 Lucia Paolini
Lucia Paolini Sajjad Hussain2
Sajjad Hussain2 Paul J. Galardy
Paul J. Galardy- 1Department of Pediatrics, University of Milano-Bicocca, San Gerardo Hospital, Monza, MI, Italy
- 2Department of Pediatric and Adolescent Medicine, Mayo Clinic, Rochester, MN, United States
- 3Division of Pediatric Hematology-Oncology, Mayo Clinic, Rochester, MN, United States
For over 100-years, genomic instability has been investigated as a central player in the pathogenesis of human cancer. Conceptually, genomic instability includes an array of alterations from small deletions/insertions to whole chromosome alterations, referred to as chromosome instability. Chromosome instability has a paradoxical impact in cancer. In most instances, the introduction of chromosome instability has a negative impact on cellular fitness whereas in cancer it is usually associated with a worse prognosis. One exception is the case of neuroblastoma, the most common solid tumor outside of the brain in children. Neuroblastoma tumors have two distinct patterns of genome instability: whole-chromosome aneuploidy, which is associated with a better prognosis, or segmental chromosomal alterations, which is a potent negative prognostic factor. Through a computational screen, we found that low levels of the de- ubiquitinating enzyme USP24 have a highly significant negative impact on survival in neuroblastoma. At the molecular level, USP24 loss leads to destabilization of the microtubule assembly factor CRMP2 - producing mitotic errors and leading to chromosome missegregation and whole-chromosome aneuploidy. This apparent paradox may be reconciled through a model in which whole chromosome aneuploidy leads to the subsequent development of segmental chromosome alterations. Here we review the mechanisms behind chromosome instability and the evidence for the progressive development of segmental alterations from existing numerical aneuploidy in support of a multi-step model of neuroblastoma progression.
Introduction
Advanced sequencing technology has produced an explosion of information regarding cancer genomes. Such efforts have generated valuable information regarding signaling networks and new therapeutic entry points. The majority of cancers, however, are characterized by variations in chromosome numbers (aneuploidy) or copy number alterations that affect small to large segments of chromosome arms (1). These relatively large genomic alterations affect the expression of many genes and perhaps understandably have a correspondingly less linear mechanistic link to malignant behavior. Aneuploidy is associated with poor outcomes in many cancer subtypes including prostate (2), breast (3–5), lung (6), and others (7). In neuroblastoma, whole chromosome aneuploidy is associated with a good prognosis, whereas segmental chromosome alterations associated with poor outcomes – even when co-existing with whole chromosome aneuploidy (8). It is notable that the type of genomic alteration (gains or losses of large chromosome segments) has a greater impact on tumor behavior than which specific chromosome region is affected. This leads us to speculate that it is the underlying mechanism that produces these alterations may itself that is the prognosis-driving event. Here we review the mechanisms that underlie whole chromosome aneuploidy as well as the mechanistic link between aneuploidy and the subsequent development of segmental chromosome alterations. We hypothesize that tumors harboring segmental alterations may have developed through a multi-step process that begins with mitotic missegregation of whole chromosomes and proceeds to segmental alterations.
Genomic instability and cancer
The term genomic instability indicates a cells tendency to accumulate new genomic alterations (3, 9). This broad term includes defects ranging from small insertions/deletion to large segmental and whole chromosomal alterations. Whole chromosome instability (CIN) refers to a state in which there is an increased incidence of cell divisions resulting in the loss of gain of whole chromosomes (10, 11). This is to be distinguished from aneuploidy – which refers to a cell state of possessing too many or few chromosomes. Otherwise normal cells occasionally undergo chromosome segregation errors in mitosis – albeit at a low rate. Frequently, the resulting daughter cells undergo apoptotic death and therefore do not lead to aneuploidy. Consequently, there is a low (but non-zero) rate of aneuploidy in otherwise normal tissues. Increased aneuploidy therefore requires there to be an increased rate of chromosome segregation errors (the state of CIN) or that there be an increased likelihood of cell survival after mitotic errors that occurred at the otherwise normal low frequency. Differently stated, aneuploid cells may result from CIN – but they do not necessarily exhibit ongoing CIN. It has been estimated that up 80% of cancer cells have abnormalities related to chromosome missegregation (12, 13). Whole chromosome instability can be induced by multiple mechanisms during cell division, in particular errors during mitosis such as: centrosome replications errors, alterations of spindle assembly checkpoint, defect in sister chromatid cohesion, damage of microtubule attachments to chromosomes (7, 14). Furthermore, anti-neoplastic therapies can be a cause of chromosome segregation errors as well (15). Many – if not most – animal models with a predisposition to CIN due to engineered alterations in mitotic regulators have an increased tumor incidence, providing strong evidence for the in vivo impact of CIN on cancer development (16, 17).
Chromosomal instability and aneuploidy are generally associated with poor prognosis in solid cancers (1, 18, 19). Gene expression signatures reflecting CIN have been applied across many cancer types. While work from yeast and mammalian systems suggests that there are hundreds of genes that may cause – or protect cells from CIN, it is noteworthy that the over-expression of some classical oncogenes [e.g. KRAS; (20)] and loss of some tumor suppressors [e.g. PTEN, (21)] also leads to CIN. In most instances, tumors that exhibit a high CIN gene expression signature have worse prognosis (18, 22). In an analysis of 2125 patients, those with a higher than the median CIN70 score had a worse outcome (18). Similarly, the analysis of TCGA (The Cancer Genome Atlas; https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) cohorts on several cancer types showed a tight inverse association between cancer cell aneuploidy and cancer-free and overall survival, not only in primary tumors but also those with metastases (19). These studies lead to the general conclusion that CIN is a cause – or marker – of aggressive cancer.
