 Shuai-Shuai Ge
Shuai-Shuai Ge Song-Bai Liu
Song-Bai Liu Sheng-Li Xue
Sheng-Li Xue- 1National Clinical Research Center for Hematologic Diseases, Jiangsu Institute of Hematology, The First Affiliated Hospital of Soochow University, Suzhou, China
- 2Institute of Blood and Marrow Transplantation, Collaborative Innovation Center of Hematology, Soochow University, Suzhou, China
- 3Suzhou Key Laboratory of Medical Biotechnology, Suzhou Vocational Health College, Suzhou, China
FLT3 mutations are one of the most common genetic alterations in acute myeloid leukemia (AML) and are identified in approximately one-third of newly diagnosed patients. Aberrant FLT3 receptor signaling has important implications for the biology and clinical management of AML. In recent years, targeting FLT3 has been a part of every course of treatment in FLT3-ITD/TKD-mutated AML and contributes to substantially prolonged survival. At the same time, wide application of next-generation sequencing (NGS) technology has revealed a series of non-canonical FLT3 mutations, including point mutations and small insertions/deletions. Some of these mutations may be able to influence downstream phosphorylation and sensitivity to FLT3 inhibitors, while the correlation with clinical outcomes remains unclear. Exploration of FLT3-targeted therapy has made substantial progress, but resistance to FLT3 inhibitors has become a pressing issue. The mechanisms underlying FLT3 inhibitor tolerance can be roughly divided into primary resistance and secondary resistance. Primary resistance is related to abnormalities in signaling factors, such as FL, CXCL12, and FGF2, and secondary resistance mainly involves on-target mutations and off-target aberrations. To overcome this problem, novel agents such as FF-10101 have shown promising potential. Multitarget strategies directed at FLT3 and anomalous signaling factors simultaneously are in active clinical development and show promising results.
1 Introduction
Acute myeloid leukemia (AML) is a clonal stem cell malignancy that is characterized by infiltration of abnormally differentiated myeloid progenitor cells (blasts) and has a historically high mortality rate (1, 2). FLT3 (FMS-like tyrosine kinase-3) is a type 3 receptor tyrosine kinase (RTK) that consists of five Ig-like domains in the extracellular region, a juxtamembrane (JM) domain, and an interrupted kinase domain in the intracellular region (3). In normal cells, FLT3 is mainly expressed in hematopoietic stem or progenitor cells and plays an important role in hematopoietic expansion by binding the FLT3 ligand (FL) (4). Most primary AML cells also express FLT3, accompanied by FL stimulation leading to proliferation and anti-apoptosis of AML cells (5–7).
FLT3 is one of the most commonly mutated genes in acute myeloid leukemia, accounting for 15%-35% of newly diagnosed patients (8). Most of these mutations are internal tandem duplication (ITD) mutations that insert into the juxtamembrane region and tyrosine kinase domain 1, while activating point mutations localized in the tyrosine kinase domain activating loop (TKD) are less frequently observed. ITD and TKD mutations occur in approximately 20% and 7% of AML patients, respectively (9, 10). FLT3 mutations in AML are of clinical significance. FLT3-ITD mutations are strongly linked to worse clinical features and poor prognosis, making FLT3-ITD an independent prognostic marker. while the presence of FLT3-TKD mutations is correlated with a favorable prognosis over FLT3-ITD-mutated patients (10–13). With the wide application of NGS, an increasing number of non-canonical activating point mutations and indel alterations have been detected, but it is unclear whether those non-canonical FLT3 alterations are associated with prognosis because of their low incidence (14–18).
In the era of targeted therapy, mutant FLT3 serves as a promising molecular target spot for the treatment of AML, and great changes have been made in the clinical management of FLT3-mutated AML due to the development of FLT3 inhibitors (13, 19, 20). However, drug resistance remains a challenge despite FLT3 inhibitors providing a dramatic therapeutic response in the frontline and relapsed/refractory settings (21–24). This review mainly focuses on the recent progress in applying FLT3 inhibitors in AML and the mechanisms of drug resistance.
2 Classification of FLT3 inhibitors
FLT3 inhibitors can be classified using two primary schemas: generation and type. The first-generation FLT3 inhibitors were tyrosine kinase inhibitors (TKIs) with multi-kinase target activity. Existing first-generation TKIs include lestaurtinib (CEP-701), sunitinib (SU11248), midostaurin (PKC412), and sorafenib (BAY43-9006) (25–28). The antileukemic effects of these multi-kinase inhibitors likely derive from the simultaneous inhibition of FLT3 and parallel pathways, but multiple off-target effects also bring about increased toxicities (29). Subsequently, second-generation FLT3 inhibitors with higher selectivity and inhibitory activity were identified. Second-generation FLT3 inhibitors can more selectively inhibit FLT3, and thus have greater clinical potential and fewer off-target effects. Second-generation FLT3 inhibitors include gilteritinib (ASP2215), quizartinib (AC220), and crenolanib (CP868596). In plasma, first-generation inhibitors have higher IC50 values and shorter half-lives than their second-generation counterparts, which explains their limited clinical potency (30–32).
Furthermore, these FLT3 inhibitors can be roughly classified into two types according to the binding mode to FLT3. Type I inhibitors bind to the ATP-binding site in the intracellular active pocket and enable downregulation of the phosphorylation of both ITD and TKD mutations. In contrast, because type II inhibitors are designed to favorably bind to the hydrophobic space of the inactive conformation of FLT3, they are made inaccessible by TKD mutations (24). The details of established FLT3 inhibitors are summarized in Table 1.
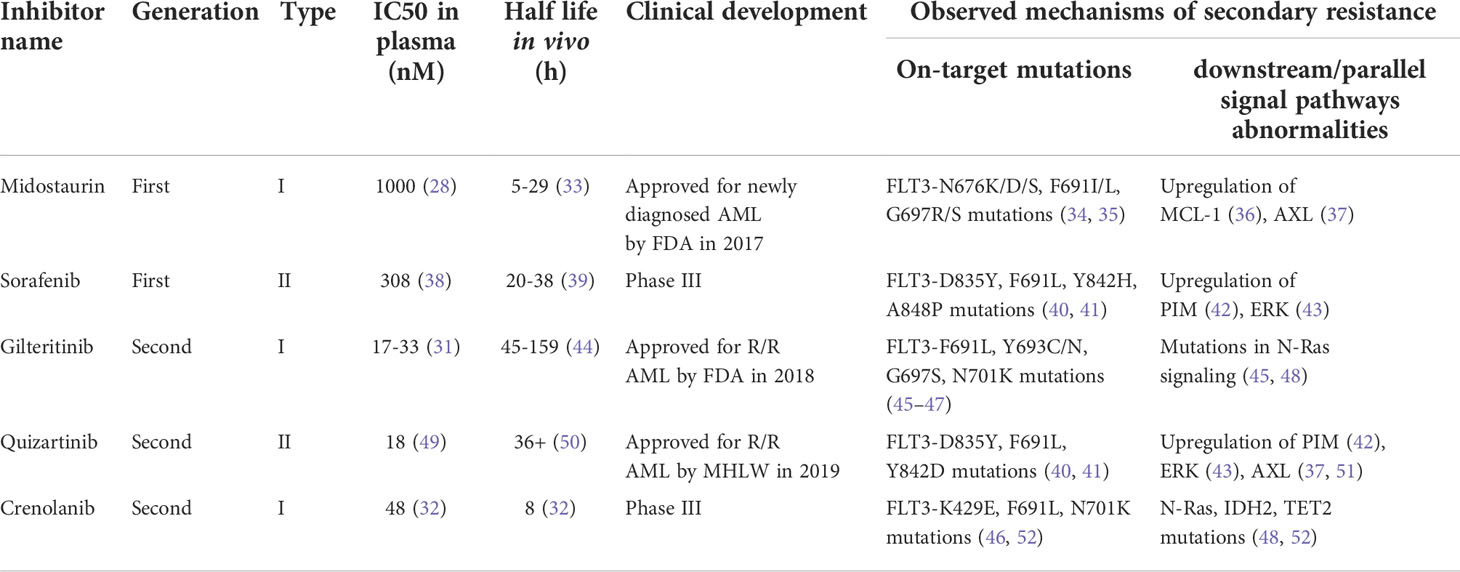
Table 1 Established FLT3 inhibitors and features.
3 FLT3 inhibitors for AML therapy
3.1 Therapy for canonical FLT3 mutated AML
Historically, except for acute promyelocytic leukemia (APL), conventional chemotherapy (“3+7”) alone was the standard strategy for FLT3-mutated AML. Although FLT3-ITD AML had a similar response to induction chemotherapy compared to WT counterparts, a shorter duration of remission and higher relapse rate attracted attention (53, 54). Preliminary results from large multicenter trials showing a survival improvement from a combination of chemotherapy and FLT3 inhibition (compared with historic controls) made this approach look promising (25, 55). After continuous optimization of treatment strategies and renewal of FLT3-targeted drugs, FLT3 inhibitors have already been widely used in the clinical treatment of FLT3-mutated AML (Figure 1).
