 Yanqing Li
Yanqing Li Ziyu Luo1,2
Ziyu Luo1,2 Xinxing Wang
Xinxing Wang Jianwu Qin
Jianwu Qin- 1Department of Thyroid and Neck, The Affiliated Cancer Hospital of Zhengzhou University, Henan Cancer Hospital, Zhengzhou, China
- 2The Medical School of Zhengzhou University, Zhengzhou, China
- 3Department of Pain and Rehabilitation and Palliative Medicine, Henan Cancer Hospital, Zhengzhou, China
Medullary thyroid carcinoma (MTC) is one of the common malignant endocrine tumors, which seriously affects human health. Although surgical resection offers a potentially curative therapeutic option to some MTC patients, most patients do not benefit from it due to the difficulty to access the tumors and tumor metastasis. The survival rate of MTC patients has improved with the recent advances in the research, which has improved our understanding of the molecular mechanism underlying MTC and enabled the development and approval of novel targeted drugs. In this article, we reviewed the molecular mechanisms related to MTC progression and the principle for the design of molecular targeted drugs, and proposed some future directions for prospective studies exploring targeted drugs for MTC.
Introduction
Medullary thyroid carcinoma (MTC) is a rare thyroid malignancy accounting for 3-5% of all thyroid malignancies (1). Thyroid cancer also includes differentiated thyroid carcinoma (DTC; including papillary, folliculous, and Hürthle cell) and anaplastic thyroid carcinoma (ATC). Different from DTC and ATC, MTC originates from the c-cells near the follicles which secrete calcitonin (2). Moreover, in comparison with DTC, MTC patients have lower morbidity and higher mortality. About 35% of MTC patients have neck metastasis and about 13% have distant metastasis (3). Similarly, the clinical manifestations of thyroid cancer are mostly neck masses. Patients with large thyroid masses may present with compression symptoms beyond the thyroid, such as dysphagia caused by the compression of esophagus, hoarseness caused by nerve compression, and dyspnea caused by the compression of the airway (4). MTC often has gene mutations, and traditional chemotherapy does not achieve satisfactory therapeutic efficacy. Therefore, the standard treatment for MTC involves surgical resection and molecular targeted therapy to improve local or systemic symptoms (5). At present, emerging targeted therapeutics have greatly alleviated the symptoms in MTC patients.
In the normal thyroid C cells, the ret proto-oncogene (RET), renin-angiotensin system (RAS), phosphatidylinositol-4,5-bisphosphate 3-kinase/protein kinase B/mechanistic target of rapamycin kinase (PI3K/AKT/mTOR), vascular endothelial growth factor (VEGF) and programmed death ligand-1 (PD-L1) are responsible for regulating the biological processes such as cell proliferation, apoptosis, differentiation, migration and immunity (6–8). However, the progression of MTC affects the normal functioning of these molecules. For example, RET mutation is frequently found in medullary carcinoma (9). Glial cell derived neurotrophic factor (GDNF) binds the GFRα receptor and the GDNF-GFRα complex binds to RET receptor resulting in the activation of tyrosine kinase domain through homo-dimerization (10). This leads to the phosphorylation of tyrosine residues in the RET protein, which mediates the activation of various intracellular signaling pathways, including PI3K/AKT/mTOR, RAS/MAPK, VEGF, etc. (11, 12). Moreover, in MTC, the related immune molecules, such as PD-L1 (13), are known to mediate the inhibition of T cell activation by binding to the receptors and their ligands, which inhibits the activation and proliferation of T cells, reducing their cytotoxic effects on the tumor cells (Figure 1; Table 1). Therefore, researchers mainly focused on the above molecules while exploring anti-tumor drugs with good efficacy.
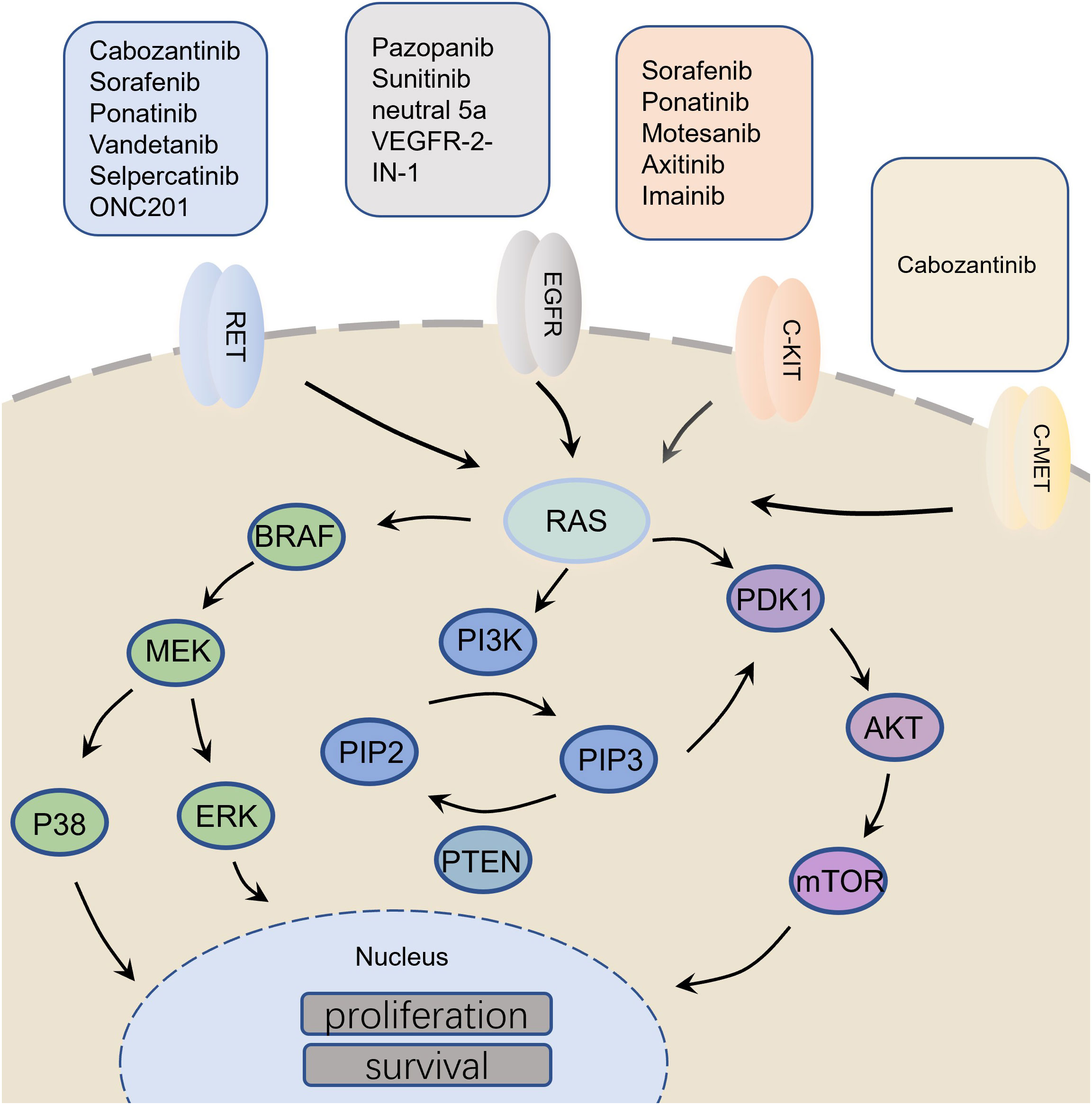
Figure 1 Molecular mechanism related to medullary thyroid carcinoma and its targeted drugs.
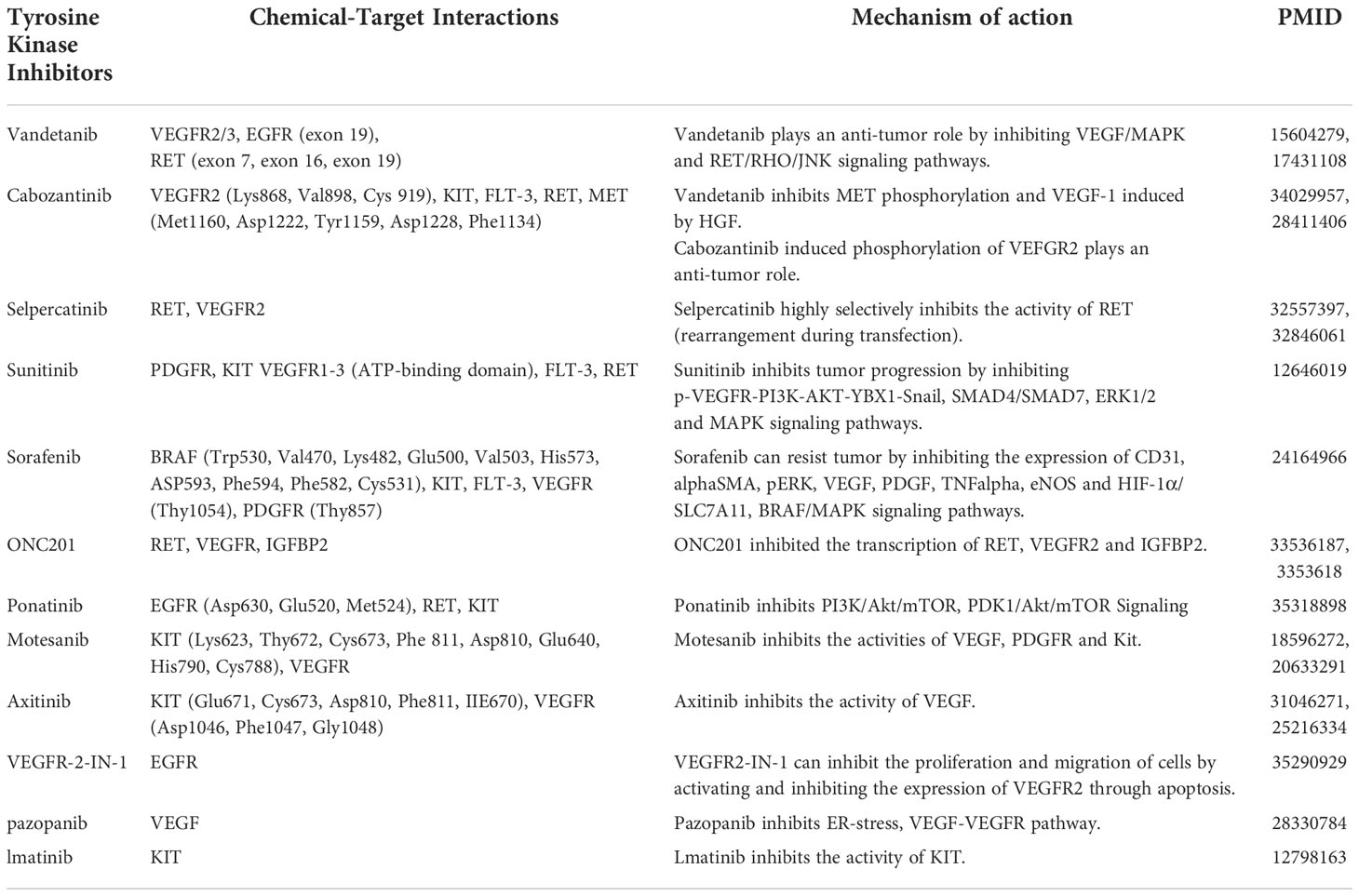
Table 1 Drug target and mechanism.
In the past decade, many novel therapeutic drug targets have been discovered and explored. Most of the anticancer drugs are known to inhibit cell proliferation, apoptosis, angiogenesis and kinase activity (14). For example, several kinase inhibitors such as imatinib, gefitinib, erlotinib, sorafenib, cetuximab and bevacizumab have been developed and approved as anticancer agents (15–17). Some of the inhibitors targeting angiogenesis and inhibitors of the RET pathway are still being used in the clinical practice. Vandetanib, selpercatinib, pralsetinib and cabozatinib have also been approved by the FDA for clinical use (15, 18). In addition, many other biological compounds are also currently in Phase 1-3 clinical trials (19). In this review, we discuss the molecular targets related to MTC therapy, as well as the design and challenges related to new anticancer drugs against these molecular targets.
