 Jiaqi Shi
Jiaqi Shi Ning Yang
Ning Yang Mingzhi Han
Mingzhi Han Chen Qiu
Chen Qiu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 22 August 2022
Sec. Cancer Metabolism
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.993316
This article is part of the Research Topic Cancer Metabolism: Molecular Insights, Metabolic Crosstalk in the Tumor Microenvironment, and Implications for Therapy View all 24 articles
Glioma is the most common primary malignant tumor in the central nervous system, and directly affects the quality of life and cognitive function of patients. Ferroptosis, is a new form of regulated cell death characterized by iron-dependent lipid peroxidation. Ferroptosis is mainly due to redox imbalance and involves multiple intracellular biology processes, such as iron metabolism, lipid metabolism, and antioxidants synthesis. Induction of ferroptosis could be a new target for glioma treatment, and ferroptosis-related processes are associated with chemoresistance and radioresistance in glioma. In the present review, we provide the characteristics, key regulators and pathways of ferroptosis and the crosstalk between ferroptosis and other programmed cell death in glioma, we also proposed the application and prospect of ferroptosis in the treatment of glioma.
Ferroptosis is an iron-dependent regulated cell death driven by the peroxidation damage of phospholipid-containing polyunsaturated fatty acyl tails (PUFA-PLs) on the cell membrane or organelle membrane and subsequent membrane rupture (1). This non-apoptotic form of cell death triggered by erastin was first named in 2012 (2). Induced ferroptosis has been shown to be efficacious in eliminating drug-resistant tumor cells in various studies (3). Glioma is characterized by rapid proliferation and treatment resistance, and studies have demonstrated that inhibition of ferroptosis promotes malignant transformation, proliferation and angiogenesis in glioma (4, 5), so induction of ferroptosis is a promising research direction.
Herein, we summarize the processes of ferroptosis in glioma, the current findings on ferroptosis in glioma, which include some of the pivotal regulators and pathways relevant to ferroptosis and the crosstalk between ferroptosis and other programmed cell death including apoptosis, autophagic cell death, necroptosis and pyroptosis. Treatment resistance is an essential contributor to poor prognosis. Therefore, we focus on the relationship between ferroptosis-related metabolic processes and treatment resistance in glioma, and present the role of ferroptosis as well as its prospects in glioma treatment. The therapies include systemic therapy (especially temozolomide chemotherapy), radiotherapy, immunotherapy, and nanotherapy.
Iron metabolism, lipid peroxidation and antioxidant system, the imbalance among these three is a trigger for ferroptosis (Figure 1) . A complex regulatory network exists within the cell to regulate iron metabolism. Research indicates that free iron abundance promotes lipid peroxidation through the accumulation of reactive oxygen species (ROS) and the activation of iron-containing enzymes (6). Antioxidant systems in cells against ferroptosis mainly include Cysteine (Cys), glutathione (GSH), and glutathione peroxidase 4 (GPX4) axis (7) (Figure 1).
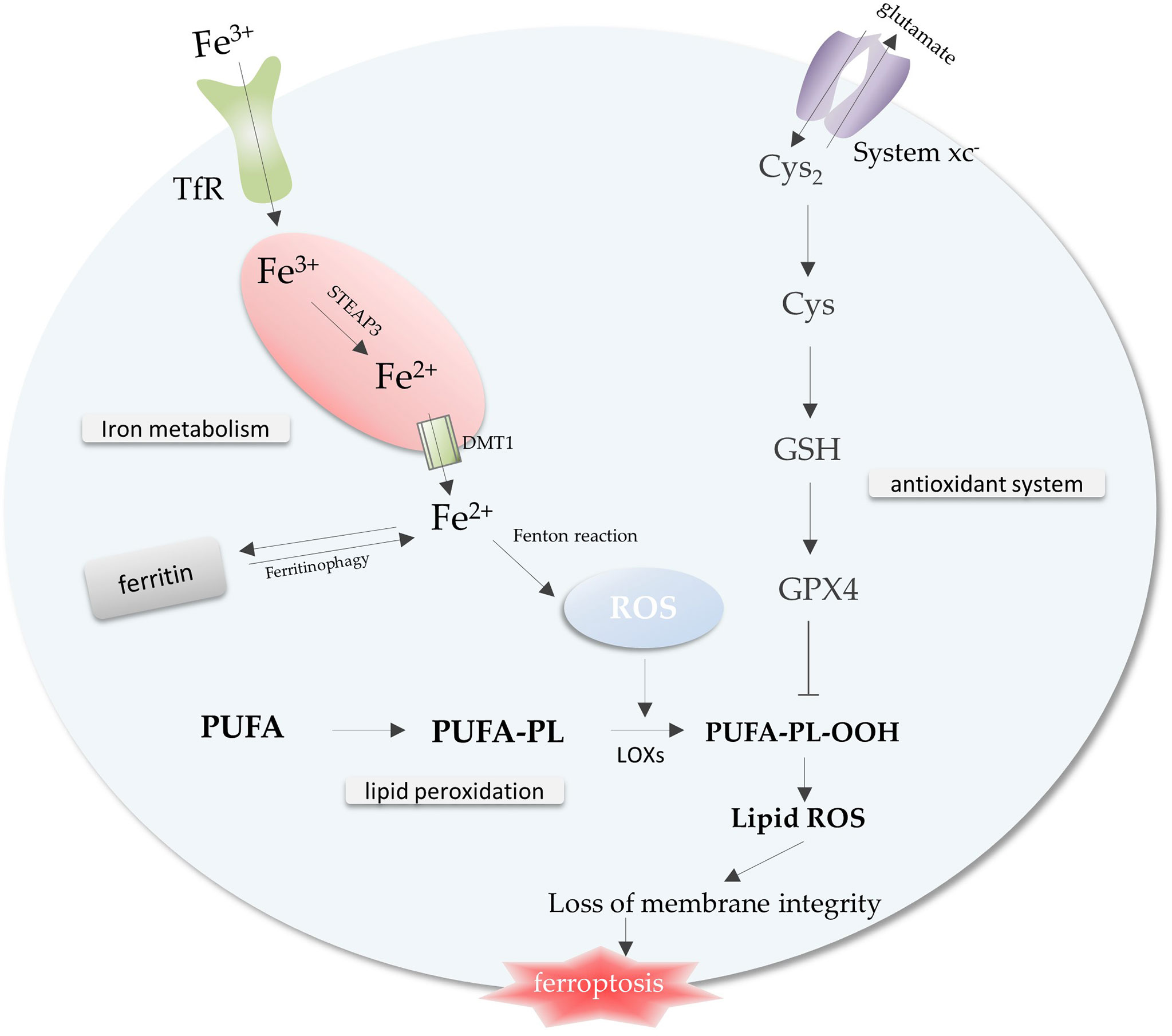
Figure 1 The core mechanisms related to ferroptosis. Free iron promotes intracellular ROS accumulation through the Fenton reaction, leading to lipid peroxidation and ferroptosis. The antioxidant system inhibits the lipid peroxidation process to prevent ferroptosis. Imbalance of iron metabolism, lipid peroxidation and antioxidant system leads to the occurrence of ferroptosis. Abbreviations: TfR, transferrin receptor; ROS, reactive oxygen species; Cys2, cystine; Cys, Cysteine; GSH, glutathione; GPX4, glutathione peroxidase 4; PUFA-PL, polyunsaturated fatty acid-containing phospholipid.
Intracellular iron is strictly regulated. Most iron in cells is ligated by heme, stored in ferritin, an iron storage protein, or exists in the form of Fe-S clusters. Nevertheless, a small amount of labile iron is present in the cells and it is inclined to catalyze the formation of ROS (8, 9) (Figure 2). Iron mediates non-enzymatic peroxidation of lipid through Fenton reaction (8). The process of enzymatic peroxidation is iron-dependent because it requires the participation of iron-containing enzymes, such as ALOXs, NOXs, and CYP (10, 11).
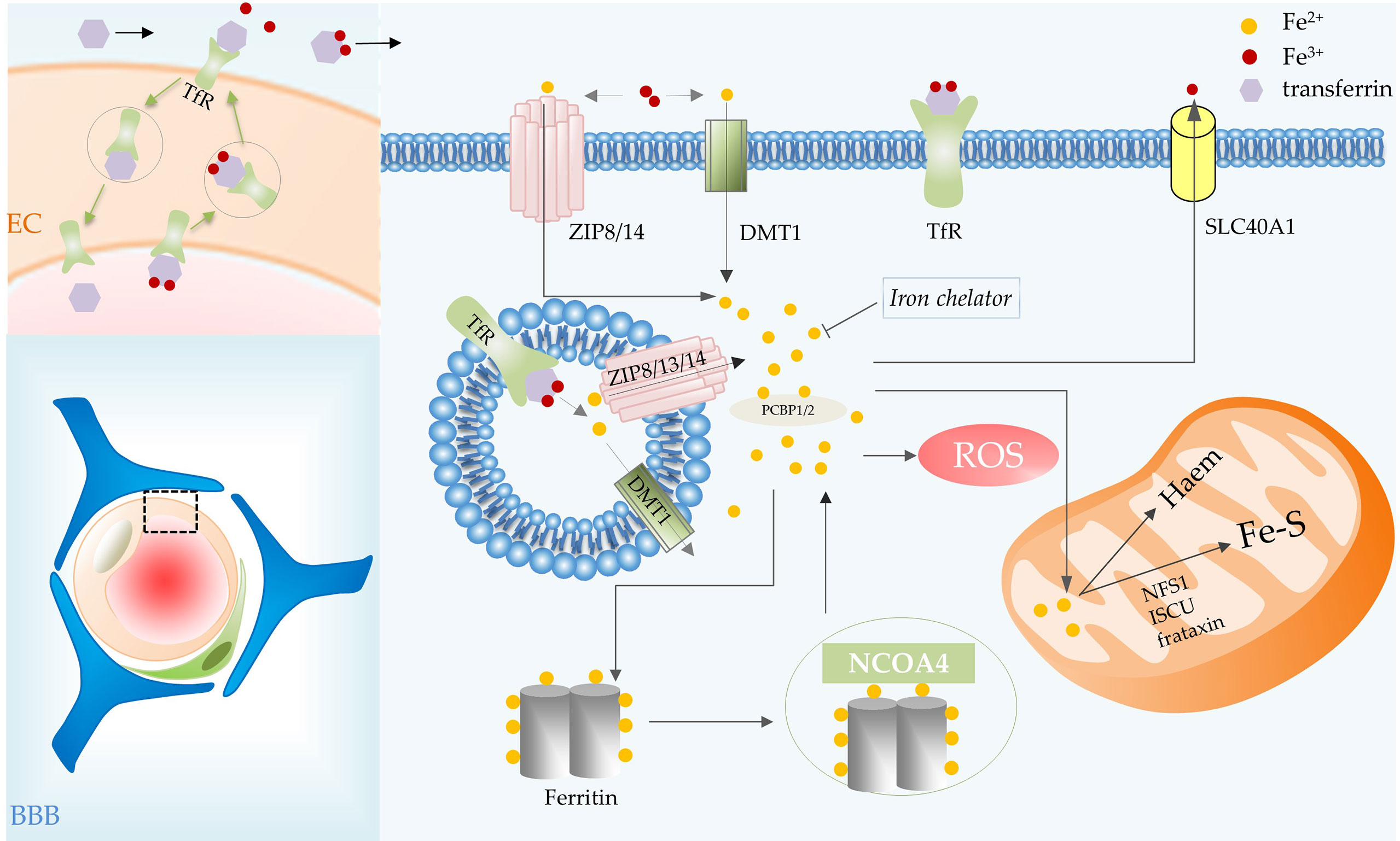
Figure 2 Iron metabolic processes involved in ferroptosis in glioma. BBB consists of ECs, basement membrane, pericytes, and astrocytic end feet. Iron transport depends on the TfR of vascular ECs in the BBB. Transferrin carries iron, the transferrin-receptor complex is internalized and then transported to the abluminal side of the endothelium. Transferrin combines with most of the iron that crosses the BBB, iron is then delivered to cells. In the cell, iron is released from transferrin in acidic endosomes. Endosomal iron can be delivered to the cytoplasm via DMT1, ZIP8, ZIP13 and ZIP14. Intracellular iron can be transported out of cells by SLC40A1, utilized by mitochondria for the synthesis of heme and Fe-S, and stored in ferritin. The iron in ferritin can be released by NCOA4-mediated ferritinophagy. Abbreviations: BBB, blood brain barrier; EC, endothelial cell; TfR, transferrin receptor; SLC40A1, solute carrier family 40 member 1; NCOA4, nuclear receptor coactivator 4.
Iron binds efficiently to extracellular transferrin, which has an important role in ferroptosis, and is released from transferrin when the iron is delivered to acidic endosomes via receptor-mediated endocytosis (10, 12). Transferrin transports iron into cells via transferrin receptors (TfR), TfR RNAi significantly inhibited cell death (12). Endosomal iron can be delivered to the cytoplasm via DMT1, ZIP8, ZIP13 and ZIP14 (13). Then PCBP1 delivers cytosolic iron to ferritin (an important iron storage protein in cells), non-heme iron enzymes and some other proteins (13, 14). PCBP2 is a DMT1-binding protein that transfers ferrous iron to the appropriate intracellular site or solute carrier family 40 member 1 (SLC40A1) (14). Nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy is a form of selective autophagy that facilitates ferritin degradation leading to Fe2+ release (15). Transferrin and receptors promote ferroptosis by increasing intracellular iron content, whereas SLC40A1-mediated iron export inhibits ferroptosis (16). Additionally, ferroptosis is regulated by the iron-regulatory proteins (such as ACO1 and IREB2) at the translational level. Some mitochondrial proteins, such as cysteine desulfurase (NFS1), iron–sulfur cluster assembly (ISCU) and frataxin (17), restrain ferroptosis by increasing the synthesis of Fe-S clusters in cells (Figure 2).