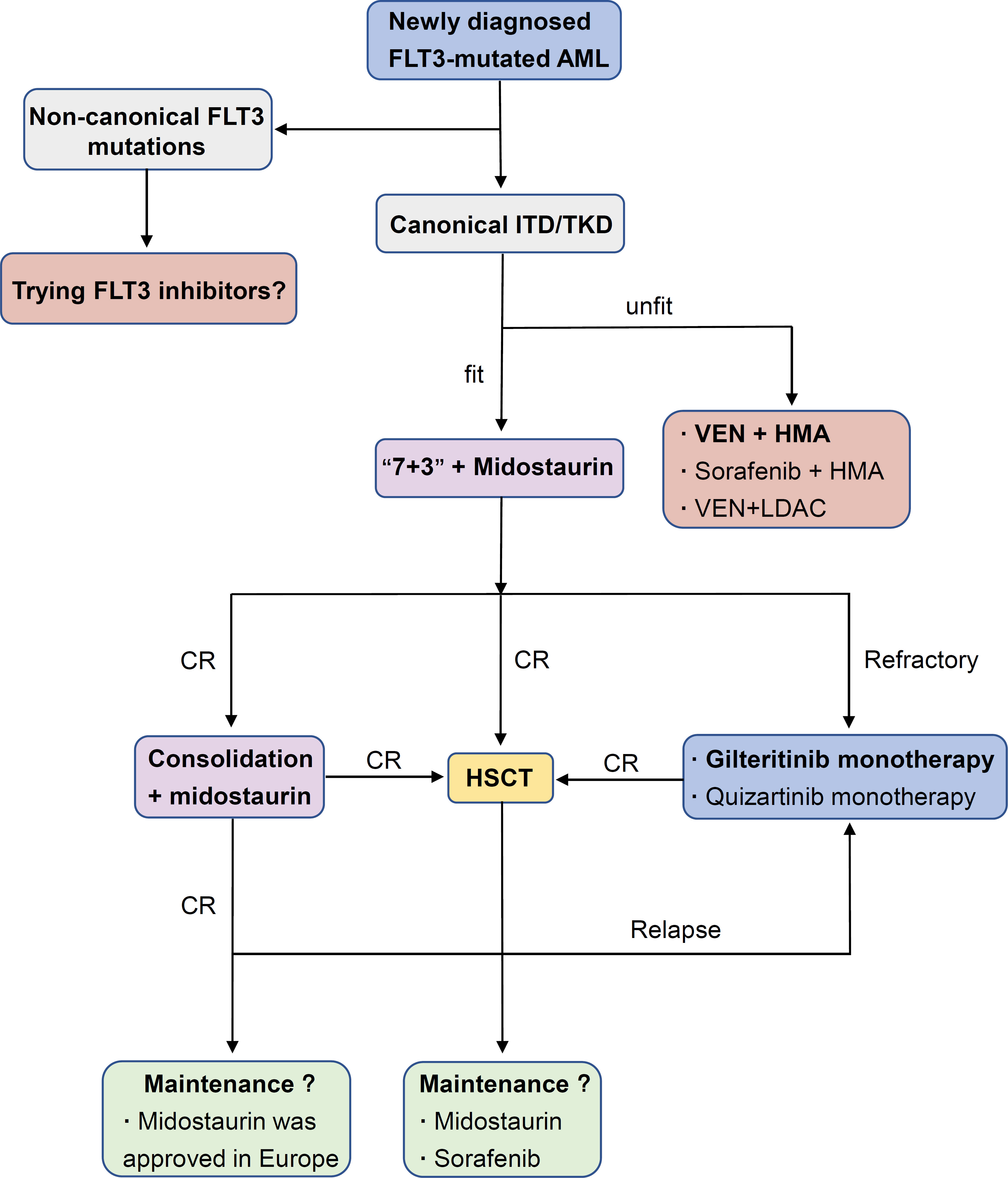
Figure 1 Applications of FLT3 inhibitors in AML.
3.1.1 Induction and consolidation therapy
The fitness and age of patients are key points in the decision for the management of newly diagnosed AML with FLT3 mutations. Midostaurin plus standard chemo-therapy is the first choice for those who are fit. In the phase III RATIFY trial, young adult patients (16-59 years old) with FLT3-ITD and FLT3-TKD were randomly assigned to receive standard induction and consolidation chemotherapy plus either midostaurin or placebo. Subsequently, those who reached remission after consolidation therapy received either midostaurin or placebo as maintenance. This combination resulted in significant improvement in event-free survival (EFS; 8.2 vs. 3.0 months, P=0.002) and median overall survival (OS; 74.7 vs. 25.6 months, P = 0.009) compared to chemotherapy alone, although a larger population with an FLT3-TKD mutation in the RATIFY study than that seen in the general population might bias the clinical outcomes toward this less aggressive subtype. Regrettably, the complete remission (CR) rate was not obviously improved (21). In 2017, midostaurin, one of the first FLT3 inhibitors to be studied in AML, was approved for use with induction and consolidation by the US Food and Drug Administration (FDA), as administered in the RATIFY trial, and com-bination therapy was recommended for the preferred strategy by the guidelines of the National Comprehensive Cancer Network (NCCN).
Older adults (>60 years old) who cannot undergo intensive chemotherapy often receive less intensive regimens, including hypomethylating agents (azacitidine or decitabine), because of limited effective treatment options. Hypomethylating agents and low-dose cytarabine were associated with poor CR plus CRi rates and median survival times (56, 57). Venetoclax, a selective small-molecule Bcl-2 inhibitor, has demonstrated an outstanding ability to induce apoptosis of AML cells in vitro (58, 59). In the phase III VIALE-A trial, treatment with azacitidine and venetoclax obviously improved the remission rate of various mutated subgroups and the median OS (60). However, the clonal evolution of FLT3-ITD loss of heterozygosity (LOH) may lead to treatment failure (12). As an alternative strategy, low-intensity chemotherapy (azacitidine or decitabine) plus sorafenib and venetoclax in combination with LDAC showed poor performance (61, 62).
However, it is important to note that the combination of venetoclax and FLT3 inhibitors shows great potential. The combination of FLT3 inhibitors (gilteritinib or sorafenib) with venetoclax could synergistically reduce cell proliferation and enhance apoptosis/cell death in FLT3/ITD cell lines and primary AML samples. Venetoclax was also able to attenuate FLT3 inhibitor-resistance of cells to gilteritinib or sorafenib treatment by inhibiting the MAPK/ERK pathway (63). In a phase II trial, 25 older patients with FLT3 mutated AML were enrolled and accepted triplet therapy combining FLT3 inhibitor, venetoclax, and HMA. The composite complete remission (CRc) rates in patients with newly diagnosed AML achieve 92% and 62% in R/R patients (64). Moreover, a retrospective analysis of their team demonstrated that older and unfit adult patients with newly diagnosed FLT3 mutated AML receiving a triplet regimen (lower intensity chemotherapy + FLT3 inhibitor + venetoclax) had a significantly higher CR/CRi rate (93% vs. 70%, P = 0.02) and longer median overall survival (NR vs. 9.5 months, P < 0.01) compared with doublet (lower intensity chemotherapy + FLT3 inhibitor) regimens (65). Overall, the combination of venetoclax and FLT3 inhibitors may be an effective frontline regimen for FLT3 mutated AML, which should be further validated prospectively.
3.1.2 Maintenance therapy
The role of FLT3 inhibitors in maintenance therapy is intriguing, either during re-mission for patients who do not accept HSCT or for those who are undergoing HSCT. Data from several clinical trials that included TKI maintenance therapy after first-line induction and consolidation suggest that it may be a promising approach (21, 66–69).
• Post-chemotherapy maintenance therapy
In the notable phase III RATIFY study, patients accepted up to one year of midostaurin maintenance following induction and consolidation chemotherapy plus midostaurin, while FLT3 inhibitor maintenance was discontinued once patients underwent HSCT. An unplanned post hoc efficacy analysis of the midostaurin maintenance phase in the RATIFY trial suggested that midostaurin maintenance might not further reduce the probability of relapse, although it was well tolerated (21). Maintenance of midostaurin after chemotherapy did not receive US FDA approval due to the result of limited efficacy shown from clinical data. In the SORAML study, maintenance therapy with sorafenib after chemotherapy ameliorated RFS, though the trial could not determine to what extent sorafenib maintenance influenced RFS (66). Recently, a long-term follow-up study validated a clear benefit in RFS; however, it did not translate into an OS benefit (67). To evaluate actual benefits from maintenance therapy of FLT3 inhibitors, separate randomized trials will compare gilteritinib or quizartinib vs. placebo maintenance for up to two and three years following chemo-therapy, respectively (NCT02927262, NCT02668653).
• Post-HSCT maintenance therapy
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is considered the most powerful method for hematopoietic malignancies. Allo-HCST improves the out-come of AML patients with FLT3-ITD AML, but leukemia relapse remains a frequent factor of failure (70–72). In the post-HSCT maintenance setting, recent evidence has shown the antileukemic synergism of FLT3 inhibitors. In a phase I hypothesis-generating trial, midostaurin was added to intensive chemotherapy followed by allogeneic hematopoietic cell transplantation (allo-HSCT) and midostaurin maintenance therapy for 12 months. This study demonstrated a significant improvement in EFS by midostaurin compared to 415 historical controls (73). Sorafenib maintenance after HSCT in a randomized, placebo-controlled, double-blind phase II trial also showed po-tential improvement in RFS (24-month RFS, sorafenib vs. placebo, 85.0% vs. 53.3%, P=0.002) and OS (24-month OS, sorafenib vs. placebo, 90.5% vs. 60.2%, P=0.007) (74). Based on the data from midostaurin and sorafenib, the efficacy of quizartinib, gilteritinib, and crenolanib in maintenance therapy after HSCT will be evaluated in clinical trials (NCT02668653, NCT02997202, NCT02400255).
