 Undraga Schagdarsurengin
Undraga Schagdarsurengin Vanessa Breiding1
Vanessa Breiding1 Maria Loose
Maria Loose Temuujin Dansranjav
Temuujin Dansranjav
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 26 September 2022
Sec. Genitourinary Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.991368
This article is part of the Research TopicEpigenetics in Prostate CancerView all 7 articles
The use of immune adjuvants such as toll-like receptor (TLR) agonists reflects a novel strategy in prostate cancer (PCa) therapy. However, interleukin-1 receptor associated kinase 1 (IRAK1), a central effector of TLR signaling, has been shown to be responsible for resistance to radiation-induced tumor cell death. In order to better understand the function and epigenetic regulation of IRAK1 in PCa, we performed in vitro cell culture experiments together with integrative bioinformatic studies using the latest single-cell RNA-sequencing data of human PCa and normal prostate (NOR), and data from The Cancer Genome Atlas. We focused on key effectors of TLR signaling, the Myddosome-complex components IRAK1, IRAK4 and MYD88 (myeloid differentiation primary response 88), and TRAF6 (tumor-necrosis-factor receptor associated factor 6). In PCa, IRAK1-mRNA was specifically enriched in luminal epithelial cells, representing 57% of all cells, whereas IRAK4 and MYD88 were predominantly expressed in leukocytes, and TRAF6, in endothelial cells. Compared to NOR, only IRAK1 was significantly overexpressed in PCa (Benjamini-Hochberg adjusted p<2x10-8), whereas the expression of IRAK4, MYD88, and TRAF6 was unchanged in PCa, and IRAK1-expression was inversely correlated with a specific differentially methylated region (IRAK1-DMR) within a predicted promoter region enriched for H3K27ac (Spearman correlation r<-0.36; Fisher’s test, p<10-10). Transcription factors with high binding affinities in IRAK1-DMR were significantly enriched for canonical pathways associated with viral infection and carcinogenic transformation in the Kyoto Encyclopedia of Gene and Genomes analysis. DU145 cells, exhibiting hypermethylated IRAK1-DMR and low IRAK1-expression, reacted with 4-fold increased IRAK1-expression upon combined treatment with 5-aza-2-deoxycytidine and trichostatin A, and were unresponsive to infection with the uropathogenic Escherichia coli strain UTI89. In contrast, PC3 and LNCaP cells, exhibiting hypomethylated IRAK1-DMR and high endogenous IRAK1-mRNA levels, responded with strong activation of IRAK1-expression to UTI89 infection. In summary, exclusive overexpression of IRAK1 was observed in luminal epithelial cells in PCa, suggesting it has a role in addition to Myddosome-dependent TLR signaling. Our data show that the endogenous epigenetic status of PCa cells within IRAK1-DMR is decisive for IRAK1 expression and should be considered as a predictive marker when selective IRAK1-targeting therapies are considered.
Prostate cancer (PCa) is the second most frequent malignancy after lung cancer, and the fifth leading cause of cancer-related deaths among men worldwide (1). Chronic inflammation is commonly observed in normal and malignant prostates and is considered to be one of the main driving mechanisms of prostate carcinogenesis (2, 3).
Toll-like receptors (TLRs) are a family of transmembrane proteins that are the main sensors of innate and adaptive immunity. They are responsible for the activation of pro-inflammatory cytokines and chemokines in inflammatory cells within tumors, as has been shown in hepatocellular, head neck, gastric, and breast cancers (4–7). In the prostate, TLR4 and TLR9 are known to be expressed in normal and malignant cells, thereby enabling prostate epithelial cells to act as immune sensors (8–10).
Under certain conditions, inflammation can counter tumor progression, and activation of effective antitumor immunity is considered a potent adjuvant therapy option (10). As recently reviewed, TLR stimulation in combination with chemotherapy or radiotherapy has been shown to be effective against tumor progression in multiple clinical trials (11). Immunotherapy with TLR agonists significantly improves the outcomes of radiation therapy (RT) in preclinical models (11, 12). The antitumor immune response, which is triggered by tumor antigens released from irradiated tumor cells, is considered to be the main mechanism underlying this therapeutic effect (13, 14). The assembly of the Myddosome complex and recruitment of tumor necrosis factor receptor-associated factor 6 (TRAF6) to the Myddosome complex are crucial events that ultimately promote Myddosome-dependent TLR immune signaling (15). The interleukin-1 receptor-associated kinase 1 (IRAK1) is a critical component of the Myddosome complex and is one of the main effectors of TLR signaling. In human cancer cells and a zebrafish model, IRAK1 was shown to be responsible for RT resistance (14). Selective IRAK1 inhibitors, such as the organic JAK2/FLT3 inhibitor pacritinib and chemical compounds, such as Jh-X-119-01, are being considered for cancer treatment (16). However, setting up individualized PCa therapy strategies may be complicated by the molecular heterogeneity of the tumors. Despite its critical role in tumorigenesis and tumor therapy, little is known about PCa-associated epigenetic and transcriptional regulation of genes encoding effectors of TLR signaling. In order to better understand and utilize the TLR-mediated anti-tumor immune response in PCa treatment, it is essential to first characterize the role of epithelial cells in immune signaling and the function of key effectors of TLR signaling.
Here, we analyzed genes encoding key components of the Myddosome complex, including IRAK1, IRAK4, and MYD88, and the TLR adaptor protein TRAF6, using recently published single-cell RNA-seq data of PCa (17), The Cancer Genome Atlas (TCGA) data bank, and in vitro cell culture experiments, with a focus on their epigenetic regulation in PCa. We showed that in PCa, IRAK1 plays an exclusive role in the luminal epithelial cell population and is tightly regulated by the epigenetic status of a specific differentially methylated region (IRAK1-DMR) within the predicted gene promoter.
Gene expression matrix files from single-cell RNA sequencing (scRNA-seq) of eight radical prostatectomy (RP) specimens from men with localized PCa were downloaded from the Gene Expression Omnibus (GEO) database GSE176031 (17) (Table S1). The data were computationally processed using the Seurat (v4.1) package (18). Cells with fewer than 200 detected genes and mitochondrial levels greater than 5% were excluded from analysis. Doublets were identified using DoubletFinder (v2.03) and were removed. Only the genes expressed in more than three cells were considered. Data were merged using the integration method based on commonly expressed anchor genes using ‘FindIntegrationAnchors’ and ‘IntegrateData’ functions in the Seurat package (19). Subsequent dimensionality reduction, clustering, and visualization were performed using the ‘FindNeighbors’ and ‘FindClusters’ functions of the Seurat package. Cells in clusters were annotated using the AUCell package (v1.16) utilizing cell-type signature gene sets, which were generated by single-cell profiling of the normal prostate (20) (Table S2).
Gene expression across the major cell types was visualized using the R package Nebulosa (v1.4) (21), which uses weighted kernel density estimation to recover gene expression signals.
To assess the role of DNA methylation in the expression of IRAK1, IRAK4, MYD88 and TRAF6, bulk RNA sequencing and 450K Illumina methylation data from 341 PCa and 35 normal prostate tissue specimens from The Cancer Genome Atlas (TCGA) were utilized. Supplemental clinical data, raw methylation data (‘IDAT’) and RNA-seq data (‘HTSeq-counts’) were extracted from TCGA using the R package ‘TCGAbiolinks’ (v2.18.0). Data pre-processing and subsequent analyses were performed using the R packages ‘minfi’ (v1.36.0) and ‘missMethyl’ (v1.28) for 450K Illumina methylation data, and ‘Biobase’ (v2.50) for RNA-seq data. Furthermore, IRAK1 expression was analyzed in castration-resistant prostate cancer metastases (mCRPC, n=99) (TCGA: Project ID WCDT-MCRPC). CRPC-related overexpressed hub genes, including TARDBP, HNRNPA2B1, MRPS25, MAPK8IP3, CCDC14 and GOLGA8B, which were also found to be associated with PCa progression and prognosis (22), were used to select non-CRPC-like PCa specimens (N=194) within TCGA. Differential expression analysis was performed using the empirical Bayes statistics (eBayes) function in the ‘limma’ package. P-values were adjusted for multiple comparisons using the Benjamini-Hochberg (BH) method. Differentially methylated regions were identified using the DMRcate package (v2.8.4), hidden Markov model and Fisher exact test (HMM-Fisher). For illustration, ChAMP graphical user interface was used (23).
