 Yuhang Zhou1,2†Chaoyou Fang3†
Yuhang Zhou1,2†Chaoyou Fang3† Houshi Xu3†Ling Yuan3Yibo Liu4
Houshi Xu3†Ling Yuan3Yibo Liu4 Xiaoyu Wang4
Xiaoyu Wang4 Anke Zhang4*
Anke Zhang4* Anwen Shao4*Danyang Zhou1*
Anwen Shao4*Danyang Zhou1*- 1Health Management Center, Tongde Hospital of Zhejiang Province, Hangzhou, China
- 2The First Clinical Medical College, Heilongjiang University of Chinese Medicine, Harbin, China
- 3Department of Neurosurgery, Shanghai General Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
- 4Department of Neurosurgery, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
Ferroptosis is a regulatory form of iron-dependent cell death caused by the accumulation of lipid-based reactive oxygen species (ROS) and differs from apoptosis, pyroptosis, and necrosis. Especially in neoplastic diseases, the susceptibility of tumor cells to ferroptosis affects prognosis and is associated with complex effects. Gliomas are the most common primary intracranial tumors, accounting for disease in 81% of patients with malignant brain tumors. An increasing number of studies have revealed the particular characteristics of iron metabolism in glioma cells. Therefore, agents that target a wide range of molecules involved in ferroptosis may regulate this process and enhance glioma treatment. Here, we review the underlying mechanisms of ferroptosis and summarize the potential therapeutic options for targeting ferroptosis in glioma.
Introduction
Glioma is the most common malignancy of the central nervous system (CNS) and manifests with highly invasive growth, neovascularization, and resistance to various combination therapies (1). Despite advanced therapeutic strategies, including aggressive surgery, radiotherapy, and chemotherapy, glioblastoma (GBM) patients still show poor prognosis and a median overall survival of less than 16 months (2). Despite aggressive treatment measures, including maximal safe surgical resection followed by external irradiation therapy accompanied with adjuvant temozolomide (TMZ) treatment, approximately 90% of grade WHO IV gliomas recur locally within 2 years (3). Gross total resection (GTR), defined as complete radiectomy of the contrast-enhanced region of high-grade glioma (HGG) and T2-weighted/fluid attenuated inversion recovery (T2/FLAIR) MRI-indicated hyperintensive nonenhancing lesions, almost always fails to completely remove all microscopic residual tumor cells (4). Similar to other malignancies, GBM exhibits a distinct anti-DNA-damage phenotype, which leads to chemoresistance (5).
Hence, therapies targeted to gliomas have not been considered sufficiently effective (6). However, ferroptosis has recently attracted considerable interest, especially because the mechanism involves downregulation and silencing of genes involved in the initiation and execution of cancer necroptosis (7). Ferroptosis is a unique iron-dependent form of nonapoptotic cell death in which the affected cells are morphologically, biochemically, and genetically distinct from apoptotic, necrotic, and autophagic cells (8). Ferroptosis is driven by the lost lipid repair enzymatic activity of glutathione peroxidase 4 (GPX4) and subsequent accumulation of lipid-based reactive oxygen species (ROS), particularly lipid hydroperoxides (9). As a common recognition feature, ferroptotic cells appear as clear and transparent round cells under the microscope, mainly composed of empty cytosol, which is called the “ballooning phenotype”. In addition, ferroptotic cells also have ultrastructural changes in mitochondria such as volume decreased, bilayer membrane density increased, outer mitochondrial membrane (OMM) destroyed, and mitochondrial cristae disappeared.
To promote tumor growth, cancer cells exhibit a higher iron demand than normal cells. This iron dependence makes cancer cells more susceptible to ferroptosis (10). Therefore, induced ferroptosis induction may offer the unique possibility of effectively eradicating certain tumor cells, especially those in a highly mesenchymal state (11) and those that evade drug treatment (12). Furthermore, ferroptosis plays a pivotal role in suppressing tumorigenesis by eliminating cells in environments that lack key nutrients or produce cellular stress or that are infected with pathogens (13). The ferroptotic sensitivity of cancer cells may be related to the activation of Ras-mitogen-activated protein kinase (MEK) (14), which contributes to the upregulation of transferrin receptor 1 and increased intracellular iron levels, as well as to the additional formation of ROS via inhibited cystine-based reactions (15). Many other molecules in different pathways have been found to be involved in ferroptosis in glioma (16), and the related content is summarized in this review.
Focused overview of ferroptosis pathways
An overview of ferroptosis pathways is shown in Figure 1.
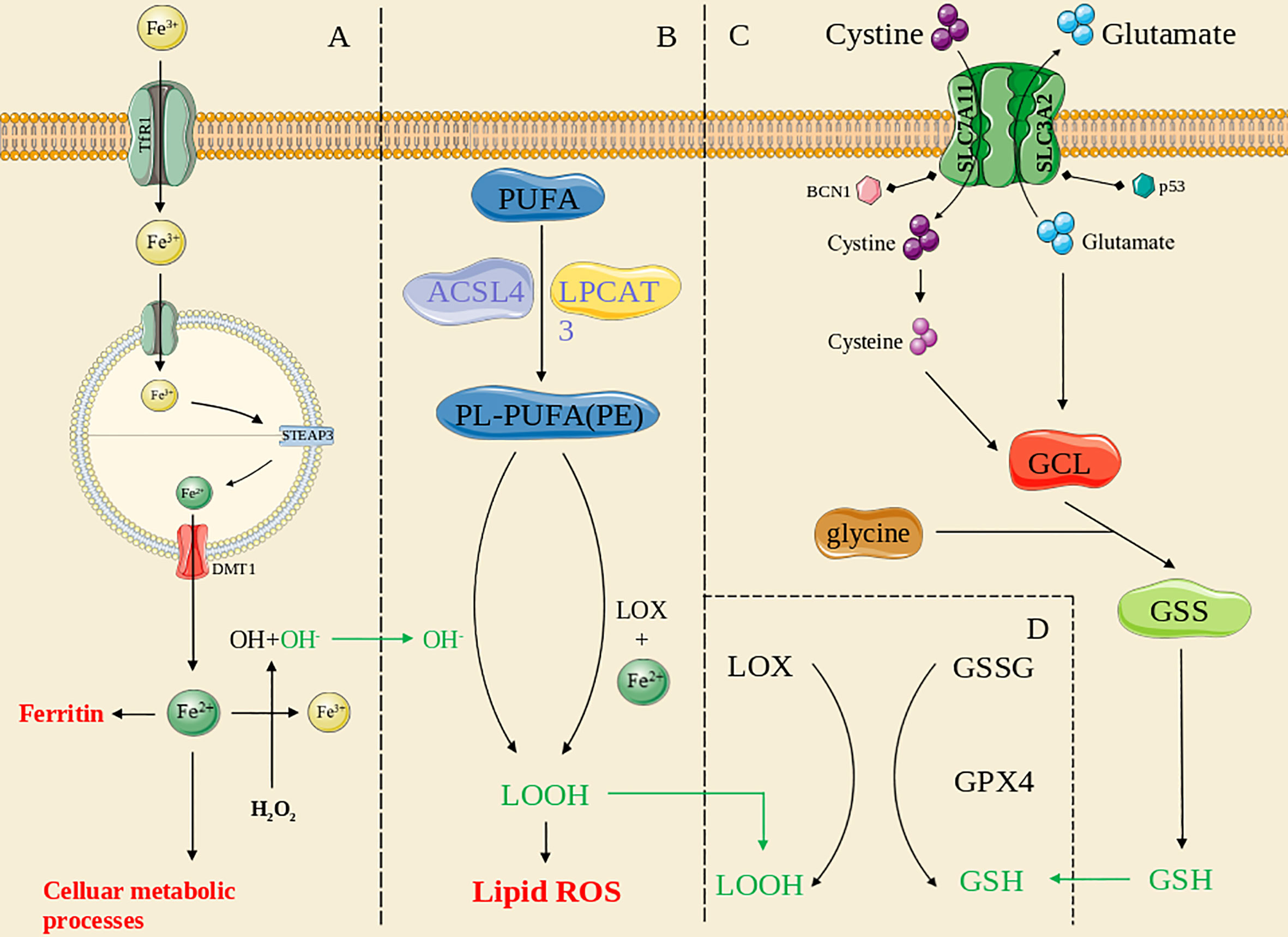
Figure 1 The overview of ferroptosis pathways. (A): the iron metabolism pathway; (B): the lipid peroxidation pathway; (C): the antioxidant systems pathway; (D): the GPX4-mediated pathway. The green line means the substance acts across pathways.