Causes of CIN in cancer
The emergence of aneuploid cells depends on multiple pathways such as 1) mis-segregation of a chromosome(s) during mitosis resulting in daughter cells with non-modal chromosome numbers and 2) survival of the aneuploid cell. Increased numbers of aneuploid cells then may result from an increase in chromosome mis-segregation events (CIN), increased survival of aneuploidy progeny, or both (23). In this review, we will focus on the origins of CIN and the potential contribution of these mechanisms to NBL pathogenesis. However, we must recognize that events that interfere with cell death pathways – such as the loss of functional TP53 or other p53 superfamily genes, or gain of anti-apoptotic proteins such as BCL2, may also play a role in the development of CIN in NBL. Indeed, genes such as CCNB1, PRC1, TPX2, AIRKB, NEK2 that are known regulators of cell cycle and accurate mitosis are over-expressed in high risk NBL tumors (22, 24).
Aberrant attachments of spindle microtubules to the kinetochores of sister chromatids (Figure 1) is the most common cause of chromosome segregation errors in mitosis (25). In an error-free metaphase, each sister chromatid is attached to microtubules originating from only one spindle pole. There are different forms of incorrect attachment of the microtubules to the kinetochores such as: absence of attachment, monotelic, syntelic, and merotelic attachment, multipolar spindle and premature loss of sister chromatid cohesion. In the monotelic attachment only one chromatid sister has the kinetochore attached to the microtubules. This error is that which is most readily corrected – as the presence of an unbound kinetochore triggers the spindle assembly checkpoint (SAC) to delay anaphase onset. In other cases – such as syntelic (both sister chromatids linked to one spindle pole) and merotelic attachments (one chromatid bound to microtubules from more than one spindle pole) – it is possible for errors to be unrecognized by the SAC and therefore persist through mitosis (13). However, these errors result in a lack of tension that normally exists when paired kinetochores are pulled towards opposing spindle poles. This lack of tension is an activation signal for the aurora B error correction machinery – that leads to detachment – and reattachment of microtubule-connections until errors are corrected (26). Merotelic attachments are some of the more frequent errors in mitosis. They are particularly dangerous as they are ‘invisible’ to the SAC due to the presence of microtubule-kinetochore attachments (13). The origins of merotelic attachments may originate due to supernumerary centrosomes, defective centrosome separation, premature anaphase due to SAC errors, or defects within the aurora B correction process (27–29).
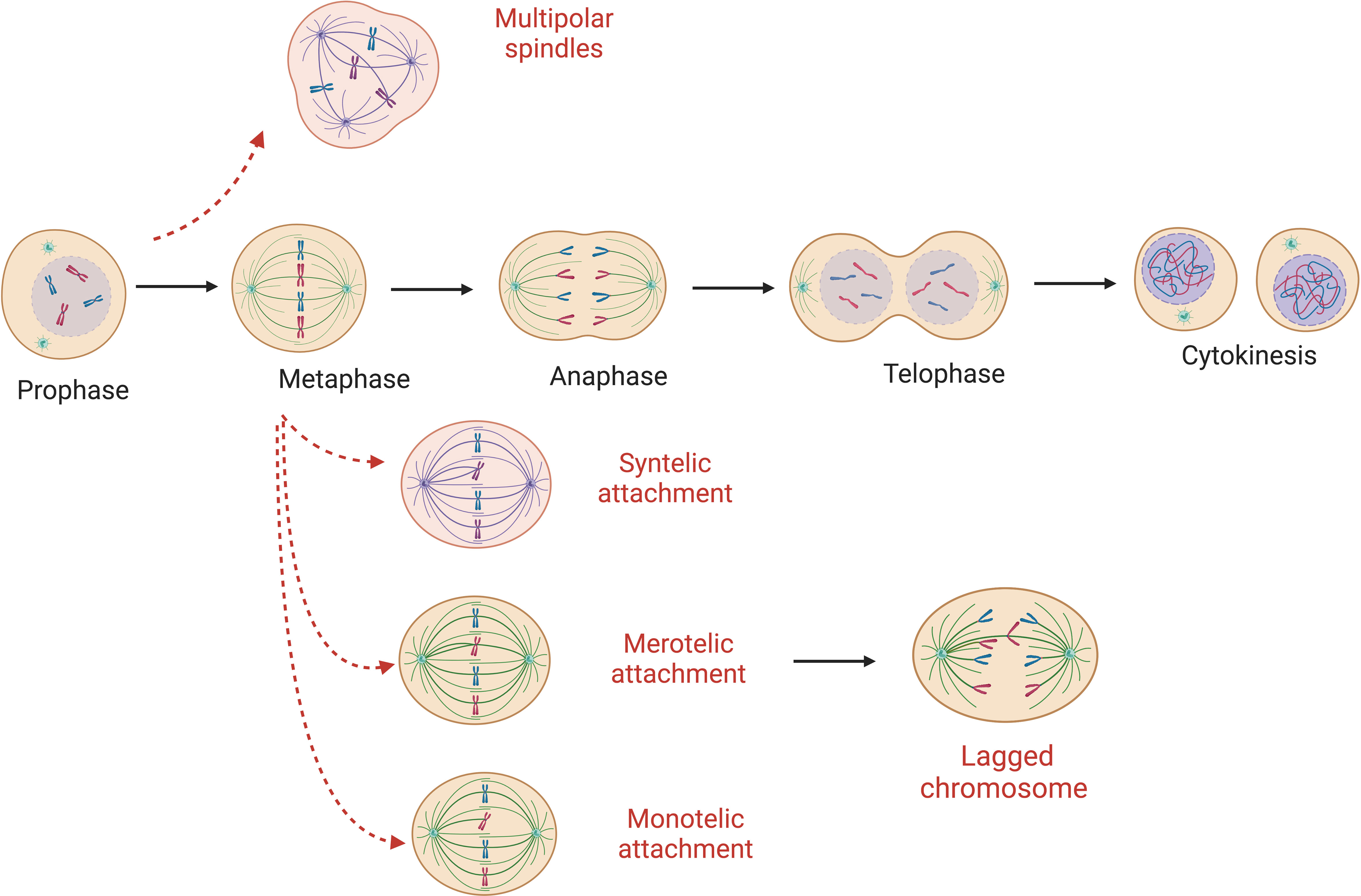
Figure 1 Abnormal microtubule attachments during mitosis. Red-dashed arrows show possible errors that can occur during cell replication ultimately leading to mis-segregated chromosomes. Here monotelic, syntelic, merotelic and multipolar spindle are represented. Absence of attachment and premature loss of sister chromatid cohesion are possible origins of W-CIN as well.
