 Daniela Magliulo
Daniela Magliulo Rosa Bernardi
Rosa Bernardi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 18 August 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.973978
This article is part of the Research TopicCellular Stress in Blood Cancer: Pathophysiology and Therapeutic OpportunitiesView all 5 articles
To cope with hypoxic stress, ancient organisms have developed evolutionally conserved programs centered on hypoxia-inducible transcriptional factors (HIFs). HIFs and their regulatory proteins have evolved as rheostats to adapt cellular metabolism to atmospheric oxygen fluctuations, but the amplitude of their transcriptional programs has tremendously increased along evolution to include a wide spectrum of physiological and pathological processes. The bone marrow represents a notable example of an organ that is physiologically exposed to low oxygen levels and where basal activation of hypoxia signaling appears to be intrinsically wired within normal and neoplastic hematopoietic cells. HIF-mediated responses are mainly piloted by the oxygen-labile α subunits HIF1α and HIF2α, and current literature suggests that these genes have a functional specification that remains to be fully defined. Since their identification in the mid 90s, HIF factors have been extensively studied in solid tumors, while their implication in leukemia has lagged behind. In the last decades however, many laboratories have addressed the function of hypoxia signaling in leukemia and obtained somewhat contradictory results. Suppression of HIFs expression in different types of leukemia has unveiled common leukemia-promoting functions such as stimulation of bone marrow neoangiogenesis, maintenance of leukemia stem cells and chemoresistance. However, genetic studies are revealing that a definition of HIF factors as bona fide tumor promoters is overly simplistic, and, depending on the leukemia subtype, the specific oncogenic event, or the stage of leukemia development, activation of hypoxia-inducible genes may lead to opposite consequences. With this article we will provide an updated summary of the studies describing the regulation and function of HIF1α and HIF2α in blood malignancies, spanning from acute to chronic, lymphoid to myeloid leukemias. In discussing these data, we will attempt to provide plausible explanations to contradictory findings and point at what we believe are areas of weakness in which further investigations are urgently needed. Gaining additional knowledge into the role of hypoxia signaling in leukemia appears especially timely nowadays, as new inhibitors of HIF factors are entering the clinical arena for specific types of solid tumors but their utility for patients with leukemia is yet to be determined.
Oxygen supply and consumption are tightly regulated processes in multicellular organisms and when oxygen demand exceeds supply, a state of reduced oxygenation called hypoxia ensues. Hypoxia-inducible factors (HIFs) are master regulators of cellular and systemic adaptation to poor oxygen availability in physiological and pathological conditions. They promote expression of a growing number of tissue-specific genes that mediate adaptation to acute decrease in oxygen supply as well as molecular functions occurring in cell compartments that are physiologically exposed to low oxygen levels (1).
Breakthrough discoveries into the molecular mechanisms of how cells sense and adapt to oxygen availability led to the 2019 Nobel Prize in Physiology or Medicine being awarded to Drs. Gregg Semenza, William Kaelin, and Peter Ratcliffe (2).
Genes that tune cellular metabolism to atmospheric oxygen availability belong to an ancient pathway that co-evolved with oxidative phosphorylation in the first metazoans (3). Orthologues of various components of the HIF pathway (PHD, FIH, VHL, HIF, described later in detail) are conserved from simple organisms like nematodes to mammals (3) and along evolution many of these genes have been subjected to events of gene duplication. Such is the case of HIF factors, the central molecules in the hypoxia adaptation pathway, with HIF1A being shared by all metazoans, and EPAS1 (the gene encoding HIF2α) and HIF3A having appeared at later branches of metazoan evolution. Additionally, oxygen sensing domains embedded within HIF molecules display increased molecular complexity throughout evolution, with primitive forms of HIFα expressing a single oxygen-sensitive regulatory domain and genes that have evolved later containing repetitions of these sites (3). Ongoing studies are directed to define the functional specification of the three mammalian HIFα genes in different physio/pathological cell states and cell types. However, these studies are at the very beginning, and further analyses in normal and pathological tissues will lead to a better understanding of the diversification and full regulatory potential of this pathway.
Importantly, although HIF factors have been initially defined as central regulators of cellular adaptation to acute oxygen scarcity, it must be highlighted that low oxygen levels are physiological in certain tissues or tissue microenvironments (4) and that, besides acute hypoxia, HIF factors are also expressed in cells ordinarily exposed to low oxygen, where they take part to the regulation of physiological mechanisms. Along these lines, a common semantic error is to consider the atmospheric oxygen partial pressure (pO2) of 160 mm Hg as normoxic, with hypoxia indicating pO2 values below 38 mm Hg. Although in vitro this is the condition where HIF factors begin to accumulate in many cell types, we must remember that most cell lines are adapted to grow at oxygen tensions as in the atmosphere, even though the tissue they originated from was probably exposed to much lower oxygen levels. Thus, the terms “hypoxia” and “normoxia” should be used attentively (5).
One typical tissue that is considered physiologically hypoxic is the bone marrow (BM), where low oxygen levels are believed to be intrinsic, especially in microenvironments devolved to the control of hematopoietic stem cells (HSCs) homeostasis (4). As such, HIF factors have been implicated in the biology of HSCs and hematopoietic cells at large and, as we will review in this manuscript, this also applies to the pathological derivatives of hematopoietic cells: leukemic cells.
Before describing the known functions and regulation of hypoxia-inducible genes in leukemia, we will use this chapter to briefly summarize the way these genes work. HIF proteins are a family of evolutionarily conserved DNA-binding transcription factors belonging to the bHLH/PAS (basic-helix-loop-helix/Per-ARNT-Sim) family. They function as heterodimers composed of an oxygen-sensitive HIFα subunit and the constitutively expressed HIF1β subunit (or aryl hydrocarbon receptor nuclear translocator, ARNT) (6). Three different HIFα genes exist in the human genome: HIF1A, EPAS1 (endothelial PAS domain protein 1 gene, encoding HIF2α), and HIF3A. HIF1α and HIF2α bind HIF1β to assemble HIF1 and HIF2 active transcription factors, which recognize hypoxia responsive elements (5’-(A/G)CGTG-3’, HREs) in the regulatory regions of HIF-target genes (7). HIF3α is less studied than its two siblings and has been suggested to act as a dominant-negative regulator of hypoxia signaling via competition for HIF1β binding by some of its splicing variants and transcriptional inhibition (8). More recently, HIF3α has been identified as a lipid sensor, with endogenous lipids stabilizing its heterodimerization with HIF1β (9), which may lead to regulation of lipolysis genes in adipocytes (10). However, because information on the function of HIF3α is still scarce, in this review we will focus on HIF1α and HIF2α.
HIF proteins share homologous amino-terminal (N-terminal) domains, whilst diverging at their carboxyl-terminal (C-terminal) region. At the N-terminus, all HIF proteins carry a bHLH domain that is required for DNA binding and PAS-A and PAS-B domains that mediate heterodimerization and formation of the transcriptional complex. In the central region, an oxygen-dependent degradation domain (ODDD) is crucial for oxygen-dependent degradation of HIFα proteins as described below, and is lacking in HIF1β, thus explaining its oxygen-independent expression. At the C-terminus, HIF1α and HIF2α have two transactivation domains (N-TAD and C-TAD) that are essential to promote transcription of HIF-target genes, while HIF3α and HIF1β have only one TAD domain (11). Importantly, the C-terminal half of HIFα and HIF1β proteins contains intrinsically disordered regions (IDRs) that exert critical functions in transcriptional regulation by mediating diffusion properties and interaction with chromatin factors (12).
As their name suggests, HIF proteins are importantly regulated by oxygen levels. However, HIFs are not oxygen sensors per se, rather they are post-translationally regulated by enzymes that use molecular oxygen to promote protein hydroxylation, thus acting as oxygen sensors (13). When oxygen is available, prolyl hydroxylases (PHD1-3) modify two conserved prolyl-residues located in the ODDD of HIFα proteins (P402/P564 in human HIF1α and P405/P531 in HIF2α) and, in so doing, promote their recognition by the von Hippel-Lindau (pVHL) E3 ubiquitin ligase that triggers their degradation by the proteasome via poly-ubiquitination (14, 15). In addition, an asparagine residue located in the C-TAD of HIF1α and HIF2α (N803 in human HIF1α and N851 in HIF2α) confers further oxygen-dependent regulation upon hydroxylation by the asparaginyl hydroxylase FIH (factor inhibiting HIF), which blocks interaction with the transcriptional coactivator complex p300/CBP and suppresses transcriptional activity (11, 16, 17) (Figure 1A). Both PHDs and FIH belong to the 2-oxoglutarate (2-OG)-dependent dioxygenase superfamily that uses oxygen and 2-OG as co-substrates together with ferrous iron (Fe2+) and ascorbate as obligate cofactors to catalyze protein hydroxylation (15). The activity of PHDs and FIH is inhibited under low oxygen tensions due to lack of the essential co-substrate. As a consequence, HIFα subunits are stabilized, dimerize with HIF1β and bind HREs on specific target genes together with several coactivators, including CBP/p300 (Figure 1B). Of note, PHD2 and PHD3 are themselves hypoxia-inducible proteins, thus providing a self-sufficient mechanism to control HIFα protein levels and ensure rapid removal of HIFα upon reoxygenation (18). Also, PHDs exhibit tissue-specific expression (for example, PHD3 is strongly expressed in the heart and PHD1 is the only isoform expressed in the testis) and differential HIFα hydroxylation, with PHD1 and PHD3 being more active on HIF2α and PHD2 hydroxylating more efficiently HIF1α (18, 19). Consequently, oxygen-dependent HIFα induction may also reflect differences in PHDs tissue distribution and activity.