Iron metabolism in ferroptosis
The regulatory mechanism that coordinates intracellular iron homeostasis is centered on iron regulatory proteins (IRPs), which exerts effects by binding to iron-responsive elements (IREs) (17, 18). Under physiological conditions, cellular iron absorption is controlled mainly by the plasma membrane protein transferrin receptor 1 (TFR1), and therefore, knocking down TFR1 expression can block transferrin-bound iron entry into a cell (19, 20), preventing ferroptosis caused by erastin or cystine deprivation (21). Diminishing ferritin expression (22) or FPN1 or ceruloplasmin depletion increases the cell sensitivity to ferroptosis (23, 24). In addition, reduced IRP2 activity, increased expression of transferrin (Tf) and the transferrin receptor (TFR) (19), and recognition of FTH1 by a specific cargo receptor (nuclear coactivator 4, NCOA4), which leads to formation of a complex that fuses with lysosomes (25), cause an abnormal increase in unstable intracellular iron stores, a critical factor in ferroptosis. Other iron metabolism-related proteins also affect cell sensitivity to ferroptosis (26), and certain genes exert the same effects. Recently, the critical role played by STEAP3 in cancer has been extensively investigated, and STEAP3 has thus been found to be a key regulator of ferroptosis by mediating iron metabolism (27, 28). Overexpression of STEAP3 contributes to iron uptake and maintains iron stores (29), supporting the proliferation of multiple types of cancer cells (30–32). Hence, dysregulation of iron metabolism is an important contributing factor to ferroptosis.
Lipid peroxidation in ferroptosis
Lipids are critical for maintaining the membrane integrity of a cell, and extensive peroxidation of lipids changes the assembly, composition, structure, and dynamics of lipid membranes (33). Polyunsaturated fatty acids (PUFAs) containing phospholipids (PLs; PUFA PLs) are substrates for lipid peroxidation (34). ROS are free radicals and/or oxygen derivatives, including superoxide anions, hydrogen peroxide, hydroxyl radicals, lipid hydroperoxides, peroxy radicals, and peroxynitrite (35). Membranes containing high levels of PUFAs are extremely sensitive to ROS effects and highly vulnerable to lipid peroxidation (36, 37). Lipid undergo peroxidation through two routes: nonenzymatic autoxidation and enzymatic PL peroxidation; the former pathway is known as “nonenzymatic PL autoxidation”.
Nonenzymatic peroxidation of lipids is mediated by carbon- and oxygen-based radicals and can be divided into three discrete stages: initiation, proliferation, and termination (33). The initial phase involves a series of reactions collectively known as “Fenton chemistry” in which labile iron reacts with endogenous hydrogen peroxide or superoxide to form oxygen-based radicals (38). Radical compounds produce new radicals, which are markers of the proliferative phase. The hydroxyl and peroxide radicals produced through a Fenton reaction can form a resonant stable carbon-based radical by extracting hydrogen from the bis allylic methylene of a membrane PUFA, which can react with molecular oxygen in solution to form the lipid peroxide radical ROO−, which can remove a hydrogen from a different bis allylic methylene to generate peroxidized lipid (ROOH) and another carbon-based radical that can react with oxygen (33, 39, 40). Finally, antioxidants terminate radical propagation (41).
Enzymatic PL peroxidation is mainly mediated by cyclooxygenases (COXs), cytochrome p450 species (CYPs), NADPH oxidase (NOX), and, especially, lipoxygenases (LOXs) (42). Arachidonic acid (C20:4) and linoleic acid (LA; C18:2) are substrates for LOX (43), and ferric iron is a cofactor of LOX (44, 45). In contrast to 5-lipoxygenases, 12- and 15-lipoxygenases exhibit incomplete regional selectivity in producing lipid peroxides (46) and are thought to respond to intact phospholipids and do not promote hydrolysis for peroxidation (47, 48). Lipid hydroperoxides (LOOHs) and the autoxidation products of PUFAs are currently markers of ferroptosis (49, 50).
Antioxidant systems in ferroptosis
In addition to lipid peroxidation, the cellular antioxidant system contributes to ferroptosis by decomposing ROS. GPX4 is a central factor in anti-ferroptosis reactions (51). This protein is expressed as several isoenzymes with different subcellular locations and distinct tissue-specific expression patterns (52, 53). GSH is a cofactor of GPX4, and GSH synthesis is maintained by the amino acid antiporter SLC7A11/xCT/system (54). Some small-molecule compounds can regulate the activity of glutamate-cysteine ligase (GCL) and xCT (8) and thus affect GSH synthesis, eventually leading to ferroptosis. Several other small-molecule compounds can directly inhibit GPX4 activity or cause GPX4 protein degradation (55, 56). Nonoxidized dopamine and activated heat shock protein family A member 5 (HSPA5) prevent GPX4 degradation (57, 58), whereas heat shock protein 90 (HSP90)-dependent chaperone-mediated autophagy promotes erastin-induced GPX4 degradation (59). Furthermore, GPX4-independent ferroptosis pathways have been identified. Ferroptosis inhibitory protein (FSP1) and CoQ10 facilitate a shuttle of reducing equivalents derived from NAD(P)H to the lipid bilayer (60). In addition, POR is involved in ML210-induced ferroptosis (61), and P53 can affect ferroptosis without GPX4 inhibition (62). The main regulatory factors are described in detail in the next section.
Critical factors of ferroptosis in glioma
Ferroptosis follows multiple pathways and involves pivotal factors that are regulated by many different regulators. Certain regulators exert valuable regulatory effects and metabolic changes in glioma cells. In this section, the regulators best characterized to date are described, and additional regulators are presented in Table 1.

Table 1 Critical factors of ferroptosis in glioma.
GPX4
GPX4, a core factor in the antioxidant system, regulates certain LOX activities by controlling cellular peroxide formation (82). LOX binds to molecular oxygen when iron is oxidized into trivalent iron and adds this molecular oxygen to a PUFA after proton extraction from the bis-allylic positions of the PUFA, leading to the enzymatic peroxidation of the PUFA (43). The GPX4-mediated antioxidant system can reduce the peroxide concentration, which may affect LOX activity, reducing the peroxidation rate of PUFAs and ultimately inhibiting ferroptosis (63). Studies have pointed out that 15-LOX and its linoleic acid (LA)-derived metabolites exerted protumorigenic effects on GBM cells in vitro (83). This report may imply that GPX4 affects ferroptosis by regulating LOX activity and can be exploited for glioma treatment.
GSH is a reducing substrate for GPX4, and its interaction with SCL7A11 plays a crucial regulatory role in ferroptosis. However, both GSH and SCLA11 activities are intricately regulated by p53 and NRF2, among other proteins., as described in detail in a subsequent section (64).
Western blot and immunohistochemistry (IHC) analyses showed relatively high expression levels of Gpx4 in glioma tissues and cell lines, and its expression was found to be augmented as the glioma grade increased. In addition, experiments showed that knocking down GPX4 expression inhibited the proliferation and migration of glioma cells (84). Previously, inhibition of GPX4 activity was thought to induce apoptosis (85), and combined with the aforementioned findings, it can be concluded that GPX4 inhibition can also induce ferroptosis, which may become a new research target.
Nrf2
Under normoxic conditions, Nrf2, a transcription factor, binds to Kelch-like ECH-associated protein 1 (Keap1) and is inactivated by proteasome degradation after ubiquitination (86). After cells contact a large number of electrophiles or cytotoxic agents or enter into an oxidative stress state, Nrf2 dissociates from Keap1 and rapidly transfers to the nucleus where interacts with antioxidant response elements (AREs) to ultimately maintain intracellular redox homeostasis (65). Nrf2 regulates ferroptosis by regulating the expression of genes related to GSH regulation (genes that encode proteins involved in GSH synthesis and, supply cysteine mediated by xCT, GSH reductase, GPX4), iron regulation (including export and storage of iron, heme synthesis, and catabolism), and NADPH regeneration (87–89). Considering recent research, we speculated that Nrf2 partially targets xCT to regulate GPX4 synthesis and function, thus regulating ferroptosis. When Keap1 activity is inhibited, Nrf2 activity increases, leading to the upregulated expression of the ATP-binding cassette (ABC)-family transporter multidrug resistance protein 1 (MRP1), which prevents GSH efflux from the cells and profoundly inhibits ferroptosis (66). The expression of Nrf2 was increased 3-fold in human GBM compared to that in normal brain tissue (67). Both the low expression of Keap1 and the overexpression of Nrf2 led to a significant increase in xCT mRNA levels (up to a 5-fold increase), which subsequently reduced ROS formation. In contrast, both the overexpression of Keap1 and the low expression of Nrf2 eventually led to a substantial increase in ROS levels (67). Thus, the levels of NRF2 are directly related to ferroptosis sensitivity, as increased NRF2 expression prevents ferroptosis, and decreased NRF2 expression enhances the sensitivity of cancer cells to ferroptosis (67, 90).
P53
The tumor suppressor p53 is a transcription factor that regulates various cellular responses through selective transcriptional regulation of various target genes or interaction with other proteins. Studies have shown that xCT is a target of p53 and that p53 sensitizes cells to ferroptosis through transcriptional inhibition of xCT expression (68). The combination of p53 with response elements in the xCT promoter region inhibited xCT expression and increased the sensitivity of cancer cells to ferroptosis inducers such as erastin; however, p533RK failed to induce cell cycle arrest, senescence, or modulation and inhibited xCT expression, ultimately promoting the response to stress induced by ROS (68). However, another acetylation-defective mutant of p53, p534KR98 (with a lysine K98 substitution), showed no ability to reduce xCT expression (91). As recently reported, p53 sensitized cells to erastin-induced ferroptosis through a comprehensive pathway. P53 promotes nuclear translocation of USP7 (a deubiquitinase) that removes the H2Bub1 mark (monoubiquitinated histone H2B on lysine 120) from the regulatory region of the xCT gene. Loss of the H2Bub1 mark inhibited the expression of xCT, leading to ferroptosis (69).
