 Xingyue Yang1
Xingyue Yang1 Jing Dai1
Jing Dai1 Shun Yao1Jiaxing An1Guorong Wen1Hai Jin1Li Zhang1Liming Zheng1Xingyue Chen1Zhiqiang Yi1
Shun Yao1Jiaxing An1Guorong Wen1Hai Jin1Li Zhang1Liming Zheng1Xingyue Chen1Zhiqiang Yi1 Biguang Tuo1,2*
Biguang Tuo1,2*- 1Department of Gastroenterology, Digestive Disease Hospital, Affiliated Hospital of Zunyi Medical University, Zunyi, China
- 2The Collaborative Innovation Center of Tissue Damage Repair and Regenerative Medicine of Zunyi Medical University, Zunyi, China
Liver cancer is one of the most common cancers in the world, and the rate of liver cancer is high due to the of its illness. The main risk factor for liver cancer is infection with the hepatitis B virus (HBV), but a considerable number of genetic and epigenetic factors are also directly or indirectly involved in the underlying pathogenesis of liver cancer. In particular, the apolipoprotein B mRNA editing enzyme, catalytic peptide-like protein (APOBEC) family (DNA or mRNA editor family), which has been the focus of virology research for more than a decade, has been found to play a significant role in the occurrence and development of various cancers, providing a new direction for the research of liver cancer. APOBEC3B is a cytosine deaminase that controls a variety of biological processes, such as protein expression, innate immunity, and embryonic development, by participating in the process of cytidine deamination to uridine in DNA and RNA. In humans, APOBEC3B has long been known as a DNA editor for limiting viral replication and transcription. APOBEC3B is widely expressed at low levels in a variety of normal tissues and organs, but it is significantly upregulated in different types of tumor tissues and tumor lines. Thus, APOBEC3B has received increasing attention in various cancers, but the role of APOBEC3B in the occurrence and development of liver cancer due to infection with HBV remains unclear. This review provides a brief introduction to the pathogenesis of hepatocellular carcinoma induced by HBV, and it further explores the latest results of APOBEC3B research in the development of HBV and liver cancer, thereby providing new directions and strategies for the treatment and prevention of liver cancer.
Introduction
Liver cancer is one of the most common cancers, and its fatality rate ranks fourth in the world (1) due to its high degree of malignancy and hidden disease symptoms. Hepatitis B virus (HBV) is the leading cause of liver cancer incidence and mortality globally, particularly in Africa and East Asia where 60% of cases are caused by HBV (2). HBV is a partially double-stranded DNA virus whose clinical outcomes of infection are influenced by viral replication and changes in the host immune system (2). The innate immune system is the first line of defense of the host immune system, and in recent years, a number of “limiting factors” with antiviral properties have also been discovered, including members of the apolipoprotein B mRNA editing enzyme, catalytic peptide-like protein (APOBEC) protein family (3, 4). The APOBEC family consists of 11 cytidine deaminase members in humans that function primarily by inducing DNA or RNA upper cytosine deamination mutations to defend against retroviral or retrotransposon transmission, and these members have been identified as widely involved in various antiviral processes. APOBEC3B is the most common member of the family, and it is involved in the development of cancer, including liver cancer (5), breast cancer (6), gastric cancer (7), chondrosarcoma (8), kidney cancer (9), colorectal cancer (10), cervical squamous cell carcinoma (11), lung cancer (12), and bladder cancer (13). In humans, APOBEC3B has also been reported to be significantly upregulated in liver cancer, but its role in liver cancer is unclear. In this article, we mainly review the latest research on the molecular mechanisms of APOBEC3B in limiting the pathogenesis of HBV infection and liver cancer to provide a new therapeutic direction or target for liver cancer treatment.
Manuscript
APOBEC3B: family, localization, structure, and function
The APOBEC genes belong to a family of cytidine deaminases that deaminize cytidine to uridine on DNA or mRNA, and their high mutagenic activity limits viral reverse transcriptase and promotes instability in the genome of cancer cells (14). In humans, the APOBEC superfamily consists of 11 members, namely activation-induced cytosine deaminase (AID), APOBEC1 (A1), APOBEC2 (A2), APOBEC3A-H (A3A, A3B, A3C, A3D, A3F, A3G, and A3H), and APOBEC4 (A4), all of which play a key role in the biological processes of cells (Table 1). The chromosomal locations of the APOBEC family members are as follows: APOBEC3A-H are concatenated on chromosome 22; AID and APOBEC1 are concatenated on chromosome 12; APOBEC2 is localized on chromosome 6; and APOBEC4 is localized on chromosome 1, respectively (16, 17) (Figure 1). The genes in this family encode a cytidine deaminase, containing one or more conserved cytidine deaminase domains (CDAs), which are zinc-dependent catalytic domains that contain the common amino acid sequence of H-X-E-X23-28-P-CX2-4-C (X represents any amino acid) (18). These enzymes transmute DNA by a zinc-mediated hydrolysis mechanism to deaminize cytosine to uracil or convert cytosine to guanine (C to G) (19, 20). This pattern of mutations is the most common in cancer, second only to those that are attributed to aging (C-to-T in the CG dinucleotide motif, most likely due to water-mediated methylcytosine deamination). A1 was firstly discovered as an RNA-editing enzyme to deaminate at specific locations in mRNA to produce early stop codons, and it has been shown that A1 has strong deamination activity at the DNA level (21, 22). The function of A2 and A4 in humans remains unclear as no enzymatic activity has been demonstrated thus far. The seven A3 members are generally considered to be important barriers to viral replication and transcription, playing an important role in acquired immunity, unlike the basic function of AIDS, which is innate immunity (23). The A3 members are closely involved in immunity in a variety of ways and are a powerful force against endogenous and exogenous viruses. For example,A3G acts as a DNA editor to resist virus replication and transcription, and A3G also facilitates CD8+ cytotoxic T lymphocyte (CTL) recognition of infected T lymphocytes and limits marginal band B cells to transform rapid immune responses into more long-lasting B cell responses in germination centers (24). In addition, a recent study has suggested that inflammation-related factors induce A3A to edit the mRNAs of viral disease-causing genes in a large number of macrophages and monocytes, suggesting that A3A also plays an important role in the immune microenvironment (25). In particular, as the most involved member of this family in the cancer pathophysiology process, APOBEC3B limits viral reverse transcriptase by editing complementary DNA (cDNA) intermediates by relying on the action of cytidine deaminase (26), and it is also involved in promoting the biological occurrence and evolution of cancer through its role as a dependent or independent cytidine deaminase in the immune microenvironment (6, 11, 27).