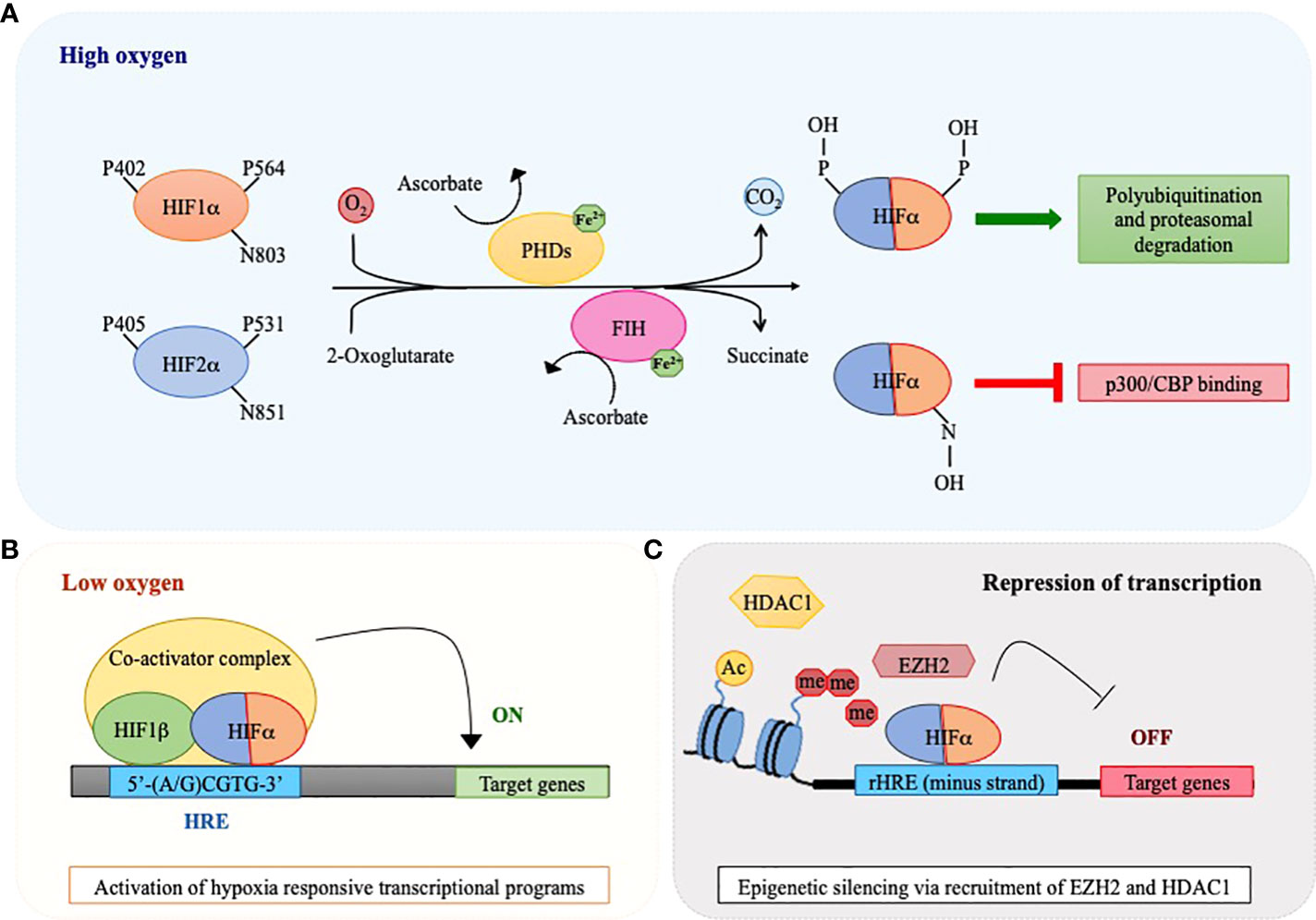
Figure 1 Oxygen-sensing regulatory pathway of HIF1α and HIF2α. A schematic view of HIF1α and HIF2α regulation via the action of oxygen sensors prolyl hydroxylases (PHDs) and factor inhibiting HIF (FIH). (A) PHD and FIH are HIFα hydroxylases that require iron (Fe2+) and ascorbate as co-factors and utilize 2-oxoglutarate and molecular oxygen as co-substrates of their enzymatic reaction, with the release of CO2 and succinate as waste products. In the presence of oxygen, these enzymes catalyze hydroxylation of HIF1α and HIF2α at proline (P) and asparagine (N) residues respectively, thus provoking a dual effect: PHDs cause HIFα polyubiquitination and degradation by the proteasome, while FIH inhibits binding of co-activators like CREB-binding protein (CBP) and p300 to the HIF transcriptional complex. (B) In conditions of oxygen scarcity, the activities of PHDs and FIH are inhibited and HIFα subunits become stabilized, dimerize with HIF1β and bind hypoxia responsive elements (HREs) within the regulatory regions of specific target genes to activate their transcription. (C) HIFα factors may also recognize HREs in the minus DNA strand (reverse HRE, rHRE) thus allowing recruitment of histone methyltransferases or deacetylases (EZH2 and HDAC1) and provoking epigenetic silencing and transcriptional repression.
Active HIF heterodimers promote transcription of an ever-growing number of genes involved in a variety of cellular functions, with important implications for both physiology and disease. Hypoxia signaling has been linked to control of cellular metabolism (with a switch from mitochondrial oxidation to glycolysis), neo-angiogenesis, regulation of cell migration and tissue invasion, regulation of cell proliferation and apoptosis, and regulation of stem cell maintenance, amongst other functions (20). Mechanistically, HIF1α and HIF2α show high sequence homology in their DNA-binding domains (21) and bind identical HRE motifs in vitro (7, 22), thus regulating common target genes (23, 24). However, emerging evidence indicates that HIF1α and HIF2α are endowed with important target selectivity, with HIF1α mainly promoting the expression of genes involved in glycolytic metabolism, pH regulation and apoptosis, whilst HIF2α regulates genes involved in stem cell maintenance, cell cycle and invasion (24), albeit this division of functions does not occur in all tissues and a strong element of tissue specificity is emerging in the HIF transcriptional program. The functional specification of HIFα factors has been connected to specific interactions with transcriptional coactivators occurring in their C-terminal transactivation domains, which have a lower degree of sequence homology (25). In addition, more recently it was proposed that HIFα proteins display different diffusion properties and DNA binding profiles thanks to the different amino acidic compositions of their IDRs (12). Specifically, it was revealed that HIF2α has slower nuclear diffusion than HIF1α and a tendency to bind negatively charged chromatin-associated RNA and proteins due to the preeminent presence of positively charged amino acids in its IDR (12). Adding to these intrinsic properties, tissue- and developmental-specific expression and different sensitivity to oxygen levels lead to an emerging pattern of differential gene regulation by HIF1α and HIF2α (23, 24, 26).
Interestingly, it has been suggested that the diversification of HIFα functions may originate from their evolutionary history (3). Having evolved to adapt cellular physiology to acute oxygen scarcity, HIF1A, which is the most ancient member of the family, is presumed to be the primary regulator of metabolic rewiring, while the later appearance of EPAS1 may have led to a wider spectrum of functions, including processes that occur within cell types residing physiologically at low oxygen levels (i.e. stem cells). However, as we will describe later, such a clear diversification of functions may not apply to all tissues or cells.
In conclusion, it is worth mentioning that, beside their role as transcriptional activators, HIFα have also been described as transcriptional repressors for certain genes (e.g. peroxisome proliferator activated receptor (PPAR)α, α-fetoprotein, leukemia inhibitory factor receptor) via binding to HREs located on the minus DNA strand (also called reverse HREs, rHREs) (27–29). Recently, binding of HIFα subunits to rHREs has been shown to repress transcription via epigenetic silencing promoted by recruitment of histone modifiers like HDAC1 and EZH2 (the catalytic subunit of Polycomb repressive complex 2, PRC2) (30–32) (Figure 1C). Because HIF factors also promote the expression of a number of epigenetic factors, epigenetic mechanisms are emerging as a relevant way of controlling gene expression downstream hypoxia signaling (33).
In humans, oxygen gradients differ greatly among tissues and tissue microenvironments. In the BM, oxygen levels are overall lower than in many other organs, and hematopoietic cells experience physiological pO2 ranging from 32 mm Hg in arteriole-rich endosteal zones to 9.9 mm Hg in deeper regions (34, 35). The BM is home to a variety of non-hematopoietic cell types that constitute specialized microenvironments, known as niches, where stem cells, progenitors and more differentiated hematopoietic cells reside and are physiologically regulated (36). Despite its extensive vascularization, the BM is considered a tissue low in O2, likely due to the extent of oxygen consumption caused by its high cellularity. Initial studies using in silico modeling of oxygen BM gradients and quantification of BM perfusion led to the assumption that HSCs reside near the poorly perfused endosteal niche and show signs of hypoxia due to their distance from oxygen-rich blood vessels (37–39). Despite this general assumption, measuring the exact oxygen tension and physical location of niches low in O2 in the BM is technically challenging, therefore indirect measurements have been often utilized, such as evaluation of HIF1α expression or incorporation of hypoxia markers like pimonidazole (Pimo) and Hoechst 33342 (35, 38, 39). However, use of chemical agents, such as Pimo and Hoechst 33342 may be misleading as they do not provide a direct assessment of O2 levels and may rather offer a readout of specific cellular conditions. As an example, Pimo, which competes with oxygen as an electron acceptor, may display increased reductive activation not only as a consequence of decreased intracellular oxygen, but also upon distinctive metabolic shifts from mitochondrial to glycolytic catabolism (40). Similarly, uptake of the fluorescent DNA intercalant Hoechst 33342, which has been used to measure blood perfusion as an indirect marker of tissue oxygenation, may vary depending on cell proliferation rates. Conversely, use of two-photon phosphorescence lifetime microscopy, which directly measures oxygen levels, revealed that the BM is overall a tissue low in O2 where intravascular oxygen tensions are quite similar in endosteal and sinusoidal vessels (21.9 and 17.7 mm Hg respectively), and a moderate extravascular oxygen gradient exists, with higher oxygen levels in the endosteal region and decreased oxygen towards the sinusoidal region (34). Of note, these experiments have been performed in the calvaria, which may not represent all BM compartments, as Pimo staining revealed that hypoxic cells are less frequent in this site compared to long bones (41). Therefore, it remains unclear whether the current knowledge on BM oxygen levels may be relevant for most bones.
