 327:1–8. doi: 10.1016/j.jbiotec.2020.12.011
2. Paul BD, Sbodio JI, Snyder SH. Cysteine metabolism in neuronal redox homeostasis. Trends Pharmacol Sci (2018) 39(5):513–24. doi: 10.1016/j.tips.2018.02.007
3. Go YM, Chandler JD, Jones DP. The cysteine proteome. Free Radic Biol Med (2015) 84:227–45. doi: 10.1016/j.freeradbiomed.2015.03.022
4. Casini A, Scozzafava A, Supuran CT. Cysteine-modifying agents: A possible approach for effective anticancer and antiviral drugs. Environ Health Perspect (2002) 110 Suppl 5:801–6. doi: 10.1289/ehp.02110s5801
5. Stepka P, Vsiansky V, Raudenska M, Gumulec J, Adam V, Masarik M. Metabolic and amino acid alterations of the tumor microenvironment. Curr Med Chem (2021) 28(7):1270–89. doi: 10.2174/0929867327666200207114658
6. Banjac A, Perisic T, Sato H, Seiler A, Bannai S, Weiss N, et al. The cystine/cysteine cycle: A redox cycle regulating susceptibility versus resistance to cell death. Oncogene. (2008) 27(11):1618–28. doi: 10.1038/sj.onc.1210796
7. Wang W, Kryczek I, Dostal L, Lin H, Tan L, Zhao L, et al. Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell. (2016) 165(5):1092–105. doi: 10.1016/j.cell.2016.04.009
8. Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system xc- in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal (2013) 18(5):522–55. doi: 10.1089/ars.2011.4391
9. Lin W, Wang C, Liu G, Bi C, Wang X, Zhou Q, et al. SLC7A11/xCT in cancer: biological functions and therapeutic implications. Am J Cancer Res (2020) 10(10):3106–26.
10. Zhong W, Weiss HL, Jayswal RD, Hensley PJ, Downes LM, St Clair DK, et al. Extracellular redox state shift: A novel approach to target prostate cancer invasion. Free Radic Biol Med (2018) 117:99–109. doi: 10.1016/j.freeradbiomed.2018.01.023
11. Lim JKM, Delaidelli A, Minaker SW, Zhang HF, Colovic M, Yang H, et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc Natl Acad Sci U S A. (2019) 116(19):9433–42. doi: 10.1073/pnas.1821323116
12. Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol (2012) 14(3):276–86. doi: 10.1038/ncb2432
13. Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, et al. Systemic depletion of l-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med (2017) 23(1):120–7. doi: 10.1038/nm.4232
14. Meister A, Anderson ME. Glutathione. Annu Rev Biochem (1983) 52:711–60. doi: 10.1146/annurev.bi.52.070183.003431
15. Hanigan MH, Ricketts WA. Extracellular glutathione is a source of cysteine for cells that express gamma-glutamyl transpeptidase. Biochemistry. (1993) 32(24):6302–6. doi: 10.1021/bi00075a026
