 Mengmeng Dong
Mengmeng Dong Lidong Cao
Lidong Cao Ranji Cui
Ranji Cui Yingjun Xie2,3*
Yingjun Xie2,3*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 13 December 2022
Sec. Gastrointestinal Cancers: Hepato Pancreatic Biliary Cancers
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.992927
This article is part of the Research TopicHeterogeneity in Pancreatic Cancer - Therapeutic Obstruction or OpportunityView all 4 articles
Pancreatic cancer is a kind of aggressive tumor famous for its lethality and intractability, and pancreatic ductal adenocarcinoma is the most common type. Patients with pancreatic cancer often suffer a rapid loss of weight and abdominal neuropathic pain in their early stages and then go through cachexia in the advanced stage. These features of patients are considered to be related to metabolic reprogramming of pancreatic cancer and abundant nerve innervation responsible for the pain. With increasing literature certifying the relationship between nerves and pancreatic ductal adenocarcinoma (PDAC), more evidence point out that innervation’s role is not limited to neuropathic pain but explore its anti/pro-tumor functions in PDAC, especially the neural–metabolic crosstalks. This review aims to unite pancreatic cancer’s innervation and metabolic rearrangements with terminated published articles. Hopefully, this article could explore the pathogenesis of PDAC and further promote promising detecting or therapeutic measurements for PDAC according to the lavish innervation in PDAC.
Cancer is the most urgent public health problem worldwide, and it was estimated that 1,806,590 people will be diagnosed as cancer patients and 606,502 people will die of this deadly disease in America in 2020. Among these numbers, 57,600 cancer cases and 47,050 cancer deaths were attributed to pancreatic cancer in the United States (1). Moreover, pancreatic cancer has the lowest 5-year overall survival (OS) rate of about 9% to 10% of any other solid tumors. The percentage of incidence and mortality of pancreatic cancer is highest among people aged 65–74 for both sexes (2). The (age-adjusted) incidence and mortality rate of pancreatic cancer was 13.1 per 100,000 persons based on the database from 2013 to 2017 and 11.0 per 100,000 persons during 2014–2018. Statistically, men have a higher incidence than women (14.9 and 11.6 annually per 100,000 persons, respectively) as were the mortality rates (12.7 and 9.6 annually per 100,000 persons, respectively). Depending on the statistical analysis, the age-adjusted rates for new pancreatic cancer cases have been stable from 2008 to 2017; however, age-adjusted death rates have been increasing on average by 0.3% per year from 2009 to 2018 (2, 3). It is predicted that pancreatic cancer will be the second leading cause of cancer-related deaths by 2030 (4). Lately, it has exceeded breast cancer as the third leading cause of cancer death in all ages for both genders and the fourth leading cause of cancer death for males and females, respectively (1, 5).
Pancreatic cancer, with the malignant neoplasm, has many histologic types, beginning with neoplasms originating from ductal and non-ductal cells. The former includes pancreatic ductal adenocarcinoma (PDAC), accounting for approximately 90% of total types and representing typical pancreatic cancer (6). Other PDAC-related carcinomas in neoplasm of ductal origin include adenosquamous carcinoma, osteoclastic giant cell carcinoma, colloid carcinoma, and medullary carcinoma. The last kind of original ductal neoplasms arises in tumoral intraepithelial neoplasms consisting of intraductal papillary mucinous neoplasm (IPMN), intraductal tubulopapillary neoplasm, and mucinous cystic neoplasm with associated invasive carcinoma. Intriguingly, a comment points out that 18% IPMNs and PDAC are found to be co-occurring in the same pancreas, which supports the concept of “field effect” (7), but neither genetically related nor representing a similar oncogenetic development (8). Acinar cell carcinoma, pancreatoblastoma, pancreatic neuroendocrine neoplasm, and solid pseudopapillary neoplasm are the other four kinds belonging to neoplasms of non-ductal origin (9, 10). Known as the most aggressive cancer, PDAC always shows enormous difficulties in diagnosis and therapy. The pancreas is located within the upper abdomen, so PDAC is extremely hard to be detected in its early stage by image measurements and resected by surgery because of the surrounding encased vessels. PDAC shows an aggressive biological nature of invasion and early metastasis. With rapid proliferation and growth limiting nutrition and energy intake, patients often suffer cachexia and pancreatic dysfunction for exocrine and endocrine (11). Moreover, PDAC has a desmoplastic stromal and flexible elasticity, and plasticity exhibits apparent resistance to chemotherapy and radiotherapy and has little target molecule (12, 13). Cigarette smoking, severe obesity (body mass index known as >35) (14), type 2 diabetes mellitus, alcohol consumption, and pancreatitis increase the risk of PDAC, and cigarette smoking is the most common one (15, 16). According to the environmental risk factors listed, genetic alterations play a more significant role in the appearance of PDAC. Among the PDAC patients with a pathogenic germline mutation, BRCA2 (associated with breast and ovarian cancer) and ATM (related to ataxia–telangiectasia) are the top two genes (17, 18). Besides PLAB2 (correlated to breast cancer), BRCA1 (connected to ovarian and breast cancer), and p16/CDKN2A (associated with risk of melanoma and familial atypical multiple mole melanoma), DNA mismatch repair genes like hMSH2, hMLH1, hPMS1, hPMS2, and hMSH6/GTBP (Lynch syndrome), PRSS1 (causing rare inherited hereditary pancreatitis), and LKB1/STK11 (explaining about 80% of Peutz–Jeghers cases) also increased the estimated risk of PDAC (17, 19, 20). Notably, familial PDAC shows a remarkably rising risk of PDAC compared with the general population (21). Based on these altered genes and mutation-related syndromes, there is an excellent opportunity to quantify the risk of PDAC, which is essential for the clinical application of early detection and timely treatment of PDAC and screening for the risk of other malignant diseases (Figure 1).
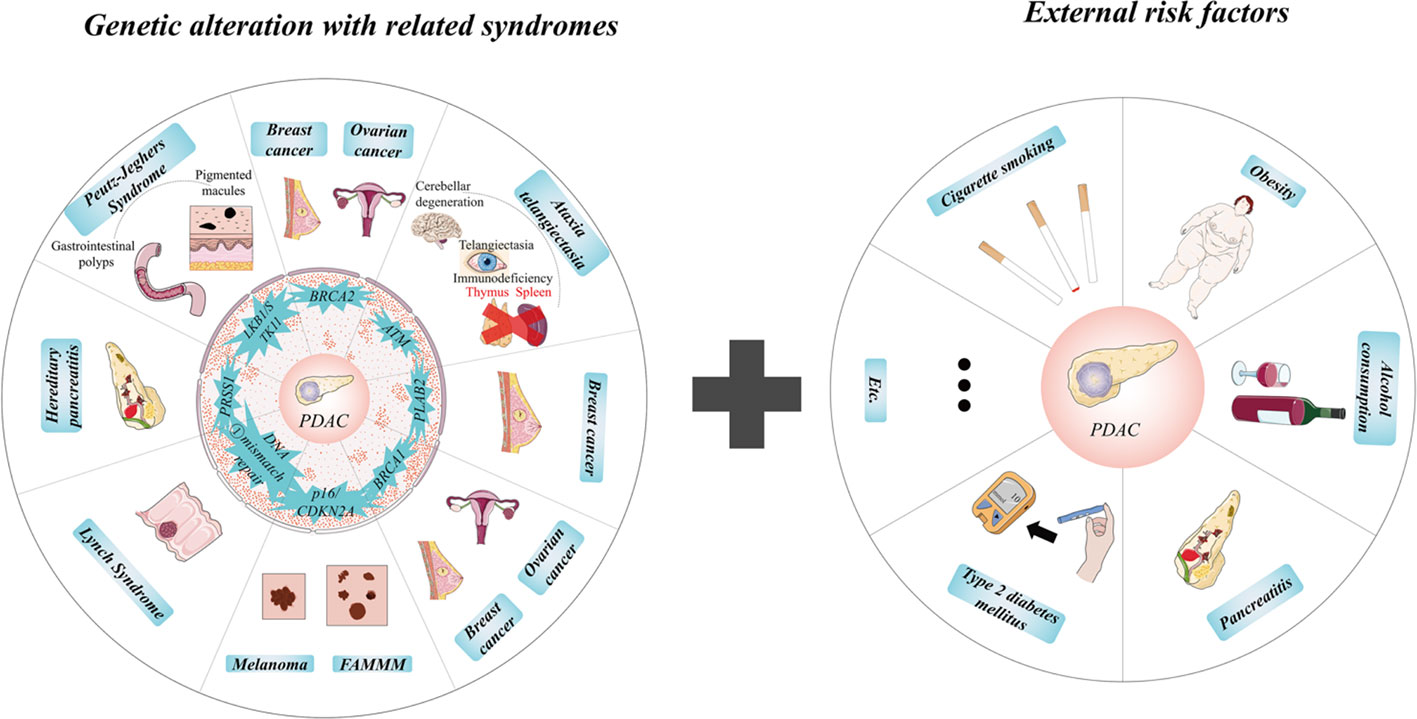
Figure 1 Internal and external risks of pancreatic ductal adenocarcinoma (PDAC). Notably, familial PDAC shows a remarkably rising risk of PDAC compared with the general population. Based on these altered genes and mutation-related syndromes, there is an excellent opportunity to quantify the risk of PDAC, which is essential for the clinical application of early detection and timely treatment of PDAC and screening for the risk of other malignant diseases. ①, DNA mismatch repair genes include hMSH2, hMLH1, hPMS1, hPMS2, and hMSH6/GTBP.
PDAC presents few clinical symptoms before it evolves into an advanced stage; if any, those symptoms often lack specificity for one to make a diagnosis. Generally, these non-specific clinical symptoms include abdominal pain, jaundice, abnormal liver functions, diabetes, dyspepsia, nausea or vomiting, back pain, and weight loss (22). The most frequent feature of PDAC is abdominal pain related to neuropathic pain mainly caused by perineural invasion (PNI), defined as cancer cells diffusing at least 33% of the surrounding nerves, including the epineural, perineural, and endoneurial space of the nerve sheath (23). Invasion of PDAC cells impairs the neural sheath and reprograms neural density, causing neuropathic and inflammatory pain, whereby PNI suggests a metastatic route for PDAC metastasis and neuropathic pain. Interestingly, a study points out that PNI has the highest prevalence in PDAC within variant gastrointestinal malignancies and shows an impactful association with poor PDAC patient outcomes (24). The genetic analysis of PDAC also confirms frequent regulators of axon guidance (25), which is consistent with the correlation between neuronal supports and axonogenesis of PDAC.
PDAC has a desmoplastic and abundantly innervated tumor microenvironment (TME). However, the dense stromal leads to a nutrient-poor environment, rendering PDAC a plastic metabolic remodeling for oxygen and metabolites (26). The metabolic rearrangements of PDAC allow the tumor to survive in the specific environment and, more importantly, increase and overgrow in human bodies. PDAC cells would choose non-classic metabolic pathways (aerobic glycolysis, hexosamine biosynthetic pathways, de novo lipogenesis, etc.) for their energy and nutrient production under some circumstances (12). Accordingly, other cellular components such as pancreatic satellite cells (“reverse Warburg effect”—pancreatic satellite cells promoting metabolites for PDAC cells to produce energy) and neurons (known to release amino acid-derived neurotransmitters like serine via peripheral axons) also provide the metabolic needs for PDAC cells under nutrient-depleted conditions (27, 28).
Organs like the pancreas are innervated by the autonomic nervous system consisting of two parts: the sympathetic nervous system (SNS) and the parasympathetic nervous system (PSNS) (29). Sensory nerves, compose of dorsal root ganglia (DRG), are responsible for receiving all nerve impulses from the pancreas. Sympathetic, sensory, and parasympathetic nerves disperse the whole pancreas and play individual roles (30, 31). Classically, the plexus leaving from the coeliac plexus and then entering the pancreas head innervates the head of the pancreas, and the plexus derived from the splenic plexus innervates the body and the tail of the pancreas (32). It has been proved that not only PDAC cells but also PanIN cells and PDAC patients demonstrate highly increased nerve penetration and neurotropism by KPC in in vivo murine and in vitro co-culture models (33–35). The abundant nerve innervation of the pancreas is the foundation of PNI, besides the precondition for interactions between nerves and PDAC cells (Figure 2).