Iron is an essential cofactor for many metabolic processes in the central nervous system (CNS), including DNA synthesis in neurons, oxidative phosphorylation, neurotransmitter production and oxygen transport (18, 19). However, brain is a very special organ in the human body, it is hidden behind the blood-brain barrier (BBB) with very low permeability, which limits its access to many substances (such as iron). Iron transport relies on the expression of TfR by vascular endothelial cells in the BBB. Transferrin binds to TfR expressed at the luminal membrane of endothelial cells. The transferrin-receptor complex will be internalized and then transported to the abluminal side of the endothelium. There, it will be exposed to the local microenvironment, which leads to the release of iron (18). Transferrin synthesized by oligodendrocytes combines with most of the iron that crosses the BBB after iron oxidation (20) (Figure 2).
Compared to normal cells, tumor cells are more dependent on iron. In glioma, reprogrammed iron metabolism is regarded as a core factor in tumorigenesis, progression and the tumor microenvironment (21, 22). Transferrin receptor 1 (TfR1) controls the rate of iron uptake by glioma cells by regulating the amount of iron delivered to cells to meet metabolic requirements. Transferrin receptor 2 (TfR2) is frequently and highly expressed in glioblastoma (GBM) (23, 24). Immunohistochemistry of some GBM tissue samples with TfR mAbs exhibited a high rate of positivity (25). TfR2 expression in normal tissues is restricted, but a frequent expression of TfR2 on cancer lineages of distinct origins can be observed. It suggests that expression of TfR2 by tumor cells, along with increased expression of TfR1, may be a strategy for tumor cells to obtain optimal iron input (23).
Changes in transferrin and receptors can affect cellular iron content and may lead to the development of ferroptosis. Ferritin, composed of ferritin heavy chain (FTH1) and ferritin light chain (FTL), is an iron storage protein in cells. Recent findings strongly support the hypothesis that glial tumors synthesize and secrete ferritin (26, 27). Iron requirements are increased in glioblastoma stem cells, so high levels of cytoplasmic ferritin may protect cells from ferroptosis by enhancing iron chelation (28).
Growing evidence suggests that iron metabolism-related processes in glioma cells contribute to tumor progression. GBM patients have elevated serum ferritin levels, probably due to the inflammatory state, and high serum ferritin levels are associated with poor prognosis (27). In glioma, high expression of TfR mediates intracellular iron accumulation and ROS formation, and promotes tumor proliferation. It also promotes an NMDA-receptor-mediated decrease in the number of neurons (29). Elimination of neurons is necessary for glioma cells to acquire space for growth. In addition, FTL is overexpressed in glioma (30). The oncogenic effect of FTL is mediated through the regulation of AKT/GSK3β/β-catenin signaling. It is proven that FTL promotes migration, invasion and chemoresistance in glioma (30).
Notably, changes in iron metabolism are probably associated with the malignant transformation of glial cells and the degree of malignancy of glioma. The higher the grade of glioma, the correspondingly stronger the treatment resistance effect it has. Purified ferritin from glioblastoma-derived cell line has marked apoptosis-stimulating activity and are inhibited by neutralizing anti-ferritin antibody Ab rH02, however, isoferrin released from cultured neonatal astrocytes did not show this activity, suggesting that the transition of astrocytes to a malignant phenotype is accompanied by alterations in iron metabolic processes (26). And research demonstrated that the extent of TfR expression in glioma is positively correlated with tumor grading (29). FTL expression is also elevated in high-grade glioma (30). The main result of these changes is increased iron uptake by glioma cells. While iron may differentially affect the effectiveness of treatment by activating ROS production and/or signaling pathways including HIF-1 or IRP-1 recruitment (31).
In recent years, an increasing number of studies have found that the inevitability of treatment resistance and recurrence in GBM may be due to the presence of cancer stem-like cells (CSCs) (32). Owing to the nature of enhanced resistance to conventional chemo/radiotherapy and metastasis, CSCs have been proposed as promising targets for cancer eradication (33). Basuli et al. found that iron uptake by GBM CSCs is 2-3 folds higher than that of non-stem cell tumor cells (22). This alteration in iron metabolism is likely to be related to the treatment resistance of GBM CSCs. Expression of stemness-related markers has been proven to be affected by iron chelators, therefore targeting iron metabolic processes in CSCs is promising for reducing tumor cell therapeutic resistance (32).
In ferroptosis, the most essential process related to lipid metabolism is lipid peroxidation. Oxidized lipid disrupts and distorts the bilayer membrane. Oxidized lipid clusters in membranes destroy their barrier function by formation of hydrophilic pores, leading to ferroptosis (34).
Polyunsaturated fatty acid (PUFA)-containing membranes are susceptible to oxidation (6, 13) (Figure 3). Long-chain fatty acid–CoA ligase 4 (ACSL4) has a preference for long-chain PUFAs such as arachidonic acid (AA) and adrenic acid (AdA). In an ATP-dependent manner, ACSL4 catalyzes the binding of AA or AdA to coenzyme A to form derived AA-CoA or AdA-CoA (35, 36), which are then esterified under the catalysis of LPCAT3 and form PUFA-PE by reaction with phosphatidylethanolamine (PE). Then lipoxygenases (LOXs) are required for ferroptosis to oxidize AA-PE and AdA-PE to hydroperoxides AA–PE-OOH and AdA–PE-OOH (13, 37). Six arachidonic acid lipoxygenase (ALOX) genes in humans (ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B and ALOXE3) have distinct expression profiles in different tissues (37). Other contributors of fatty acids include Acetyl-CoA carboxylase (ACC)-mediated fatty acid synthesis (38), and lipophagy-mediated fatty acid releasing (6).
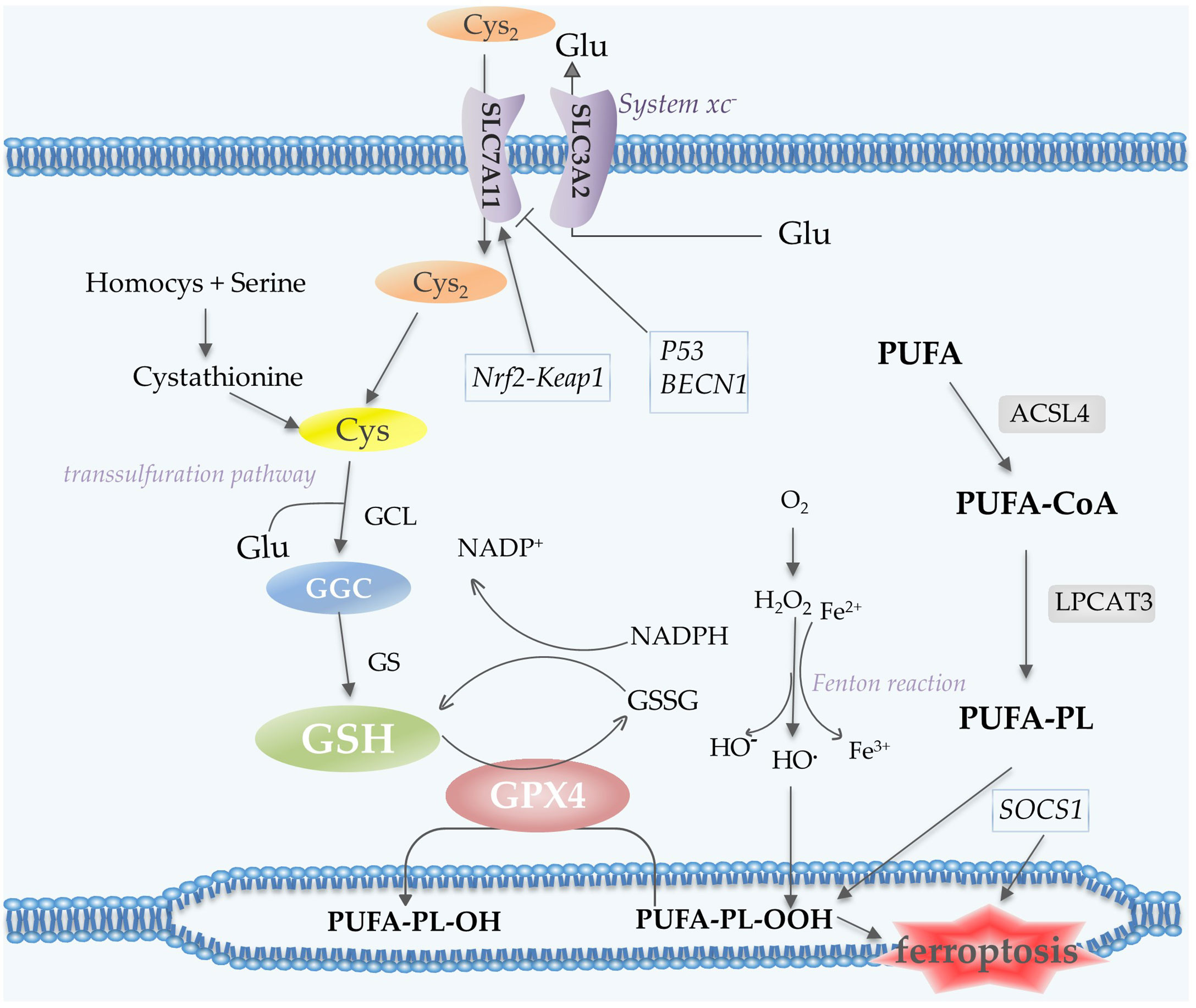
Figure 3 Lipid peroxidation and antioxidant processes in ferroptosis. System xc- contains two subunits, SLC7A11 and SLC3A2, which mediate the transport of Cys2 and Glu. Cys2 is an ingredient for the synthesis of GSH, which together with GPX4 acts as the reductant for the inhibition of ferroptosis. Cys can also be generated via the transsulfuration pathway. PUFA undergoes a series of oxidation reactions and the product PUFA-PL-OOH leads to the occurrence of ferroptosis. Fenton reaction contributes to lipid peroxidation. P53 and BECN1 function as inhibitors of SLC7A11, while Nrf2-Keap1 pathway promotes the expression of this subunit. Abbreviations: Cys2, cystine; Glu, glutamate; GSH, glutathione; GPX4, glutathione peroxidase 4; Cys, Cysteine; PUFA-PL, polyunsaturated fatty acid- containing phospholipid; Nrf2, nuclear factor erythroid 2-related factor 2.
Increased fatty acid synthesis and increased cholesterol uptake are considered to be features of malignant glioma. The altered lipid metabolism may mediate the resistance to chemotherapy and radiotherapy in GBM (39). Lipid peroxidation also contributes significantly to treatment resistance (40). Therefore, targeting glioma lipid regulation is one of the therapeutic strategies. The levels of PUFA are much higher in glioma than in normal brain tissue (41). However, glioma cells can appropriately reduce lipid peroxidation during lipid metabolism to avoid ferroptosis. It is notable that ACSL4 protein expression level was found to be decreased in glioma cells and BAO et al. found that knockdown of ACSL4 reduces ferroptosis and stimulates cell proliferation in glioma cells. In contrast, ACSL4 overexpression decreased the expression of GPX4, while upregulating the expression of ferroptosis indicators such as 5-HETE (5). Liu et al. reported a significant difference in the expression of ALOX5 in glioma and normal brain tissue (42). The above studies prove that many differences in lipid metabolism exist between glioma and normal tissues.
The Cys2/Glu antiporter system xc-, which is composed of two subunits SLC7A11 and SLC3A2, is needed to import cystine (Cys2) into cells for subsequent GSH synthesis (6). In the cell, Cys2 is oxidized to Cys, which is then synthesized with glutamate (Glu) by glutamate-cysteine ligase (GCL) to form GGC, and subsequently synthesize GSH catalyzed by glutathione synthetase (GS) (43). A cycle can be envisaged in which Glu can enter the cell via its transporter protein and be exported via the xc- reverse transport system, thus supporting the cellular uptake of Cys2 (44). Cys for GSH synthesis can also be obtained from protein degradation within the lysosome and transsulfuration pathway (44). Transsulfuration promotes the sulfur oxidation of homocysteine/methionine. Methionine is converted to homocysteine via the transsulfuration pathway, which is then converted to cystathionine and finally to Cys under the catalysis of cystathionine-γ-lyase (45, 46). GPX4 exhibits excellent resistance to irreversible peroxidation in the utilization of GSH to reduce hydrogen peroxide (H2O2) or organic hydroperoxides to water or the corresponding alcohols, while GSH is oxidized to glutathione disulfide (GSSG) (47, 48) (Figure 3).
System xc- is vital in the survival of glioma cells. In glioblastoma cells, most of the Cys is derived from the reduction of Cys2 imported by the system xc- (46). Cancer cells exhibit higher ROS levels compared to normal cells (49), and this leads to higher expression of NOXs and GPX4. Cancer cells can use GSH to reduce oxidation products and inhibit cell death, causing resistance to treatment (50). In GBM cells, high GSH/GPX4 levels induce epithelial-mesenchymal transition, leading to tumor progression, metastasis and chemoresistance (50).
Several studies have documented that glioma cells upregulate the expression of SLC7A11 (xCT). Regulation of SLC7A11 does not alter cell proliferation, but its overexpression increases the growth of anchorage-independent cells (51). Increased SLC7A11 expression correlates with tumor invasion and prognosis in patients with GBM. SLC7A11 is an independent predictive factor in GBM (52).