3.1.3 The relapsed/refractory setting
Response rates are low in adult patients with relapsed/refractory (R/R) AML, and no standard strategy has emerged for treating primary refractory or relapsed AML (13). Patients with relapsed or refractory FLT3-mutated AML in the phase III ADMIRAL trial were randomly assigned to the subgroups of single-agent gilteritinib or salvage chemotherapy. Compared to salvage chemotherapy, gilteritinib monotherapy resulted in a higher percentage of patients with complete remission and full or partial hematologic recovery (34% vs. 15.3%) and improved OS with or without censoring at HSCT (9.3 months vs. 5.3 months, P<0.001) (23). The promising results of the established trial prompted FDA approval of gilteritinib in the R/R setting. Similarly, in the phase III QUANTUM-R trial, patients in the subgroup of single-agent quizartinib had an improved OS of 6.2 months versus 4.7 months (P=0.02) and a hazard ratio for death of 0.76 (95% CI: 0.58–0.98). However, complete remission rate did not improve from single-agent quizartinib (23). A post hoc analysis of the ADMIRAL and QUANTUM-R trials demonstrated that quizartinib treatment achieved remission faster and response may be more durable and survival potentially longer with gilteritinib regardless of substantial limitations in cross-study comparisons (75). Furthermore, the combination of venetoclax with gilteritinib showed a potential for molecular clearance, which seemed to be associated with increased OS in a recent trial (76). The promising latent capacity of FLT3 inhibitors in the re-lapsed/refractory setting has been demonstrated, and further exploration is ongoing (NCT03989713, NCT04140487, NCT05010122).
3.2 Therapy for non-canonical FLT3 mutated AML
With the clinical application of NGS methods over recent years, an increased number of FLT3 mutations outside of the ITD and D835/I836 regions have been de-scribed (16, 18, 77, 78). These non-canonical FLT3 mutations, including point mutations and small insertions/deletions, occur in every FLT3 protein domain and frequently in the juxtamembrane (JM) domain and the KD domain adjacent to D835/I836. Several studies have validated that many of the mutations acquire increased phosphorylation activity, and some of them might confer resistance or high sensitivity to specific FLT3 inhibitors (16, 29, 78, 79).
A series of point mutations located in the non-TKD region tend to result in aberrant phosphorylation of FLT3 and an enhanced ability to activate STAT5. Several point mutations in the TKDs were less sensitive to the type II inhibitor quizartinib than to the type I TKI crenolanib (80). In contrast, small insertions/deletions of FLT3, such as p.Glu598_Tyr599del and p. Phe590_Asp593delinsLeuTyr did not show an increase in Y589/Y591 phosphorylation but led to constitutive activation of STAT3, ERK1/2, SFK (Src family kinases), SHP2 and AKT. Interestingly, p.Phe590_Asp593delinsLeuTyr-transduced cells showed a higher sensitivity toward PKC412 and AC220 than FLT3-ITD-transduced cells (17).
The clinical characteristics of non-canonical mutations remain unclear because of their low incidence. In a brief report, a patient with mutation of p. Glu598_Tyr599del, which is described in the above study, is likely to be responsive to targeted therapy with tyrosine kinase inhibitors (77). Another report demonstrated that patients har-boring activating non-canonical FLT3 mutations (V592G and N676K) may benefit from TKI therapy in the relapsed/refractory setting (81). However, a recent study revealed that FLT3 non-canonical mutations in the ITD region have a higher rate of concomitant mutation of KTM2A-PTD, and the patients with dual FLT3 non-ITD and KMT2A-PTD mutations may indicate a trend for inferior outcome (82). The phosphorylation activity and clinical characteristics of non-ITD and TKD FLT3 mutations vary widely, but most of them may be sensitive to FLT3 TKIs.
4 Resistance to FLT3 inhibitors
Although multiple small molecule inhibitors of FLT3 have rapidly improved the outcomes of FLT3-mutated AML, resistance to FTL3 inhibitors has become increasingly prominent. Frequent short-lived responses and therapeutic resistance pose an ongoing issue. Resistance mechanisms, including primary resistance and secondary resistance, vary due to drug type. Primary resistance is considered innate. For example, overexpression of FL and the abnormal status of the marrow microenvironment induce resistance when FLT3 inhibitors are used for the first time. Secondary resistance defines the resistance that occurs after using FLT3 inhibitors, including the second mutation of FLT3 (on-target) and a non-FLT3 abnormality (off-target), such as acquiring an alteration of downstream and parallel signal pathways (Figure 2).
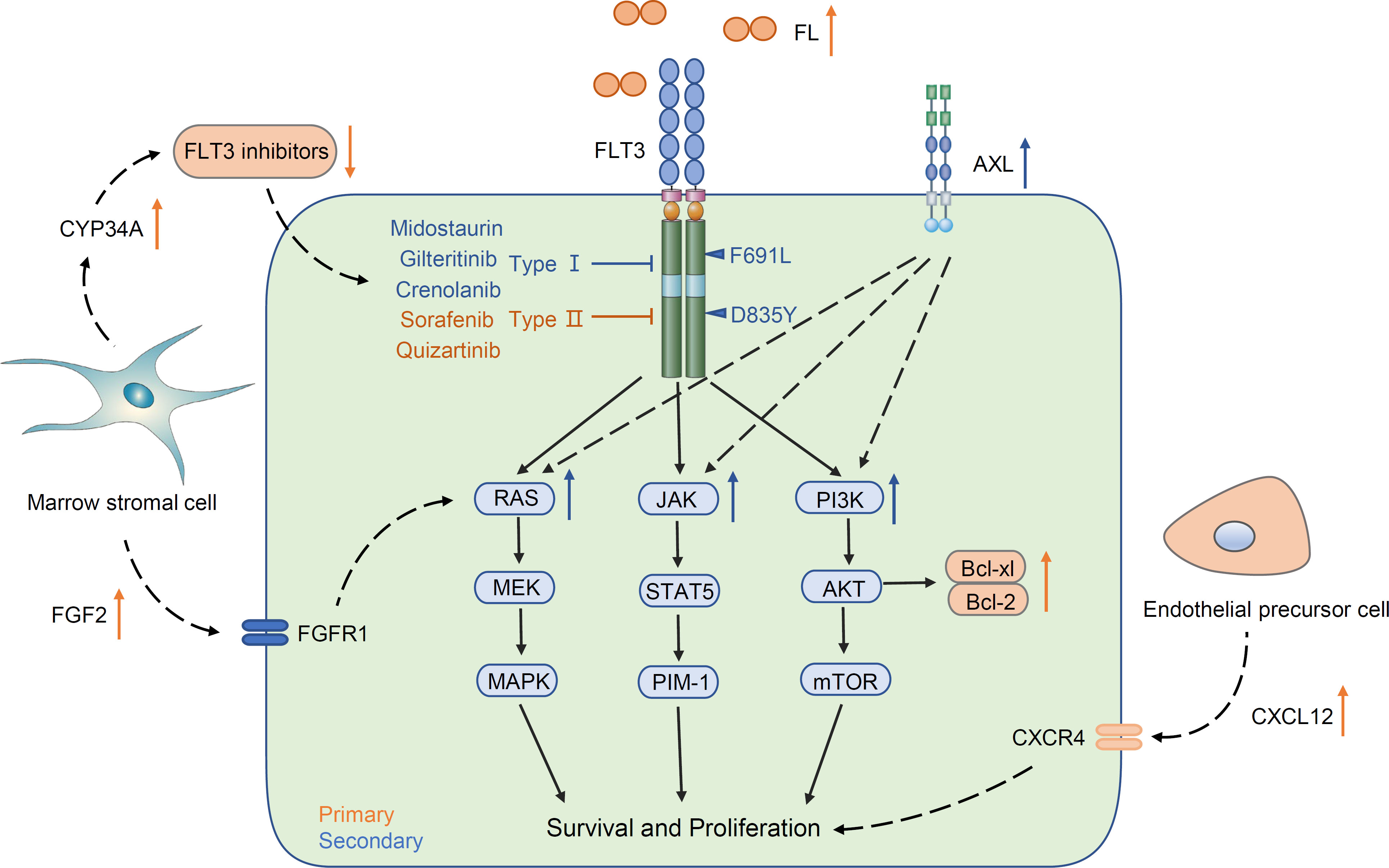
Figure 2 Common mechanisms of primary and secondary resistance.
4.1 Primary resistance
One common mechanism of primary resistance is the increased FLT3 ligand. FLT3 ligand is an important regulator in hematopoiesis. FL is expressed by most leukemic cells and promotes proliferation through an autocrine process (83, 84). The soluble FL concentrations increase with the courses of chemotherapy and maintain a high level and increase more rapidly in relapse patients. Furthermore, increased plasma FL levels are tightly correlated with decreased efficacy of FLT3 inhibitors in vitro and in vivo (85). It is connected to increased FL that overexpression of FLT3 in AML blast cells can clearly impair the efficacy of FLT3 inhibitors. In addition, Bcl-2 is located down-stream of the FLT3/PI3K pathway and plays a significant anti-apoptotic role. The ab-normal elevation of Bcl-2 in FLT3-ITD-positive blasts protects tumor cells from apoptosis, and the Bcl-2 protein level is not decreased when FLT3 inhibitors attenuate the phosphorylation of FLT3 (86–88). Similar to Bcl-2, activation of other anti-apoptotic proteins, such as Mcl-1 and Bcl-xl, mediates resistance (36, 88).
