 Qingbo Feng
Qingbo Feng Hancong Li
Hancong Li Guoteng Qiu1†
Guoteng Qiu1† Zhaolun Cai
Zhaolun Cai Jiaxin Li
Jiaxin Li Yong Zeng
Yong Zeng Jiwei Huang
Jiwei Huang- 1Department of Liver Surgery and Liver Transplantation Centre, West China Hospital, Sichuan University, Chengdu, China
- 2West China School of Medicine, West China Hospital, Sichuan University, Chengdu, China
- 3Department of Gastrointestinal Surgery, West China Hospital, Sichuan University, Chengdu, China
Introduction: Pheochromocytoma is a neuroendocrine tumor originating from chromaffin cells in the adrenal medulla. Giant pheochromocytomas with a maximum diameter of over 20 cm are particularly rare.
Case presentation: We present a case of giant cystic pheochromocytoma in a 64-year-old woman who was found to have a right abdominal mass during an ultrasound examination, which is the largest pheochromocytoma ever documented in China. Meanwhile, obvious atrophy of the right lobe of the liver was found in preoperative CT and during the operation. Our literature review identified 20 cases with a diameter of over 20 cm. The average age at diagnosis was 51.7 (range 17–85), and 35% of cases did not exhibit classic symptoms.
Conclusion: Giant pheochromocytoma is an uncommon neoplasm. It can be discovered late due to a lack of clinical manifestations. Diagnosis is dependent on imaging recognition together with catecholamine secretion. Surgical resection is the only curative treatment for such tumors.
Introduction
Pheochromocytoma is an infrequent catecholamine-secreting neoplasm that arises from chromaffin tissues, which tends to occur in the adrenal medulla (1). The estimated annual incidence of pheochromocytoma is 0.4 to 9.5 per million (2). Hypertension, which can be sustained or paroxysmal, is the most common sign (3). Episodic headache, palpitations, and diaphoresis are the classical triad of clinical features, which can be seen in <25% of patients. At least one component of the triad occurs in slightly less than 50% of patients. These episodes are connected with the catecholamine excess produced by the tumor (4). In addition, local symptoms caused by the giant tumor include stomachache, backache, abdominal distension, and some atypical gastrointestinal symptoms. Commonly, it is a solid tumor and histologically benign (5). Nevertheless, cystic pheochromocytoma is a particular rare entity and is usually asymptomatic but can be fatal due to cardiovascular complications (6). The most accurate metabolic testing for the biochemical diagnosis of this tumor is the elevated plasma-free or 24-h urinary fractionated metanephrines (7). Computed tomography (CT) or magnetic resonance imaging (MRI) is the preference for the initial anatomical localization of the tumor due to its high sensitivity (90%–100%) and reasonable specificity (70%–80%) (4). Once the pheochromocytoma is diagnosed, patients are referred to surgical extirpation, which is the only curative strategy (5). Currently, just a few cases concerning giant cystic pheochromocytomas have been reported worldwide (8). Here, a case of cystic pheochromocytoma with enormous size, a 20 × 15 × 10 cm tumor, is described. The study is reported in agreement with the principles of the CAse REport (CARE) guidelines (9). Additionally, we have performed a literature review on pheochromocytoma measuring 20 cm or greater to update the clinical features of this rare disease. To our best knowledge, this is the largest cystic pheochromocytoma in China as per the available indexed literature.
Case presentation
On 20 April 2022, a 64-year-old woman was referred to the West China Hospital after an abdominal mass was found on ultrasound examination for mild right upper abdominal discomfort. The patient reported no significant symptoms or past medical history, while physical examination indicated a large mass measuring 20 × 15 cm was palpated at the right upper quadrant. On admission, her blood pressure was 127/89 mmHg, and her heart rate was 78 beats per minute. Initial laboratory investigations concerning biochemistry and hematology examinations were within normal limits.
Abdominal enhanced CT revealed obvious compression atrophy of the liver and a 20 cm × 13 cm mixture of cystic and solid lesions between the right lobe of the retroperitoneal liver and the right kidney (Figures 1A, B). Plasma metanephrine and catecholamine measurements were performed, and evidence of pheochromocytoma was increased upon observation of elevated metanephrines and their metabolites in serum (Table 1). The patient did not have underlying diseases such as hypertension or diabetes. Before the metabolic testing, she did not take blood pressure medications, acetaminophen, beta- and alpha-adreno receptor blocking drugs, psychotropic medications, or any drugs that might interfere with her metabolism.