A number of studies have reported that the system xc- is related to many properties of glioma cells. For example, system xc- is the main pathway of the release of glutamate, glutamate excitotoxicity kills surrounding neurons, thus enhancing tumor invasion and growth. Glutamate release from glioma is also considered to be associated with tumor-associated seizures (53). High expression of SLC7A11 becomes an independent biomarker of seizures (54). Overexpression of SLC7A11 in anti-VEGF-treated GBM cells results in elevated extracellular glutamate. Glutamate promotes regulatory T (Treg) cells proliferation, activation, and suppressive function (55). This immunosuppression can be alleviated by reducing Treg cells to enhance the antitumor effect. In recent years, the relationship between SLC7A11 and cellular ferroptosis has also been elucidated. SLC7A11 promotes the absorption of Cys2, which in turn supports GSH synthesis and inhibits ferroptosis in tumor cells. This has also resulted in the birth of many drugs. In addition, although Cys2 uptake is essential for antioxidant protection of cancer cells against ferroptosis, Cys2 transport through SLC7A11 can also induce oxidative stress and cell death in glucose-deprived glioblastoma cells (56). Moreover, some research reported that cell survival under glucose deprivation conditions also depends on cell density. High cell density inactivates mTOR and promotes lysosomal degradation of SLC7A11, enhancing the viability of GBM cells under glucose-restricted conditions (57). While EGF contributes to cell death under glucose-deprived conditions by upregulating SLC7A11 at the transcriptional and protein levels (58). SLC7A11 overexpressing U251 glioma cells exhibit actin cytoskeletal changes reminiscent of epithelial-like cells and display an increased CSC-like phenotype, which might cause tumor drug resistance and recurrence (59).
The p53 gene has been identified as the most commonly mutated tumor suppressor gene in human cancers. It can transcriptionally regulate a range of genes to modulate DNA damage repair, cell cycle arrest, senescence, apoptosis and ferroptosis (60). Previous results showed that almost 50% of glioma samples have tumor protein p53 (TP53) mutations. This number is even higher when alterations in the p53 pathway are taken into consideration. The p53 gene or pathway is more frequently mutated in astrogliomas and GBM (60, 61). The regulatory network for p53 expression in glioma cells is very complex. The latest report has verified that p53 in glioma reduces MDM2 levels by inducing expression of miR-29a, thus reducing the degradation of p53 by MDM2, forming a feedback loop (62).
The role of p53 has a double-sided regulation mode in cells. p53, the first gene to be studied for increased susceptibility to ferroptosis, can inhibit the transcription of SLC7A11, leading to Cys deprivation. It was demonstrated that p53 downmodulates the level of histone H2B monoubiquitination (H2Bub1), which is involved in ferroptosis regulation by controlling the expression of the downstream gene SLC7A11 (63). P53 acts on glutaminase 2 (GLS2) to increase GSH hydrolysis, causing GXP4 inactivation. It also acts on lipid peroxide synthase to increase cellular susceptibility to ferroptosis (63, 64). SAT1, as a transcriptional target of p53, induces lipid peroxidation and allows cells to undergo ferroptosis in response to ROS-induced stress (65). It has been reported that TP53-induced glycolysis and apoptosis regulator (TIGAR) are direct targets of ferroptosis mediated by p53. TIGAR and cytochrome c oxidase 2 (SCO2) promote the pentose phosphate pathway (PPP) which is engaged in the production of NADPH, a reducing agent during ferroptosis. Additionally, PVT1 may mediate the role of p53 in promoting ferroptosis (63). MDM2 and MDMX, negative regulators of p53, normally promote ferroptosis (66). TP53 can prevent ferroptosis by inhibiting dipeptidyl-peptidase-4 (DPP4) activity and relevant findings have suggested that DPP4 is a key co-ordinator of lipid metabolism in colorectal cancer (67). Nucleotide synthesis is an essential cellular metabolic process, and a recent report indicates that the p53 pathway can inhibit the expression of ribonucleotide reductase, leading to GSH accumulation and the avoidance of ferroptosis (68). P53 could positively and negatively regulate ferroptosis (69), the role of p53 in ferroptosis needs to be further elucidated. Nevertheless, the role of p53 is influenced by several factors, such as cell type and p53 mutation. In normal tissues, wild-type p53 exhibits positive regulation of ferroptosis to prevent the accumulation of genetic mutations but inhibits ferroptosis to protect cells under stressed conditions (63, 70). In tumors, other ferroptosis regulators supersede the role of p53, and the effect of wild-type p53 appears to be limited. Conversely, mutant p53 renders tumor cells sensitive to ferroptosis (70). In summary, studies on the ferroptosis-related pathways involved in p53 in glioma and their distinction from other tumors are not numerous and deserve further study in the future.
BECN1, a haploinsufficient tumor suppressor gene (71), its dysfunction is correlated with many diseases, including carcinoma and neurodegeneration (72). BECN1 is a core autophagy protein essential for the autophagosome nucleation phase in mammals. ROS levels are higher in cancer cells compared to normal cells, and it is widely believed that high ROS levels induce autophagy. Recent studies have demonstrated that hTERT in GBM can reduce autophagy levels by inhibiting BECN1, leading to increased ROS and ultimately cell death (73). High expression of autophagy-related proteins such as BECN1 is more pronounced in high-grade glioma than in low-grade, so BECN1 might be a prognostic marker for glioma patients (74).
The role of BECN1 in ferroptosis has also received attention in recent years. Experimental data demonstrated that BECN1 does not influence intracellular iron accumulation or expression related to iron metabolism (75). BECN1 promotes ferroptosis by the direct blockade of the SLC7A11 subunit of system xc-. AMPK-mediated phosphorylation of the BECN1 Ser90/93/96 sites is essential for BECN1 to form a complex with SLC7A11 (76).
Suppressor of cytokine signaling 1 (SOCS1) has been demonstrated to be a tumor suppressor capable of bridging p53 and ATM at sites of DNA damage, leading to p53 phosphorylation and consequently increasing its transcriptional activity (77). It has been proven that the expression of SOCS1 decreases the levels of SLC7A11 and GSH in cells, suggesting its ability to sensitize cells to ferroptosis (78). Ferroptosis-related gene SOCS1, has become a biomarker for the diagnosis or prognosis of many diseases, such as tuberculosis (79), AML (80) and head and neck squamous cell carcinoma (HNSCC) (81).
The effects of SOCS proteins in GBM have recently become a research hotspot. SOCS inhibits proliferation and angiogenesis of GBM through the negative regulation of the JAK/STAT3 signaling pathway and SOCS proteins can also control the invasion and metastasis of GBM through multiple pathways (82). Evidence indicated that SOCS1 and SOCS3 might be involved in tumor aggressiveness and radiation tolerance (83). It has been implicated that SOCS1 tends to be repressed in GBM as a result of CpG island-mediated epigenetic silencing of the SOCS1 locus. Reintroduction of SOCS1 can sensitize cells to radiation (84). Mutation status of p53 may have a regulatory role in the transcriptional plasticity of the SOCS1 promoter (85). Increasing SOCS1 expression appears to simultaneously induce ferroptosis and improve radiotherapy sensitivity, and SOCS1 is a possible therapeutic target for glioma. However, SOCS1 is involved in a complex regulatory network, and the role of SOCS1 in inducing ferroptosis in glioma cells is currently less known.
Frataxin, a highly conserved protein, is localized in the mitochondrial matrix and is involved in the biosynthesis of Fe-S clusters. Frataxin is a key regulator of ferroptosis via the regulation of iron homeostasis and mitochondrial function (86). Frataxin can accelerate the rate of persulfide formation on NFS1, promoting Fe-S cluster synthesis (87). Suppression of frataxin significantly inhibits proliferation, disrupts mitochondrial morphology, blocks Fe-S cluster assembly and exacerbates iron accumulation (86). Insufficient maintenance of Fe-S clusters strongly activates the iron starvation response and combines with inhibition of GSH biosynthesis (88). It has been shown that frataxin has a tumor suppressive effect, but it has a dual, pro-proliferative role in astrocytic tumors (89).
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a critical transcription factor in the cellular response to oxidative stress. Nrf2-dependent transcription is repressed by Keap1 under basal conditions, when cells are exposed to oxidative stress, Nrf2 escapes repression and activates antioxidant responsive element (ARE)-dependent gene expression (90). Nrf2 is overexpressed in GBM cells and associated with poor prognosis (91). Nrf2-Keap1 pathway is involved in ferroptosis in glioma. Activation of Nrf2-Keap1 signaling can upregulate SLC7A11. It was also found that fostering Nrf2 expression and inhibiting Keap1 both increase the resistance to ferroptosis in glioma cells.
COPZ1 expression is upregulated in GBM cell lines and it has a tendency to negatively regulate the activity of NCOA4. Knockdown of COPZ1 leads to an increase of NCOA4, contributing to the degradation of ferritin, which leads to increased intracellular ferrous iron levels and eventually to ferroptosis. Thus, the COPZ1/NCOA4/FTH1 axis is a novel therapeutic target for the treatment of GBM (92). Targeting pathways including NCOA4 may be a promising approach for the treatment of glioma.
Apoptosis, autophagic cell death, necroptosis, pyroptosis and ferroptosis represent a group of highly ordered programmed cell death (PCD) events that can eliminate cells that are running chaotically or destined to die. In tumor cells, survival signaling and programmed death resistance (such as apoptosis resistance) are two complementary aspects. Targeting increased survival may not be effective without also addressing cellular PCD resistance (93, 94). Therefore, it is especially necessary to explain the process of PCD and the crosstalk between them, which might provide new strategies for tumor treatment.
Apoptosis is a relatively early discovery and well-studied PCD, and here we will present some findings about the intersections of ferroptosis and apoptosis in glioma (Figure 4).
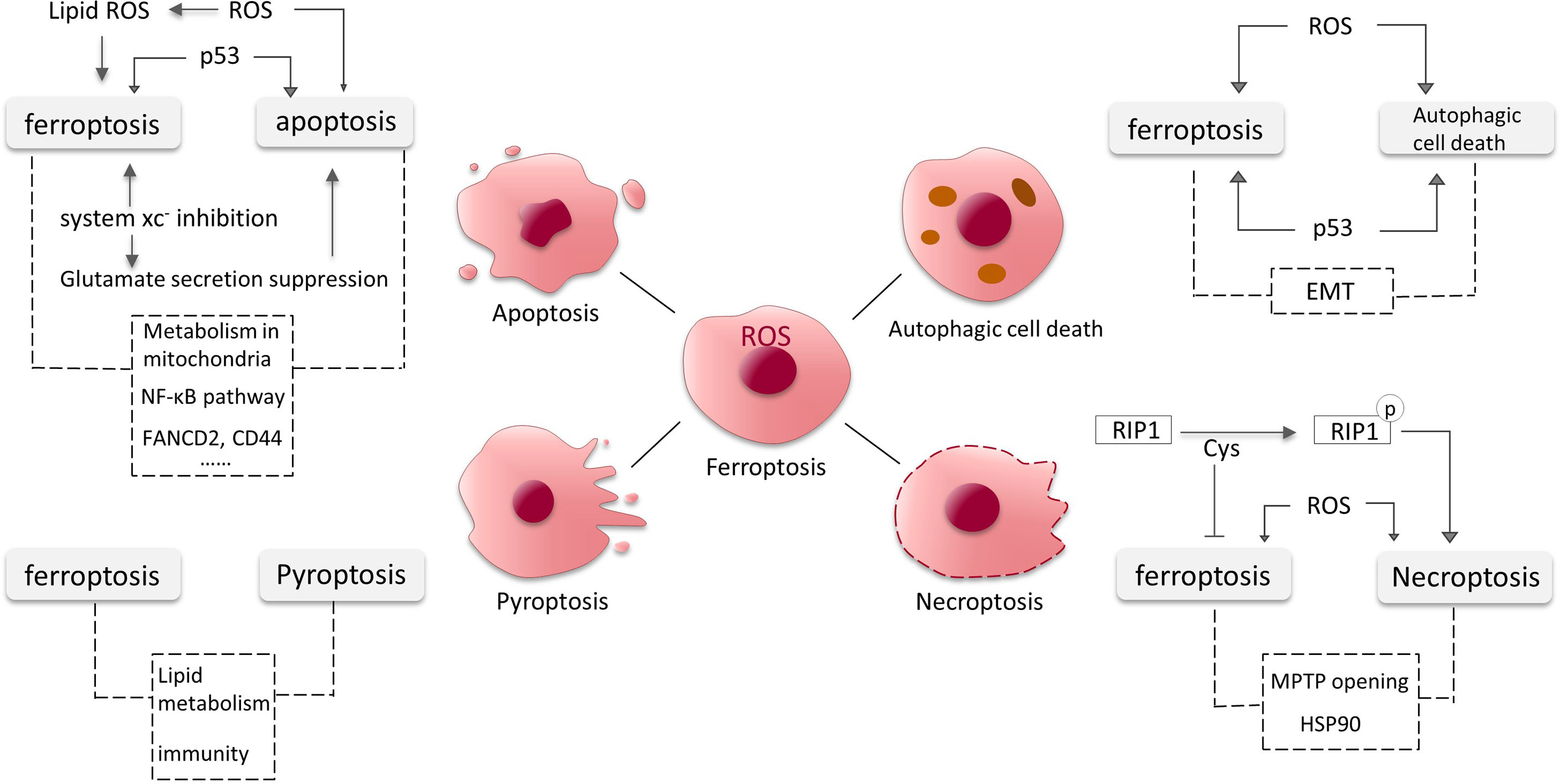
Figure 4 Crosstalk between ferroptosis, apoptosis, autophagic cell death, necroptosis and pyroptosis in glioma. The dotted lines represent possible intersections and require more research to elucidate.