Mutation of RET gene and designing drugs targeting the RET gene
Mechanism associated with RET gene mutation in MTC
Currently, medullary thyroid cancer is mainly thought to arise due to gene mutation. Among them, 98% of the familial cases harbor mutations in the RET gene, 44% cases have sporadic mutations, and 13% of the patients have mutations in the RAS gene (mainly HRAS and KRAS) (20). The RET gene is located at 10q11.2, and contains 21 exons, which encodes a Receptor Tyrosine Kinase (RTK). The RET protein transmits intracellular signals when growth factors bind to the receptor, thus regulating the growth, survival, differentiation and migration of neural crest-derived cells (21). RET mutation usually occurs in the cysteine-rich regions of the exons 10 and 11, but may also occur in the tyrosine kinase domain (22). In a few cases, papillary thyroid carcinoma has been shown to harbor rearrangements in the RET gene. On the contrary, MTC is characterized by point mutation in the RET gene. These mutations are concentrated in the exons 5, 8, 10, 11 and 13-16 (9). Among them, M918T mutation in exon 16 is the most common one in sporadic MTC, which is usually associated with the highest risk of invasive cancer (23).
RET targeted drug design
Compound library screening and computer simulation are one of the main approaches for designing drugs. For example, ONC201, a small molecule anticancer agent (imipridone), is currently under phase II clinical trials for advanced thyroid cancer. ONC201 (Animidazo [1,2-a] Pyrido [4,3-d] Pyrimine derivative) was identified for the first time from NCI chemical library screening, and has a regular [3,4-e] structure. ONC201 has been reported to trigger cell death in various tumor types by inducing integrated stress response involving the transcription factor ATF4, transactivator DDIT3 (CHOP), the pro-apoptotic protein TRAIL encoded by TNFSF10 and the TRAIL receptor DR5 encoded by TNFSF10B (24). Moreover, with the recent advances in the drug design concept, computer based simulation has attracted great attention. Some studies have used the computer to generate receptor-ligand pharmacophore model for identifying small molecule inhibitors effective against the wild-type (WT) or mutant RET kinases. Following this, small molecule inhibitors consistent with the model were selected from the natural product database. Then, the obtained drugs were subjected to molecular docking with the RET WT kinase domain, and drugs with better RET binding free energy scores were screened out (25). For example, Hit1 (ZINC02123418), Hit2 (ZINC02113839) and Hit3 (ZINC04030012) were small molecule inhibitors that were identified as the hits against WT RET, and V804M and V804 RET. However, the potency of the above drugs has not yet been tested in vitro and in vivo.
ATP competitive kinase inhibitors have attracted much attention due to their high affinity and clear action sites, and are also the most studied type of kinase inhibitors. ATP competitive kinase inhibitors compete with the substrate ATP to bind to the ATP binding pocket of the kinase, thereby inhibiting the kinase activity in the signaling pathway. According to the ligand protein binding pattern, ATP competitive kinase inhibitors are generally classified into type I and type II (26). Type I inhibitors bind to active kinases with a “DFG (Asp-Phe-Gly) - in” conformation, while type II inhibitors bind to inactive kinases with a “DFG - out” conformation. Type II inhibitors have been reported to have better cellular potency and kinase selectivity than the type I inhibitors. Because the type II inhibitors occupy the non-conservative hydrophobic pockets produced by DFG flip, they have an expanded chemical space for drug molecule discovery (27). Finally, type II inhibitors show lower affinity for ATP than the for the active conformation of the RET kinase. Common type II RET inhibitors against RET-positive cancers include cabozantinib, sorafenib, ponatinib etc., all of which show good efficacy (Figure 2). Additionally, FDA has also approved multi-kinase inhibitors targeting RET kinase such as vandetinib (inhibits multiple receptors: RET, EGFR and VEGF2) and cabozantinib (inhibits RET, C-Met, VEGFR2 and AXL), for the treatment of MTC (28, 29). The above drugs were efficacious in MTC patients, especially those harbouring the RET M918T mutation, according to ZETA and EXAM trials. However, the “functional acquisition” of mutation in the RET gate-keeper residue Val804, confers resistance to most of the known RET inhibitors, such as vandetanib (15). There is still controversy about the correlation between RET V804 mutation and vandetanib resistance. Carlomagno F et al. proved in vitro that V804 mutation caused cell resistance to vandetanib (30). They explained that V804 is the gatekeeper in ATP-binding pocket. Its mutation affects the stability of protein, thus endowing it with resistance to vandetanib. However, Samuel A et al. found no correlation between V804 mutation and vandetanib (31). They think that in their experiment, there are a large number of patients with unknown RET mutation status, because their paraffin blocks or slides don’t have enough quantity or quality of DNA for complete analysis. The small number of RET negative patients means that subgroup analysis of PFS and objective remission rate according to RET mutation status is uncertain. Therefore, the difference of curative effect of vandetanib on RET V804 mutation patients needs to be included in more patients for further analysis. To overcome this drug resistance, researchers developed selpercatinib (LOXO-292) and pralsetinib (BLU-667) against the mutant forms of the RET gate-keeper-V804M/L (32). Selpercatinib and pralsetinib were designed not only to target the gatekeeper mutations (such as V804) but also to minimize the serious adverse events recorded with multi TKIs. This was achieved due to the higher affinity of these drugs for the RET receptor. It is worth mentioning that even RET selective inhibitors that overcome the gatekeeper mutation are unable to target the acquired solvent front mutation (RET G810S/R/C). TPX-0046, a new drug, has shown promising results in inhibiting the solvent frontier mutation G810R (33).
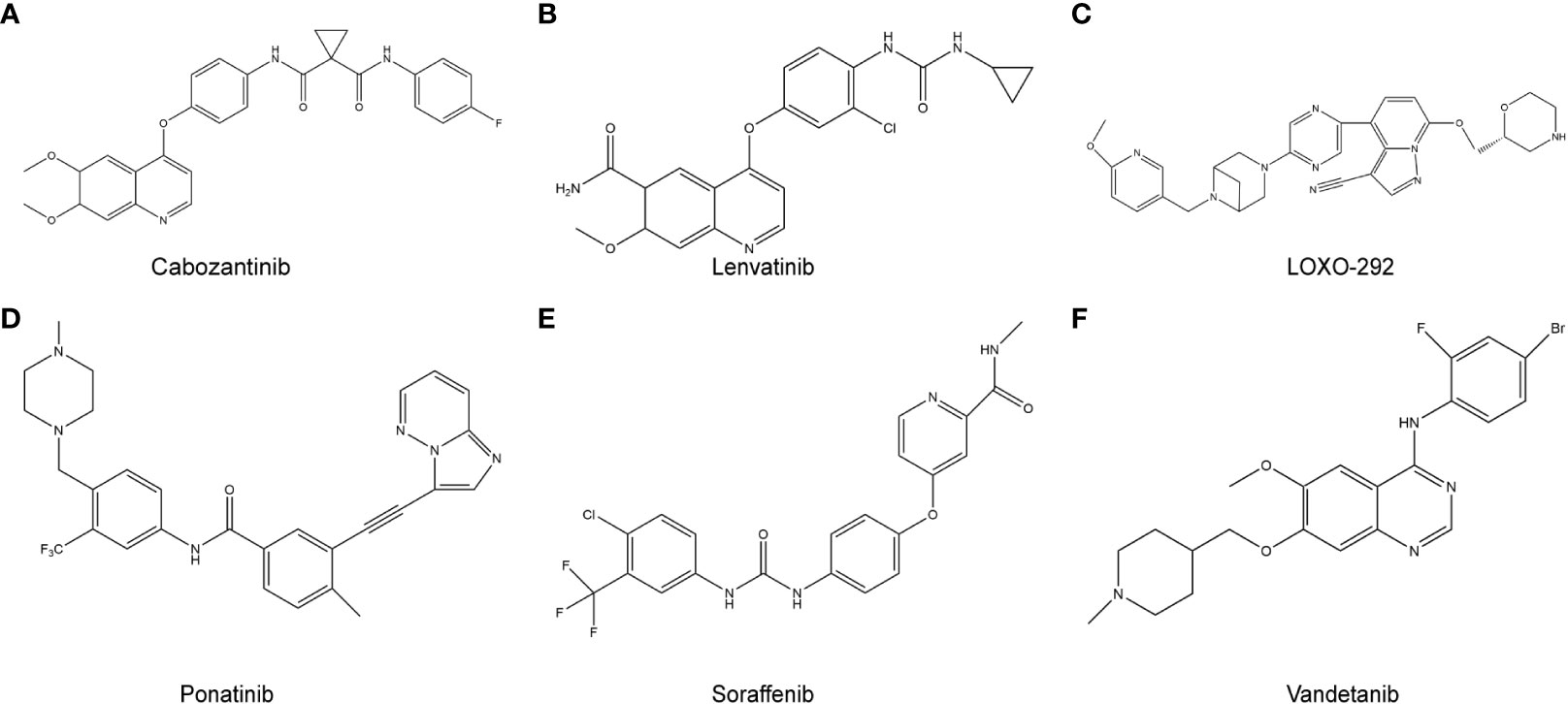
Figure 2 RET target-related chemotherapy drugs. (A), cabozantinib; (B), Lenvatinib; (C), LOXO-292; (D), Ponatinib; (E), Sorafenib; (F), Vandetanib.
The RAS signaling pathway and designing targeted drugs against this pathway
The role of RAS signaling pathway in MTC
About 70% of MTC cases without recognizable RET mutation are attributed to mutations in the RAS oncogene. The RAS gene encodes GTP enzymes, which regulates a variety of downstream pathways including the MEK/ERK and PI3K/AKT pathways, and plays important roles in a variety of processes including cell growth and proliferation, apoptosis and differentiation (34). About 20% of cancers harbor activating mutations in the RAS oncogene. Activating mutation in HRAS is the most common in MTC, and the proportion of KRAS and rare NRAS mutations is low (35). RAS and RET mutations are usually mutually exclusive in MTC, except for a few anecdotal reports of mutations in both genes (36). As with other RAS-driven cancers, mutations in MTC mainly occur in the exons 2 and 3. The most common activating mutations in RAS occur in the G12, G13 and Q61 residues (37).
Design of RAS inhibitors
For several decades since the discovery of RAS, no drugs were developed that directly targeted RAS and inhibited its abnormal function (38). It was not until May 2021 that the FDA approved the first KRAS inhibitor Sotorasib for clinical use, which opened the doors for the clinical application of KRAS inhibitors. There are several reasons that hamper the development of drugs that directly target the RAS oncogene. Firstly, the RAS protein has a strong binding affinity to GTP, but the concentration of GTP in normal cells is high, therefore, it is difficult to develop competitive inhibitors that can directly target GTP. Secondly, the surface of the RAS protein is similar to a smooth sphere, lacking the pocket for binding to drugs (39). Finally, RAS is an essential protein that regulates normal cellular activity besides having oncogenic role in different types of tumors (40). Targeted small molecule inhibitors not only inhibit the activity of mutant RAS, but also inhibit the activity of the WT RAS, thus causing toxic side effects and adverse reactions in cancer patients. Therefore, it is difficult to develop competitive inhibitors that directly target RAS. However, with the analysis of the crystal structure of RAS, researchers found that there were two switches on its surface, namely the switch I and switch II. Among them, the switch II pocket in the protein showed high variability (41). Switch II induced conformational change of the protein in the switch II region, which inactivated RAS and blocked signal transmission through the RAS pathway. The small molecule RAS inhibitor sotorasib, bound to the switch II pocket and only engaged with the inactive GDP state of KRAS G12C. Sotorasib was shown to promote therapeutic effects in lung cancer (42), however, similar studies have not yet been conducted in MTC patients to evaluate the therapeutic effects of sorafenib in MTC.