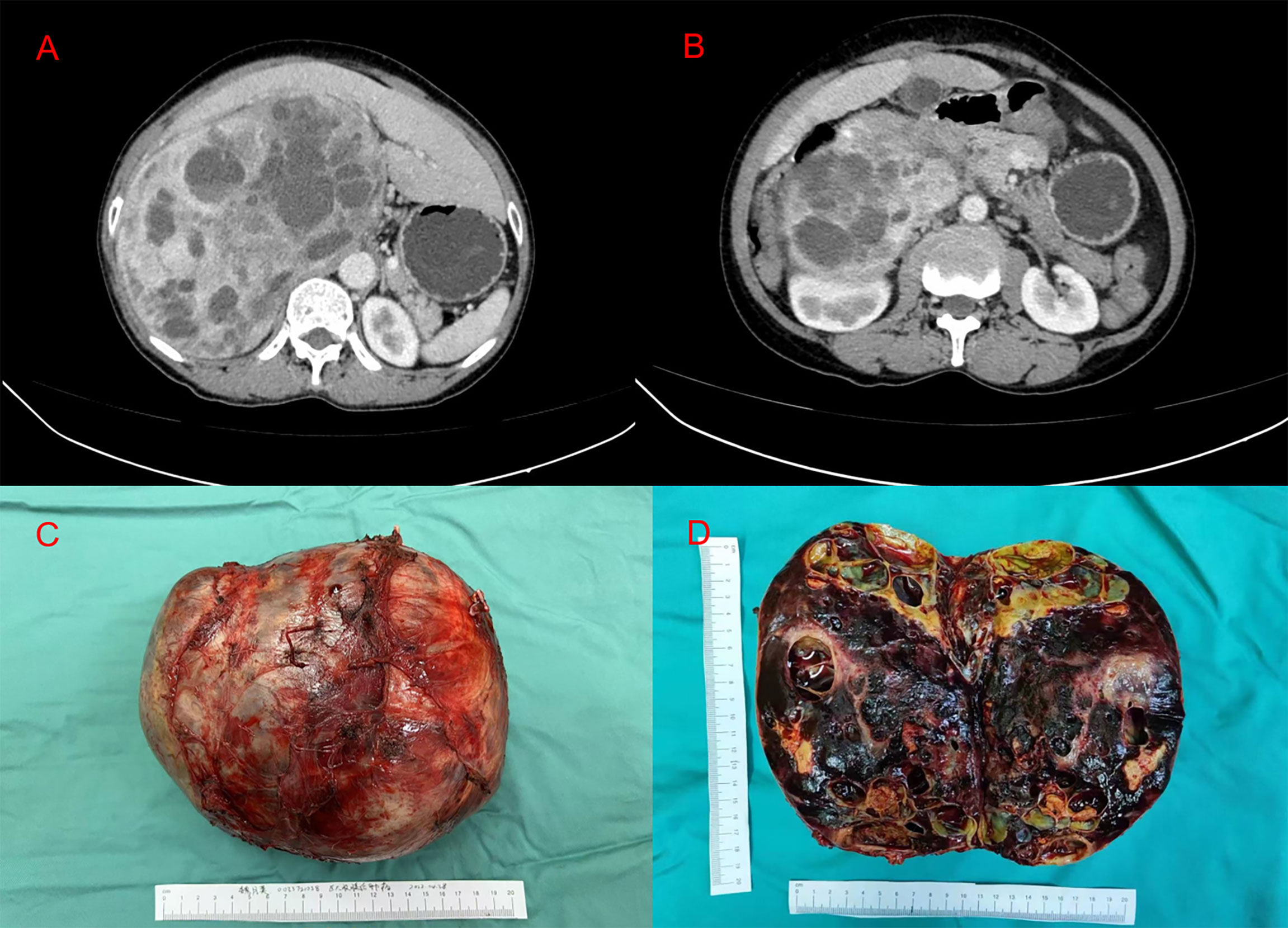
Figure 1 (A, B) Abdominal enhanced computed tomography imaging representing the huge tumor between the right lobe of the retroperitoneal liver and the right kidney. (C) A macroscopic image of the post-section cystic pheochromocytoma. (D) Excised tumor, measuring 20 × 15 × 10 cm.

Table 1 Plasma metanephrines measurements confirming the pheochromocytoma diagnosis.
Based on endocrine consultation, an alpha receptor blocker (phenoxybenzamine) was used for 2 weeks preoperatively. Three days after that, we encouraged the patient to start a salt-rich diet. In addition, an adequate fluid replacement was performed preoperatively. Blood pressure and heart rate of 139/79 mmHg and 77 beats per minute were recorded prior to surgery. The resection of the giant right adrenal pheochromocytoma was completed through a right subcostal incision under general anesthesia (Figure 1C). During the operation, the omentum majus widely adhered to the small intestine, abdominal wall, and liver. The right lobe of the liver was squeezed and atrophied by the retroperitoneal tumor, which was located behind the inferior vena cava. Blood pressure fluctuated intraoperatively, between 83 and 157 mmHg for the systolic pressure and between 55 and 92 mmHg for the diastolic pressure. Heart rate was measured between 57 and 87 beats per minute. After surgery, her blood pressure was stable at 112/75 mmHg, and her heart rate was 74 beats per minute. Subsequently, the patient was admitted to an intensive care unit (ICU) for 2 days and then transferred to the ward. Symptomatic supportive treatment was given while the patient was in ICU, including hemodynamic monitoring, oxygen inhalation, analgesia, infection prevention, fluid replacement, and liver function protection. On postoperative day 4, oral feeding was resumed, and the patient was discharged on the eighth day after surgery with no complications. We suggested genetic testing and developed a collaborative, multidisciplinary long-term follow-up plan. With the first outpatient visit 1 month after surgery, an annual follow-up with clinical and biochemical assessment was recommended. The individualized follow-up plan could be flexibly adjusted based on the gene detection and monitoring results.