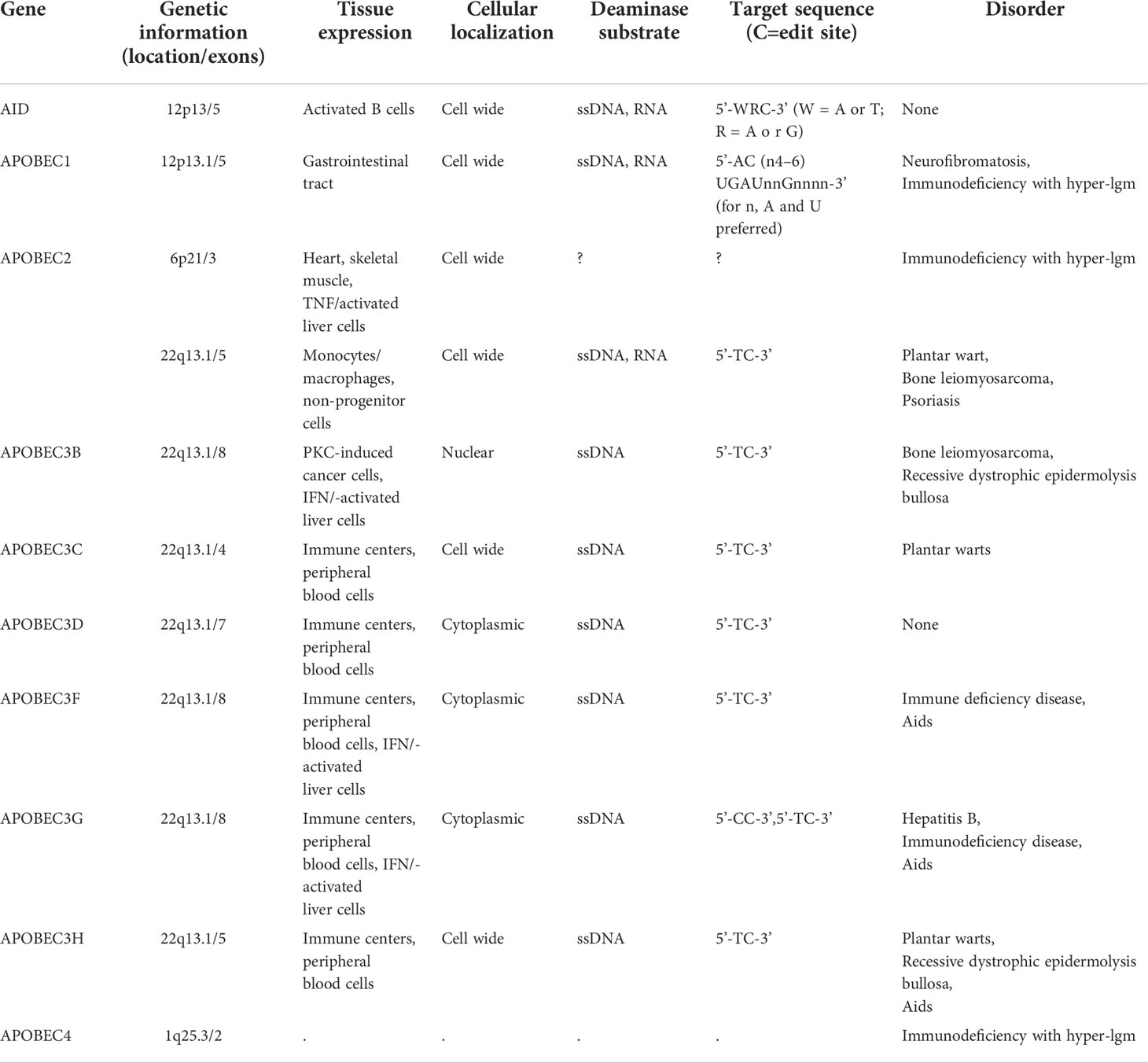
Table 1 Genomic structure, function and sequence specificity of APOBE3C family (1, 15) (Partial data source: genecard).
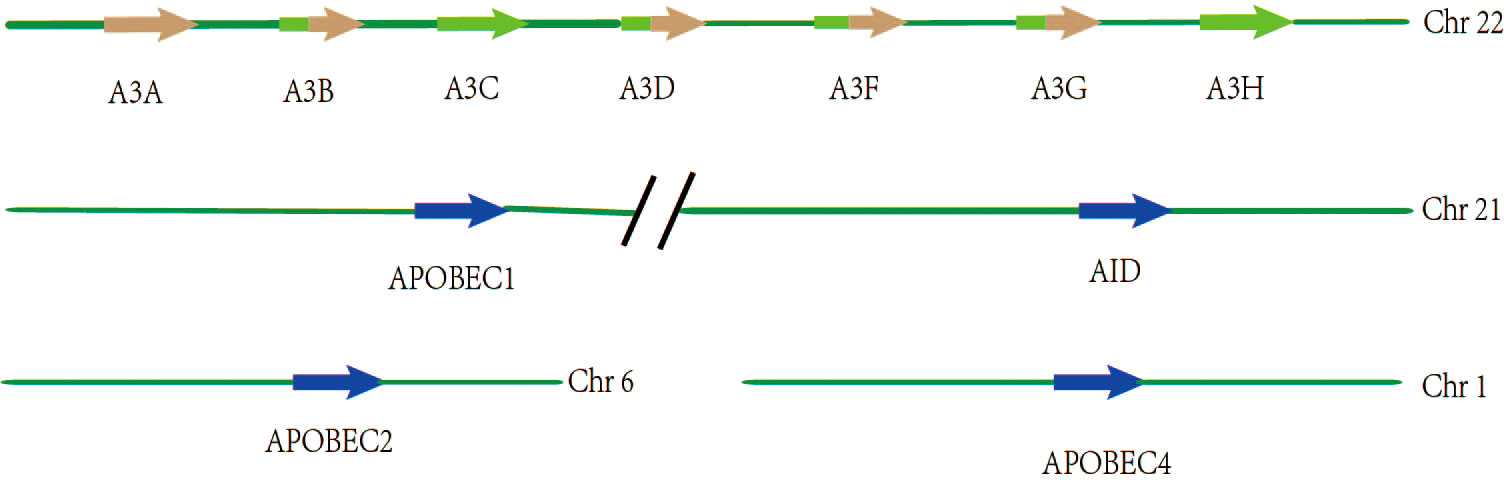
Figure 1 Spatial location distribution of genes among members of the APOBEC3 family. The seven members of APOBEC3 are arranged in tandem on chromosome 22, of which APOBEC3B, APOBEC3D, APOBEC3F, and APOBEC3G have two zinc catalytic enzyme structures, while the others have only one. APOBEC1 and AID are connected in tandem on chromosome 21, respectively. APOBEC2 and APOBEC4 are present on chromosome 6 and chromosome 1, respectively.