The human PCa cell lines PC3, LNCaP, and DU145 were obtained from German Collection of Microorganisms and Cell Cultures GmbH (DSMZ, Braunschweig, Germany). DU145 and LNCaP cells were cultured in RPMI 1640 medium (Gibco), and PC3 in DMEM (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Gibco). Cells were cultured in 10 cm dishes in 5% CO2 at 37°C to 70–90% confluency and then used for subsequent analyses.
The effects of the DNA methyltransferase 1 (DNMT1) inhibitor 5-aza-2′-deoxycytidine (5-AZA) (Sigma Aldrich Corp, St. Louis, MS, USA) and the histone deacetylase (HDAC) inhibitor trichostatin A (TSA) (Upstate Biotechnology, Lake Placid, NY, USA) on the expression of IRAK1 were examined in PCa cell lines. Cells were grown to 40% confluency and treated with 5µM 5-AZA for 96 h and/or with 0.3µM TSA for 24 h. Total RNA was extracted from untreated and treated PC3, DU145, and LNCaP cells using peqGOLD TriFast reagent (VWR, Radnor, PA, USA) according to the manufacturer’s protocol. Reverse transcription (RT) was performed on 1 µg of total RNA using M-MLV transcriptase and an adjusted buffer system (Promega, Madison, WI, USA), random hexamers, and poly-dT primers for 1 h at 42°C. Quantitative PCR (qPCR) was performed using 50 ng of cDNA per PCR in a Rotor-Gene Q PCR Cycler (Qiagen, Hilden, Germany) with IRAK1 and GAPDH, as reference genes (Table S3). Relative IRAK1 expression was calculated by normalizing to GAPDH using the 2 -ΔΔCt method.
IRAK1 promoter methylation was analyzed in the PCa cell lines. DNA was isolated from proteinase K-treated cells using a standard phenol-chloroform extraction method. In total, 2µg of DNA was bisulfite-treated using an EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA). Specific DNA fragments representing IRAK1-DMR with six CpG sites were amplified using the appropriate primer sets (Table S3). The PCR products were separated on a 2% agarose gel according to product size and pyrosequenced using the PyroMark Q24 System (Qiagen). Methylation values were analyzed using the PyroMark Q24 Software (Qiagen). The degree of methylation for each CpG site was calculated in a program as the percentage of methylated cytosines over the sum of the total cytosines at the indicated single CpG site.
The uropathogenic Escherichia coli strain UTI89 was obtained from DSMZ and used to infect PCa cell lines PC3, LNCaP, and DU145. In total, 1×106 PCa cells cultured in 6-well plates were infected with UTI89 at a multiplicity of infection (MOI) of 10 for two and four hours. Untreated and treated cells were washed twice with phosphate-buffered saline and lysed with peqGOLD TriFast reagent (VWR). RNA extraction and RT-qPCR were performed as previously described.
The regulatory function of IRAK1-DMR was analyzed using the Encyclopedia of DNA Elements (ENCODE) and taking into consideration the enrichment of histone 3 acetylated at lysine 27 (H3K27ac) in the predicted IRAK1 enhancer and promoter region (gathered from Segway and ChromHMM data) and JASPAR-2022 data, a core database of transcription factor binding sites (TF-BSs). Prediction of TF-BSs was made according to the position weight matrix (PWM), and relative scores ≥ 0.8 with a p-value < 0.001 were considered as true TF-BSs. Functional characterization of the identified TFs was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG).
The expression of genes encoding the main components of the Myddosome complex, IRAK1, IRAK4 and MYD88, and the adaptor protein TRAF6 were assigned to specific cell types within PCa using scRNA-seq data generated from eight radical prostatectomy (RP) specimens from men with localized PCa (17) (Figure 1 and Table S1). Overall, 9643 cells underwent clustering analysis using principal component analysis (PCA) in the Seurat package (18). Using signature gene sets generated from single-cell profiling of normal prostate (20), PCa cells were classified as luminal (57%), basal (11%), hillock (1%), and club (2%) cells as the main epithelial cell lineage; endothelial (4%), fibroblast (2%), and smooth muscle (3%) cells as the main stromal lineage; and leukocytes (20%) as immune cells (Figures 1A–C; Table S2). The identities of the generated 16 cell clusters were assigned in compliance with the major (>80%) cell type represented in a cluster (Figures 1B, C). IRAK1 is specifically enriched in luminal epithelial cell clusters. In comparison, IRAK4 and MYD88 were predominantly expressed in the leukocyte cluster and TRAF6 was expressed in the endothelial cell cluster (Figure 1D).
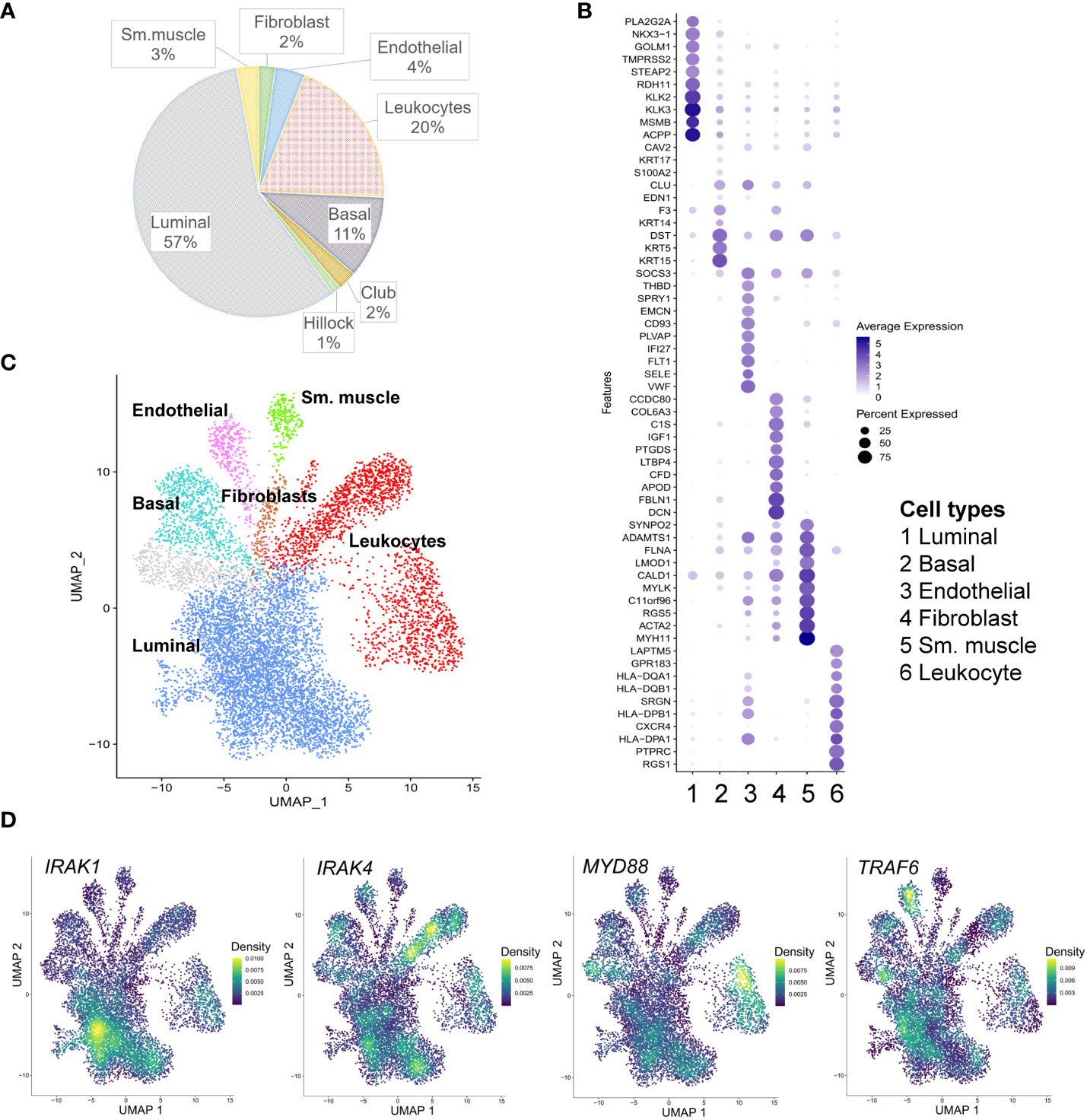
Figure 1 Single cell RNA-sequencing (scRNA-seq) analysis of IRAK1, IRAK4, Myd88 and TRAF6 expression in prostate cancer. (A) Cell type proportions within the merged scRNA-seq dataset from eight radical prostatectomy specimens (17). (B) Dot plot of top 10 marker genes for each of the displayed cell type clusters. Dot size and color represent the percentage of marker gene expression and average scaled expression value (minimum percentage expression 0.2%). (C) Cell type clusters detected in prostate cancer are presented in the uniform manifold approximation and projection (UMAP). (D) Density plots of IRAK1, IRAK4, Myd88 and TRAF6 expression in prostate cancer sm. muscle: smooth muscle cells.