Pathological examination of the surgical specimen indicated a necrotic change measuring 20 × 15 × 10 cm and weighing 2,240 g located in the retroperitoneum and right adrenal gland (Figure 1D). Hematoxylin–eosin (H&E) staining showed a well-circumscribed tumor surrounded by a fibrous capsule (Figures 2A, B). Immunohistochemistry showed marked pleomorphism within a few tumor cells, CD56(+), CgA (+), Syn(+), CK (Pan)(−), MART-1(−), inhibitA(−), S-100(+), INI1(+), Desmin(−), and Myogenin(−) (Figures 2C, D). According to the immunohistochemical results, the diagnosis of pheochromocytoma was made. Based on the staging system introduced in the 8th edition of the American Joint Committee on Cancer (AJCC) staging system, the tumor was determined as T2M0N0.

Figure 2 H&E staining in (A) ×10 view and (B) ×40 view. (C, D) CD56 staining immunohistochemistry of the tumor.
Discussion
Our case reported a rare cystic pheochromocytoma with a lack of evident endocrine symptoms, resulting in a large size and late diagnosis. The extensive necrosis of the adrenal gland, resulting in reduced production of catecholamines, and the retention of these hormones in the capsule mass after secretion may be the explanation for the absence of symptoms. As a result, the time to diagnosis was delayed, and the tumor size was larger once it was detected.
Since serious complications such as myocardial infarction, arrhythmias, dissection aortic aneurysms, heart failure, malignant hypertension, and sudden death can be caused by pheochromocytoma, it might give rise to premature mortality if overlooked (10, 11). In addition to this, cases of misdiagnosis of cystic pheochromocytoma such as pancreatic cystic tumor (12, 13), hepatic cystic tumor (14), simple adrenal cyst (15), and liver abscess (16) under CT scan have also been previously documented. Hence, it is essential to investigate the characteristics of this tumor.
To date, a total of 20 cases (5, 8, 17–33) of giant pheochromocytoma ≥20 cm were documented since the tumor was first described in 1886 by Fränke (34). We reviewed previous reports and summarized the clinical characteristics (Table 2). The average age at the discovery of giant pheochromocytoma was 51 (range 17–85) years. No significant difference was observed in gender, with slightly more women (11 cases). Additionally, similar findings were revealed in tumor histopathology (five and six cases manifested as malignant and benign, respectively). More cases had tumors that occurred on the left (11 cases) side of the abdomen than on the right (seven cases), and the rest of the articles did not explicitly state the site. Most clinical manifestations were asymptomatic (seven cases) and were often discovered occasionally during a physical examination. In addition, backache (three cases), abdominal pain (two cases), and chest pain (two cases) are relatively common chief complaints. What is more, non-endocrine manifestations, such as abdominal swelling, nausea, constipation, fatigue, and weight loss, could also be presented as the patient’s chief complaints.