CIN, aneuploidy, and neuroblastoma
Neuroblastoma (NBL) is a pediatric solid tumor originating from the sympathetic nervous system (30) that represents 7-8% of pediatric malignancies (31). It can originate from any sympathetic ganglia such as in the adrenal medulla (the most common site involved), the chest and neck (along the sympathetic nervous system chain), or the pelvis (para- aortic body; organ of Zuckerkandl) (31–33). According to the International Neuroblastoma Risk Group (INRG) Staging System (INRGSS), NBL is divided into locoregional disease (L1 and L2 determined respectively by the absence or presence of imaging defined risk-factors), or stage M in the presence of metastatic disease (34). Common locations of metastatic spread include the bone marrow, bone, lymph nodes, and liver. Lastly, is stage MS (metastatic special) described as a tumor with metastatic lesions of the skin, liver and bone marrow in patients aged from 0 to 18 months (35). What makes this ‘special’ is the tendency of stage MS tumor to undergo spontaneous resolution.
NBL is characterized by a wide molecular and clinical diversity (31, 36–39). Prognosis depends not only by the conventional risk markers such as age, stage and histopathology (40), but also on gene expression (41–43) and genomic signature of the tumor (31, 40, 44). CGH array studies have furthermore helped to classify neuroblastoma tumors in different groups depending on their genomic alteration patterns (37, 45, 46). Neuroblastoma exhibit recurrent genomic alterations that may be characterized as numerical (whole chromosome aneuploidy), or segmental. Common segmental chromosomal alterations include deletion of 1p (47); 3p; 11q (36, 48, 49); 4p; 9p and 14q chromosomes and gain of 1q; 2p and 17q (40, 50–54). In contrast to the frequent macro-genomic alterations, NBL has a generally low mutation burden, and few recurrently altered genes (55, 56). The two of best studied gene alterations are: MYCN (57), which is amplified from 20% to 25% of NBL and is correlated with a poor prognosis (58–60), and ALK mutations occurring in 6-9% of sporadic tumors. Germline mutation in ALK is also a major cause of familial neuroblastoma (61).
Numerical and structural alterations are correlated with specific clinical features and prognosis. Younger patients, usually less than 1 year old (38), are found to havetumors that exhibit mostly numerical genomic alterations (62), and are associated with localized stages of the disease and excellent survival rates. In contrast, patients with tumors harboring structural chromosome aberrations are usually older, present with advanced stage disease (52), and have tumors with more aggressive growth patterns (63, 64). In large study comparing various genomic patterns, Janoueix- Loresey and colleagues analyzed 493 tumors using array CGH with accompanying outcomes data (8). The 4-year progression free survival was over 90% in those tumors with only numerical alterations, compared with 37-45% for those whose tumors had either MYCN amplification or segmental alterations (8). Similar results were seen in an independent study of 556 tumors – in which a higher overall number of chromosome breakpoints also was significantly associated with poor outcomes. Notably, in both studies those whose tumors have both segmental and numerical alterations behave as those with segmental defects – losing the apparent benefit of the numerical alterations. Similarly, with the exception of a novel chromosome 6q loss, the specific nature of the segmental alteration was less important than the presence of any segmental change. From this, it is evident that numerical genomic alterations are linked to a better prognosis, while on the contrary structural alterations are linked to poorer outcome and increased incidence of relapse (8, 56, 65). The mechanisms that lead to those states, however, is not clear. We propose an intermediate state where the tumor, or subclones within, become aneuploid due to the loss or gain of whole chromosomes. This state then predisposes the cells to acquire further genomic insults such as DNA damage, chromosome fragmentation, or chromothripsis, that result in segmental chromosome alterations.