Low-molecular-weight polyamines such as putrescine, spermidine, and spermine are amino acid-derived polycationic alkylamines involved in the regulation of cell growth, proliferation, and differentiation (92). Spermidine/spermine N1-acetyltransferase 1 (SAT1) is a rate-limiting enzyme that controls polyamine catabolism in cells by acetylating spermidine and spermine mediated through acetyl-coenzyme A (93). Overexpression of SAT1 causes a rapid depletion of spermidine and spermidine levels and an increase in putrescine abundance, which causes significant cellular growth inhibition and mitochondrial pathway apoptosis (94). SAT1 has been confirmed to be a transcriptional target of p53, and only the ferroptosis inhibitor ferrostatin-1 was able to inhibit ROS-induced cell death in SAT1-overexpressing cells. In contrast to its effect on conventional pathways, SAT1 exerted no effect on xCT or GPX4 expression or activity but induced an increase in ALOX15 level, which in turn led to ferroptosis mediated via the oxidation of PUFAs (70).
Glutamine metabolism affects ferroptosis and exerts a particularly high effect on serum-dependent pathways after amino acid deficiency (19). GSL2 (glutaminase 2) in mitochondria is a transcriptional target of p53 and is the core glutaminase in the glutamine-to-glutamate metabolic pathway (95). the GSL2 is transcribed by p53 and mediates the generation of GSH in LN-2024 cells (a human glioblastoma cell line) to enhance their antioxidant capacity (96).
In addition to the aforementioned effects, p53 inhibited ferroptosis in some tumor cells. For example, studies showed that binding of p53 to dipeptidyl peptidase-4 (DPP4) inhibited ferroptosis in colorectal cancer cells, and certain DPP4 inhibitors completely blocked erastin-induced cell death in p53-deficient colorectal cancer cells (97). These studies suggest that the inhibition of p53 activity is specific to ferroptosis inducers (98). The tumor suppressor CDKN1A/p21 induces cell cycle arrest and senescence (99, 100). Although the cell cycle arrest mediated by CDKN1A is insufficient to inhibit ferroptosis (101), the induction of p53 increases GSH synthesis and thus inhibits ferroptosis (102).
According to The Cancer Genome Atlas (TCGA) data, 78% of GBM cases present with mutations in the p53 pathway (103), including direct mutations in the p53 gene (in secondary GBM) and a loss of the INK4A/ARF (CDKN2A) gene locus, PTEN mutations and EGFR amplification/loss (in primary GBM) (104). Since p53 is involved in various cellular responses involving the cell cycle or leading to apoptosis, differentiation and DNA damage, the regulatory effect of p53 on ferroptosis needs to be assessed on the basis of the situation, and further research is required (105).
BAP1
BRCA1-associated protein 1 (BAP1) is a tumor suppressor with functions such as tumor suppression, cell cycle control, DNA damage repair, and differentiation (106–109) that is widely recognized as a deubiquitinating enzyme (DUB) (110). Study results have suggested that wild-type (WT) BAP1 significantly decreased H2Aub occupancy on the promoter and gene body of xCT, but the C91A mutant did not exert this effect (71). Because WT BAP1 exhibited DUB activity and BAP1 C91A did not in this experiment, WT BAP1 was the clear cause of inhibited xCT expression (71). Therefore, BAP1 may be recruited by other proteins in the PR-DUB complex, such as ASXL1, which also strongly bind to the xCT promoter (111). BAP1 has been frequently shown to inactivate the expression of genes with mutations or deletions in tumor cells (77), but its behavior in glioma is abnormal. For example, although BAP1 is generally considered to be a chromatin-associated protein and thus to reside within the nucleus (112), recent studies have found it in both the nucleus and cytoplasm of glioma cells, suggesting BAP1 protein is differentially distributed in glioma cells (113, 114). Notably, high cytoplasmic abundance of BAP1 was significantly associated with low overall survival, and nuclear abundance of BAP1 cells was not correlated with overall survival (114). Since BAP1 shows aberrant cytosolic abundance in glioma and because the BAP1-related pathway inhibiting ferroptosis is located in the nucleus, the abnormal distribution of BAP1 in glioma cells, compared to that in other cancer cells, and the BAP1 regulatory pathway in the nucleus can be new research targets.
OTUB1
The ubiquitin hydrolase OTUB1 was previously thought to regulate the p53 pathway by regulating the activities of Mdm2 and Mdmx (72, 73), but OTUB1 has been found to interact directly with xCT to regulate xCT independent of p53 (74). The expression of OTUB1 in glioma compared to adjacent tissues and its expression level was correlated with the low survival of glioma patients (115). Coimmunoprecipitation assays showed that the endogenous OTUB1 protein was coprecipitated with an anti-xCT-specific antibody, and endogenous xCT was coprecipitated with an anti-OTUB1-specific antibody. In vitro GST pull-down assays confirmed that OTUB1 is a binding partner of xCT (74). Inactivation of OTUB1 directly led to a substantial reduction in the xCT level, and this effect was confirmed to sensitize cells to erastin and the ferroptosis inhibitor ferrostatin-1 (8, 74). However, the sensitization effect caused by OTUB1 knockdown, which affected both cysteine and glutathione levels in glioma, was rescued by the overexpression of xCT (115). Notably, the ectopic overexpression of xCT is evident occurs in many cancers (68, 116–118). Hence, xCT levels may be stabilized by the absence of OTUB1, promoting ferroptosis and ultimately inhibiting tumor growth (74).
ATF4
Activating transcription factor 4 (ATF4) is another key transcriptional regulator and mediator of metabolism and oxidative homeostasis (76, 119) that can be activated by several stress signals, such as those triggered by anoxia, hypoxia, endoplasmic reticulum (ER) stress, oxidative stress and amino acid deprivation (120). ATF4 expression is significantly higher in malignant gliomas than in untransformed human brain tissue; moreover, ATF4 can promote the proliferation and migration of glioma cells, and patients with high ATF4 expression exhibit a relatively short overall survival time (76). ATF4 expression resulted in a significant increase in xCT mRNA levels in human glioma specimens compared to that in normal brain tissue (a 5-fold increase in gliomas with a WHO° II classification and 19-fold in gliomas with a WHO° IV classification), and xCT protein levels were increased with AFT4 levels. xCT antiporter activity is determined on the basis of extracellular glutamate levels, and ATF4 knockout significantly reduced glutamate release and cystine uptake, which in turn significantly reduced xCT transporter activity (75, 76). These data suggest that ATF4 deficiency increases ROS levels in cells, but the accumulation of ROS has been shown to prevented by chelation of the iron internalized by cells, and the effects produced by ATF4 overexpression can be inhibited by sorafenib and erastin (76, 77). In addition, the growth-promoting effect of ATF4 on cells is mediated by xCT.
Pathological vessels constitute a the specific microenvironmental niche in primary brain tumors (121, 122). The expression level of ATF4 affected the growth of tumor vessels; specifically, ATF4 overexpression increased the number and length of tumor vessels, and ATF4 knockdown led to the opposite effect (76). The effects of ATF4 activity on tumor vessels were regulated by ferroptosis; moreover, erastin and RSL3 inhibited angiogenesis in glioma, and this inhibitory effect was attenuated with increased expression of ATF4 expression, although the outcome was not notable (76). ATF4 is thought to interact with components associated with ER stress (123) and to prevent cellular resistance to partial ferroptosis inducers, such as TMZ and dihydroartemisinin (124). Therefore, ATF4 is involved in multiple pathways and thus presents possibilities for ferroptosis regulation, which may lead to new research prospects.
NCOA4
Nuclear receptor coactivator 4 (NCOA4) is a selective cargo receptor for autophagic turnover that binds to ferritin to mediate its delivery to autophagosomes and subsequently to the lysosome for ferritin degradation and concomitant iron release (78, 125, 126). When the cellular iron content is high, iron-bound NCOA4 interacts with the ubiquitin E3 ligase HERC2 to target NCOA4 for proteasomal degradation, which subsequently reduces ferritinophagy. However, when the cellular iron content is low, this interaction is inhibited, stabilizing NCOA4, which in turn increases ferritinophagic flux and iron release in lysosomes (78). This mechanism enables NCOA4 to regulate cellular iron homeostasis, determine the ferritin energy flux, and affect the sensitivity of ferroptosis-inducing agents (127–129).
Previous studies reported that NCOA4 activity led to inhibited FTH1 activity levels and that decreased FTH1 levels caused cells to respond to several ferroptosis-inducing agents, such as erastin (79, 130). Cystine deprivation led to ferroptosis, which decreased FTH1 protein levels in control glioblastoma cells (carrying NCOA4 T98G). In NCOA4-deficient GBM cells (NCOA4-knockout [KO] cells), cystine deprivation exerted little effect on the FTH1 level, and therefore, cystine removal did not cause cell death (79). Furthermore, cystine deprivation caused increases in the amount of microtubule-associated protein light chain 3 (LC3)-II (which is related to autophagosome formation) in NCOA4 T98G-mutant cells (79, 131, 132). This finding suggests that cystine deprivation induces NCOA4-mediated ferritin iron release, which in turn leads to the ferroptosis of GBM cells (79).