Amgen developed sotorasib using a fragment-based approach for their drug design. After analyzing the crystal structure of RAS with X-rays or magnetic resonance imaging, the researchers used molecular simulation and covalent binding methods to find allosteric binding sites in the target protein. The designed drug covalently bound to the Cys 12 site, leaving KRAS in an irreversible inactive conformation. To achieve this goal, they first screened 380 disulfide compounds with weak electrophilic properties and identified compound 1 (structural formula is shown in Figure 3A) as the specific compound with a strong binding affinity to the KRAS-GDP complex (43). The eutectic structure showed that compound 1 bound to KRASG12C at the switch II region, and the disulfide bond of compound 1 was substituted and bound to the mutant Cys 12 residue. Researchers then replaced the disulfide structure with acrylamide, which was more electrophilic to enhance the binding strength to the Cys 12 residue. It is necessary to couple the compound containing the reactant group and the drug skeleton to generate a single molecule. Therefore, Amgen used the Chemotype Evolution platform of Carmot Therapeutics to identify such compounds. Compound 2 (structural formula is shown in Figure 3A) was identified by rapid screening and analyzed by several biochemical assays and mass spectrometry, and after a series of skeleton fusions, structural optimization, and rigorous testing of its in vitro and in vivo potency, sotorasib was finally developed (44).
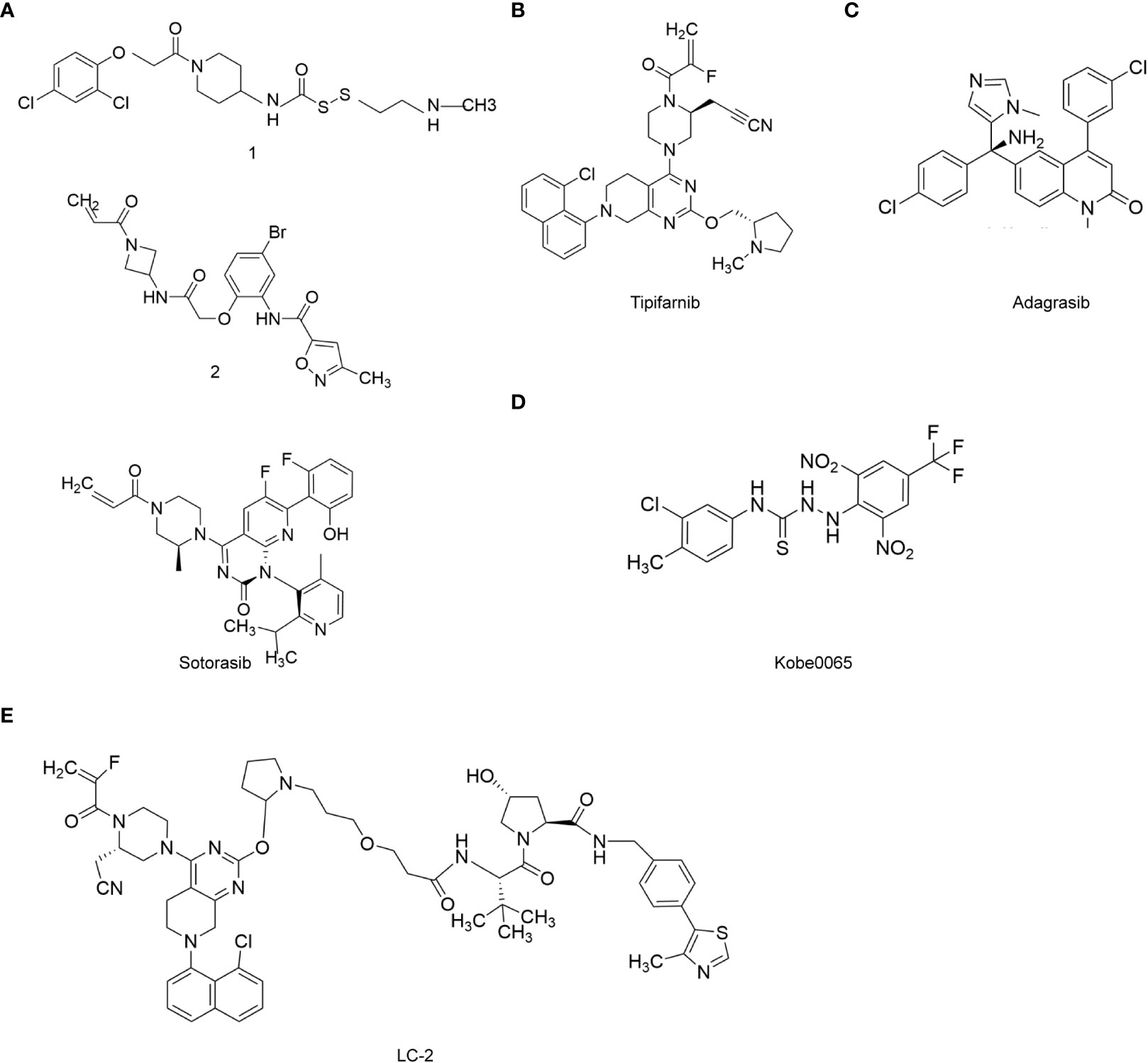
Figure 3 RAS target-related chemotherapy drugs. (A), Sotorasib; (B), Tipifarnib; (C), Adagrasib; (D), Kobe0065; (E), PROTAC LC-2.
Adagrasib (MRTX849) was developed as a drug with a similar mechanism of action as sotorasib. Adagrasib also inhibited KRASG12C and was developed by Mirati Therapeutics. Adagrasib showed good efficacy in clinical trials against patients with advanced/metastatic NSCLC harboring KRASG12C mutation. Adagrasib monotherapy showed an objective response rate (ORR) of 45% and a disease control rate of 96% (45). Therefore, it may be a potential drug for MTC patients with KRAS mutation. The molecular formula of Adagrasib is C32H35ClFN7O2, the relative molecular weight is 604.12, and its structural formula is shown in Figure 3C. The design concept and synthetic process of adagrasib are similar to that of sotorasib.
HRAS mutation is also a common type of mutation in MTC patients, however, there are relatively few studies reporting targeted drugs for HRAS as compared with KRAS. Among them, tipifarnib is an FTase inhibitor showed good efficacy in patients with HRAS mutations (46). The molecular formula of tipifarnib is C27H22Cl2N4O and the molecular weight is 489.40. The structural formula of tipifarnib is shown in Figure 3B.
It is worth mentioning that in the conventional synthesis process of tipifarnib and active R-configuration drugs involves obtaining active pharmaceutical ingredients through chiral resolution, which results in a large amount of wastage of intermediates. The intermediates can be racemized by using appropriate acidation during the synthetic process, thus improving the synthesis efficiency of the drug. Tipifarnib was first developed by Johnson & Johnson and submitted to the FDA for new drug application in 2004. In April 2021, tipifarnib was approved by the FDA for the treatment of head and neck squamous cell carcinoma patients who failed to respond to platinum based chemotherapy and had HRAS mutation ≥20% (47). Also, Kobe0065, is also a small molecule drug targeting HRAS, which has not yet been used for treating MTC patients in the clinic. This drug holds a great promise for clinical use in MTC patients. It is known to inhibit the interaction between HRAS and cRaf1 (48), and effectively prevents the binding of HRAS-GTP to cRaf1-RBD. The molecular formula of Kobe0065 is C15H11ClF3N5O4S, and the molecular weight is 449.79. The structural formula of Kobe0065 is shown in Figure 3D.
All targeted drugs face the problem of drug resistance, and drugs targeting RAS are no exception. Degradation of chimeras with targeted proteins is a solution to overcome drug resistance, for example, PROTAC LC-2 (structural formula shown in Figure 3E), was developed by CREW et al., which was the first drug that degraded endogenous KRASG12C mutants (49). The precursor of LC-2 was LC-1, which contained the adagrasib molecule, the E3 ubiquitin ligand and the ligand for both of them. Due to the presence of hydrolyzable amides in LC-1, KRAS could not be degraded effectively. Thus, LC-2 was optimized to solve this problem and its ability to degrade KRAS was greatly enhanced. Firstly, the adagrasib fragment in LC-2 covalently bound to KRASG12C, then the E3 ubiquitin ligase protein was recruited to degrade the KRASG12C mutant. PROTAC drugs are independent of the active site and do not require continuous binding to the target. Although it has not been applied to MTC patients, PROTACs may be an effective strategy for treating MTC patients and to overcome the problem of RAS inhibitor resistance.
The PI3K/AKT mTOR signaling pathway and designing drugs against this pathway
The role of PI3K/AKT/mTOR signaling pathway in MTC
The success of RET inhibition in MTC therapy suggests that the downstream signaling pathway activated by RET may also be an effective therapeutic target. It was found that the proteins in the PI3K/AKT/mTOR pathway were preferentially activated in hereditary RET positive cases (50). PI3K is activated after its phosphorylation and AKT is activated by phosphorylation on the cell membrane. AKT regulates downstream processes including the inhibition of p27, translocation of FOXO in the cytoplasm, activation of PtdIns-3ps, mediating the effects of p70 or 4EBP1 on transcriptional regulation and activation of the mTOR signaling pathway. The mTOR gene is located on chromosome 1p36.22 and contains 60 exons. mTOR activation is an early event in the transformation of C cells, which plays a role in the early stage of MTC tumorigenesis. mTOR encodes serine/threonine kinase belonging to the phosphatidylinositol kinase-related kinase family, which is involved in the regulation of cell proliferation, apoptosis, cell cycle, angiogenesis, metabolism and protein synthesis.
Design of PI3K/AKT/mTOR inhibitors
PI3K inhibitors can be divided into three categories. The first category consists of broad spectrum PI3K inhibitors, such as buparlisib (BKM120). In a clinical trial on thyroid cancer patients, buparlisib showed good therapeutic effects, but 63% of the patients developed serious adverse reactions (51). The second type is PI3K subtype selective inhibitors, such as PI3Kα selective inhibitor piqray (Alpelisib) and PI3Kδ selective inhibitor ukoniq (Umbralisib). So far, the efficacy of these drugs have not been tested in MTC patients. The third category consists of PI3K and mTOR dual-target inhibitors, such as dactolisib (BEZ235) (52). Dactolisib was reported to significantly inhibit the proliferation of thyroid cancer cells in vitro and in mice (53). However, it has not been studied in MTC patients. The clinical use of broad-spectrum PI3K inhibitors is limited due to their severe adverse effects. In contrast, the PI3K subtype selective inhibitors have greatly reduced the incidence of adverse events and have shown good therapeutic efficacy. Umbralisib (54) is a selective PI3Kδ inhibitor with a molecular formula of C38H32F3N5O6S, and molecular weight of 743.75, and its synthetic route is depicted in Figure 4A. In view of its excellent therapeutic effect in relapsed/refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia, umbralisib may be a potential new therapeutic drug for MTC (55). Table 2 shows the list of PI3K/AKT/mTOR pathway inhibitors that are currently under investigation.