Nonetheless, the notion that hematopoietic cells thrive in an environment that is low in O2 has been validated over the years by a number of in vitro or ex vivo experiments demonstrating that low oxygen levels: i) promote HSCs quiescence and maintenance (42–48), while at the same time supporting a balanced differentiation program towards many hematopoietic lineages (49–52); ii) favor a metabolic switch to (anaerobic) glycolysis thus exerting protection from oxidative stress that may cause DNA damage (35). Also, hypoxia efficiently enhances in vitro expansion and in vivo engraftment of HSCs (53–55).
More recently, it was revealed that, besides external oxygen availability, HSCs display an inherent hypoxic state that is independent of their exact location within the BM and may be rather dictated by internal metabolic activity (40). In this respect, it must be emphasized that expression and activity of HIF factors is not only regulated by oxygen levels but also by a variety of oxygen-independent mechanisms that promote their expression and transcriptional activity. Providing a list of these mechanisms is beyond the scope of this review, as it has been discussed elsewhere (56, 57), but suffice it to say that they include extracellular stimuli like growth factors, cytokines and chemokines. Thus, the molecular milieu of different BM niches may participate considerably to the regulation of hypoxia signaling within hematopoietic cells. Whatever the triggering event, one certain finding is that HIF factors are highly expressed in hematopoietic stem and progenitor cells (HSPCs), where they play important functions. This was clearly demonstrated in knock-out mice, where it was shown that HIF1α exerts a critical cell-autonomous functions in promoting HSCs quiescence (58), and HIF2α promotes HSCs maintenance predominantly via regulation of the BM microenvironment (59).
In the case of hematological malignancies, leukemic cells are great oxygen consumers, but at the same time promote BM neoangiogenesis, which increases nutrients and oxygen availability (60). Thus, it is not clear if abundance of proliferating leukemic cells and high oxygen and nutrient consumption may further decrease oxygen availability in the leukemic BM. By measuring BM blood gas levels in healthy volunteers and AML patients, two independent reports observed similar pO2 of around 47 mm Hg (61, 62). Conversely, in vivo intracellular hypoxia labeling with the indirect marker 2-nitroimidazole in a rat model of AML showed increased reactivity during disease progression and in comparison with healthy animals (63). More recently, direct measurement of oxygen tension in leukemic BM was obtained in a BCR-ABL B-ALL mouse model by intravital fast scanning two-photon phosphorescence lifetime imaging microscopy, which showed that oxygen levels vary from the initial to the final stages of leukemia, with a transient increase in oxygen levels at intermediate leukemia burden, correlating with expansion of the vasculature network, and a later significant decrease of BM oxygenation as leukemia cellularity increased at disease end-stage (64). By suggesting that different oxygen levels characterize different stages of leukemia development, these findings may provide an explanation to the apparently contradictory findings described above. However, as previously discussed for normal BM, also and particularly in a leukemic BM where the cytokine milieu is perturbed towards pro-inflammatory and leukemia-sustaining cytokines, intracellular hypoxic pathways may be activated via additional routes. In this respect, work from our and other laboratories has shown that HIF1α is transcriptionally upregulated when leukemic cells are cocultured with BM mesenchymal cells (65–67). Also, a number of studies that we will describe in the next sections have reported that upregulation of HIF factors in leukemic blasts does not necessarily occur at the post-translational level, thus implying oxygen-independent mechanisms. In the following chapter, we will provide a summary of the regulation and function of HIF1α and HIF2α across blood malignancies.
Since their cloning and molecular characterization, the function of HIF factors has been intensely investigated in solid tumors. More recently, increasing research efforts have also focused on the involvement of hypoxia signaling in leukemia, albeit with somewhat controversial results that fail to provide a unanimous consensus. Depending on the type/subtype of leukemia, or the experimental approach that has been utilized (knock-out animals versus knock-down experiments in cell lines/primary cells), HIFs have been described as tumor suppressors, oncogenes, or neutral genes even within the same leukemia. In the next paragraphs, we will summarize current knowledge on the expression and activity of HIF factors in different leukemic contexts (acute and chronic, myeloid and lymphoid) and discuss possible explanations to controversial evidence recently published. Of note, the majority of the studies that we will describe have focused on defining the functions of HIF1α, whereas HIF2α has been much less investigated and detailed analyses on the functions of this factor in leukemia establishment and progression are still lacking.
Expression of HIFα factors in leukemia has been extensively studied at both mRNA and protein levels and increased expression of HIFα subunits has been found in various blood malignancies compared to normal cells (Table 1) (77). One of the first reports of HIF1α upregulation in leukemia came from studies in chronic myeloid leukemia (CML), where it was observed that BCR-ABL induces HIF1α mRNA and protein expression downstream PI3K/mTOR activation (68). Later on, this finding was confirmed in primary CML cells where the HIF1α transcript was found more expressed in leukemic cells compared to BM from healthy volunteers (69). In acute myeloid leukemia (AML), a disease characterized by broad genetic and morphological heterogeneity, a general increase in HIF1α and HIF2α protein levels was reported in mouse and human AML cells when compared to normal BM leukocytes (70). In addition, expression of HIFα transcripts was reported to vary in morphological and molecular subtypes, and this may echo functional cooperation with specific oncoproteins. For example, HIF1α mRNA was found particularly elevated in AML with the t(8;21) translocation (encoding RUNX1-RUNX1T1, or AML1-ETO), where it associates with unfavorable prognosis and tumor aggressiveness (71). Functionally, HIF1α and AML1-ETO were found to engage in a positive regulatory circuit where they stimulate their reciprocal expression and cooperate to alter DNMT3a levels and global DNA methylation towards increased AML proliferation (71). In another study, HIF2α expression was found elevated in M3 and M6 AMLs, as defined by FAB morphological classification, and in AMLs with t(15;17) translocation (generating the PML-RARα oncoprotein), inv(16) and complex karyotype (70), although a functional cooperation of HIF2α with the oncogenic drivers of these AML subtypes was not tested.

Table 1 Expression of HIF1α and HIF2α in leukemia.
In addition to these studies, recent work has reported that HIF1α-target genes are upregulated in AML cells carrying TP53 mutations, although HIF1α levels were not evaluated (78). A connection between HIF1α and mutant p53 was reported also in chronic lymphocytic leukemia (CLL). In this disease, it was first observed that HIF1α is highly expressed at the mRNA and protein level compared to normal B cells (72–74), which correlates with leukemia progression (79). In addition, more recently it was reported that HIF1α expression is higher in patients with TP53 mutations compared to wild-type TP53 (66). Taken together, these data suggest that a connection between TP53 mutational status and HIF1α expression and/or function exists in different leukemic contexts. However, it remains to be elucidated whether HIF1α specifically cooperates with gain of function TP53 mutants in leukemia, as it has been established in some solid tumors and lymphoma (80).
Of note, constitutive expression of the HIF1α protein in CLL cells is also driven by post-translational stabilization due to miR-92-1-mediated pVHL downregulation (73), thus indicating that multiple mechanisms converge into elevating HIF1α expression in this disease. Although HIF2α expression has not been measured in CLL cells, some of these mechanisms may also promote HIF2α upregulation (e.g. pVHL suppression).
In acute lymphoblastic leukemia (ALL), HIF1α expression has been measured via immunostaining of BM biopsies, which revealed that HIF1α is overexpressed in BM of childhood ALL (75) and correlates with worse overall survival (76). In addition, HIF2α protein expression was found increased in a subset of primary ALL cells compared to normal hematopoietic cells (70).
Besides increased basal expression of HIF factors compared to normal cells across different leukemias, as mentioned above, expression of HIF1α within the CLL and ALL leukemic compartment increases upon co-culture of leukemic blasts with BM stromal cells (65, 66, 76). Consistently, transcriptomic analyses of primary CLL cells cultured on human stromal cells revealed that hypoxic signatures are amongst the most upregulated (81). A crosstalk between the BM microenvironment and leukemic cells towards increased hypoxia signaling was also found in AML, where recent evidence obtained in ex vivo and in vivo models demonstrated that AML cells impair normal hematopoiesis by rewiring the transcriptome of mesenchymal stromal cells via increased HIF1α expression (82).
In conclusion, current literature indicates that upregulation of HIFα factors and their target genes may be a general phenomenon in leukemia with respect to normal hematopoietic tissue, occasionally with further accumulation in specific genetic backgrounds or upon not yet fully characterized environmental cues provided by BM microenvironments. Of note, many of these studies describe upregulation of HIFα factors both at the mRNA and protein levels, thus revealing that increased HIFs expression in leukemia is not only caused by hypoxic post-translational stabilization.
A note of caution in the interpretation of these studies is that whole BM hematopoietic tissue has often been used as the normal counterpart of leukemic cells, although BM aspirates contain a variety of cell types that are much more heterogeneous than leukemic blasts. Thus, defining the real extent of HIFs overexpression in leukemia remains an open question. Nonetheless, functional studies have provided strong evidence of the involvement of hypoxia signaling in leukemogenesis and drug resistance. These will be described next, along with contradictory studies suggesting that HIF factors may be endowed with tumor-suppressive functions in some leukemias.
In the last decades, several studies have provided evidence that HIFα factors exert relevant tumor-promoting functions in leukemia (Figure 2). At first, hypoxia-regulated genes were implicated in fostering VEGF production and BM neo-angiogenesis in CLL and ALL (73, 75). Following these reports, HIFα factors, prevalently HIF1α, have been attributed other important tumor-promoting functions, with some leukemia-specific nuances.