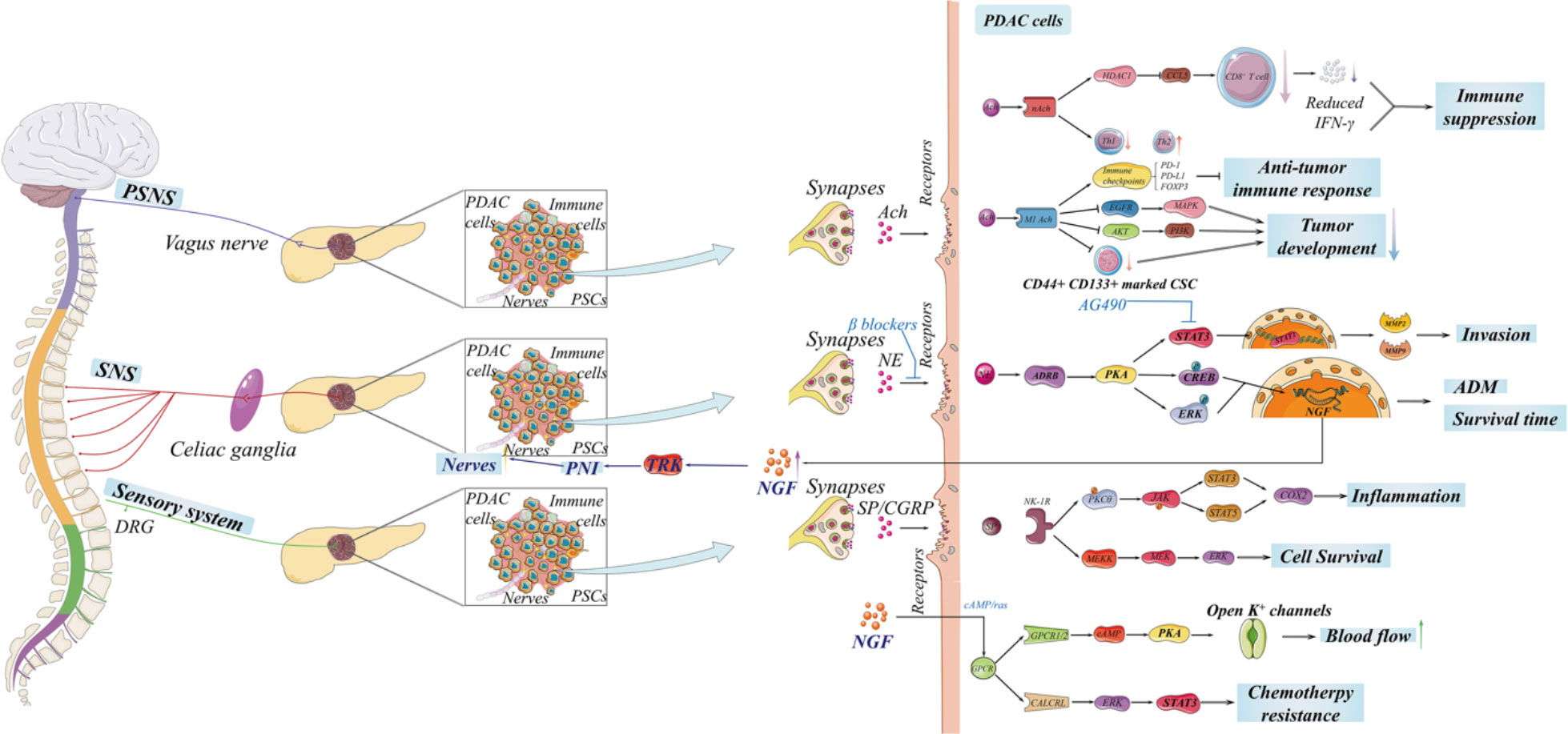
Figure 2 The innervation of the pancreas is complex, including the autonomic nervous system and sensory nerves, which is significant for tumorigenesis and the development of pancreatic ductal adenocarcinoma (PDAC). The sympathetic nervous and sensory systems promote the development of PDAC through neurotransmitters. Notably, neuropathic pain induced by PDAC activates sensory nerves in nerve impulses and then triggers transient receptor potential vanilloid 1 (TRPV1) to secrete substance P and calcitonin gene-related peptide. However, there is an extensive debate on the complicated role of parasympathetic nervous system in PDAC, and the entire signaling network remains to be discovered.
Sympathetic innervation in the pancreas comes from the sympathetic preganglionic neurons from the thoracic 5–10 segments of the spinal cord. They leave as sympathetic ganglia, become splanchnic nerves, and end in celiac ganglia (36). Sympathetic preganglionic neurons release acetylcholine (Ach) as functional neurotransmitters comprising cholinergic neurons. However, sympathetic postganglionic neurons releasing norepinephrine (NE) linking to adrenergic receptors on its downstream tissues as their functional neurotransmitters suggest that sympathetic postganglionic neurons should be classified as noradrenergic neurons (31, 37). It is widely accepted that catecholamines produced by sympathetic nerves as stress molecules could stimulate the proliferation of PDAC. An orthotopic mouse model, non-invasively tracked by in vivo optical imaging, is applied to evaluate the growth and proliferation of PDAC. This model greatly reappears the interlaced association of PDAC cells and microenvironment, and it indicates that pharmacological β-adrenergic is ample to reverse the activated β-adrenergic signaling caused by chronic stress responsible for the development of PDAC. After treating PDAC cells with isoproterenol, invasion-related genes like matrix metalloprotease 2 (MMP2) and MMP9 were assayed. These accelerating expressions collectively suggested that β-adrenergic signaling impacts the growth, progression, and invasion of PDAC in vivo (38). Another in vitro mouse model implanted a DRG nearby a colony of PDAC cells (including MIA PaCa-2 and BxPC-3 cell lines) in Matrigel, whose growth factors are depleted, and then applied with different concentrations of NE for 7 days (39). The model strongly shows that NE is critical in the PNI development of PDAC. Mechanistically, NE activates the STAT3 pathway through β-adrenergic receptors (ADRB) and its downstream PKA. After blocking the effect of STAT3 by phosphorylation inhibitor AG490, the expressions of STAT3 were attenuated, accompanied by the invasion-related protein levels of MMP9 and MMP2 and the secretion of neural growth factors (NGF) induced by NE. The in vitro experiments demonstrate that STAT3 blockade could inhibit NE-induced PDAC growth, migration, and PNI (39). It is well studied that sympathetic innervation impacts tumorigenesis and the growth of PDAC (mainly associated with NE produced by sympathetic postganglionic neurons) and breast cancers (associated with the expression of immune checkpoint molecules) (38–41). Additionally, a recent work aims to determine whether sympathetic neural signaling would affect the progression of PanIN (41). They utilized the genetically modified LSL-Kras+/LSL-G12D;Pdx1- [(KC) mice to simulate murine PanIN. After being treated by daily repeated immobilization, chronic restraint stress accelerated the systemic epinephrine levels and promoted PDAC carcinogenesis in the KC murine models. The common mediator of chronic stress is reported as β2-adrenergic receptor (ADRB2), which was also increasing in the pancreas of KC mice. Then, the LSL-Kras+/LSL-G12D;LSL-Trp53+/R172H;Pdx1-Cre (KPC) model, which could develop an advanced stage of PanIN, is established; these results claimed that ADRB2 blockade tremendously prolonged OS in KPC mice and that ADRB2 signaling is crucial for nerve growth in PDAC carcinogenesis. Mechanistically, adrenergic signaling related to catecholamine, specifically isoproterenol, induces PDAC development through acinar to ductal metaplasia (ADM), which is also described as the first step of PDAC carcinogenesis (42, 43), and promotes the secretion of neurotrophins (NT) through ADRB2 and the PKA/ERK pathways. Moreover, after assessing the expression of several NT, including NGF, brain-derived neurotrophic factor (BDNF), neurotrophic factor 3 (NTF3), NTF4, glial cell line-derived neurotrophic factor (GDNF), and Netrin-1 (encoded by NTN1), NGF demonstrated the highest expression in PDAC cell lines, KC mice, and KPC mice. Significantly, the blockade of the NGF/Trk pathway could inhibit proliferation, PNI, and invasion of PDAC, prolonging the OS for KPC mice (41, 44, 45). That hints us in utilizing β-blocker to suppress adrenergic signaling for highly innervated PDAC patients. Clinically speaking, treating postoperative PDAC patients of stages II and III with non-selective β-blocker reveals attenuated BDNF levels, decreased nerve density, and extended OS (41). In summary, sympathetic neuron-expressed NE promotes carcinogenesis and the development of PDAC while facilitating the secretion of NT, which conversely stimulates innervation in PDAC, followed by increased sympathetic related NE and PDAC growth. There is a feed-forward loop among sympathetic neurons, ADRB2 pathway, PDAC cells, and NT, and this loop could render PDAC sufficient neuroplasticity for innervation, proliferation, and growth. In that case, targeting this specific pathway gives us a novel strategy to treat the neoneurogenesis of PDAC. Coincidentally, this paper testified the possibility of targeting ADRB2 signaling to influence innervation in human PDAC to further improve the clinical outcomes of PDAC patients (41). Sahni and colleagues collected a cohort of PDAC patients who underwent surgical resection for their stage II and III tumors, and they were treated with non-selective β-blockers (NSBB), β-1 selective blocker (SB1B), and no β blockers (NBB). Then, the patients were retrospectively analyzed to compare their OS. Interestingly, patients with NSBB have no significant difference from those with SB1B in OS. However, they had a nearly double OS compared with patients with NBB. The immunohistochemistry analysis also confirms that there were fewer innervation (presented by neuronal marker S-100) (46) and decreased BDNF expressions in the NSBB group than in the NBB and SB1B groups. Notably, stages II and III PDAC patients who undergo surgical resection would benefit from NSBB and improve their clinical outcomes. Nevertheless, PDAC patients can barely benefit from β-blocker (NB) usage before diagnosis for their survival advantage. However, they acquire a clear survival advantage through constant β-blocker usage before and after diagnosis (n = 2,564) (47). Furthermore, another latest clinical research claims that NB usage is not correlated to accumulated PDAC risk but reduces the risk in long-term usage, especially NSBB usage (n = 4,113) (48). To date, these publications confirm the therapeutic effect of NSBB for the blockade of ADRB signaling associated with sympathetic relevant NE in PDAC and guarantee reliability and security in PDAC patients.
Although sympathetic preganglionic neurons are concluded into cholinergic neurons, sympathetic nerves have typically been considered to exert NE/ADRB signaling in PDAC. There is little literature to investigate sympathetic Ach in PDAC and other cancers to date. Ach, as a chemical messenger secreted from the ganglion synapse, directly binds to nicotinic acetylcholine receptors (nAchRs) at postganglionic neurons to induce the release of NE from postganglionic neurons. We believe that the cholinergic parts of sympathetic nerves have a little direct impact on the oncogenesis and neoneurogenesis of PDAC. Hypothetically, we deem that the secretion of sympathetic preganglionic Ach is straightly binding to nAchRs at postganglionic neurons, and superfluous Ach is sabotaged and devitalized by cholinesterase which barely leaks into the microenvironment nor has direct connections with cancer cells in PDAC or other cancers.