By analyzing bulk RNA-seq data from TCGA using ebayes for differential expression, we found that IRAK1 was significantly overexpressed in prostate tumor tissue (TU) compared to normal prostate tissue (TU, N=341; NOR, N=35; Benjamini-Hochberg adjusted p<2x10-8) (Figures 2A, B). Further differential analysis of IRAK1 expression in non-CRPC and mCRPC specimens showed no difference between these two groups (non-CRPC, N=194; CRPC, N=99; Benjamini-Hochberg adjusted p>0.05) (data not shown). In contrast, IRAK4, TRAF6 and MYD88 were not differentially expressed in PCa and exhibited relatively low mRNA levels (Figures 2A, B). By analyzing 450K Illumina methylation array data from TCGA, we detected a PCa-associated differentially methylated region (DMR) within the predicted IRAK1-promoter (hg19 chrX:153283694-153284103) (TU, N=341; NOR, N=35; HMM-Fischer test, p<1.8x10-10, Figure 3A). Two CpG sites flanking this DMR were significantly hypomethylated in PCa (cg23604959 and cg02742918), and their methylation status showed an inverse correlation with IRAK1 mRNA levels in PCa (n=341, Spearman coefficient =-0.36 and -0.37, respectively) (Figure 3B). The region including cg23604959 and cg02742918 (hg19 chrX:153283646-153284152) was determined as IRAK1-DMR in PCa (Figures 3, 4).
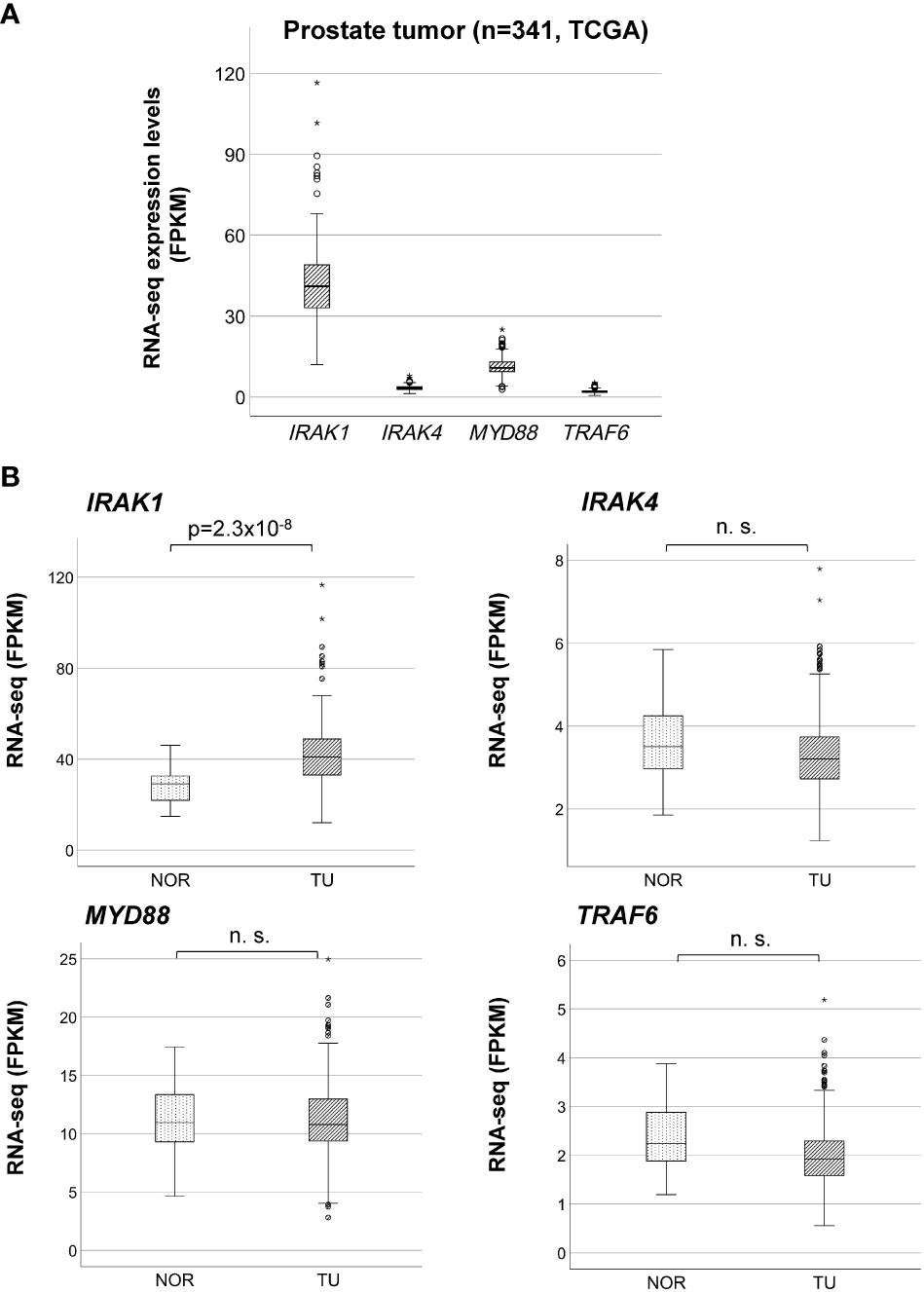
Figure 2 Overexpression of IRAK1 in prostate cancer. (A) Comparison of bulk mRNA expression levels of IRAK1, IRAK4, Myd88 and TRAF6 in prostate cancer. Relative RNA expression is given in fragments per kilo base per million mapped reads (FPKM). (B) Differential mRNA expression analysis of IRAK1, IRAK4, Myd88 and TRAF6 in normal (NOR) and cancer (TU) prostate tissue. Significant difference was found only for IRAK1 (eBayes, Benjamini-Hochberg adjusted p-value). n. s., not significant (p>0.05).