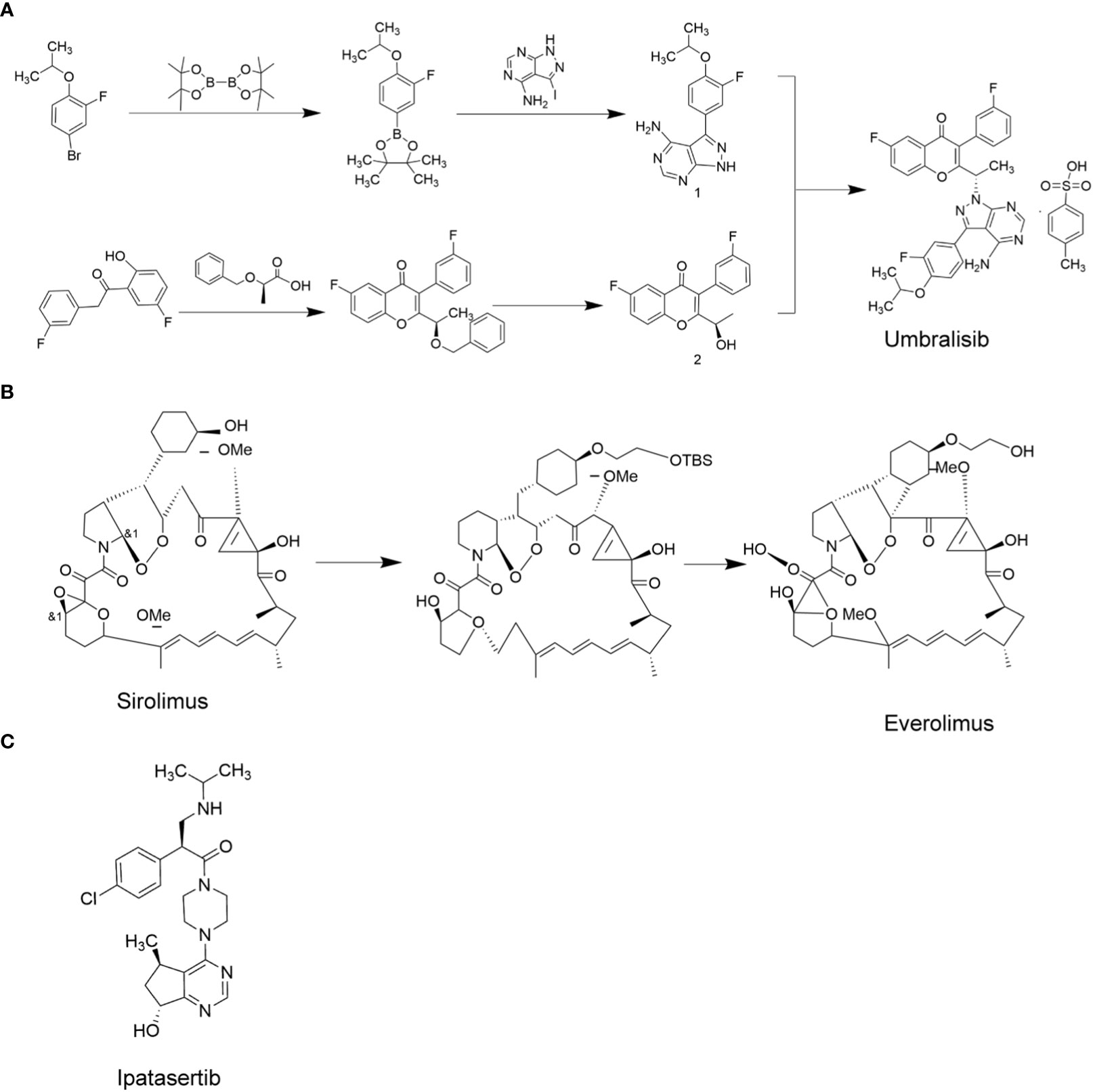
Figure 4 PI3K/AKT/mTOR target-related chemotherapy drugs. (A), Umbralisib; (B), Everolimus; (C), Ipatasertib.
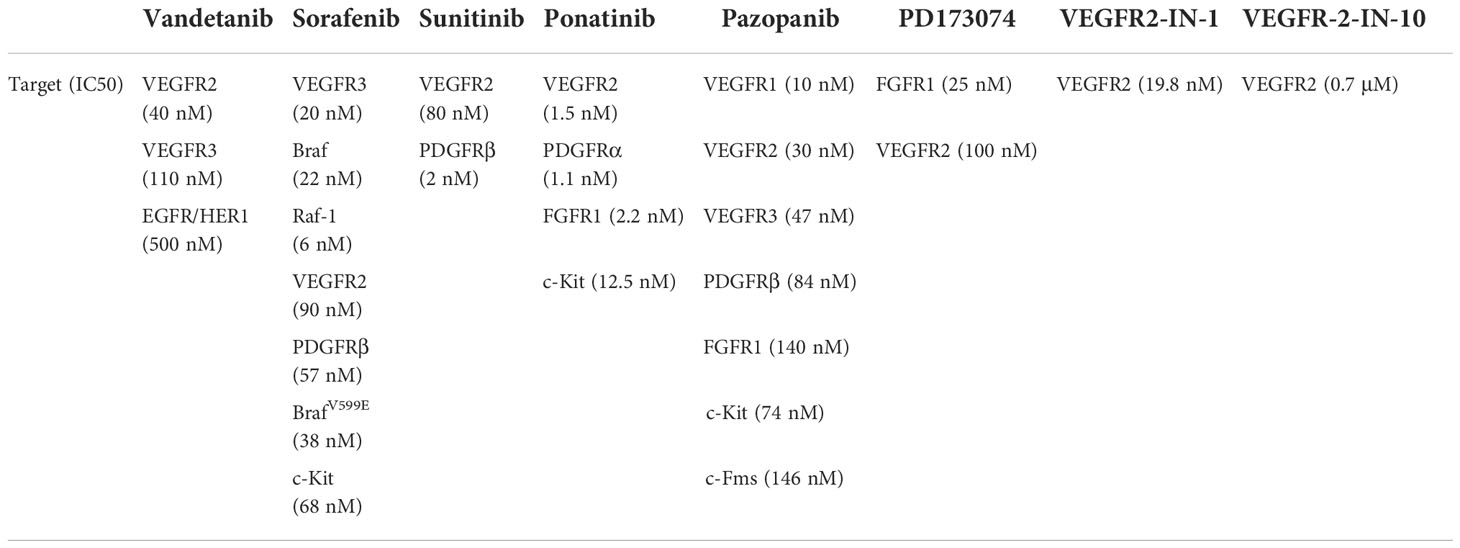
Table 2 Specific targets of Protein Tyrosine Kinase/RTK drugs acting on VEGFR.
AKT is an effector in the PI3K/AKT/mTOR pathway, and is a promising target for cancer treatment. The AKT kinase family includes AKT1, AKT2, and AKT3 (56). AKT inhibitors are classified into three categories based on how they inhibit the activity of AKT. ATP competitive inhibitors are the first category of AKT inhibitors that suppress AKT phosphorylation by competing with ATP. The second category consists of allosteric inhibitors, which inhibit the interaction between AKT and its substrate through different conformations. Lastly, the PH domain inhibitors inhibit AKT by competing with PIP3 for the binding sites on the PH domain, preventing PIP3 induced migration of AKT to the cell membrane (57). ATP competitive inhibitors are currently the focus of research for AKT inhibitors. Some of the related small molecule drugs include ipatasertib, uprosertib, etc. The allosteric inhibitor (MK-2206) has been reported to show stronger AKT inhibition in the laboratory than the ATP competitive inhibitors, but the clinical data for MK-2206 is not yet available. The PH domain inhibitor perifosine, did not show satisfactory results in clinical trials. A phase III randomized, double-blind, placebo-controlled trial of perifosine for rectal cancer showed no overall survival benefit (58). However, in vitro experiments showed that perifosine could effectively inhibit the growth of thyroid cancer cells (59). However, it has not yet been tested in MTC patients. Among all the AKT inhibitors, ipatasertib (structural formula shown in Figure 4C) is the most advanced compound in clinical research. Due to the unique chiral pyrimidine cyclopentanol structure of ipatasertib, its inhibitory effect on AKT was found to be 620 times that of its homologous protein PKA, and its IC50 values against AKT1, AKT2 and AKT3 in in vitro assays were 5, 18 and 8nmol/L, respectively (60). Ipatasertib has not yet been tested in MTC patients, which may be a new direction for treating MTC. Ipatasertib was developed by Array BioPharma, and currently it is promoted by Roche.
The mTOR kinase family includes mTOR1, mTOR2 and mTOR3, of which mTOR1 and mTOR2 are associated with tumorigenesis (61). mTOR1 is a downstream effector of several common signaling pathways, including the PI3K/AKT and MAPK signaling pathways (62). mTOR is overactive in a variety of cancers, making it an ideal target for cancer therapy (63). mTOR inhibitors can be divided into two types. The first category consists of rapamycin and its analogs, and the second category consists of ATP-competitive inhibitors (64). The former inhibits mTORC1 and the latter inhibits mTORC1/2. Rapamycin has not been approved for cancer therapy due to its immunosuppressive effects. However, its analogue, everolimus, was reported to promote relatively low immunosuppressive effects and has been approved by the FDA for treating renal cell carcinoma and pancreatic derived neuroendocrine tumors, advanced hormone receptor-positive HER-2 negative breast cancer, gastrointestinal neuroendocrine tumor, and giant cell astrocytoma (65, 66). In addition, in MTC patients, everolimus was reported to induce significant anti-tumor effects (67). Everolimus is currently the most widely used mTOR inhibitor. The synthesis of everolimus uses sirolimus as raw material, and everolimus precursor is obtained through etherification reaction with the sulfonate side chain. The everolimus precursor is then processed by desilication ether to obtain everolimus. Figure 4B depicts the synthetic process for everolimus.
Moreover, broad-spectrum PI3K and mTOR dual-target inhibitors are also the major focus of cancer research, but currently, drugs belonging to this category have not yet been approved by the FDA for clinical use. For example, it was found that dactolisib (BEZ235) inhibited the growth of thyroid cancer through p53-dependent/independent p21 up-regulation (53). However, the development of PI3K and mTOR dual-target inhibitors is an effective strategy to overcome resistance to PI3K or mTOR single-target inhibitors. On the basis of the existing structures of the dual-target inhibitor compounds, different series of compounds can be designed and synthesized by computer simulation, using skeleton transition strategies. Through the study of structure-activity relationship and the effect of the drugs on in vitro proliferation of cancer cells, dual-target inhibitors with good activity and low toxicity can be selected for broad therapeutic applications.
Angiogenesis and drugs targeting the angiogenesis pathway
The role of angiogenesis in MTC
Angiogenesis and lymphangiogenesis are indispensable for tumor occurrence and metastasis. Angiogenesis and lymphangiogenesis are distinct cellular processes. Angiogenesis refers to the re-growth of capillaries from existing blood vessels, while lymphangiogenesis refers to the development of new lymphatic vessels. The VEGFR2 receptor tyrosine kinase is expressed by vascular endothelial cells and is activated by VEGF-A produced by the immune cells in the tumor and the tumor microenvironment (TME) (68). VEGFR1, -2, -3, VEGF-A and VEGF-C have been reported to be highly expressed in MTC patients. Moreover, the overexpression of VEGFR2 is related to the prognosis of metastatic MTC patients (14). VEGF-A also regulates lymphangiogenesis through the participation of VEGFR3. VEGF-C and VEGF-D, produced by several immune cells in the TME, are known to stimulate lymphangiogenesis and metastasis of tumors through the combination of VEGFR3 and lymphatic endothelial cells. EGFR is a cell surface protein belonging to the ErbB receptor family, and about 30% of epithelial cancers have dysregulated EGFR signaling, amplification or mutation. EGFR is involved in cell growth stimulation and signal transduction, as well as in the activation of RET kinase, which is related to thyroid cancer progression and invasion (69).
Drug design for targeting angiogenesis in MTC
Many kinds of targeted drugs have been developed for MTC in the past 20 years. Tyrosine kinase receptors are involved in cancer cell proliferation, angiogenesis and lymphangiogenesis. Several tyrosine kinase inhibitors (TKIs) have been developed as new drugs for the treatment of MTC, which were successful in suppressing disease progression. At present, FDA has approved vandetanib and cabozatinib for MTC treatment. Vandetanib was synthesized by adding a basic side chain at the C-7 position of quinazoline nucleus of 4- anilino quinazoline compounds (70). The study found that early use of vandetanib could improve the prognosis of MTC patients (71). The chemical structural formula of Cabozatinib is 1-n-[4-(6,7-dimethylquinolin-4-yl) oxyphenyl]-1-n’-(4-fluorophenyl) cyclopropane-1,1-dicarboxyamide, and it has obtained 7579473 patents (WO 2005030140 A2) in the United States (72). In a study on 330 patients with metastatic MTC, it was found that 140 mg/d Cabozantinib could significantly improve the PFS of patients (73). Previously, studies have reported that targeting the C-4’ position of VEGF-R2 and PDGF-Rβ alters the inhibitory properties of drugs against VEGF-R2 and PDGF-Rβ. For example, neutral 5a is an effective inhibitor of VEGF-R2, while acidic 5b is an effective inhibitor of PDGF-Rβ. In order to improve the solubility of indoline -2- one and broaden its kinase inhibition spectrum, researchers introduced various basic side chains at the C-4’ position of 5a. As a kind of indoline -2- one drug, sunitinib is a multi-target tyrosine kinase inhibitor. Phase II clinical trials showed that sunitinib had obvious curative effects in patients with advanced, locally advanced or metastatic medullary thyroid cancer (74). Also, ponatinib was reported to effectively inhibit Abl, PDGFRα, VEGFR2, FGFR1 and Src, thus suppressing metastasis in MTC patients. Ponatinib mainly formed five hydrogen bonds with the target protein, one skeleton with M318 in the hinge region, one skeleton with D381, one side chain with E286, and two from methylpiperazine groups (75, 76).