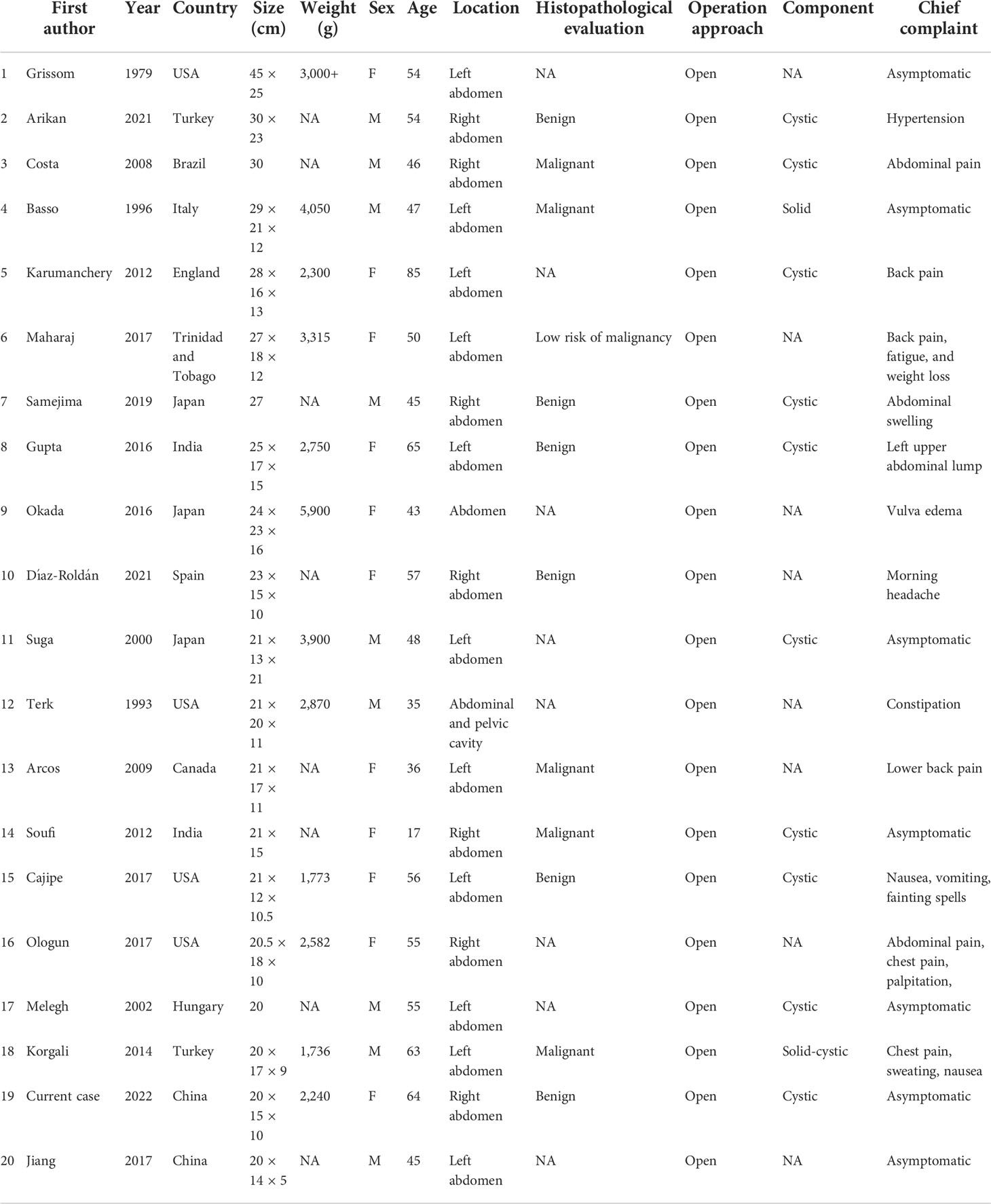
Table 2 A summary of reported giant pheochromocytomas with a maximal diameter greater than 20 cm, arranged by the largest to smallest maximum diameter.
Till now, the largest pheochromocytoma in the world was recorded to be 45 × 20 cm originally by Grissom et al. in 1979 (17). The current case turned out to be the largest cystic pheochromocytoma in China.
One of the unusual features of the present case is the cystic components. Most of the time, pheochromocytoma is a solid neoplasm originating from the adrenal medulla (29, 35), while cystic pheochromocytoma is a rare neuroendocrine tumor, with only a few described in former studies (5, 18, 19, 21, 22, 25, 29, 31). Goldberg et al. and Samejimaonce et al. successively found that the cystic fluid contains high concentrations of catecholamines and metanephrines (22, 35). Extensive hemorrhage followed by necrosis with cyst formation and subsequent resorption of the contents may be the postulated mechanism for this cystic degeneration in the tumor. Furtherly, extensive hemorrhage was likely triggered by the tumor outgrowing its vascular supply (36). This cystic change has also been demonstrated in other primary adrenal neoplasms, either benign cortical adenomas and hemangioma or primary and metastatic malignant adrenal lesions (37).
Notably, despite the surgical margin being histologically negative, the nature of the cystic pheochromocytoma should be interpreted with caution.
As with many other neuroendocrine tumors, it is almost impossible to confirm whether a tumor is benign or malignant only by histological criteria (5, 38). According to 2017 WHO classification of endocrine tumors, all pheochromocytomas are deemed to have metastatic potential, replacing the previous term “malignant” (39). This approach is maintained in the 2022 WHO classification (40). Thus, many researchers are attempting to develop scoring or classification systems that would predict the future behavior of pheochromocytoma. Multifactorial assessment, including tumor size (≥5 cm), Ki-67 index, SDHB mutation, and the dopaminergic phenotype have been suggested to assess the metastatic potential (40–42). These methods for screening for SDHx mutations are reasonable for rapid identification of patients at high risk of metastasis. However, accurate genetic testing remains essential (40). In addition to this, the Pheochromocytoma of the Adrenal gland Scaled Score (PASS) system has carried out a meaningful attempt (43). This is a risk-stratification system entirely based on histological features. However, the PASS score seems to be more reliable concerning the negative predictive value for the absence of metastatic behavior (44). The Grading system for Adrenal Pheochromocytoma and Para-ganglioma (GAPP) system, based on the growth pattern, cellularity, comedo-type necrosis, and vascular or capsular invasion, complemented by the Ki-67 index and catecholamine type demonstrates good predictive performance (45).