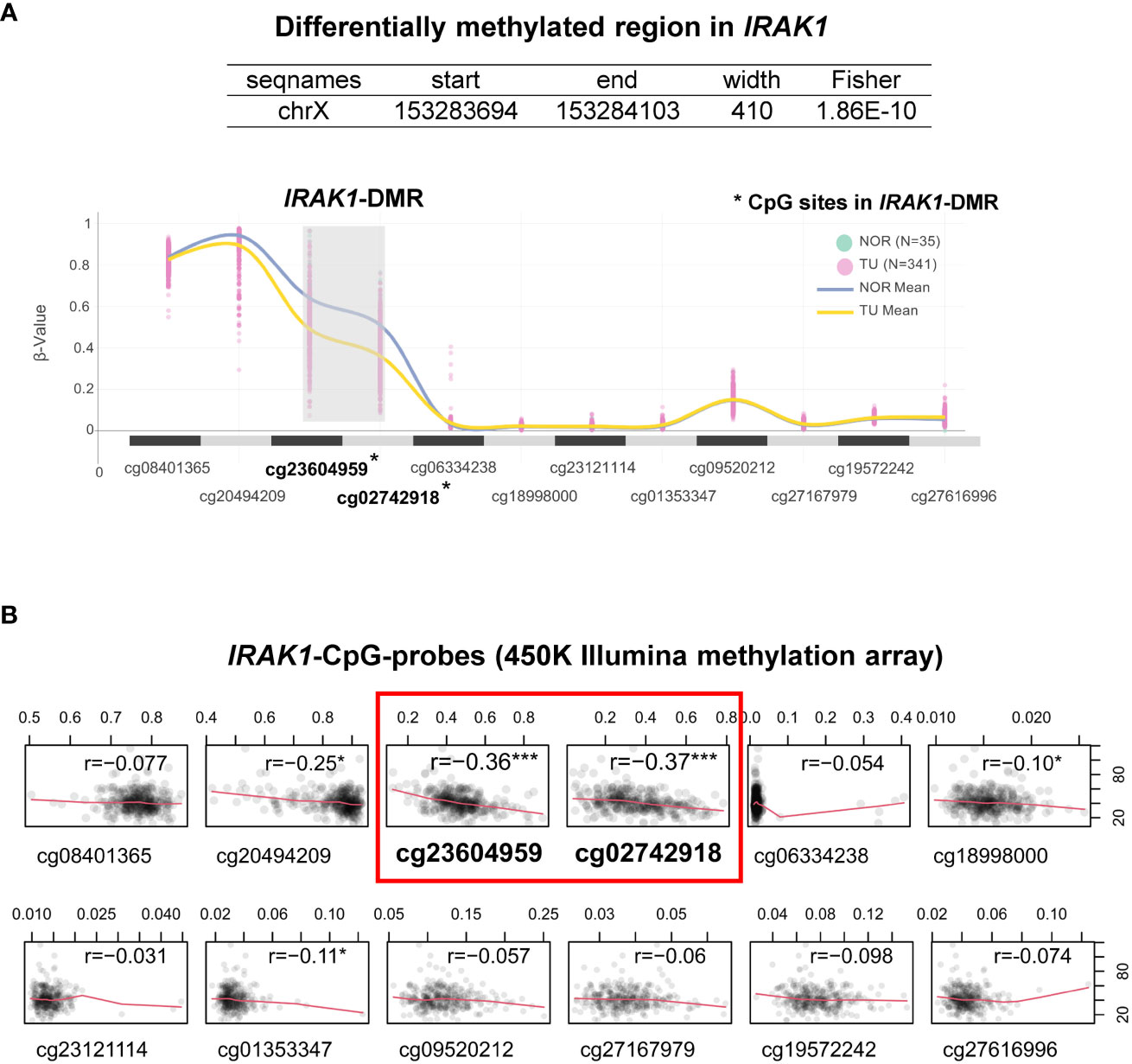
Figure 3 Characterization of the Differentially Methylated Region in IRAK1 (IRAK1-DMR). (A) A snapshot of graphical user interface from ChAMP (Chip Analysis Methylation Pipeline) package showing a trend of DNA methylation levels (β-values) at different CpG-sites (presented as CpG-probes cg-number) in normal prostate (NOR) and prostate cancer tissue (TU). Lines represent mean methylation levels of analyzed CpG-probes (NOR, blue; TU, yellow). (B) Correlation plots of IRAK1-mRNA levels and IRAK1-methylation values in different CpG-probes (Spearman’s correlation coefficient r is indicated for each correlation analysis). *CpG-sites located within IRAK1-DMR. Significance levels: ***p<0.001, *p<0.05.
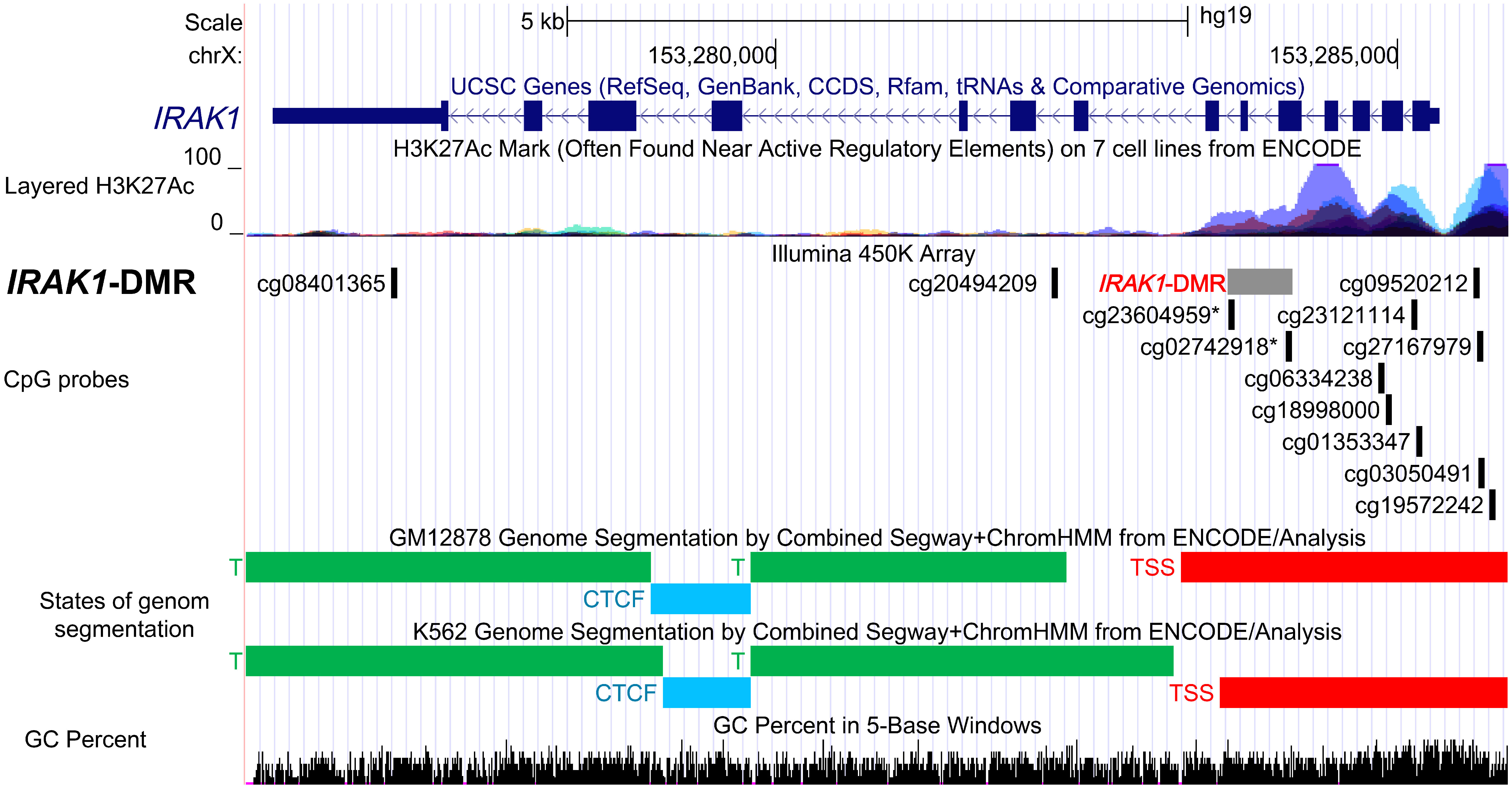
Figure 4 Functional characterization of the differentially methylated region in IRAK1 gene (IRAK1-DMR) using ENCODE Data in the UCSC Genome Browser. Genomic position of IRAK1-DMR is shown in the USCS genome browser with reference to positions of CpG-probes (Illumina 450K Methylation array), enrichment of histone 3 lysine 27 acetylation (H3K27ac) peaks (layered H3K27ac peaks generated on seven human cell lines), predicted functional genomic states assembled by ChromHMM and Segway segmentation analysis in GM12878 and K562 cell lines (red, predicted promoter region including transcription start site, TSS). The GC percent track shows the densities of G (guanine) and C (cytosine).