In GBM cells, ROS controls cellular stability by affecting different signaling pathways. It is a pivotal participant in the occurrence of ferroptosis. It has been found that excess ROS can also induce apoptosis (95). ROS may act as a key substance in the onset of apoptosis and ferroptosis in GBM cells. As a tumor suppressor molecule, p53 increases ferroptosis susceptibility and can also induce cell cycle arrest and apoptosis. Dysregulated p53 pathway is relevant to apoptosis evasion (96). We have previously mentioned that system xc- mediates the toxic secretion of most glutamate from GBM cells, and its inhibition induces ferroptosis. A recent study revealed that the suppression of extracellular glutamate release promotes apoptosis and autophagy in GBM cells (97). Moreover, RSL3 (a GPX4 inhibitor) was found to drive ferroptosis via NF-κB pathway in GBM cells (98), and NF-κB pathway is also involved in apoptosis (99, 100). Down-regulation of FANCD2 and CD44 expression by sponging hsa-miR-27a-3p promotes apoptosis and ferroptosis in glioma cells, the pathways involved might also be intersections of apoptosis and ferroptosis (101). Mitochondria are known to be involved in various PCD processes including apoptosis, and the role of mitochondria in ferroptosis is gradually being discovered and the connections deserve further studies (102, 103).
Autophagic cell death, independent of caspase, can be defined as cell demise with strict requirements for autophagy (104, 105). In tumors, autophagy has both pro-survival and pro-death functions (106). Nevertheless, autophagic cell death exhibits extensive autophagic degradation (104). GBM cells have lower levels of autophagy-related proteins compared to low-grade astrocytomas (107). Autophagy is an attractive target for anti-cancer therapy (108).
Similar to apoptosis, p53 and excess ROS also induce autophagy in GBM cells (95, 107). NF-κB is released from the BNIP3 promoter and permits the action of E2F1 to induce autophagy under hypoxic conditions (107). Epithelial mesenchymal transition (EMT) promotes aggressive migration, immunosuppression and drug/radiotherapy resistance of cancer cells (109). EMT processes might also be linked to both autophagy and ferroptosis in glioma. An increasing number of studies have shown links between ferroptosis and EMT in tumor cells. For instance, increased levels of H2O2 associated with EMT confers susceptibility to ferroptosis (110). The crosstalk between autophagy and EMT processes is complex. In the early stages of metastasis, autophagy primarily inhibits the EMT programme, and later, metastatic cells may require sustained autophagy for survival under environmental and metabolic stress conditions (111) (Figure 4).
Necroptosis is a newly found PCD that combines necrosis and apoptosis. It is regulated by caspase-independent pathway and has morphological characteristics of necrosis (112, 113). Necroptosis is regulated by receptor-interacting protein (RIP) 1 activation (114).
Cys plays important roles in ferroptosis and necroptosis. The three cysteines in RIP1 form intermolecular disulfide bonds, which induce ROS generation and consequently RIP1 autophosphorylation, promoting necroptosis (114). Necroptosis can also be induced by high levels of intracellular ROS (115). Meanwhile, Cys is involved in the synthesis of GSH to inhibit ferroptosis. Mitochondrial permeability transition pore (MPTP) opening and heat shock protein 90 (HSP90) also could be the intersections between ferroptosis and necroptosis (114) (Figure 4).
Pyroptosis is a novel PCD mediated by gasdermin D protein and triggered by certain inflammasomes (116, 117). Pyroptosis can affect tumor proliferation, invasion and metastasis (117). Recent studies have revealed its role in gastrointestinal cancer, hepatocellular carcinoma, breast cancer and other cancers (118–121). Pyroptosis is a vital regulator of the immune microenvironment and a prognostic predictor in glioma (122), and relevant studies are currently limited.
Lipid also seems to be associated with pyroptosis induction. Substantial lipid aggregation induces activation of pyroptosis signaling pathways in the formation of vulnerable atherosclerotic plaques (123). Lipid peroxidation can drive pyroptosis in lethal polymicrobial sepsis (124). Lipid levels are elevated in glioma cells and the metabolism of lipid is critical in ferroptosis, but it is unclear whether lipid in glioma contributes to pyroptosis. Nitric oxide (NO) is involved in cell proliferation, cardiovascular formation and apoptosis in glioma (125, 126). Many recent studies have indicated that inhibition of NO mediates the processes of ferroptosis and pyroptosis (127). It has also been mentioned that CD8+ T cells can suppress tumor growth by triggering ferroptosis and pyroptosis (128). The intersections and shared pathways of ferroptosis and pyroptosis in glioma require more research to be elucidated (Figure 4).
The applications of ferroptosis in the treatment of glioma are promising, and the induction of ferroptosis or ferroptosis inducers in combination with other treatments have proven to be effective in a variety of glioma cell lines, tissues, and animal models (Table 1), but the clinical applications currently have yet to advance.

Table 1 The promising applications of ferroptosis in the treatment of glioma.
Glioma comprises 40% of all primary brain tumors and is a serious threat to human life. In particular, GBM is the most common and aggressive primary CNS malignancy, with a median survival of only 15 months despite various treatments including surgery, temozolomide (TMZ) chemotherapy, and radiotherapy (136, 137). The resistance of tumor cells to these therapies is an important reason for the poor prognosis.
The drivers of chemoresistance in glioma can be simply attributed to the influence of genetic aspects and the effects of the external environment. Altered expression of multidrug resistance (MDR)-related genes correlates with reduced treatment responsiveness (138). It has been observed that glioma can exhibit overexpression of ABC transporter proteins, which decrease therapeutic drug accumulation in tumor cells and are directly related to the chemoresistance (139). Further, this resistance is also associated with the DNA damage response of tumor cells (140), the mismatch repair system (141, 142), MGMT status (143, 144), and the regulation of a large number of microRNAs which involves a complex regulatory network consisting of various intracellular molecular signaling pathways (145–150). Tumor microenvironment containing endothelial cells, immune cells, stromal cells, noncellular factors and special conditions, also supports chemoresistance of tumor cells especially CSCs (140, 151). BBB prevents almost all large molecules and more than 95% of small molecules from entering the brain, resulting in unsatisfactory chemotherapy for glioma (152).
With some similarities to chemoresistance, the tumor microenvironment also plays an important role in radioresistance, and multiple signaling pathways (AKT pathway, notch pathway, Wnt/β-catenin pathway, STAT3 pathway and other pathways), proteins and microRNAs in differentiated glioma cells or CSCs have been shown to affect radiation resistance (153). Tumor cell networks with high cell density also have a significant resistance function. Research has revealed that the perivascular niche (the preferential location of quiescent glioma cells) and the formation of a multicellular network by tumor microtubules are involved in radiotherapy and chemotherapy resistance (154). Heterogeneity within GBM (regional genetic variance and cellular hierarchies often regulated by different CSC niches) is accepted as the basis for resistance to multiple treatments (155, 156). Moreover, the close association between hypoxia and resistance to radiotherapy in glioma, especially in GBM, deserves attention. Severe hypoxia is more common in GBM than in lower-grade glioma (157). Oxygenation is essential to the effectiveness of radiotherapy. Hypoxia also stimulates enzymes responsible for cancer survival under hypoxic stress via upregulation of HIF (158).
GBM is highly immunosuppressive and has multiple immune evasion mechanisms (159). Due to the special structure of the brain such as BBB, its immune environment is unique (160, 161). In brain tumor patients, immunological dysfunction is a major obstacle to immunotherapy (155). Some infiltrating immune cells in the tumor microenvironment such as Treg cells (162), myeloid-derived suppressor cells (163), etc. are also engaged in immunotherapy resistance. And the molecular heterogeneity of GBM hinders efforts to identify high-quality clonal neoantigens (155). In addition, glioma cells have potent adaptive and acquired resistance mechanisms, which involve genetic alterations shaped by immunological pressure (164). Probably the use of combined immune checkpoint blockade to overcome adaptive resistance is one solution (165).
Since therapeutic resistance is a major impediment to glioma treatment, we will focus on the relationship between ferroptosis and treatment resistance in various therapeutic approaches in the following introduction. In addition, we will also present the role and potential applications of ferroptosis in the treatment of glioma.
Chemotherapy and targeted therapy are critical aspects of malignancy treatment. In glioma, TMZ remains the mainstay of chemotherapy (166). However, drug resistance in glioma cells is currently a major challenge. A growing number of studies suggested that ferroptosis may be related to this resistance and that the efficacy of many drugs might also be associated with the ferroptosis induction.
TMZ was approved by the US Food and Drug Administration (FDA) for the treatment of adult refractory anaplastic astrocytoma in 1999 and newly diagnosed glioblastoma in adults in 2005 (167). TMZ, the most effective drug for the treatment of glioma, has the advantages of oral administration, easy penetration of BBB, and acidic environment stability. DNA methylation is regarded as the principal mechanism of cytotoxicity of TMZ to malignant cells (168). But its clinical efficacy is not ideal and glioma resistance to TMZ is the most important reason for chemotherapy failure. Ferroptosis has been shown to be linked to drug resistance of TMZ, and clarifying this relationship facilitates the application of ferroptosis to the clinical practice of glioma treatment.
Decreased GSH levels and GPX4 levels and inhibition of SLC7A11 can induce ferroptosis through the production of excess ROS. Chen et al. demonstrated that down-regulation of GSH levels could sensitize GBM cells to TMZ. TMZ significantly induces the expression of Nrf2 and ATF4 (Figure 5). Transcription factor Nrf2 can mediate TMZ resistance via the synthesis and utilization of GSH, and inhibition of Nrf2 increases the TMZ sensitivity of glioma cells (169, 170). ATF4 promotes the expression of GSH and SLC7A11 to avoid ferroptosis in glioma cells, inhibition of ATF4 can reduce the resistance of glioma cells to TMZ (171–173). SLC7A11 expression is enhanced by TMZ via Nrf2 and ATF4 activation pathway, and erastin-inhibited SLC7A11 enhances TMZ toxicity (173). It has also been observed that anti-treatment cells are GPX4-dependent and that loss of GPX4 function causes ferroptosis (174). Cystathionine γ-lyase (an enzyme involved in the transsulfuration pathway) is induced by TMZ to increase the supply of Cys (175). Overproduction of ROS is likely to be important in enhancing TMZ sensitivity (Figure 5), sorafenib alters TMZ sensitivity via autophagy and the JAK2/STAT3-AIF axis, and this alteration can be reversed by ROS clearance (176). It was also reported that the effectiveness of TMZ treatment is related to p53 status (177).

Figure 5 The association between drugs such as temozolomide (TMZ) and ferroptosis in glioma cells and some factors influencing TMZ sensitivity. TMZ increases the levels of Nrf2 and ATF4 and thus induces the expression of SLC7A11 and GSH via multiple mechanisms. Silibinin and sulfasalazine inhibit the SLC7A11 subunit. TMZ facilitates Cys synthesis through the transsulfuration pathway. SLC7A11, GSH, and GPX4 suppress ROS formation, while ROS promotes ferroptosis. ROS overproduction is likely to be important in enhancing TMZ sensitivity. Sorafenib, deferiprone, and curcumin also increase the sensitivity of glioma cells to TMZ.
Some agents that function as iron chelators to suppress ferroptosis have been demonstrated to be associated with the reversal of TMZ resistance. Deferiprone (also known as ferriprox) is an orally active, brain-permeable drug. TMZ and deferiprone combination therapy significantly reduces cell viability in glioma cells (178). Curcumin, a component of the Indian spice turmeric, is able to sensitize GBM cells to TMZ treatment. The effect is achieved by enhancing apoptosis. The combination treatment of curcumin and TMZ was observed to have a synergistic effect in generating ROS, which may contribute to therapeutic sensitization (131, 179).
TMZ resistance is also associated with iron metabolic processes in glioma cells. In GBM patients treated with radiotherapy and temozolomide, a highly significant correlation was found between the level of TfR2 and overall survival (OS). One of the reasons is that TfR2-positive cells are more sensitive to TMZ (180, 181). Fluorescence density of PAMAM-PEG-Tf/TMZ in TfR+ glioma stem cells (GSCs) was significantly higher than that of matched non-stem cells and active apoptosis of tumor cells could be observed after the uptake of PAMAM-PEG-Tf/TMZ, suggesting that targeting transferrin receptors to deliver TMZ is a potential GSC-mediated treatment method (182).
Sulfasalazine is a drug widely used to treat a number of chronic inflammatory conditions (183). It is also an established inhibitor of system xc- (Figure 5). Sulfasalazine impacts on ferroptotic cell death of tumors and has also been proven to alleviate glioma-related brain edema and epileptic events (129, 183).
Sulfasalazine did not show significant benefit in a small, discontinued phase I study, but it was not concluded to be ineffective given the patients’ health status, etc. Sulfasalazine might be used as an adjuvant treatment for malignant glioma (183). The combination of TMZ and sulfasalazine was shown to be cytotoxic to T98G and A172 cells, and sulfasalazine was found to enhance the cytotoxicity of TMZ to human GBM cells (132). Sulfasalazine and valproic acid drive GBM cell death through an imbalance in the intracellular oxidative response, making this drug combination a hopeful therapeutic strategy (184).
Many derivatives of sulfasalazine have been synthesized, and further studies on the molecular structure of system xc- and its combination mode with inhibitors may help guide the design of potential inhibitors. SLC7A11 ligand models can be further optimized to find powerful lead molecules for the discovery of new drugs (185).
Silibinin has been shown to be effective in removing tumor cells from breast cancer, colorectal cancer, glioma, etc. Silibinin is believed to result in glioma cell death through the induction of lethal autophagy, which is through the induction of oxidative stress-mediated BNIP3-dependent AIF nuclear translocation (186). Recent studies have found that silibinin leads to downregulation of SLC7A11 and also depletes Cys in a time-dependent manner, resulting in depletion of GSH and accumulation of ROS. BNIP3 plays an essential role in the functional performance of silibinin. Reduction of ROS with the antioxidant GSH significantly prevents silibinin-induced DNA double-strand breaks and glioma cell death (130). Growing evidence suggests that silibinin-induced cell death is likely to be associated with ferroptosis, but more studies are needed to prove it.