Afferent sensory (afferent) nerves transmit sensory information from the pancreas to the central nervous system (CNS) and company with splanchnic nerves and vagi, whose neural bodies lie in DRG, the spinal afferents and nodose ganglia (NG), vagal afferents. Sensory nerves consist of unmyelinated fibers and have sensory and secretory functions (49, 50). It was observed that there is augmented axon density in PanIN lesions in vivo. Moreover, a microfluidic device was utilized to co-culture DRG neurons with PDAC cells and separate the axons (33). This co-cultured system allows axonal interactions and simulates intrapancreatic sensory innervation in human PDAC. Undoubtedly, accelerated sensory axons were recruited through PDAC cells, and an increased number of axons expressed synapsin proteins, suggesting functional axons with enhanced neurotransmitter release and transport. Intriguingly, the genetic and functional aberration of axons has a strong association with the outcome of PDAC patients and cancer progression in murine PDAC models (51). It was found that semaphorin exhibits an augmented expression, which is correlated to the poor OS of PDAC patients on univariate analysis. The same change occurred in early pancreatic tumorigenesis modeling by in vivo pancreatic injury and in vitro ADM, in contrast to normal pancreas (25). Taken together, utilizing antagonists against aberration of axon guidance genes and expression would significantly inhibit the initiation and progress of PDAC. DRG and NG contain sensory neurons sensitive to capsaicin, and capsaicin activates TRPV1 on the unmyelinated sensory fibers (52). Activated TRPV1 has a calcium preference, mainly leading to the release of substance P (SP) and calcitonin gene-related peptide (CGRP) (53). SP belongs to tachykinins encoded by TAC-1, also known as pre-pro-tachykinin-A (Ppt-a) (54). Neurokinin receptor 1 (NK-1R), encoded by TACR1, is one of the families of G-protein coupled receptors (GPCR) combined with SP, which then promotes several signaling pathways for the progress of cancer (55)—for instance, SP, along with its high-affinity target NK-1R, was augmented and activated HER2 in breast cancer cell lines (HER2+) and primary cell culture from breast cancer patients (HER2+). SP acted as a pro-inflammatory factor to modulate the expression of EGFR and HER2 for tumor malignancy. This transactivation is significantly reduced by the inhibition of NK-1R RNA expression, a chemical inhibitor of NK-1R, and inhibition of GPCR-induced signaling (56). In conclusion, activated sensory nerves in the pancreas release SP, and the SP–NK-1R axis could stimulate many pathways like JAK/STAT, MAPK/ERK, and HER2/EGFR (mentioned before) signaling for oncogenesis and evolution of PDAC (56–59). While CGRP (encoded by the calcitonin gene) is a relatively novel neuropeptide first isolated in 1982, human-type CGRP is isolated from medullary thyroid carcinoma (60, 61). The release of CGRP by sensory neurons regulated by NGF via a cAMP/ras manner and activation of mitogen-activated protein kinase (MAPK) binds it to its target receptors CGRP1 and CGRP2, facilitating the concentration of cAMP followed by the activation of protein kinase A (PKA), which then unfolds K+ channels leading to the relaxation of smooth muscles in the vascular endothelium (62–64). Therefore, blood flow in PDAC is accelerated by the generation of CGRP, which is important for transporting trophic substances and modulation of metabolic plasticity. It has been published that CGRP has chemotherapy resistance in acute myeloid leukemia (AML) cell lines via the CGRP/CALCRL axis and promotes prostate tumor growth in murine models, possibly via ERKs/STAT3 signaling (65, 66). By the end of the article, there are few experimental articles about the specific impacts of CGRP and its receptors in PDAC. Hence, we promote a possible mechanism of sensory released CGRP based on the existing theories: secreted CGRP binds to CGRP1/CGRP2, activates cAMP/PKA signaling, K+ channel, and expansionary blood flow potentially accompanied with another bypass, which renders PDAC abundant nutrition, metabolic materials, and neural/metabolic plasticity. Hopefully, this article could provoke more related experiments to unveil the secretory characteristics of CGRP in PDAC. Taken together, neuropathic pain caused by PDAC induces sensory nerve impulses and then evokes TRPV1 to release neuropeptides, including CGRP and SP, which directly or indirectly activates individual signaling pathways for the tumorigenesis and evolution of PDAC. Recent research claims that the neuropathic pain of PDAC results from the abnormally expressed sonic hedgehog (sHH) signaling and is mediated by SP and CGRP in DRGs in an NGF-dependent manner (67). Furthermore, PanIN organoid colonies are directly promoted by sensory neurons via SP/NK-1R signaling and phosphorylated STAT3 (33, 68). Similarly, the NK-1R+ neuroendocrine cells in PanIN shows trophic effects to potentiate global organoid growth and provide developmental signals for NK-1R‐ cells to accelerate the growth rate of organoid colonies. Inversely, sensory denervation in KPCPdx1 mice greatly reduced the growth and progression of PanIN. Meanwhile, the subpopulation of NK-1R+ cells possibly correlated to impaired STAT3 phosphorylation and neuroendocrine cell maintenance (33). Interestingly, capsaicin purified from the pepper plant has a dose-dependent effect on its target TRPV1; the appropriate dose of capsaicin binds to TRPV1 and activates sensory nerves with the release of SP and CGRP, whereas an accumulating dose of capsaicin could achieve capsaicin desensitization which reversibly or permanently silences whole neurons for the treatment proposed (69, 70). According to these mechanisms, the application of TRPV1 antagonist (AG1529 has a partially inhibitory effect) has been approved to prevent the transition from acute pancreatitis to chronic pancreatitis (CP), along with the development of CP and inflammation-related pain in a murine model (71, 72). Similar treatments targeting the sensory innervation signaling in PDAC should be established only after they have been well tested to guarantee safety and validity.
Parasympathetic innervation in the pancreas originates from the dorsal motor nucleus of the vagus nerve as parasympathetic preganglionic fibers. It substitutes neurons with parasympathetic postganglionic neurons in intrapancreatic ganglia, which innervate the pancreas (36, 73–75). The parasympathetic nervous system is sorted into cholinergic nerves because its preganglionic and postganglionic neurons release Ach as neurotransmitters. When the vagal nerve is activated, the parasympathetic preganglionic nerve secrets Ach to activate parasympathetic postganglionic nerves via nAchRs about the ligand-gated ion channel family, and then postganglionic neurons also release Ach which binds to muscarinic acetylcholine receptors (mAchRs) at target tissues (76). There are five subtypes of mAchRs on target tissue cells, differing from M1 to M5 (encoded by CHRM1 to CHRM5), and all five belong to the GPCR family (77, 78). It is illustrated that Ach, along with its acetylcholine receptor ligands, overexpresses several human cancers like colon, gastric, lung, and PDAC (44, 79–82). Cholinergic signaling induces an immune-suppressive microenvironment for the neoplastic part in PDAC to support tumor growth. Mechanistically, the Ach/nAchRs pathway impairs CD8+ T cell accumulation, attenuates IFN-γ generation in CD8+ T cells, and switches Th1/Th2 balance to Th2 guiding phenotype through both in vivo orthotopic PDAC model and in vitro high-performance liquid chromatography (83). The proximate discovery about cholinergic signaling reveals an aspect of the secret why PDAC is considered a “cold tumor” and lacks reactions to immunotherapy. In other words, diffuse cholinergic signaling renders PDAC a privilege to escape the immune system and indirectly mediate the microenvironment of PDAC to promote its growth. In addition, nicotine, the agonist of nAchRs, simultaneously suppresses GABA signaling and accumulates ARDB signaling to activate the SHH pathway collectively and then directly induces the self-renewal of pancreatic cancer stem cells, a mass of which initializes oncogenesis and the progression of PDAC (84). Moreover, clinical statistics display that overexpressed M3 is associated with pejorative development and unfavorable prognosis of PDAC patients (n = 58), in whom the cells are usually located at invasive tumor budding cells, metastatic lymph nodes, and parasympathetic nerve fibers (79). This article suggests that M3 should be a novel marker to predict the prognosis of PDAC. However, it seldom excavates the molecular mechanisms of the Ach/M3 axis in PDAC, and a small specimen leads to its limitation for clinical usage. Intriguingly, increasing publications also show that PSNS has recently decelerated cancer evolvement (40, 82, 85). Experiments in breast cancers reveal the innervation of SNS and PSNS, illustrating opposite effects in the tumor microenvironment. The denervation of sympathetic nerves and the neurostimulation of parasympathetic nerves in breast tumor decelerate the expression of immune checkpoint molecules probably mediated by M1 cholinergic mechanisms including PD-1, PD-L1, and FOXP3, which significantly suppress the anti-cancer immune response and become a potential treatment for breast cancer (40, 86, 87). However, whether parasympathetic nervous neurostimulation (M1-associated) also decumulates the immune checkpoint molecules in PDAC (like breast cancer) is not discovered up to the present, and the relational theoretical blank remains as both mystery and opportunity for researchers. As mentioned before, PSNS is also indicated to impair immune response by regulating immune cells to induce an immune-suppressive microenvironment for PDAC growth (83). Meanwhile, Renz and colleagues investigated the parasympathetic system’s specific effects and the possible mechanisms in PDAC. They performed subdiaphragmatic vagotomy on genetically engineered KC mice to disclose parasympathetic denervation in PDAC, which appeared to accelerate PDAC tumorigenesis and augment the expression of M1 under KRAS mutation. Along with vagotomy, utilizing the antagonist (pilocarpine) and the agonist (scopolamine) of M1 receptors has a similar and opposite result for cell viability and expression of M1. Mechanistically, cholinergic signaling suppresses PDAC oncogenesis and development in primary PDAC and metastatic lesions through the activation of M1-related signaling, then reduces the CD44+ CD133+-marked CSC subpopulation, and suppresses the downstream EGFR/MAPK and PI3K/AKT pathway, which indirectly and directly inhibits PDAC development, respectively (85). Overall, this article reveals that PSNS, the cholinergic nerves, suppress the initial PDAC. Still it did not clarify the process of cholinergic signaling activation and what form of M1-related signaling stimulates it downstream for restraining the development of PDAC. Given the existing evidence of neurotransmitters mediating the tumorous functions of automatic innervation in PDAC, we consider that activated cholinergic signaling secretes Ach through parasympathetic postganglionic neurons binding to M1 receptors on the membrane of target tissue cells, followed by the inhibition of EGFR/MAPK and AKT/PI3K signaling and the reduction of CSC subpopulation stimulated by Ach/M1 receptor axis. Furthermore, treatment with pilocarpine reveals a lower M1 expression, but scopolamine shows a higher M1 expression consistent with vagotomy, which suggests that the expression of M1 muscarinic receptors has a potential feedback loop with M1-related signaling activation.
To date, there is an extensive debate on the complicated role of the parasympathetic nervous system during the tumorigenesis of PDAC. Abundant innervations in the pancreas and complex cholinergic neurotransmitters and receptors result in the externally inconsistent conclusion of PSNS in PDAC. Here we propose a possible unified explanation for the two distinct opposite mechanisms of cholinergic signaling: tumorous plasticity of PDAC. PDAC, the most aggressive cancer, has incomparable adaption and high plasticity for its aggressive proliferation, growth, invasion, and metastasis. Take metabolic plasticity, for instance, PDAC could transfer its respiration manner from oxidative phosphorylation (OXPHOS)-dependent manner into aerobic glycolysis, leading to a style under a hypoxic microenvironment (emerging of the Warburg effect) (88); glycolysis and OXPHOS can also co-exist in the same lesions (89). Moreover, glycolytic PDAC cells even transform pyruvate in the mitochondria for OXPHOS when glycolysis is inhibited to promote energy and metabolites for proliferation and invasion (90). Taken together, we deem that the only famous principle about PDAC is that such aggressive cancer cells would take whatever it takes to invade and proliferate. Besides metabolic plasticity, angiogenesis, neurogenesis, immune suppression, systematic cell death (like apoptosis, autophagy, pyroptosis, and ferroptosis), chemo/radiotherapy resistance, etc., generally confirm the proposed principle. The flexible plasticity weaves a complex and gigantic signaling network for tumor initialization and development. It is pretty arbitrary to consider that there is only one independent signaling pathway like PSNS for PDAC progression, which has a dichotomous “all or none” impact without any other possibility. We forecast that PSNS might have a plastic impact on PDAC, while the entire signaling network remains obscure and needs to be thoroughly excavated in PDAC. Moreover, targeting or suppressing associated signaling should be affirmatively verified and experimented with for clinical application in PDAC patients because the deformability and plasticity might switch the PDAC cells into more invasive types.
The latest research suggests the interaction between neuronal innervation and metabolic remodeling in PDAC (28, 91). Notably, metabolic plasticity renders PDAC cells to adapt to nutrient-deprived microenvironment via increasing scavenging pathways like autophagy, apoptosis, and reverse Warburg effects associated with pancreatic stellate cells (PSCs) or cancer-associated fibroblasts (CAFs) (92–94). Highly innervated PDAC has a strong interaction between PDAC cells and innervation from the microenvironment, and neurons also release amino acids (AA) as neurotransmitters, like D-serine, for long-term potentiation at the synapses or neurodevelopment like synapse maturation and axonal stabilization (95, 96). Given that serine is a non-essential AA and the little evidence about the metabolic association with tumor growth, the authors investigated the possibility of whether neurons have metabolic support to PDAC cells under a nutrient-deprived condition. In summary, this paper innovatively proves that peripheral axons secrete serine to metabolically sustain PDAC growth under a nutrient-deprived microenvironment (28).