To assess the possible regulatory function of the IRAK1-DMR that was identified, we used data from ENCODE, which includes ChromHMM and Segway segmentation that predict and classify the functions of different gene regions (Figure 4). For the prediction of enhancer and promoter regions, Segway segmentation tools use features such as DNase-1 hypersensitivity and epigenetic marks. We found that IRAK1-DMR is located within the active promoter region and is enriched for histone 3 acetylated at lysine 27 (H3K27ac), a histone mark that is often found near active gene promoters (Figure 4).
Next, using the Transcription Factors Track database (JASPAR-2022), we identified 211 TFs with predicted strong binding sites in IRAK1-DMR (PWM relative score ≥ 0.8, p < 0.001) (Table S4). The biological function of TFs that bind specifically to IRAK1-DMR was further analyzed using KEGG enrichment analysis (Figures 5A, B). A substantial number of TFs were enriched in pathways associated with carcinogenic transformation and viral infection (Figure 5A). The top three enriched KEGG pathways were transcriptional dysregulation in cancer, herpes simplex virus 1 infection, and human T-cell leukemia virus 1 infection. Notably, 15 TFs were identified as members of the E26 transformation-specific (ETS) family of transcription factors, and genes encoding these TFs are known to fuse with hormone-regulated genes such as TMPRSS2 (transmembrane protease serine 2), SLC45A3 (solute carrier family 45 member 3, also known as prostate cancer-associated protein), and DDX5 (DEAD-Box Helicase 5) (24) (Figure 5B).
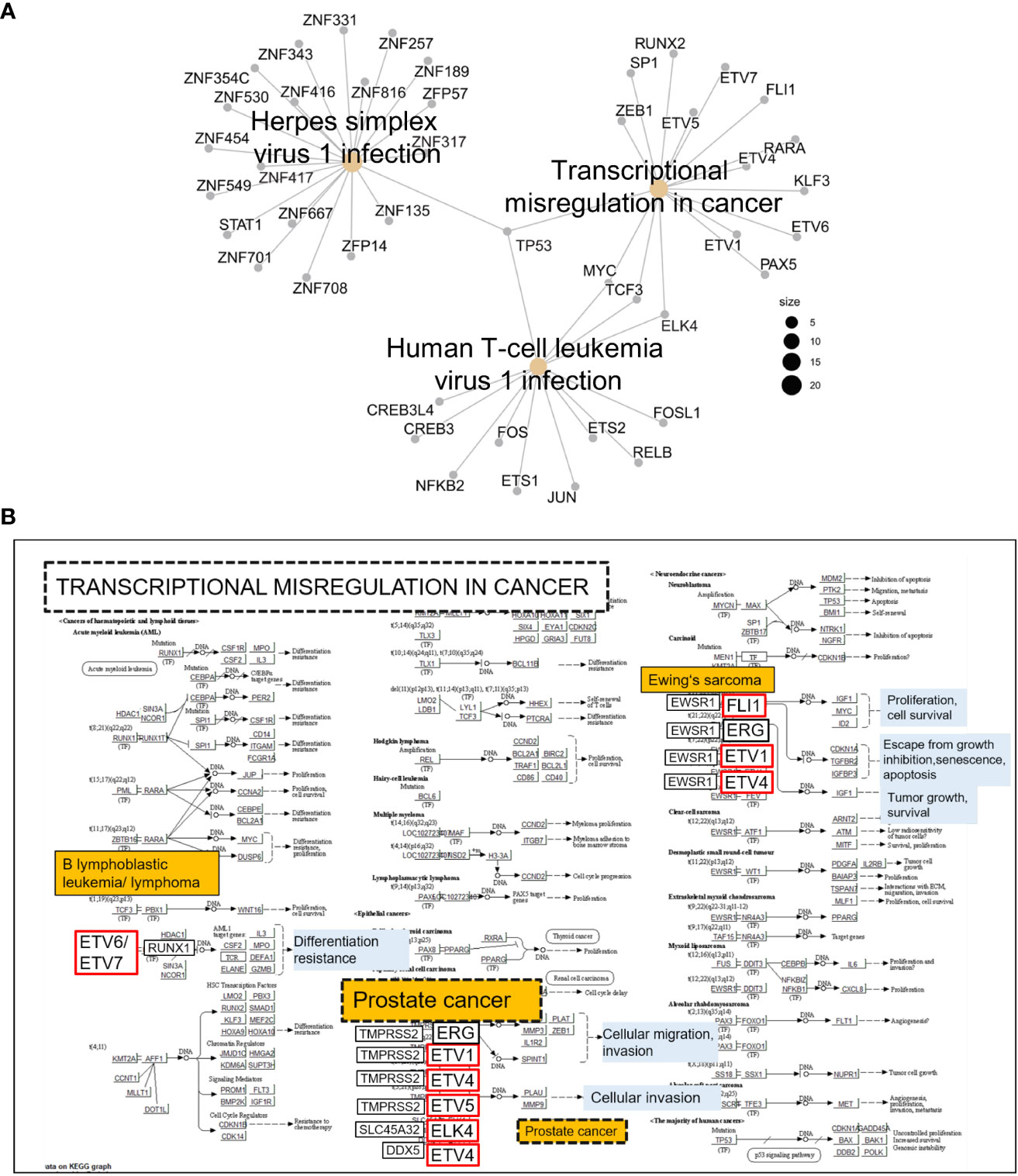
Figure 5 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of transcription factors (TFs) with predicted binding sites in IRAK1-DMR. TFs with binding scores ≥300 were considered (JASPAR-2022). (A) Cnet plot showing top three enriched KEGG pathways among TFs with predicted strong binding sites in IRAK1-DMR. (B) The enriched KEGG pathway ‘Transcriptional misregulation in cancer’ is shown together with implicated TFs exhibiting predicted binding sites in IRAK1-DMR (red framed).
The decisive role of DNA methylation in IRAK1-DMR in IRAK1 activation was analyzed and confirmed in cell culture experiments using bacterial infection. First, the methylation status of IRAK1-DMR was analyzed in DU145, LNCaP, and PC3 cell lines and was correlated with IRAK1 expression (Figure 6). In DU145 cells, IRAK1-DMR hypermethylation (62% by pyrosequencing) was associated with markedly low endogenous gene expression (Figures 6A, B). A strong increase in IRAK1 expression (>400%) was achieved by the combined treatment of DU145 cells with the DNMT1 inhibitor 5-AZA and HDAC inhibitor TSA (Figure 6B). 5-AZA treatment alone had no effect on IRAK1 expression in DU145 cells (Figure 6B). In contrast, PC3 and LNCaP cells exhibited weak methylation in IRAK1-DMR (<14.5% by pyrosequencing) and showed >2-fold higher endogenous IRAK1 expression than DU145 cells (Figures 6A, B). Treatment of PC3 and LNCaP cells with 5-AZA led to a moderate increase in IRAK1 expression (146% and 248%, respectively) (Figure 6B). In PC3 and LNCaP, no further increase of IRAK1 expression was achieved by combined treatment of cells with 5-AZA and TSA (Figure 6B).