Analysis of The Cancer Genome Atlas (TCGA) mutation data has shown that APOBEC cytidine deaminase mutation patterns lead to genomic instability and are thus involved in carcinogenic somatic mutations (28, 29). In this family, all APOBEC3s, except for APOBEC3B, APOBEC3D, APOBEC3F, and APOBEC3G, contain only one CDA, which may explain why APOBEC3B is an epigenetic factor in many cancers (Figure 1) (20, 30). It remains controversial whether the two CDA structures in APOBEC3B are deaminated. Recent studies have suggested that only the end of the carboxyl group in the CDA show deamination activity, while the CDA at the N-terminus does not have deamination activity (31). There is increasing evidence that APOBEC3B-catalyzed genomic cytidine deaminase mutation patterns are the leading cause of dispersion and aggregation mutations in variouscancers (11). In addition, the expression level of APOBEC3B in normal human organ tissues is relatively low, but the expression level of APOBEC3B in different tumors tissue and cell lines is abnormally elevated (32, 33). Moreover, the unique nuclear localization of APOBEC3B also facilitates mutations to promote tumor development (34). Previous studies have indicated that APOBEC3B is uniquely enriched in cervical, breast, lung, and bladder cancers (35). In particular, APOBEC3B has been shown to be a prognostic marker in estrogen receptor (ER)+ breast cancer, and that it plays a key role in the progression of breast cancer (7). Furthermore, APOBEC3B has been reported to be significantly elevated in liver cancer tissues relative to normal tissues, suggesting that it also plays an essential role in the occurrence and development of liver cancer (5). However, the role of APOBEC3B in liver cancer remains unclear, thereby requiring further investigation.
HBV and liver cancer
Liver cancer is one of the most prevalent malignant tumors, and it has characteristics of late detection and high fatality rate (36). The progression of liver cancer is driven by a combination of factors including genetic predisposition, lifestyle and environmental factors, as well as chronic HBV and hepatitis C virus (HCV) infections. In particular, HBV infection is the most important cause of the high incidence of liver cancer in Asian populations (37, 38). There are approximately 240 million long-term HBV infections in the world, and uncontrolled chronic HBV infection can further evolve into life-threatening end-stage chronic liver diseases such as cirrhosis and liver cancer (39, 40). Previous studies have suggested that normal hepatocytes are be converted into hepatocellular carcinoma cells through chronic inflammation, DNA damage, chromosomal instability, epigenetic modification, and early neovascularization after HBV (41).
HBV, which belongs to the Hepadnaviridae family, is a relaxed circular double chain DNA virus with a molecular weight of 3.2Kb, and it replicates mainly through RNA intermediates (42, 43). In the human liver, 90% of the liver is comprised of hepatocytes, and HBV enters liver cells through Na+/taurocholate cotransporter polypeptide (NTCP) (44). With entry into hepatocytes, the relaxed circular DNA (RC DNA) of HBV is converted into covalent closed circular DNA (cccDNA) within the nucleus, which in turn evolves into a template for transcription of all viral RNAs (pregenomic RNA, pgRNA; and subgenomic RNA) (41). HBV synthesizes proteins, including P, core, precore, and X proteins (45). It is worth mentioning that P protein, also known as DNA polymerase, is translated from pgRNA and can function as reverse transcriptase, DNA-dependent DNA polymerase, and RNaseH (46). PgRNA is important in viral replication, and it can be reversed by the P protein into new viral rcDNA, which is then retransmitted into the nucleus to form a pool of cccDNA or secreted as an infectious virion with envelope proteins (47). In contrast, HBX (the X protein), as a key oncovirus gene protein, promotes the occurrence and development of hepatocellular carcinoma by regulating the cell cycle, cell signaling pathways, and DNA repair (48, 49). Surprisingly, HBX also participates in the replication of HBV by recruiting CBP, P300, and PCAF proteins into the hidden pathway of cccDNA (50).
HBV mainly drives the occurrence and development of liver cancer through indirect inflammatory liver damage and direct carcinogenic potential. Because HBV is a non-cytopathic virus, the specific immunity mediated by CD4+ and CD8+ T cells as well as the non-specific immunity produced by B cells are used to control the virus after invading the body, of which CD8+ T cells play a leading role (51–53). In addition, some cytokine responses (upregulation of IL-10 or TGF-β) regulatory T cells (Tregs) also play a role in liver damage (52, 54, 55). In acute injury, the liver can restore its initial liver function and size through regeneration of normal hepatocytes; however, chronic inflammatory liver injury and repeated compensatory proliferation of liver cells associated with persistent viral infection in chronic hepatitis B can lead to hepatic fibrosis or hepatic sclerosis and hepatocellular carcinoma (56, 57). Previous experiments have provided evidence for compensatory proliferation (58), chronic inflammation (52), and concomitant changes in molecular signaling pathways (59) associated with HBVinfection that drive the occurrence and development of liver cancer. However, occult HBV (the virus persists in the state of rcDNA or cccDNA in HBs Ag-negative patients) with the absence of inflammation, severe liver damage, and direct carcinogenic viral factors, or cancer supports the hypothesis of the direct carcinogenic potential of the HBV (60). The existing mainstream hypothesis about the direct carcinogenic mechanism of HBV mainly includes the following three aspects: a) HBV integrates tumor genes of the host through its own reverse transcription pathway; b) genomic is instability caused by HBV retroreplication when integrating the host genome group; and c) HBV directly affects cellular function or activates oncogenic signaling pathways through its own oncovirus gene proteins (HBx and PreS) (37). Hence, controlling HBV infection is important in the prevention and treatment of liver cancer.
A molecular mechanism of APOBEC3B restriction in HBV virus infection
HBV is a partial double-stranded DNA virus that is replicated by retrotransmission within the cytoplasmic viral core particles, and its continuous replication within the host is a high-risk factor for the development of liver cancer. In the past, the treatment of HBV infection has involved the inhibition of its replication, resulting in reduced risk of liver sclerosis or liver cancer, but, cccDNA remains in the nucleus. Therefore, these treatments do not completely eliminate the infection of the HBV. Recent reports have suggested that the APOBEC3B cytidine deaminase family member is the “limiting factor” of HBV because it edits HBV cccDNA within the nuclear DNA, resulting in its degradation, which completely eliminates the HBV (61). Moreover, reports have suggested that lymphotoxin-β receptors upregulate APOBEC3B, resulting in cytidine deamination-dependent cccDNA degradation (62). Hence, it is important to explore the molecular mechanism of APOBEC3B in the degradation of ccc DNA to provide a new therapeutic direction for the treatment of hepatitis B.