USP24: a CIN gene associated with poor outcomes in neuroblastoma
We performed an on online computational screen using the PREdiction of Clinical Outcomes from Genomic Profiles tool (PRECOG; https://precog.stanford.edu/) to identify novel therapeutic targets in NBL (66). This tool analyzes the expression of genes in correlation with survival statistics and calculates a z-score to represent its impact on outcomes. Scores near zero are predicted to have little impact on disease outcomes, whereas significantly low, or high, z-scores suggest that reduced expression or elevated expression respectively is associated with por outcomes. Our screen focussed on the gene family encoding de-ubiquitinating enzymes (DUBs) – as these are emerging as novel therapeutic targets. When examining the impact of 95 DUBs on outcomes in NBL, we found that reduced expression of USP24 had the largest impact on outcomes, with a z-score of -10.14 (0.38 percentile) suggesting that USP24 reductions have a powerful negative impact in this disease (67). For comparison, the canonical NBL oncogene MYCN has a z-score of +8.18 (97.34 percentile) - in line with the known relationship between high expression and aggressive disease. We further found that patients whose tumors had reduced expression of USP24, defined as less than the 20th percentile, were significantly more likely to suffer disease progression and to die of any cause. USP24 is encoded on chromosome 1p32.3 – and area of the genome that is frequently lost in neuroblastoma tumors (68, 69). It is notable then, that cells lacking USP24 have a significant increase in erroneous mitosis, with a particular increase in lagging chromosomes (67). These cells and tissues from mice missing even 1 copy of the Usp24 gene have a significant increase in numerical chromosome aneuploidy. We identified that USP24 is required to preserve expression of CRMP2 – a microtubule assembly factor encoded by the DPYSL2 gene. We found CRMP2 to co-localize with spindle microtubules. Cells lacking USP24 had a significant reduction in spindle associated CRMP2 – a defect that was corrected by proteasome inhibition. Furthermore, restoring the expression of CRMP2 was able to correct the mitotic defect seen in cells lacking USP24. Looking again at human NBL, we also found that low USP24 was highly correlated with a high CIN25 score, supportive of the connection between USP24 and CIN in these tumors. Importantly, the poor survival seen in patients with tumors with low levels of USP24 is significantly improved by higher levels of CRMP2 (DPYSL2). Taken in aggregate, this data strongly implicates USP24 as a novel tumor suppressor gene in NBL that, when lost, promotes CIN and disease aggression.
Connecting the dots between numerical and segmental chromsome alterations
The association of USP24 reduction and aggressive NBL is seemingly at odds with the general association between numerical chromosome instability and favorable disease outcomes. However, the mutually exclusive nature of the literature that separates numerical from segmental chromosome alterations may be an oversimplification when it comes to the underlying biology. As will be discussed further below, recently studies have demonstrated that whole chromosome missegregation leads to the subsequent development of structural chromosome damage. We propose a model by which aggressive NBL with segmental chromosome alterations may arise from an intermediate stage characterized by whole chromosome aneuploidy (Figure 2).
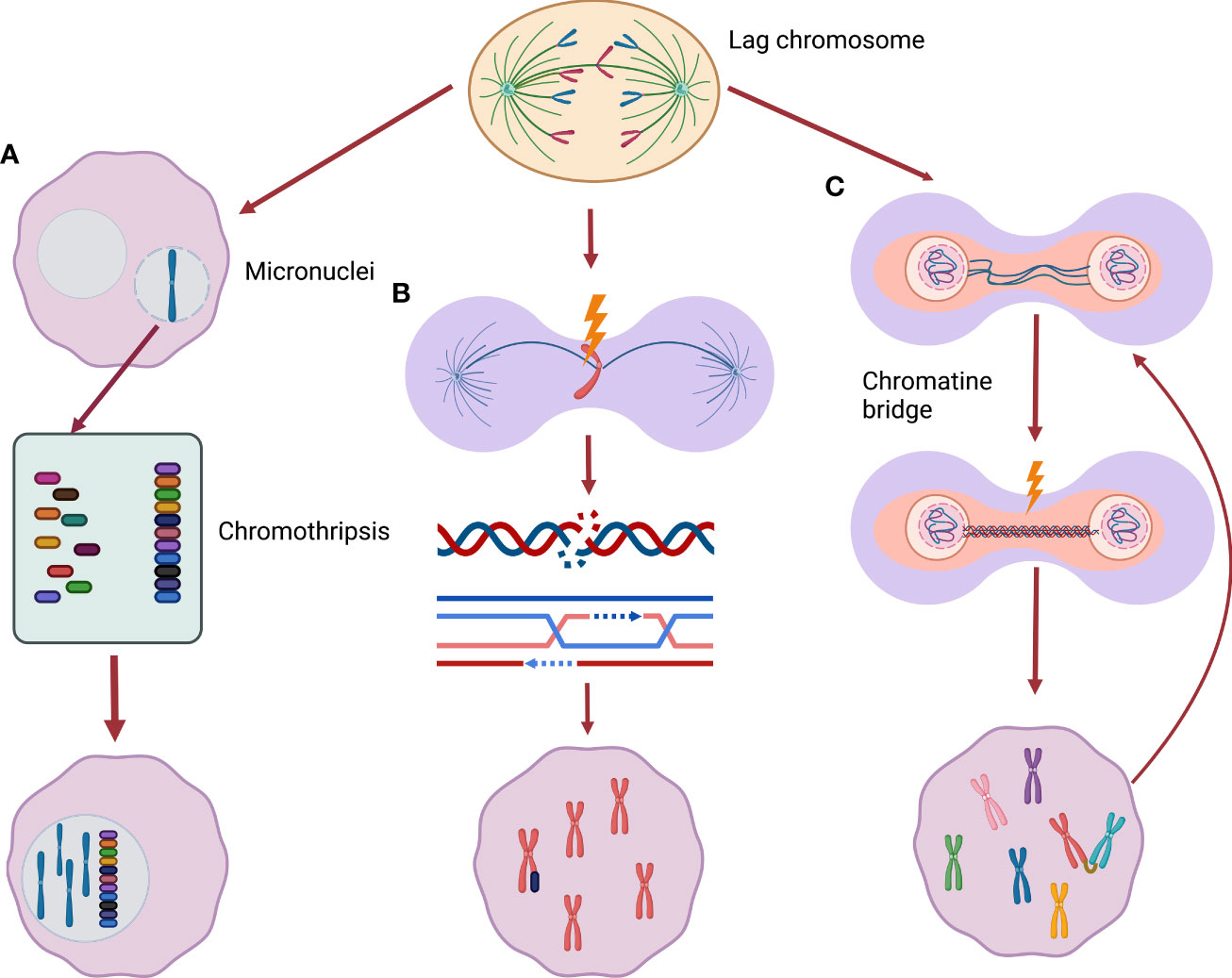
Figure 2 From whole chromosome mis-segregation to segmental alterations. (A) Lagging chromosomes may be incorporate into micronuclei, encountering chromothripsis (a phenomenon of massive chromatin fragmentation) during the following cell replication cycle. These severe rearranged chromosomes can be subsequently included back in the main nucleus. (B) Missegregated chromosomes can be frequently damaged during cytokinesis activating DNA double-strand break response leading to new segmental alterations. (C) Chromatin bridges can cause chromatin breakage triggering breakage-fusion-bridge cycle, a well described origin of segmental alterations.