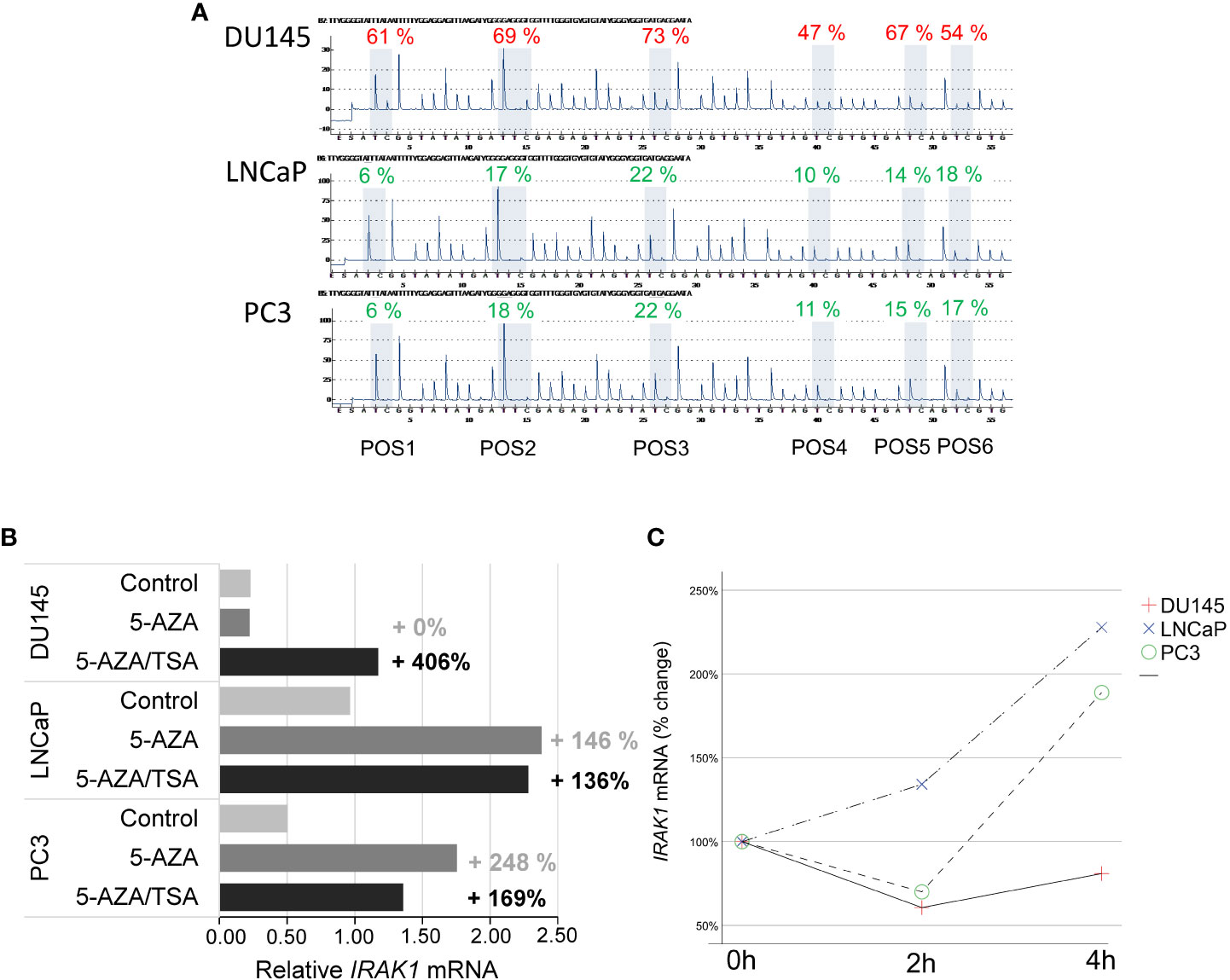
Figure 6 Analysis of DNA methylation in IRAK1-DMR and its impact on IRAK1 expression using prostate cancer cell lines. (A) Pyrosequencing of IRAK1-DMR showed strong methylation in DU145 (47-69%) and weak methylation in LNCaP and PC3 cells (8-22%). (B) Effect of the DNA demethylating agent 5-Aza-2-deoxycytidine (5-AZA) and the histone-deacetylase inhibitor Trichostatin A (TSA) on IRAK1 expression. In DU145, endogenous IRAK1-mRNA levels were low and increased over 400% after combined 5-AZA/TSA-treatment. LNCaP and PC3 cell lines showed higher endogenous IRAK1 expression and lesser response to 5-AZA/TSA treatment than DU145. (C) Treatment with the uropathogenic E. coli strain UTI89 for 4 hours activated IRAK1 expression in LNCaP and PC3, and had no effect in DU145 cells. Expression of IRAK1 in untreated cells is set as 100%.
Four hours after infection of PCa cell lines with uropathogenic E. coli UTI89 at a multiplicity of infection of 10, PC3 and LNCaP cells had a 2-fold increase of IRAK1 expression. In contrast, in DU145 cells, which possess strongly hypermethylated IRAK1-DMR and low endogenous IRAK1 expression, UTI89 infection did not induce IRAK1 activation (Figure 6C).
In the scRNA-seq data analysis, IRAK1 was mainly enriched in luminal prostate epithelial cells, representing the majority of cells detected in PCa. In contrast, in PCa, IRAK4 and MYD88 were predominantly expressed in leukocytes, and TRAF6 was predominantly expressed in endothelial cells. This result implies a distinctive role for IRAK1 in epithelial cells in PCa in a Myddosome-independent manner. Prior studies have demonstrated that IRAK1 is biologically active in a Myddosome-independent manner by interacting with alternative proteins and even independent of its catalytic function. Several substrates that interact with IRAK1 have been identified, including IRAK1 itself, Tollip, E3 ligase Pellino, transcription factor IRF7 (interferon regulatory factor 7), and intracellular adaptor protein TRIF (TIR-domain-containing adapter-inducing interferon-β (TRIF) (25–29). It has been shown that IRAK1 does not necessarily need catalytic activity to exert its function. Catalytically inactive IRAK1 with a point mutation at the catalytic site and a splice variant lacking part of the kinase domain were able to induce NF-kB activation (30, 31).
Consistent with the results of the scRNA-seq data analysis, bulk RNA sequencing data from TCGA showed in comparison to normal prostate a PCa-associated overexpression of IRAK1. No difference in IRAK1 expression has been found between non-CRPC and mCRPC specimens. The remaining TLR signaling genes, IRAK4, TRAF6 and Myd88, were not differentially expressed in PCa and exhibited relatively low mRNA levels. Overexpression of IRAK1 has been found in several human carcinomas, including breast, endometrial, lung, and liver cancers, and is significantly associated with poor survival and unfavorable clinical parameters (32–36). Androgen deprivation therapy induces in PCa cells apoptosis and cell death. However, IRAK1 has been shown to have an antiapoptotic effect in cancer cells, and to contribute to developing resistance to radiotherapy as well as to paclitaxel and methotrexate treatment (14, 37, 38). It is known that androgens enhance the glycolytic metabolism and lactate export in PCa cells. Here, the conversion from pyruvate to lactate, which is catalyzed by lactate dehydrogenase (LDH) proteins including the androgen receptor target LDHA1 presents a critical step (39). Current evidence, gathered primarily from studies on beta-cells in pancreatic islets, suggests that LDH release is reduced in the absence of IRAK1 (40). In cell culture and xenograft tumor models, gain of IRAK1 promotes tumorigenic growth and metastasis of breast, head neck, and hepatocellular carcinoma. In accordance with that, specific inhibition of IRAK1 using endogenous inhibitors, e.g. mir-146a or shRNA, has an anti-tumorigenic effect on cells (34, 41, 42).