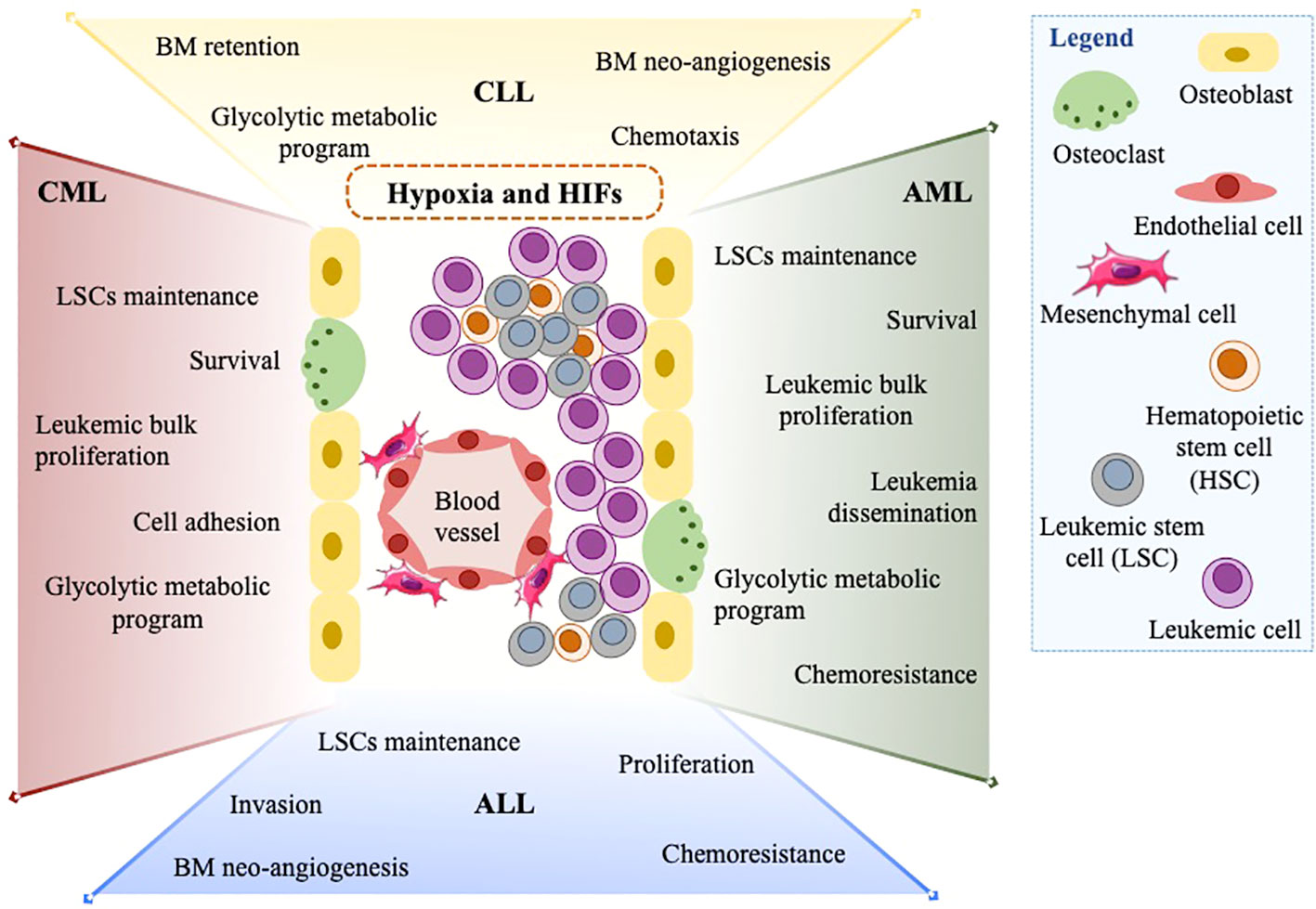
Figure 2 The assortment of HIF1α and HIF2α functions in leukemias. In the leukemic BM microenvironment, high cellularity and oxygen consumption expose leukemic cells to hypoxia and promote the expression of HIF1α and HIF2α. HIFs and hypoxia exert a wide range of tumor-promoting functions in many leukemia types (CML, CLL, ALL, and AML). Amongst others: maintenance of LSCs, increased leukemic cell proliferation, metabolic switch to glycolysis, and chemoresistance are summarized in this figure.
Perhaps the most concordant role of HIF1α across diverse types of leukemia is to promote maintenance of leukemia stem cells (LSCs), a function that HIF1α also exerts in the non-malignant hematopoietic stem cell compartment (58). Different groups have shown that hypoxia conditioning prompts decreased proliferation and longer maintenance of leukemia initiating cells in primary CML and AML (83, 84). Microarray analysis of CD34+ CML progenitor cells exposed to hypoxia revealed increased expression of cell adhesion and survival genes, concordantly with decreased apoptosis and improved colony-forming potential, thereby suggesting that targeting HIF1α along with BCR-ABL may represent a therapeutic opportunity for eradication of LSCs in CML patients (85). The crucial role of HIF1α in promoting maintenance of CML LSCs was also validated in a BCR-ABL transgenic mouse model, where HIF1α conditional deletion resulted in impaired LSCs propagation caused by delayed proliferation and induction of apoptosis via expression of p16Ink4a, p19Arf and p53 (86). In AML, implication of HIF1α in LSCs maintenance was first described via activation of Notch1 and expression of Hes1, a transcription factor with essential stem cell-promoting roles (87). In addition, a context-specific function of HIF1α was described in AML cells carrying the AML1-ETO oncoprotein, where HIF1α sustains LSCs maintenance by transcriptional cooperation with AML1-ETO (71). In line with these data, it was recently suggested that inhibiting HIF1α with compounds like echinomycin may represent a new therapeutic opportunity to impair maintenance of the LSCs population, and this may be particularly relevant in the genetic background of TP53-mutated AMLs, where echinomycin showed a potent cytotoxic effect and affected leukemia propagation in xenograft mouse models (78). Similarly, the HIF1α inhibitor acriflavine impaired the stem cell potential of primary CML cells and murine BM cells transduced with BCR-ABL, thus suggesting that acriflavine may add therapeutic value to currently used tyrosine kinase inhibitors that target BCR-ABL by preventing CML relapse (88).
Besides promoting maintenance of a subpopulation of leukemic cells characterized by specific surface markers or functional properties (LSCs), HIF signaling also drives proliferation of bulk leukemic cells in CML and AML. For example, knockdown of HIF1α led to a marked reduction in cell proliferation in the CML cell line K562 (69) and hypoxia conditioning promoted proliferation of AML KG-1 cells (89). Also, pharmacologic inhibition of HIF1α in AML cells carrying the AML1-ETO or PML-RARα oncoproteins suppressed leukemia expansion in vivo (71, 90).
A dual activity of HIF1α in supporting proliferation of leukemia bulk and maintenance of leukemia-repopulating cells was also observed in T-ALL. Mechanistically, it was demonstrated that HIF1α activates Notch1 signaling and T-ALL cell proliferation (91), while also promoting Wnt signaling via upregulation of β-catenin, thus supporting LSCs maintenance (92).
In T-ALL, HIF1α-mediated Notch1 activation was also implicated in promoting leukemia cell invasion via expression of metalloproteases MMP2 and MMP9 (91). Interestingly, a similar role of stimulating leukemia dissemination was reported in AML sub-types including acute promyelocytic and monocytic leukemia, where HIF1α induces chemokine-dependent cell migration and transcriptional programs of epithelial to mesenchymal transition (90, 93, 94). Consistently, in the AML cell line KG-1, hypoxia conditioning promotes epithelial-mesenchymal transition via activation of PI3K/Akt (95).
In summary, across different types of leukemia, HIF1α has been implicated in the regulation of LSCs maintenance as well as in promoting leukemia dissemination, at times via transcriptional programs reminiscent of epithelial to mesenchymal transition in neoplastic epithelial cells. These results are in line with a recognized link between epithelial to mesenchymal transition programs and formation of cancer stem cells in solid tumors (96), and suggest that a similar connection may also occur in hematopoietic malignancies via activation of HIF factors within other mechanisms.
In apparent contrast to these findings, in CLL HIF1α regulates the expression of genes involved in cell adhesion and BM homing, with the ultimate effect of promoting BM retention and chemoresistance (65). These studies highlight the diversity of HIF-dependent transcriptional programs and cellular functions in different leukemic contexts, a concept that is beginning to be widely appreciated and is probably due in large part to chromatin accessibility and tissue-specific epigenetic landscapes (33).
Another function of HIF factors that appears conserved in distinct leukemias and is in line with their ancestral evolutionary function is modulation of metabolic activity. In CLL, hypoxia conditioning causes a metabolic shift from mitochondrial metabolism to glycolysis that occurs via activation of classical HIF-target genes (65, 97, 98). Interestingly, in AML and CML cell lines, as well as normal hematopoietic progenitor cells, it was reported that both HIF1α and HIF2α promote the expression of genes belonging to the glycolytic program (99). Surprisingly, knock out of either gene or their obligate heterodimeric partner HIF1β did not result in impaired glucose consumption and lactate production, albeit abolishing expression of glycolytic genes. Accordingly, the proliferation capacity of K562 cells was not impacted by HIF-1α, HIF-2α or HIF-1β knock-out (99). This study suggests that the oncogenic properties of HIF genes in leukemia may not rely on their metabolic functions or that adaptive metabolic rewiring compensates for deficiency in hypoxia signaling. In addition, this work provides evidence of a strong contribution of HIF2α in regulating the expression of glycolytic genes, albeit this function may be tissue-specific.
Few other studies have investigated the role of HIF2α in leukemia, and they focused specifically on AML. Silencing of HIF2α in primary AML cells cultured ex vivo resulted in impaired proliferation and reduced engraftment in recipient mice. Mechanistically, this phenotype was linked to HIF2α-mediated protection from apoptosis induced by ER stress and UPR response (100). In line with these results, ectopic expression of HIF2α accelerated leukemia progression in mice, while its knockdown in a human AML cell line reduced proliferation and prolonged survival of transplanted mice (70). As of today, the function of HIF2α in other types of leukemia has not been reported.