16. Meister A. Glutathione, ascorbate, and cellular protection. Cancer Res (1994) 54(7 Suppl):1969s–75s.
17. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
18. Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta (2013) 1830(5):3289–303. doi: 10.1016/j.bbagen.2012.11.020
19. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
20. Fujii J, Homma T, Kobayashi S. Ferroptosis caused by cysteine insufficiency and oxidative insult. Free Radic Res (2020) 54(11-12):969–80. doi: 10.1080/10715762.2019.1666983
21. Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun (2021) 12(1):1589. doi: 10.1038/s41467-021-21841-w
22. Yu X, Long YC. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci Rep (2016) 6:30033. doi: 10.1038/srep30033
23. Fu D, Wang C, Yu L, Yu R. Induction of ferroptosis by ATF3 elevation alleviates cisplatin resistance in gastric cancer by restraining Nrf2/Keap1/xCT signaling. Cell Mol Biol Lett (2021) 26(1):26. doi: 10.1186/s11658-021-00271-y
24. Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. (2017) 6(8):e371. doi: 10.1038/oncsis.2017.65
25. Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett (2016) 381(1):96–103. doi: 10.1016/j.canlet.2016.07.035
26. Bhattacharyya S, Saha S, Giri K, Lanza IR, Nair KS, Jennings NB, et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS One (2013) 8(11):e79167. doi: 10.1371/journal.pone.0079167
27. Szabo C, Coletta C, Chao C, Modis K, Szczesny B, Papapetropoulos A, et al. Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci U S A. (2013) 110(30):12474–9. doi: 10.1073/pnas.1306241110
28. Hellmich MR, Coletta C, Chao C, Szabo C. The therapeutic potential of cystathionine beta-synthetase/hydrogen sulfide inhibition in cancer. Antioxid Redox Signal (2015) 22(5):424–48. doi: 10.1089/ars.2014.5933
29. Pupo E, Pla AF, Avanzato D, Moccia F, Cruz JE, Tanzi F, et al. Hydrogen sulfide promotes calcium signals and migration in tumor-derived endothelial cells. Free Radic Biol Med (2011) 51(9):1765–73. doi: 10.1016/j.freeradbiomed.2011.08.007
30. Cao X, Bian JS. The role of hydrogen sulfide in renal system. Front Pharmacol (2016) 7:385. doi: 10.3389/fphar.2016.00385
31. Wang M, Guo Z, Wang S. Regulation of cystathionine gamma-lyase in mammalian cells by hypoxia. Biochem Genet (2014) 52(1-2):29–37. doi: 10.1007/s10528-013-9624-7
32. Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, et al. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol Cell (2012) 45(1):13–24. doi: 10.1016/j.molcel.2011.10.021
33. Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, et al. Hydrogen sulfide protects against cellular senescence via s-sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal (2013) 18(15):1906–19. doi: 10.1089/ars.2012.4645
34. Ma Z, Bi Q, Wang Y. Hydrogen sulfide accelerates cell cycle progression in oral squamous cell carcinoma cell lines. Oral Dis (2015) 21(2):156–62. doi: 10.1111/odi.12223
35. Stipanuk MH, Jurkowska H, Roman HB, Niewiadomski J, Hirschberger LL. Insights into taurine synthesis and function based on studies with cysteine dioxygenase (CDO1) knockout mice. Adv Exp Med Biol (2015) 803:29–39. doi: 10.1007/978-3-319-15126-7_3
36. Lambert IH, Kristensen DM, Holm JB, Mortensen OH. Physiological role of taurine–from organism to organelle. Acta Physiol (Oxf). (2015) 213(1):191–212. doi: 10.1111/apha.12365
37. Marcinkiewicz J, Kontny E. Taurine and inflammatory diseases. Amino Acids (2014) 46(1):7–20. doi: 10.1007/s00726-012-1361-4
38. Liu Z, Xia Y, Zhang X, Liu L, Tu S, Zhu W, et al. Roles of the MST1-JNK signaling pathway in apoptosis of colorectal cancer cells induced by taurine. Libyan J Med (2018) 13(1):1500346. doi: 10.1080/19932820.2018.1500346
39. Tu S, Zhang XL, Wan HF, Xia YQ, Liu ZQ, Yang XH, et al. Effect of taurine on cell proliferation and apoptosis human lung cancer A549 cells. Oncol Lett (2018) 15(4):5473–80. doi: 10.3892/ol.2018.8036
40. Park SH, Lee H, Park KK, Kim HW, Park T. Taurine-responsive genes related to signal transduction as identified by cDNA microarray analyses of HepG2 cells. J Med Food (2006) 9(1):33–41. doi: 10.1089/jmf.2006.9.33
41. Yu J, Kim AK. Effect of taurine on antioxidant enzyme system in B16F10 melanoma cells. Adv Exp Med Biol (2009) 643:491–9. doi: 10.1007/978-0-387-75681-3_51
42. Choi EJ, Tang Y, Lee CB, Cheong SH, Sung SH, Oh MR, et al. Effect of taurine on in vitro migration of MCF-7 and MDA-MB-231 human breast carcinoma cells. Adv Exp Med Biol (2015) 803:191–201. doi: 10.1007/978-3-319-15126-7_17
43. El Agouza IM, Eissa SS, El Houseini MM, El-Nashar DE, Abd El Hameed OM. Taurine: a novel tumor marker for enhanced detection of breast cancer among female patients. Angiogenesis. (2011) 14(3):321–30. doi: 10.1007/s10456-011-9215-3
44. He YU, Li QQ, Guo SC. Taurine attenuates dimethylbenz[a]anthracene-induced breast tumorigenesis in rats: A plasma metabolomic study. Anticancer Res (2016) 36(2):533–43.
45. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
46. Nunes SC, Ramos C, Santos I, Mendes C, Silva F, Vicente JB, et al. Cysteine boosts fitness under hypoxia-mimicked conditions in ovarian cancer by metabolic reprogramming. Front Cell Dev Biol (2021) 9:722412. doi: 10.3389/fcell.2021.722412
47. Nunes SC, Lopes-Coelho F, Gouveia-Fernandes S, Ramos C, Pereira SA, Serpa J. Cysteine boosters the evolutionary adaptation to CoCl2 mimicked hypoxia conditions, favouring carboplatin resistance in ovarian cancer. BMC Evol Biol (2018) 18(1):97. doi: 10.1186/s12862-018-1214-1
48. Nunes SC, Ramos C, Lopes-Coelho F, Sequeira CO, Silva F, Gouveia-Fernandes S, et al. Cysteine allows ovarian cancer cells to adapt to hypoxia and to escape from carboplatin cytotoxicity. Sci Rep (2018) 8(1):9513. doi: 10.1038/s41598-018-27753-y
49. Zhang HF, Klein Geltink RI, Parker SJ, Sorensen PH. Transsulfuration, minor player or crucial for cysteine homeostasis in cancer. Trends Cell Biol (2022) 32(9):800–14. doi: 10.1016/j.tcb.2022.02.009
50. Zhu J, Berisa M, Schworer S, Qin W, Cross JR, Thompson CB. Transsulfuration activity can support cell growth upon extracellular cysteine limitation. Cell Metab (2019) 30(5):865–76.e5. doi: 10.1016/j.cmet.2019.09.009
51. Liu N, Lin X, Huang C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br J Cancer. (2020) 122(2):279–92. doi: 10.1038/s41416-019-0660-x
52. Erdelyi K, Ditroi T, Johansson HJ, Czikora A, Balog N, Silwal-Pandit L, et al. Reprogrammed transsulfuration promotes basal-like breast tumor progression via realigning cellular cysteine persulfidation. Proc Natl Acad Sci U.S.A. (2021) 118(45):e2100050118. doi: 10.1073/pnas.2100050118
53. Floros KV, Chawla AT, Johnson-Berro MO, Khatri R, Stamatouli AM, Boikos SA, et al. MYCN upregulates the transsulfuration pathway to suppress the ferroptotic vulnerability in MYCN-amplified neuroblastoma. Cell Stress. (2022) 6(2):21–9. doi: 10.15698/cst2022.02.264
54. Bonifacio VDB, Pereira SA, Serpa J, Vicente JB. Cysteine metabolic circuitries: druggable targets in cancer. Br J Cancer. (2021) 124(5):862–79. doi: 10.1038/s41416-020-01156-1
55. Schiliro C, Firestein BL. Mechanisms of metabolic reprogramming in cancer cells supporting enhanced growth and proliferation. Cells. (2021) 10(5):1056. doi: 10.3390/cells10051056
56. Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol (2006) 7(7):473–83. doi: 10.1038/nrm1960
57. Akabas MH. Cysteine modification: Probing channel structure, function and conformational change. Adv Exp Med Biol (2015) 869:25–54. doi: 10.1007/978-1-4939-2845-3_3
58. Bak DW, Bechtel TJ, Falco JA, Weerapana E. Cysteine reactivity across the subcellular universe. Curr Opin Chem Biol (2019) 48:96–105. doi: 10.1016/j.cbpa.2018.11.002
59. Gu L, Robinson RA. Proteomic approaches to quantify cysteine reversible modifications in aging and neurodegenerative diseases. Proteomics Clin Appl (2016) 10(12):1159–77. doi: 10.1002/prca.201600015
60. Held JM, Gibson BW. Regulatory control or oxidative damage? proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol Cell Proteomics. (2012) 11(4):R111 013037. doi: 10.1074/mcp.R111.013037
61. Hoffman S, Nolin J, McMillan D, Wouters E, Janssen-Heininger Y, Reynaert N. Thiol redox chemistry: Role of protein cysteine oxidation and altered redox homeostasis in allergic inflammation and asthma. J Cell Biochem (2015) 116(6):884–92. doi: 10.1002/jcb.25017
62. Wang Z. Protein S-nitrosylation and cancer. Cancer Lett (2012) 320(2):123–9. doi: 10.1016/j.canlet.2012.03.009
63. Azad N, Vallyathan V, Wang L, Tantishaiyakul V, Stehlik C, Leonard SS, et al. S-nitrosylation of bcl-2 inhibits its ubiquitin-proteasomal degradation. a novel antiapoptotic mechanism that suppresses apoptosis. J Biol Chem (2006) 281(45):34124–34. doi: 10.1074/jbc.M602551200
64. Chanvorachote P, Nimmannit U, Stehlik C, Wang L, Jiang BH, Ongpipatanakul B, et al. Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through s-nitrosylation and inhibition of bcl-2 ubiquitination. Cancer Res (2006) 66(12):6353–60. doi: 10.1158/0008-5472.CAN-05-4533
65. Calmels S, Hainaut P, Ohshima H. Nitric oxide induces conformational and functional modifications of wild-type p53 tumor suppressor protein. Cancer Res (1997) 57(16):3365–9.
66. Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, et al. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A. (2009) 106(15):6297–302. doi: 10.1073/pnas.0901043106
67. Yu CX, Li S, Whorton AR. Redox regulation of PTEN by S-nitrosothiols. Mol Pharmacol (2005) 68(3):847–54. doi: 10.1124/mol.104.010504
68. Rahman MA, Senga T, Ito S, Hyodo T, Hasegawa H, Hamaguchi M. S-nitrosylation at cysteine 498 of c-src tyrosine kinase regulates nitric oxide-mediated cell invasion. J Biol Chem (2010) 285(6):3806–14. doi: 10.1074/jbc.M109.059782
69. da Costa PE, Batista WL, Moraes MS, Stern A, Monteiro HP. Src kinase activation by nitric oxide promotes resistance to anoikis in tumour cell lines. Free Radic Res (2018) 52(5):592–604. doi: 10.1080/10715762.2018.1455095
70. Jin L, Cao Y, Zhang T, Wang P, Ji D, Liu X, et al. Effects of ERK1/2 s-nitrosylation on ERK1/2 phosphorylation and cell survival in glioma cells. Int J Mol Med (2018) 41(3):1339–48. doi: 10.3892/ijmm.2017.3334
71. Fhu CW, Ali A. Protein lipidation by palmitoylation and myristoylation in cancer. Front Cell Dev Biol (2021) 9:673647. doi: 10.3389/fcell.2021.673647
72. Bollu LR, Ren J, Blessing AM, Katreddy RR, Gao G, Xu L, et al. Involvement of de novo synthesized palmitate and mitochondrial EGFR in EGF induced mitochondrial fusion of cancer cells. Cell Cycle (2014) 13(15):2415–30. doi: 10.4161/cc.29338
73. Rossi S, Graner E, Febbo P, Weinstein L, Bhattacharya N, Onody T, et al. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol Cancer Res (2003) 1(10):707–15.
74. Takahiro T, Shinichi K, Toshimitsu S. Expression of fatty acid synthase as a prognostic indicator in soft tissue sarcomas. Clin Cancer Res (2003) 9(6):2204–12.
75. Visca P, Sebastiani V, Botti C, Diodoro MG, Lasagni RP, Romagnoli F, et al. Fatty acid synthase (FAS) is a marker of increased risk of recurrence in lung carcinoma. Anticancer Res (2004) 24(6):4169–73.