APOBEC3 is involved primarily in antiviral molecular mechanisms through both dependent and independent deaminases (4). APOBEC3B has been reported to integrate HIV-1 particles in HIV viruses as a nucleoplasmic shuttle protein to limit its replication (63), but its mechanism remains unclear. Lucifora et al. proposed a mechanistic model of APOBEC3B degradation of ccc DNA, in which APOBEC3B participates in the degradation of cccDNA by co-localizing or interacting with HBV cores in the nucleus (62). ccc DNA is deaminated by APOBEC3B in a state of temporary single-stranded cc DNA, and DNA glycosyl deaminase then recognizes the uracils that excise ccc DNA to produce AP sites, which are then are recognized and degraded by AP endonucleases (64) (Figure 2). However, the APOBEC3B protein contains two cytidine deaminase structures, namely, CD1 and CD2, at the C terminus, and the antiviral activity of these two structures has been explored. Fu et al. demonstrated that H66 mutations in CD1 have no effect on deamination and antiviral activity, and they also demonstrated that H253 and D316 in the CD2 region are primarily responsible for this process (31, 65). Furthermore, Yanmeng et al. explored whether the APOBEC3B protein encodes HBV-related DNAs during reverse transcription in HBVs (65); they found that the antiviral mechanism of APOBEC3B differs from the activation-induced cytidine deaminase (AID), which primarily edits HBV RNA and single-stranded DNA during reverse transcription, while APOBEC3B primarily edits negative and positive stranded DNA of HBV (excluding pgRNA) (66). This protein can be in the nucleus or in the cytoplasm by interacting with the core protein or capsidprotein during the deamination process of DNA in the capsid, and DNA after deamination has more exposed AP sites, which are then recognized by DNA glycosylases and degraded by the base excision repair (BER) pathway (67). Thus, these findings suggest a new antiviral mechanism of APOBEC3B, involving core correlation in addition to targeting the degradation of ccc DNA within nuclear DNA.
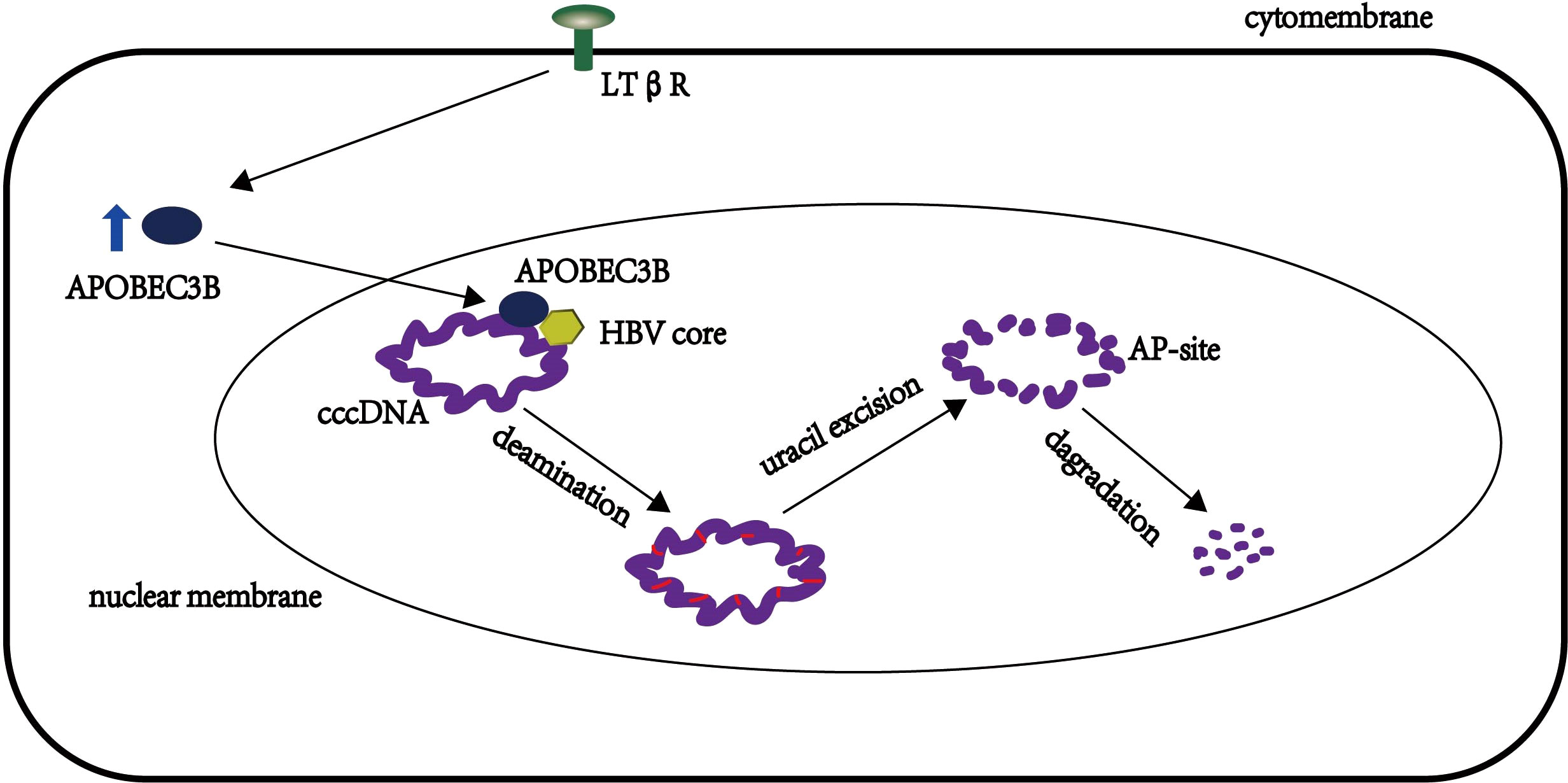
Figure 2 APOBEC3B-induced molecular mechanism model of degradation of HBV cccDNA.
There are still some unknown aspects of APOBEC3B in the mechanism of limiting HBV replication. For example, it is unknown why ccc DNA or associated DNA is degraded rather than repaired by cellular DNA repair mechanisms. Studies have reported that APOBEC3B also mutates the HBX protein of HBV to promote the development of liver cancer. Thus, it remains unclear whether, the mutation effect of APOBEC3B on HBV promotes liver cancer or inhibits liver cancer. Previous studies have suggested that cellular DNA repair mechanisms, such as UNG (uracil DNA glycosylase), resist the degradation mechanisms of ccc DNA by some APOBEC3B proteins but that these repair mechanisms do not affect the overall process of APOBEC3B restriction of HBV replication (68). In contrast, the basis for APOBEC3B mutation of the HBX protein of HBV is to promote the development of liver cancer in liver cancer cells (i.e., after liver cancer has occurred) (69), which does not contradict our hypothesis that APONEC3B inhibits the occurrence of liver cancer at the stage of HBV infection by degrading ccc DNA and DNA.
APOBEC3B role in liver cancer and its molecular mechanisms
APOBEC3B, as a cytidine deaminase, has significant contributions to variouscancers (70). However, relatively little has been reported in highly fatal liver cancers, but there are still reports of APOBEC3B upregulation in liver cancer patients, contradicting the restriction of HBV infection and liver cancer by APOBEC3B via degrading the HBV genome (69, 71). Therefore, it is important to fully understand the role and molecular mechanisms of APOBEC3B in liver cancer. Scientists have also speculated whether APOBEC3B promotes the development of liver cancer by mutating the genome of liver cells, but previous studies have reported lack of mutation patterns of APOBEC3B in liver cancer (72). Although recent reported mutational signals for APOBEC3B during replication and transcription, overexpression of APOBEC3B does not affect genomic DNA (62, 73, 74). Therefore, further exploration of the role of APOBEC3B in liver cancer is required.