As mentioned previously, the most common chromosome segregation error observed in cancer cells is chromosome lagging. This error, the result of mal-attachment of spindle microtubules to sister chromatid kinetochores, often results in genetic material separate from the main chromatin mass as cells enter interphase following cytokinesis (70). Consequentially as the nuclear membrane reforms, the erroneous segregation the lagged chromosomes can be surrounded by a distinct nuclear membrane forming a micronucleus. This phenomenon has led to micronuclei (MNi) being used as a marker of CIN. The Pellman laboratory observed that these MNi are highly susceptible to DNA damage in the next cell division cycle (71, 72). By following the fate of cultured cells with MNi, the group found that chromatin in MNi are subject to defective DNA replication during subsequent S-phases – with resulting DNA damage that may be catastrophic. Some cells encountered severe fragmentation of the chromatin within the MNi, a process known as chromothripsis. Moreover, the resulting fragmented chromatin was often re- incorporated into the main nuclear genome (71). In such way, the new rearrangements can be merged into the new daughter cells genome. The labs of Kops and Medema also demonstrated that missegregated chromosomes commonly develop DNA damage – with the subsequent double-strand break response leading to the development of chromosome translocations (73). While these studies were not performed specifically in NBL cells, it provides clear evidence that numerical chromosome missegregation may lead to severe DNA damage and subsequent structural chromosome rearrangements.
In addition to the consequences resulting from lagging chromosomes, another error that may produce segmental chromosome alterations is the chromatin bridge (74, 75). Chromatin bridges are thin chromatin strings that connect the separating chromosome masses during anaphase. In many instances, these bridges persist through telophase with chromosome breaks introduced during cytokinesis. Anaphase bridges often originate from incomplete DNA replication or fusion of telomeric end regions of two chromosomes which are later pulled to different poles during anaphase (dicentrinc chromosomes). Some bridges, in particular ultrafine DNA bridges can resolve without permanent DNA damage (75), others can lead to chromatin breakage causing DNA damage (73). Chromosomes harboring chromatin bridges often develop a variety of structural rearrangements (76) and often break into multiple fragments (74, 77). These breaks may trigger chromosome breakage-fusion-bridge (BFB) cycles – a known contributor to chromosomal structural rearrangements (76). Notably, recent evidence suggests that – as with lagging chromosomes – the attempted resolution of dicentric-chromosome induced chromatin bridges may result in severe chromosome fragmentation – including chromothripsis.
Discussion
CIN, aneuploidy, and segmental chromosome alterations are a pan-cancer feature. The apparent impact of these changes on prognosis – differs widely across tumor types – and in neuroblastoma numerical changes are often associated with good outcomes (Bilke S et al., 2005; Janoueix-Lerosey I et al., 2009; Fusco P et al., 2018). Here we review evidence that aneuploidy can be a first step towards the development of segmental alterations and propose a model in which segmental alterations in neuroblastoma may arise through this intermediate step of numerical aneuploidy. As summarized above there are several scientific studies that demonstrate an association between these two phenomena. The mechanisms that may connect whole-chromosome missegregation, aneuploidy, and the subsequent development of segmental chromosome alterations provide a potential explanation for our observation that the loss of USP24 may promote aggressive NBL. We have found that reduced expression of USP24 leads to abnormalities in the mitotic spindle and a significant increase in chromosome missegregation and aneuploidy in a mouse model. Surprisingly, there are no data involving the intercross of aneuploidy mouse models with those predisposed to neuroblastoma. Our Usp24 deficient mouse model may be a useful tool to help further explore this question. Such experimental models may shed light on the role of aneuploidy in initial tumor development, tumor progression, tumor evolution and of course in therapy responses. As there are hundreds of genes required for accurate cell division – it is unclear whether any cause of CIN may contribute to neuroblastoma development – or whether cell, tissue, or developmentally specific gene expression influences how the alteration of any one CIN gene impacts downstream events. We predict that specific mechanisms that produce CIN in this context will have important prognostic effects – since disease outcomes vary more by the presence or absence of segmental anomalies rather than the presence of any selected event.