The bone marrow microenvironment can also directly contribute to primary FLT3 inhibitor resistance. The upregulation of CXCL12 and fibroblast growth factor 2 (FGF2), which are secreted by endothelial precursor cells and bone marrow stromal cells in the bone marrow, respectively, can shield AML blasts from FLT3 inhibitor injury (89, 90). Hepatic CYP3A4 has also been shown to inactivate all TKIs and provide chemoprotection. Enhanced CYP34A expression on BM stromal cells will weaken the drug availability of FLT3-TKIs, leading to limited efficacy (91).
4.2 Secondary resistance
4.2.1 On-target resistance
Secondary resistance can be broadly subdivided into on-target and off-target mechanisms according to where the alteration occurs. The most common on-target resistance is the development of the second FLT3 mutation, often in the KD (Table 1). Acquisition of point mutations at four residues (F691, D835, Y842, E608) in the kinase domain of FLT3-ITD confers resistance to quizartinib by disrupting binding (92). The gatekeeper F691L mutation and D835 mutation confer resistance not only to quizartinib but also to sorafenib, gilteritinib, and crenolanib (93). After using midostaurin, the TKD1 mutations N676D/S, F691I/L, and G697R/S were screened for efficacy (34). In these acquiring point mutations, D835 alterations tend to confer drug tolerance to type II FLT3 inhibitors by disrupting the binding and keeping the A-loop in a DFG-out state (94, 95). In contrast, the acquisition of the gatekeeper F691L and G697R/S mutations often means tolerance to most FLT3 inhibitors (34).
4.2.2 Off-target resistance
On-target mutations only partly explain FLT3 inhibitor resistance, and upregulation or emergence of non-FLT3 mutant clones (off-target) is a key resistance mechanism (Table 1). Recent studies have demonstrated obvious differentiation in paired patients be-tween previous and posttreatment gilteritinib and crenolanib resistance. Upregulation of the Ras/MAPK pathway occurs frequently, causing resistance generation (48, 52). Using patient-derived cell lines, Lindblad O et al. and Knapper S et al. revealed an enrichment of the PI3K/mTOR and JAK/STAT5 signaling pathways in resistant cells (96, 97). The above evidence clearly shows that the aberrant activation of down-stream signaling pathways of FLT3 enables AML cells to become tolerant to FLT3 inhibitors.
Abnormal upregulation of parallel AXL tyrosine kinase signaling is another mechanism of FLT3 inhibitor resistance. AXL is a member of the TAM family with the high-affinity ligand growth arrest-specific protein 6 (GAS6). Activation of the GAS6/AXL signaling axis serves as an important pathway driving cancer cell survival and proliferation, which is similar and parallel to the FLT3 signaling pathway (98). In one study, AML cell lines and primary blasts from FLT3-ITD-mutated AML patients were treated with PKC412 and AC220 concomitantly, and the expression of phospho-AXL and AXL was detected. Enhanced phosphorylation of AXL by treatment with PKC412 and AC220 occurred not only in AML cell lines but also in primary blasts (37). Similarly, the results from another study validated that the increased level of AXL dampened the response to the FLT3 inhibitor quizartinib. In a xenograft mouse model of this study, inhibition of AXL significantly enhanced the response of FLT3-ITD cells to quizartinib exclusively within a bone marrow environment (51). These data high-light a new bypass mechanism that attenuated the response to FLT3 inhibitors through upregulation of AXL activity.
4.2.3 Other mechanisms of secondary resistance
The emergence of metabonomics has prompted the exploration of the mechanisms driving FLT3-ITD acute myeloid leukemia resistance to FLT3 inhibitors from a new perspective. Metabolic reprogramming mediated the evolution of gilteritinib resistance (Figure 3A). Sunil K Joshi et al. demonstrated dramatic differences in the metabolome of early and late gilteritinib-resistant AML cell lines. Metabolic profiling of these cells affirmed a trend toward upregulation of sphingolipid/phospholipid or fatty acid/carnitine metabolites relative to MOLM14 parental cells. Early resistant cells undergo metabolic reprogramming with a slow proliferation rate, while expansion of pre-existing NRAS mutant subclones is dominant in late resistant cells (99). The role of autophagy in targeted therapy has gradually been revealed. Enhanced autophagy activity was observed in sorafenib-resistant AML cell lines bearing FLT3-TKD mutations and FLT3-ITD cells participating in AML progression and drug resistance (100, 101). Although the molecular mechanisms remain a mystery, it is highly probable that the transcription factor ATF4 (activating transcription factor 4) stimulated by FLT3-ITD is crucial to the upregulation of autophagy (102). Mitophagy is a cellular process for the degradation of mitochondria by the autophagic machinery and is regulated by ceramide on the outer mitochondrial membrane (103). FLT3-ITD mutations rescue AML cells from mitophagy by suppressing pro-cell death lipid ceramide generation, and FLT3-ITD inhibition mediates ceramide-dependent mitophagy, leading to AML blast death. The abnormality of mitochondrial ceramide arresting mitophagy results in resistance to FLT3-ITD inhibition (104) (Figure 3B).
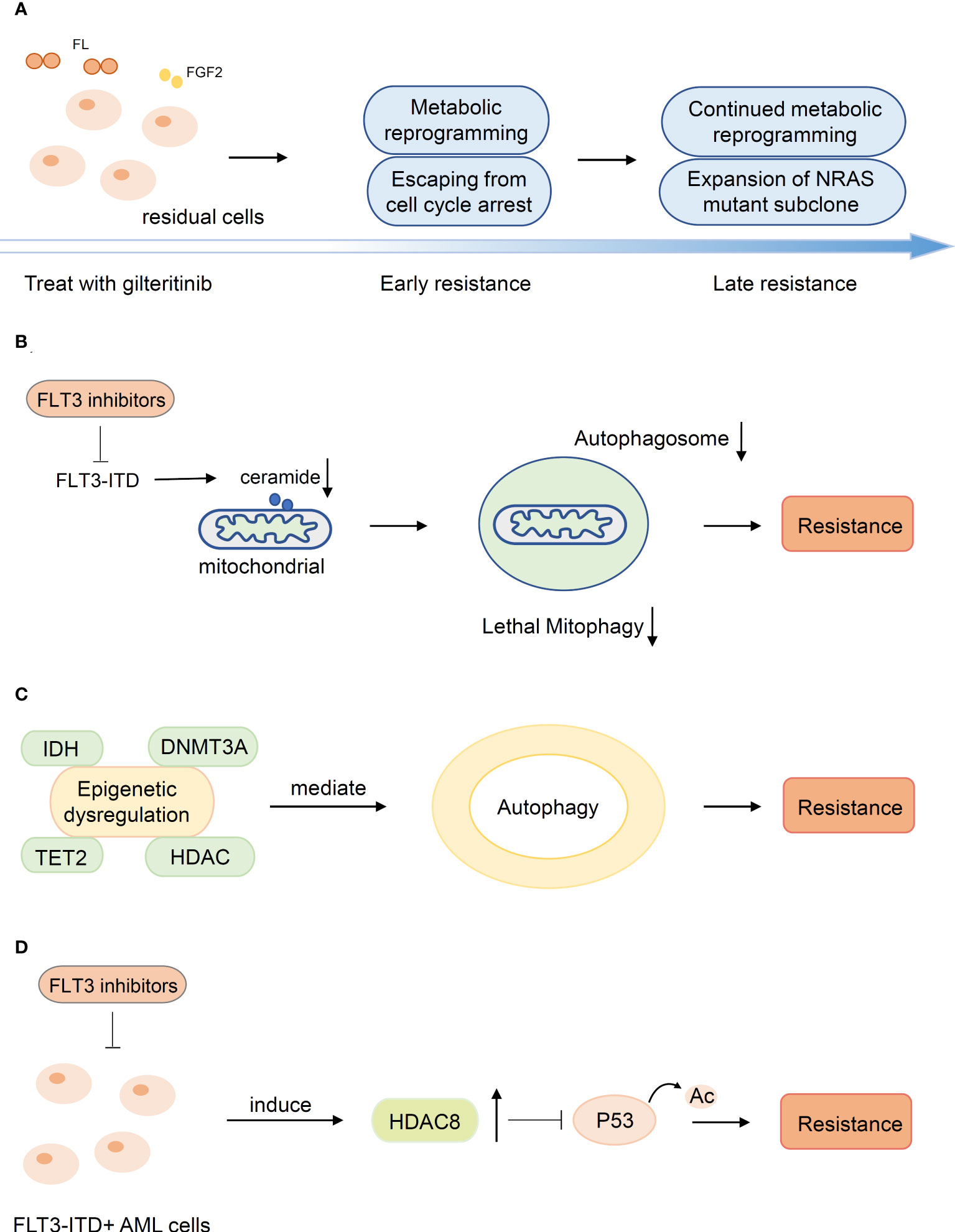
Figure 3 Other mechanisms of secondary resistance. (A) Metabolic reprogramming mediated the evolution of resistance. (B) The abnormality of mitochondrial ceramide results in resistance to FLT3-inhibitors by arresting mitophagy. (C) Mutations in epigenetic modification genes suggests potential associations between alterations and autophagy associated with secondary resistance (D) HDAC8 was upregulated upon FLT3 inhibitor exposure and enhanced HDAC8 binding and deacetylation of p53 to promote resistance.