Radiotherapy is a highly effective and targeted treatment for cancers (181). The main molecular target of ionizing radiation (IR) is DNA, leading to a whole range of DNA damage, including double-strand breaks, cross-links and complex chromosomal rearrangements (187). It also leads to an increase in intracellular ROS by eliciting radiolysis of water (187, 188). IR induces apoptosis, senescence, methuosis and other cellular outcomes (189). IR-induced DNA damage is initially recognized by ataxia telangiectasia mutated (ATM), and after a complex signaling cascade, this damage may eventually be corrected by DNA repair mechanisms (190, 191). Tumor cells also inhibit apoptosis, and these mechanisms also contribute to radiotherapy resistance (134). Sensitizing cancer cells to radiation through alternative cell death pathways (such as ferroptosis) is a promising way to improve radiotherapy outcomes.
It was found that the antitumor efficacy of radiation may be driven by triggering ferroptosis in some contexts, and that ferroptosis inducers may effectively lead to radiosensitization (134). Iron-containing water prior to radiotherapy has been proven to stimulate glioma cell death through apoptosis and ferroptosis, thereby increasing treatment efficiency (192). In U87 cell line of glioma, synergistic effects of erastin and RSL3 with radiation promote clonogenic ferroptosis (134). A recent study found that IR promotes the expression of ACSL4 in addition to inducing ROS, suggesting a strong induction of ferroptosis by IR. The researchers suggested that IR also induces adaptive responses involving SLC7A11 or GPX4 induction to promote tumor cell survival during radiotherapy, which is one of the reasons for radioresistance. Inhibition of SLC7A11 or GPX4 induces resensitization of radiation-resistant cancer cells to IR-induced ferroptosis, leading to radiosensitization (193). The specific mechanisms of these alterations need to be explored. Further, the effect of in vivo radiation therapy is thought to be dependent on the presence of CD8+ T cells, and Wang et al. found that CD8+ T cells regulate tumor ferroptosis via IFNγ (194, 195).
Current immunotherapy trials in glioma are focused on immune checkpoint inhibitors, vaccines designed to induce immune responses by increasing the recruitment of antigen-specific effector T cells to tumor sites, chimeric antigen receptor (CAR)-T cells, and oncolytic viruses (196). These approaches have had some achievements, although there are currently many obstacles to immunotherapy in glioma. In GBM, the upregulation of immunosuppressive factors and recruitment of Treg cells can be detected after CART-EGFRvIII infusion (197). This suggests that multiple immune escape mechanisms in GBM are challenging to overcome. We consider that identifying the intersections of immunotherapy and ferroptosis in glioma may be expected to improve therapeutic effectiveness.
Some association exists between immunotherapy and ferroptosis. IFNγ derived from CD8+ T cells activated by immunotherapy and ATM activated by radiotherapy synergistically inhibit SLC7A11, inducing ferroptosis in tumor cells (194). Recent studies have shown that the ferroptosis suppressors CD44, HSPB1, and SLC40A1 are significantly associated with prognosis in GBM and correlated with immunosuppression. Acetaminophen might have an antitumor function in GBM by regulating CD44, HSPB1, and SLC40A1 to induce ferroptosis (198). Since most tumors including glioma are much more immunoreactive to TfR1 than normal brain tissue, this component may have the necessary properties to be a target for brain tumor immunotherapy (23). In glioma microenvironment, enhanced ferroptosis was shown to induce immune cell activation and infiltration, but weakened anti-tumor cytotoxic killing (199).
Technological advances promote new nanoscale diagnostic and therapeutic approaches in cancer medicine. We have mentioned above that the BBB is one of the reasons for resistance to drug treatment such as chemotherapy, while nanomaterials rely on their favorable physicochemical properties to be excellent transport vehicles capable of crossing the BBB (200). Several methods using nanocarriers, such as liposomes, micelles, metal ions, and nanoparticles, have been investigated for intracerebral drug delivery (201–203). Nanotechnology can be used to improve direct local treatment of glioma by extending the half-life of encapsulated drugs or providing a sustained release system (201). Nanotechnology may improve the efficacy of ferroptosis inducers, which is expected to develop a promising new approach for the treatment of glioma.
Gold nanocages (AuNCs) as carriers loaded with doxorubicin (DOX) and L-buthionine sulfoximine (BSO) evoke ferroptosis and immune responses in cancer therapy. DOX increases ROS levels, BSO restrains GSH levels, and the ROS production is further amplified by the photothermal effect mediated by AuNCs under laser irradiation (204). In glioma therapy, a recent paper reported a new nanoparticle-mediated drug delivery system that patches heparin-based nanoparticles loaded with DOX to the surface of natural grapefruit extracellular vesicles (205), however, it didn’t mention whether this system could trigger ferroptosis in glioma cells. Nevertheless, the design and successful application of these nanoparticle-dependent delivery systems show us the prospect of nanotechnology in the delivery of ferroptosis inducers for the therapy of glioma.
In this review, we discuss the mechanisms and metabolic features associated with ferroptosis in glioma, and summarize other advances including regulatory targets and pathways and the intersections between ferroptosis and different forms of programmed cell death. We also provide perspectives on the application of ferroptosis in different therapeutic modalities. However, ferroptosis, as a new form of cell death, has not been extensively studied in glioma.
Many questions remain to be explored in depth. Ferroptosis is iron-dependent, iron can be involved in the Fenton reaction and also act as a cofactor to promote oxidative enzymes to engage in reactive oxygen species generation, but the role of executive factors downstream of lipid oxidation has not been elucidated by current studies (206). Moreover, what other genes and metabolic processes are involved in the occurrence of ferroptosis in glioma cells, and if there are other key regulators? Although we have concluded the crosstalk between ferroptosis and other forms of death in glioma, more discoveries are needed to address the question of how to combine different types of cell death with ferroptosis to improve killing of cancer cells.
Several studies show a remarkable potential of ferroptosis in eliminating aggressive malignancies resistant to traditional therapies. Erastin, a ferroptosis inducer, has been used to sensitize GBM cells to TMZ by blocking SLC7A11 and reducing cystathionine-γ-lyase activity. It has been shown that certain human gliomas may be sensitive to the combination therapy of a ferroptosis inducer and radiation (134). Furthermore, how to target ferroptosis induction to eliminate drug-resistant glioma cells while minimizing the impact on normal tissues is a critical therapeutic issue. And the treatment of CNS tumors such as glioma differs from other tumors, the use of ferroptosis inducers requires consideration of BBB penetration for optimal drug concentrations. Possibly nanotherapy be an ideal approach, with the development of nanomaterial technology, the combination inducing ferroptosis with nanotechnology enhances the stability, biosecurity, targeting, and controlled release of drugs to glioma cells. Research on ferroptosis provides new biomarkers and prospective targets for glioma treatment, the potential clinical application remains to be further investigated. In the future, many issues need to be clarified in more epigenetic molecules and detailed mechanistic insights to design the effective cancer therapy strategies based on ferroptosis.
CQ is the corresponding author and organized this review. JS, NY, and MH analyzed the articles and drafted the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of China (81803045, 82103277), the China Postdoctoral Science Foundation (2020M672072), the Research Project of Jinan Microecological Biomedicine Shandong Laboratory (JNL-2022041C), Shandong Excellent Young Scientists Fund Program (2022HWYQ-035), Shandong Provincial Natural Science Foundation (ZR2021QH030), and the Qilu Young Scholar Program of Shandong University, China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
2. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
3. Xu H, Ye D, Ren M, Zhang H, Bi F. Ferroptosis in the tumor microenvironment: perspectives for immunotherapy. Trends Mol Med (2021) 27(9):856–67. doi: 10.1016/j.molmed.2021.06.014
4. Liu HJ, Hu HM, Li GZ, Zhang Y, Wu F, Liu X, et al. Ferroptosis-related gene signature predicts glioma cell death and glioma patient progression. Front Cell Dev Biol (2020) 8:538. doi: 10.3389/fcell.2020.00538
5. Cheng J, Fan YQ, Liu BH, Zhou H, Wang JM, Chen QX. Acsl4 suppresses glioma cells proliferation via activating ferroptosis. Oncol Rep (2020) 43(1):147–58. doi: 10.3892/or.2019.7419
6. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol (2021) 18(5):280–96. doi: 10.1038/s41571-020-00462-0
7. Lu B, Chen XB, Ying MD, He QJ, Cao J, Yang B. The role of ferroptosis in cancer development and treatment response. Front Pharmacol (2017) 8:992. doi: 10.3389/fphar.2017.00992
8. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun (2017) 482(3):419–25. doi: 10.1016/j.bbrc.2016.10.086
9. Breuer W, Shvartsman M, Cabantchik ZI. Intracellular labile iron. Int J Biochem Cell Biol (2008) 40(3):350–4. doi: 10.1016/j.biocel.2007.03.010
10. Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol (2020) 8:590226. doi: 10.3389/fcell.2020.590226
11. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol (2020) 16(3):302–9. doi: 10.1038/s41589-020-0472-6
12. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell (2015) 59(2):298–308. doi: 10.1016/j.molcel.2015.06.011
13. Huang R, Dong R, Wang N, He Y, Zhu P, Wang C, et al. Adaptive changes allow targeting of ferroptosis for glioma treatment. Cell Mol Neurobiol (2021). doi: 10.1007/s10571-021-01092-5
14. Yanatori I, Yasui Y, Tabuchi M, Kishi F, Iron. Biochem J. (2014) 462(1):25–37. doi: 10.1042/BJ20140225
15. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ. 3rd, et al. Autophagy Promotes Ferroptosis by Degradation Ferritin Autophagy (2016) 12(8):1425–8. doi: 10.1080/15548627.2016.1187366
16. Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci (2018) 22(12):3826–36. doi: 10.26355/eurrev_201806_15267
17. Lill R, Freibert SA. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu Rev Biochem (2020) 89:471–99. doi: 10.1146/annurev-biochem-013118-111540
18. Crichton RR, Dexter DT, Ward RJ. Brain iron metabolism and its perturbation in neurological diseases. J Neural Transm (Vienna) (2011) 118(3):301–14. doi: 10.1007/s00702-010-0470-z
19. Mandel S, Grunblatt E, Riederer P. Iron in brain function and neurodegenerative disorders. Editorial J Neural Transm (Vienna) (2011) 118(3):299–300. doi: 10.1007/s00702-011-0618-5
20. Rouault TA, Cooperman S. Brain iron metabolism. Semin Pediatr Neurol (2006) 13(3):142–8. doi: 10.1016/j.spen.2006.08.002
21. Xu S, Wang Z, Ye J, Mei S, Zhang J. Identification of iron metabolism-related genes as prognostic indicators for lower-grade glioma. Front Oncol (2021) 11:729103. doi: 10.3389/fonc.2021.729103
22. Park KJ, Kim J, Testoff T, Adams J, Poklar M, Zborowski M, et al. Quantitative characterization of the regulation of iron metabolism in glioblastoma stem-like cells using magnetophoresis. Biotechnol Bioeng (2019) 116(7):1644–55. doi: 10.1002/bit.26973
23. Voth B, Nagasawa DT, Pelargos PE, Chung LK, Ung N, Gopen Q, et al. Transferrin receptors and glioblastoma multiforme: current findings and potential for treatment. J Clin Neurosci (2015) 22(7):1071–6. doi: 10.1016/j.jocn.2015.02.002
24. Calzolari A, Larocca LM, Deaglio S, Finisguerra V, Boe A, Raggi C, et al. Transferrin receptor 2 is frequently and highly expressed in glioblastomas. Transl Oncol (2010) 3(2):123–34. doi: 10.1593/tlo.09274