Author contributions
LP, SH, and PG co-authored, edited, and approved the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
Figures were created with Biorender.com. The work was supported by grants to P.J. Galardy including from the Howard Hughes Medical Institute (HHMI), Fraternal Order of Eagles, and a Mayo Clinic Career Development award.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ben-David U, Amon A. Context is everything: aneuploidy in cancer. Nat Rev Genet (2020) 21:44–62. doi: 10.1038/s41576-019-0171-x
2. Stopsack KH, Whittaker CA, Gerke TA, Loda M, Kantoff PW, Mucci LA, et al. Aneuploidy drives lethal progression in prostate cancer. Proc Natl Acad Sci USA. (2019) 116:11390–5. doi: 10.1073/pnas.1902645116
3. Duijf PHG, Nanayakkara D, Nones K, Srihari S, Kalimutho M, Khanna KK. Mechanisms of genomic instability in breast cancer. Trends Mol Med (2019) 25:595–611. doi: 10.1016/j.molmed.2019.04.004
4. Oltmann J, Heselmeyer-Haddad K, Hernandez LS, Meyer R, Torres I, Hu Y, et al. Aneuploidy, TP53 mutation, and amplification of MYC correlate with increased intratumor heterogeneity and poor prognosis of breast cancer patients. Genes Chromosomes Cancer (2018) 57:165–75. doi: 10.1002/gcc.22515
5. Xu J, Huang L, Li J. DNA Aneuploidy and breast cancer: a meta-analysis of 141,163 cases. Oncotarget (2016) 7:60218–29. doi: 10.18632/oncotarget.11130
6. Choma D, Daures JP, Quantin X, Pujol JL. Aneuploidy and prognosis of non- small-cell lung cancer: a meta-analysis of published data. Br J Cancer (2001) 85:14–22. doi: 10.1054/bjoc.2001.1892
7. Sansregret L, Vanhaesebroeck B, Swanton C. Determinants and clinical implications of chromosomal instability in cancer. Nat Rev Clin Oncol (2018) 15:139–50. doi: 10.1038/nrclinonc.2017.198
8. Janoueix-Lerosey I, Schleiermacher G, Michels E, Mosseri V, Ribeiro A, Lequin D, et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol (2009) 27:1026–33. doi: 10.1200/JCO.2008.16.0630
9. Moon JJ, Lu A, Moon C. Role of genomic instability in human carcinogenesis. Exp Biol Med (Maywood) (2019) 244:227–40. doi: 10.1177/1535370219826031
10. Giam M, Rancati G. Aneuploidy and chromosomal instability in cancer: a jackpot to chaos. Cell Div (2015) 10:3. doi: 10.1186/s13008-015-0009-7
11. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol (2010) 11:220–8. doi: 10.1038/nrm2858
12. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol (2012) 30:413–21. doi: 10.1038/nbt.2203
13. Cimini D. Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim Biophys Acta (2008) 1786:32–40. doi: 10.1016/j.bbcan.2008.05.003
14. Bakhoum SF, Cantley LC. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell (2018) 174:1347–60. doi: 10.1016/j.cell.2018.08.027
15. Bakhoum SF, Silkworth WT, Nardi IK, Nicholson JM, Compton DA, Cimini D. The mitotic origin of chromosomal instability. Curr Biol (2014) 24:R148–149. doi: 10.1016/j.cub.2014.01.019
16. Funk LC, Zasadil LM, Weaver BA. Living in CIN: Mitotic infidelity and its consequences for tumor promotion and suppression. Dev Cell (2016) 39:638–52. doi: 10.1016/j.devcel.2016.10.023
17. Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends Genet (2008) 24:457–66. doi: 10.1016/j.tig.2008.07.002
18. Birkbak NJ, Eklund AC, Li Q, McClelland SE, Endesfelder D, Tan P, et al. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res (2011) 71:3447–52. doi: 10.1158/0008-5472.CAN-10-3667
19. Hieronymus H, Murali R, Tin A, Yadav K, Abida W, Moller H, et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. Elife (2018) 7:e37294. doi: 10.7554/eLife.37294.027
20. Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, et al. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol (2013) 15:96–102. doi: 10.1038/ncb2643
21. van Ree JH, Nam HJ, Jeganathan KB, Kanakkanthara A, van Deursen JM. Pten regulates spindle pole movement through Dlg1-mediated recruitment of Eg5 to centrosomes. Nat Cell Biol (2016) 18:814–21. doi: 10.1038/ncb3369
22. Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet (2006) 38:1043–8. doi: 10.1038/ng1861
23. Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol (2007) 179:255–67. doi: 10.1083/jcb.200706015
24. Fusco P, Esposito MR, Tonini GP. Chromosome instability in neuroblastoma. Oncol Lett (2018) 16:6887–94. doi: 10.3892/ol.2018.9545
25. Bastians H. Causes of chromosomal instability. Recent Results Cancer Res (2015) 200:95–113. doi: 10.1007/978-3-319-20291-4_5
26. McVey SL, Cosby JK, Nannas NJ. Aurora b tension sensing mechanisms in the kinetochore ensure accurate chromosome segregation. Int J Mol Sci (2021) 22:8818. doi: 10.3390/ijms22168818
27. Bakhoum SF, Thompson SL, Manning AL, Compton DA. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol (2009) 11:27–35. doi: 10.1038/ncb1809