However, the problem concerning the label “poorly differentiated” for these neoplasms that are not poorly differentiated in the context used for high-grade neuroendocrine tumors has been noted by the WHO/International Agency for Research on Cancer (IARC) classification. Currently, COmposite Pheochromocytoma/para-ganglioma (COPPS) Prognostic Score classification systems, with a sensitivity of 100% and specificity of 95%, are proposed. This score focuses on clinicopathological criteria, including tumor size (>7 cm), necrosis, vascular invasion, and SDHB immunohistochemical staining. As the study was only recently published, it has not been independently verified (40). At present, the latest version of WHO classification does not recognize any of these systems but at the same time does not discourage their use in individual practices (40). Thereby, subsequent follow-up and metastasis monitoring are still critical to patient care.
Upon suspicion of pheochromocytoma, verification can be performed by biochemical testing (46). Among hormonal assay tests, elevated levels of metanephrines in 24-h urine or plasma are of the highest suggestibility (47). Anatomical imaging should be followed as the first modality if biochemical tests indicate the presence of pheochromocytomas. Abdominal or pelvic CT scans are helpful and highly recommended (47, 48). Other imaging studies, such as abdominal/pelvic multiphasic CT or MRI scans or nuclear medicine imaging, including meta-iodobenzylguanidine (MIBG) scintigraphy, FDG-PET/CT scans, and DOTA-SSA PET/CT scans, should be performed as appropriate if metastatic or multifocal disease is suspected (47). Functional imaging enables accurate diagnosis of tumor recurrence or metastasis that might go undetected by anatomical imaging (49). Regarding differential diagnosis, [18F]-FDG can be helpful to determine whether the tumors were benign or indicate primary malignant adrenal diseases in non-functioning adrenal masses with inconclusive CT/MRI imaging (50). Further, the latest review recommends specific nuclear imaging for the different clusters of pheochromocytoma when conducting personalized surveillance and management (39, 50). Based on genetic testing, [68Ga]-DOTA-SSA PET/CT is demonstrated as the most sensitive functional imaging modality for cluster 1A, while [18F] FDOPA PET/CT is more sensitive for cluster 1B and cluster 2 tumors (39). Our CT image presents a sporadic isolated adrenal mass, and pheochromocytoma was highly suspected based on the imaging features. In addition, given the patient’s financial situation and her wishes, we finally had no nuclear medical examination performed.
Once a pheochromocytoma is diagnosed, surgery is the preferred treatment whenever possible, regardless of its nature, component, and size. In addition to the conventional open approach, laparoscopic surgery has emerged as a favorable approach due to its ability to decrease hospitalizations, transfusion, and analgesia requirements. However, considering tumor size and location, the open operation is still recommended when tumors are with malignant potential, bilateral, and larger than 8 cm. Currently, there are four reports of laparoscopic excision of pheochromocytoma larger than 10 cm, and the maximum is 14 cm (51–54). If metastatic disease is present, primary tumor removal/debulking surgery may also be performed to alleviate symptoms and signs of catecholamine overdose or local symptoms.
Intraoperative hemodynamic monitoring, especially management of the hypertensive crisis by an experienced anesthesiologist, is essential for optimal surgical outcomes. Arrhythmias are common during the procedure, and intravenous administration of esmolol and lidocaine are effective measures. After removal of the origin of excess catecholamines in the circulation, postoperative hypotension may be intractable and is usually managed with intravenous fluid replacement (sometimes with vasopressors). Intravenous glucose prevents hypoglycemia, which occurs in 10%–15% of patients due to the elimination of the inhibitory effect of catecholamines on insulin secretion.
Postoperatively, although there has been no accurate consensus on follow-up, long-term multidisciplinary follow-up is indispensable. Since genotype–phenotype presentations have been proved to be associated with pheochromocytoma personalized management, gene detection is encouraged for patients. So far, 14 different susceptibility genes of pheochromocytomas have been documented: NF1, RET, VHL, SDHD, SDHC, SDHB, EGLN1/PHD2, KIF1β, SDH5/SDHAF2, IDH1, TMEM127, SDHA, MAX, and HIF2α (55). Among them, mutations of SDHB lead to tumor metastasis in 40% or more of affected patients (56). The 5th edition of the WHO Classification encourages routine use of SDHB immunohistochemistry (40). Vigilant imaging examinations should be performed as required.
Collectively, we reported a fairly rare case of a giant cystic pheochromocytoma and provided an up-to-date literature review of patients with such tumors larger than 20 cm. Large pheochromocytomas are usually asymptomatic and require individualized surgery. The prognosis is generally well, but long-term follow-up is required to monitor the tumor for metastasis or recurrence.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual for the publication of any potentially identifiable images or data included in this article.