It is well known that the inflammatory process in the tumor microenvironment and the complex interaction between immune cells and cancer cells are considered to be the drivers and determinants of tumor disease outcome. In addition, the most common cause of hepatocellular carcinoma is underlying chronic liver inflammation and altered immune response (75). Recent studies have found that APOBEC3B provides a potential link among inflammation, cancer, and immune processes, which may be a novel finding for the non-enzyme-dependent function of APOBEC3B (76). There are many myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) in the microenvironment of liver cancer. These cells release oxidative molecules and stimulate other immunosuppressive cells to inhibit the function of CD8+ T cells, resulting in a better prognosis of liver cancer (77). Importantly, it has been reported that there is a positive feedback loop in the presence of chemotaxis between IL-6 and APOBEC3B in hepatoma cells (78), i.e., APOBEC3B promotes IL-6 to recruit MDSCs and TAMs to promote the development of liver cancer. Regarding molecular mechanisms of interaction between chronic hepatitis and liver cancer, it has been reported that triggering the classical and non-classical NF-κB signaling pathways leads to the promotion of chronic hepatitis to liver cancer (79, 80), such as the lymphatic toxin, LTα1β2, which is significantly upregulated in liver cancer. Moreover, Duowei et al. discovered and identified that nonclassical NF-κB signaling pathways stimulate the APOBEC3B-binding promoter through the RelB/p52 complex to increase the transcriptional expression of APOBEC3B, while an increase in the expression of APOBEC3B significantly increases the CCL2 chemokine, thus recruiting MDSCs and TAMs to contribute to the development of liver cancer (81). The role of APOBEC3B in inflammation, cancer, and immunity suggests that– APOBEC3B may be used as an immunomodulatory factor to promote tumor progression by altering the immune microenvironment (76).
Duowei et al. explored how APOBEC3B promotes the upregulation of chemokine expression and thus the recruitment of MDSCs and TAMs to promote the occurrence and development of liver cancer; they reviewed previous studies and found that chemokine expression is regulated by genetic and epigenetic mechanisms inherent in cancer, such as DNA methylation and polycomb repressive complex 2 (PRC 2) (82). Furthermore, PRC2 regulates the expression of certain genes by participating in H3K27 methylation (83). In addition, studies have also shown that inhibiting the expression of histone H3 lysine 27 trimethylation (H3K27me3) promotes the upregulation of chemokines such as CCL2, IL-8, and CCL-2, in breast cancer (84), thereby establishing and validating a molecular mechanism for APOBEC3B as an immunomodulatory factor to regulate chemokine expression (81). In this mechanism, APOBEC3B binds to the three core proteins of PRC2, namely, EED, EZH2, and SUZ12, to inhibit PRC2 HMT activity, thereby inhibiting H3K27me3 to upregulate the expression of chemokines, including CCL2, IL-34, and BMP7 (81) (Figure 3). Among these chemokines, CCL2 has been established to play a key role in the occurrence and development of liver cancer by aggregating monocytes and macrophages into tumor tissue as well as stimulating tumor cell survival and immune escape (85). In addition, previous studies have shown that EZH2 deficiency promotes cancer formation and increases inflammatory factors to exacerbate the inflammatory response (86, 87). Therefore, the non-enzyme-dependent function that interferes with APOBEC3B inhibits the immunosuppressive microenvironment mediated by a variety of chemokines, thereby inhibiting the occurrence and development of liver cancer.
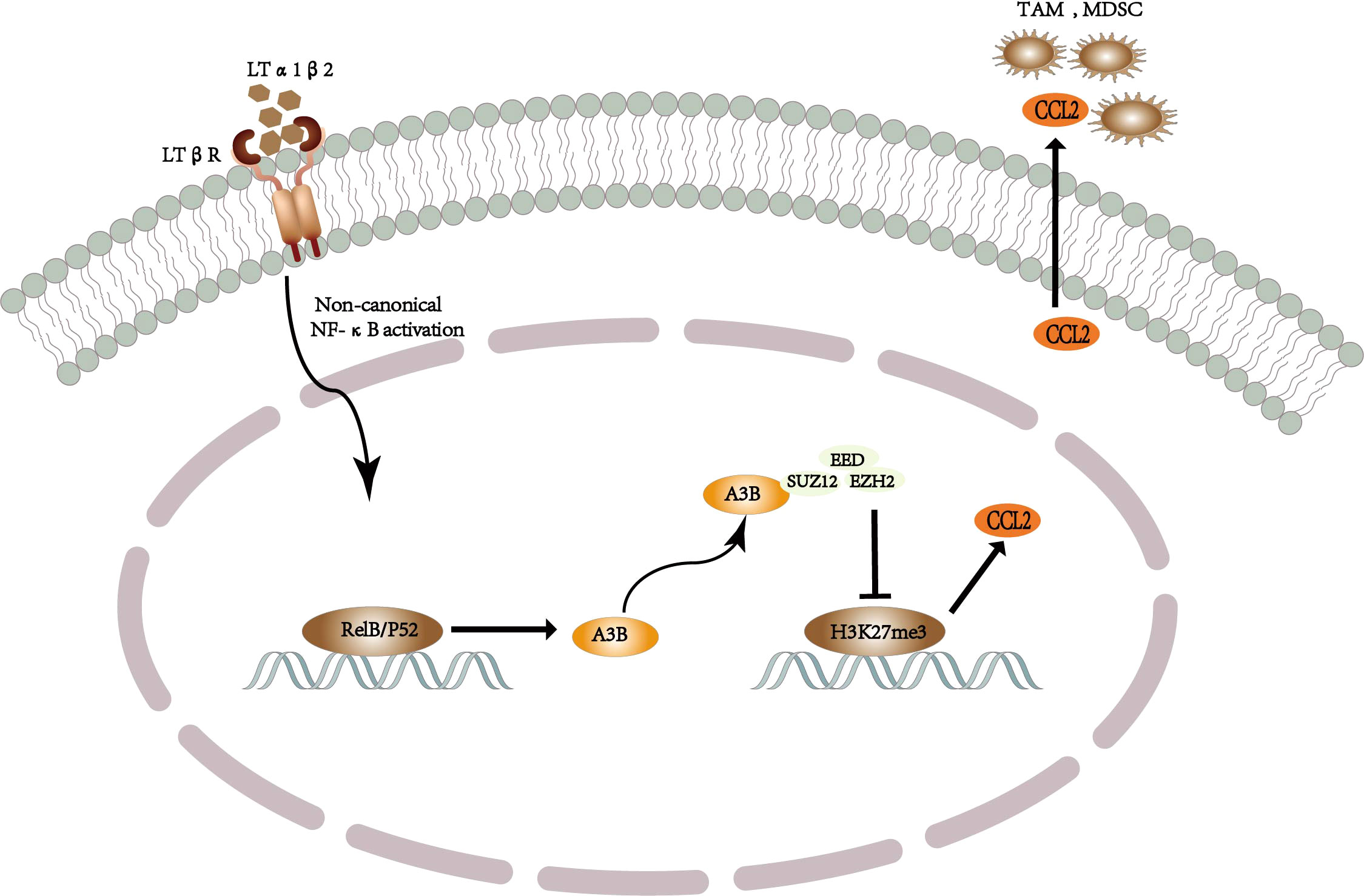
Figure 3 Schematic diagram of the mechanism by which APOBEC3B-driven TAM and MDSC recruit to promote the development of liver cancer.