Hypoxia and HIFα factors have been implicated in mechanisms of resistance to chemotherapeutic agents in different leukemias. In B-ALL, hypoxia conditioning was found to increase expression of anti-apoptotic genes thus dampening the effect of chemotherapeutic compounds (101). Similarly, low oxygen levels lowered T-ALL cell sensitivity to chemotherapy and preserved their ability to initiate leukemia progression in vivo, while silencing of HIF1α sensitized leukemic cells to treatment, thus pointing to HIF1α as an important regulator of T-ALL chemoresistance (102). In line with these data, activation of Notch1 by HIF1α also resulted in protection of leukemic T-ALL cells from dexamethasone-induced apoptosis (91).
Similar results have been reported for AML, albeit via different molecular mechanisms. Hypoxic exposure of AML LSCs promotes chemoresistance to cytarabine arabinoside (Ara-C) via upregulation of the Polycomb transcriptional repressors BMI-1, which in turn supports malignancy via activation of PI3K/Akt signaling and EMT programs (95). The influence of hypoxia exposure on Ara-C susceptibility via HIF1α expression was also confirmed in a panel of leukemic cell lines, and this was suggested to represent a possible mechanism of minimal residual disease maintenance in the bone marrow after chemotherapy (103).
Besides promoting resistance to general chemotherapeutic drugs, hypoxia signaling has also been implicated in resistance to targeted therapy, specifically to the BCR-ABL tyrosine kinase inhibitor imatinib (104). Imatinib has become the main therapeutic opportunity for CML patients, but resistance often occurs even after prolonged treatment exposure (105). Interestingly, imatinib-resistant CML cells were shown to exhibit non-hypoxic upregulation of HIF1α and its target genes, resulting in upregulation of glycolytic processes, increased glucose uptake and lactate production (106). Recently, it was suggested that activation of glycolytic metabolism in CML cells may be facilitated by reduced expression of miR-18a-5p, which targets the 3’-UTR of HIF1α and leads to HIF1α downregulation in normal hematopoietic cells (107). In line with a relevant function of HIF1α in resistance to imatinib, it was shown that although imatinib partly inhibits HIF1α expression and transcriptional activity, HIF1α residual function is sufficient to suppress imatinib-induced apoptosis of CML cells (85). This may be particularly true in bone marrow microenvironments low in O2, where hypoxia signaling may effectively overcome BCR-ABL inhibition.
In conclusion, hypoxia and activation of HIF signaling take an important part to resistance mechanisms to chemotherapeutic agents and targeted therapy in different types of leukemia. As a consequence, a combination of chemotherapy and compounds targeting the hypoxic pathway may represent a valuable therapeutic approach for some types of leukemia. Along these lines, we and others have recently demonstrated that in CLL targeting HIF1α with different compounds increases response to current CLL therapeutic strategies including fludarabine and ibrutinib (66, 108). In more detail, HIF1α inhibition with BAY87-2243 causes downregulation of the HIF1α targets CXCL12 and CXCR4 thus abrogating the pro-survival effect exerted by stromal cells and promoting fludarabine-induced apoptosis (66, 67). Concordantly, inhibition of HIF1α with the camptothecin derivative EZN-2208 exerts anti-tumor activities and acts as a chemosensitizer by interrupting protective microenvironmental interactions of CLL cells both in vitro and in vivo (108). Therefore, we posit that this may be an interesting direction for future investigations into improving leukemia treatment, also in view of the availability of novel small molecule inhibitors of HIF2α (109), whose function should be investigated in leukemia.
As stated at the beginning of this chapter, studies on the role of HIF1α and HIF2α in leukemia failed to produce universal consent, especially in AML and CLL where contrasting evidence has been reported (Table 2). This is particularly evident in AML, where the function of HIFα factors has been studied by various groups. Beside the implication of HIF1α and HIF2α in LSCs maintenance, leukemia proliferation and survival that was previously described, other studies have revealed that HIF1α and HIF2α may display tumor-suppressive roles or no relevant functions in AML development and progression. For instance, deletion of the HIF1α gene in mouse hematopoietic stem and progenitor cells alongside retroviral transfer of AML oncogenic drivers (MLL-AF9, AML1-ETO, or MEIS1 and HOXA9) failed to impact leukemia initiation, progression and LSCs self-renewal by serial transplantation experiments (111). Rather, in the case of MLL-AF9-driven leukemia, HIF1α deletion accelerated leukemia progression by increasing cell proliferation (111). In addition, inducible HIF1α deletion in the MLL-AF9 model resulted in increased recovery upon withdrawal of chemotherapeutic regimens (112), thus suggesting that HIF1α inhibition cannot be expected to improve chemotherapy sensitivity in all leukemia. Of note, in the AML1-ETO mouse model it was reported that genetic deletion of the HIF1α gene resulted in compensatory expression of HIF2α (111), which led the authors to suggest that HIF2α upregulation may lead to increased leukemia aggressiveness, at least in some genetic backgrounds of AML. However, other authors reported that HIF2α gene deletion in MEIS1/HOXA9 and MLL-AF9 mouse models also shortened AML latency and seemed dispensable for LSCs maintenance, an effect that was even potentiated by HIF1α co-deletion (113).
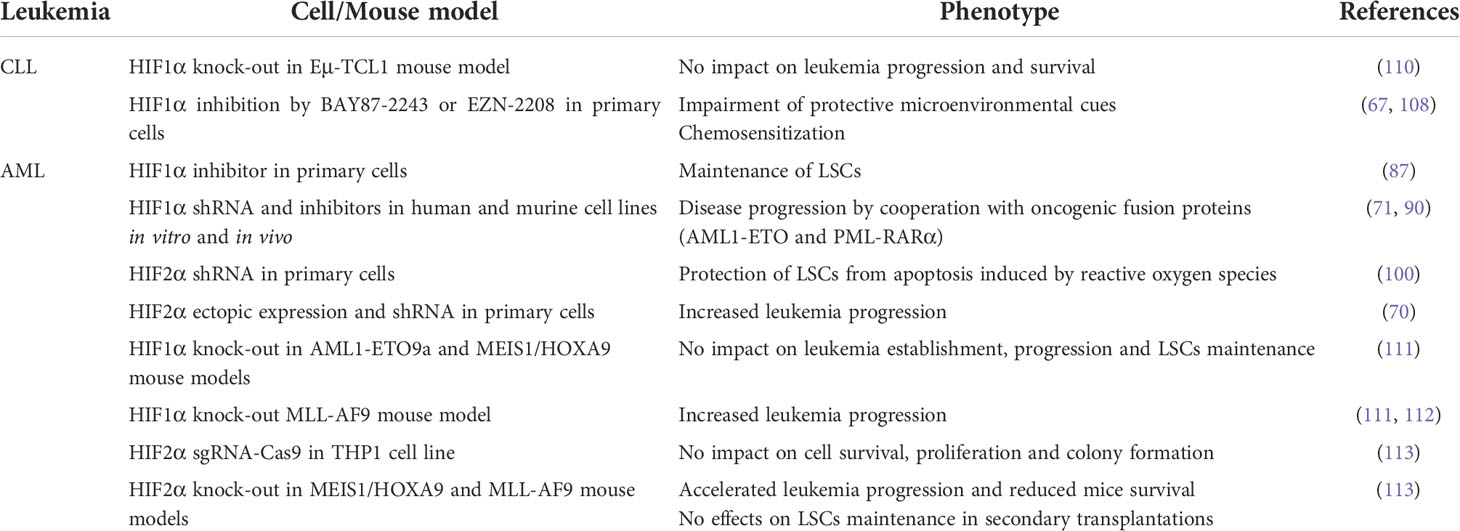
Table 2 Contrasting reports on HIFα functions in leukemia.
Similar head-scratching results have been recently published for CLL, where it was shown that knock-out of the HIF1α gene in the Eμ-TCL1 mouse model did not impact CLL progression or increase mice survival, suggesting that HIF1α is not essential for CLL leukemogenesis (110). This genetic experiment is particularly puzzling in view of the significant upregulation of hypoxic gene signatures that was reported in leukemic cells from Eμ-TCL1 mice (110), thus confirming data obtained by other laboratories of augmented hypoxia signaling in CLL (65–67). However, HIF1α knock-out did not affect the transcriptional program of Eμ-TCL1 leukemic cells (110). Therefore, the authors of this paper speculated that compensatory mechanisms may occur that render HIF1α inactivation of no consequence. Alternatively, dependency on HIF factors may be triggered by specific leukemogenic mutations and not others, an explanation that may very well apply also to AML.
These explanations are equally plausible, but we would like to argue that another possible explanation is that HIF factors exert different functions at distinct stages of leukemia development and progression. Thus, genetic inactivation in hematopoietic cells before leukemia onset may not necessarily recapitulate inhibition of HIF functions in overt leukemia (24). These are all interesting open questions that will need to be addressed in the future to better understand the biology of leukemia and the targetability of this pathway for leukemia treatment.
Hypoxia-responsive transcription factors are being increasingly implicated in the regulation of many normal and pathological cellular processes that expand well beyond mediating metabolic adaptation to oxygen deprivation, which led to their evolutionary emergence. Their increasing pervasiveness in cell biology is probably due to a number of reasons. First and foremost, their constitutive expression in conditions of physiological hypoxia. Most cell lines in use in laboratories around the world have been adapted to in vitro culture at pO2 of ambient air (160 mm Hg). However, direct measurement of oxygen levels in vivo reveals that much lower oxygen tensions are homeostatic in many tissues. The BM is a typical example of an organ where low oxygen levels appear directly implicated in maintenance of both normal hematopoietic and leukemic cells. This is particularly true in tissue microenvironments such as stem cell niches, where HIF factors promote a condition of metabolic “dormancy” based on a shift from mitochondrial metabolism to glycolysis that lowers production of DNA-damaging reactive oxygen species (ROS), and at the same time activate molecular programs implicated in stem cell maintenance. In addition to local hypoxia, growing evidence indicates that HIF1α and HIF2α expression is also promoted by extracellular stimuli or cell adhesion molecules provided by stromal cells, which probably reinforce HIF activity in specific BM microenvironments. As a consequence, hypoxic responses may be intrinsically wired in some hematopoietic and leukemic cells and occur at least in part independently of oxygen levels.