In recent years, few novel targeted drugs against VEGRF have been reported. In 2021, Cho et al. synthesized VEGFR-2-IN-10 by directly promoting the interaction between the indole core of voacangine (Voa) and the active residues of the kinase domain of VEGFR2 (Leu840, Val848, Ala866 and Leu1035) through hydrophobic interaction. Firstly, VEGFR-2-IN-10 showed no cytotoxicity. Secondly, it had a strong inhibitory effect on VEGF mediated angiogenesis. This was mainly because two oxygen atoms of Voa formed hydrogen bonds with Asn923 and Cys919 in the hydrophobic pockets promoting higher affinity and direct interaction with VEGFR2. In order to replicate the interaction between Voa and VEGFR2, researchers designed indole core (5- methoxy -1H- indole) and various functional groups that were different from cycloheptane (77). In 2022, Nafie and Boraei et al. conducted the reaction between 4- amino -5-(1H- indole -2- yl) -2,4-dihydro-3h-1,2,4-triazole -3- thione 1 and indole -3- formaldehyde 2, pyridine -2- formaldehyde 3, pyridine -3- formaldehyde 4 and pyridine-1, respectively, leading to the synthesis of the drug VEGFR2-IN-1. VEGFR2-IN-1 inhibited the proliferation and migration of cancer cells by activating apoptosis and inhibiting the expression of VEGFR2 (78).
Immunotherapy and targeted drug design
Immune mechanisms associated with MTC
The TME in TC is rich in immune cells, therefore immunotherapy is a potential strategy for treating TC (79). Immunotherapy enhances antitumor immunity in patients, and through its inherent specificity, exerts cytotoxic effects on cancer cells with minimal side effects (80). Immunotherapy mediates its effects mainly by increasing the tumor specific immune response, and by suppressing the immunosuppressive nature of the tumor (Figure 5). For example, the dendritic cell (DC) vaccine with heterologous calcitonin polypeptide (in mice) and allogeneic tumor cell lysate (in humans) has been demonstrated to increase the tumor specific immunity in MTC patients (81). Moreover, some stromal cells in the TME are known to induce immune dysfunction, such as inhibiting DC differentiation and maturation, and further inducing CD4+ T cells to differentiate into immunosuppressive regulatory T cells (Treg). Treg express the cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed death receptor-1 (PD-1), which are known to inhibit antitumor immune response. These inhibitory molecules were identified as “immune checkpoints” (82). CTLA-4, also known as CD152, is expressed by activated CD4 + and CD8+ T cells. CTLA-4 is a member of the immunoglobulin superfamily, which inhibits T cell activation after binding with its ligand B7. Therefore, blocking CTLA-4 stimulates the activation and proliferation of immune cells, inducing or enhancing the antitumor immune response. In addition, T cell immunotherapy using chimeric antigen receptor and macrophage targeting therapy have shown significant therapeutic efficacy in MTC patients (83).
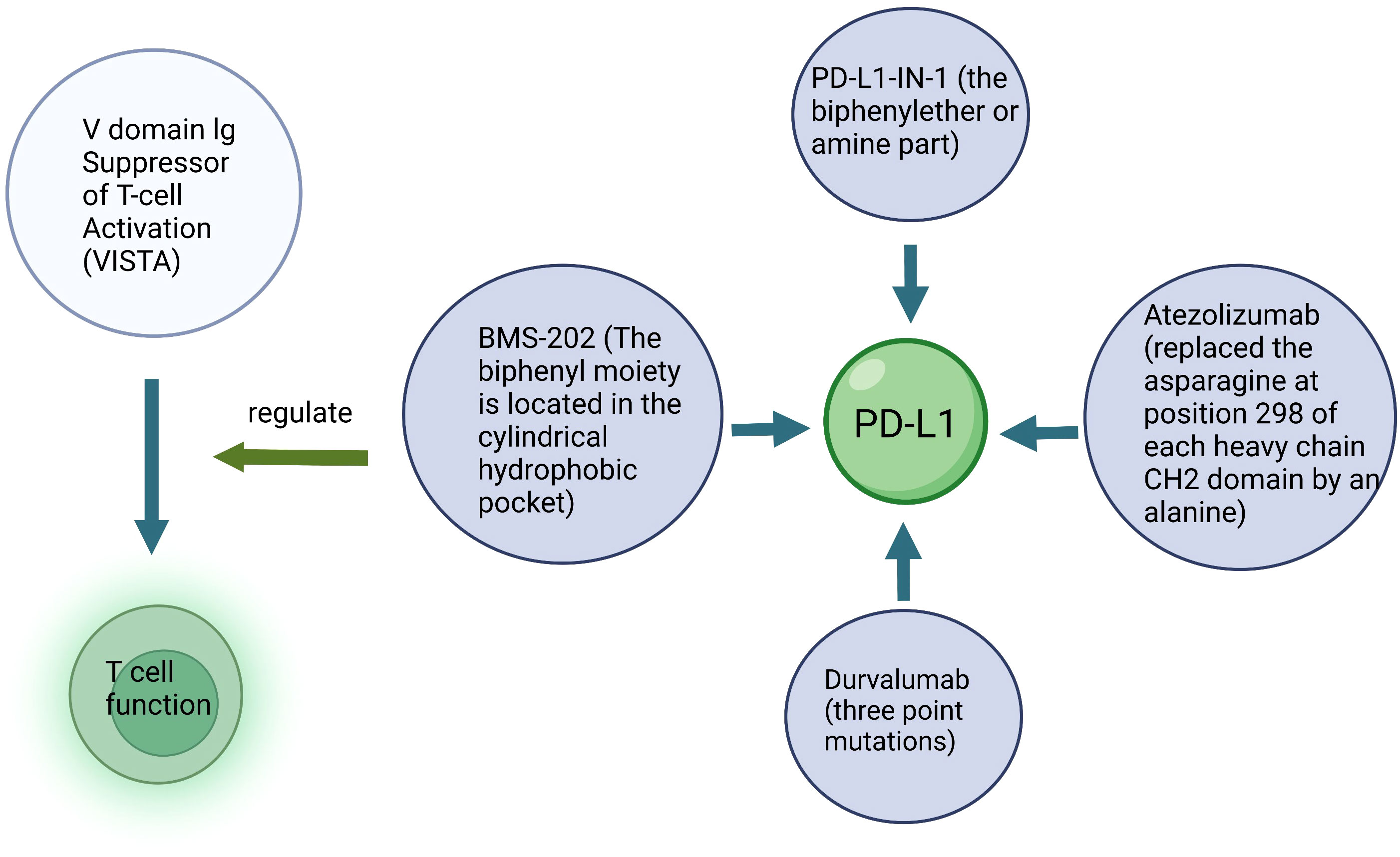
Figure 5 Schematic diagram of immunotherapy. Key structure of a specific drug and the target cells are described.
PD-1, a member of the CD28 superfamily, is expressed by activated T cells, B cells and myeloid cells. It has two ligands, namely, PD-L1 and PD-L2. The combination of PD-1 and PD-L1 mediates the inhibitory signal of T cell activation, which inhibits the activation and proliferation of T cells and plays a negative regulatory role similar to that of CTLA-4. PD-L1 expression has also been reported as one of the important factors associated with T cell failure during chronic viral infection and cancer. Therefore, blocking PD-1/PD-L1 helps to rebuild T cell function, which is of great significance for tumor growth inhibition. Anti-PD-1/PD-L1 inhibitors have been reported to enhance the immune response in certain patients with malignant tumors such as melanoma, lung cancer and renal cell carcinoma.
In most cases, tumors evade the surveillance of immune system through many mechanisms, including the down-regulation of MHC molecules, suppressing immune response and inducing tolerance to tumor antigens (84). However, little is known about the immune escape mechanism of MTC (85). Mitsiades et al. reported a potential role of FasL in immune evasion and the progression of thyroid cancer (86). FasL is a transmembrane protein belonging to the tumor necrosis factor (TNF) family, which induces apoptosis by binding and activating the Fas (APO-1/CD95) receptor. Fas/FasL system plays an important role in immune homeostasis and participates in T cell-mediated cytotoxicity. FasL has been reported to be expressed in five thyroid cancer cell lines and induces apoptosis of lymphocytes expressing FasL. Although its expression is low or absent in MTC, the above studies indicate a potential role of FasL in the progression and immune evasion of thyroid cancer.
Designing drugs for cancer immunotherapy
Because little is known about the immune escape process of MTC, the current focus of drug development is to identify the molecular pathways leading to the formation of new antigens, so as to initiate the immune response. Such new antigens may serve as effective therapeutic targets. Currently, most of the drugs target PD-L1. In order to reduce the antibody-dependent cytotoxicity (ADCC) of traditional anti-PD-L1 agents, researchers designed three classic immunotherapeutic agents for MTC, namely durvalumab, atezolizumab and nivolumab (87). Durvalumab is a selective, high-affinity human IgG1 mAb designed to prevent ADCC (88). Stewart et al. altered the constant domain of durvalumab by three point mutations, and reduced the likelihood of triggering effector functions that could lead to non-specific cytotoxic effects on healthy cells expressing PD-L1 (89). Atezolizumab is a fully humanized IgG1mAb designed to interfere with the binding of PD-L1 ligands to their two receptors, PD-1 and B7.1. Deng et al. replaced the asparagine at position 298 of each heavy chain CH2 domain by an alanine (90). This FcgR-binding-deficient structure meant that atezolizumab could not bind to the Fc receptors on phagocytes, thereby suppressing ADCC. Wang et al. found the desired drug from a transfected CHO cell line by sequencing the variable region and grafting it onto human kappa and IgG4 constant region sequences containing the S228P mutation (91).
Small molecule inhibitors targeting PD-L1 are still being explored (Figure 6). PD-1/PD-L1-IN-23 (BMS-202) is an ester prodrug of L7. The biphenyl moiety of BMS-202 is located in the cylindrical hydrophobic pocket of the PD-L1 dimer protein to block the PD-1/PD-L1 interaction. In addition, combination of the benzoxadiazole compound L1 and BMS-1016 had a good arrangement pattern. The slip docking fractions of BMS-1016 and L1 were 13.085 and 13.195 kcal/mol, respectively, which indicated that L1 could be a new lead molecule for future PD-L1 inhibitor design (92). The combination of PD-L1-IN-1 and PD-L1 proteins enhances the antitumor immune activity of cells. PD-L1-IN-1 was shown to significantly increase the release of interferon-γ and induce apoptosis in cancer cells, and induce low cytotoxicity towards healthy cells. Researchers speculated that the biphenyl ether or amine part of the compound was the driving group that bound to the surface of PD-L1. As a result, they retained this key model, replaced the polar chain with a small number of polar groups, designed some triazines7-21, and obtained patent approval (BMS-202) (93). Another study showed that PD-1/PD-L1-IN-14 (compound 17) inhibited PD-1/PD-L1 interaction, and promoted the dimerization and degradation of PD-L1 (94). In addition, PD-1/PD-L1-IN-27 had the ability to activate CD8+T cells and reduce T cell depletion. Targeting PD-1/PD-L1-IN-27 has been proposed as a new strategy to develop PD-L1 inhibitors (95).