Author contributions
QF and HL drafted and revised the manuscript. GQ and ZC collected the data and revised the manuscript. JL revised the manuscript for content. YZ and JH designed the study and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Key Technologies R&D Program (2018YFC1106800), the Natural Science Foundation of China (82170621, 82070644, 81800564, and 81770615), and the 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYJC18008).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Curfman KR, Di Como JA, Chung TR, Dumire RD. Functionally silent, giant pheochromocytoma presenting with varicocele. Am Surg (2021) 87(1):97–100. doi: 10.1177/0003134820945274
2. Lam AK. Update on adrenal tumours in 2017 world health organization (who) of endocrine tumours. Endocr Pathol (2017) 28(3):213–27. doi: 10.1007/s12022-017-9484-5
3. Wang X, Zhao Q, Sang H, Dong J, Bai M. Research on the damage of adrenal pheochromocytoma to patients’ cardiovascular vessels and its correlation with hypertension. J Oncol (2022) 2022:3644212. doi: 10.1155/2022/3644212
4. Pappachan JM, Raskauskiene D, Sriraman R, Edavalath M, Hanna FW. Diagnosis and management of pheochromocytoma: a practical guide to clinicians. Curr Hypertens Rep (2014) 16(7):442. doi: 10.1007/s11906-014-0442-z
5. Gupta A, Bains L, Agarwal MK, Gupta R. Giant cystic pheochromocytoma: A silent entity. Urol Ann (2016) 8(3):384–6. doi: 10.4103/0974-7796.184886
6. Ferreira VM, Marcelino M, Piechnik SK, Marini C, Karamitsos TD, Ntusi NAB, et al. Pheochromocytoma is characterized by catecholamine-mediated myocarditis, focal and diffuse myocardial fibrosis, and myocardial dysfunction. J Am Coll Cardiol (2016) 67(20):2364–74. doi: 10.1016/j.jacc.2016.03.543
7. Chen H, Sippel RS, O’Dorisio MS, Vinik AI, Lloyd RV, Pacak K. The north American neuroendocrine tumor society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. (2010) 39(6):775–83. doi: 10.1097/MPA.0b013e3181ebb4f0
8. Maharaj R, Parbhu S, Ramcharan W, Baijoo S, Greaves W, Harnanan D, et al. Giant cystic pheochromocytoma with low risk of malignancy: A case report and literature review. Case Rep Oncol Med (2017) 2017:4638608. doi: 10.1155/2017/4638608
9. Riley DS, Barber MS, Kienle GS, Aronson JK, von Schoen-Angerer T, Tugwell P, et al. CARE guidelines for case reports: explanation and elaboration document. J Clin Epidemiol. (2017) 89:218–35. doi: 10.1016/j.jclinepi.2017.04.026
10. Andreoni C, Krebs RK, Bruna PC, Goldman SM, Kater CE, Alves MT, et al. Cystic phaeochromocytoma is a distinctive subgroup with special clinical, imaging and histological features that might mislead the diagnosis. BJU Int (2008) 101(3):345–50. doi: 10.1111/j.1464-410X.2007.07370.x
11. Schmid H, Mussack T, Wörnle M, Pietrzyk MC, Banas B. Clinical management of large adrenal cystic lesions. Int Urol Nephrol. (2005) 37(4):767–71. doi: 10.1007/s11255-005-4662-7
12. Antedomenico E, Wascher RA. A case of mistaken identity: giant cystic pheochromocytoma. Curr Surg (2005) 62(2):193–8. doi: 10.1016/j.cursur.2004.08.015
13. Yagnik VD, Joshipura V, Sadhu R. Cystic pheochromocytoma masquerading as a cystic pancreatic tumour. ANZ J Surg (2019) 89(5):E195–e7. doi: 10.1111/ans.14213
14. Wu JS, Ahya SN, Reploeg MD, Singer GG, Brennan DC, Howard TK, et al. Pheochromocytoma presenting as a giant cystic tumor of the liver. Surgery. (2000) 128(3):482–4. doi: 10.1067/msy.2000.104113
15. Kumar S, Parmar KM, Aggarwal D, Jhangra K. Simple adrenal cyst masquerading clinically silent giant cystic pheochromocytoma. BMJ Case Rep (2019) 12(9). doi: 10.1136/bcr-2019-230730
16. Sarveswaran V, Kumar S, Kumar A, Vamseedharan M. A giant cystic pheochromocytoma mimicking liver abscess an unusual presentation - a case report. Clin Case Rep (2015) 3(1):64–8. doi: 10.1002/ccr3.149
17. Grissom JR, Yamase HT, Prosser PR. Giant pheochromocytoma with sarcoidosis. South Med J (1979) 72(12):1605–7. doi: 10.1097/00007611-197912000-00035
18. Arikan S, Tatar C, Emre Nayci A, Ersoz F, Baki Dogan M, Gunver F. Giant composite pheochromocytoma and gastrointestinal stromal tumor in a patient with neurofibromatosis: A case report. North Clin Istanb. (2021) 8(6):629–33. doi: 10.14744/nci.2020.37431