HIF1α and HIF2α share substantial homology and a similar structural organization, yet emerging literature is revealing diverging transcriptional outputs. The growing complexity of HIFα specific functions appears to be governed at different levels: i) tissue-specific expression of HIFα genes; ii) different sensitivity to oxygen concentrations of HIFα proteins; iii) different oxygen-independent regulation; iv) different nuclear diffusion properties and DNA binding profiles; v) binding to specific partners that promote cooperative transcriptional activation; vi) tissue-specific epigenetic modulation of target genes. The sum of all of these events leads to a diversification of HIF1α and HIF2α functions with non-overlapping consequences that are constantly emerging in physiological and pathological conditions.
With this in mind, it is to be expected that the importance of this pathway in biology will continue to grow. As per the role of hypoxia signaling in oncology, at current time a large amount of work has described the implication of HIFα factors in solid tumors, where in most cases they promote features of tumor aggressiveness, such as metastatic spread and relapse after treatment. Conversely, although hypoxia is a physiological hallmark of hematopoietic organs, the contribution of HIF1α and HIF2α to hematological malignancies has been underestimated for a long time. In recent years, many research groups have started to look into the function of hypoxia-responsive genes in blood malignancies. As we have summarized in this manuscript, important leukemia-promoting functions of HIF factors have emerged across distinct types of leukemia, including protection of LSCs, leukemia dissemination, leukemia expansion and impaired sensitivity to apoptosis and chemotherapy. Nonetheless, parallel work suggests that HIFα factors may not be essential or rather play tumor-suppressive functions in leukemia. Explanations to these contradictory results may rely on divergence of functions in distinct leukemia subtypes or at the preleukemic stage versus overt leukemia. In this respect, most of the studies that describe tumor-suppressive functions of HIFα factors in leukemia are based on genetic inactivation of these genes at a preleukemic stage, or concomitantly to leukemia initiation by mutated oncoproteins, a circumstance where it may be difficult to disentangle the function of HIFα genes in hematopoiesis or leukemia. In conclusion, further investigation is required to fully elucidate the extent and relevance of hypoxia-responsive gene activation in leukemia. In so doing, we should also aim to address the translational implications of inhibiting these pathways for the treatment of blood malignancies.
In this respect, an important note is that the vast majority of studies performed in leukemic contexts have focused on the HIF1α gene, with much less effort into elucidating the functions of HIF2α. Because HIF2α is specifically expressed in some cell types and its transcriptional output may differ from that of HIF1α, additional work may uncover specific context-dependent functions of HIF2α that are different from those exerted by HIF1α. Of relevance, because a specific small molecule inhibitor of HIF2α (belzutifan) has been recently approved by the U.S. FDA (Food and Drugs Administration) for the treatment of adult patients with VHL disease associated with renal cell carcinoma or pancreatic tumors, future studies into this pathway may pave the way for the use of this compound in other malignancies including blood cancers.
Both authors collected and reviewed relevant literature and wrote this manuscript.
This work was supported by the Italian Ministry of Health (Ricerca Finalizzata, RF-2019-12369841).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Kaelin WG, Ratcliffe PJ, Semenza GL, Kaelin WG Jr, Ratcliffe PJ, Semenza GL. For their discoveries of how cells sense and adapt to oxygen availability. Nobel Prize. (2019) 1(1):9.
3. Taylor CT, McElwain JC. Ancient atmospheres and the evolution of oxygen sensing via the hypoxia-inducible factor in metazoans. Physiol (Bethesda). (2010) 25(5):272–9. doi: 10.1152/physiol.00029.2010
4. Morikawa T, Takubo K. Hypoxia regulates the hematopoietic stem cell niche. Pflugers Arch Eur J Physiol (2016) 468(1):13–22. doi: 10.1007/s00424-015-1743-z
5. Ivanovic Z. Hypoxia or in situ normoxia: The stem cell paradigm. J Cell Physiol (2009) 219(2):271–5. doi: 10.1002/jcp.21690
6. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci (2012) 33(4):207–14. doi: 10.1016/j.tips.2012.01.005
7. Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood (2011) 117(23):e207–17. doi: 10.1182/blood-2010-10-314427
8. Duan C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am J Physiol Cell Physiol (2016) 310(4):C260–9. doi: 10.1152/ajpcell.00315.2015
9. Diao X, Ye F, Zhang M, Ren X, Tian X, Lu J, et al. Identification of oleoylethanolamide as an endogenous ligand for HIF-3α. Nat Commun (2022) 13(1):1–12. doi: 10.1038/s41467-022-30338-z
10. Kulyté A, Lundbäck V, Lindgren CM, Luan J, Lotta LA, Langenberg C, et al. Genome-wide association study of adipocyte lipolysis in the GENetics of adipocyte lipolysis (GENiAL) cohort. Mol Metab (2020) 34:85–96. doi: 10.1016/j.molmet.2020.01.009
11. Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol (2014) 49(1):1–15. doi: 10.3109/10409238.2013.838205
12. Chen Y, Cattoglio C, Dailey G, Zhu Q, Tjian R, Darzacq X. Mechanisms governing target search and binding dynamics of hypoxia-inducible factors. bioRxiv (2021). doi: 10.1101/2021.10.27.466110
13. Liao C, Zhang Q. Understanding the oxygen-sensing pathway and its therapeutic implications in diseases. Am J Pathol (2020) 190(8):1584–95. doi: 10.1016/j.ajpath.2020.04.003
14. Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von hippel-lindau protein. Nat Cell Biol (2000) 2(7):423–7. doi: 10.1038/35017054
15. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von hippel-lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292(5516):468–72. doi: 10.1126/science.1059796
16. Hewitson KS, McNeill LA, Riordan MV, Tian Y-M, Bullock AN, Welford RW, et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem (2002) 277(29):26351–5. doi: 10.1074/jbc.C200273200
17. Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science (2002) 295(5556):858–61. doi: 10.1126/science.1068592
18. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol (2004) 5(5):343–54. doi: 10.1038/nrm1366
19. Appelhoff RJ, Tian Y-M, Raval RR, Turley H, Harris AL, Pugh CW, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem (2004) 279(37):38458–65. doi: 10.1074/jbc.M406026200
20. Schito L, Semenza GL. Hypoxia-inducible factors: Master regulators of cancer progression. Trends cancer (2016) 2(12):758–70. doi: 10.1016/j.trecan.2016.10.016
21. Wu D, Potluri N, Lu J, Kim Y, Rastinejad F. Structural integration in hypoxia-inducible factors. Nature (2015) 524(7565):303–8. doi: 10.1038/nature14883
22. Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE (2005) 2005(306):re12. doi: 10.1126/stke.3062005re12
23. Smythies JA, Sun M, Masson N, Salama R, Simpson PD, Murray E, et al. Inherent DNA-binding specificities of the HIF-1α and HIF-2α transcription factors in chromatin. EMBO Rep (2019) 20(1):e46401.doi: 10.15252/embr.201846401.
24. Magliulo D, Bernardi R. HIF-α factors as potential therapeutic targets in leukemia. Expert Opin Ther Targets (2018) 22(11):917–28. doi: 10.1080/14728222.2018.1538357
25. Hu C-J, Sataur A, Wang L, Chen H, Simon MC. The n-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol Biol Cell (2007) 18(11):4528–42. doi: 10.1091/mbc.e06-05-0419
26. Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. (2011) 12(1):9–22.doi: 10.1038/nrc3183
27. Narravula S, Colgan SP. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J Immunol (2001) 166(12):7543–8. doi: 10.4049/jimmunol.166.12.7543
28. Mazure NM, Chauvet C, Bois-Joyeux B, Bernard M-A, Nacer-Chérif H, Danan J-L. Repression of alpha-fetoprotein gene expression under hypoxic conditions in human hepatoma cells: Characterization of a negative hypoxia response element that mediates opposite effects of hypoxia inducible factor-1 and c-myc. Cancer Res (2002) 62(4):1158–65.