By analyzing 450K Illumina methylation array data generated in PCa, we discovered a differentially methylated region, IRAK1-DMR. The potential regulatory function of the identified IRAK1-DMR was assessed using ENCODE data, and supported by its localization within the active IRAK1 promoter region and enrichment of the activating histone mark H3K27ac. It is well known that over 70% of regulatory DNA elements marked by H3K27ac are active and positively affect transcription in vivo (43). Importantly, the presence of H3K27ac and low DNA methylation preserves the accessibility of transcription factor-binding sites at the enhancer and promoter regions (44). In our study, TFs with strong binding affinity for IRAK1-DMR were found to represent key components of canonical pathways associated with carcinogenic transformation and virus infection, such as transcriptional misregulation in cancer, herpes simplex virus 1 infection, and human T-cell leukemia virus 1 infection. Notably, a considerable number of members of the ETS family of TFs were found among TFs with strong binding sites on IRAK1-DMR. In approximately 50% of PCa, ETS members are known to fuse with hormone-regulated genes such as TMPRSS2, prostate cancer-associated protein (SLC45A3), and RNA helicase DDX5, as reviewed by Nicholas and colleagues (24). Our results from integrative bioinformatic analyses provide convincing evidence for a decisive role of the DNA methylation status of IRAK1-DMR in transcriptional activation of IRAK1. These findings were further supported by our in vitro cell culture experiments, showing in the PCa cell line DU145 that weak endogenous IRAK1-mRNA expression is correlated with hypermethylation of IRAK1-DMR and can be dramatically increased (>400%) by combined treatment with the DNMT1 inhibitor 5-AZA and the HDAC inhibitor TSA. In contrast, weak DNA methylation in IRAK1-DMR, as was found in the PCa cell lines PC3 and LNCaP, was accompanied by a 2-3-fold higher endogenous IRAK1-mRNA expression than in DU145 cells, and with only a moderate activation of IRAK1 expression upon 5-AZA and TSA treatment (maximum 169%). Importantly, different endogenous methylation statuses in IRAK1-DMR and expression levels of IRAK1, detected in DU145, LNCaP, and PC3 cells, respectively, were decisive determinants for the degree of IRAK1 activation upon uropathogenic E. coli infection. LNCaP and PC3 cells were able to activate IRAK1 4 h after infection, but DU145 cells did not show any response to IRAK1 expression.
The use of immune adjuvants is of growing importance in PCa therapy. Studies on IRAK1 knockout mice and experiments with a specific endogenous IRAK1 inhibitor, miR-146 indicate a clinical advantage for selective IRAK1 inhibition (45, 46). To date, only a few compounds, including the recently described chemical compound Jh-X-119-01 and the already established organic compound pacritinib (known as a potent tyrosine kinase inhibitor of JAK2/FLT3) are known to selectively inhibit IRAK1 (47, 48). Our data emphasize that determining the endogenous DNA methylation status of IRAK1-DMR and expression in PCa is of potential value in predicting the ability of PCa cells to activate IRAK1 and should be considered in setting up individualized PCa treatment algorithms in the clinic.
Publicly available datasets were analyzed in this study. The datasets presented in this study can be found in Gene Expression Omnibus (GEO) database: GSE176031. The accession numbers of used data can be found in the Table S1.
TD and US designed the study. VB performed PCR assays. ML performed E. coli infection experiments. TD performed PCa cell culture experiments and bioinformatic data analysis. FW provided resources. TD and US wrote the paper. All authors contributed to the article and approved the submitted version.
The study was supported by a Research Grant from the University Medical Center Giessen and Marburg to TD and FW (UKGM, project Nr. 6/2012 GI), and the German Research Foundation (DFG) to US and FW (GRK1871 “Molecular pathogenesis of male reproductive disorders”, project 5).
We would like to thank Editage (www.editage.com) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.991368/full#supplementary-material
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. Dennis LK, Lynch CF, Torner JC. Epidemiologic association between prostatitis and prostate cancer. Urology (2002) 60:78–83. doi: 10.1016/S0090-4295(02)01637-0
3. Nelson WG, DeWeese TL, DeMarzo AM. The diet, prostate inflammation, and the development of prostate cancer. Cancer Metastasis Rev (2002) 21:3–16. doi: 10.1023/a:1020110718701
4. Tye H, Kennedy CL, Najdovska M, McLeod L, McCormack W, Hughes N, et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell (2012) 22:466–78. doi: 10.1016/j.ccr.2012.08.010
5. Shi W, Su L, Li Q, Sun L, Lv J, Li J, et al. Suppression of toll-like receptor 2 expression inhibits the bioactivity of human hepatocellular carcinoma. Tumour Biol (2014) 35:9627–37. doi: 10.1007/s13277-014-2268-3
6. Farnebo L, Shahangian A, Lee Y, Shin JH, Scheeren FA, Sunwoo JB. Targeting toll-like receptor 2 inhibits growth of head and neck squamous cell carcinoma. Oncotarget (2015) 6:9897–907. doi: 10.18632/oncotarget.3393
7. Dajon M, Iribarren K, Cremer I. Toll-like receptor stimulation in cancer: A pro- and anti-tumor double-edged sword. Immunobiology (2017) 222:89–100. doi: 10.1016/j.imbio.2016.06.009
8. Kundu SD, Lee C, Billips BK, Habermacher GM, Zhang Q, Liu V, et al. The toll-like receptor pathway: A novel mechanism of infection-induced carcinogenesis of prostate epithelial cells. Prostate (2008) 68:223–9. doi: 10.1002/pros.20710
9. González-Reyes S, Fernández JM, González LO, Aguirre A, Suárez A, González JM, et al. Study of TLR3, TLR4, and TLR9 in prostate carcinomas and their association with biochemical recurrence. Cancer Immunol Immunother (2011) 60:217–26. doi: 10.1007/s00262-010-0931-0
10. Gambara G, de Cesaris P, de Nunzio C, Ziparo E, Tubaro A, Filippini A, et al. Toll-like receptors in prostate infection and cancer between bench and bedside. J Cell Mol Med (2013) 17:713–22. doi: 10.1111/jcmm.12055
11. Ding R, Jiao A, Zhang B. Targeting toll-like receptors on T cells as a therapeutic strategy against tumors. Int Immunopharmacol (2022) 107:108708. doi: 10.1016/j.intimp.2022.108708
12. Roses RE, Xu M, Koski GK, Czerniecki BJ. Radiation therapy and toll-like receptor signaling: Implications for the treatment of cancer. Oncogene (2008) 27:200–7. doi: 10.1038/sj.onc.1210909
13. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13:1050–9. doi: 10.1038/nm1622
14. Liu PH, Shah RB, Li Y, Arora A, Ung PM-U, Raman R, et al. An IRAK1-PIN1 signalling axis drives intrinsic tumour resistance to radiation therapy. Nat Cell Biol (2019) 21:203–13. doi: 10.1038/s41556-018-0260-7
15. Wang JQ, Jeelall YS, Ferguson LL, Horikawa K. Toll-like receptors and cancer: MYD88 mutation and inflammation. Front Immunol (2014) 5:367. doi: 10.3389/fimmu.2014.00367
16. Singer JW;, Fleischman A, Al-Fayoumi S, Mascarenhas JO, Yu Q, Agarwal A. Inhibition of interleukin-1 receptor-associated kinase 1 (IRAK1) as a therapeutic strategy. Oncotarget (2018) 9(70):33416–39. doi: 10.18632/oncotarget.26058