Epigenetic dysregulation is a significant cause of secondary resistance. Mutations in epigenetic modification genes (DNMT3A, TET2, IDH1/2, HDAC) are related to diverse processes of epigenetic regulation and suggest potential associations between alterations and autophagy associated with secondary resistance (48, 102, 105) (Figure 3C). In addition, HDAC8 was upregulated upon FLT3 inhibitor exposure in FLT3-mutated AML cells and subsequently enhanced HDAC8 binding and deacetylation of p53 to promote AML cell survival and TKI resistance (106, 107) (Figure 3D).
5 Strategies to overcome resistance
Numerous efforts have been made to attenuate the effect of FLT3 TKI drug tolerance. To overcome primary and off-target resistance, doctors attempt to combine different types of agents and use multitarget inhibitors to avert limited response. Due to on-target resistance, it is necessary to develop new FLT3 inhibitors that are effective against primary and secondary mutations via an original molecular design rationale.
5.1 Development of novel FLT3 inhibitors
As described above, type II inhibitors exhibit high selectivity but no response to FLT3-TKD mutation because of a persistent state of DFG-out (95). Although type I inhibitors show efficacy in overcoming FLT3-TKD, several secondary FLT3 mutations, for example, the gatekeeper mutation (F691L), confer limited efficacy to all existing FLT3 inhibitors (95). To tackle this challenge, the discovery of novel agents that are sensitive to secondary mutations conferring resistance is essential. The development of new type II inhibitors seemed to bring promising prospects. Pexidartinib (PLX3397) is a triple-kinase inhibitor of FLT3, KIT, and CSF1R and shows excellent inhabitation of FLT3-ITD (half-maximal inhibitory concentration [IC50]: 11 nM). Pexidartinib has a shorter linker between the middle and tail pyridine rings than other type II inhibitors and forms two hydrogen bonds with D829, which sequesters pexidartinib away from F691, thus avoiding the adverse influence of the F691L mutation. Extraordinary efficacy in overcoming the secondary F691L mutation has been shown due to the above mechanism (108, 109). Ge et al. discovered a novel type II FLT3 inhibitor, MZH29, which can stably bind to the FLT3-F691L model by four hydrogen bonds formed between MZH29 and E661, C694, and D829 and a π−π stacking interaction formed between MZH29 and F830 (110). Another novel FLT3 inhibitor, cabozantinib, potently inhibited FLT3 phosphorylation in FLT3-ITD-positive and FLT3-ITD-F691L-positive cells under both medium and plasma conditions, and the result of the docking binding model in which cabozantinib maintained a remote distance from F691 and formed a hydrogen bond with D829 opposite F691 explains the potent inhibitory activity (111). All these novel type II inhibitors, which noncovalently bind to the FLT3-ITD-F691L model, show stable binding ability and excellent efficiency in vitro. However, they were vulnerable to other residue mutations, particularly the residue located in the activation loop (108, 110, 111). Consequently, targeted covalent inhibitors have attracted the attention of many researchers. FF-10101, the first reported covalent FLT3 inhibitor, made great progress in enhancing the binding affinity and response to secondary on-target mutations by targeting conserved amino acids. A covalent bond was generated between the warhead of FF-10101 and the “SH” of C695.70 Based on its high affinity for FLT3 kinase, FF-10101 showed outstanding efficacy against not only the FLT3-ITD-TKD mutation but also the uncommon K663Q, N841I, R834Q, and F691L mutations in vitro. Furthermore, oral administration in NOD/SCID mice also showed potent inhibitory effects (112). FF-10101 is now in a phase I/II clinical trial (NCT03194685) to treat refractory or relapsed AML patients in the US.
5.2 Therapies of multitarget inhibition
Monotherapy with FLT3 inhibitors frequently causes drug resistance, a short duration of remission, and poor effects due to complex pathological changes, such as an abnormal microenvironment using FLT3 inhibitors and compensatory off-target alterations. That many non-FLT3 abnormalities are involved in the occurrence of FLT3 inhibitor tolerance indicates that simultaneously inhibiting multiple synergetic signaling molecules might contribute to improving the efficacy and overcoming resistance. Much evidence has demonstrated that the combination of different kinds of agents according to various abnormal states results in promising outcomes.
The combination of midostaurin or gilteritinib with the Bcl-2 inhibitor venetoclax contributed to the simultaneous downregulation of Mcl-1 and Bcl-2, resulting in the synergistic induction of apoptosis and attenuating the adverse impact of increased Bcl-2 (113, 114). Similarly, the concurrent inhibition of CXCL12 and FGF2 could have a favorable effect (89, 90). Furthermore, a synergistic effect has been shown in therapies combining FLT3-TKIs with agents targeting downstream or parallel signaling pathways, such as JAK/STAT5 pathway inhibitors (115, 116) and PI3K/mTOR pathway inhibitors (99, 117). In addition to combination therapies, dual inhibitors can achieve good results by reaching the identical goal of multitarget inhabitation, such as dual Pim kinase/FLT3 inhibitors, dual FLT3/JAK2 inhibitors, and dual FLT3/CDK4 inhibitors (118–122).
Moreover, addressing metabolic abnormalities is also a potential strategy to drive AML cells to return to a susceptible state (99, 104).
6 Conclusions and future directions
In the era of individual diagnosis and treatment, FLT3-activating mutations are a marker of poor prognosis and specific therapeutic needs. Although the emergence of FLT3 inhibitors has provided us with numerous powerful creative treatment tools, survival remains poor in FLT3-mutated AML, and new strategies need to be explored. In addition to FLT3-ITD/TKD, FLT3 non-canonical alterations also deserve attention with the wide use of NGS. At the same time, the influence of FLT3 inhibitor tolerance has become an inevitable issue. To solve this problem, a deeper exploration of the underlying mechanisms and resolvents from many different angles are essential.
Author contributions
S-SG wrote the manuscript. S-BL and S-LX conducted review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (grant No. 81970138), Translational Research Grant of NCRCH (grant No. 2020ZKMB05), Jiangsu Province “333” project, Social Development Project of the Science and Technology Department of Jiangsu (Grant No. BE2021649) and Gusu Key Medical Talent Program (grant No. GSWS2019007). Key technology program of Suzhou people's livelihood technology projects (Grant No. SKY2021029).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med (2015) 373:1136–52. doi: 10.1056/NEJMra1406184
2. Newell LF, Cook RJ. Advances in acute myeloid leukemia. BMJ (2021) 375:n2026. doi: 10.1136/bmj.n2026
3. Rosnet O, Schiff C, Pebusque MJ, Marchetto S, Tonnelle C, Toiron Y, et al. Human FLT3/FLK2 gene: cDNA cloning and expression in hematopoietic cells. Blood (1993) 82:1110–9. doi: 10.1182/blood.V82.4.1110.1110
4. Small D, Levenstein M, Kim E, Carow C, Amin S, Rockwell P, et al. STK-1, the human homolog of flk-2/Flt-3, is selectively expressed in CD34+ human bone marrow cells and is involved in the proliferation of early progenitor/stem cells. Proc Natl Acad Sci (1994) 91:459–63. doi: 10.1073/pnas.91.2.459
5. Carow CE, Levenstein M, Kaufmann SH, Chen J, Small D. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood (1996) 87:1089–96. doi: 10.1182/blood.V87.3.1089.bloodjournal8731089
6. Lisovsky M, Estrov Z, Zhang X, Consoli U, SanchezWilliams G, Snell V, et al. Flt3 ligand stimulates proliferation and inhibits apoptosis of acute myeloid leukemia cells: Regulation of bcl-2 and bax. Blood (1996) 88:3987–97. doi: 10.1182/blood.V88.10.3987.bloodjournal88103987