25. Recht L, Torres CO, Smith TW, Raso V, Griffin TW. Transferrin receptor in normal and neoplastic brain tissue: implications for brain-tumor immunotherapy. J Neurosurg (1990) 72(6):941–5. doi: 10.3171/jns.1990.72.6.0941
26. Jaksch-Bogensperger H, Spiegl-Kreinecker S, Arosio P, Eckl P, Golaszewski S, Ebner Y, et al. Ferritin in glioblastoma. Br J Cancer (2020) 122(10):1441–4. doi: 10.1038/s41416-020-0808-8
27. Sato Y, Honda Y, Asoh T, Oizumi K, Ohshima Y, Honda E. Cerebrospinal fluid ferritin in glioblastoma: evidence for tumor synthesis. J Neurooncol (1998) 40(1):47–50. doi: 10.1023/a:1006078521790
28. Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, et al. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell (2015) 28(4):441–55. doi: 10.1016/j.ccell.2015.09.002
29. Chirasani SR, Markovic DS, Synowitz M, Eichler SA, Wisniewski P, Kaminska B, et al. Transferrin-receptor-mediated iron accumulation controls proliferation and glutamate release in glioma cells. J Mol Med (Berl) (2009) 87(2):153–67. doi: 10.1007/s00109-008-0414-3
30. Liu J, Gao L, Zhan N, Xu P, Yang J, Yuan F, et al. Hypoxia induced ferritin light chain (ftl) promoted epithelia mesenchymal transition and chemoresistance of glioma. J Exp Clin Cancer Res (2020) 39(1):137. doi: 10.1186/s13046-020-01641-8
31. Legendre C, Garcion E. Iron metabolism: a double-edged sword in the resistance of glioblastoma to therapies. Trends Endocrinol Metab (2015) 26(6):322–31. doi: 10.1016/j.tem.2015.03.008
32. Szymonik J, Wala K, Gornicki T, Saczko J, Pencakowski B, Kulbacka J. The impact of iron chelators on the biology of cancer stem cells. Int J Mol Sci (2021) 23(1):89. doi: 10.3390/ijms23010089
33. Taga T, Tabu K. Glioma progression and recurrence involving maintenance and expansion strategies of glioma stem cells by organizing self-advantageous niche microenvironments. Inflammation Regener (2020) 40:33. doi: 10.1186/s41232-020-00142-7
34. Boonnoy P, Karttunen M, Wong-Ekkabut J. Alpha-tocopherol inhibits pore formation in oxidized bilayers. Phys Chem Chem Phys (2017) 19(8):5699–704. doi: 10.1039/c6cp08051k
35. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic pes navigate cells to ferroptosis. Nat Chem Biol (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
36. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. Acsl4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
37. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA (2016) 113(34):E4966–75. doi: 10.1073/pnas.1603244113
38. Brownsey RW, Boone AN, Elliott JE, Kulpa JE, Lee WM. Regulation of acetyl-coa carboxylase. Biochem Soc Trans (2006) 34(Pt 2):223–7. doi: 10.1042/bst20060223
39. Guo D, Bell EH, Chakravarti A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol (2013) 2(3):289–99. doi: 10.2217/cns.13.20
40. Wu W, Wu Y, Mayer K, von Rosenstiel C, Schecker J, Baur S, et al. Lipid peroxidation plays an important role in chemotherapeutic effects of temozolomide and the development of therapy resistance in human glioblastoma. Transl Oncol (2020) 13(3):100748. doi: 10.1016/j.tranon.2020.100748
41. Gopal K, Grossi E, Paoletti P, Usardi M. Lipid composition of human intracranial tumors: a biochemical study. Acta Neurochir (Wien) (1963) 11:333–47. doi: 10.1007/bf01402012
42. Liu Y, Xu Z, Jin T, Xu K, Liu M, Xu H. Ferroptosis in low-grade glioma: a new marker for diagnosis and prognosis. Med Sci Monit (2020) 26:e921947. doi: 10.12659/MSM.921947
43. Lu SC. Glutathione synthesis. Biochim Biophys Acta (2013) 1830(5):3143–53. doi: 10.1016/j.bbagen.2012.09.008
44. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of gsh and gpx4. Free Radic Biol Med (2020) 152:175–85. doi: 10.1016/j.freeradbiomed.2020.02.027
45. Stipanuk MH, Ueki I. Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis (2011) 34(1):17–32. doi: 10.1007/s10545-009-9006-9
46. McBean GJ. The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids (2012) 42(1):199–205. doi: 10.1007/s00726-011-0864-8
47. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by gpx4 is required to prevent hydroperoxide-induced ferroptosis. Cell (2018) 172(3):409–22 e21. doi: 10.1016/j.cell.2017.11.048
48. Xie X, Chen M, Zhu A. Molecular characterization and functional analysis of two phospholipid hydroperoxide isoforms from larimichthys crocea under vibrio parahaemolyticus challenge. Fish Shellfish Immunol (2018) 78:259–69. doi: 10.1016/j.fsi.2018.04.052
49. Nakamura H, Takada K. Reactive oxygen species in cancer: current findings and future directions. Cancer Sci (2021) 112(10):3945–52. doi: 10.1111/cas.15068
50. Shaw P, Kumar N, Privat-Maldonado A, Smits E, Bogaerts A. Cold atmospheric plasma increases temozolomide sensitivity of three-dimensional glioblastoma spheroids via oxidative stress-mediated dna damage. Cancers (Basel) (2021) 13(8):1780. doi: 10.3390/cancers13081780
51. Polewski MD, Reveron-Thornton RF, Cherryholmes GA, Marinov GK, Cassady K, Aboody KS. Increased expression of system xc- in glioblastoma confers an altered metabolic state and temozolomide resistance. Mol Cancer Res (2016) 14(12):1229–42. doi: 10.1158/1541-7786.Mcr-16-0028
52. Takeuchi S, Wada K, Toyooka T, Shinomiya N, Shimazaki H, Nakanishi K, et al. Increased xct expression correlates with tumor invasion and outcome in patients with glioblastomas. Neurosurgery (2013) 72(1):33–41. doi: 10.1227/NEU.0b013e318276b2de
53. Robert SM, Buckingham SC, Campbell SL, Robel S, Holt KT, Ogunrinu-Babarinde T, et al. Slc7a11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci Transl Med (2015) 7(289):289ra86. doi: 10.1126/scitranslmed.aaa8103
54. Sorensen MF, Heimisdottir SB, Sorensen MD, Mellegaard CS, Wohlleben H, Kristensen BW, et al. High expression of cystine-glutamate antiporter xct (slc7a11) is an independent biomarker for epileptic seizures at diagnosis in glioma. J Neurooncol (2018) 138(1):49–53. doi: 10.1007/s11060-018-2785-9
55. Long Y, Tao H, Karachi A, Grippin AJ, Jin L, Chang YE, et al. Dysregulation of glutamate transport enhances treg function that promotes vegf blockade resistance in glioblastoma. Cancer Res (2020) 80(3):499–509. doi: 10.1158/0008-5472.CAN-19-1577
56. Goji T, Takahara K, Negishi M, Katoh H. Cystine uptake through the cystine/glutamate antiporter xct triggers glioblastoma cell death under glucose deprivation. J Biol Chem (2017) 292(48):19721–32. doi: 10.1074/jbc.M117.814392
57. Yamaguchi I, Yoshimura SH, Katoh H. High cell density increases glioblastoma cell viability under glucose deprivation via degradation of the cystine/glutamate transporter xct (slc7a11). J Biol Chem (2020) 295(20):6936–45. doi: 10.1074/jbc.RA119.012213
58. Yamamoto M, Teramoto K, Katoh H. Epidermal growth factor promotes glioblastoma cell death under glucose deprivation via upregulation of xct (slc7a11). Cell Signal (2021) 78:109874. doi: 10.1016/j.cellsig.2020.109874
59. Polewski MD, Reveron-Thornton RF, Cherryholmes GA, Marinov GK, Aboody KS. Slc7a11 overexpression in glioblastoma is associated with increased cancer stem cell-like properties. Stem Cells Dev (2017) 26(17):1236–46. doi: 10.1089/scd.2017.0123
60. Xiong Y, Zhang Y, Xiong S, Williams-Villalobo AE. A glance of P53 functions in brain development, neural stem cells, and brain cancer. Biol (Basel) (2020) 9(9):285. doi: 10.3390/biology9090285
61. Guo CF, Zhuang Y, Chen Y, Chen S, Peng H, Zhou S. Significance of tumor protein p53 mutation in cellular process and drug selection in brain lower grade (who grades ii and iii) glioma. biomark Med (2020) 14(12):1139–50. doi: 10.2217/bmm-2020-0331
62. Chen Q, Wang W, Chen S, Chen X, Lin Y. Mir-29a sensitizes the response of glioma cells to temozolomide by modulating the p53/mdm2 feedback loop. Cell Mol Biol Lett (2021) 26(1):21. doi: 10.1186/s11658-021-00266-9
63. Ji H, Wang W, Li X, Han X, Zhang X, Wang J, et al. P53: A double-edged sword in tumor ferroptosis. Pharmacol Res (2022) 177:106013. doi: 10.1016/j.phrs.2021.106013
64. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (Gls2), a P53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U.S.A. (2010) 107(16):7461–6. doi: 10.1073/pnas.1002459107
65. Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of sat1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U.S.A. (2016) 113(44):E6806–E12. doi: 10.1073/pnas.1607152113
66. Venkatesh D, O’Brien NA, Zandkarimi F, Tong DR, Stokes ME, Dunn DE, et al. Mdm2 and mdmx promote ferroptosis by pparalpha-mediated lipid remodeling. Genes Dev (2020) 34(7-8):526–43. doi: 10.1101/gad.334219.119
67. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking dpp4 activity. Cell Rep (2017) 20(7):1692–704. doi: 10.1016/j.celrep.2017.07.055
68. Tarangelo A, Rodencal J, Kim JT, Magtanong L, Long JZ, Dixon SJ. Nucleotide biosynthesis links glutathione metabolism to ferroptosis sensitivity. Life Sci Alliance (2022) 5(4):e202101157. doi: 10.26508/lsa.202101157
69. Kuganesan N, Dlamini S, Tillekeratne LMV, Taylor WR. Tumor suppressor p53 promotes ferroptosis in oxidative stress conditions independent of modulation of ferroptosis by p21, cdks, rb, and e2f. J Biol Chem (2021) 297(6):101365. doi: 10.1016/j.jbc.2021.101365
70. Gnanapradeepan K, Basu S, Barnoud T, Budina-Kolomets A, Kung CP, Murphy ME. The P53 tumor suppressor in the control of metabolism and ferroptosis. Front Endocrinol (Lausanne) (2018) 9:124. doi: 10.3389/fendo.2018.00124
71. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U.S.A. (2003) 100(25):15077–82. doi: 10.1073/pnas.2436255100
72. Kang R, Zeh HJ, Lotze MT, Tang D. The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ (2011) 18(4):571–80. doi: 10.1038/cdd.2010.191
73. Ding X, Nie Z, She Z, Bai X, Yang Q, Wang F, et al. The regulation of ros- and becn1-mediated autophagy by human telomerase reverse transcriptase in glioblastoma. Oxid Med Cell Longev (2021) 2021:6636510. doi: 10.1155/2021/6636510
74. Cj P, Hv E, Vijayakurup V, RM G, Nair S, Gopala S. High Lc3/beclin expression correlates with poor survival in glioma: a definitive role for autophagy as evidenced by in vitro autophagic flux. Pathol Oncol Res (2019) 25(1):137–48. doi: 10.1007/s12253-017-0310-7
75. Kang R, Zhu S, Zeh HJ, Klionsky DJ, Tang D. Becn1 is a new driver of ferroptosis. Autophagy (2018) 14(12):2173–5. doi: 10.1080/15548627.2018.1513758
76. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. Ampk-mediated becn1 phosphorylation promotes ferroptosis by directly blocking system xc(-) activity. Curr Biol (2018) 28(15):2388–99.e5. doi: 10.1016/j.cub.2018.05.094
77. Lessard F, Saint-Germain E, Mignacca L, Ferbeyre G. Socs1: phosphorylation, dimerization and tumor suppression. Oncoscience (2019) 6(11-12):386–9. doi: 10.18632/oncoscience.495
78. Saint-Germain E, Mignacca L, Vernier M, Bobbala D, Ilangumaran S, Ferbeyre G. Socs1 regulates senescence and ferroptosis by modulating the expression of p53 target genes. Aging (Albany NY) (2017) 9(10):2137–62. doi: 10.18632/aging.101306
79. Liang T, Chen J, Xu G, Zhang Z, Xue J, Zeng H, et al. Ferroptosis-related gene socs1, a marker for tuberculosis diagnosis and treatment, involves in macrophage polarization and facilitates bone destruction in tuberculosis. Tuberculosis (Edinb) (2022) 132:102140. doi: 10.1016/j.tube.2021.102140
80. Zhou F, Chen B. Prognostic significance of ferroptosis-related genes and their methylation in aml. Hematology (2021) 26(1):919–30. doi: 10.1080/16078454.2021.1996055
81. Li Q, Jin Y, Shen Z, Liu H, Shen Y, Wu Z. Construction of a ferroptosis-related gene signature for head and neck squamous cell carcinoma prognosis prediction. Int J Gen Med (2021) 14:10117–29. doi: 10.2147/IJGM.S343233
82. Dai L, Li Z, Liang W, Hu W, Zhou S, Yang Z, et al. Socs proteins and their roles in the development of glioblastoma. Oncol Lett (2022) 23(1):5. doi: 10.3892/ol.2021.13123
83. Ventero MP, Fuentes-Baile M, Quereda C, Perez-Valeciano E, Alenda C, Garcia-Morales P, et al. Radiotherapy resistance acquisition in glioblastoma. Role Socs1 Socs3 PloS One (2019) 14(2):e0212581. doi: 10.1371/journal.pone.0212581
84. Zhou H, Miki R, Eeva M, Fike FM, Seligson D, Yang L, et al. Reciprocal regulation of socs 1 and socs3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin Cancer Res (2007) 13(8):2344–53. doi: 10.1158/1078-0432.CCR-06-2303