29. Jeong C-H, Lee H-J, Cha J-H, Kim JH, Kim KR, Kim J-H, et al. Hypoxia-inducible factor-1 alpha inhibits self-renewal of mouse embryonic stem cells in vitro via negative regulation of the leukemia inhibitory factor-STAT3 pathway. J Biol Chem (2007) 282(18):13672–9. doi: 10.1074/jbc.M700534200
30. Lee KJ, Lee KY, Lee YM. Downregulation of a tumor suppressor RECK by hypoxia through recruitment of HDAC1 and HIF-1alpha to reverse HRE site in the promoter. Biochim Biophys Acta (2010) 1803(5):608–16. doi: 10.1016/j.bbamcr.2010.01.004
31. Xu J, Wang B, Xu Y, Sun L, Tian W, Shukla D, et al. Epigenetic regulation of HIF-1α in renal cancer cells involves HIF-1α/2α binding to a reverse hypoxia-response element. Oncogene (2012) 31(8):1065–72. doi: 10.1038/onc.2011.305
32. Li X, Zhang X, Xia J, Zhang L, Chen B, Lian G, et al. Macrophage HIF-2α suppresses NLRP3 inflammasome activation and alleviates insulin resistance. Cell Rep (2021) 36(8):109607.doi: 10.1016/j.celrep.2021.109607
33. Kindrick JD, Mole DR. Hypoxic regulation of gene transcription and chromatin: Cause and effect. Int J Mol Sci (2020) 21(21):1–27. doi: 10.3390/ijms21218320
34. Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature (2014) 508(7495):269–73. doi: 10.1038/nature13034
35. Nombela-Arrieta C, Silberstein LE. The science behind the hypoxic niche of hematopoietic stem and progenitors. Hematol Am Soc Hematol Educ Progr (2014) 2014(1):542–7. doi: 10.1182/asheducation-2014.1.542
36. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature (2014) 505(7483):327–34. doi: 10.1038/nature12984
37. Chow DC, Wenning LA, Miller WM, Papoutsakis ET. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. i. krogh’s model. Biophys J (2001) 81(2):675–84.doi: 10.1016/S0006-3495(01)75732-3
38. Parmar K, Mauch P, Vergilio J-A, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A (2007) 104(13):5431–6. doi: 10.1073/pnas.0701152104
39. Winkler IG, Barbier V, Wadley R, Zannettino ACW, Williams S, Lévesque J-P. Positioning of bone marrow hematopoietic and stromal cells relative to blood flow in vivo: Serially reconstituting hematopoietic stem cells reside in distinct nonperfused niches. Blood (2010) 116(3):375–85. doi: 10.1182/blood-2009-07-233437
40. Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol (2013) 15(5):533–43. doi: 10.1038/ncb2730
41. Lassailly F, Foster K, Lopez-Onieva L, Currie E, Bonnet D. Multimodal imaging reveals structural and functional heterogeneity in different bone marrow compartments: Functional implications on hematopoietic stem cells. Blood (2013) 122(10):1730–40.doi: 10.1182/blood-2012-11-467498
42. Shima H, Takubo K, Tago N, Iwasaki H, Arai F, Takahashi T, et al. Acquisition of G₀ state by CD34-positive cord blood cells after bone marrow transplantation. Exp Hematol (2010) 38(12):1231–40.doi: 10.1016/j.exphem.2010.08.004
43. Shima H, Takubo K, Iwasaki H, Yoshihara H, Gomei Y, Hosokawa K, et al. Reconstitution activity of hypoxic cultured human cord blood CD34-positive cells in NOG mice. Biochem Biophys Res Commun (2009) 378(3):467–72.doi: 10.1016/j.bbrc.2008.11.056
44. Moirangthem RD, Singh S, Adsul A, Jalnapurkar S, Limaye L, Kale VP. Hypoxic niche-mediated regeneration of hematopoiesis in the engraftment window is dominantly affected by oxygen tension in the milieu. Stem Cells Dev (2015) 24(20):2423–36.doi: 10.1089/scd.2015.0112
45. Hermitte F, Brunet de la Grange P, Belloc F, Praloran V, Ivanovic Z. Very low O2 concentration (0.1%) favors G0 return of dividing CD34+ cells. Stem Cells (2006) 24(1):65–73.doi: 10.1634/stemcells.2004-0351
46. Danet GH, Pan Y, Luongo JL, Bonnet DA, Simon MC. Expansion of human SCID-repopulating cells under hypoxic conditions. J Clin Invest. (2003) 112(1):126–35. doi: 10.1172/JCI17669
47. Hammoud M, Vlaski M, Duchez P, Chevaleyre J, Lafarge X, Boiron J-M, et al. Combination of low O(2) concentration and mesenchymal stromal cells during culture of cord blood CD34(+) cells improves the maintenance and proliferative capacity of hematopoietic stem cells. J Cell Physiol (2012) 227(6):2750–8. doi: 10.1002/jcp.23019
48. Eliasson P, Rehn M, Hammar P, Larsson P, Sirenko O, Flippin LA, et al. Hypoxia mediates low cell-cycle activity and increases the proportion of long-term-reconstituting hematopoietic stem cells during in vitro culture. Exp Hematol (2010) 38(4):301–310.e2. doi: 10.1016/j.exphem.2010.01.005
49. Bapat A, Schippel N, Shi X, Jasbi P, Gu H, Kala M, et al. Hypoxia promotes erythroid differentiation through the development of progenitors and proerythroblasts. Exp Hematol (2021) 97:32–46.e35. doi: 10.1016/j.exphem.2021.02.012
50. Mostafa SS, Miller WM, Papoutsakis ET. Oxygen tension influences the differentiation, maturation and apoptosis of human megakaryocytes. Br J Haematol (2000) 111(3):879–89. doi: 10.1046j.1365-2141.2000.02457.x
51. Chen S, Su Y, Wang J. ROS-mediated platelet generation: a microenvironment-dependent manner for megakaryocyte proliferation, differentiation, and maturation. Cell Death Dis (2013) 4(7):e722. doi: 10.1038/cddis.2013.253
52. Taneja R, Rameshwar P, Upperman J, Wang MT, Livingston DH. Effects of hypoxia on granulocytic-monocytic progenitors in rats. Role of bone marrow stroma. Am J Hematol (2000) 64(1):20–5.doi: 10.1002/(sici)1096-8652(200005)64:1<20::aid-ajh4>3.0.co;2-#
53. Katahira J, Mizoguchi H. Improvement of culture conditions for human megakaryocytic and pluripotent progenitor cells by low oxygen tension. Int J Cell Cloning. (1987) 5(5):412–20. doi: 10.1002/stem.5530050506
54. Koller MR, Bender JG, Miller WM, Papoutsakis ET. Reduced oxygen tension increases hematopoiesis in long-term culture of human stem and progenitor cells from cord blood and bone marrow. Exp Hematol (1992) 20(2):264–70.
55. LaIuppa JA, Papoutsakis ET, Miller WM. Oxygen tension alters the effects of cytokines on the megakaryocyte, erythrocyte, and granulocyte lineages. Exp Hematol (1998) 26(9):835–43.
56. Iommarini L, Porcelli AM, Gasparre G, Kurelac I. Non-canonical mechanisms regulating hypoxia-inducible factor 1 alpha in cancer. Front Oncol (2017) 7:286. doi: 10.3389/fonc.2017.00286
57. Albadari N, Deng S, Li W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin Drug Discov (2019) 14(7):667–82. doi: 10.1080/17460441.2019.1613370
58. Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell (2010) 7(3):391–402. doi: 10.1016/j.stem.2010.06.020
59. Scortegagna M, Morris MA, Oktay Y, Bennett M, Garcia JA. The HIF family member EPAS1/HIF-2alpha is required for normal hematopoiesis in mice. Blood (2003) 102(5):1634–40. doi: 10.1182/blood-2003-02-0448
60. Méndez-Ferrer S, Bonnet D, Steensma DP, Hasserjian RP, Ghobrial IM, Gribben JG, et al. Bone marrow niches in haematological malignancies. Nat Rev Cancer (2020) 20(5):285–98. doi: 10.1038/s41568-020-0245-2
61. Fiegl M, Samudio I, Clise-Dwyer K, Burks JK, Mnjoyan Z, Andreeff M. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood (2009) 113(7):1504–12. doi: 10.1182/blood-2008-06-161539
62. Harrison JS, Rameshwar P, Chang V, Bandari P. Oxygen saturation in the bone marrow of healthy volunteers. Blood (2002) 99:394. doi: 10.1182/blood.V99.1.394
63. Jensen PO, Mortensen BT, Hodgkiss RJ, Iversen PO, Christensen IJ, Helledie N, et al. Increased cellular hypoxia and reduced proliferation of both normal and leukaemic cells during progression of acute myeloid leukaemia in rats. Cell Prolif (2000) 33(6):381–95. doi: 10.1046/j.1365-2184.2000.00183.x
64. Rytelewski M, Harutyunyan K, Baran N, Mallampati S, Zal MA, Cavazos A, et al. Inhibition of oxidative phosphorylation reverses bone marrow hypoxia visualized in imageable syngeneic b-ALL mouse model. Front Oncol (2020) 10:991. doi: 10.3389/fonc.2020.00991
65. Valsecchi R, Coltella N, Belloni D, Ponente M, Ten Hacken E, Scielzo C, et al. HIF-1α regulates the interaction of chronic lymphocytic leukemia cells with the tumor microenvironment. Blood (2016) 127(16):1987–97. doi: 10.1182/blood-2015-07-657056
66. Griggio V, Vitale C, Todaro M, Riganti C, Kopecka J, Salvetti C, et al. HIF-1α is over-expressed in leukemic cells from TP53-disrupted patients and is a promising therapeutic target in chronic lymphocytic leukemia. Haematologica (2020) 105(4):1042–54. doi: 10.3324/haematol.2019.217430
67. Vitale C, Griggio V, Riganti C, Todaro M, Kopecka J, Jones R, et al. Targeting HIF-1α regulatory pathways as a strategy to hamper tumor-microenvironment interactions in CLL. Cancers (Basel). (2021) 13(12):2883.doi: 10.3390/cancers13122883
68. Mayerhofer M, Valent P, Sperr WR, Griffin JD, Sillaber C. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1alpha, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood (2002) 100(10):3767–75. doi: 10.1182/blood-2002-01-0109
69. Chen H, Shen Y, Gong F, Jiang Y, Zhang R. HIF-α promotes chronic myelogenous leukemia cell proliferation by upregulating p21 expression. Cell Biochem Biophys (2015) 72(1):179–83. doi: 10.1007/s12013-014-0434-2
70. Forristal CE, Brown AL, Helwani FM, Winkler IG, Nowlan B, Barbier V, et al. Hypoxia inducible factor (HIF)-2α accelerates disease progression in mouse models of leukemia and lymphoma but is not a poor prognosis factor in human AML. Leukemia (2015) 29(10):2075–85. doi: 10.1038/leu.2015.102