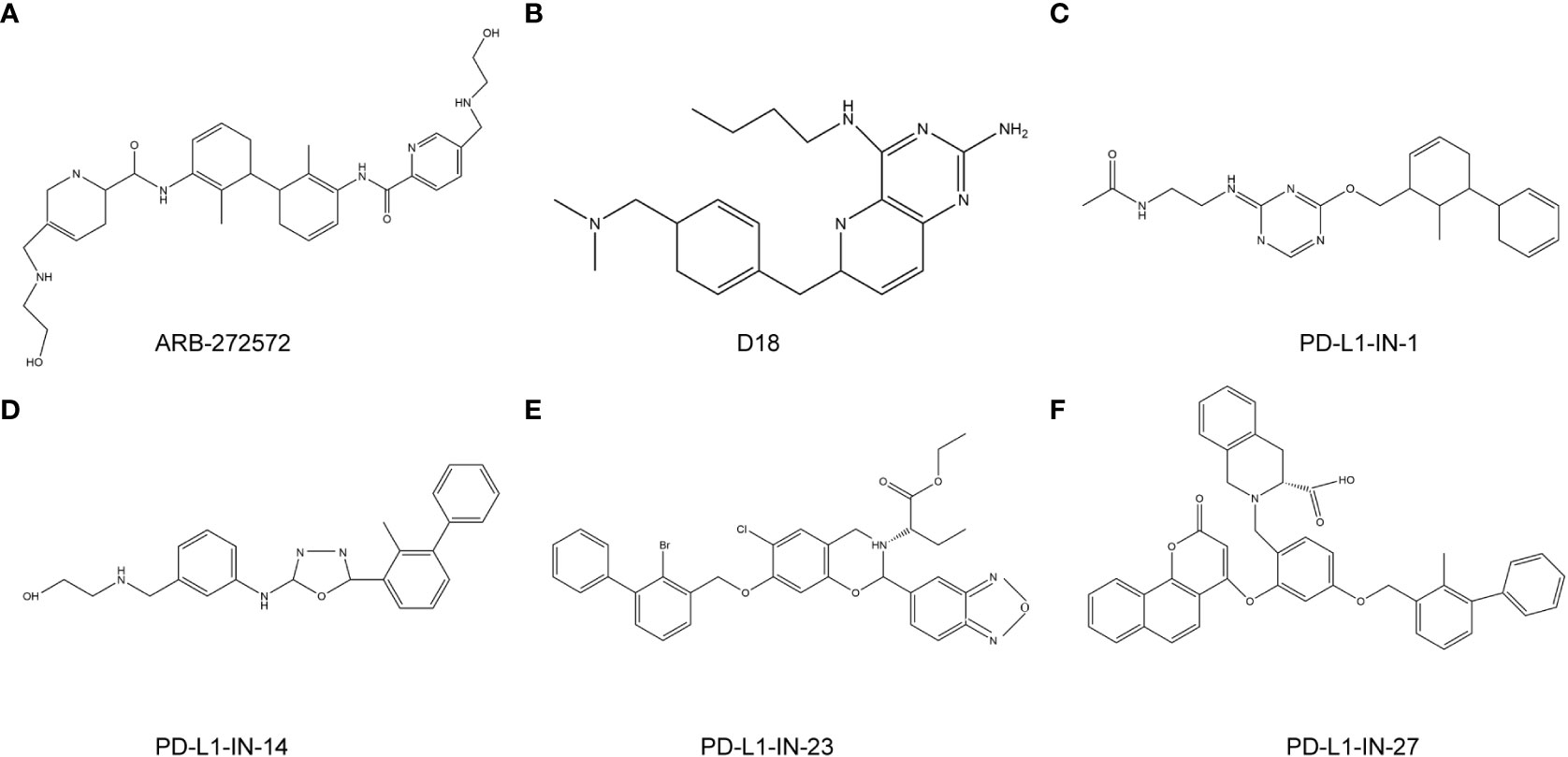
Figure 6 anti-PD-L1 targeted chemotherapy drugs. (A), ARB-272572; (B), D18; (C), PD-L1-IN-1; (D), PD-L1-IN-14; (E), PD-L1-IN-23; (F), PD-L1-IN-27.
Drugs targeting other immune molecules are currently being investigated for treating MTC. V-domain Ig Suppressor of T-cell Activation (VISTA) membrane receptors are expressed not only by myeloid cells, but also by T cells. VISTA is expressed in tumor cells and regulates T cell function, thus promoting antitumor effects. In 2019, Marhelava et al. developed Onvatilimab for targeting the membrane receptor VISTA (96).
Conclusion
Although the 10-year survival rate of early stage MTC patients has reached more than 70%, the survival rate of late stage patients has reduced to less than 40% (4, 97, 98). Although several targeted drugs have been developed for the clinical treatment of MTC patients, their efficacy is still unsatisfactory. Therefore, it is urgent to further optimize the drug development strategy. Gene targeted therapy is the most effective method to treat tumors at present. For example, RET, RAS, PI3Kk/AKT/mTOR, VEGF and PD-L1 play major roles in promoting MTC tumorigenesis (Figure 7). Development of targeted drugs has emerged as a hot spot in recent years. Increasing number of studies have identified the fusion or mutation sites of RET and RAS as potential targets for drugs. However, targeted inhibitors for these sites are yet to be developed. In the future, in addition to exploring and designing inhibitors of these new fusion and mutation sites, it is also necessary to develop small molecule inhibitors that can target and inhibit other signal transduction pathways, such as RET-PTC1 and RAS-PI3K. Moreover, the PI3K/AKT/mTOR pathway is a classic signaling pathway that promotes tumorigenesis. The mTOR inhibitor everolimus, has been approved for clinical use for more than a decade, but drug resistance has always been a problem, and dual-target inhibitor has emerged as a potential solution. Moreover, classical TKIs inhibit cancer metastasis by introducing basic side chains at different positions of the TK and by binding to the hydrogen bonds. Moreover, VOA was shown to have an inhibitory effect on cancer cells through the formation of hydrogen bonds between the oxygen atom in the indole core of VOA and asn923 and cys919. The idea of directly combining the active residues of VEGFR2 kinase domain to develop new drugs may also be explored in the future. In addition, the design and development of traditional immunotherapeutic drugs follow the concept of suppressing ADCC. Immunotherapy agents have shown remarkable efficacy both in animal studies and in the clinic. It is a promising strategy to synthesize new immune drugs that enhance the antitumor activity of MTC by adjusting the sliding docking fraction of PD-L1 and diphenyl ether or amine. All the above findings have provided new direction for future drug discovery for treating MTC.
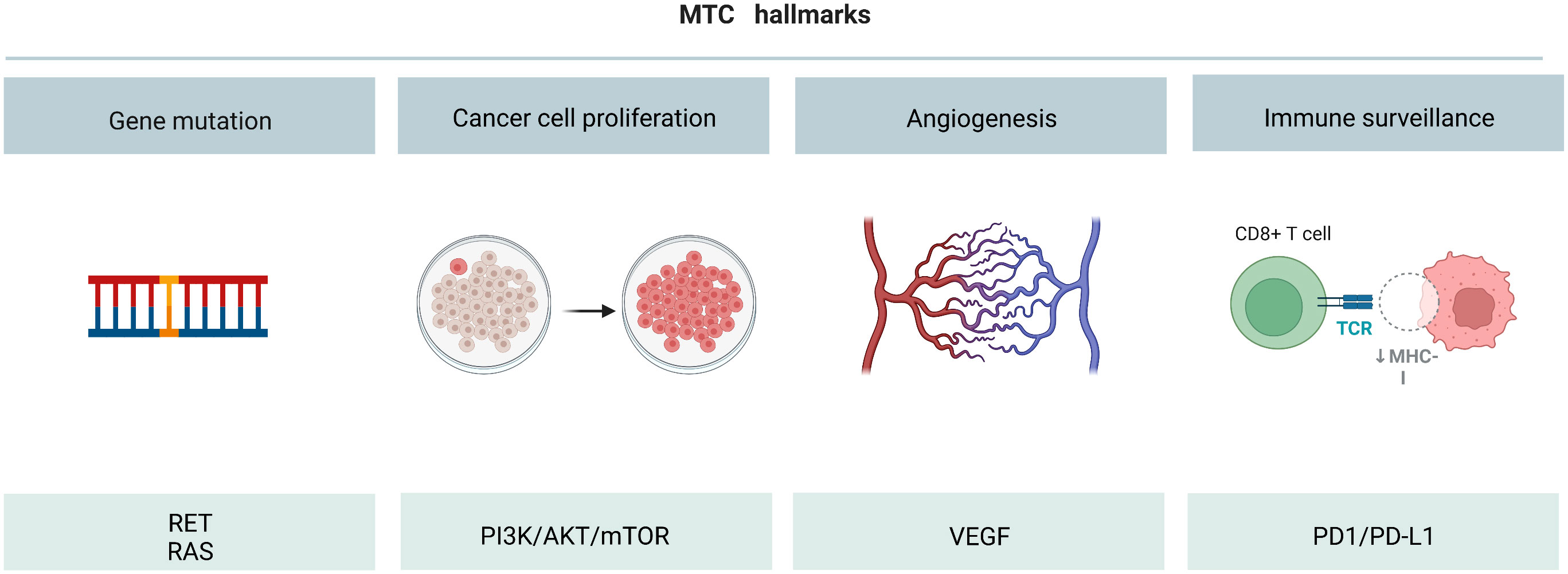
Figure 7 Overall diagram of MTC initiation, cell survival increase, angiogenesis and immune surveillance.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Author contributions
Conceptualization, YL and JQ. Methodology, HH. Software, XW. Validation, SZ, YL and JQ. Formal analysis, ZL. Investigation, KQ. Resources, SZ. Data curation, SZ. Writing—original draft preparation, YL. Writing—review and editing, JQ. Visualization, YL. Supervision, YL. Project administration, YL. Funding acquisition, YL. All authors contributed to the article and approved the submitted version.
Funding
This research is funded by the Henan Science and Technology Key Project (222102310612).