17. Song H, Weinstein HNW, Allegakoen P, Wadsworth MH, Xie J, Yang H, et al. Single-cell analysis of human primary prostate cancer reveals the heterogeneity of tumor-associated epithelial cell states. Nat Commun (2022) 13:141. doi: 10.1038/s41467-021-27322-4
18. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol (2018) 36:411–20. doi: 10.1038/nbt.4096
19. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single-cell data. Cell (2019) 177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031
20. Henry GH, Malewska A, Joseph DB, Malladi VS, Lee J, Torrealba J, et al. A cellular anatomy of the normal adult human prostate and prostatic urethra. Cell Rep (2018) 25:3530–3542.e5. doi: 10.1016/j.celrep.2018.11.086
21. Alquicira-Hernandez J, Powell JE. Nebulosa recovers single-cell gene expression signals by kernel density estimation. Bioinformatics (2021) 37:2485–7. doi: 10.1093/bioinformatics/btab003
22. Cheng Y, Li L, Qin Z, Li X, Qi F. Identification of castration-resistant prostate cancer-related hub genes using weighted gene co-expression network analysis. J Cell Mol Med (2020) 24:8006–17. doi: 10.1111/jcmm.15432
23. Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, et al. ChAMP: 450k chip analysis methylation pipeline. Bioinf (Oxford. England). (2014) 30(3):428–30. doi: 10.1093/bioinformatics/btt684
24. Nicholas TR, Strittmatter BG, Hollenhorst PC. Oncogenic ETS factors in prostate cancer. Adv Exp Med Biol (2019) 1210:409–36. doi: 10.1007/978-3-030-32656-2_18
25. Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, et al. Cutting edge: A novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the toll-like receptor signaling. J Immunol (2002) 169:6668–72. doi: 10.4049/jimmunol.169.12.6668
26. Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by tollip. J Biol Chem (2002) 277:7059–65. doi: 10.1074/jbc.M109537200
27. Strelow A, Kollewe C, Wesche H. Characterization of Pellino2, a substrate of IRAK1 and IRAK4. FEBS Lett (2003) 547:157–61. doi: 10.1016/S0014-5793(03)00697-5
28. Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J Exp Med (2005) 201:915–23. doi: 10.1084/jem.20042372
29. Wang Le, Lu B, He M, Wang Y, Wang Z, Du L. Prostate cancer incidence and mortality: Global status and temporal trends in 89 countries from 2000 to 2019. Front Public Health (2022) 10:811044. doi: 10.3389/fpubh.2022.811044
30. Li X, Commane M, Burns C, Vithalani K, Cao Z, Stark GR. Mutant cells that do not respond to interleukin-1 (IL-1) reveal a novel role for IL-1 receptor-associated kinase. Mol Cell Biol (1999) 19:4643–52. doi: 10.1128/MCB.19.7.4643
31. Jensen LE, Whitehead AS. IRAK1b, a novel alternative splice variant of interleukin-1 receptor-associated kinase (IRAK), mediates interleukin-1 signaling and has prolonged stability. J Biol Chem (2001) 276:29037–44. doi: 10.1074/jbc.M103815200
32. Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene (2008) 27:5643–7. doi: 10.1038/onc.2008.171
33. Behrens C, Feng L, Kadara H, Kim H-J, Lee JJ, Mehran R, et al. Expression of interleukin-1 receptor-associated kinase-1 in non-small cell lung carcinoma and preneoplastic lesions. Clin Cancer Res (2010) 16:34–44. doi: 10.1158/1078-0432.CCR-09-0650
34. Wee ZN, Yatim SMJM, Kohlbauer VK, Feng M, Goh JY, Bao Y, et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat Commun (2015) 6:8746. doi: 10.1038/ncomms9746
35. Ye Z-H, Gao L, Wen D-Y, He Y, Pang Y-Y, Chen G. Diagnostic and prognostic roles of IRAK1 in hepatocellular carcinoma tissues: An analysis of immunohistochemistry and RNA-sequencing data from the cancer genome atlas. Onco. Targets Ther (2017) 10:1711–23. doi: 10.2147/OTT.S132120
36. Wang Y, Wang Y, Duan X, Wang Y, Zhang Z. Interleukin-1 receptor-associated kinase 1 correlates with metastasis and invasion in endometrial carcinoma. J Cell Biochem (2018) 119:2545–55. doi: 10.1002/jcb.26416
37. Liu L, Liu S, Deng P, Liang Y, Xiao R, Tang L-Q, et al. Targeting the IRAK1-S100A9 axis overcomes resistance to paclitaxel in nasopharyngeal carcinoma. Cancer Res (2021) 81:1413–25. doi: 10.1158/0008-5472.CAN-20-2125
38. Rahemi S, Nematollahi-Mahani SN, Rajaie A, Fallah H. Inhibitor of interleukin-1 receptor-associated kinases 1/4, can increase the sensitivity of breast cancer cells to methotrexate. Int J Mol Cell Med (2019) 8:200–9. doi: 10.22088/IJMCM.BUMS.8.3.200
39. Uo T, Sprenger CC, Plymate SR. Androgen receptor signaling and metabolic and cellular plasticity during progression to castration resistant prostate cancer. Front Oncol (2020) 10:580617. doi: 10.3389/fonc.2020.580617
40. Huang Y-N, Chiang S-L, Lin Y-J, Liu S-C, Li Y-H, Liao Y-C, et al. Long, noncoding RNA SRA induces apoptosis of β-cells by promoting the IRAK1/LDHA/Lactate pathway. Int J Mol Sci (2021) 22:1720–38. doi: 10.3390/ijms22041720
41. Adams AK, Bolanos LC, Dexheimer PJ, Karns RA, Aronow BJ, Komurov K, et al. IRAK1 is a novel DEK transcriptional target and is essential for head and neck cancer cell survival. Oncotarget (2015) 6:43395–407. doi: 10.18632/oncotarget.6028
42. Li N, Jiang J, Fu J, Yu T, Wang B, Qin W, et al. Targeting interleukin-1 receptor-associated kinase 1 for human hepatocellular carcinoma. J Exp Clin Cancer Res (2016) 35:140. doi: 10.1186/s13046-016-0413-0
43. Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell (2013) 155:1521–31. doi: 10.1016/j.cell.2013.11.033
44. Charlet J, Duymich CE, Lay FD, Mundbjerg K, Dalsgaard Sørensen K, Liang G, et al. Bivalent regions of cytosine methylation and H3K27 acetylation suggest an active role for DNA methylation at enhancers. Mol Cell (2016) 62:422–31. doi: 10.1016/j.molcel.2016.03.033
45. Guo F, Wu S. Antisense IRAK-1 oligonucleotide blocks activation of NF-κB and AP-1 induced by IL-18. Immunopharmacology (2000) 49(3):S. doi: 10.1016/S0162-3109(00)00200-9
46. Thomas JA, Allen JL, Tsen M, Dubnicoff T, Danao J, Liao XC, et al. Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J Immunol (Baltimore. Md. 1950) (1999) 163(2):S. 978–984. ISSN.1550-6606
47. Hatcher JM, Yang G, Wang Li, Ficarro SB, Buhrlage S, Wu H, et al. Discovery of a selective, covalent IRAK1 inhibitor with antiproliferative activity in MYD88 mutated b-cell lymphoma. ACS medicinal. Chem Lett (2020) 11(11):S.2238–2243. doi: 10.1021/acsmedchemlett.0c00378
Keywords: prostate cancer, IRAK1, Myddosome complex, epigenetic regulation, DNA methylation
Citation: Schagdarsurengin U, Breiding V, Loose M, Wagenlehner F and Dansranjav T (2022) Interleukin-1 receptor associated kinase 1 (IRAK1) is epigenetically activated in luminal epithelial cells in prostate cancer. Front. Oncol. 12:991368. doi: 10.3389/fonc.2022.991368
Received: 11 July 2022; Accepted: 01 September 2022;
Published: 26 September 2022.
Edited by:
Mustafa Ozen, Baylor College of Medicine, United StatesReviewed by:
Eswar Shankar, The Ohio State University, United StatesCopyright © 2022 Schagdarsurengin, Breiding, Loose, Wagenlehner and Dansranjav. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Temuujin Dansranjav, VGVtdXVqaW4uZGFuc3JhbmphdmluQGNoaXJ1Lm1lZC51bmktZ2llc3Nlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.