YAP/TAZ
Yes-associated protein 1 (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) are two dominant effectors of the Hippo pathway. The Hippo pathway is a potent tumor suppression pathway, and its core kinases include mammalian STE20-like protein kinase 1/2 (MST1/2) and large tumor suppressor ½ (LATS1/2), which inhibit proliferation by inhibiting YAP and TAZ (133, 134). After receiving an activation signal, MST1/2 associates with Salvador 1 (SAV1) to activate the Hippo pathway and to phosphorylate LATS1/2 and its coenzyme factor MOB1. The latter then phosphorylates the transcription cofactor YAP/TAZ, and phosphorylated YAP/TAZ is isolated in the cytoplasm and not translocated to the nucleus (80). Moreover, cell density and cellular communications can influence the regulation of ferroptosis induced by YAP/TAZ (81). For example, Yang et al. showed that TAZ, but not YAP, was abundantly expressed in several cancer cell lines and underwent density-dependent nuclear translocation (135, 136). TAZ depletion led to cell resistance to various ferroptosis inducers, while overexpression of the constitutively active form of TAZ, TAZS89A, sensitized cells to ferroptosis (137).
Additionally, YAP/TAZ regulates autophagy, and overexpression of MST1/2 or contact inhibition caused by high cell density inactivates YAP/TAZ activity, suppressing the transport of autophagosome components mediated by actin-myosin complexes and reducing LC3 levels (134). In contrast, knocking down LATS1/2 activities promotes YAP/TAZ activity and autophagy, which in turn induces ferroptosis (81). Compared to that of TAZ knockdown, the inhibitory effect of YAP knockdown on ferroptosis inducers (erastin, etc.) was more significant, and the knockdown of both YAP and TAZ induced the most significant inhibitory effect (138). The expression of both YAP and TAZ was elevated in multiple tumor types, including glioma cells, and was associated with the grade of malignancy, which was highest in GBM patients (139). YAP is also regarded as an independent prognostic factor for low-grade gliomas, and studies have shown that YAP/TAZ can control GBM cell plasticity (140), which may indicate a high value for YAP and TAZ in glioma and ferroptosis research.
Therapeutic drugs for glioma based on targeting ferroptosis
Compared with widely used ferroptosis drugs, particularly the few drugs used to treat glioma, many drugs are used to treat other malignancies, but these drugs induce drug resistance and fail to cross the blood–brain barrier, making them ineffective glioma treatments (150). TMZ is a widely used chemotherapeutic drug, but the resistance it causes is a very serious problem. Recently, research has been focused on weakening the resistance of malignant tumor cells to TMZ, and to this end, combinations of drugs and molecular hybridization are being tested (151). In addition, photodynamic therapies for ferroptosis may be used to overcome the blood–brain barrier in glioma treatment (152). Some newly tested drugs, such as dihydroartemisinin (DHA) and sulfasalazine (SAS), have shown obvious ferroptosis-inducing effects on glioma cells, and most of these drugs have been previously used to treat other malignancies. In this section, we provide an overview of the dominant therapeutic drugs used for glioma treatment that target ferroptosis. A list of these drugs is also provided in Table 2.
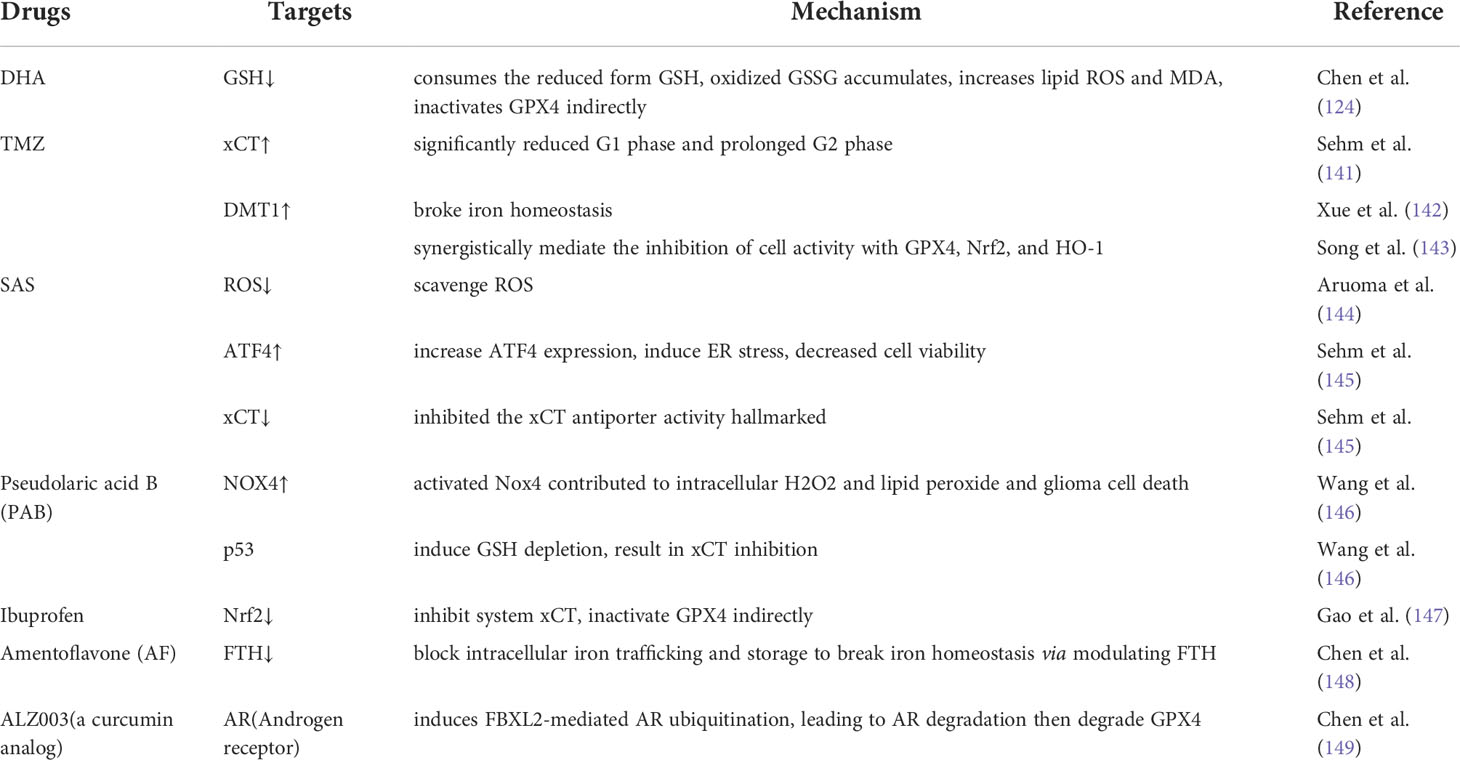
Table 2 Therapeutic Drugs towards Glioma Treatment by targeting Ferroptosis.
Dihydroartemisinin (DHA)
Artemisinin (ART) is the active component extracted from Artemisia annua, and DHA, its main active derivative, has been shown to exert desired cytotoxic effects on various human malignancies (153–156).
Studies showed that the DHA-activiated pathway consumed the reduced form of glutathione (GSH) and that the oxidized form (GSSG) accumulated in glioma cells, leading to increasing levels of lipid ROS and malondialdehyde (MDA, the end product of lipid peroxidation) in glioma cells (124). In addition, transmission electron microscopy showed that the size of mitochondria was decreased, the number of mitochondrial ridges was decreased, and the bilayer membrane density was increased in DHA-treated cells, which was consistent with the ultramorphological features of cells undergoing ferroptosis (63, 157, 158). These observations also prove that DHA induced ferroptosis in glioma cells (159). To determine the targets of regulated by DHA in ferroptosis, the expression of GPX4, xCT and ACSL-4 was determined. GPX4 expression was downregulated and decreased with increasing DHA concentrations in DHA-treated groups compared to controls, while the levels of xCT and ACSL-4 were unchanged (159).
The effect of DHA on the induction of ferroptosis depended on multiple factors. Inhibition of the PERK/ATF4 signaling pathway enhanced the ferroptosis rate in DHA-induced glioma cells, and ATF4-induced HSPA5 expression was induced by increasing the GPX4 level in glioma cells undergoing DHA-induced ferroptosis (124). Thus, HSPA5 inhibitors synergistically enhanced the antitumor effects of DHA. Both the iron chelator deferoxamine (DFO) and lipid peroxidation were shown to inhibit ferrostatin-1 (Fer-1) activity, and liproxstatin-1 (Lip-1) inhibited the DHA-induced production of ROS, lipid ROS and MDA (159). Thus, both Fer-1 and Lip-1 reversed DHA-induced ferroptosis. Because DHA affects many high-impact targets and since these effects are regulated by multiple factors, studies into its selective killing effect on glioma cells are promising research directions.