Acknowledgments
The authors would like to thank all the reviewers who participated in the review, as well as MJEditor (www.mjeditor.com) for providing English editing services during the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hu J, Yuan IJ, Mirshahidi S, Simental A, Lee SC, Yuan X. Thyroid carcinoma: Phenotypic features, underlying biology and potential relevance for targeting therapy. Int J Mol Sci (2021) 22(4):1950. doi: 10.3390/ijms22041950
2. Wong A, Nabata K, Wiseman S. Medullary thyroid carcinoma: a narrative historical review. Expert Rev Anticancer Ther (2022) 22(8):823–34. doi: 10.1080/14737140.2022.2089118
3. Angelousi A, Hayes AR, Chatzellis E, Kaltsas GA, Grossman AB. Metastatic medullary thyroid carcinoma: a new way forward. Endocrine-related Cancer (2022) 29:R85–r103. doi: 10.1530/ERC-21-0368
4. Kuo EJ, Sho S, Li N, Zanocco KA, Yeh MW, Livhits MJ. Risk factors associated with reoperation and disease-specific mortality in patients with medullary thyroid carcinoma. JAMA Surg (2018) 153:52–9. doi: 10.1001/jamasurg.2017.3555
5. Walgama E, Busaidy N, Zafereo M. Novel therapeutics and treatment strategies for medullary thyroid cancer. Endocrinol Metab Clinics North America (2022) 51:379–89. doi: 10.1016/j.ecl.2022.02.001
6. Refardt J, Hofland J, Wild D, Christ E. Molecular imaging of neuroendocrine neoplasms. J Clin Endocrinol Metab (2022) 107:e2662–70. doi: 10.1210/clinem/dgac207
7. Klein M, Catargi B. VEGF in physiological process and thyroid disease. Annales d'endocrinol (2007) 68:438–48. doi: 10.1016/j.ando.2007.09.004
8. Zhang S, Chen S, Wang Y, Zhan Y, Li J, Nong X, et al. Association of a novel prognosis model with tumor mutation burden and tumor-infiltrating immune cells in thyroid carcinoma. Front Genet (2021) 12:744304. doi: 10.3389/fgene.2021.744304
9. Gemignani F, Romei C, Ciampi R, Corrado A, Melaiu O, Figlioli G, et al. Polymorphisms within the RET proto-oncogene and risk of sporadic medullary thyroid carcinoma. Thyroid Off J Am Thyroid Assoc (2020) 30:1579–88. doi: 10.1089/thy.2019.0352
10. Roskoski R Jr., Sadeghi-Nejad A. Role of RET protein-tyrosine kinase inhibitors in the treatment RET-driven thyroid and lung cancers. Pharmacol Res (2018) 128:1–17. doi: 10.1016/j.phrs.2017.12.021
11. Rosen EY, Won HH, Zheng Y, Cocco E, Selcuklu D, Gong Y, et al. The evolution of RET inhibitor resistance in RET-driven lung and thyroid cancers. Nat Commun (2022) 13:1450. doi: 10.1038/s41467-022-28848-x
12. Rodríguez-Antona C, Muñoz-Repeto I, Inglada-Pérez L, de Cubas AA, Mancikova V, Cañamero M, et al. Influence of RET mutations on the expression of tyrosine kinases in medullary thyroid carcinoma. Endocrine-related Cancer (2013) 20:611–9. doi: 10.1530/ERC-12-0316
13. Naoum GE, Morkos M, Kim B, Arafat W. Novel targeted therapies and immunotherapy for advanced thyroid cancers. Mol Cancer (2018) 17:51. doi: 10.1186/s12943-018-0786-0
14. Fallahi P, Ferrari SM, Galdiero MR, Varricchi G, Elia G, Ragusa F, et al. Molecular targets of tyrosine kinase inhibitors in thyroid cancer. Semin Cancer Biol (2022) 79:180–96. doi: 10.1016/j.semcancer.2020.11.013
15. Ma W, Zhu M, Wang B, Gong Z, Du X, Yang T, et al. Vandetanib drives growth arrest and promotes sensitivity to imatinib in chronic myeloid leukemia by targeting ephrin type-b receptor 4. Mol Oncol (2022) 16(14):2747–65. doi: 10.21203/rs.3.rs-633064/v1
16. Guan S, Chen X, Chen Y, Xie W, Liang H, Zhu X, et al. FOXM1 variant contributes to gefitinib resistance via activating wnt/β-catenin signal pathway in non-small cell lung cancer patients. Clin Cancer Res an Off J Am Assoc Cancer Res (2022) 28(17):3770–84. doi: 10.1158/1078-0432.CCR-22-0791
17. Xu GQ, Gong XQ, Zhu YY, Yao XJ, Peng LZ, Sun G, et al. Novel 1,2,3-triazole erlotinib derivatives as potent IDO1 inhibitors: Design, drug-target interactions prediction, synthesis, biological evaluation, molecular docking and ADME properties studies. Front Pharmacol (2022) 13:854965. doi: 10.3389/fphar.2022.854965
18. Lee SY, Doh I, Lee DW. A high throughput apoptosis assay using 3D cultured cells. Mol (Basel Switzerland) (2019) 24(18):3362. doi: 10.3390/molecules24183362
19. Boccaccino A, Borelli B, Intini R, Antista M, Bensi M, Rossini D, et al. Encorafenib plus cetuximab with or without binimetinib in patients with BRAF V600E-mutated metastatic colorectal cancer: real-life data from an Italian multicenter experience. ESMO Open (2022) 7:100506. doi: 10.1016/j.esmoop.2022.100506
20. Prete A, Borges de Souza P, Censi S, Muzza M, Nucci N, Sponziello M. Update on fundamental mechanisms of thyroid cancer. Front Endocrinol (2020) 11:102. doi: 10.3389/fendo.2020.00102
21. Ma X, Ma X, Chin L, Zhu Z, Han H. A novel germline deletion of p.C630 in RET causes MTC and promotes cell proliferation and sensitivity to pralsetinib. J Clin Endocrinol Metab (2022) 107(9):2636–43. doi: 10.1210/clinem/dgac352
22. Román-Gil MS, Pozas J, Rosero-Rodríguez D, Chamorro-Pérez J, Ruiz-Granados Á., Caracuel IR, et al. Resistance to RET targeted therapy in thyroid cancer: Molecular basis and overcoming strategies, Cancer Treat Rev (2022) 105:102372.
23. Zhao L, Yang KQ, Fan P, Gong DX, Zhang L, Lu YT, et al. RET c.1901G<A and novel SLC12A3 mutations in familial pheochromocytomas. Genes 13 (2022) 13(5):864. doi: 10.3390/genes13050864
24. Bagheri-Yarmand R, Dadu R, Ye L, Jebaraj YS, Martinez JA, Ma J, et al. ONC201 shows potent anticancer activity against medullary thyroid cancer via transcriptional inhibition of RET, VEGFR2, and IGFBP2. Mol Cancer Ther (2021) 20:665–75. doi: 10.1158/1535-7163.MCT-20-0386
25. Parate S, Kumar V, Chan Hong J, Lee KW. Investigating natural compounds against oncogenic RET tyrosine kinase using pharmacoinformatic approaches for cancer therapeutics. RSC Adv (2021) 12:1194–207. doi: 10.1039/D1RA07328A
26. Hu H, Laufkötter O, Miljković F, Bajorath J. Systematic comparison of competitive and allosteric kinase inhibitors reveals common structural characteristics. Eur J medicinal Chem (2021) 214:113206. doi: 10.1016/j.ejmech.2021.113206
27. Agius MP, Ko K, Johnson TK, Phadke S, Soellner MB. Conformation-tunable ATP-competitive kinase inhibitors. Chem Commun (Cambridge England) (2022) 58:3541–4. doi: 10.1039/D1CC06893H
28. Gruber JJ, Colevas AD. Differentiated thyroid cancer: focus on emerging treatments for radioactive iodine-refractory patients. oncologist (2015) 20:113–26. doi: 10.1634/theoncologist.2014-0313
29. Schlumberger M, Elisei R, Müller S, Schöffski P, Brose M, Shah M, et al. Overall survival analysis of EXAM, a phase III trial of cabozantinib in patients with radiographically progressive medullary thyroid carcinoma. Ann Oncol Off J Eur Soc Med Oncol (2017) 28:2813–9. doi: 10.1093/annonc/mdx479
30. Carlomagno F, Guida T, Anaganti S, Vecchio G, Fusco A, Ryan AJ, et al. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene (2004) 23:6056–63. doi: 10.1038/sj.onc.1207810
31. Wells SA Jr., Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol Off J Am Soc Clin Oncol (2012) 30:134–41. doi: 10.1200/JCO.2011.35.5040
32. Subbiah V, Shen T, Terzyan SS, Liu X, Hu X, Patel KP, et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann Oncol Off J Eur Soc Med Oncol (2021) 32:261–8. doi: 10.1016/j.annonc.2020.10.599
33. Repetto M, Crimini E, Ascione L, Boscolo Bielo L, Belli C, Curigliano G. The return of RET GateKeeper mutations? an in-silico exploratory analysis of potential resistance mechanisms to novel RET macrocyclic inhibitor TPX-0046. Investigat New Drugs (2022) 40:1133–6. doi: 10.1007/s10637-022-01259-x
34. Ryan MB, Coker O, Sorokin A, Fella K, Barnes H, Wong E, et al. KRAS(G12C)-independent feedback activation of wild-type RAS constrains KRAS(G12C) inhibitor efficacy. Cell Rep (2022) 39:110993. doi: 10.1016/j.celrep.2022.110993
35. Ciampi R, Romei C, Pieruzzi L, Tacito A, Molinaro E, Agate L, et al. Classical point mutations of RET, BRAF and RAS oncogenes are not shared in papillary and medullary thyroid cancer occurring simultaneously in the same gland. J endocrinol Invest (2017) 40:55–62. doi: 10.1007/s40618-016-0526-5
36. Cordero-Barreal A, Caleiras E, López de Maturana E, Monteagudo M, Martínez-Montes Á M, Letón R, et al. CD133 expression in medullary thyroid cancer cells identifies patients with poor prognosis. J Clin Endocrinol Metab (2020) 105(11):dgaa527. doi: 10.1210/clinem/dgaa527
37. Yao Z, Su WJ, Yaeger R, Son J, Adachi Y, Han SW, et al. ARAF activates RAS through competition with the RASGAP NF1. Cancer Discovery (2022), 12(8):Of1. doi: 10.1158/2159-8290.CD-RW2022-106
38. Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci United States America (2019) 116:15823–9. doi: 10.1073/pnas.1904529116
39. McCarthy MJ, Pagba CV, Prakash P, Naji AK, van der Hoeven D, Liang H, et al. Discovery of high-affinity noncovalent allosteric KRAS inhibitors that disrupt effector binding. ACS omega (2019) 4:2921–30. doi: 10.1021/acsomega.8b03308
40. Herbst RS, Schlessinger J. Small molecule combats cancer-causing KRAS protein at last. Nature (2019) 575:294–5. doi: 10.1038/d41586-019-03242-8
41. Lu J, Harrison RA, Li L, Zeng M, Gondi S, Scott D, et al. KRAS G12C drug development: Discrimination between switch II pocket configurations using Hydrogen/Deuterium-exchange mass spectrometry. Structure (London Engl 1993) (2017) 25:1442–1448.e3. doi: 10.1016/j.str.2017.07.003
42. Sidaway P. Sotorasib effective in KRAS-mutant NSCLC. Nat Rev Clin Oncol (2021) 18:470. doi: 10.1038/s41571-021-00533-w
43. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature (2013) 503:548–51. doi: 10.1038/nature12796
44. Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. Discovery of a covalent inhibitor of KRAS(G12C) (AMG 510) for the treatment of solid tumors. J medicinal Chem (2020) 63:52–65. doi: 10.1021/acs.jmedchem.9b01180
45. Sabari JK, Park H, Tolcher AW, Ou S, Heist RS. KRYSTAL-2: A phase I/II trial of adagrasib (MRTX849) in combination with TNO155 in patients with advanced solid tumors with KRAS G12C mutation. journal of clinical oncology (2021) 39:TPS146–6. doi: 10.1200/JCO.2021.39.3_suppl.TPS146
46. Burrows F, Malik S, Wang Z, Chan S, Gutkind S. Antitumor activity of tipifarnib and PI3K pathway inhibitors in HRAS-associated HNSCC. journal of clinical oncology (2020) 38:e15658–8. doi: 10.1200/JCO.2020.38.15_suppl.e15658
47. Ho AL, Brana I, Haddad R, Bauman J, Bible K, Oosting S, et al. Tipifarnib in head and neck squamous cell carcinoma with HRAS mutations. J Clin Oncol Off J Am Soc Clin Oncol (2021) 39:1856–64. doi: 10.1200/JCO.20.02903
48. Yoshikawa Y, Takano O, Kato I, Takahashi Y, Shima F, Kataoka T. Ras inhibitors display an anti-metastatic effect by downregulation of lysyl oxidase through inhibition of the ras-PI3K-Akt-HIF-1α pathway. Cancer Lett (2017) 410:82–91. doi: 10.1016/j.canlet.2017.09.017
49. Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM. Targeted degradation of oncogenic KRAS(G12C) by VHL-recruiting PROTACs. ACS Cent Sci (2020) 6:1367–75. doi: 10.1021/acscentsci.0c00411