19. Costa SR, Cabral NM, Abhrão AT, Costa RB, Silva LM, Lupinacci RA. Giant cystic malignant pheochromocytoma invading right hepatic lobe: report on two cases. Sao Paulo Med J (2008) 126(4):229–31. doi: 10.1590/S1516-31802008000400008
20. Basso L, Lepre L, Melillo M, Fora F, Mingazzini PL, Tocchi A. Giant phaeochromocytoma: case report. Ir J Med Sci (1996) 165(1):57–9. doi: 10.1007/BF02942808
21. Karumanchery R, Nair JR, Hakeem A, Hardy R. An unusual case of back pain: A large pheochromocytoma in an 85 year old woman. Int J Surg Case Rep (2012) 3(1):16–8. doi: 10.1016/j.ijscr.2011.10.006
22. Samejima M, Taguchi S, Miyagawa S, Matsumoto R, Omura S, Ninomiya N, et al. Acute hypotension induced by suction of cystic fluid containing extremely high concentrations of catecholamines during resection of giant pheochromocytoma. IJU Case Rep (2019) 2(4):218–20. doi: 10.1002/iju5.12087
23. Okada R, Shimura T, Tsukida S, Ando J, Kofunato Y, Momma T, et al. Concomitant existence of pheochromocytoma in a patient with multiple endocrine neoplasia type 1. Surg Case Rep (2016) 2(1):84. doi: 10.1186/s40792-016-0214-x
24. Díaz-Roldán J, Díaz-Ramírez J, Franch-Arcas G, Alemán-Martín A. A new trigger in pheochromocytoma crisis: Giant leiomyoma. Ann Endocrinol (Paris). (2021) 82(2):124–6. doi: 10.1016/j.ando.2021.01.002
25. Suga K, Motoyama K, Hara A, Kume N, Ariga M, Matsunaga N. Tc-99m MIBG imaging in a huge clinically silent pheochromocytoma with cystic degeneration and massive hemorrhage. Clin Nucl Med (2000) 25(10):796–800. doi: 10.1097/00003072-200010000-00009
26. Terk MR, de Verdier H, Colletti PM. Giant extra-adrenal pheochromocytoma: magnetic resonance imaging with gadolinium-DTPA enhancement. Magn Reson Imaging. (1993) 11(1):47–50. doi: 10.1016/0730-725X(93)90410-F
27. Arcos CT, Luque VR, Luque JA, García PM, Jiménez AB, Muñoz MM. Malignant giant pheochromocytoma: a case report and review of the literature. Can Urol Assoc J (2009) 3(6):E89–91. doi: 10.5489/cuaj.1189
28. Soufi M, Lahlou MK, Benamr S, Massrouri R, Mdaghri J, Essadel A, et al. Giant malignant cystic pheochromocytoma: a case report. Indian J Surg (2012) 74(6):504–6. doi: 10.1007/s12262-012-0719-x
29. Cajipe KM, Gonzalez G, Kaushik D. Giant cystic pheochromocytoma. BMJ Case Rep (2017) 2017. doi: 10.1136/bcr-2017-222264
30. Ologun GO, Patel ZM, Adeboye A, Guduru M, Trostle D, Vandermeer T, et al. A giant adrenal mass in a super obese patient. Cureus. (2017) 9(8):e1572. doi: 10.7759/cureus.1572
31. Melegh Z, Rényi-Vámos F, Tanyay Z, Köves I, Orosz Z. Giant cystic pheochromocytoma located in the renal hilus. Pathol Res Pract (2002) 198(2):103–6. doi: 10.1078/0344-0338-00194
32. Korgali E, Dundar G, Gokce G, Kilicli F, Elagoz S, Ayan S, et al. Giant malignant pheochromocytoma with palpable rib metastases. Case Rep Urol. (2014) 2014:354687. doi: 10.1155/2014/354687
33. Jiang A, Zhang H, Liu X, Zhao H. Perioperative anesthetic management of a case of rare ectopic pheochromocytoma. World J Oncol (2017) 8(6):191–5. doi: 10.14740/wjon1065e
34. Classics in oncology. A case of bilateral completely latent adrenal tumor and concurrent nephritis with changes in the circulatory system and retinitis: Felix fränkel, 1886. CA Cancer J Clin (1984) 34(2):93–106. doi: 10.3322/canjclin.34.2.93
35. Goldberg A, Pautler SE, Harle C, Dennis A, Rachinsky I, Dhir A, et al. Giant cystic pheochromocytoma containing high concentrations of catecholamines and metanephrines. J Clin Endocrinol Metab (2011) 96(8):2308–9. doi: 10.1210/jc.2011-0465
36. Erickson LA, Lloyd RV, Hartman R, Thompson G. Cystic adrenal neoplasms. Cancer. (2004) 101(7):1537–44. doi: 10.1002/cncr.20555
37. Galatola R, Romeo V, Simeoli C, Guadagno E, De Rosa I, Basso L, et al. Characterization with hybrid imaging of cystic pheochromocytomas: correlation with pathology. Quant Imaging Med Surg (2021) 11(2):862–9. doi: 10.21037/qims-20-490
38. Adler JT, Meyer-Rochow GY, Chen H, Benn DE, Robinson BG, Sippel RS, et al. Pheochromocytoma: current approaches and future directions. Oncologist. (2008) 13(7):779–93. doi: 10.1634/theoncologist.2008-0043
39. Nölting S, Bechmann N, Taieb D, Beuschlein F, Fassnacht M, Kroiss M, et al. Personalized management of pheochromocytoma and paraganglioma. Endocr Rev (2022) 43(2):199–239. doi: 10.1210/endrev/bnab019