Temozolomide (TMZ)
TMZ is widely used as the first-line treatment of malignant gliomas, but its antitumor effects have not been clearly identified. Ferroptosis is considered one of the pathways targeted by TMZ, and TMZ affects ferroptosis in glioma cells in several ways. The efficacy of TMZ in human glioma depends on xCT expression, and xCT expression in cells is increased after TMZ treatment (141). TMZ induced toxicity in both xCT-silenced and xCT-overexpressing glioma cells, and the toxicity increased with increasing TMZ concentration. Significantly fewer TMZ-treated cells were found to be in the G1 or prolonged G2 phase, and xCT-silenced cells were more sensitive to TMZ than xCT-overexpressing cells (141). Astrocytes and neurons were less susceptible than glioma cells to TMZ, suggesting special implications for TMZ treatment of glioma. Moreover, the effect of TMZ was enhanced when it was combined with erastin or sorafenib (141).
TMZ induces ferroptosis through the divalent metal transporter DMT1, which regulates iron levels and maintains iron homeostasis (8, 142). Both DMT1 mRNA and protein expression levels were significantly increased in glioma cells treated with TMZ (143). When DMT1 activity was inhibited, GPX4, Nrf2, and HO-1 activity was also inhibited, and the ability of TMZ to reduce cell viability was diminished (143). These results suggest that TMZ induces the ferroptosis of glioma cells and that this effect was associated with xCT and DMT1 expression.
Sulfasalazine (SAS)
SAS has been shown to scavenge ROS (144), induce cancer apoptosis (160), and attenuate glioma-induced epilepsy (161, 162). Recent studies showed that SAS significantly increased ATF4 expression in glioma cells and induced ER stress, decreasing cell viability (145). Cell death was prevented by treatment with iron chelators and ferroptosis inhibitors, and high concentrations of SAS specifically inhibited the expression of an xCT antiporter activity marker (145), confirming that high concentrations of SAS inhibited xCT activity and induced ferroptosis in glioma cells. In experiments with a rat model, SAS significantly reduced glioma cell proliferation, exerted no significant toxic effects on normal neurons (163) and mild toxicity on astrocytes, and did not affect brain cell viability (145). However, due to low brain penetration, SAS showed poor efficacy in newly diagnosed and recurrent malignant glioma (150, 164). This problem is expected to be improved by convection-enhanced delivery (CED) (163).
In addition, SAS is likely to be used in several drug combinations. For example, molecular hybridization product of SAS and DHA, called AC254, showed significantly higher effects on glioma cells than either drug administered separately or in other drug combinations (165). AC254 led to changes in glioma cell shape and activity and terminated cell division, which were significantly better outcomes than those induced by the parent drugs and their mixture with other drugs (165). SAS enhanced the ability of TMZ to reduce human GBM cell activity (151), which may solve the problem of TMZ resistance.
Conclusions and perspectives
As a recently discovered form of cell death, ferroptosis shows many potential applications to glioma treatment. Recent studies have revealed three major pathways of ferroptosis, namely, iron metabolism, lipid peroxidation, and antioxidant system pathways (26). Ferroptosis is primarily regulated by the inhibition of xCT, accumulation of ROS, inhibition of GPX and GSH, which are mediators of many secondary regulatory pathways. In addition to these findings, increasing evidence links ferroptosis with autophagy, which has led to multiple research directions (166). The regulatory pathways of ferroptosis and the relationship of these pathways between ferroptosis and other forms of cell death remain to be further investigated.
Glioma cells show sensitivity to multiple types of specific ferroptosis inducers. Several critical factors inducing ferroptosis show different degrees of abnormal manifestation in glioma cells; for example, GPX4, Nrf2 and ATF4 show high expression compared with normal cells, and p53 shows complex regulatory effects. These findings provide therapeutic targets for glioma. However, few studies have focused on the specific activities of ferroptosis-related factors in glioma, and to identify more factors and their complex roles, more experiments need to be conducted.
Ferroptosis provides potential targets for further glioma treatment. Due to the complex regulatory mechanism of ferroptosis, many drugs show completely different effects in vivo than in vitro or show varying degrees of antagonistic effects in different pathways. In summary, the specific mechanism of ferroptosis remains unclear, and the indicators of ferroptosis are not obvious. Therefore, research on ferroptosis-related drugs needs to be conducted based on information obtained through additional detailed studies.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Author contributions
YZ, CF, and HX designed the review and wrote the manuscript. YL, XW, and LY conceived the artwork and performed the bibliographical research. AZ, AS, and DZ supervised the writing. All the authors revised and approved the final version of the manuscript.
Funding
This work was supported by grants from the Project Supported by Zhejiang Provincial Natural Science Foundation of China (LY22H090020).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ma Q, Long W, Xing C, Chu J, Luo M, Wang HY, et al. Cancer stem cells and immunosuppressive microenvironment in glioma. Front Immunol (2018) 9:2924. doi: 10.3389/fimmu.2018.02924
2. Walid MS, Smisson HF3, Robinson JS Jr. Long-term survival after glioblastoma multiforme. South Med J (2008) 101(9):971–2. doi: 10.1097/SMJ.0b013e31818005e5
3. Minniti G, Niyazi M, Alongi F, Navarria P, Belka C. Current status and recent advances in reirradiation of glioblastoma. Radiat Oncol (London England) (2021) 16(1):36. doi: 10.1186/s13014-021-01767-9
4. Kelly PJ, Daumas-Duport C, Kispert DB, Kall BA, Scheithauer BW, Illig JJ. Imaging-based stereotaxic serial biopsies in untreated intracranial glial neoplasms. J Neurosurg (1987) 66(6):865–74. doi: 10.3171/jns.1987.66.6.0865
5. Han B, Meng X, Wu P, Li Z, Li S, Zhang Y, et al. ATRX/EZH2 complex epigenetically regulates FADD/PARP1 axis. contributing to TMZ resistance glioma Theranostics (2020) 10(7):3351–65. doi: 10.7150/thno.41219
6. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma subclassifications and their clinical significance. Neurotherapeutics (2017) 14(2):284–97. doi: 10.1007/s13311-017-0519-x
7. Chen D, Yu J, Zhang L. Necroptosis: an alternative cell death program defending against cancer. Biochim Biophys Acta (2016) 1865(2):228–36. doi: 10.1016/j.bbcan.2016.03.003
8. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
9. Yang WS, Stockwell BR. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol (2016) 26(3):165–76. doi: 10.1016/j.tcb.2015.10.014
10. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell (2019) 35(6):830–49. doi: 10.1016/j.ccell.2019.04.002
11. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature (2017) 547(7664):453–7. doi: 10.1038/nature23007
12. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature (2017) 551(7679):247–50. doi: 10.1038/nature24297
13. Fearnhead HO, Vandenabeele P, Vanden Berghe T. How do we fit ferroptosis in the family of regulated cell death? Cell Death Differ (2017) 24(12):1991–8. doi: 10.1038/cdd.2017.149
14. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol (2008) 15(3):234–45. doi: 10.1016/j.chembiol.2008.02.010
15. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci (2016) 73(11-12):2195–209. doi: 10.1007/s00018-016-2194-1
16. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab (2020) 32(6):920–37. doi: 10.1016/j.cmet.2020.10.011
17. Chifman J, Laubenbacher R, Torti SV. A systems biology approach to iron metabolism. Adv Exp Med Biol (2014) 844:201–25. doi: 10.1007/978-1-4939-2095-2_10
18. Hentze MW, Kühn LC. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc Natl Acad Sci USA (1996) 93(16):8175–82. doi: 10.1073/pnas.93.16.8175
19. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell (2015) 59(2):298–308. doi: 10.1016/j.molcel.2015.06.011
20. Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J (2016) 473(6):769–77. doi: 10.1042/BJ20150658
21. Anderson GJ, Vulpe CD. Mammalian iron transport. Cell Mol Life Sci CMLS (2009) 66(20):3241–61. doi: 10.1007/s00018-009-0051-1