7. Drexler H. Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia (1996) 10:588–99.
8. Bullinger L, Döhner K, Döhner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol (2017) 35:934–46. doi: 10.1200/JCO.2016.71.2208
9. Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease: Presented in part at the 42nd annual meeting of the American society of hematology, December 1-5, 2000, San Francisco, CA (abstract 3569). Blood J Am Soc Hematol (2002) 100:59–66. doi: 10.1182/blood.v100.1.59
10. Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood J Am Soc Hematol (2001) 97:2434–9. doi: 10.1182/blood.V97.8.2434
11. Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood (2007) 110:1262–70. doi: 10.1182/blood-2006-04-015826
12. DiNardo C, Tiong I, Quaglieri A, MacRaild S, Loghavi S, Brown F, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood (2020) 135:791–803. doi: 10.1182/blood.2019003988
13. Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood (2017) 129:424–47. doi: 10.1182/blood-2016-08-733196
14. Spiekermann K, Bagrintseva K, Schoch C, Haferlach T, Hiddemann W, Schnittger S. A new and recurrent activating length mutation in exon 20 of the FLT3 gene in acute myeloid leukemia. Blood (2002) 100:3423–5. doi: 10.1182/blood-2002-03-0953
15. Reindl C, Bagrintseva K, Vempati S, Schnittger S, Ellwart JW, Wenig K, et al. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood (2006) 107:3700–7. doi: 10.1182/blood-2005-06-2596
16. Fröhling S, Scholl C, Levine RL, Loriaux M, Boggon TJ, Bernard OA, et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell (2007) 12:501–13. doi: 10.1016/j.ccr.2007.11.005
17. Chatain N, Perera R, Rossetti G, Rossa J, Carloni P, Schemionek M, et al. Rare FLT3 deletion mutants may provide additional treatment options to patients with AML: An approach to individualized medicine. Leukemia (2015) 29:2434–8. doi: 10.1038/leu.2015.131
18. Zhang Y, Zhang Y, Wang F, Wang M, Liu H, Chen X, et al. The mutational spectrum of FLT3 gene in acute lymphoblastic leukemia is different from acute myeloid leukemia. Cancer Gene Ther (2020) 27:81–8. doi: 10.1038/s41417-019-0120-z
19. Daver N, Venugopal S, Ravandi F. FLT3 mutated acute myeloid leukemia: 2021 treatment algorithm. Blood Cancer J (2021) 11:104. doi: 10.1038/s41408-021-00495-3
20. Perl AE. Availability of FLT3 inhibitors: how do we use them? Blood (2019) 134:741–5. doi: 10.1182/blood.2019876821
21. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. New Engl J Med (2017) 377:454–64. doi: 10.1056/NEJMoa1614359
22. Cortes JE, Khaled S, Martinelli G, Perl AE, Ganguly S, Russell N, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-r): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol (2019) 20:984–97. doi: 10.1016/S1470-2045(19)30150-0
23. Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med (2019) 381(18):1728–40. doi: 10.1056/NEJMoa1902688
24. Almatani MF, Ali A, Onyemaechi S, Zhao Y, Gutierrez L, Vaikari VP, et al. Strategies targeting FLT3 beyond the kinase inhibitors. Pharmacol Ther (2021) 225:107844. doi: 10.1016/j.pharmthera.2021.107844
25. Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood (2004) 103:3669–76. doi: 10.1182/blood-2003-11-3775
26. Fiedler W, Serve H, Döhner H, Schwittay M, Ottmann OG, O'Farrell A-M, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood (2005) 105:986–93. doi: 10.1182/blood-2004-05-1846
27. Ravandi F, Cortes JE, Jones D, Faderl S, Garcia-Manero G, Konopleva MY, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol (2010) 28:1856. doi: 10.1200/JCO.2009.25.4888
28. Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood (2005) 105:54–60. doi: 10.1182/blood-2004-03-0891
29. Kennedy VE, Smith CC. FLT3 mutations in acute myeloid leukemia: key concepts and emerging controversies. Front Oncol (2020) 10:612880. doi: 10.3389/fonc.2020.612880
30. Kampa-Schittenhelm KM, Heinrich MC, Akmut F, Döhner H, Döhner K, Schittenhelm MM. Quizartinib (AC220) is a potent second generation class III tyrosine kinase inhibitor that displays a distinct inhibition profile against mutant-FLT3,-PDGFRA and-KIT isoforms. Mol Cancer (2013) 12:1–15. doi: 10.1186/1476-4598-12-19
31. Lee LY, Hernandez D, Rajkhowa T, Smith SC, Raman JR, Nguyen B, et al. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood (2017) 129:257–60. doi: 10.1182/blood-2016-10-745133
32. Galanis A, Ma H, Rajkhowa T, Ramachandran A, Small D, Cortes J, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood (2014) 123:94–100. doi: 10.1182/blood-2013-10-529313
33. Propper DJ, McDonald AC, Man A, Thavasu P, Balkwill F, Braybrooke JP, et al. Phase I and pharmacokinetic study of PKC412, an inhibitor of protein kinase c. J Clin Oncol (2001) 19:1485–92. doi: 10.1200/jco.2001.19.5.1485
34. Cools J, Mentens N, Furet P, Fabbro D, Clark JJ, Griffin JD, et al. Prediction of resistance to small molecule FLT3 inhibitors: Implications for molecularly targeted therapy of acute leukemia. Cancer Res (2004) 64:6385–9. doi: 10.1158/0008-5472.CAN-04-2148
35. Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede M, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of asn-676 in the FLT3 tyrosine kinase domain. Blood (2006) 107:293–300. doi: 10.1182/blood-2005-06-2469
36. Breitenbuecher F, Markova B, Kasper S, Carius B, Stauder T, Böhmer FD, et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood J Am Soc Hematol (2009) 113:4063–73. doi: 10.1182/blood-2007-11-126664
37. Park I, Mundy-Bosse B, Whitman S, Zhang X, Warner S, Bearss D, et al. Receptor tyrosine kinase axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia (2015) 29:2382–9. doi: 10.1038/leu.2015.147
38. Pratz KW, Cho E, Levis MJ, Karp JE, Gore SD, McDevitt M, et al. A pharmacodynamic study of sorafenib in patients with relapsed and refractory acute leukemias. Leukemia (2010) 24:1437–44. doi: 10.1038/leu.2010.132
39. Clark JW, Eder JP, Ryan D, Lathia C, Lenz HJ. Safety and pharmacokinetics of the dual action raf kinase and vascular endothelial growth factor receptor inhibitor, BAY 43-9006, in patients with advanced, refractory solid tumors. Clin Cancer Res (2005) 11:5472–80. doi: 10.1158/1078-0432.Ccr-04-2658
40. Smith CC, Lin K, Stecula A, Sali A, Shah NP. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia (2015) 29:2390–2. doi: 10.1038/leu.2015.165
41. Smith CC, Wang Q, Chin C-S, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature (2012) 485:260–3. doi: 10.1038/nature11016
42. Green AS, Maciel TT, Hospital M-A, Yin C, Mazed F, Townsend EC, et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci Adv (2015) 1:e1500221. doi: 10.1126/sciadv.1500221
43. Yang X, Sexauer A, Levis M. Bone marrow stroma-mediated resistance to FLT 3 inhibitors in FLT 3-ITD AML is mediated by persistent activation of extracellular regulated kinase. Br J haematol (2014) 164:61–72. doi: 10.1111/bjh.12599
44. Smith CC, Levis MJ, Litzow MR, Perl AE, Altman JK, Gill S, et al. Pharmacokinetics and pharmacodynamics of gilteritinib in patients with relapsed or refractory acute myeloid leukemia. J Clin Oncol (2016) 34:4. doi: 10.1200/JCO.2016.34.15_suppl.7026
45. McMahon CM, Canaani J, Rea B, Sargent RL, Morrissette JJ, Lieberman DB, et al. Mechanisms of acquired resistance to gilteritinib therapy in relapsed and refractory FLT3-mutated acute myeloid leukemia. Blood (2017) 130:295. doi: 10.1182/blood.V130.Suppl_1.295.295
46. Joshi SK, Sharzehi S, Pittsenbarger J, Bottomly D, Tognon CE, McWeeney SK, et al. A noncanonical FLT3 gatekeeper mutation disrupts gilteritinib binding and confers resistance. Am J Hematol (2021) 96:E226–9. doi: 10.1002/ajh.26174
47. Tarver TC, Hill JE, Rahmat L, Perl AE, Bahceci E, Mori K, et al. Gilteritinib is a clinically active FLT3 inhibitor with broad activity against FLT3 kinase domain mutations. Blood Adv (2020) 4:514–24. doi: 10.1182/bloodadvances.2019000919