40. Mete O, Asa SL, Gill AJ, Kimura N, de Krijger RR, Tischler A. Overview of the 2022 WHO classification of paragangliomas and pheochromocytomas. Endocr Pathol (2022) 33(1):90–114. doi: 10.1007/s12022-022-09704-6
41. Lenders JWM, Kerstens MN, Amar L, Prejbisz A, Robledo M, Taieb D, et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: a position statement and consensus of the working group on endocrine hypertension of the European society of hypertension. J Hypertens (2020) 38(8):1443–56. doi: 10.1097/HJH.0000000000002438
42. Fassnacht M, Assie G, Baudin E, Eisenhofer G, de la Fouchardiere C, Haak HR, et al. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2020) 31(11):1476–90. doi: 10.1016/j.annonc.2020.08.2099
43. Thompson LD. Pheochromocytoma of the adrenal gland scaled score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol (2002) 26(5):551–66. doi: 10.1097/00000478-200205000-00002
44. Stenman A, Zedenius J, Juhlin CC. The value of histological algorithms to predict the malignancy potential of pheochromocytomas and abdominal paragangliomas-a meta-analysis and systematic review of the literature. Cancers (Basel). (2019) 11(2):225. doi: 10.3390/cancers11020225
45. Kimura N, Takayanagi R, Takizawa N, Itagaki E, Katabami T, Kakoi N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer. (2014) 21(3):405–14. doi: 10.1530/ERC-13-0494
46. Fassnacht M, Arlt W, Bancos I, Dralle H, Newell-Price J, Sahdev A, et al. Management of adrenal incidentalomas: European society of endocrinology clinical practice guideline in collaboration with the European network for the study of adrenal tumors. Eur J Endocrinol (2016) 175(2):G1–g34. doi: 10.1530/EJE-16-0467
47. Shah MH, Goldner WS, Benson AB, Bergsland E, Blaszkowsky LS, Brock P, et al. Neuroendocrine and adrenal tumors, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw (2021) 19(7):839–68. doi: 10.6004/jnccn.2021.0032
48. Hallin Thompson L, Makay Ö, Brunaud L, Raffaelli M, Bergenfelz A. Adrenalectomy for incidental and symptomatic phaeochromocytoma: retrospective multicentre study based on the eurocrine® database. Br J Surg (2021) 108(10):1199–206. doi: 10.1093/bjs/znab199
49. Taïeb D, Hicks RJ, Hindié E, Guillet BA, Avram A, Ghedini P, et al. European Association of nuclear medicine practice Guideline/Society of nuclear medicine and molecular imaging procedure standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. (2019) 46(10):2112–37. doi: 10.1007/s00259-019-04398-1
50. Lorusso M, Rufini V C, Pennestrì F, Bellantone R, Raffaelli M. Integration of molecular imaging in the personalized approach of patients with adrenal masses. Q J Nucl Med Mol Imaging. (2022) 66(2):104–15. doi: 10.23736/S1824-4785.22.03449-5
51. Clements HA, Wilson MS, Smith DM. Incidental giant cystic pheochromocytoma: a case report and review of the literature. Scott Med J (2020) 65(2):64–70. doi: 10.1177/0036933019900339
52. Mishra AK, Agarwal G, Agarwal A, Mishra SK. Laparoscopic adrenalectomy of large cystic pheochromocytoma. Surg Endosc. (2001) 15(2):220. doi: 10.1007/s004640040036
53. Costa Almeida CE, Silva M, Carvalho L, Costa Almeida CM. Adrenal giant cystic pheochromocytoma treated by posterior retroperitoneoscopic adrenalectomy. Int J Surg Case Rep (2017) 30:201–4. doi: 10.1016/j.ijscr.2016.12.018
54. Martins D, Rodrigues D, Melo M, Carrilho F. Laparoscopic adrenalectomy as an effective approach to massive bilateral pheochromocytomas. BMJ Case Rep (2017) 2017. doi: 10.1136/bcr-2017-221009
55. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2014) 99(6):1915–42. doi: 10.1210/jc.2014-1498
Keywords: giant cystic pheochromocytoma, liver atrophy, case report, literature review, surgery
Citation: Feng Q, Li H, Qiu G, Cai Z, Li J, Zeng Y and Huang J (2022) Case report: Significant liver atrophy due to giant cystic pheochromocytoma. Front. Oncol. 12:987705. doi: 10.3389/fonc.2022.987705
Received: 06 July 2022; Accepted: 08 August 2022;
Published: 30 August 2022.
Edited by:
Francesco Pennestrì, Università Cattolica del Sacro Cuore, ItalyReviewed by:
Pietro Locantore, Catholic University of the Sacred Heart, ItalyJoseph M. Pappachan, Lancashire Teaching Hospitals NHS Foundation Trust, United Kingdom
Copyright © 2022 Feng, Li, Qiu, Cai, Li, Zeng and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiwei Huang, aHVhbmdqaXdlaW1kQGhvdG1haWwuY29t
†These authors have contributed equally to this work