Aggressive cancer cells experience extensive metabolic rewiring during initial development. Dysregulated metabolism of glucose, lipid, and amino acid is confirmed in both primary and metastatic PDAC; those products and intermediates from those processes are essential for energy generation and material syntheses like cellular membranes, nucleotide, extracellular matrix (ECM), and cell cytoskeletons, which are fundamental for proliferation, invasion, and metastasis (12, 97–100). In PDAC, cancer cells uptake augmented glucose for metabolism and energy generation, which is utilized in positron emission tomography-computed tomography (101, 102). Glucose transfers into the cytoplasm through glucose transporter 1 (GLUT1) in the cell membrane, also highly expressed in PDAC associated with worse prognosis, and hexokinase catalyzes glucose into glucose-6-phosphate (G-6-P) where it divides into a bypass pentose phosphate pathway (PPP) contributing to the synthesis of DNA or RNA (103, 104). Another side path destination of G-6-P is synthesizing glycogen. During the course, glycogen synthase kinase 3 (GSK-3), an important kinase for glycogen synthesis, is reported to induce chemo-resistance for PDAC cells by sustaining the TopBP1/ATR/Chk1 DNA damage response pathway (105). Then, G-6-P is converted into fructose-6-phosphate (F-6-P) through the catalysis of phosphohexose isomerase, under the name of autocrine motility factor, phosphoglucose isomerase, and glucose-6-phosphate isomerase, which leads to another route, the hexosamine biosynthetic pathway (HBP). The end product of HBP is uridine diphosphate N-acetyl glucosamine (UDP-GlcNAc), generated by glucose, glutamine, and glucosamine and utilized in O-GlcNAcylation which is a post-translational modification, mainly glycosylation of serine and threonine (106). Moreover, HBP promotes the progression and survival pathways in PDAC and induces hyaluronan synthesis in the ECM. Furthermore, a study points out that they target glutamine-utilizing enzyme glutamine-fructose aminotransferase 1 via a small molecular glutamine analog, 6-diazo-5-oxo-I-norleucine, demonstrating reduced metastasis and self-renewal potential, along with decreased hyaluronan and collagen in the ECM, and sensitivity to immune checkpoint therapy (anti-PD1 therapy), thus resulting in better prognosis (107). HBP is significantly interlinked together with other metabolic pathways, including glucose, lipid, protein, and nucleotide, which exceedingly support the survival and progression of PDAC. Next, F-6-P is turned into 3-phosphoglycerate (3-PG) through the catalysis of a series of glycolytic enzymes, including phosphofructokinase (PFK), triose phosphate isomerase (TPI), etc. 3-PG directs to a shunt pathway, the serine biosynthesis pathway (SBP), employing 3-PG as a substrate for serine production. Such suggests that glycolytic intermediates could link to SBP from 3-PG and vice versa; serine can be catabolized into pyruvate. To start, 3-PG is transformed into 3-phosphohydroxypyruvate by phosphoglycerate dehydrogenase (PHGDH), which is the first enzyme in SBP, followed by phosphoserine aminotransferase 1 (PSAT1), thus inducing the transition to phosphoserine (p-serine). Subsequently, phosphoserine phosphatase (PSPH) mediates the generation of serine from p-serine, which is the last step of SBP. Serine, the second most abundant amino acid in human proteins, participates in many biosynthetic pathways, such as lipid synthesis, protein synthesis, and the one-carbon cycle, which supports nucleotide synthesis, methylation reactions, and antioxidant defense (108, 109). Interestingly, PHGDH, PSAT1, and PSPH overexpression is found in PDAC and correlated to shorter OS (110–112). When serine is limiting, augmented SBP, especially PHGDH, provides a growth advantage for tumor proliferation via sufficient serine in breast cancer and melanoma compared with normal environmental serine levels (113). Besides the metabolic aspect, SBP enzymes promote PDAC development by regulating molecular signaling. Specifically, PHGDH interlinks with translation initiation factors eIF4A1 and eIF4E while assembling translation initiation complex eIF4F to directly regulate protein synthesis, thus further promoting the progression and development of PDAC (110). In addition, overexpressed enzymes like PHGDH and PSAT1 promote the MYC/miR-494/enhancer of zeste homolog 2 (EZH2) feed-forward loop and are at least partially driven by MYC/ATF4 signaling in Burkitt lymphoma cells to sustain metabolic reprogramming. Notably, one carbon unit (including folate and methionine) produced from serine induces the production of S-adenosylmethionine, which facilitates the reaction of histone H3 trimethylation (H3K27me3), subsequently repressing miR-494 expression and thus decreasing the expression of MYC. MYC indirectly induces the methylation of H3K27me3 through regulating EZH2; the regulators combine into a feed-forward loop to sustain metabolic rearrangements (114). Clinically, the expression of PHGDH has been linked with lymph node metastasis in PDAC patients (115). Similarly, the selective loss of PSAT1 also abrogates migration, invasion, and metastasis in triple-negative breast cancer, suggesting that PSAT1 induces the migratory potential for metastasis without synthesizing serine (116). PSPH, the last enzyme in TSBP, also shows serine-expectant function via suppressing 2-hydroxyglutarate to liberate DNA 5-hydroxymethylcytosine and nuclear receptor NR4A1 expression to facilitate pro-oncogenic gene expression for melanoma growth and metastasis (117). Serine catabolism in the mitochondria is indicated by serine hydroxymethyltransferase 2 (SHMT2) to generate glycine and formate. Inversely, SHMT1 can also promote the cytosol transition from glycine to serine (118). Chemical pyrrolo[3,2-d]pyrimidine compounds targeting SHMT2 inhibitors have been investigated for serine catabolism. Small molecular compounds AGF291, AGF320, and AGF347 were established for antitumor efficiency toward lung, colon, and PDAC in vitro, respectively. AGF327 also demonstrates a potent effect in vivo models exerting a therapeutic potential for SHMT2 suppression for PDAC (119). Afterward, glycine is used to synthesize glutathione (GSH); high levels of GSH have an anti-apoptosis function for chemotherapy resistance in PDAC, and GSH/GSSG ratios regulate reactive oxygen species damage (120). Following on the heels of 3-PG, pyruvate is produced from a three-step catalytic reaction based on 3-PG, which is the initial alanine synthesis. The reversible reaction of converting pyruvate to alanine catalyzes alanine aminotransferase 1, also known as glutamate pyruvic transaminase, in the cytoplasm and vice versa. Finally, pyruvate enters the mitochondria for the tricarboxylic acid (TCA) cycle or is directly catalyzed by lactate dehydrogenase for glycolysis in the cytoplasm, severally. The TCA cycle in the mitochondria unifies the urea cycle, de novo lipogenesis, and amino acid metabolism via its intermediates. Among the intermediates, citrate is transformed into acetyl-coenzyme A via ATP-citrate lyase and finally produces fatty acid for proliferative cellular membranes and energy production signaling molecules’ second messenger generation. In addition, fumarate, as an intermediate from the TCA and urea cycles, acts as the bridge between the two cycles. Moreover, another nutrient fuel for biosynthesis other than glucose glutamine, a dispensable amino acid, enters cells through the alanine/serine/cysteine-preferring transporter 2 (ASCT2) and turns into glutamate via glutaminase, which is subsequently transferred into α-ketoglutarate (α-KG) via ALT2 as carbon donors (121). Notably, glutamine also provides nitrogen for other non-essential amino acid synthesis like serine, glycine, alanine, etc. (Figure 3).
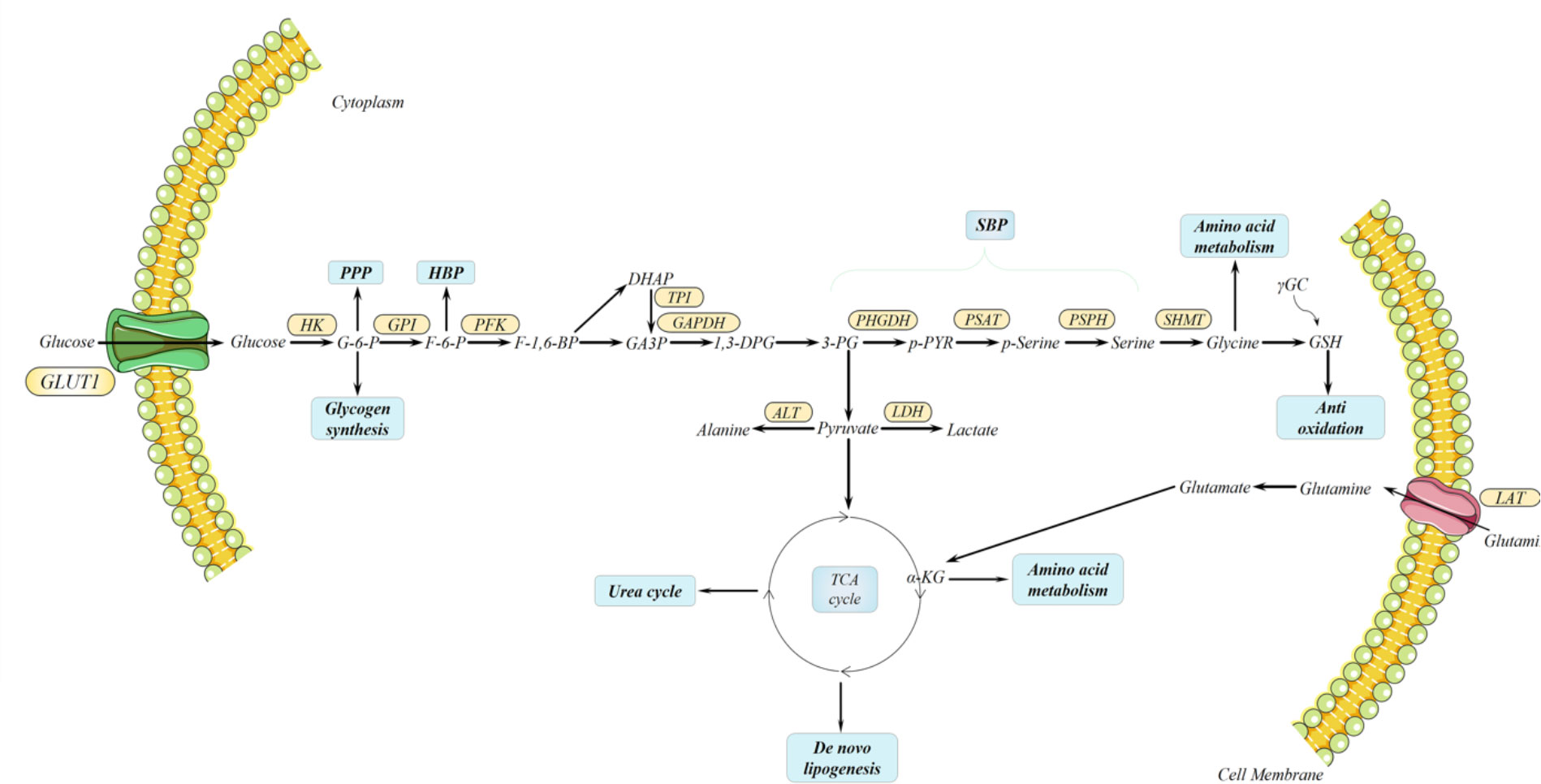
Figure 3 Metabolic rearrangements in pancreatic ductal adenocarcinoma (PDAC). In both primary and metastatic PDAC, metabolic rearrangements render PDAC extraordinary plasticity for the metabolites deprived. Among these rearrangements, mitochondrial respiration unifies several important bioenergetics; it produces precursors for lipid, amino acid, and nucleotide biosynthesizing and contributing to glutamine catabolism. Moreover, serine biosynthesis pathway is derived from glycolysis to produce serine for lipid synthesis, protein synthesis, and one-carbon cycle supporting nucleotide synthesis, methylation reactions, and antioxidant defense. Serine is catalyzed into glycine by serine hydroxymethyltransferase, which is converted to glutathione subsequently.
Until recently, increasing evidence supports the dynamically metabolic transition in metastatic cancers during the changing microenvironment. Interestingly, an article claims that aggressive cancer adjusts its metabolism to adapt to a shifty microenvironment and support growth at every stage during its metastatic cascade (98). Moreover, the authors define two novel concepts about metabolic adjustment: metabolic plasticity and flexibility. Notably, metabolic plasticity refers to the same metabolite used by metastatic cells during its different stages of metastasis, and metabolic flexibility (established on nutrient flexibility) describes different metabolites that can all fulfill the exact metabolic requirement of metastatic cascade in metastatic cells (98). Notably, PDAC, the most aggressive cancer, undergoes complicated metabolic rearrangements. Although metastasis in PDAC is a rare process, distant metastasis and cachexia of PDAC are significant causes of the death of cancer patients. Understanding the metabolic reprogramming of PDAC cells in every stage during its metastasis may promote measurements to destroy its metabolic process via targeting the metabolic vulnerabilities for potential therapy (Table 1).
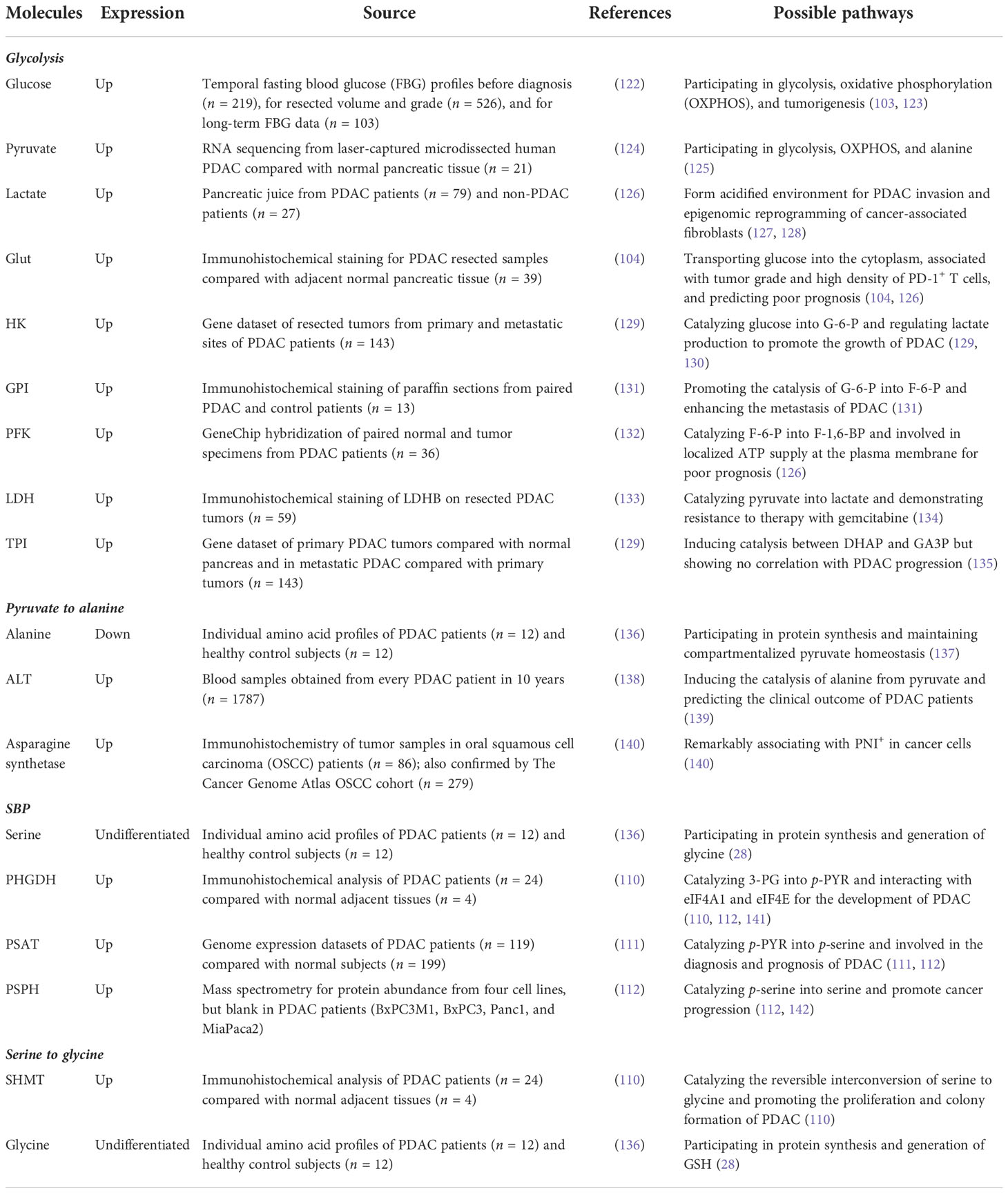
Table 1 Clinical evidence of metabolic expression for detection in pancreatic ductal adenocarcinoma (PDAC).