The high expression of APOBEC3B in liver cancer has been validated, but it remains unclear whether it is upregulated in liver cancer as a lagging upregulation. It has been established that APOBEC3B plays a key role in liver cancer. APOBEC3B overexpression promotes the development of liver cancer in vivo and in vitro, while APOBEC3B knockout inhibits the development of liver cancer (76). Thus, regulating APOBEC3B is a potential treatment strategy for liver cancer. Moreover, in the APOBEC family, APOBEC1, AID, and APOBEC3B are also closely related to cancer (1), but their related molecular mechanisms still need to be further explored. The present, review of the molecular mechanisms associated with APOBEC3B in liver cancer provides a new possibility for these mechanisms.
Discussion
In summary, APOBEC3B plays different roles at various stages of liver cancer formation, either by restricting viral replication and transcription through enzyme-dependent functions at the stage of viral infection, or by recruiting MDSCs and TAMs in the tumor microenvironment through non-enzyme-dependent functions in terms of cancer drivers and initiators to stimulate tumor cell survival and immune escape. Although there are still few reports of APOBEC3B in the occurrence and development of liver cancer, it has been established that APOBEC3B plays a key role in liver cancer. Thus the occurrence of liver cancer may be mediated by treatment with APOBEC3B agonist therapy at the HBV stage by restricting viral replication or inhibiting the development of liver cancer by interfering with the non-enzyme-dependent function of APOBEC3B in the early stages of liver cancer. In addition, specific mutation signals and the role of APOBEC3B, as a cytidine deaminase, have been established in the occurrence and development of cervical cancer, breast cancer, lung cancer, and other cancers. Although the mutation signals found in liver cancer do not appear to affect genomic DNA, there may be other roles of the nuclear-localized APOBEC3B cytidine deaminase in hepatocellular carcinoma cells, thus requiring additional exploration.
Author contributions
XY and JD made substantial contributions to the conception and design of the manuscript. SY, JA, HJ, LZ, LMZ, XC, ZY and BT were involved in revising the manuscript critically for important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
The collaborative Innovation Center of Chinese Ministry of Education (2020-39), the Science and Technology Plan Project of Guizhou Province (QIAN KE HE JI CHU-ZK (2021) YI BAN 451), the National Natural Science Foundation of China (NSFC, 82160112), the National Natural Science Foundation of China (grant nos.81960507), the Science and Technology Bureau fund of Zunyi city (grant no. ZUN SHI KE HE HZ ZI (2019)93-HAO)
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Salter JD, Bennett RP, Smith HC. The APOBEC protein family: United by structure, divergent in function. Trends Biochem Sci (2016) 41:578–94. doi: 10.1016/j.tibs.2016.05.001
2. Singal AG, Lampertico P, Nahon P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J Hepatol (2020) 72:250–61. doi: 10.1016/j.jhep.2019.08.025
3. Cullen BR. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J Virol (2006) 80:1067–76. doi: 10.1128/JVI.80.3.1067-1076.2006
4. Janahi EM, McGarvey MJ. The inhibition of hepatitis b virus by APOBEC cytidine deaminases. J Viral Hepat. (2013) 20:821–8. doi: 10.1111/jvh.12192
5. Jin Z, Han YX, Han XR. The role of APOBEC3B in chondrosarcoma. Oncol Rep (2014) 32:1867–72. doi: 10.3892/or.2014.3437
6. Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature (2013) 494:366–70. doi: 10.1038/nature11881
7. Sieuwerts AM, Willis S, Burns MB, Look MP, Meijer-Van Gelder ME, Schlicker A, et al. Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Horm Cancer (2014) 5:405–13. doi: 10.1007/s12672-014-0196-8
8. Xia S, Gu Y, Zhang H, Fei Y, Cao Y, Fang H, et al. Immune inactivation by APOBEC3B enrichment predicts response to chemotherapy and survival in gastric cancer. Oncoimmunology (2021) 10:1975386. doi: 10.1080/2162402X.2021.1975386
9. Wu PF, Chen YS, Kuo TY, Lin HH, Liu CW, Chang LC. APOBEC3B: A potential factor suppressing growth of human hepatocellular carcinoma cells. Anticancer Res (2015) 35:1521–7.
10. Xu L, Chang Y, An H, Zhu Y, Yang Y, Xu J. High APOBEC3B expression is a predictor of recurrence in patients with low-risk clear cell renal cell carcinoma. Urol Oncol (2015) 33:340.e341–348. doi: 10.1016/j.urolonc.2015.05.009
11. Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet (2013) 45:977–83. doi: 10.1038/ng.2701
12. Lamy P, Nordentoft I, Birkenkamp-Demtröder K, Thomsen MB, Villesen P, Vang S, et al. Paired exome analysis reveals clonal evolution and potential therapeutic targets in urothelial carcinoma. Cancer Res (2016) 76:5894–906. doi: 10.1158/0008-5472.CAN-16-0436
13. Gwak M, Choi YJ, Yoo NJ, Lee S. Expression of DNA cytosine deaminase APOBEC3 proteins, a potential source for producing mutations, in gastric, colorectal and prostate cancers. Tumori. (2014) 100:112e–7e. doi: 10.1177/1636.17922
14. Fox EJ, Prindle MJ, Loeb LA. Do mutator mutations fuel tumorigenesis? Cancer Metastasis Rev (2013) 32:353–61. doi: 10.1007/s10555-013-9426-8
15. Revathidevi S, Murugan AK, Nakaoka H, Inoue I, Munirajan AK. APOBEC: A molecular driver in cervical cancer pathogenesis. Cancer Lett (2021) 496:104–16. doi: 10.1016/j.canlet.2020.10.004
16. Macduff DA, Harris RS. Directed DNA deamination by AID/APOBEC3 in immunity. Curr Biol (2006) 16:R186–189. doi: 10.1016/j.cub.2006.02.035
17. Conticello SG. The AID/APOBEC family of nucleic acid mutators. Genome Biol (2008) 9:229. doi: 10.1186/gb-2008-9-6-229
18. Vieira VC, Soares MA. The role of cytidine deaminases on innate immune responses against human viral infections. BioMed Res Int (2013) 2013:683095. doi: 10.1155/2013/683095
19. Henderson S, Fenton T. APOBEC3 genes: retroviral restriction factors to cancer drivers. Trends Mol Med (2015) 21:274–84. doi: 10.1016/j.molmed.2015.02.007