76. Chen B, Zheng B, DeRan M, Jarugumilli GK, Fu J, Brooks YS, et al. ZDHHC7-mediated S-palmitoylation of scribble regulates cell polarity. Nat Chem Biol (2016) 12(9):686–93. doi: 10.1038/nchembio.2119
77. Hernandez JL, Davda D, Cheung See Kit M, Majmudar JD, Won SJ, Gang M, et al. APT2 inhibition restores scribble localization and S-palmitoylation in snail-transformed cells. Cell Chem Biol (2017) 24(1):87–97. doi: 10.1016/j.chembiol.2016.12.007
78. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. (2018) 556(7699):113–7. doi: 10.1038/nature25986
79. Weiss JM, Davies LC, Karwan M, Ileva L, Ozaki MK, Cheng RY, et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J Clin Invest. (2018) 128(9):3794–805. doi: 10.1172/JCI99169
80. Zarkovic K, Jakovcevic A, Zarkovic N. Contribution of the HNE-immunohistochemistry to modern pathological concepts of major human diseases. Free Radic Biol Med (2017) 111:110–26. doi: 10.1016/j.freeradbiomed.2016.12.009
81. Sharma R, Sharma A, Dwivedi S, Zimniak P, Awasthi S, Awasthi YC. 4-hydroxynonenal self-limits Fas-mediated DISC-independent apoptosis by promoting export of Daxx from the nucleus to the cytosol and its binding to Fas. Biochemistry. (2008) 47(1):143–56. doi: 10.1021/bi701559f
82. Lennicke C, Cocheme HM. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol Cell (2021) 81(18):3691–707. doi: 10.1016/j.molcel.2021.08.018
83. Turdo A, D'Accardo C, Glaviano A, Porcelli G, Colarossi C, Colarossi L, et al. Targeting phosphatases and kinases: How to checkmate cancer. Front Cell Dev Biol (2021) 9:690306. doi: 10.3389/fcell.2021.690306
84. Neel BG, Tonks NK. Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol (1997) 9(2):193–204. doi: 10.1016/S0955-0674(97)80063-4
85. Zhang ZY. Protein tyrosine phosphatases: structure and function, substrate specificity, and inhibitor development. Annu Rev Pharmacol Toxicol (2002) 42:209–34. doi: 10.1146/annurev.pharmtox.42.083001.144616
86. Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. (1998) 37(16):5633–42. doi: 10.1021/bi973035t
87. Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. (2005) 121(5):667–70. doi: 10.1016/j.cell.2005.05.016
88. Chen YY, Chu HM, Pan KT, Teng CH, Wang DL, Wang AH, et al. Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J Biol Chem (2008) 283(50):35265–72. doi: 10.1074/jbc.M805287200
89. Chen K, Kirber MT, Xiao H, Yang Y, Keaney JF Jr. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J Cell Biol (2008) 181(7):1129–39. doi: 10.1083/jcb.200709049
90. Fang Y, Han SI, Mitchell C, Gupta S, Studer E, Grant S, et al. Bile acids induce mitochondrial ROS, which promote activation of receptor tyrosine kinases and signaling pathways in rat hepatocytes. Hepatology. (2004) 40(4):961–71. doi: 10.1002/hep.1840400427
91. Bottini N, Vang T, Cucca F, Mustelin T. Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin Immunol (2006) 18(4):207–13. doi: 10.1016/j.smim.2006.03.008
92. Fousteri G, Liossis SN, Battaglia M. Roles of the protein tyrosine phosphatase PTPN22 in immunity and autoimmunity. Clin Immunol (2013) 149(3):556–65. doi: 10.1016/j.clim.2013.10.006
93. James J, Chen Y, Hernandez CM, Forster F, Dagnell M, Cheng Q, et al. Redox regulation of PTPN22 affects the severity of T-cell-dependent autoimmune inflammation. Elife (2022) 11:e74549. doi: 10.7554/eLife.74549
94. Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol (2013) 20(2):146–59. doi: 10.1016/j.chembiol.2012.12.006
95. Schirmer A, Kennedy J, Murli S, Reid R, Santi DV. Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. Proc Natl Acad Sci U S A. (2006) 103(11):4234–9. doi: 10.1073/pnas.0600445103
96. Tan L, Wang J, Tanizaki J, Huang Z, Aref AR, Rusan M, et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc Natl Acad Sci U S A. (2014) 111(45):E4869–77. doi: 10.1073/pnas.1403438111
Keywords: cysteine, cancer, resistance, chemotherapy, post-translational modification
Citation: Min J-Y, Chun K-S and Kim D-H (2023) The versatile utility of cysteine as a target for cancer treatment. Front. Oncol. 12:997919. doi: 10.3389/fonc.2022.997919
Received: 19 July 2022; Accepted: 28 November 2022;
Published: 19 January 2023.
Edited by:
Cinzia Domenicotti, Università di Genova, ItalyReviewed by:
Barbara Marengo, University of Genoa, ItalyPaul Dent, Virginia Commonwealth University, United States
Copyright © 2023 Min, Chun and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Do-Hee Kim, ZG9oZWVAa3lvbmdnaS5hYy5rcg==