It has been accepted that PDAC has a strong tendency for innervation and abundant PNI. These features indicate the worse prognosis of PDAC (143). Early major research about neuron–tumor interlinks points tumor-inducing neurons in PDAC (144). Neurons in PDAC express abundant neurotrophic factors, including NGF, GDNF, artemin, and neuronal chemokines, promoting the malignancy of PDAC via signaling activation (145). As we described earlier, the New York research team identified the unique group of neoplastic neuroendocrine cells in PanINs of the mouse and human models. Neuropeptide receptor NK1-R is detected in the cells, and NK-1R+ cells functionally promote PanIN nutrition utilization, thus assisting PanIN organoid growth. Furthermore, denervation of sensory neurons in murine PDAC reduces NK-1R+ cell number and retards PanIN-PDAC progression (33). In this publication, they believe that sensory neurons facilitate PDAC formation and metabolic signaling via the SP–NK1R–STAT3 pathways. To date, other direct shreds of evidence of neurons and cancer development are discovered in PDAC from the metabolic aspect (28, 146). Although PSCs and macrophages in TME can supply PDAC with some metabolites or nutrition, it is insufficient for PDAC to get growth requirements with such metabolites (147). Thus, 5-HT (serotonin), a neuro-regulator, grows in PDAC tissues and cell lines compared with normal pancreas. Incubation of PDAC with serotonin or activator of HTR2B renders the proliferative capacity and inhibition of apoptosis. Moreover, serotonin activation leads to a complex of HTR2B-LYN-p85 that activates PI3K–Akt– mTOR signaling and then enhances the Warburg effect (glycolysis) via overexpressed MYC and HIF-1α. The neurotransmitter serotonin released by axons affects metabolic rearrangements, including glycolytic flux, PPP, and HBP, through the expression of enzymes (146). This work suggests that neurotransmitters enable signaling pathways in PDAC and then incite metabolic rearrangements, influencing the malignancy or survival of PDAC afterward. To sum up, the previous research connects neural–metabolic crosstalk in PDAC through chemokines, neurotrophins, and neurotransmitters to investigate the role of innervation during PDAC development. Innovatively, Hindson and colleagues hypothesized that neurons could directly promote metabolic support for PDAC cells under nutrient deprivation in PDAC (91). To assess, micro-fluidic devices that could isolate axons from the neuronal body of DRGs were applied to stimulate an in vivo situation where PDAC cells in nutrient-deprived TME are only exposed to peripheral axons, but not the neuronal bodies (148). Surprisingly, the detection of the conditional axonal media exhibits AA levels, including serine and glycine, suggesting that axons release AA into a nutrient-poor environment. There is a heterogeneity of human PDAC about their dependence on exogenous serine. Approximately 40% of human PDAC cell lines concretely depend on exogenous serine for proliferation due to a lack of expression or ability to augment PHGDH and PSAT1, while mouse PDAC cells can promote serine synthesis and are insensitive to serine starvation. However, the repressed growth of PDAC could be rescued by a co-culture with axons in the microfluidic devices (149). Mechanistically, they noticed reduced mitochondrial activity in serine-dependent PDAC under serine deprivation and a rapidly increased oxygen consumption rate (OCR). This change is similar to utilizing an inhibitor of protein synthesis, cycloheximide (150), which indicates that serine starvation selectively suppresses the mRNA translation rates for protein synthesis, causing decreased mitochondrial activity and increased OCR. More specifically, the authors explored the concrete effects through destabilized green fluorescent proteins (151), which replace all serine codons with one of the six serine codons. Among them, the translation efficiency of TCT and TCC is repressed because of ribosome stalling as revealed by ribosome profiling (152). Interestingly, NGF is intensely expressed by serine-dependent PDAC cells under serine deprivation because the selective translation allows the synthesis of TCC and TCT-low coded proteins, exhibiting enhanced nerve infiltration for serine and glycine secretion to restore PDAC growth. These observations are consistent with in vivo models. Exogenous serine-dependent PDAC under serine-starved conditions shows a significantly smaller tumor mass, accelerating NGF secretion and accumulating sympathetic and sensory innervation. In treating mice with LOXO-101, the NGF receptor inhibitor adversely reduces tumor mass and innervation under a serine-free diet compared with the serine-free-diet mice or control diet with LOXO-101 mice (153). Moreover, PDAC tumors with augmented levels of PHGDH typically have attenuated NGF, tumor innervation, and shorter OS, which is also consistent with other results (28). In summary, this article successfully testifies the metabolic assistance of neurons in PDAC, rendering great metabolic plasticity via neural innervation, which also hints us to concentrate on other aspects in TME for metabolic arrangements. However, as the first published document implicating the function of serine and innervation, there are still unclear aspects of this article, mainly about the mechanisms allowing neurons to release amino acid to support PDAC cells metabolically, mechanisms about the selective translation efficiency of codons, and why PDAC would lose the ability to synthesize serine? More importantly, clinical transformation based on the observed phenomenon would promote novel therapeutic potential targeting PDAC metabolic and neural crosstalk for PDAC patients (Figure 4).
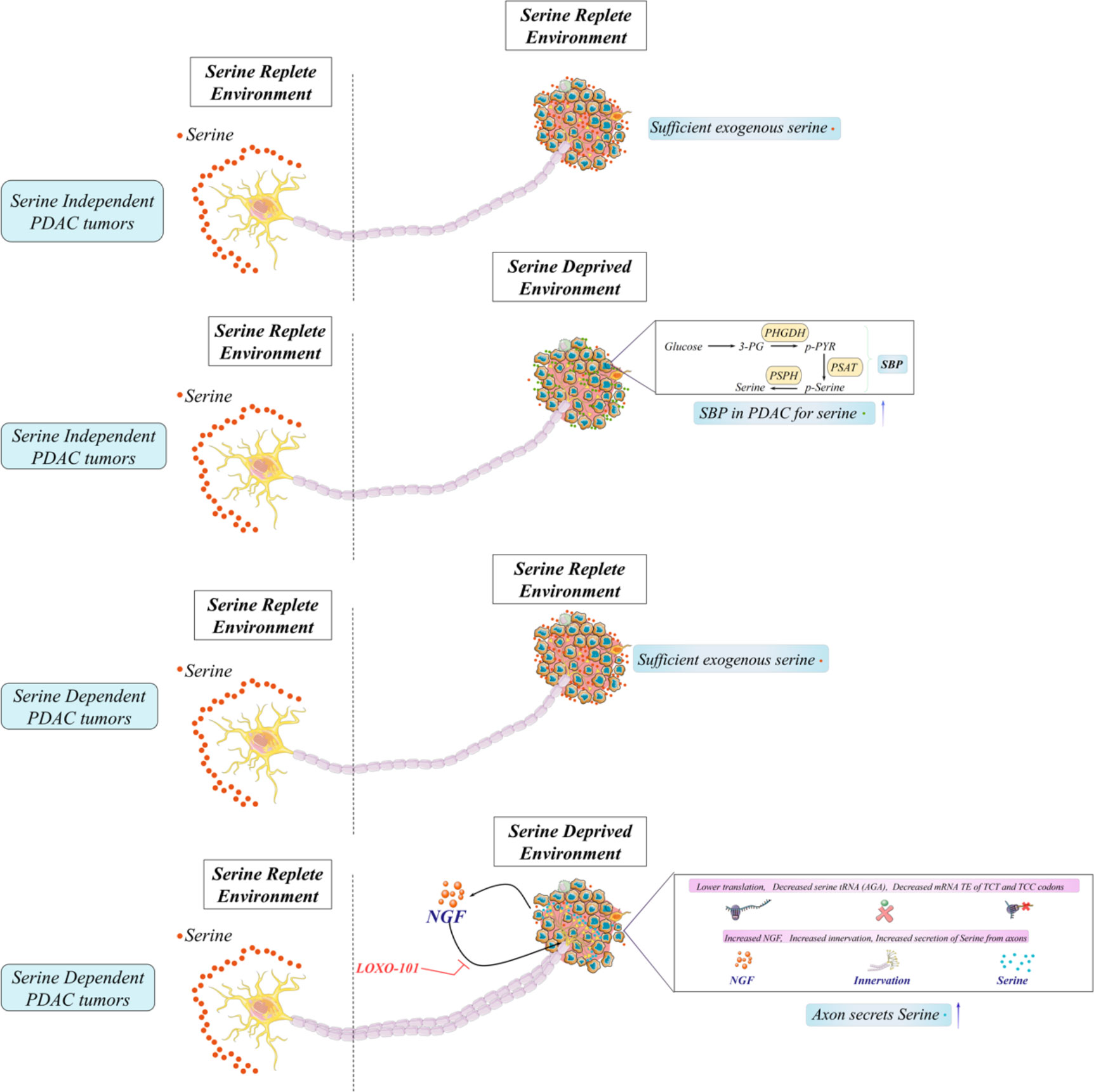
Figure 4 Neural and metabolic interactions in pancreatic ductal adenocarcinoma (PDAC). A large fraction of human PDAC cells lack SBP enzymes (PHGDH and PSAT-1) and cannot generate serine, and other pathways like autophagy and pancreatic stellate cells cannot generate sufficient levels of serine for growth in PDAC cells under serine deprivation. Therefore, these PDAC cells which must import serine for growth are exogenous serine-dependent PDAC. Moreover, those with SBP enzymes (PHGDH and PSAT-1) for the generation of serine from glucose are exogenous serine-independent PDAC. To date, neurons could directly promote metabolic support (releasing serine) for PDAC cells under the serine-deprived conditions in PDAC.
Recently, some documents gave an overview on serine and other metabolites in neural–cancer progress (154). Serine is considered to not only facilitate cancer progress but also maintain neurites’ formation—for example, NGF induces the phosphorylation of RhoA on serine 188 and reduces RhoA–Rho-related kinase for neurite growth (155); another in vitro experiment declaims that NGF-associated phosphorylated STAT3 at serine (727) is involved in neurite growth (156). Apart from serine, additional metabolites are associated with neural innervation in cancers. For a start, solute carrier family 2 member 3 (SLC2A3), asparagine, and asparagine synthetase are all remarkably expressed in PNI+ cancer. Overexpressed SLC2A3, increased glucose, and reduced vitamin C indicate a diminished OS in colorectal cancer patients and AML patients (157, 158). However, the concrete part of SLC2A3 in neural–metabolic crosstalk is not excavated. The immunohistochemistry of OSCC clinical study (n = 86), together with The Cancer Genome Atlas database (n = 267), presents that asparagine synthetase is notably added in PNI+ tumors, and L-asparagine is the only amino acid able to indicate PNI with satisfactory sensitivity and specificity (140). Regrettably, these studies fail to trench the concrete mechanisms of the metabolic–neural functions during cancer development. Banh and colleagues introduce an excellent method to excavate metabolic–neural interactions in cancer through microfluidic devices to isolate neurons and cancer cells in nutrition-deprived conditions to stimulate an in vivo environment, and we believe that utilizing the unique method could elaborate the specific mechanisms of metabolic–neural interlinks during cancer evolution, especially metabolites like serine and glycine.