48. McMahon CM, Ferng T, Canaani J, Wang ES, Morrissette JJ, Eastburn DJ, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid LeukemiaSecondary resistance to selective FLT3 inhibition in AML. Cancer Discovery (2019) 9:1050–63. doi: 10.1158/2159-8290.CD-18-1453
49. Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood (2009) 114:2984–92. doi: 10.1182/blood-2009-05-222034
50. Cortes JE, Kantarjian H, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol (2013) 31:3681–7. doi: 10.1200/JCO.2013.48.8783
51. Dumas P-Y, Naudin C, Martin-Lannerée S, Izac B, Casetti L, Mansier O, et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5-and hypoxia-dependent upregulation of AXL. Haematologica (2019) 104:2017. doi: 10.3324/haematol.2018.205385
52. Zhang H, Savage S, Schultz AR, Bottomly D, White L, Segerdell E, et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat Commun (2019) 10:1–13. doi: 10.1038/s41467-018-08263-x
53. Stirewalt DL, Kopecky KJ, Meshinchi S, Appelbaum FR, Slovak ML, Willman CL, et al. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood (2001) 97:3589–95. doi: 10.1182/blood.v97.11.3589
54. Levis M. FLT3/ITD AML and the law of unintended consequences. Blood (2011) 117:6987–90. doi: 10.1182/blood-2011-03-340273
55. Stone RM, Fischer T, Paquette R, Schiller G, Schiffer CA, Ehninger G, et al. Phase 1b study of midostaurin (PKC412) in combination with daunorubicin and cytarabine induction and high-dose cytarabine consolidation in patients under age 61 with newly diagnosed de novo acute myeloid leukemia: Overall survival of patients whose blasts have FLT3 mutations is similar to those with wild-type FLT3. Blood (2009) 114:634. doi: 10.1182/blood.V114.22.634.634
56. Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with> 30% blasts. Blood J Am Soc Hematol (2015) 126:291–9. doi: 10.1182/blood-2015-01-621664
57. Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol (2012) 30:2670. doi: 10.1200/JCO.2011.38.9429
58. Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discovery (2017) 16:273–84. doi: 10.1038/nrd.2016.253
59. Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid LeukemiaBCL-2 inhibition by ABT-199 in AML. Cancer Discovery (2014) 4:362–75. doi: 10.1158/2159-8290.CD-13-0609
60. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. New Engl J Med (2020) 383:617–29. doi: 10.1056/NEJMoa2012971
61. Wei AH, Strickland JSA, Hou J-Z, Fiedler W, Lin TL, Walter RB, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J Clin Oncol (2019) 37:1277. doi: 10.1200/JCO.18.01600
62. Ravandi F, Alattar ML, Grunwald MR, Rudek MA, Rajkhowa T, Richie MA, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood J Am Soc Hematol (2013) 121:4655–62. doi: 10.1182/blood-2013-01-480228
63. Zhu R, Li L, Nguyen B, Seo J, Wu M, Seale T, et al. FLT3 tyrosine kinase inhibitors synergize with BCL-2 inhibition to eliminate FLT3/ITD acute leukemia cells through BIM activation. Signal Transduct Target Ther (2021) 6:186. doi: 10.1038/s41392-021-00578-4
64. Maiti A, DiNardo CD, Daver NG, Rausch CR, Ravandi F, Kadia TM, et al. Triplet therapy with venetoclax, FLT3 inhibitor and decitabine for FLT3-mutated acute myeloid leukemia. Blood Cancer J (2021) 11:25. doi: 10.1038/s41408-021-00410-w
65. Yilmaz M, Kantarjian H, Short NJ, Reville P, Konopleva M, Kadia T, et al. Hypomethylating agent and venetoclax with FLT3 inhibitor "triplet" therapy in older/unfit patients with FLT3 mutated AML. Blood Cancer J (2022) 12:77. doi: 10.1038/s41408-022-00670-0
66. Röllig C, Serve H, Hüttmann A, Noppeney R, Müller-Tidow C, Krug U, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol (2015) 16:1691–9. doi: 10.1016/S1470-2045(15)00362-9
67. Röllig C, Serve H, Noppeney R, Hanoun M, Krug U, Baldus CD, et al. Sorafenib or placebo in patients with newly diagnosed acute myeloid leukaemia: Long-term follow-up of the randomized controlled SORAML trial. Leukemia (2021) 35:2517–25. doi: 10.1038/s41375-021-01148-x
68. Serve H, Krug U, Wagner R, Sauerland MC, Heinecke A, Brunnberg U, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol (2013) 31:3110–8. doi: 10.1200/JCO.2012.46.4990
69. Metzelder S, Schroeder T, Lübbert M, Ditschkowski M, Götze K, Scholl S, et al. Long-term survival of sorafenib-treated FLT3-ITD–positive acute myeloid leukaemia patients relapsing after allogeneic stem cell transplantation. Eur J Cancer (2017) 86:233–9. doi: 10.1016/j.ejca.2017.09.016
70. Bornhäuser M, Illmer T, Schaich M, Soucek S, Ehninger G, Thiede C. Improved outcome after stem-cell transplantation in FLT3/ITD-positive AML. Blood (2007) 109:2264–5. doi: 10.1182/blood-2006-09-047225
71. DeZern AE, Sung A, Kim S, Smith BD, Karp JE, Gore SD, et al. Role of allogeneic transplantation for FLT3/ITD acute myeloid leukemia: outcomes from 133 consecutive newly diagnosed patients from a single institution. Biol Blood Marrow Transplant (2011) 17:1404–9. doi: 10.1016/j.bbmt.2011.02.003
72. Brunet S, Labopin M, Esteve J, Cornelissen J, Socié G, Iori AP, et al. Impact of FLT3 internal tandem duplication on the outcome of related and unrelated hematopoietic transplantation for adult acute myeloid leukemia in first remission: a retrospective analysis. J Clin Oncol (2012) 30:735–41. doi: 10.1200/JCO.2011.36.9868
73. Schlenk RF, Weber D, Fiedler W, Salih HR, Wulf G, Salwender H, et al. Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood J Am Soc Hematol (2019) 133:840–51. doi: 10.1182/blood-2018-08-869453
74. Burchert A, Bug G, Fritz LV, Finke J, Stelljes M, Röllig C, et al. Sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with FLT3–internal tandem duplication mutation (SORMAIN). J Clin Oncol (2020) 38:2993–3002. doi: 10.1200/JCO.19.03345
75. Perl AE, Lu Q, Fan A, Hasabou N, Berrak E, Tiu RV. Clinical outcomes following treatment with gilteritinib or quizartinib in patients with relapsed/refractory FLT3-ITD+ acute myeloid leukemia. Blood (2020) 136:42–3. doi: 10.1182/blood-2020-136118
76. Daver N, Perl AE, Maly J, Levis M, Ritchie E, Litzow MR, et al. Venetoclax in combination with gilteritinib demonstrates molecular clearance of FLT3 mutation in relapsed/refractory FLT3-mutated acute myeloid leukemia. Blood (2021) 138:691. doi: 10.1182/blood-2021-150743
77. Higgins A, Mangaonkar AA, Hefazi M, Viswanatha D, Horna P, Foran J, et al. Non-internal tandem duplication (ITD), non-tyrosine kinase domain (TKD) FLT3 mutations in myeloid malignancies: A brief report of 10 patients. Leuk Res (2018) 72:1–4. doi: 10.1016/j.leukres.2018.07.018
78. Kindler T, Breitenbuecher F, Kasper S, Estey E, Giles F, Feldman E, et al. Identification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML). Blood (2005) 105:335–40. doi: 10.1182/blood-2004-02-0660
79. Ambinder AJ, Levis M. Potential targeting of FLT3 acute myeloid leukemia. Haematologica (2021) 106:671. doi: 10.3324/haematol.2019.240754
80. Tarlock K, Hylkema TA, Pollard JA, Hansen ME, Ries R, Sweat R, et al. FUNCTIONAL ASSESSMENT OF NOVEL DIAGNOSTIC FLT3 MUTATIONS AND INHIBITION BY KINASE INHIBITORS. Haematologica (2017) 102:40–1. doi: 10.3324/%25x
81. Daver N, Price A, Benton CB, Patel K, Zhang W, Konopleva M, et al. First report of sorafenib in patients with acute myeloid leukemia harboring non-canonical FLT3 mutations. Front Oncol (2020) 10:1538. doi: 10.3389/fonc.2020.01538
82. Stasik S, Kramer M, Zukunft S, Röllig C, Baldus CD, Platzbecker U, et al. Point mutations in the FLT3-ITD region are rare but recurrent alterations in adult AML and associated with concomitant KMT2A-PTD. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.862991
83. Brasel K, Escobar S, Anderberg R, De Vries P, Gruss H, Lyman S. Expression of the flt3 receptor and its ligand on hematopoietic cells. Leukemia (1995) 9:1212–8.