50. Manfredi GI, Dicitore A, Gaudenzi G, Caraglia M, Persani L, Vitale G. PI3K/Akt/mTOR signaling in medullary thyroid cancer: a promising molecular target for cancer therapy. Endocrine (2015) 48:363–70. doi: 10.1007/s12020-014-0380-1
51. Borson-Chazot F, Dantony E, Illouz F, Lopez J, Niccoli P, Wassermann J, et al. Effect of buparlisib, a pan-class I PI3K inhibitor, in refractory follicular and poorly differentiated thyroid cancer. Thyroid Off J Am Thyroid Assoc (2018) 28:1174–9. doi: 10.1089/thy.2017.0663
52. Simon JA. Using isoform-specific inhibitors to target lipid kinases. Cell (2006) 125:647–9. doi: 10.1016/j.cell.2006.05.008
53. Ruan B, Liu W, Chen P, Cui R, Li Y, Ji M, et al. NVP-BEZ235 inhibits thyroid cancer growth by p53- dependent/independent p21 upregulation. Int J Biol Sci (2020) 16:682–93. doi: 10.7150/ijbs.37592
54. Dhillon S, Keam SJ. Umbralisib: First approval. Drugs (2021) 81:857–66. doi: 10.1007/s40265-021-01504-2
55. Lunning M, Vose J, Nastoupil L, Fowler N, Burger JA, Wierda WG, et al. Ublituximab and umbralisib in relapsed/refractory b-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood (2019) 134:1811–20. doi: 10.1182/blood.2019002118
56. Manning BD, Toker A. AKT/PKB signaling: Navigating the network. Cell (2017) 169:381–405. doi: 10.1016/j.cell.2017.04.001
57. Gills JJ, Dennis PA. The development of phosphatidylinositol ether lipid analogues as inhibitors of the serine/threonine kinase, akt. Expert Opin investigat Drugs (2004) 13:787–97. doi: 10.1517/13543784.13.7.787
58. Richardson PG, Eng C, Kolesar J, Hideshima T, Anderson KC. Perifosine , an oral, anti-cancer agent and inhibitor of the akt pathway: mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert Opin Drug Metab Toxicol (2012) 8:623–33. doi: 10.1517/17425255.2012.681376
59. Liu R, Liu D, Xing M. The akt inhibitor MK2206 synergizes, but perifosine antagonizes, the BRAF(V600E) inhibitor PLX4032 and the MEK1/2 inhibitor AZD6244 in the inhibition of thyroid cancer cells. J Clin Endocrinol Metab (2012) 97:E173–82. doi: 10.1210/jc.2011-1054
60. Blake JF, Xu R, Bencsik JR, Xiao D, Kallan NC, Schlachter S, et al. Discovery and preclinical pharmacology of a selective ATP-competitive akt inhibitor (GDC-0068) for the treatment of human tumors. J medicinal Chem (2012) 55:8110–27. doi: 10.1021/jm301024w
61. Meyer LA, Slomovitz BM, Djordjevic B, Westin SN, Iglesias DA, Munsell MF, et al. The search continues: looking for predictive biomarkers for response to mammalian target of rapamycin inhibition in endometrial cancer. Int J gynecol Cancer Off J Int Gynecol Cancer Soc (2014) 24:713–7. doi: 10.1097/IGC.0000000000000118
62. Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Sci (New York N.Y.) (2008) 320:1496–501. doi: 10.1126/science.1157535
63. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. nature reviews. Mol Cell Biol (2011) 12:21–35. doi: 10.1038/nrm3025
64. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem (2009) 284:8023–32. doi: 10.1074/jbc.M900301200
65. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. New Engl J Med (2011) 364:514–23. doi: 10.1056/NEJMoa1009290
66. Dorris 3JR, Jones S. Everolimus in breast cancer: The role of the pharmacist. Ann pharmacother (2014) 48:1194–201. doi: 10.1177/1060028014542415
67. Hanna GJ, Busaidy NL, Chau NG, Wirth LJ, Barletta JA, Calles A, et al. Genomic correlates of response to everolimus in aggressive radioiodine-refractory thyroid cancer: A phase II study. Clin Cancer Res an Off J Am Assoc Cancer Res (2018) 24:1546–53. doi: 10.1158/1078-0432.CCR-17-2297
68. Song YS, Kim MJ, Sun HJ, Kim HH, Shin HS, Kim YA, et al. Aberrant thyroid-stimulating hormone receptor signaling increases VEGF-a and CXCL8 secretion of thyroid cancer cells, contributing to angiogenesis and tumor growth. Clin Cancer Res an Off J Am Assoc Cancer Res (2019) 25:414–25. doi: 10.1158/1078-0432.CCR-18-0663
69. Liang J, Jin Z, Kuang J, Feng H, Zhao Q, Yang Z, et al. The role of anlotinib-mediated EGFR blockade in a positive feedback loop of CXCL11-EGF-EGFR signalling in anaplastic thyroid cancer angiogenesis. Br J Cancer (2021) 125:390–401. doi: 10.1038/s41416-021-01340-x
70. Hennequin LF, Stokes ES, Thomas AP, Johnstone C, Plé PA, Ogilvie DJ, et al. Novel 4-anilinoquinazolines with c-7 basic side chains: design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors. J medicinal Chem (2002) 45:1300–12. doi: 10.1021/jm011022e
71. Valerio L, Bottici V, Matrone A, Piaggi P, Viola D, Cappagli V, et al. Medullary thyroid cancer treated with vandetanib: predictors of a longer and durable response. Endocrine-related Cancer (2020) 27:97–110. doi: 10.1530/ERC-19-0259
72. Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther (2011) 10:2298–308. doi: 10.1158/1535-7163.MCT-11-0264
73. Elisei R, Schlumberger MJ, Müller SP, Schöffski P, Brose MS, Shah MH, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol Off J Am Soc Clin Oncol (2013) 31:3639–46. doi: 10.1200/JCO.2012.48.4659
74. Sun L, Liang C, Shirazian S, Zhou Y, Miller T, Cui J, et al. Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J medicinal Chem (2003) 46:1116–9. doi: 10.1021/jm0204183
75. De Falco V, Buonocore P, Muthu M, Torregrossa L, Basolo F, Billaud M, et al. Ponatinib (AP24534) is a novel potent inhibitor of oncogenic RET mutants associated with thyroid cancer. J Clin Endocrinol Metab (2013) 98:E811–9. doi: 10.1210/jc.2012-2672
76. O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell (2009) 16:401–12. doi: 10.1016/j.ccr.2009.09.028
77. Cho SM, Kim Y, Jung Y, Ko M, Marko-Varga G, Kwon HJ. Development of novel VEGFR2 inhibitors originating from natural product analogues with antiangiogenic impact. J medicinal Chem (2021) 64:15858–67. doi: 10.1021/acs.jmedchem.1c01168
78. Nafie MS, Boraei ATA. Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole schiff bases for targeting breast cancer. Bioorganic Chem (2022) 122:105708. doi: 10.1016/j.bioorg.2022.105708
79. Imam S, Dar P, Paparodis R, Almotah K, Al-Khudhair A, Hasan SA, et al. Nature of coexisting thyroid autoimmune disease determines success or failure of tumor immunity in thyroid cancer. J immunother Cancer (2019) 7:3. doi: 10.1186/s40425-018-0483-y
80. Ferreira MN, Choe JH. Guiding immunotherapy combinations: Who gets what? Adv Drug delivery Rev (2021) 178:113962. doi: 10.1016/j.addr.2021.113962
81. Pointer KB, Pitroda SP, Weichselbaum RR. Radiotherapy and immunotherapy: open questions and future strategies. Trends Cancer (2022) 8:9–20. doi: 10.1016/j.trecan.2021.10.003
82. Vesely MD, Zhang T, Chen L. Resistance mechanisms to anti-PD cancer immunotherapy. Annu Rev Immunol (2022) 40:45–74. doi: 10.1146/annurev-immunol-070621-030155
83. Gupta R, Mehta A, Wajapeyee N. Transcriptional determinants of cancer immunotherapy response and resistance. Trends Cancer (2022) 8:404–15. doi: 10.1016/j.trecan.2022.01.008
84. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of "tumor escape" phenotypes. Nat Immunol (2002) 3:999–1005. doi: 10.1038/ni1102-999
85. Hińcza-Nowak K, Kowalik A, Walczyk A, Pałyga I, Gąsior-Perczak D, Płusa A, et al. Immune profiling of medullary thyroid cancer-an opportunity for immunotherapy. Genes (2021) 12(10):1534. doi: 10.3390/genes12101534
86. Mitsiades N, Poulaki V, Mastorakos G, Tseleni-Balafouta ST, Kotoula V, Koutras DA, et al. Fas ligand expression in thyroid carcinomas: a potential mechanism of immune evasion. J Clin Endocrinol Metab (1999) 84:2924–32. doi: 10.1210/jcem.84.8.5917
87. Rouquette I, Taranchon-Clermont E, Gilhodes J, Bluthgen MV, Perallon R, Chalabreysse L, et al. Immune biomarkers in thymic epithelial tumors: expression patterns, prognostic value and comparison of diagnostic tests for PD-L1. biomark Res (2019) 7:28. doi: 10.1186/s40364-019-0177-8
88. Goldman JW, Piha-Paul SA, Curti B, Pedersen KS, Bauer TM, Groenland SL, et al. Safety and tolerability of MEDI0562, an OX40 agonist monoclonal antibody, in combination with durvalumab or tremelimumab in adult patients with advanced solid tumors. Clin Cancer Res an Off J Am Assoc Cancer Res (2022) 28(17):3709–19. doi: 10.1158/1078-0432.CCR-21-3016
89. Stewart R, Morrow M, Hammond SA, Mulgrew K, Marcus D, Poon E, et al. Identification and characterization of MEDI4736, an antagonistic anti-PD-L1 monoclonal antibody. Cancer Immunol Res (2015) 3:1052–62. doi: 10.1158/2326-6066.CIR-14-0191
90. Deng R, Bumbaca D, Pastuskovas CV, Boswell CA, West D, Cowan KJ, et al. Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti-PD-L1 monoclonal antibody, an immune checkpoint inhibitor. mAbs (2016) 8:593–603. doi: 10.1080/19420862.2015.1136043
91. Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res (2014) 2:846–56. doi: 10.1158/2326-6066.CIR-14-0040
92. Liu L, Yao Z, Wang S, Xie T, Wu G, Zhang H, et al. Syntheses, biological evaluations, and mechanistic studies of Benzo[c][1,2,5]oxadiazole derivatives as potent PD-L1 inhibitors with In vivo antitumor activity. J medicinal Chem (2021) 64:8391–409. doi: 10.1021/acs.jmedchem.1c00392
93. Russomanno P, Assoni G, Amato J, D'Amore VM, Scaglia R, Brancaccio D, et al. Interfering with the tumor-immune interface: Making way for triazine-based small molecules as novel PD-L1 inhibitors. J medicinal Chem (2021) 64:16020–45. doi: 10.1021/acs.jmedchem.1c01409
94. Wang T, Cai S, Cheng Y, Zhang W, Wang M, Sun H, et al. Discovery of small-molecule inhibitors of the PD-1/PD-L1 axis that promote PD-L1 internalization and degradation. J medicinal Chem (2022) 65:3879–93. doi: 10.1021/acs.jmedchem.1c01682
95. Zhang M, Liu J, Wang Y, Wang P, Morris-Natschke S, Lee KH. Molecular hybridization used to design and synthesize neo-tanshinlactone derivatives as PD-1/PD-L1 inhibitors. Bioorganic medicinal Chem (2022) 54:116579. doi: 10.1016/j.bmc.2021.116579
96. Marhelava K, Pilch Z, Bajor M, Graczyk-Jarzynka A, Zagozdzon R. Targeting negative and positive immune checkpoints with monoclonal antibodies in therapy of cancer. Cancers (2019) 11(11):1756. doi: 10.3390/cancers11111756
97. Thiesmeyer JW, Limberg J, Ullmann TM, Stefanova D, Bains S, Beninato T, et al. Impact of multikinase inhibitor approval on survival and physician practice patterns in advanced or metastatic medullary thyroid carcinoma. Surgery (2021) 169:50–7. doi: 10.1016/j.surg.2020.03.021
Keywords: medullary thyroid carcinoma, targeted drug, molecular mechanism, designed principle, progression
Citation: Li Y, Luo Z, Wang X, Zhang S, Hei H and Qin J (2022) Design of new drugs for medullary thyroid carcinoma. Front. Oncol. 12:993725. doi: 10.3389/fonc.2022.993725
Received: 14 July 2022; Accepted: 18 November 2022;
Published: 05 December 2022.
Edited by:
Francesco Pepe, University of Naples Federico II, ItalyReviewed by:
Alessandro Prete, University of Pisa, ItalyGeorge Simeakis, 401 General Military Hospital of Athens, Greece
Copyright © 2022 Li, Luo, Wang, Zhang, Hei and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Songtao Zhang, emhzdDE0M0AxMjYuY29t; Hu Hei, dGlnZXJibGFja0AxMjYuY29t; Jianwu Qin, emx5eXFpbmppYW53dTAzOTdAenp1LmVkdS5jbg==; Xinxing Wang, emx5eXdhbmd4aW54aW5nNDY0OEB6enUuZWR1LmNu