28. Chunduri NK, Storchova Z. The diverse consequences of aneuploidy. Nat Cell Biol (2019) 21:54–62. doi: 10.1038/s41556-018-0243-8
29. Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, Salmon ED. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol (2001) 153:517–27. doi: 10.1083/jcb.153.3.517
30. Hanemaaijer ES, Margaritis T, Sanders K, Bos FL, Candelli T, Al-Saati H, et al. Single-cell atlas of developing murine adrenal gland reveals relation of schwann cell precursor signature to neuroblastoma phenotype. Proc Natl Acad Sci U.S.A. (2021) 118:e2022350118. doi: 10.1073/pnas.2022350118
31. Mlakar V, Jurkovic Mlakar S, Lopez G, Maris JM, Ansari M, Gumy-Pause F. 11q deletion in neuroblastoma: a review of biological and clinical implications. Mol Cancer (2017) 16:114. doi: 10.1186/s12943-017-0686-8
32. Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet (2007) 369:2106–20. doi: 10.1016/S0140-6736(07)60983-0
33. Zobel M, Zamora A, Sura A, Wang L, Stein J, Marachelian A, et al. The clinical management and outcomes of pelvic neuroblastic tumors. J Surg Res (2020) 249:8–12. doi: 10.1016/j.jss.2019.12.009
34. Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, et al. The international neuroblastoma risk group (INRG) staging system: an INRG task force report. J Clin Oncol (2009) 27:298–303. doi: 10.1200/JCO.2008.16.6876
35. Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The international neuroblastoma risk group (INRG) classification system: an INRG task force report. J Clin Oncol (2009) 27:289–97. doi: 10.1200/JCO.2008.16.6785
36. Guo C, White PS, Weiss MJ, Hogarty MD, Thompson PM, Stram DO, et al. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene (1999) 18:4948–57. doi: 10.1038/sj.onc.1202887
37. Ohira M, Nakagawara A. Global genomic and RNA profiles for novel risk stratification of neuroblastoma. Cancer Sci (2010) 101:2295–301. doi: 10.1111/j.1349-7006.2010.01681.x
38. Schleiermacher G, Michon J, Huon I, d'Enghien CD, Klijanienko J, Brisse H, et al. Chromosomal CGH identifies patients with a higher risk of relapse in neuroblastoma without MYCN amplification. Br J Cancer (2007) 97:238–46. doi: 10.1038/sj.bjc.6603820
39. Vandesompele J, Baudis M, De Preter K, Van Roy N, Ambros P, Bown N, et al. Unequivocal delineation of clinicogenetic subgroups and development of a new model for improved outcome prediction in neuroblastoma. J Clin Oncol (2005) 23:2280–99. doi: 10.1200/JCO.2005.06.104
40. Weinstein JL, Katzenstein HM, Cohn SL. Advances in the diagnosis and treatment of neuroblastoma. Oncologist (2003) 8:278–92. doi: 10.1634/theoncologist.8-3-278
41. Asgharzadeh S, Pique-Regi R, Sposto R, Wang H, Yang Y, Shimada H, et al. Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification. J Natl Cancer Inst (2006) 98:1193–203. doi: 10.1093/jnci/djj330
42. Ohira M, Oba S, Nakamura Y, Isogai E, Kaneko S, Nakagawa A, et al. Expression profiling using a tumor-specific cDNA microarray predicts the prognosis of intermediate risk neuroblastomas. Cancer Cell (2005) 7:337–50. doi: 10.1016/j.ccr.2005.03.019
43. Vermeulen J, De Preter K, Naranjo A, Vercruysse L, Van Roy N, Hellemans J, et al. Predicting outcomes for children with neuroblastoma using a multigene-expression signature: a retrospective SIOPEN/COG/GPOH study. Lancet Oncol (2009) 10:663–71. doi: 10.1016/S1470-2045(09)70154-8
44. Look AT, Hayes FA, Nitschke R, McWilliams NB, Green AA. Cellular DNA content as a predictor of response to chemotherapy in infants with unresectable neuroblastoma. N Engl J Med (1984) 311:231–5. doi: 10.1056/NEJM198407263110405
45. Brinkschmidt C, Poremba C, Christiansen H, Simon R, Schafer KL, Terpe HJ, et al. Comparative genomic hybridization and telomerase activity analysis identify two biologically different groups of 4s neuroblastomas. Br J Cancer (1998) 77:2223–9. doi: 10.1038/bjc.1998.370
46. Plantaz D, Vandesompele J, Van Roy N, Lastowska M, Bown N, Combaret V, et al. Comparative genomic hybridization (CGH) analysis of stage 4 neuroblastoma reveals high frequency of 11q deletion in tumors lacking MYCN amplification. Int J Cancer (2001) 91:680–6. doi: 10.1002/1097-0215(200002)9999:9999<::AID-IJC1114>3.0.CO;2-R
47. Fong CT, Dracopoli NC, White PS, Merrill PT, Griffith RC, Housman DE, et al. Loss of heterozygosity for the short arm of chromosome 1 in human neuroblastomas: correlation with n-myc amplification. Proc Natl Acad Sci U.S.A. (1989) 86:3753–7. doi: 10.1073/pnas.86.10.3753
48. Mertens F, Johansson B, Hoglund M, Mitelman F. Chromosomal imbalance maps of malignant solid tumors: a cytogenetic survey of 3185 neoplasms. Cancer Res (1997) 57:2765–80.