22. Mumbauer S, Pascual J, Kolotuev I, Hamaratoglu F. Ferritin heavy chain protects the developing wing from reactive oxygen species and ferroptosis. PloS Genet (2019) 15(9):e1008396. doi: 10.1371/journal.pgen.1008396
23. Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, et al. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death differentiation (2020) 27(3):1008–22. doi: 10.1038/s41418-019-0393-7
24. Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci (2018) 22(12):3826–36. doi: 10.26355/eurrev_201806_15267
25. Muhoberac BB, Vidal R. Iron, ferritin, hereditary ferritinopathy, and neurodegeneration. Front Neurosci (2019) 13:1195. doi: 10.3389/fnins.2019.01195
26. Huang R, Dong R, Wang N, He Y, Zhu P, Wang C, et al. Adaptive changes allow targeting of ferroptosis for glioma treatment. Cell Mol Neurobiol (2021) 42(7):2055–74. doi: 10.1007/s10571-021-01092-5
27. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
28. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol (2019) 12(1):34. doi: 10.1186/s13045-019-0720-y
29. Yan Y, Liang Q, Xu Z, Huang J, Chen X, Cai Y, et al. Downregulated ferroptosis-related gene STEAP3 as a novel diagnostic and prognostic target for hepatocellular carcinoma and its roles in immune regulation. Front Cell Dev Biol (2021) 9:743046. doi: 10.3389/fcell.2021.743046
30. Barresi V, Trovato-Salinaro A, Spampinato G, Musso N, Castorina S, Rizzarelli E, et al. Transcriptome analysis of copper homeostasis genes reveals coordinated upregulation of SLC31A1,SCO1, and COX11 in colorectal cancer. FEBS Open Bio (2016) 6(8):794–806. doi: 10.1002/2211-5463.12060
31. Kim SH, Ho JN, Jin H, Lee SC, Lee SE, Hong SK, et al. Upregulated expression of BCL2, MCM7, and CCNE1 indicate cisplatin-resistance in the set of two human bladder cancer cell lines: T24 cisplatin sensitive and T24R2 cisplatin resistant bladder cancer cell lines. Invest Clin Urol (2016) 57(1):63–72. doi: 10.4111/icu.2016.57.1.63
32. Yu Z, Wang H, Fang Y, Lu L, Li M, Yan B, et al. Molecular chaperone HspB2 inhibited pancreatic cancer cell proliferation via activating p53 downstream gene RPRM, BAI1, and TSAP6. J Cell Biochem (2020) 121(3):2318–29. doi: 10.1002/jcb.29455
33. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun (2017) 482(3):419–25. doi: 10.1016/j.bbrc.2016.10.086
34. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell (2021) 81(2):355–369.e10. doi: 10.1016/j.molcel.2020.11.024
35. Guerriero G, Trocchia S, Abdel-Gawad FK, Ciarcia G. Roles of reactive oxygen species in the spermatogenesis regulation. Front Endocrinol (2014) 5:56. doi: 10.3389/fendo.2014.00056
36. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
37. Xiao M, Zhong H, Xia L, Tao Y, Yin H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radical Biol Med (2017) 111:316–27. doi: 10.1016/j.freeradbiomed.2017.04.363
38. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol (2019) 15(12):1137–47. doi: 10.1038/s41589-019-0408-1
39. Gardner HW. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radical Biol Med (1989) 7(1):65–86. doi: 10.1016/0891-5849(89)90102-0
40. Bang S, Park S, Lee YM, Hong S, Cho KB, Nam W. Demonstration of the heterolytic O-O bond cleavage of putative nonheme iron(II)-OOH(R) complexes for fenton and enzymatic reactions. Angewandte Chemie (International Ed English) (2014) 53(30):7843–7. doi: 10.1002/anie.201404556
41. Pratt DA, Tallman KA, Porter NA. Free radical oxidation of polyunsaturated lipids: New mechanistic insights and the development of peroxyl radical clocks. Accounts Chem Res (2011) 44(6):458–67. doi: 10.1021/ar200024c
42. Zhang L, Wang X, Cueto R, Effi C, Zhang Y, Tan H, et al. Biochemical basis and metabolic interplay of redox regulation. Redox Biol (2019) 26:101284. doi: 10.1016/j.redox.2019.101284
43. Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta (2015) 1851(4):308–30. doi: 10.1016/j.bbalip.2014.10.002
44. Shintoku R, Takigawa Y, Yamada K, Kubota C, Yoshimoto Y, Takeuchi T, et al. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci (2017) 108(11):2187–94. doi: 10.1111/cas.13380
45. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA (2016) 113(34):E4966–75. doi: 10.1073/pnas.1603244113
46. Brash AR, Boeglin WE, Chang MS. Discovery of a second 15S-lipoxygenase in humans. Proc Natl Acad Sci USA (1997) 94(12):6148–52. doi: 10.1073/pnas.94.12.6148
47. Jung G, Yang DC, Nakao A. Oxygenation of phosphatidylcholine by human polymorphonuclear leukocyte 15-lipoxygenase. Biochem Biophys Res Commun (1985) 130(2):559–66. doi: 10.1016/0006-291X(85)90453-X
48. Takahashi Y, Glasgow WC, Suzuki H, Taketani Y, Yamamoto S, Anton M, et al. Investigation of the oxygenation of phospholipids by the porcine leukocyte and human platelet arachidonate 12-lipoxygenases. Eur J Biochem (1993) 218(1):165–71. doi: 10.1111/j.1432-1033.1993.tb18362.x
49. Michalski MC, Calzada C, Makino A, Michaud S, Guichardant M. Oxidation products of polyunsaturated fatty acids in infant formulas compared to human milk–a preliminary study. Mol Nutr Food Res (2008) 52(12):1478–85. doi: 10.1002/mnfr.200700451
50. Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PloS Biol (2018) 16(5):e2006203. doi: 10.1371/journal.pbio.2006203
51. Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis. Autophagy Ferroptosis Oxid Med Cell Longevity (2019) 2019:5080843. doi: 10.1155/2019/5080843
52. Matoušková P, Hanousková B, Skálová L. MicroRNAs as potential regulators of glutathione peroxidases expression and their role in obesity and related pathologies. Int J Mol Sci (2018) 19(4):1199. doi: 10.3390/ijms19041199
53. Bela K, Horváth E, Gallé Á., Szabados L, Tari I, Csiszár J. Plant glutathione peroxidases: emerging role of the antioxidant enzymes in plant development and stress responses. J Plant Physiol (2015) 176:192–201. doi: 10.1016/j.jplph.2014.12.014
54. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res (2021) 31(2):107–25. doi: 10.1038/s41422-020-00441-1
55. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol (2016) 12(7):497–503. doi: 10.1038/nchembio.2079
56. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
57. Wang D, Peng Y, Xie Y, Zhou B, Sun X, Kang R, et al. Antiferroptotic activity of non-oxidative dopamine. Biochem Biophys Res Commun (2016) 480(4):602–7. doi: 10.1016/j.bbrc.2016.10.099
58. Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res (2017) 77(8):2064–77. doi: 10.1158/0008-5472.CAN-16-1979
59. Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci United States America (2019) 116(8):2996–3005. doi: 10.1073/pnas.1819728116
60. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
61. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol (2020) 16(3):302–9. doi: 10.1038/s41589-020-0472-6
62. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol (2019) 21(5):579–91. doi: 10.1038/s41556-019-0305-6
63. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radical Biol Med (2019) 133:144–52. doi: 10.1016/j.freeradbiomed.2018.09.014
64. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radical Biol Med (2020) 152:175–85. doi: 10.1016/j.freeradbiomed.2020.02.027
65. Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev (2006) 38(4):769–89. doi: 10.1080/03602530600971974
66. Cao JY, Poddar A, Magtanong L, Lumb JH, Mileur TR, Reid MA, et al. A genome-wide haploid genetic screen identifies regulators of glutathione abundance and ferroptosis sensitivity. Cell Rep (2019) 26(6):1544–1556.e8. doi: 10.1016/j.celrep.2019.01.043
67. Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis (2017) 6(8):e371. doi: 10.1038/oncsis.2017.65
68. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature (2015) 520(7545):57–62. doi: 10.1038/nature14344
69. Wang Y, Yang L, Zhang X, Cui W, Liu Y, Sun QR, et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep (2019) 20(7):e47563. doi: 10.15252/embr.201847563
70. Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci United States America (2016) 113(44):E6806–12. doi: 10.1073/pnas.1607152113
71. Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol (2018) 20(10):1181–92. doi: 10.1038/s41556-018-0178-0
72. Sun XX, Dai MS. Deubiquitinating enzyme regulation of the p53 pathway: A lesson from Otub1. World J Biol Chem (2014) 5(2):75–84. doi: 10.4331/wjbc.v5.i2.75
73. Chen Y, Wang YG, Li Y, Sun XX, Dai MS. Otub1 stabilizes MDMX and promotes its proapoptotic function at the mitochondria. Oncotarget (2017) 8(7):11053–62. doi: 10.18632/oncotarget.14278