85. Sheikh T, Sen E. P53 affects epigenetic signature on socs1 promoter in response to tlr4 inhibition. Cytokine (2021) 140:155418. doi: 10.1016/j.cyto.2020.155418
86. Du J, Zhou Y, Li Y, Xia J, Chen Y, Chen S, et al. Identification of frataxin as a regulator of ferroptosis. Redox Biol (2020) 32:101483. doi: 10.1016/j.redox.2020.101483
87. Patra S, Barondeau DP. Mechanism of activation of the human cysteine desulfurase complex by frataxin. Proc Natl Acad Sci U.S.A. (2019) 116(39):19421–30. doi: 10.1073/pnas.1909535116
88. Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. Nfs1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature (2017) 551(7682):639–43. doi: 10.1038/nature24637
89. Kirches E, Andrae N, Hoefer A, Kehler B, Zarse K, Leverkus M, et al. Dual role of the mitochondrial protein frataxin in astrocytic tumors. Lab Invest (2011) 91(12):1766–76. doi: 10.1038/labinvest.2011.130
90. Zhang DD. Mechanistic studies of the nrf2-keap1 signaling pathway. Drug Metab Rev (2006) 38(4):769–89. doi: 10.1080/03602530600971974
91. Ji XJ, Chen SH, Zhu L, Pan H, Zhou Y, Li W, et al. Knockdown of nf-E2-related factor 2 inhibits the proliferation and growth of u251mg human glioma cells in a mouse xenograft model. Oncol Rep (2013) 30(1):157–64. doi: 10.3892/or.2013.2476
92. Zhang Y, Kong Y, Ma Y, Ni S, Wikerholmen T, Xi K, et al. Loss of copz1 induces ncoa4 mediated autophagy and ferroptosis in glioblastoma cell lines. Oncogene (2021) 40(8):1425–39. doi: 10.1038/s41388-020-01622-3
93. Dai X, Wang D, Zhang J. Programmed cell death, redox imbalance, and cancer therapeutics. Apoptosis (2021) 26(7-8):385–414. doi: 10.1007/s10495-021-01682-0
94. Krakstad C, Chekenya M. Survival signalling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Mol Cancer (2010) 9:135. doi: 10.1186/1476-4598-9-135
95. Olivier C, Oliver L, Lalier L, Vallette FM. Drug s. Resistance in glioblastoma: the two faces of oxidative stres.Front Mol Biosci (2020) 7:620677. doi: 10.3389/fmolb.2020.620677
96. Zhang Y, Dube C, Gibert M Jr., Cruickshanks N, Wang B, Coughlan M, et al. The P53 pathway in glioblastoma. Cancers (Basel) (2018) 10(9):297. doi: 10.3390/cancers10090297
97. Mollazadeh H, Mohtashami E, Mousavi SH, Soukhtanloo M, Vahedi MM, Hosseini A, et al. Deciphering the role of glutamate signaling in glioblastoma multiforme: current therapeutic modalities and future directions. Curr Pharm Des (2020) 26(37):4777–88. doi: 10.2174/1381612826666200603132456
98. Li S, He Y, Chen K, Sun J, Zhang L, He Y, et al. Rsl3 drives ferroptosis through nf-kappab pathway activation and gpx4 depletion in glioblastoma. Oxid Med Cell Longev (2021) 2021:2915019. doi: 10.1155/2021/2915019
99. Uddin MS, Kabir MT, Mamun AA, Sarwar MS, Nasrin F, Emran TB, et al. Natural small molecules targeting nf-kappab signaling in glioblastoma. Front Pharmacol (2021) 12:703761. doi: 10.3389/fphar.2021.703761
100. Xia Y, Shen S, Verma IM. Nf-kappab, an active player in human cancers. Cancer Immunol Res (2014) 2(9):823–30. doi: 10.1158/2326-6066.CIR-14-0112
101. Chen Q, Wang W, Wu Z, Chen S, Chen X, Zhuang S, et al. Over-expression of lncrna tmem161b-as1 promotes the malignant biological behavior of glioma cells and the resistance to temozolomide via up-regulating the expression of multiple ferroptosis-related genes by sponging hsa-mir-27a-3p. Cell Death Discovery (2021) 7(1):311. doi: 10.1038/s41420-021-00709-4
102. Wu H, Wang F, Ta N, Zhang T, Gao W. The multifaceted regulation of mitochondria in ferroptosis. Life (Basel) (2021) 11(3):222. doi: 10.3390/life11030222
103. Griguer CE, Oliva CR. Bioenergetics pathways and therapeutic resistance in gliomas: emerging role of mitochondria. Curr Pharm Des (2011) 17(23):2421–7. doi: 10.2174/138161211797249251
104. Lefranc F, Kiss R. Autophagy, the trojan horse to combat glioblastomas. Neurosurg Focus (2006) 20(4):E7. doi: 10.3171/foc.2006.20.4.4
105. Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ (2019) 26(4):605–16. doi: 10.1038/s41418-018-0252-y
106. Paskeh MDA, Entezari M, Clark C, Zabolian A, Ranjbar E, Farahani MV, et al. Targeted regulation of autophagy using nanoparticles: new insight into cancer therapy. Biochim Biophys Acta Mol Basis Dis (2022) 1868(3):166326. doi: 10.1016/j.bbadis.2021.166326
107. Escamilla-Ramirez A, Castillo-Rodriguez RA, Zavala-Vega S, Jimenez-Farfan D, Anaya-Rubio I, Briseno E, et al. Autophagy as a potential therapy for malignant glioma. Pharm (Basel) (2020) 13(7):156. doi: 10.3390/ph13070156
108. Buccarelli M, Marconi M, Pacioni S, De Pascalis I, D’Alessandris QG, Martini M, et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis (2018) 9(8):841. doi: 10.1038/s41419-018-0864-7
109. Greaves D, Calle Y. Epithelial mesenchymal transition (emt) and associated invasive adhesions in solid and haematological tumours. Cells (2022) 11(4):649. doi: 10.3390/cells11040649
110. Milton AV, Konrad DB. Epithelial-mesenchymal transition and h(2)o(2) signaling - a driver of disease progression and a vulnerability in cancers. Biol Chem (2022) 403(4):377–90. doi: 10.1515/hsz-2021-0341
111. Colella B, Faienza F, Di Bartolomeo S. Emt regulation by autophagy: A new perspective in glioblastoma biology. Cancers (Basel) (2019) 11(3):312. doi: 10.3390/cancers11030312
112. Huangfu M, Wei R, Wang J, Qin J, Yu D, Guan X, et al. Osthole induces necroptosis via ros overproduction in glioma cells. FEBS Open Bio (2021) 11(2):456–67. doi: 10.1002/2211-5463.13069
113. Park MY, Ha SE, Vetrivel P, Kim HH, Bhosale PB, Abusaliya A, et al. Differences of key proteins between apoptosis and necroptosis. BioMed Res Int (2021) 2021:3420168. doi: 10.1155/2021/3420168
114. Zhou Y, Liao J, Mei Z, Liu X, Ge J. Insight into crosstalk between ferroptosis and necroptosis: novel therapeutics in ischemic stroke. Oxid Med Cell Longev (2021) 2021:9991001. doi: 10.1155/2021/9991001
115. Galadari S, Rahman A, Pallichankandy S, Thayyullathil F. Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic Biol Med (2017) 104:144–64. doi: 10.1016/j.freeradbiomed.2017.01.004
116. Li XY, Zhang LY, Li XY, Yang XT, Su LX. A pyroptosis-related gene signature for predicting survival in glioblastoma. Front Oncol (2021) 11:697198. doi: 10.3389/fonc.2021.697198
117. Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: A new frontier in cancer. BioMed Pharmacother (2020) 121:109595. doi: 10.1016/j.biopha.2019.109595
118. Zhou CB, Fang JY. The role of pyroptosis in gastrointestinal cancer and immune responses to intestinal microbial infection. Biochim Biophys Acta Rev Cancer (2019) 1872(1):1–10. doi: 10.1016/j.bbcan.2019.05.001
119. Zhang X, Zhang P, An L, Sun N, Peng L, Tang W, et al. Miltirone induces cell death in hepatocellular carcinoma cell through gsdme-dependent pyroptosis. Acta Pharm Sin B (2020) 10(8):1397–413. doi: 10.1016/j.apsb.2020.06.015
120. Fu XW, Song CQ. Identification and validation of pyroptosis-related gene signature to predict prognosis and reveal immune infiltration in hepatocellular carcinoma. Front Cell Dev Biol (2021) 9:748039. doi: 10.3389/fcell.2021.748039
121. Xu D, Ji Z, Qiang L. Molecular characteristics, clinical implication, and cancer immunity interactions of pyroptosis-related genes in breast cancer. Front Med (Lausanne) (2021) 8:702638. doi: 10.3389/fmed.2021.702638
122. Lin J, Lai X, Liu X, Yan H, Wu C. Pyroptosis in glioblastoma: a crucial regulator of the tumour immune microenvironment and a predictor of prognosis. J Cell Mol Med (2022) 26(5):1579–93. doi: 10.1111/jcmm.17200
123. Yang Z, Shi J, Chen L, Fu C, Shi D, Qu H. Role of pyroptosis and ferroptosis in the progression of atherosclerotic plaques. Front Cell Dev Biol (2022) 10:811196. doi: 10.3389/fcell.2022.811196
124. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin d-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe (2018) 24(1):97–108 e4. doi: 10.1016/j.chom.2018.05.009
125. Hwang SY, Yoo BC, Jung JW, Oh ES, Hwang JS, Shin JA, et al. Induction of glioma apoptosis by microglia-secreted molecules: the role of nitric oxide and cathepsin b. Biochim Biophys Acta (2009) 1793(11):1656–68. doi: 10.1016/j.bbamcr.2009.08.011
126. Lam-Himlin D, Espey MG, Perry G, Smith MA, Castellani RJ. Malignant glioma progression and nitric oxide. Neurochem Int (2006) 49(8):764–8. doi: 10.1016/j.neuint.2006.07.001
127. He Q, Qu M, Xu C, Shi W, Hussain M, Jin G, et al. The emerging roles of nitric oxide in ferroptosis and pyroptosis of tumor cells. Life Sci (2022) 290:120257. doi: 10.1016/j.lfs.2021.120257
128. Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol (2020) 13(1):110. doi: 10.1186/s13045-020-00946-7
129. Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget (2016) 7(24):36021–33. doi: 10.18632/oncotarget.8651
130. Hua C, Wang X, Liang S, Chen X, Li C, You G, et al. Bnip3 contributes to silibinin-induced dna double strand breaks in glioma cells via inhibition of mtor. Biochem Biophys Res Commun (2022) 589:1–8. doi: 10.1016/j.bbrc.2021.11.110
131. Chen TC, Chuang JY, Ko CY, Kao TJ, Yang PY, Yu CH, et al. Ar ubiquitination induced by the curcumin analog suppresses growth of temozolomide-resistant glioblastoma through disrupting gpx4-mediated redox homeostasis. Redox Biol (2020) 30:101413. doi: 10.1016/j.redox.2019.101413
132. Ignarro RS, Facchini G, Vieira AS, De Melo DR, Lopes-Cendes I, Castilho RF, et al. Sulfasalazine intensifies temozolomide cytotoxicity in human glioblastoma cells. Mol Cell Biochem (2016) 418(1-2):167–78. doi: 10.1007/s11010-016-2742-x
133. Villalpando-Rodriguez GE, Blankstein AR, Konzelman C, Gibson SB. Lysosomal destabilizing drug siramesine and the dual tyrosine kinase inhibitor lapatinib induce a synergistic ferroptosis through reduced heme oxygenase-1 (ho-1) levels. Oxid Med Cell Longev (2019) 2019:9561281. doi: 10.1155/2019/9561281
134. Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol (2020) 15(2):469–84. doi: 10.1021/acschembio.9b00939
135. Sleire L, Skeie BS, Netland IA, Førde HE, Dodoo E, Selheim F, et al. Drug repurposing: Sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system xc-, leading to glutathione depletion. Oncogene (2015) 34(49):5951–9. doi: 10.1038/onc.2015.60
136. Poonan P, Agoni C, Ibrahim MAA, Soliman MES. Glioma-targeted therapeutics: computer-aided drug design prospective. Protein J (2021) 40(5):601–55. doi: 10.1007/s10930-021-10021-w
137. Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev (2014) 23(10):1985–96. doi: 10.1158/1055-9965.EPI-14-0275
138. Bredel M. Anticancer drug resistance in primary human brain tumors. Brain Res Brain Res Rev (2001) 35(2):161–204. doi: 10.1016/s0165-0173(01)00045-5
139. Gomez-Zepeda D, Taghi M, Scherrmann JM, Decleves X, Menet MC. Abc transporters at the blood-brain interfaces, their study models, and drug delivery implications in gliomas. Pharmaceutics (2019) 12(1):20. doi: 10.3390/pharmaceutics12010020
140. Da Ros M, De Gregorio V, Iorio AL, Giunti L, Guidi M, de Martino M, et al. Glioblastoma chemoresistance: the double play by microenvironment and blood-brain barrier. Int J Mol Sci (2018) 19(10):2879. doi: 10.3390/ijms19102879
141. Ortiz R, Perazzoli G, Cabeza L, Jimenez-Luna C, Luque R, Prados J, et al. Temozolomide: An updated overview of resistance mechanisms, nanotechnology advances and clinical applications. Curr Neuropharmacol (2021) 19(4):513–37. doi: 10.2174/1570159X18666200626204005
142. Natsume A, Hirano M, Ranjit M, Aoki K, Wakabayashi T. Aberrant transcriptional regulation of super-enhancers by ret finger protein-histone deacetylase 1 complex in glioblastoma: chemoresistance to temozolomide. Neurol Med Chir (Tokyo) (2019) 59(8):293–8. doi: 10.2176/nmc.ra.2019-0049
143. Jiapaer S, Furuta T, Tanaka S, Kitabayashi T, Nakada M. Potential strategies overcoming the temozolomide resistance for glioblastoma. Neurol Med Chir (Tokyo) (2018) 58(10):405–21. doi: 10.2176/nmc.ra.2018-0141
144. Butler M, Pongor L, Su YT, Xi L, Raffeld M, Quezado M, et al. Mgmt status as a clinical biomarker in glioblastoma. Trends Cancer (2020) 6(5):380–91. doi: 10.1016/j.trecan.2020.02.010
145. Liu H, Chen C, Zeng J, Zhao Z, Hu Q. Microrna-210-3p is transcriptionally upregulated by hypoxia induction and thus promoting emt and chemoresistance in glioma cells. PloS One (2021) 16(7):e0253522. doi: 10.1371/journal.pone.0253522