84. Kazi JU, Rönnstrand L. FMS-like tyrosine kinase 3/FLT3: from basic science to clinical implications. Physiol Rev (2019) 99:1433–66. doi: 10.1152/physrev.00029.2018
85. Sato T, Yang X, Knapper S, White P, Smith BD, Galkin S, et al. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood J Am Soc Hematol (2011) 117:3286–93. doi: 10.1182/blood-2010-01-266742
86. Campos L, Rouault J-P, Sabido O, Oriol P, Roubi N, Vasselon C, et al. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood (1993) 81(11):3091–6. doi: 10.1182/blood.V81.11.3091.3091
87. Bagrintseva K, Geisenhof S, Kern R, Eichenlaub S, Reindl C, Ellwart JW, et al. FLT3-ITD-TKD dual mutants associated with AML confer resistance to FLT3 PTK inhibitors and cytotoxic agents by overexpression of bcl-x (L). Blood (2005) 105:3679–85. doi: 10.1182/blood-2004-06-2459
88. Kohl T, Hellinger C, Ahmed F, Buske C, Hiddemann W, Bohlander S, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia (2007) 21:1763–72. doi: 10.1038/sj.leu.2404776
89. Traer E, Martinez J, Javidi-Sharifi N, Agarwal A, Dunlap J, English I, et al. FGF2 from marrow microenvironment promotes resistance to FLT3 inhibitors in acute myeloid LeukemiaFGF2 promotes resistance in FLT3 AML. Cancer Res (2016) 76:6471–82. doi: 10.1158/0008-5472.CAN-15-3569
90. Zeng Z, Xi Shi Y, Samudio IJ, Wang R-Y, Ling X, Frolova O, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood J Am Soc Hematol (2009) 113:6215–24. doi: 10.1182/blood-2008-05-158311
91. Chang Y-T, Hernandez D, Alonso S, Gao M, Su M, Ghiaur G, et al. Role of CYP3A4 in bone marrow microenvironment–mediated protection of FLT3/ITD AML from tyrosine kinase inhibitors. Blood Adv (2019) 3:908–16. doi: 10.1182/bloodadvances.2018022921
92. Daver N, Cortes J, Ravandi F, Patel KP, Burger JA, Konopleva M, et al. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood J Am Soc Hematol (2015) 125:3236–45. doi: 10.1182/blood-2014-10-605808
93. Gebru MT, Wang H-G. Therapeutic targeting of FLT3 and associated drug resistance in acute myeloid leukemia. J Hematol Oncol (2020) 13:1–13. doi: 10.1186/s13045-020-00992-1
94. Klug LR, Kent JD, Heinrich MC. Structural and clinical consequences of activation loop mutations in class III receptor tyrosine kinases. Pharmacol Ther (2018) 191:123–34. doi: 10.1016/j.pharmthera.2018.06.016
95. Kiyoi H, Kawashima N, Ishikawa Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci (2020) 111:312–22. doi: 10.1111/cas.14274
96. Lindblad O, Cordero E, Puissant A, Macaulay L, Ramos A, Kabir N, et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene (2016) 35:5119–31. doi: 10.1038/onc.2016.41
97. Knapper S, Mills KI, Gilkes AF, Austin SJ, Walsh V, Burnett AK. The effects of lestaurtinib (CEP701) and PKC412 on primary AML blasts: The induction of cytotoxicity varies with dependence on FLT3 signaling in both FLT3-mutated and wild-type cases. Blood (2006) 108:3494–503. doi: 10.1182/blood-2006-04-015487
98. Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer (2019) 18:1–22. doi: 10.1186/s12943-019-1090-3
99. Joshi SK, Nechiporuk T, Bottomly D, Piehowski PD, Reisz JA, Pittsenbarger J, et al. The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance. Cancer Cell (2021) 39:999–1014.e1018. doi: 10.1016/j.ccell.2021.06.003
100. Heydt Q, Larrue C, Saland E, Bertoli S, Sarry J, Besson A, et al. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene (2018) 37:787–97. doi: 10.1038/onc.2017.376
101. Qiu S, Paterson AJ, Abraham A, He J, Shah M, Agarwal P, et al. Role of enhanced autophagy in resistance of FLT3-ITD AML stem cells to FLT3 TKI treatment. Blood (2018) 132:1358. doi: 10.1182/blood-2018-99-115702
102. Du W, Xu A, Huang Y, Cao J, Zhu H, Yang B, et al. The role of autophagy in targeted therapy for acute myeloid leukemia. Autophagy (2021) 17:2665–79. doi: 10.1080/15548627.2020.1822628
103. Baechler BL, Bloemberg D, Quadrilatero J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy (2019) 15:1606–19. doi: 10.1080/15548627.2019.1591672
104. Dany M, Gencer S, Nganga R, Thomas RJ, Oleinik N, Baron KD, et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood J Am Soc Hematol (2016) 128:1944–58. doi: 10.1182/blood-2016-04-708750
105. Wakita S, Yamaguchi H, Omori I, Terada K, Ueda T, Manabe E, et al. Mutations of the epigenetics-modifying gene (DNMT3a, TET2, IDH1/2) at diagnosis may induce FLT3-ITD at relapse in de novo acute myeloid leukemia. Leukemia (2013) 27:1044–52. doi: 10.1038/leu.2012.317
106. Long J, Jia M-Y, Fang W-Y, Chen X-J, Mu L-L, Wang Z-Y, et al. FLT3 inhibition upregulates HDAC8 via FOXO to inactivate p53 and promote maintenance of FLT3-ITD+ acute myeloid leukemia. Blood (2020) 135:1472–83. doi: 10.1182/blood.2019003538
107. Hua W-K, Qi J, Cai Q, Carnahan E, Ayala Ramirez M, Li L, et al. HDAC8 regulates long-term hematopoietic stem-cell maintenance under stress by modulating p53 activity. Blood J Am Soc Hematol (2017) 130:2619–30. doi: 10.1182/blood-2017-03-771386
108. Smith CC, Zhang C, Lin KC, Lasater EA, Zhang Y, Massi E, et al. Characterizing and overriding the structural mechanism of the quizartinib-resistant FLT3 “Gatekeeper” F691L mutation with PLX3397PLX3397 overrides AC220 resistance due to the F691L mutation. Cancer Discovery (2015) 5:668–79. doi: 10.1158/2159-8290.CD-15-0060
109. Wang Z, Cai J, Cheng J, Yang W, Zhu Y, Li H, et al. FLT3 inhibitors in acute myeloid leukemia: Challenges and recent developments in overcoming resistance. J Medicin Chem (2021) 64:2878–900. doi: 10.1021/acs.jmedchem.0c01851
110. Xu B, Zhao Y, Wang X, Gong P, Ge W. MZH29 is a novel potent inhibitor that overcomes drug resistance FLT3 mutations in acute myeloid leukemia. Leukemia (2017) 31:913–21. doi: 10.1038/leu.2016.297
111. Fathi AT, Blonquist TM, Hernandez D, Amrein PC, Ballen KK, McMasters M, et al. Cabozantinib is well tolerated in acute myeloid leukemia and effectively inhibits the resistance-conferring FLT3/tyrosine kinase domain/F691 mutation. Cancer (2018) 124:306–14. doi: 10.1002/cncr.31038
112. Yamaura T, Nakatani T, Uda K, Ogura H, Shin W, Kurokawa N, et al. A novel irreversible FLT3 inhibitor, FF-10101, shows excellent efficacy against AML cells with FLT3 mutations. Blood J Am Soc Hematol (2018) 131:426–38. doi: 10.1182/blood-2017-05-786657
113. Ma J, Zhao S, Qiao X, Knight T, Edwards H, Polin L, et al. Inhibition of bcl-2 synergistically enhances the antileukemic activity of midostaurin and gilteritinib in preclinical models of FLT3-mutated acute myeloid LeukemiaJoint FLT3 and BCL-2 inhibition in FLT3-mutated AML. Clin Cancer Res (2019) 25:6815–26. doi: 10.1158/1078-0432.CCR-19-0832
114. Brinton LT, Zhang P, Williams K, Canfield D, Orwick S, Sher S, et al. Synergistic effect of BCL2 and FLT3 co-inhibition in acute myeloid leukemia. J Hematol Oncol (2020) 13:1–10. doi: 10.1186/s13045-020-00973-4
115. Weisberg E, Liu Q, Nelson E, Kung AL, Christie AL, Bronson R, et al. Using combination therapy to override stromal-mediated chemoresistance in mutant FLT3-positive AML: synergism between FLT3 inhibitors, dasatinib/multi-targeted inhibitors and JAK inhibitors. Leukemia (2012) 26:2233–44. doi: 10.1038/leu.2012.96
116. Kapoor S, Natarajan K, Baldwin PR, Doshi KA, Lapidus RG, Mathias TJ, et al. Concurrent inhibition of pim and FLT3 kinases enhances apoptosis of FLT3-ITD acute myeloid leukemia cells through increased mcl-1 proteasomal DegradationPim and FLT3 kinase coinhibition in FLT3-ITD AML. Clin Cancer Res (2018) 24:234–47. doi: 10.1158/1078-0432.CCR-17-1629
117. Weisberg E, Liu Q, Zhang X, Nelson E, Sattler M, Liu F, et al. Selective akt inhibitors synergize with tyrosine kinase inhibitors and effectively override stroma-associated cytoprotection of mutant FLT3-positive AML cells. PloS One (2013) 8:e56473. doi: 10.1371/journal.pone.0056473
118. Czardybon W, Windak R, Gołas A, Gałęzowski M, Sabiniarz A, Dolata I, et al. A novel, dual pan-PIM/FLT3 inhibitor SEL24 exhibits broad therapeutic potential in acute myeloid leukemia. Oncotarget (2018) 9:16917. doi: 10.18632/oncotarget.24747
119. Hart S, Goh K, Novotny-Diermayr V, Tan Y, Madan B, Amalini C, et al. Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer J (2011) 1:e44–4. doi: 10.1038/bcj.2011.43
120. Jeon JY, Zhao Q, Buelow DR, Phelps M, Walker AR, Mims AS, et al. Preclinical activity and a pilot phase I study of pacritinib, an oral JAK2/FLT3 inhibitor, and chemotherapy in FLT3-ITD-positive AML. Investig New Drugs (2020) 38:340–9. doi: 10.1007/s10637-019-00786-4
121. Li Y, Ye T, Xu L, Dong Y, Luo Y, Wang C, et al. Discovery of 4-piperazinyl-2-aminopyrimidine derivatives as dual inhibitors of JAK2 and FLT3. Eur J Medicin Chem (2019) 181:111590. doi: 10.1016/j.ejmech.2019.111590
Keywords: AML, FLT3 inhibitors, targeted therapy, drug resistance, mechanisms of resistance
Citation: Ge S-S, Liu S-B and Xue S-L (2022) Developments and challenges of FLT3 inhibitors in acute myeloid leukemia. Front. Oncol. 12:996438. doi: 10.3389/fonc.2022.996438
Received: 17 July 2022; Accepted: 15 August 2022;
Published: 14 September 2022.
Edited by:
Keqiang Zhang, City of Hope National Medical Center, United StatesReviewed by:
Zheng Zhu, Cedars Sinai Medical Center, United StatesXiong Ni, Changhai Hospital, China
Chunrui Li, Huazhong University of Science and Technology, China
Copyright © 2022 Ge, Liu and Xue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Song-Bai Liu, bGl1c29uZ2JhaUAxMjYuY29t; Sheng-Li Xue, c2x4dWVAc3VkYS5lZHUuY24=