To sum up, we first summarized the epidemiology of PDAC, especially risk factors and clinical symptoms, and then introduced a classification for PDAC and related genomic alterations. Notably, the pancreas is a highly innervated organ, so we also discussed three different nerve innervation and pro/anti-oncogenic functions in PDAC. Lastly, we mainly reviewed metabolic rearrangements in PDAC and commented on a recent article about neural and metabolic crosstalk. This review concludes the relationship between innervation and PDAC and also recommends a novel method to excavate metabolic–neural interactions in cancer through microfluidic devices to isolate neurons and cancer cells introduced by Banh and colleagues. We believe that a more direct proof of metabolic–neural crosstalk in PDAC remains unclear, especially metabolites like amino acids. Hopefully, this article could further explore the pathogenic mechanisms of PDAC and then induce the transformation of detection and treatment for PDAC considering the innervation.
RC and MD contributed to the conception and design of the study. LC wrote the first draft of the manuscript. YX and MD wrote sections of the manuscript and provided the critical revisions. All authors contributed to the article and approved the submitted version.
This article is funded by following projects: Special Medical and Health Personnel Program of Jilin Province Finance Department (2019SCZT015) and Health Technology Innovative Program of Jilin Province (2018J053).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: Cancer J Clin (2020) 70(1):7–30. doi: 10.3322/caac.21590
2. Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science (2017) 357(6348):eaal2380. doi: 10.1126/science.aal2380
3. Vitale I, Shema E, Loi S, Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med (2021) 27(2):212–24. doi: 10.1038/s41591-021-01233-9
4. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res (2014) 74(11):2913–21. doi: 10.1158/0008-5472.CAN-14-0155
5. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA: Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
6. Zaccari P, Cardinale V, Severi C, Pedica F, Carpino G, Gaudio E, et al. Common features between neoplastic and preneoplastic lesions of the biliary tract and the pancreas. World J Gastroenterol (2019) 25(31):4343–59. doi: 10.3748/wjg.v25.i31.4343
7. Felsenstein M, Noë M, Masica DL, Hosoda W, Chianchiano P, Fischer CG, et al. IPMNs with co-occurring invasive cancers: neighbours but not always relatives. Gut (2018) 67(9):1652–62. doi: 10.1136/gutjnl-2017-315062
8. Scarpa A, Real FX, Luchini C. Genetic unrelatedness of co-occurring pancreatic adenocarcinomas and IPMNs challenges current views of clinical management. Gut (2018) 67(9):1561–3. doi: 10.1136/gutjnl-2018-316151
9. Mostafa ME, Erbarut-Seven I, Pehlivanoglu B, Adsay V. Pathologic classification of "pancreatic cancers": current concepts and challenges. Chin Clin Oncol (2017) 6(6):59. doi: 10.21037/cco.2017.12.01
10. Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet (London England) (2020) 395(10242):2008–20. doi: 10.1016/S0140-6736(20)30974-0
11. Bachmann J, Heiligensetzer M, Krakowski-Roosen H, Büchler MW, Friess H, Martignoni ME. Cachexia worsens prognosis in patients with resectable pancreatic cancer. J gastrointestinal Surg (2008) 12(7):1193–201. doi: 10.1007/s11605-008-0505-z
12. Cao L, Wu J, Qu X, Sheng J, Cui M, Liu S, et al. Glycometabolic rearrangements–aerobic glycolysis in pancreatic cancer: causes, characteristics and clinical applications. J Exp Clin Cancer Res (2020) 39(1):267. doi: 10.1186/s13046-020-01765-x
13. Grossberg AJ, Chu LC, Deig CR, Fishman EK, Hwang WL, Maitra A, et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA: Cancer J Clin (2020) 70(5):375–403. doi: 10.3322/caac.21626
14. Arslan AA, Helzlsouer KJ, Kooperberg C, Shu XO, Steplowski E, Bueno-de-Mesquita HB, et al. Anthropometric measures, body mass index, and pancreatic cancer: a pooled analysis from the pancreatic cancer cohort consortium (PanScan). Arch Internal Med (2010) 170(9):791–802. doi: 10.1001/archinternmed.2010.63
15. Bosetti C, Lucenteforte E, Silverman DT, Petersen G, Bracci PM, Ji BT, et al. Cigarette smoking and pancreatic cancer: an analysis from the international pancreatic cancer case-control consortium (Panc4). Ann Oncol (2012) 23(7):1880–8. doi: 10.1093/annonc/mdr541
16. Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, et al. Pancreatitis and the risk of pancreatic cancer. international pancreatitis study group. New Engl J Med (1993) 328(20):1433–7. doi: 10.1056/NEJM199305203282001
17. Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. Jama (2018) 319(23):2401–9. doi: 10.1001/jama.2018.6228
18. Petersen GM. Familial pancreatic cancer. Semin Oncol (2016) 43(5):548–53. doi: 10.1053/j.seminoncol.2016.09.002
19. Lowenfels AB, Maisonneuve P, DiMagno EP, Elitsur Y, Gates LK Jr., Perrault J, et al. Hereditary pancreatitis and the risk of pancreatic cancer. international hereditary pancreatitis study group. J Natl Cancer Institute (1997) 89(6):442–6. doi: 10.1093/jnci/89.6.442
20. Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, et al. Very high risk of cancer in familial peutz-jeghers syndrome. Gastroenterology (2000) 119(6):1447–53. doi: 10.1053/gast.2000.20228
21. Brune KA, Lau B, Palmisano E, Canto M, Goggins MG, Hruban RH, et al. Importance of age of onset in pancreatic cancer kindreds. J Natl Cancer Institute (2010) 102(2):119–26. doi: 10.1093/jnci/djp466
22. Schmidt-Hansen M, Berendse S, Hamilton W. Symptoms of pancreatic cancer in primary care: A systematic review. Pancreas (2016) 45(6):814–8. doi: 10.1097/MPA.0000000000000527
23. Bapat AA, Hostetter G, Von Hoff DD, Han H. Perineural invasion and associated pain in pancreatic cancer, nature reviews. Cancer (2011) 11(10):695–707. doi: 10.1038/nrc3131
24. Liebl F, Demir IE, Mayer K, Schuster T, Dʼ;Haese JG, Becker K, et al. The impact of neural invasion severity in gastrointestinal malignancies: a clinicopathological study. Ann Surg (2014) 260(5):900–7; discussion 907-8. doi: 10.1097/SLA.0000000000000968
25. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature (2012) 491(7424):399–405. doi: 10.1038/nature11547
26. Harris NLE, Vennin C, Conway JRW, Vine KL, Pinese M, Cowley MJ, et al. SerpinB2 regulates stromal remodelling and local invasion in pancreatic cancer. Oncogene (2017) 36(30):4288–98. doi: 10.1038/onc.2017.63
27. Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the "Reverse warburg effect" in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, alzheimer's disease, and "Neuron-glia metabolic coupling". Aging (2010) 2(4):185–99. doi: 10.18632/aging.100134
28. Banh RS, Biancur DE, Yamamoto K, Sohn ASW, Walters B, Kuljanin M, et al. Neurons release serine to support mRNA translation in pancreatic cancer. Cell (2020) 183(5):1202–1218.e25. doi: 10.1016/j.cell.2020.10.016
29. Borden P, Houtz J, Leach SD, Kuruvilla R. Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Rep (2013) 4(2):287–301. doi: 10.1016/j.celrep.2013.06.019
30. Bockman DE. Nerves in the pancreas: what are they for? Am J Surg (2007) 194(4, Supplement):S61–4. doi: 10.1016/j.amjsurg.2007.05.028
31. Waxenbaum JA, Reddy V, Varacallo M. Anatomy, autonomic nervous system. In: StatPearls. Treasure Island (FL: StatPearls Publishing LLC. (2021).
32. Yi SQ, Miwa K, Ohta T, Kayahara M, Kitagawa H, Tanaka A, et al. Innervation of the pancreas from the perspective of perineural invasion of pancreatic cancer. Pancreas (2003) 27(3):225–9. doi: 10.1097/00006676-200310000-00005
33. Sinha S, Fu YY, Grimont A, Ketcham M, Lafaro K, Saglimbeni JA, et al. PanIN neuroendocrine cells promote tumorigenesis via neuronal cross-talk. Cancer Res (2017) 77(8):1868–79. doi: 10.1158/0008-5472.CAN-16-0899
34. Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science (2013) 341(6142):1236361. doi: 10.1126/science.1236361
35. Stopczynski RE, Normolle DP, Hartman DJ, Ying H, DeBerry JJ, Bielefeldt K, et al. Neuroplastic changes occur early in the development of pancreatic ductal adenocarcinoma. Cancer Res (2014) 74(6):1718–27. doi: 10.1158/0008-5472.CAN-13-2050
36. Gasparini G, Pellegatta M, Crippa S, Lena MS, Belfiori G, Doglioni C, et al. Nerves and pancreatic cancer: New insights into a dangerous relationship. Cancers (2019) 11(7). doi: 10.3390/cancers11070893
37. Tan X, Sivakumar S, Bednarsch J, Wiltberger G, Kather JN, Niehues J, et al. Nerve fibers in the tumor microenvironment in neurotropic cancer-pancreatic cancer and cholangiocarcinoma. Oncogene (2021) 40(5):899–908. doi: 10.1038/s41388-020-01578-4
38. Kim-Fuchs C, Le CP, Pimentel MA, Shackleford D, Ferrari D, Angst E, et al. Chronic stress accelerates pancreatic cancer growth and invasion: a critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain behavior Immun (2014) 40:40–7. doi: 10.1016/j.bbi.2014.02.019
39. Guo K, Ma Q, Li J, Wang Z, Shan T, Li W, et al. Interaction of the sympathetic nerve with pancreatic cancer cells promotes perineural invasion through the activation of STAT3 signaling. Mol Cancer Ther (2013) 12(3):264–73. doi: 10.1158/1535-7163.MCT-12-0809
40. Kamiya A, Hayama Y, Kato S, Shimomura A, Shimomura T, Irie K, et al. Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat Neurosci (2019) 22(8):1289–305. doi: 10.1038/s41593-019-0430-3
41. Renz BW, Takahashi R, Tanaka T, Macchini M, Hayakawa Y, Dantes Z, et al. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell (2018) 33(1):75–90.e7. doi: 10.1016/j.ccell.2017.11.007
42. Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JPT, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 22(6):737–50. doi: 10.1016/j.ccr.2012.10.025
43. Storz P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma, nature reviews. Gastroenterol Hepatol (2017) 14(5):296–304. doi: 10.1038/nrgastro.2017.12
44. Hayakawa Y, Sakitani K, Konishi M, Asfaha S, Niikura R, Tomita H, et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell (2017) 31(1):21–34. doi: 10.1016/j.ccell.2016.11.005
45. Zhu Z, Friess H, diMola FF, Zimmermann A, Graber HU, Korc M, et al. Nerve growth factor expression correlates with perineural invasion and pain in human pancreatic cancer. J Clin Oncol (1999) 17(8):2419–28. doi: 10.1200/JCO.1999.17.8.2419
46. Sahni S, Moon EA, Howell VM, Mehta S, Pavlakis N, Chan D, et al. Tissue biomarker panel as a surrogate marker for squamous subtype of pancreatic cancer. Eur J Surg Oncol (2020) 46(8):1539–42. doi: 10.1016/j.ejso.2020.02.001
47. Yang A, Zylberberg HM, Rustgi SD, Amin SP, Bar-Mashiah A, Boffetta P, et al. Beta-blockers have no impact on survival in pancreatic ductal adenocarcinoma prior to cancer diagnosis. Sci Rep (2021) 11(1):1038. doi: 10.1038/s41598-020-80570-0
48. Saad A, Goldstein J, Margalit O, Shacham-Shmueli E, Lawrence YR, Yang YX, et al. Assessing the effects of beta-blockers on pancreatic cancer risk: A nested case-control study. Pharmacoepidemiology Drug Saf (2020) 29(5):599–604. doi: 10.1002/pds.4993
49. Fasanella KE, Christianson JA, Chanthaphavong RS, Davis BM. Distribution and neurochemical identification of pancreatic afferents in the mouse. J Comp Neurol (2008) 509(1):42–52. doi: 10.1002/cne.21736
50. Li Q, Peng J. Sensory nerves and pancreatitis. Gland Surg (2014) 3(4):284–92. doi: 10.3978/j.issn.2227-684X.2013.10.08
51. Mehlen P, Delloye-Bourgeois C, Chédotal A. Novel roles for slits and netrins: axon guidance cues as anticancer targets? Nat Rev Cancer (2011) 11(3):188–97. doi: 10.1038/nrc3005
52. Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature (1997) 389(6653):816–24. doi: 10.1038/39807
53. Akiba Y, Kato S, Katsube K, Nakamura M, Takeuchi K, Ishii H, et al. Transient receptor potential vanilloid subfamily 1 expressed in pancreatic islet beta cells modulates insulin secretion in rats. Biochem Biophys Res Commun (2004) 321(1):219–25. doi: 10.1016/j.bbrc.2004.06.149
54. Steinhoff MS, von Mentzer B, Geppetti P, Pothoulakis C, Bunnett NW. Tachykinins and their receptors: contributions to physiological control and the mechanisms of disease. Physiol Rev (2014) 94(1):265–301. doi: 10.1152/physrev.00031.2013
55. Singh D, Joshi DD, Hameed M, Qian J, Gascón P, Maloof PB, et al. Increased expression of preprotachykinin-I and neurokinin receptors in human breast cancer cells: implications for bone marrow metastasis. Proc Natl Acad Sci United States America (2000) 97(1):388–93. doi: 10.1073/pnas.97.1.388
56. Garcia-Recio S, Fuster G, Fernandez-Nogueira P, Pastor-Arroyo EM, Park SY, Mayordomo C, et al. Substance p autocrine signaling contributes to persistent HER2 activation that drives malignant progression and drug resistance in breast cancer. Cancer Res (2013) 73(21):6424–34. doi: 10.1158/0008-5472.CAN-12-4573
57. Koon HW, Zhao D, Zhan Y, Rhee SH, Moyer MP, Pothoulakis C. Substance p stimulates cyclooxygenase-2 and prostaglandin E2 expression through JAK-STAT activation in human colonic epithelial cells. J Immunol (2006) 176(8):5050–9. doi: 10.4049/jimmunol.176.8.5050
58. Pierce KL, Luttrell LM, Lefkowitz RJ. New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene (2001) 20(13):1532–9. doi: 10.1038/sj.onc.1204184
59. Hu CP, Feng JT, Tang YL, Zhu JQ, Lin MJ, Yu ME. LIF upregulates expression of NK-1R in NHBE cells. Mediators Inflammation (2006) 2006(5):84829. doi: 10.1155/MI/2006/84829
60. Morris HR, Panico M, Etienne T, Tippins J, Girgis SI, MacIntyre I. Isolation and characterization of human calcitonin gene-related peptide. Nature (1984) 308(5961):746–8. doi: 10.1038/308746a0
61. Brain SD, Grant AD. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol Rev (2004) 84(3):903–34. doi: 10.1152/physrev.00037.2003
62. Nelson MT, Huang Y, Brayden JE, Hescheler J, Standen NB. Arterial dilations in response to calcitonin gene-related peptide involve activation of k+ channels. Nature (1990) 344(6268):770–3. doi: 10.1038/344770a0
63. Reed WR, Little JW, Lima CR, Sorge RE, Yarar-Fisher C, Eraslan M, et al. Spinal mobilization prevents NGF-induced trunk mechanical hyperalgesia and attenuates expression of CGRP. Front Neurosci (2020) 14:385. doi: 10.3389/fnins.2020.00385
64. Gao X, Zhang D, Xu C, Li H, Caron KM, Frenette PS. Nociceptive nerves regulate haematopoietic stem cell mobilization. Nature (2021) 589(7843):591–6. doi: 10.1038/s41586-020-03057-y
65. Zhu W, Sheng D, Shao Y, Zhang Q, Peng Y. Neuronal calcitonin gene-related peptide promotes prostate tumor growth in the bone microenvironment. Peptides (2021) 135:170423. doi: 10.1016/j.peptides.2020.170423
66. Gluexam T, Grandits AM, Schlerka A, Nguyen CH, Etzler J, Finkes T, et al. CGRP signaling via CALCRL increases chemotherapy resistance and stem cell properties in acute myeloid leukemia. Int J Mol Sci (2019) 20(23):5826. doi: 10.3390/ijms20235826
67. Han L, Jiang J, Xue M, Qin T, Xiao Y, Wu E, et al. Sonic hedgehog signaling pathway promotes pancreatic cancer pain via nerve growth factor. Regional Anesth Pain Med (2020) 45(2):137–44. doi: 10.1136/rapm-2019-100991
68. Boj SF, Hwang CI, Baker LA, Chio II, D.D. Engle, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell (2015) 160(1-2):324–38. doi: 10.1016/j.cell.2014.12.021
69. Fischer MJM, Ciotu CI, Szallasi A. The mysteries of capsaicin-sensitive afferents. Front Physiol (2020) 11:554195. doi: 10.3389/fphys.2020.554195
70. Cabezón-Gutiérrez L, Custodio-Cabello S, Palka-Kotlowska M, Khosravi-Shahi P. High-dose 8% capsaicin patch in treatment of chemotherapy-induced peripheral neuropathy. a systematic review. J Pain symptom Manage (2020) 60(5):1047–1054.e1. doi: 10.1016/j.jpainsymman.2020.06.026
71. Schwartz ES, La JH, Scheff NN, Davis BM, Albers KM, Gebhart GF. TRPV1 and TRPA1 antagonists prevent the transition of acute to chronic inflammation and pain in chronic pancreatitis. J Neurosci (2013) 33(13):5603–11. doi: 10.1523/JNEUROSCI.1806-12.2013
72. Nikolaeva-Koleva M, Butron L, González-Rodríguez S, Devesa I, Valente P, Serafini M, et al. A capsaicinoid-based soft drug, AG1529, for attenuating TRPV1-mediated histaminergic and inflammatory sensory neuron excitability. Sci Rep (2021) 11(1):246. doi: 10.1038/s41598-020-80725-z
73. Ahrén B. Autonomic regulation of islet hormone secretion–implications for health and disease. Diabetologia (2000) 43(4):393–410. doi: 10.1007/s001250051322
74. Lin EE, Scott-Solomon E, Kuruvilla R. Peripheral innervation in the regulation of glucose homeostasis. Trends Neurosci (2021) 44(3):189–202. doi: 10.1016/j.tins.2020.10.015
75. Rodriguez-Diaz R, Abdulreda MH, Formoso AL, Gans I, Ricordi C, Berggren PO, et al. Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab (2011) 14(1):45–54. doi: 10.1016/j.cmet.2011.05.008
76. Paleari L, Grozio A, Cesario A, Russo P. The cholinergic system and cancer. Semin Cancer Biol (2008) 18(3):211–7. doi: 10.1016/j.semcancer.2007.12.009
77. Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol (2003) 63(6):1256–72. doi: 10.1124/mol.63.6.1256
78. Kruse AC, Kobilka BK, Gautam D, Sexton PM, Christopoulos A, Wess J. Muscarinic acetylcholine receptors: novel opportunities for drug development, nature reviews. Drug Discovery (2014) 13(7):549–60. doi: 10.1038/nrd4295
79. Zhang L, Xiu D, Zhan J, He X, Guo L, Wang J, et al. High expression of muscarinic acetylcholine receptor 3 predicts poor prognosis in patients with pancreatic ductal adenocarcinoma. OncoTargets Ther (2016) 9:6719–26. doi: 10.2147/OTT.S111382
80. Park YS, Liu Z, Vasamsetti BM, Cho NJ. The ERK1/2 and mTORC1 signaling pathways are involved in the muscarinic acetylcholine receptor-mediated proliferation of SNU-407 colon cancer cells. J Cell Biochem (2016) 117(12):2854–63. doi: 10.1002/jcb.25597
81. Lin G, Sun L, Wang R, Guo Y, Xie C. Overexpression of muscarinic receptor 3 promotes metastasis and predicts poor prognosis in non-small-cell lung cancer. J Thorac Oncol (2014) 9(2):170–8. doi: 10.1097/JTO.0000000000000066
82. Hutchings C, Phillips JA, Djamgoz MBA. Nerve input to tumours: Pathophysiological consequences of a dynamic relationship, biochimica et biophysica acta. Rev Cancer (2020) 1874(2):188411. doi: 10.1124/mol.63.6.1256
83. Yang MW, Tao LY, Jiang YS, Yang JY, Huo YM, Liu DJ, et al. Perineural invasion reprograms the immune microenvironment through cholinergic signaling in pancreatic ductal adenocarcinoma. Cancer Res (2020) 80(10):1991–2003. doi: 10.1158/0008-5472.CAN-19-2689
84. Al-Wadei MH, Banerjee J, Al-Wadei HA, Schuller HM. Nicotine induces self-renewal of pancreatic cancer stem cells via neurotransmitter-driven activation of sonic hedgehog signalling. Eur J Cancer (2016) 52:188–96. doi: 10.1016/j.ejca.2015.10.003
85. Renz BW, Tanaka T, Sunagawa M, Takahashi R, Jiang Z, Macchini M, et al. Cholinergic signaling via muscarinic receptors directly and indirectly suppresses pancreatic tumorigenesis and cancer stemness. Cancer Discovery (2018) 8(11):1458–73. doi: 10.1158/2159-8290.CD-18-0046
86. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27(1):109–18. doi: 10.1038/cr.2016.151
87. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest (2015) 125(9):3384–91. doi: 10.1172/JCI80011
88. Humpton TJ, Alagesan B, DeNicola GM, Lu D, Yordanov GN, Leonhardt CS, et al. Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discovery (2019) 9(9):1268–87. doi: 10.1158/2159-8290.CD-18-1409
89. Siska PJ, Singer K, Evert K, Renner K, Kreutz M. The immunological warburg effect: Can a metabolic-tumor-stroma score (MeTS) guide cancer immunotherapy? Immunol Rev (2020) 295(1):187–202. doi: 10.1111/imr.12846
90. Shiratori R, Furuichi K, Yamaguchi M, Miyazaki N, Aoki H, Chibana H, et al. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci Rep (2019) 9(1):18699. doi: 10.1038/s41598-019-55296-3
91. Hindson J. Neuronal innervation supports PDAC growth via release of serine, nature reviews. Gastroenterol Hepatol (2021) 18(1):5. doi: 10.1038/s41575-020-00394-1
92. Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature (2015) 524(7565):361–5. doi: 10.1038/nature14587
93. Hong X, Zhong L, Xie Y, Zheng K, Pang J, Li Y, et al. Matrine reverses the warburg effect and suppresses colon cancer cell growth via negatively regulating HIF-1α. Front Pharmacol (2019) 10:1437. doi: 10.3389/fphar.2019.01437
94. Andersen JL, Kornbluth S. The tangled circuitry of metabolism and apoptosis. Mol Cell (2013) 49(3):399–410. doi: 10.1016/j.molcel.2012.12.026
95. Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of d-serine from astrocytes. Nature (2010) 463(7278):232–6. doi: 10.1038/nature08673
96. Van Horn MR, Strasser A, Miraucourt LS, Pollegioni L, Ruthazer ES. The gliotransmitter d-serine promotes synapse maturation and axonal stabilization In vivo. J Neurosci (2017) 37(26):6277–88. doi: 10.1523/JNEUROSCI.3158-16.2017
97. Satriano L, Lewinska M, Rodrigues PM, Banales JM, Andersen JB. Metabolic rearrangements in primary liver cancers: cause and consequences, nature reviews. Gastroenterol Hepatol (2019) 16(12):748–66. doi: 10.1038/s41575-019-0217-8
98. Bergers G, Fendt SM. The metabolism of cancer cells during metastasis, nature reviews. Cancer (2021) 21(3):162–80. doi: 10.1038/s41568-020-00320-2
99. Tadros S, Shukla SK, King RJ, Gunda V, Vernucci E, Abrego J, et al. De novo lipid synthesis facilitates gemcitabine resistance through endoplasmic reticulum stress in pancreatic cancer. Cancer Res (2017) 77(20):5503–17. doi: 10.1158/0008-5472.CAN-16-3062
100. Abrego J, Gunda V, Vernucci E, Shukla SK, King RJ, Dasgupta A, et al. GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett (2017) 400:37–46. doi: 10.1016/j.canlet.2017.04.029
101. Fox RG, Lytle NK, Jaquish DV, Park FD, Ito T, Bajaj J, et al. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature (2016) 534(7607):407–11. doi: 10.1038/nature17988
103. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell (2012) 149(3):656–70. doi: 10.1016/j.cell.2012.01.058
104. Achalandabaso Boira M, Di Martino M, Gordillo C, Adrados M, Martín-Pérez E. GLUT-1 as a predictor of worse prognosis in pancreatic adenocarcinoma: immunohistochemistry study showing the correlation between expression and survival. BMC Cancer (2020) 20(1):909. doi: 10.21203/rs.3.rs-18270/v1
105. Ding L, Madamsetty VS, Kiers S, Alekhina O, Ugolkov A, Dube J, et al. Glycogen synthase kinase-3 inhibition sensitizes pancreatic cancer cells to chemotherapy by abrogating the TopBP1/ATR-mediated DNA damage response. Clin Cancer Res (2019) 25(21):6452–62. doi: 10.1158/1078-0432.CCR-19-0799
106. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions, nature reviews. Mol Cell Biol (2017) 18(7):452–65. doi: 10.1038/nrm.2017.22
107. Sharma NS, Gupta VK, Garrido VT, Hadad R, Durden BC, Kesh K, et al. Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. J Clin Invest (2020) 130(1):451–65. doi: 10.1172/JCI127515
108. Yang M, Vousden KH. Serine and one-carbon metabolism in cancer, nature reviews. Cancer (2016) 16(10):650–62. doi: 10.1038/nrc.2016.81