146. Deng Y, Zhu H, Xiao L, Liu C, Meng X. Circ_0005198 enhances temozolomide resistance of glioma cells through mir-198/trim14 axis. Aging (Albany NY) (2020) 13(2):2198–211. doi: 10.18632/aging.202234
147. Li C, Feng S, Chen L. Msc-As1 knockdown inhibits cell growth and temozolomide resistance by regulating mir-373-3p/cpeb4 axis in glioma through pi3k/akt pathway. Mol Cell Biochem (2021) 476(2):699–713. doi: 10.1007/s11010-020-03937-x
148. Peng Y, Wu W, Shang Z, Li W, Chen S. Inhibition of lncrna linc00461/mir-216a/aquaporin 4 pathway suppresses cell proliferation, migration, invasion, and chemoresistance in glioma. Open Life Sci (2020) 15(1):532–43. doi: 10.1515/biol-2020-0048
149. Xu JX, Yang Y, Zhang X, Luan XP. Microrna-29b promotes cell sensitivity to temozolomide by targeting stat3 in glioma. Eur Rev Med Pharmacol Sci (2020) 24(4):1922–31. doi: 10.26355/eurrev_202002_20370
150. Li B, Zhao H, Song J, Wang F, Chen M. Linc00174 down-regulation decreases chemoresistance to temozolomide in human glioma cells by regulating mir-138-5p/sox9 axis. Hum Cell (2020) 33(1):159–74. doi: 10.1007/s13577-019-00281-1
151. Zhang X, Ding K, Wang J, Li X, Zhao P. Chemoresistance caused by the microenvironment of glioblastoma and the corresponding solutions. BioMed Pharmacother (2019) 109:39–46. doi: 10.1016/j.biopha.2018.10.063
152. Balca-Silva J, Matias D, Carmo AD, Sarmento-Ribeiro AB, Lopes MC, Moura-Neto V. Cellular and molecular mechanisms of glioblastoma malignancy: implications in resistance and therapeutic strategies. Semin Cancer Biol (2019) 58:130–41. doi: 10.1016/j.semcancer.2018.09.007
153. Han X, Xue X, Zhou H, Zhang G. A molecular view of the radioresistance of gliomas. Oncotarget (2017) 8(59):100931–41. doi: 10.18632/oncotarget.21753
154. Jung E, Osswald M, Ratliff M, Dogan H, Xie R, Weil S, et al. Tumor cell plasticity, heterogeneity, and resistance in crucial microenvironmental niches in glioma. Nat Commun (2021) 12(1):1014. doi: 10.1038/s41467-021-21117-3
155. Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol (2019) 20(9):1100–9. doi: 10.1038/s41590-019-0433-y
156. Schonberg DL, Lubelski D, Miller TE, Rich JN. Brain tumor stem cells: molecular characteristics and their impact on therapy. Mol Aspects Med (2014) 39:82–101. doi: 10.1016/j.mam.2013.06.004
157. Gerard M, Corroyer-Dulmont A, Lesueur P, Collet S, Cherel M, Bourgeois M, et al. Hypoxia imaging and adaptive radiotherapy: a state-of-the-art approach in the management of glioma. Front Med (Lausanne) (2019) 6:117. doi: 10.3389/fmed.2019.00117
158. Mudassar F, Shen H, O’Neill G, Hau E. Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J Exp Clin Cancer Res (2020) 39(1):208. doi: 10.1186/s13046-020-01724-6
159. Medikonda R, Dunn G, Rahman M, Fecci P, Lim M. A review of glioblastoma immunotherapy. J Neurooncol (2021) 151(1):41–53. doi: 10.1007/s11060-020-03448-1
160. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the cns. Nat Immunol (2017) 18(2):123–31. doi: 10.1038/ni.3666
161. Najem H, Khasraw M, Heimberger AB. Immune microenvironment landscape in cns tumors and role in responses to immunotherapy. Cells (2021) 10(8):2032. doi: 10.3390/cells10082032
162. Amoozgar Z, Kloepper J, Ren J, Tay RE, Kazer SW, Kiner E, et al. Targeting treg cells with gitr activation alleviates resistance to immunotherapy in murine glioblastomas. Nat Commun (2021) 12(1):2582. doi: 10.1038/s41467-021-22885-8
163. Di Ianni N, Musio S, Pellegatta S. Altered metabolism in glioblastoma: Myeloid-derived suppressor cell (mdsc) fitness and tumor-infiltrating lymphocyte (til) dysfunction. Int J Mol Sci (2021) 22(9):4460. doi: 10.3390/ijms22094460
164. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov (2017) 7(3):264–76. doi: 10.1158/2159-8290.CD-16-0828
165. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic pd-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
166. Daniel P, Sabri S, Chaddad A, Meehan B, Jean-Claude B, Rak J, et al. Temozolomide induced hypermutation in glioma: evolutionary mechanisms and therapeutic opportunities. Front Oncol (2019) 9:41. doi: 10.3389/fonc.2019.00041
167. Lee SY. Temozolomide resistance in glioblastoma multiforme. Genes Dis (2016) 3(3):198–210. doi: 10.1016/j.gendis.2016.04.007
168. García M, Clopés A, Bruna J, Martínez M, Fort E, Gil M. Critical appraisal of temozolomide formulations in the treatment of primary brain tumors: patient considerations. Cancer Manag Res (2009) 1:137–50. doi: 10.2147/cmr.s5598
169. Zhang L, Wang H. Fty720 inhibits the nrf2/are pathway in human glioblastoma cell lines and sensitizes glioblastoma cells to temozolomide. Pharmacol Rep (2017) 69(6):1186–93. doi: 10.1016/j.pharep.2017.07.003
170. Rocha CR, Kajitani GS, Quinet A, Fortunato RS, Menck CF. Nrf2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget (2016) 7(30):48081–92. doi: 10.18632/oncotarget.10129
171. Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. Atf4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xct-dependent manner. Oncogene (2017) 36(40):5593–608. doi: 10.1038/onc.2017.146
172. Chen D, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. The oxido-metabolic driver atf4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget (2017) 8(31):51164–76. doi: 10.18632/oncotarget.17737
173. Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xct and cystathionine-γ-lyase function. Oncol Rep (2015) 33(3):1465–74. doi: 10.3892/or.2015.3712
174. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to gpx4 inhibition. Nature (2017) 551(7679):247–50. doi: 10.1038/nature24297
175. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol (2019) 12(1):34. doi: 10.1186/s13045-019-0720-y
176. Wei J, Wang Z, Wang W, Liu X, Wan J, Yuan Y, et al. Oxidative stress activated by sorafenib alters the temozolomide sensitivity of human glioma cells through autophagy and jak2/stat3-aif axis. Front Cell Dev Biol (2021) 9:660005. doi: 10.3389/fcell.2021.660005
177. Lee SW, Kim HK, Lee NH, Yi HY, Kim HS, Hong SH, et al. The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells. Cancer Lett (2015) 360(2):195–204. doi: 10.1016/j.canlet.2015.02.012
178. Alexiou GA, Gerogianni P, Vartholomatos E, Kyritsis AP. Deferiprone enhances temozolomide cytotoxicity in glioma cells. Cancer Invest (2016) 34(10):489–95. doi: 10.1080/07357907.2016.1233424
179. Yin H, Zhou Y, Wen C, Zhou C, Zhang W, Hu X, et al. Curcumin sensitizes glioblastoma to temozolomide by simultaneously generating ros and disrupting akt/mtor signaling. Oncol Rep (2014) 32(4):1610–6. doi: 10.3892/or.2014.3342
180. Pantopoulos K. Iron metabolism and the ire/irp regulatory system: an update. Ann N Y Acad Sci (2004) 1012:1–13. doi: 10.1196/annals.1306.001
181. Jaffray DA. Image-guided radiotherapy: from current concept to future perspectives. Nat Rev Clin Oncol (2012) 9(12):688–99. doi: 10.1038/nrclinonc.2012.194
182. Sun T, Wu H, Li Y, Huang Y, Yao L, Chen X, et al. Targeting transferrin receptor delivery of temozolomide for a potential glioma stem cell-mediated therapy. Oncotarget (2017) 8(43):74451–65. doi: 10.18632/oncotarget.20165
183. Sontheimer H, Bridges RJ. Sulfasalazine for brain cancer fits. Expert Opin Investig Drugs (2012) 21(5):575–8. doi: 10.1517/13543784.2012.670634
184. Garcia CG, Kahn SA, Geraldo LHM, Romano I, Domith I, Silva D, et al. Combination therapy with sulfasalazine and valproic acid promotes human glioblastoma cell death through imbalance of the intracellular oxidative response. Mol Neurobiol (2018) 55(8):6816–33. doi: 10.1007/s12035-018-0895-1
185. Patel D, Kharkar PS, Gandhi NS, Kaur E, Dutt S, Nandave M. Novel analogs of sulfasalazine as system xc (-) antiporter inhibitors: insights from the molecular modeling studies. Drug Dev Res (2019) 80(6):758–77. doi: 10.1002/ddr.21557
186. Wang C, He C, Lu S, Wang X, Wang L, Liang S, et al. Autophagy activated by silibinin contributes to glioma cell death via induction of oxidative stress-mediated bnip3-dependent nuclear translocation of aif. Cell Death Dis (2020) 11(8):630. doi: 10.1038/s41419-020-02866-3
187. Baidoo KE, Yong K, Brechbiel MW. Molecular pathways: targeted alpha-particle radiation therapy. Clin Cancer Res (2013) 19(3):530–7. doi: 10.1158/1078-0432.CCR-12-0298
188. Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett (2012) 327(1-2):48–60. doi: 10.1016/j.canlet.2011.12.012
189. Adjemian S, Oltean T, Martens S, Wiernicki B, Goossens V, Vanden Berghe T, et al. Ionizing radiation results in a mixture of cellular outcomes including mitotic catastrophe, senescence, methuosis, and iron-dependent cell death. Cell Death Dis (2020) 11(11):1003. doi: 10.1038/s41419-020-03209-y
190. Lei G, Mao C, Yan Y, Zhuang L, Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell (2021) 12(11):836–57. doi: 10.1007/s13238-021-00841-y
191. Huang RX, Zhou PK. DNA Damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther (2020) 5(1):60. doi: 10.1038/s41392-020-0150-x
192. Ivanov SD, Semenov AL, Kovan’ko EG, Yamshanov VA. Effects of iron ions and iron chelation on the efficiency of experimental radiotherapy of animals with gliomas. Bull Exp Biol Med (2015) 158(6):800–3. doi: 10.1007/s10517-015-2865-1
193. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res (2020) 30(2):146–62. doi: 10.1038/s41422-019-0263-3
194. Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of slc7a11. Cancer Discovery (2019) 9(12):1673–85. doi: 10.1158/2159-8290.CD-19-0338
195. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. Cd8(+) t cells regulate tumour ferroptosis during cancer immunotherapy. Nature (2019) 569(7755):270–4. doi: 10.1038/s41586-019-1170-y
196. Wang H, Xu T, Huang Q, Jin W, Chen J. Immunotherapy for malignant glioma: current status and future directions. Trends Pharmacol Sci (2020) 41(2):123–38. doi: 10.1016/j.tips.2019.12.003
197. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused egfrviii-directed car t cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
198. Deng S, Zheng Y, Mo Y, Xu X, Li Y, Zhang Y, et al. Ferroptosis suppressive genes correlate with immunosuppression in glioblastoma. World Neurosurg (2021) 152:e436–e48. doi: 10.1016/j.wneu.2021.05.098
199. Liu T, Zhu C, Chen X, Guan G, Zou C, Shen S, et al. Ferroptosis, as the most enriched programmed cell death process in glioma, induces immunosuppression and immunotherapy resistance. Neuro Oncol (2022) 24(7):1113–25. doi: 10.1093/neuonc/noac033
200. Zottel A, Videtic Paska A, Jovcevska I. Nanotechnology meets oncology: Nanomaterials in brain cancer research, diagnosis and therapy. Materials (Basel) (2019) 12(10) 12(10):1588. doi: 10.3390/ma12101588
201. Saenz del Burgo L, Hernandez RM, Orive G, Pedraz JL. Nanotherapeutic approaches for brain cancer management. Nanomedicine (2014) 10(5):905–19. doi: 10.1016/j.nano.2013.10.001
202. Iturrioz-Rodriguez N, Bertorelli R, Ciofani G. Lipid-based nanocarriers for the treatment of glioblastoma. Adv Nanobiomed Res (2021) 1(2):2000054. doi: 10.1002/anbr.202000054
203. Glaser T, Han I, Wu L, Zeng X. Targeted nanotechnology in glioblastoma multiforme. Front Pharmacol (2017) 8:166. doi: 10.3389/fphar.2017.00166
204. Wei Y, Wang Z, Yang J, Xu R, Deng H, Ma S, et al. Reactive oxygen species/photothermal therapy dual-triggered biomimetic gold nanocages nanoplatform for combination cancer therapy via ferroptosis and tumor-associated macrophage repolarization mechanism. J Colloid Interface Sci (2022) 606(Pt 2):1950–65. doi: 10.1016/j.jcis.2021.09.160
205. Niu W, Xiao Q, Wang X, Zhu J, Li J, Liang X, et al. A biomimetic drug delivery system by integrating grapefruit extracellular vesicles and doxorubicin-loaded heparin-based nanoparticles for glioma therapy. Nano Lett (2021) 21(3):1484–92. doi: 10.1021/acs.nanolett.0c04753
Keywords: ferroptosis, glioma, programmed cell death, iron metabolism, therapy resistance
Citation: Shi J, Yang N, Han M and Qiu C (2022) Emerging roles of ferroptosis in glioma. Front. Oncol. 12:993316. doi: 10.3389/fonc.2022.993316
Received: 13 July 2022; Accepted: 28 July 2022;
Published: 22 August 2022.
Edited by:
Balkrishna Chaube, Yale University, United StatesReviewed by:
Suyasha Roy, National Institutes of Health (NIH), United StatesCopyright © 2022 Shi, Yang, Han and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chen Qiu, cmFjaGVscWl1QHNkdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.