74. Liu T, Jiang L, Tavana O, Gu W. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res (2019) 79(8):1913–24. doi: 10.1158/0008-5472.CAN-18-3037
75. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife (2014) 3:e02523. doi: 10.7554/eLife.02523
76. Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene (2017) 36(40):5593–608. doi: 10.1038/onc.2017.146
77. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol (2014) 16(12):1180–91. doi: 10.1038/ncb3064
78. Mancias JD, Pontano Vaites L, Nissim S, Biancur DE, Kim AJ, Wang X, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife (2015) 4:e10308. doi: 10.7554/eLife.10308
79. Hayashima K, Kimura I, Katoh H. Role of ferritinophagy in cystine deprivation-induced cell death in glioblastoma cells. Biochem Biophys Res Commun (2021) 539:56–63. doi: 10.1016/j.bbrc.2020.12.075
80. Masliantsev K, Karayan-Tapon L, Guichet PO. Hippo signaling pathway in gliomas. Cells (2021) 10(1):184. doi: 10.3390/cells10010184
81. Sun T, Chi JT. Regulation of ferroptosis in cancer cells by YAP/TAZ and hippo pathways: The therapeutic implications. Genes Dis (2021) 8(3):241–9. doi: 10.1016/j.gendis.2020.05.004
82. Conrad M, Schneider M, Seiler A, Bornkamm GW. Physiological role of phospholipid hydroperoxide glutathione peroxidase in mammals. Biol Chem (2007) 388(10):1019–25. doi: 10.1515/BC.2007.130
83. Souza FDC, Ferreira MT, Colquhoun A. Influence of lipoxygenase inhibition on glioblastoma cell biology. Int J Mol Sci (2020) 21(21):626–33. doi: 10.3390/ijms21218395
84. Zhao H, Ji B, Chen J, Huang Q, Lu X. Gpx 4 is involved in the proliferation, migration and apoptosis of glioma cells. Pathology Res Pract (2017) 213(6):626–33. doi: 10.1016/j.prp.2017.04.025
85. Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radical Biol Med (2003) 34(2):145–69. doi: 10.1016/S0891-5849(02)01197-8
86. Song X, Long D. Nrf2 and ferroptosis: A new research direction for neurodegenerative diseases. Front Neurosci (2020) 14:267. doi: 10.3389/fnins.2020.00267
87. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
88. Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxidants Redox Signaling (2018) 29(17):1756–73. doi: 10.1089/ars.2017.7176
89. Abdalkader M, Lampinen R, Kanninen KM, Malm TM, Liddell JR. Targeting Nrf2 to suppress ferroptosis and mitochondrial dysfunction in neurodegeneration. Front Neurosci (2018) 12:466. doi: 10.3389/fnins.2018.00466
90. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells, hepatology (Baltimore, md. ) (2016) 63(1):173–84. doi: 10.1002/hep.28251
91. Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, et al. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep (2016) 17(2):366–73. doi: 10.1016/j.celrep.2016.09.022
92. Gerner EW, Meyskens FL Jr. Polyamines and cancer: old molecules, new understanding, nature reviews. Cancer (2004) 4(10):781–92. doi: 10.1038/nrc1454
93. Casero RA Jr., Murray Stewart T, Pegg AE. Polyamine metabolism and cancer: treatments, challenges and opportunities, nature reviews. Cancer (2018) 18(11):681–95. doi: 10.1038/s41568-018-0050-3
94. Mandal S, Mandal A, Johansson HE, Orjalo AV, Park MH. Depletion of cellular polyamines, spermidine and spermine, causes a total arrest in translation and growth in mammalian cells. Proc Natl Acad Sci USA (2013) 110(6):2169–74. doi: 10.1073/pnas.1219002110
95. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA (2010) 107(16):7455–60. doi: 10.1073/pnas.1001006107
96. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA (2010) 107(16):7461–6. doi: 10.1073/pnas.1002459107
97. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep (2017) 20(7):1692–704. doi: 10.1016/j.celrep.2017.07.055
98. Liu J, Zhang C, Wang J, Hu W, Feng Z. The regulation of ferroptosis by tumor suppressor p53 and its pathway. Int J Mol Sci (2020) 21(21):8387. doi: 10.3390/ijms21218387
99. Kastenhuber ER, Lowe SW. Putting p53 in context. Cell (2017) 170(6):1062–78. doi: 10.1016/j.cell.2017.08.028
100. Levine AJ. The many faces of p53: something for everyone. J Mol Cell Biol (2019) 11(7):524–30. doi: 10.1093/jmcb/mjz026
101. Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep (2018) 22(3):569–75. doi: 10.1016/j.celrep.2017.12.077
102. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities, nature reviews. Cancer (2009) 9(6):400–14. doi: 10.1038/nrc2657
103. Mao H, Lebrun DG, Yang J, Zhu VF, Li M. Deregulated signaling pathways in glioblastoma multiforme: molecular mechanisms and therapeutic targets. Cancer Invest (2012) 30(1):48–56. doi: 10.3109/07357907.2011.630050
104. Ludwig K, Kornblum HI. Molecular markers in glioma. J neuro-oncology (2017) 134(3):505–12. doi: 10.1007/s11060-017-2379-y
105. Karsy M, Neil JA, Guan J, Mahan MA, Colman H, Jensen RL. A practical review of prognostic correlations of molecular biomarkers in glioblastoma. Neurosurgical Focus (2015) 38(3):E4. doi: 10.3171/2015.1.FOCUS14755
106. Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene (1998) 16(9):1097–112. doi: 10.1038/sj.onc.1201861
107. Misaghi S, Ottosen S, Izrael-Tomasevic A, Arnott D, Lamkanfi M, Lee J, et al. Association of c-terminal ubiquitin hydrolase BRCA1-associated protein 1 with cell cycle regulator host cell factor 1. Mol Cell Biol (2009) 29(8):2181–92. doi: 10.1128/MCB.01517-08
108. Nishikawa H, Wu W, Koike A, Kojima R, Gomi H, Fukuda M, et al. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Res (2009) 69(1):111–9. doi: 10.1158/0008-5472.CAN-08-3355
109. Okino Y, Machida Y, Frankland-Searby S, Machida YJ. BRCA1-associated protein 1 (BAP1) deubiquitinase antagonizes the ubiquitin-mediated activation of FoxK2 target genes. J Biol Chem (2015) 290(3):1580–91. doi: 10.1074/jbc.M114.609834
110. Han A, Purwin TJ, Aplin AE. Roles of the BAP1 tumor suppressor in cell metabolism. Cancer Res (2021) 81(11):2807–14. doi: 10.1158/0008-5472.CAN-20-3430
111. Zhang Y, Zhuang L, Gan B. BAP1 suppresses tumor development by inducing ferroptosis upon SLC7A11 repression. Mol Cell Oncol (2019) 6(1):1536845. doi: 10.1080/23723556.2018.1536845
112. Arzt L, Quehenberger F, Halbwedl I, Mairinger T, Popper HH. BAP1 protein is a progression factor in malignant pleural mesothelioma. Pathol Oncol Res POR (2014) 20(1):145–51. doi: 10.1007/s12253-013-9677-2
113. Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer, nature reviews. Cancer (2013) 13(3):153–9. doi: 10.1038/nrc3459
114. Zhang XK, Xi SY, Sai K, Chen HD, Zhong ZH, Wu QL, et al. Cytoplasmic expression of BAP1 as an independent prognostic biomarker for patients with gliomas. Int J Clin Exp Pathol (2015) 8(5):5035–43.
115. Zhao X, Zhou M, Yang Y, Luo M. The ubiquitin hydrolase OTUB1 promotes glioma cell stemness via suppressing ferroptosis through stabilizing SLC7A11 protein. Bioengineered (2021) 12(2):12636–45. doi: 10.1080/21655979.2021.2011633
116. Guo W, Zhao Y, Zhang Z, Tan N, Zhao F, Ge C, et al. Disruption of xCT inhibits cell growth via the ROS/autophagy pathway in hepatocellular carcinoma. Cancer Lett (2011) 312(1):55–61. doi: 10.1016/j.canlet.2011.07.024
117. Huang Y, Dai Z, Barbacioru C, Sadée W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res (2005) 65(16):7446–54. doi: 10.1158/0008-5472.CAN-04-4267
118. Liu XX, Li XJ, Zhang B, Liang YJ, Zhou CX, Cao DX, et al. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett (2011) 585(9):1363–7. doi: 10.1016/j.febslet.2011.04.018
119. Singleton DC, Harris AL. Targeting the ATF4 pathway in cancer therapy. Expert Opin Ther Targets (2012) 16(12):1189–202. doi: 10.1517/14728222.2012.728207
120. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell (2000) 6(5):1099–108. doi: 10.1016/S1097-2765(00)00108-8
121. Mao XY, Tokay T, Zhou HH, Jin WL. Long-range and short-range tumor-stroma networks synergistically contribute to tumor-associated epilepsy. Oncotarget (2016) 7(22):33451–60. doi: 10.18632/oncotarget.7962
122. Eyüpoglu IY, Buchfelder M, Savaskan NE. Surgical resection of malignant gliomas-role in optimizing patient outcome, nature reviews. Neurology (2013) 9(3):141–51. doi: 10.1038/nrneurol.2012.279
123. Chevet E, Hetz C, Samali A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov (2015) 5(6):586–97. doi: 10.1158/2159-8290.CD-14-1490
124. Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang L, et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res CR (2019) 38(1):402. doi: 10.1186/s13046-019-1413-7
125. Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol (2014) 16(11):1069–79. doi: 10.1038/ncb3053
126. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature (2014) 509(7498):105–9. doi: 10.1038/nature13148
127. Santana-Codina N, Gikandi A, Mancias JD. The role of NCOA4-mediated ferritinophagy in ferroptosis. Adv Exp Med Biol (2021) 1301:41–57. doi: 10.1007/978-3-030-62026-4_4
128. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res (2016) 26(9):1021–32. doi: 10.1038/cr.2016.95
129. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy (2016) 12(8):1425–8. doi: 10.1080/15548627.2016.1187366
130. Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radical Biol Med (2019) 131:356–69. doi: 10.1016/j.freeradbiomed.2018.12.011
131. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J (2000) 19(21):5720–8. doi: 10.1093/emboj/19.21.5720
132. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy (2007) 3(6):542–5. doi: 10.4161/auto.4600
133. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev (2007) 21(21):2747–61. doi: 10.1101/gad.1602907
134. Pavel M, Renna M, Park SJ, Menzies FM, Ricketts T, Füllgrabe J, et al. Contact inhibition controls cell survival and proliferation via YAP/TAZ-autophagy axis. Nat Commun (2018) 9(1):2961. doi: 10.1038/s41467-018-05388-x
135. Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep (2019) 28(10):2501–2508.e4. doi: 10.1016/j.celrep.2019.07.107
136. Yang WH, Huang Z, Wu J, Ding CC, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol Cancer Res MCR (2020) 18(1):79–90. doi: 10.1158/1541-7786.MCR-19-0691
137. Yang WH, Chi JT. Hippo pathway effectors YAP/TAZ as novel determinants of ferroptosis. Mol Cell Oncol (2020) 7(1):1699375. doi: 10.1080/23723556.2019.1699375
138. Yang WH, Lin CC, Wu J, Chao PY, Chen K, Chen PH, et al. The hippo pathway effector YAP promotes ferroptosis via the E3 ligase SKP2. Mol Cancer Res (2021) 19(6):1005–14. doi: 10.1158/1541-7786.MCR-20-0534
139. Zhou X, Lei QY. Regulation of TAZ in cancer. Protein Cell (2016) 7(8):548–61. doi: 10.1007/s13238-016-0288-z
140. Castellan M, Guarnieri A, Fujimura A, Zanconato F, Battilana G, Panciera T, et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in glioblastoma. Nat Cancer (2021) 2(2):174–88. doi: 10.1038/s43018-020-00150-z
141. Sehm T, Rauh M, Wiendieck K, Buchfelder M, Eyüpoglu IY, Savaskan NE. Temozolomide toxicity operates in a xCT/SLC7a11 dependent manner and is fostered by ferroptosis. Oncotarget (2016) 7(46):74630–47. doi: 10.18632/oncotarget.11858
142. Xue X, Ramakrishnan SK, Weisz K, Triner D, Xie L, Attili D, et al. Iron uptake via DMT1 integrates cell cycle with JAK-STAT3 signaling to promote colorectal tumorigenesis. Cell Metab (2016) 24(3):447–61. doi: 10.1016/j.cmet.2016.07.015
143. Song Q, Peng S, Sun Z, Heng X, Zhu X. Temozolomide drives ferroptosis via a DMT1-dependent pathway in glioblastoma cells. Yonsei Med J (2021) 62(9):843–9. doi: 10.3349/ymj.2021.62.9.843
144. Aruoma OI, Wasil M, Halliwell B, Hoey BM, Butler J. The scavenging of oxidants by sulphasalazine and its metabolites A possible contribution to their anti-inflammatory effects? Biochem Pharmacol (1987) 36(21):3739–42. doi: 10.1016/0006-2952(87)90028-1
145. Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget (2016) 7(24):36021–33. doi: 10.18632/oncotarget.8651
146. Wang Z, Ding Y, Wang X, Lu S, Wang C, He C, et al. Pseudolaric acid b triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett (2018) 428:21–33. doi: 10.1016/j.canlet.2018.04.021
147. Gao X, Guo N, Xu H, Pan T, Lei H, Yan A, et al. Ibuprofen induces ferroptosis of glioblastoma cells via downregulation of nuclear factor erythroid 2-related factor 2 signaling pathway. Anti-cancer Drugs (2020) 31(1):27–34. doi: 10.1097/CAD.0000000000000825
148. Chen Y, Li N, Wang H, Wang N, Peng H, Wang J, et al. Amentoflavone suppresses cell proliferation and induces cell death through triggering autophagy-dependent ferroptosis in human glioma. Life Sci (2020) 247:117425. doi: 10.1016/j.lfs.2020.117425
149. Chen TC, Chuang JY, Ko CY, Kao TJ, Yang PY, Yu CH, et al. AR ubiquitination induced by the curcumin analog suppresses growth of temozolomide-resistant glioblastoma through disrupting GPX4-mediated redox homeostasis. Redox Biol (2020) 30:101413. doi: 10.1016/j.redox.2019.101413
150. Robe PA, Martin DH, Nguyen-Khac MT, Artesi M, Deprez M, Albert A, et al. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer (2009) 9:372. doi: 10.1186/1471-2407-9-372
151. Ignarro RS, Facchini G, Vieira AS, De Melo DR, Lopes-Cendes I, Castilho RF, et al. Sulfasalazine intensifies temozolomide cytotoxicity in human glioblastoma cells. Mol Cell Biochem (2016) 418(1-2):167–78. doi: 10.1007/s11010-016-2742-x
152. Shui S, Zhao Z, Wang H, Conrad M, Liu G. Non-enzymatic lipid peroxidation initiated by photodynamic therapy drives a distinct ferroptosis-like cell death pathway. Redox Biol (2021) 45:102056. doi: 10.1016/j.redox.2021.102056
153. Singh NP, Lai H. Selective toxicity of dihydroartemisinin and holotransferrin toward human breast cancer cells. Life Sci (2001) 70(1):49–56. doi: 10.1016/S0024-3205(01)01372-8
154. Lu JJ, Chen SM, Zhang XW, Ding J, Meng LH. The anti-cancer activity of dihydroartemisinin is associated with induction of iron-dependent endoplasmic reticulum stress in colorectal carcinoma HCT116 cells. Investigational New Drugs (2011) 29(6):1276–83. doi: 10.1007/s10637-010-9481-8
155. Mi YJ, Geng GJ, Zou ZZ, Gao J, Luo XY, Liu Y, et al. Dihydroartemisinin inhibits glucose uptake and cooperates with glycolysis inhibitor to induce apoptosis in non-small cell lung carcinoma cells. PloS One (2015) 10(3):e0120426. doi: 10.1371/journal.pone.0120426
156. Våtsveen TK, Myhre MR, Steen CB, Wälchli S, Lingjærde OC, Bai B, et al. Artesunate shows potent anti-tumor activity in b-cell lymphoma. J Hematol Oncol (2018) 11(1):23. doi: 10.1186/s13045-018-0561-0
157. Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med (2017) 21(4):648–57. doi: 10.1111/jcmm.13008
158. Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radical Biol Med (2019) 133:130–43. doi: 10.1016/j.freeradbiomed.2018.09.043
159. Yi R, Wang H, Deng C, Wang X, Yao L, Niu W, et al. Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Bioscience Rep (2020) 40(6):BSR20193314. doi: 10.1042/BSR20193314
160. Bus PJ, Nagtegaal ID, Verspaget HW, Lamers CB, Geldof H, Van Krieken JH, et al. Mesalazine-induced apoptosis of colorectal cancer: on the verge of a new chemopreventive era? Alimentary Pharmacol Ther (1999) 13(11):1397–402. doi: 10.1046/j.1365-2036.1999.00652.x
161. Buckingham SC, Campbell SL, Haas BR, Montana V, Robel S, Ogunrinu T, et al. Glutamate release by primary brain tumors induces epileptic activity. Nat Med (2011) 17(10):1269–74. doi: 10.1038/nm.2453
162. Robert SM, Buckingham SC, Campbell SL, Robel S, Holt KT, Ogunrinu-Babarinde T, et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci Trans Med (2015) 7(289):289ra86. doi: 10.1126/scitranslmed.aaa8103
163. Haryu S, Saito R, Jia W, Shoji T, Mano Y, Sato A, et al. Convection-enhanced delivery of sulfasalazine prolongs survival in a glioma stem cell brain tumor model. J neuro-oncology (2018) 136(1):23–31. doi: 10.1007/s11060-017-2621-7
164. Takeuchi S, Wada K, Nagatani K, Otani N, Osada H, Nawashiro H. Sulfasalazine and temozolomide with radiation therapy for newly diagnosed glioblastoma. Neurol India (2014) 62(1):42–7. doi: 10.4103/0028-3886.128280
165. Ackermann A, Çapcı A, Buchfelder M, Tsogoeva SB, Savaskan N. Chemical hybridization of sulfasalazine and dihydroartemisinin promotes brain tumor cell death. Sci Rep (2021) 11(1):20766. doi: 10.1038/s41598-021-99960-z
Keywords: glioma, ferroptosis, targeting treatment, reactive oxygen species, iron metabolism
Citation: Zhou Y, Fang C, Xu H, Yuan L, Liu Y, Wang X, Zhang A, Shao A and Zhou D (2022) Ferroptosis in glioma treatment: Current situation, prospects and drug applications. Front. Oncol. 12:989896. doi: 10.3389/fonc.2022.989896
Received: 09 July 2022; Accepted: 14 September 2022;
Published: 29 September 2022.
Edited by:
Eduard Yakubov, Paracelsus Medical Private University, Nuremberg, GermanyReviewed by:
Anna Martina Battaglia, Magna Græcia University of Catanzaro, ItalyMarc Dahlmanns, Friedrich-Alexander-University Erlangen-Nuremberg, Germany
Copyright © 2022 Zhou, Fang, Xu, Yuan, Liu, Wang, Zhang, Shao and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anke Zhang, dGhlYW5rZUAxNjMuY29t; Anwen Shao, c2hhb2Fud2VuQHpqdS5lZHUuY24=; Danyang Zhou, MTI1NjU1MzQzQHFxLmNvbQ==
†These authors have contributed equally to this work