109. Mauro VP, Chappell SA. A critical analysis of codon optimization in human therapeutics. Trends Mol Med (2014) 20(11):604–13. doi: 10.1016/j.molmed.2014.09.003
110. Ma X, Li B, Liu J, Fu Y, Luo Y. Phosphoglycerate dehydrogenase promotes pancreatic cancer development by interacting with eIF4A1 and eIF4E. J Exp Clin Cancer Res (2019) 38(1):66. doi: 10.1186/s13046-019-1053-y
111. Zhou YY, Chen LP, Zhang Y, Hu SK, Dong ZJ, Wu M, et al. Integrated transcriptomic analysis reveals hub genes involved in diagnosis and prognosis of pancreatic cancer. Mol Med (2019) 25(1):47. doi: 10.1186/s10020-019-0113-2
112. Ross KC, Andrews AJ, Marion CD, Yen TJ, Bhattacharjee V. Identification of the serine biosynthesis pathway as a critical component of BRAF inhibitor resistance of melanoma, pancreatic, and non-small cell lung cancer cells. Mol Cancer Ther (2017) 16(8):1596–609. doi: 10.1158/1535-7163.MCT-16-0798
113. Sullivan MR, Mattaini KR, Dennstedt EA, Nguyen AA, Sivanand S, Reilly MF, et al. Increased serine synthesis provides an advantage for tumors arising in tissues where serine levels are limiting. Cell Metab (2019) 29(6):1410–1421.e4. doi: 10.1016/j.cmet.2019.02.015
114. Bialopiotrowicz E, Noyszewska-Kania M, Kachamakova-Trojanowska N, Loboda A, Cybulska M, Grochowska A, et al. Serine biosynthesis pathway supports MYC-miR-494-EZH2 feed-forward circuit necessary to maintain metabolic and epigenetic reprogramming of burkitt lymphoma cells. Cancers (2020) 12(3):580. doi: 10.3390/cancers12030580
115. Song Z, Feng C, Lu Y, Lin Y, Dong C. PHGDH is an independent prognosis marker and contributes cell proliferation, migration and invasion in human pancreatic cancer. Gene (2018) 642:43–50. doi: 10.1016/j.gene.2017.11.014
116. Metcalf S, Dougherty S, Kruer T, Hasan N, Biyik-Sit R, Reynolds L, et al. Selective loss of phosphoserine aminotransferase 1 (PSAT1) suppresses migration, invasion, and experimental metastasis in triple negative breast cancer. Clin Experiment Metastasis (2020) 37(1):187–97. doi: 10.1007/s10585-019-10000-7
117. Rawat V, Malvi P, Della Manna D, Yang ES, Bugide S, Zhang X, et al. PSPH promotes melanoma growth and metastasis by metabolic deregulation-mediated transcriptional activation of NR4A1. Oncogene (2021) 40(13):2448–62. doi: 10.1038/s41388-021-01683-y
118. Dekhne AS, Ning C, Nayeen MJ, Shah K, Kalpage H, Frühauf J, et al. Cellular pharmacodynamics of a novel Pyrrolo[3,2-d]pyrimidine inhibitor targeting mitochondrial and cytosolic one-carbon metabolism. Mol Pharmacol (2020) 97(1):9–22. doi: 10.1124/mol.119.117937
119. Dekhne AS, Shah K, Ducker GS, Katinas JM, Wong-Roushar J, Nayeen MJ, et al. Novel Pyrrolo[3,2-d]pyrimidine compounds target mitochondrial and cytosolic one-carbon metabolism with broad-spectrum antitumor efficacy. Mol Cancer Ther (2019) 18(10):1787–99. doi: 10.1158/1535-7163.MCT-19-0037
120. Wang JQ, Wang LY, Li SJ, Tong T, Wang L, Huang CS, et al. Histone methyltransferase G9a inhibitor-loaded redox-responsive nanoparticles for pancreatic ductal adenocarcinoma therapy. Nanoscale (2020) 12(29):15767–74. doi: 10.1039/D0NR03138K
121. Sun HW, Yu XJ, Wu WC, Chen J, Shi M, Zheng L, et al. GLUT1 and ASCT2 as predictors for prognosis of hepatocellular carcinoma. PloS One (2016) 11(12):e0168907. doi: 10.1371/journal.pone.0168907
122. Sharma A, Smyrk TC, Levy MJ, Topazian MA, Chari ST. Fasting blood glucose levels provide estimate of duration and progression of pancreatic cancer before diagnosis. Gastroenterology (2018) 155(2):490–500.e2. doi: 10.1053/j.gastro.2018.04.025
123. Yan L, Raj P, Yao W, Ying H. Glucose metabolism in pancreatic cancer. J Biol Chem (2003) 11(10).
124. Penheiter AR, Deelchand DK, Kittelson E, Damgard SE, Murphy SJ, O'Brien DR, et al. Identification of a pyruvate-to-lactate signature in pancreatic intraductal papillary mucinous neoplasms. Pancreatology (2018) 18(1):46–53. doi: 10.1016/j.pan.2017.11.006
125. McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, Kletzien RF, et al. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metab (2015) 22(4):682–94. doi: 10.1016/j.cmet.2015.07.028
126. Cortese N, Capretti G, Barbagallo M, Rigamonti A, Takis PG, Castino GF, et al. Metabolome of pancreatic juice delineates distinct clinical profiles of pancreatic cancer and reveals a link between glucose metabolism and PD-1(+) cells. Cancer Immunol Res (2020) 8(4):493–505. doi: 10.1158/2326-6066.CIR-19-0403
127. Bhagat TD, Von Ahrens D, Dawlaty M, Zou Y, Baddour J, Achreja A, et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. eLife (2019) 8:e50663. doi: 10.7554/eLife.50663
128. Brockmueller A, Sameri S, Liskova A, Zhai K, Varghese E, Samuel SM, et al. Resveratrol's anti-cancer effects through the modulation of tumor glucose metabolism. Cancers (2021) 13(2):188. doi: 10.3390/cancers13020188
129. Anderson M, Marayati R, Moffitt R, Yeh JJ. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget (2017) 8(34):56081–94. doi: 10.18632/oncotarget.9760
130. Amendola CR, Mahaffey JP, Parker SJ, Ahearn IM, Chen WC, Zhou M, et al. KRAS4A directly regulates hexokinase 1. Nature (2019) 576(7787):482–6. doi: 10.1038/s41586-019-1832-9
131. Tsutsumi S, Yanagawa T, Shimura T, Kuwano H, Raz A. Autocrine motility factor signaling enhances pancreatic cancer metastasis. Clin Cancer Res (2004) 10(22):7775–84. doi: 10.1158/1078-0432.CCR-04-1015
132. Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepato-gastroenterology (2008) 55(88):2016–27.
133. Moir JAG, Long A, Haugk B, French JJ, Charnley RM, Manas DM, et al. Therapeutic strategies toward lactate dehydrogenase within the tumor microenvironment of pancreatic cancer. Pancreas (2020) 49(10):1364–71. doi: 10.1097/MPA.0000000000001689
134. Maftouh M, Avan A, Sciarrillo R, Granchi C, Leon LG, Rani R, et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br J Cancer (2014) 110(1):172–82. doi: 10.1038/bjc.2013.681
135. Ponsuksili S, Trakooljul N, Hadlich F, Methling K, Lalk M, Murani E, et al. Genetic regulation of liver metabolites and transcripts linking to biochemical-clinical parameters. Front Genet (2019) 10:348. doi: 10.3389/fgene.2019.00348
136. Schrader H, Menge BA, Belyaev O, Uhl W, Schmidt WE, Meier JJ. Amino acid malnutrition in patients with chronic pancreatitis and pancreatic carcinoma. Pancreas (2009) 38(4):416–21. doi: 10.1097/MPA.0b013e318194fc7a
137. Parker SJ, Amendola CR, Hollinshead KER, Yu Q, Yamamoto K, Encarnacion-Rosado J, et al. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discovery (2020) 10(7):1018–37. doi: 10.1158/2159-8290.CD-19-0959
138. Dong S, Wang L, Guo YB, Ying HF, Shen XH, Meng ZQ, et al. Risk factors of liver metastasis from advanced pancreatic adenocarcinoma: a large multicenter cohort study. World J Surg Oncol (2017) 15(1):120. doi: 10.1186/s12957-016-1064-5
139. Riedl JM, Posch F, Prager G, Eisterer W, Oehler L, Sliwa T, et al. The AST/ALT (De ritis) ratio predicts clinical outcome in patients with pancreatic cancer treated with first-line nab-paclitaxel and gemcitabine: post hoc analysis of an Austrian multicenter, noninterventional study. Ther Adv Med Oncol (2020) 12:1758835919900872. doi: 10.1177/1758835919900872
140. Fu Y, Ding L, Yang X, Ding Z, Huang X, Zhang L, et al. Asparagine synthetase-mediated l-asparagine metabolism disorder promotes the perineural invasion of oral squamous cell carcinoma. Front Oncol (2021) 11:637226. doi: 10.3389/fonc.2021.637226
141. Zheng M, Guo J, Xu J, Yang K, Tang R, Gu X, et al. Ixocarpalactone a from dietary tomatillo inhibits pancreatic cancer growth by targeting PHGDH. Food Funct (2019) 10(6):3386–95. doi: 10.1039/C9FO00394K
142. Yan Y, Huang P, Mao K, He C, Xu Q, Zhang M, et al. Anti-oncogene PTPN13 inactivation by hepatitis b virus X protein counteracts IGF2BP1 to promote hepatocellular carcinoma progression. Oncogene (2021) 40(1):28–45. doi: 10.1038/s41388-020-01498-3
143. Ceyhan GO, Bergmann F, Kadihasanoglu M, Altintas B, Demir IE, Hinz U, et al. Pancreatic neuropathy and neuropathic pain–a comprehensive pathomorphological study of 546 cases. Gastroenterology (2009) 136(1):177–186.e1. doi: 10.1053/j.gastro.2008.09.029
144. Hessmann E, Buchholz SM, Demir IE, Singh SK, Gress TM, Ellenrieder V, et al. Microenvironmental determinants of pancreatic cancer. Physiol Rev (2020) 100(4):1707–51. doi: 10.1152/physrev.00042.2019
145. Demir IE, Friess H, Ceyhan GO. Nerve-cancer interactions in the stromal biology of pancreatic cancer. Front Physiol (2012) 3:97. doi: 10.3389/fphys.2012.00097
146. Jiang SH, Li J, Dong FY, Yang JY, Liu DJ, Yang XM, et al. Increased serotonin signaling contributes to the warburg effect in pancreatic tumor cells under metabolic stress and promotes growth of pancreatic tumors in mice. Gastroenterology (2017) 153(1):277–291.e19. doi: 10.1053/j.gastro.2017.03.008
147. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016) 536(7617):479–83. doi: 10.1038/nature19084
148. Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods (2005) 2(8):599–605. doi: 10.1038/nmeth777
149. Harjes U. The neuronal-metabolic interface, nature reviews. Cancer (2021) 21(2):68. doi: 10.1038/s41568-020-00324-y
150. Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, et al. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol (2010) 6(3):209–17. doi: 10.1038/nchembio.304
151. Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, et al. Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem (1998) 273(52):34970–5. doi: 10.1074/jbc.273.52.34970
152. Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc (2012) 7(8):1534–50. doi: 10.1038/nprot.2012.086
153. Ghilardi JR, Freeman KT, Jimenez-Andrade JM, Mantyh WG, Bloom AP, Kuskowski MA, et al. Administration of a tropomyosin receptor kinase inhibitor attenuates sarcoma-induced nerve sprouting, neuroma formation and bone cancer pain. Mol Pain (2010) 6:87. doi: 10.1186/1744-8069-6-87
154. Gu I, Gregory E, Atwood C, Lee SO, Song YH. Exploring the role of metabolites in cancer and the associated nerve crosstalk. Nutrients (2022) 14(9):1722. doi: 10.3390/nu14091722
155. Nusser N, Gosmanova E, Makarova N, Fujiwara Y, Yang L, Guo F, et al. Serine phosphorylation differentially affects RhoA binding to effectors: implications to NGF-induced neurite outgrowth. Cell signalling (2006) 18(5):704–14. doi: 10.1016/j.cellsig.2005.06.010
156. Zhou L, Too HP. Mitochondrial localized STAT3 is involved in NGF induced neurite outgrowth. PloS One (2011) 6(6):e21680. doi: 10.1371/journal.pone.0021680
157. Kim E, Jung S, Park WS, Lee JH, Shin R, Heo SC, et al. Upregulation of SLC2A3 gene and prognosis in colorectal carcinoma: analysis of TCGA data. BMC Cancer (2019) 19(1):302. doi: 10.1186/s12885-019-5475-x
Keywords: malignant tumors, pancreatic, innervation, metabolic rearrangements, cancer
Citation: Dong M, Cao L, Cui R and Xie Y (2022) The connection between innervation and metabolic rearrangements in pancreatic cancer through serine. Front. Oncol. 12:992927. doi: 10.3389/fonc.2022.992927
Received: 13 July 2022; Accepted: 31 October 2022;
Published: 13 December 2022.
Edited by:
Direna Alonso Curbelo, Institute for Research in Biomedicine, SpainReviewed by:
Ludi Yang, Shanghai Jiao Tong University, ChinaCopyright © 2022 Dong, Cao, Cui and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ranji Cui, Y3VpcmFuamlAamx1LmVkdS5jbg==; Yingjun Xie, eGlleno1NEBqbHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.