49. Vandesompele J, Van Roy N, Van Gele M, Laureys G, Ambros P, Heimann P, et al. Genetic heterogeneity of neuroblastoma studied by comparative genomic hybridization. Genes Chromosomes Cancer (1998) 23:141–52. doi: 10.1002/(SICI)1098-2264(199810)23:2<141::AID-GCC7>3.0.CO;2-2
50. Attiyeh EF, London WB, Mosse YP, Wang Q, Winter C, Khazi D, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med (2005) 353:2243–53. doi: 10.1056/NEJMoa052399
51. Bown N, Cotterill S, Lastowska M, O'Neill S, Pearson AD, Plantaz D, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med (1999) 340:1954–61. doi: 10.1056/NEJM199906243402504
52. Lastowska M, Cullinane C, Variend S, Cotterill S, Bown N, O'Neill S, et al. Comprehensive genetic and histopathologic study reveals three types of neuroblastoma tumors. J Clin Oncol (2001) 19:3080–90. doi: 10.1200/JCO.2001.19.12.3080
53. Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol (1999) 17:2264–79. doi: 10.1200/JCO.1999.17.7.2264
54. Pezzolo A, Rossi E, Gimelli S, Parodi F, Negri F, Conte M, et al. Presence of 1q gain and absence of 7p gain are new predictors of local or metastatic relapse in localized resectable neuroblastoma. Neuro Oncol (2009) 11:192–200. doi: 10.1215/15228517-2008-086
55. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature (2018) 555:321–7. doi: 10.1038/nature25480
56. Nakagawara A, Li Y, Izumi H, Muramori K, Inada H, Nishi M. Neuroblastoma. Jpn J Clin Oncol (2018) 48:214–41. doi: 10.1093/jjco/hyx176
57. Seeger RC, Brodeur GM, Sather H, Dalton A, Seigel S, Wong KY, et al. Association of Multiple Copies of the N-myc Oncogene with Rapid Progression of Neuroblastomas. New Engl J Medicine (1985) 313:1111–6. doi: 10.1056/NEJM198510313131802
58. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature (2008) 455:967–70. doi: 10.1038/nature07398
59. Matsunaga T, Takahashi H, Ohnuma N, Tanabe M, Yoshida H, Iwai J, et al. Expression of n-myc and c-src protooncogenes correlating to the undifferentiated phenotype and prognosis of primary neuroblastomas. Cancer Res (1991) 51:3148–52.
60. Slavc I, Ellenbogen R, Jung WH, Vawter GF, Kretschmar C, Grier H, et al. Myc gene amplification and expression in primary human neuroblastoma. Cancer Res (1990) 50:1459–1463.
61. Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature (2008) 455:930–5. doi: 10.1038/nature07261
62. Bilke S, Chen QR, Westerman F, Schwab M, Catchpoole D, Khan J. Inferring a tumor progression model for neuroblastoma from genomic data. J Clin Oncol (2005) 23:7322–31. doi: 10.1200/JCO.2005.03.2821
63. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer (2003) 3:203–16. doi: 10.1038/nrc1014
64. Schleiermacher G, Mosseri V, London WB, Maris JM, Brodeur GM, Attiyeh E, et al. Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the INRG project. Br J Cancer (2012) 107:1418–22. doi: 10.1038/bjc.2012.375
65. Schleiermacher G, Janoueix-Lerosey I, Ribeiro A, Klijanienko J, Couturier J, Pierron G, et al. Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol (2010) 28:3122–30. doi: 10.1200/JCO.2009.26.7955
66. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med (2015) 21:938–45. doi: 10.1038/nm.3909
67. Bedekovics T, Hussian S, Zhang Y, Ali A, Jeon YJ, Galardy PJ, et al. USP24 Is a Cancer-Associated Ubiquitin Hydrolase, Novel Tumor Suppressor, and Chromosome Instability Gene Deleted in Neuroblastoma. Cancer Res (2021) 81:1321–31. doi: 10.1158/0008-5472.CAN-20-1777
68. Brodeur GM, Sekhon G, Goldstein MN. Chromosomal aberrations in human neuroblastomas. Cancer (1977) 40:2256–63. doi: 10.1002/1097-0142(197711)40:5<2256::AID-CNCR2820400536>3.0.CO;2-1
69. Depuydt P, Boeva V, Hocking TD, Cannoodt R, Ambros IM, Ambros PF, et al. Genomic amplifications and distal 6q loss: Novel markers for poor survival in high-risk neuroblastoma patients. J Natl Cancer Inst (2018) 110:1084–93. doi: 10.1093/jnci/djy022
70. Cimini D, Cameron LA, Salmon ED. Anaphase spindle mechanics prevent mis- segregation of merotelically oriented chromosomes. Curr Biol (2004) 14:2149–55. doi: 10.1016/j.cub.2004.11.029
71. Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, et al. DNA Breaks and chromosome pulverization from errors in mitosis. Nature (2012) 482:53–8. doi: 10.1038/nature10802
72. Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, et al. Chromothripsis from DNA damage in micronuclei. Nature (2015) 522:179–84. doi: 10.1038/nature14493
73. Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science (2011) 333:1895–8. doi: 10.1126/science.1210214
74. Hoffelder DR, Luo L, Burke NA, Watkins SC, Gollin SM, Saunders WS. Resolution of anaphase bridges in cancer cells. Chromosoma (2004) 112:389–97. doi: 10.1007/s00412-004-0284-6
75. Petsalaki E, Zachos G. Building bridges between chromosomes: novel insights into the abscission checkpoint. Cell Mol Life Sci (2019) 76:4291–307. doi: 10.1007/s00018-019-03224-z
76. Gisselsson D, Pettersson L, Hoglund M, Heidenblad M, Gorunova L, Wiegant J, et al. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci U.S.A. (2000) 97:5357–62. doi: 10.1073/pnas.090013497
Keywords: neuroblastoma, chromosome instability (CIN), aneuploidy, deubuquitylases, mitosis, chromosome missegregation, mitotic spindle
Citation: Paolini L, Hussain S and Galardy PJ (2022) Chromosome instability in neuroblastoma: A pathway to aggressive disease. Front. Oncol. 12:988972. doi: 10.3389/fonc.2022.988972
Received: 07 July 2022; Accepted: 03 October 2022;
Published: 20 October 2022.
Edited by:
Rafael Pulido, IKERBASQUE Basque Foundation for Science, SpainReviewed by:
Nevim Aygun, Ege University, TurkeyCopyright © 2022 Paolini, Hussain and Galardy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul J. Galardy, Z2FsYXJkeS5wYXVsQG1heW8uZWR1