20. Swanton C, Mcgranahan N, Starrett GJ, Harris RS. APOBEC enzymes: Mutagenic fuel for cancer evolution and heterogeneity. Cancer Discovery (2015) 5:704–12. doi: 10.1158/2159-8290.CD-15-0344
21. Navaratnam N, Morrison JR, Bhattacharya S, Patel D, Funahashi T, Giannoni F, et al. The p27 catalytic subunit of the apolipoprotein b mRNA editing enzyme is a cytidine deaminase. J Biol Chem (1993) 268:20709–12. doi: 10.1016/S0021-9258(19)36836-X
22. Severi F, Chicca A, Conticello SG. Analysis of reptilian APOBEC1 suggests that RNA editing may not be its ancestral function. Mol Biol Evol (2011) 28:1125–9. doi: 10.1093/molbev/msq338
23. Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol (2008) 26:317–53. doi: 10.1146/annurev.immunol.26.021607.090350
24. Beck-Engeser GB, Winkelmann R, Wheeler ML, Shansab M, Yu P, Wünsche S, et al. APOBEC3 enzymes restrict marginal zone b cells. Eur J Immunol (2015) 45:695–704. doi: 10.1002/eji.201445218
25. Sharma S, Patnaik SK, Taggart RT, Kannisto ED, Enriquez SM, Gollnick P, et al. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat Commun (2015) 6:6881. doi: 10.1038/ncomms7881
26. Refsland EW, Harris RS. The APOBEC3 family of retroelement restriction factors. Curr Top Microbiol Immunol (2013) 371:1–27. doi: 10.1007/978-3-642-37765-5_1
27. Kuong KJ, Loeb LA. APOBEC3B mutagenesis in cancer. Nat Genet (2013) 45:964–5. doi: 10.1038/ng.2736
28. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science (2011) 333:1157–60. doi: 10.1126/science.1208130
29. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet (2012) 44:685–9. doi: 10.1038/ng.2279
30. LaRue RS, Jónsson SR, Silverstein KA, Lajoie M, Bertrand D, El-Mabrouk N, et al. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol Biol (2008) 9:104. doi: 10.1186/1471-2199-9-104
31. Fu Y, Ito F, Zhang G, Fernandez B, Yang H, Chen XS. DNA Cytosine and methylcytosine deamination by APOBEC3B: enhancing methylcytosine deamination by engineering APOBEC3B. Biochem J (2015) 471:25–35. doi: 10.1042/BJ20150382
32. Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, et al. An anthropoid-specific locus of orphan c to U RNA-editing enzymes on chromosome 22. Genomics (2002) 79:285–96. doi: 10.1006/geno.2002.6718
33. Mussil B, Suspène R, Aynaud MM, Gauvrit A, Vartanian JP, Wain-Hobson S. Human APOBEC3A isoforms translocate to the nucleus and induce DNA double strand breaks leading to cell stress and death. PloS One (2013) 8:e73641. doi: 10.1371/journal.pone.0073641
34. Lackey L, Demorest ZL, Land AM, Hultquist JF, Brown WL, Harris RS. APOBEC3B and AID have similar nuclear import mechanisms. J Mol Biol (2012) 419:301–14. doi: 10.1016/j.jmb.2012.03.011
35. Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet (2013) 45:970–6. doi: 10.1038/ng.2702
36. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol (2019) 16:589–604. doi: 10.1038/s41575-019-0186-y
37. Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol (2016) 64:S84–s101. doi: 10.1016/j.jhep.2016.02.021
38. Liang HW, Wang N, Wang Y, Wang F, Fu Z, Yan X, et al. Hepatitis b virus-human chimeric transcript HBx-LINE1 promotes hepatic injury via sequestering cellular microRNA-122. J Hepatol (2016) 64:278–91. doi: 10.1016/j.jhep.2015.09.013
39. Venook AP, Papandreou C, Furuse J, De Guevara LL. The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist (2010) 15 Suppl 4:5–13. doi: 10.1634/theoncologist.2010-S4-05
40. Chen S, Li J, Wang D, Fung H, Wong LY, Zhao L. The hepatitis b epidemic in China should receive more attention. Lancet (2018) 391:1572. doi: 10.1016/S0140-6736(18)30499-9
41. Levrero M, Pollicino T, Petersen J, Belloni L, Raimondo G, Dandri M. Control of cccDNA function in hepatitis b virus infection. J Hepatol (2009) 51:581–92. doi: 10.1016/j.jhep.2009.05.022
42. Quasdorff M, Protzer U. Control of hepatitis b virus at the level of transcription. J Viral Hepat. (2010) 17:527–36. doi: 10.1111/j.1365-2893.2010.01315.x
43. Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis b virus cccDNA. Science (2014) 343:1221–8. doi: 10.1126/science.1243462
44. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis b and d virus. Elife (2012) 1:e00049. doi: 10.7554/eLife.00049
45. Chan DW, Ng IO. Knock-down of hepatitis b virus X protein reduces the tumorigenicity of hepatocellular carcinoma cells. J Pathol (2006) 208:372–80. doi: 10.1002/path.1901
46. Nassal M. Hepatitis b viruses: reverse transcription a different way. Virus Res (2008) 134:235–49. doi: 10.1016/j.virusres.2007.12.024
47. Bruss V. Hepatitis b virus morphogenesis. World J Gastroenterol (2007) 13:65–73. doi: 10.3748/wjg.v13.i1.65
48. Tang H, Oishi N, Kaneko S, Murakami S. Molecular functions and biological roles of hepatitis b virus x protein. Cancer Sci (2006) 97:977–83. doi: 10.1111/j.1349-7006.2006.00299.x
49. Zhang WY, Cai N, Ye LH, Zhang XD. Transformation of human liver l-O2 cells mediated by stable HBx transfection. Acta Pharmacol Sin (2009) 30:1153–61. doi: 10.1038/aps.2009.99
50. Cougot D, Wu Y, Cairo S, Caramel J, Renard CA, Lévy L, et al. The hepatitis b virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J Biol Chem (2007) 282:4277–87. doi: 10.1074/jbc.M606774200
51. Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, et al. Molecular pathogenesis of hepatocellular carcinoma in hepatitis b virus transgenic mice. Cell (1989) 59:1145–56. doi: 10.1016/0092-8674(89)90770-8
52. Nakamoto Y, Guidotti LG, Kuhlen CV, Fowler P, Chisari FV. Immune pathogenesis of hepatocellular carcinoma. J Exp Med (1998) 188:341–50. doi: 10.1084/jem.188.2.341
53. Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol (2006) 1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230
54. Stoop JN, van der Molen RG, Baan CC, van der Laan LJ, Kuipers EJ, Kusters JG, et al. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis b virus infection. Hepatology (2005) 41:771–8. doi: 10.1002/hep.20649
55. Manigold T, Racanelli V. T-Cell regulation by CD4 regulatory T cells during hepatitis b and c virus infections: Facts and controversies. Lancet Infect Dis (2007) 7:804–13. doi: 10.1016/S1473-3099(07)70289-X
56. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology (2004) 127:S35–50. doi: 10.1053/j.gastro.2004.09.014
57. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet (2012) 379:1245–55. doi: 10.1016/S0140-6736(11)61347-0
58. Weber A, Boger R, Vick B, Urbanik T, Haybaeck J, Zoller S, et al. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology (2010) 51:1226–36. doi: 10.1002/hep.23479
59. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature (2004) 431:461–6. doi: 10.1038/nature02924
60. Shi Y, Wu YH, Wu W, Zhang WJ, Yang J, Chen Z. Association between occult hepatitis b infection and the risk of hepatocellular carcinoma: A meta-analysis. Liver Int (2012) 32:231–40. doi: 10.1111/j.1478-3231.2011.02481.x
61. Ringelhan M, O'connor T, Protzer U, Heikenwalder M. The direct and indirect roles of HBV in liver cancer: prospective markers for HCC screening and potential therapeutic targets. J Pathol (2015) 235:355–67. doi: 10.1002/path.4434
62. Xia Y, Lucifora J, Reisinger F, Heikenwalder M, Protzer U. Virology. response to comment on "Specific and nonhepatotoxic degradation of nuclear hepatitis b virus cccDNA". Science (2014) 344(6189):1237. doi: 10.1126/science.1254083
63. Doehle BP, Schäfer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 vif. Virology (2005) 339:281–8. doi: 10.1016/j.virol.2005.06.005
64. Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat Struct Mol Biol (2010) 17:222–9. doi: 10.1038/nsmb.1744
65. Chen Y, Hu J, Cai X, Huang Y, Zhou X, Tu Z, et al. APOBEC3B edits HBV DNA and inhibits HBV replication during reverse transcription. Antiviral Res (2018) 149:16–25. doi: 10.1016/j.antiviral.2017.11.006
66. Liang G, Kitamura K, Wang Z, Liu G, Chowdhury S, Fu W, et al. RNA Editing of hepatitis b virus transcripts by activation-induced cytidine deaminase. Proc Natl Acad Sci U.S.A. (2013) 110:2246–51. doi: 10.1073/pnas.1221921110
67. Krokan HE, Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol (2013) 5:a012583. doi: 10.1101/cshperspect.a012583
68. Liu W, Wu J, Yang F, Ma L, Ni C, Hou X, et al. Genetic polymorphisms predisposing the interleukin 6-induced APOBEC3B-UNG imbalance increase HCC risk via promoting the generation of APOBEC-signature HBV mutations. Clin Cancer Res (2019) 25:5525–36. doi: 10.1158/1078-0432.CCR-18-3083
69. Xu R, Zhang X, Zhang W, Fang Y, Zheng S, Yu XF. Association of human APOBEC3 cytidine deaminases with the generation of hepatitis virus b x antigen mutants and hepatocellular carcinoma. Hepatology (2007) 46:1810–20. doi: 10.1002/hep.21893
70. Zou J, Wang C, Ma X, Wang E, Peng G. APOBEC3B, a molecular driver of mutagenesis in human cancers. Cell Biosci (2017) 7:29. doi: 10.1186/s13578-017-0156-4
71. Vartanian JP, Henry M, Marchio A, Suspène R, Aynaud MM, Guétard D, et al. Massive APOBEC3 editing of hepatitis b viral DNA in cirrhosis. PloS Pathog (2010) 6:e1000928. doi: 10.1371/journal.ppat.1000928
72. Henderson S, Chakravarthy A, Su X, Boshoff C, Fenton TR. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep (2014) 7:1833–41. doi: 10.1016/j.celrep.2014.05.012
73. Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell (2016) 164:538–49. doi: 10.1016/j.cell.2015.12.050
74. Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, Nikolaev SI. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res (2016) 26:174–82. doi: 10.1101/gr.197046.115
75. Elsharkawy AM, Mann DA. Nuclear factor-kappaB and the hepatic inflammation-fibrosis-cancer axis. Hepatology (2007) 46:590–7. doi: 10.1002/hep.21802
76. Ma W, Ho DW, Sze KM, Tsui YM, Chan LK, Lee JM, et al. APOBEC3B promotes hepatocarcinogenesis and metastasis through novel deaminase-independent activity. Mol Carcinog. (2019) 58:643–53. doi: 10.1002/mc.22956
77. Wan S, Kuo N, Kryczek I, Zou W, Welling TH. Myeloid cells in hepatocellular carcinoma. Hepatology (2015) 62:1304–12. doi: 10.1002/hep.27867
78. Li S, Bao X, Wang D, You L, Li X, Yang H, et al. APOBEC3B and IL-6 form a positive feedback loop in hepatocellular carcinoma cells. Sci China Life Sci (2017) 60:617–26. doi: 10.1007/s11427-016-9058-6
79. Greten FR, Karin M. The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett (2004) 206:193–9. doi: 10.1016/j.canlet.2003.08.029
80. Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell (2009) 16:295–308. doi: 10.1016/j.ccr.2009.08.021
81. Wang D, Li X, Li J, Lu Y, Zhao S, Tang X, et al. APOBEC3B interaction with PRC2 modulates microenvironment to promote HCC progression. Gut (2019) 68:1846–57. doi: 10.1136/gutjnl-2018-317601
82. van Mierlo G, Veenstra GJC, Vermeulen M, Marks H. The complexity of PRC2 subcomplexes. Trends Cell Biol (2019) 29:660–71. doi: 10.1016/j.tcb.2019.05.004
83. Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer (2006) 6:846–56. doi: 10.1038/nrc1991
84. Sun F, Chan E, Wu Z, Yang X, Marquez VE, Yu Q. Combinatorial pharmacologic approaches target EZH2-mediated gene repression in breast cancer cells. Mol Cancer Ther (2009) 8:3191–202. doi: 10.1158/1535-7163.MCT-09-0479
85. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell (2016) 30:533–47. doi: 10.1016/j.ccell.2016.09.003
86. Sashida G, Harada H, Matsui H, Oshima M, Yui M, Harada Y, et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun (2014) 5:4177. doi: 10.1038/ncomms5177
Keywords: liver cancer, hbv, APOBEC3B, APOBEC family, NF-κB signaling pathways
Citation: Yang X, Dai J, Yao S, An J, Wen G, Jin H, Zhang L, Zheng L, Chen X, Yi Z and Tuo B (2022) APOBEC3B: Future direction of liver cancer research. Front. Oncol. 12:996115. doi: 10.3389/fonc.2022.996115
Received: 17 July 2022; Accepted: 22 August 2022;
Published: 20 September 2022.
Edited by:
Yuen Gao, Michigan State University, United StatesReviewed by:
Lexiang Yu, Columbia University Irving Medical Center, United StatesYongqiang Gao, Harvard Medical School, United States
Copyright © 2022 Yang, Dai, Yao, An, Wen, Jin, Zhang, Zheng, Chen, Yi and Tuo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Biguang Tuo, dHVvYmlndWFuZ0BhbGl5dW4uY29t