71. Gao XN, Yan F, Lin J, Gao L, Lu XL, Wei SC, et al. AML1/ETO cooperates with HIF1α to promote leukemogenesis through DNMT3a transactivation. Leukemia (2015) 29(8):1730–40. doi: 10.1038/leu.2015.56
72. Kontos CK, Papageorgiou SG, Diamantopoulos MA, Scorilas A, Bazani E, Vasilatou D, et al. mRNA overexpression of the hypoxia inducible factor 1 alpha subunit gene (HIF1A): An independent predictor of poor overall survival in chronic lymphocytic leukemia. Leuk Res (2017) 53:65–73. doi: 10.1016/j.leukres.2016.11.014
73. Ghosh AK, Shanafelt TD, Cimmino A, Taccioli C, Volinia S, Liu C, et al. Aberrant regulation of pVHL levels by microRNA promotes the HIF/VEGF axis in CLL b cells. Blood (2009) 113(22):5568–74. doi: 10.1182/blood-2008-10-185686
74. Manso BA, Zhang H, Mikkelson MG, Gwin KA, Secreto CR, Ding W, et al. Bone marrow hematopoietic dysfunction in untreated chronic lymphocytic leukemia patients. Leukemia (2019) 33(3):638–52. doi: 10.1038/s41375-018-0280-0
75. Wellmann S, Guschmann M, Griethe W, Eckert C, von Stackelberg A, Lottaz C, et al. Activation of the HIF pathway in childhood ALL, prognostic implications of VEGF. Leukemia (2004) 18(5):926–33. doi: 10.1038/sj.leu.2403332
76. Frolova O, Samudio I, Benito JM, Jacamo R, Kornblau SM, Markovic A, et al. Regulation of HIF-1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol Ther (2012) 13(10):858–70. doi: 10.4161/cbt.20838
77. Schito L, Rey S, Konopleva M. Integration of hypoxic HIF-α signaling in blood cancers. Oncogene (2017) 36(38):5331–40. doi: 10.1038/onc.2017.119
78. Wang Y, Liu Y, Bailey C, Zhang H, He M, Sun D, et al. Therapeutic targeting of TP53-mutated acute myeloid leukemia by inhibiting HIF-1α with echinomycin. Oncogene (2020) 39(14):3015–27. doi: 10.1038/s41388-020-1201-z
79. Ijabi R, Roozehdar P, Afrisham R, Moradi-Sardareh H, Kaviani S, Ijabi J, et al. Association of GRP78, HIF-1α and BAG3 expression with the severity of chronic lymphocytic leukemia. Anticancer Agents Med Chem (2020) 20(4):429–36. doi: 10.2174/1871520619666191211101357
80. Zhang C, Liu J, Wang J, Zhang T, Xu D, Hu W, et al. The interplay between tumor suppressor p53 and hypoxia signaling pathways in cancer. Front Cell Dev Biol (2021) 9:648808. doi: 10.3389/fcell.2021.648808
81. Yosifov DY, Idler I, Bhattacharya N, Reichenzeller M, Close V, Ezerina D, et al. Oxidative stress as candidate therapeutic target to overcome microenvironmental protection of CLL. Leukemia (2020) 34(1):115–27. doi: 10.1038/s41375-019-0513-x
82. Waclawiczek A, Hamilton A, Rouault-Pierre K, Abarrategi A, Albornoz MG, Miraki-Moud F, et al. Mesenchymal niche remodeling impairs hematopoiesis via stanniocalcin 1 in acute myeloid leukemia. J Clin Invest (2020) 130(6):3038–50. doi: 10.1172/JCI133187
83. Desplat V, Faucher J-L, Mahon FX, Dello Sbarba P, Praloran V, Ivanovic Z. Hypoxia modifies proliferation and differentiation of CD34(+) CML cells. Stem Cells (2002) 20(4):347–54. doi: 10.1634/stemcells.20-4-347
84. Griessinger E, Anjos-Afonso F, Pizzitola I, Rouault-Pierre K, Vargaftig J, Taussig D, et al. A niche-like culture system allowing the maintenance of primary human acute myeloid leukemia-initiating cells: A new tool to decipher their chemoresistance and self-renewal mechanisms. Stem Cells Transl Med (2014) 3(4):520–9. doi: 10.5966/sctm.2013-0166
85. Ng KP, Manjeri A, Lee KL, Huang W, Tan SY, Chuah CTH, et al. Physiologic hypoxia promotes maintenance of CML stem cells despite effective BCR-ABL1 inhibition. Blood (2014) 123(21):3316–26. doi: 10.1182/blood-2013-07-511907
86. Zhang H, Li H, Xi HS, Li S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood (2012) 119(11):2595–607. doi: 10.1182/blood-2011-10-387381
87. Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell (2011) 8(4):399–411. doi: 10.1016/j.stem.2011.02.006
88. Cheloni G, Tanturli M, Tusa I, Ho DeSouza N, Shan Y, Gozzini A, et al. Targeting chronic myeloid leukemia stem cells with the hypoxia-inducible factor inhibitor acriflavine. Blood (2017) 130(5):655–65. doi: 10.1182/blood-2016-10-745588
89. He P, Lei J, Zou L-X, Zhou G-Z, Peng L, Deng Q, et al. Effects of hypoxia on DNA hydroxymethylase tet methylcytosine dioxygenase 2 in a KG-1 human acute myeloid leukemia cell line and its mechanism. Oncol Lett (2021) 22(4):692. doi: 10.3892/ol.2021.12953
90. Coltella N, Percio S, Valsecchi R, Cuttano R, Guarnerio J, Ponzoni M, et al. HIF factors cooperate with PML-RARα to promote acute promyelocytic leukemia progression and relapse. EMBO Mol Med (2014) 6(5):640–50. doi: 10.1002/emmm.201303065
91. Zou J, Li P, Lu F, Liu N, Dai J, Ye J, et al. Notch1 is required for hypoxia-induced proliferation, invasion and chemoresistance of T-cell acute lymphoblastic leukemia cells. J Hematol Oncol (2013) 6:3. doi: 10.1186/1756-8722-6-3
92. Giambra V, Jenkins CE, Lam SH, Hoofd C, Belmonte M, Wang X, et al. Leukemia stem cells in T-ALL require active Hif1α and wnt signaling. Blood (2015) 125(25):3917–27. doi: 10.1182/blood-2014-10-609370
93. Migliavacca J, Percio S, Valsecchi R, Ferrero E, Spinelli A, Ponzoni M, et al. Hypoxia inducible factor-1α regulates a pro-invasive phenotype in acute monocytic leukemia. Oncotarget (2016) 7(33):53540–57. doi: 10.18632/oncotarget.10660
94. Percio S, Coltella N, Grisanti S, Bernardi R, Pattini L. A HIF-1 network reveals characteristics of epithelial-mesenchymal transition in acute promyelocytic leukemia. Genome Med (2014) 6(12):84. doi: 10.1186/s13073-014-0084-4
95. Jiang M, He G, Li J, Li J, Guo X, Gao J. Hypoxic exposure activates the b cell-specific moloney murine leukaemia virus integration site 1/PI3K/Akt axis and promotes EMT in leukaemia stem cells. Oncol Lett (2021) 21(2):98.doi: 10.3892/ol.2020.12359
96. Lambert AW, Weinberg RA. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer (2021) 21(5):325–38. doi: 10.1038/s41568-021-00332-6
97. Serra S, Vaisitti T, Audrito V, Bologna C, Buonincontri R, Chen S-S, et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv (2016) 1(1):47–61. doi: 10.1182/bloodadvances.2016000984
98. Rigoni M, Riganti C, Vitale C, Griggio V, Campia I, Robino M, et al. Simvastatin and downstream inhibitors circumvent constitutive and stromal cell-induced resistance to doxorubicin in IGHV unmutated CLL cells. Oncotarget (2015) 6(30):29833–46. doi: 10.18632/oncotarget.4006
99. Wierenga ATJ, Cunningham A, Erdem A, Lopera NV, Brouwers-Vos AZ, Pruis M, et al. HIF1/2-exerted control over glycolytic gene expression is not functionally relevant for glycolysis in human leukemic stem/progenitor cells. Cancer Metab (2019) 7:11. doi: 10.1186/s40170-019-0206-y
100. Rouault-Pierre K, Lopez-Onieva L, Foster K, Anjos-Afonso F, Lamrissi-Garcia I, Serrano-Sanchez M, et al. HIF-2α protects human hematopoietic Stem/Progenitors and acute myeloid leukemic cells from apoptosis induced by endoplasmic reticulum stress. Cell Stem Cell (2013) 13(5):549–63. doi: 10.1016/j.stem.2013.08.011
101. Petit C, Gouel F, Dubus I, Heuclin C, Roget K, Vannier JP. Hypoxia promotes chemoresistance in acute lymphoblastic leukemia cell lines by modulating death signaling pathways. BMC Cancer. (2016) 16(1):746. doi: 10.1186/s12885-016-2776-1
102. Fahy L, Calvo J, Chabi S, Renou L, Le Maout C, Poglio S, et al. Hypoxia favors chemoresistance in T-ALL through an HIF1α-mediated mTORC1 inhibition loop. Blood Adv (2021) 5(2):513–26. doi: 10.1182/bloodadvances.2020002832
103. Matsunaga T, Imataki O, Torii E, Kameda T, Shide K, Shimoda H, et al. Elevated HIF-1α expression of acute myelogenous leukemia stem cells in the endosteal hypoxic zone may be a cause of minimal residual disease in bone marrow after chemotherapy. Leuk Res (2012) 36:e122–4. doi: 10.1016/j.leukres.2012.02.028
104. Moen MD, McKeage K, Plosker GL, Siddiqui MAA. Imatinib: a review of its use in chronic myeloid leukaemia. Drugs (2007) 67(2):299–320. doi: 10.2165/00003495-200767020-00010
105. Chu S, McDonald T, Lin A, Chakraborty S, Huang Q, Snyder DS, et al. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood (2011) 118(20):5565–72. doi: 10.1182/blood-2010-12-327437
106. Zhao F, Mancuso A, Bui TV, Tong X, Gruber JJ, Swider CR, et al. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming. Oncogene (2010) 29(20):2962–72. doi: 10.1038/onc.2010.67
107. Wu K, Guo C, Li Y, Yang J, Zhou Q, Cheng S, et al. MicroRNA-18a-5p regulates the warburg effect by targeting hypoxia-inducible factor 1α in the K562/ADM cell line. Exp Ther Med (2021) 22(4):1069.doi: 10.3892/etm.2021.10503
108. Valsecchi R, Coltella N, Magliulo D, Bongiovanni L, Scarfò L, Ghia P, et al. EZN-2208 treatment suppresses chronic lymphocytic leukaemia by interfering with environmental protection and increases response to fludarabine. Open Biol (2020) 10(5):190262. doi: 10.1098/rsob.190262
109. Zhou J, Gong K. Belzutifan: a novel therapy for von hippel–lindau disease. Nat Rev Nephrol. (2022) 18(4):205–6. doi: 10.1038/s41581-022-00544-5
110. Gonder S, Largeot A, Gargiulo E, Pierson S, Fernandez Botana I, Pagano G, et al. The tumor microenvironment-dependent transcription factors AHR and HIF-1α are dispensable for leukemogenesis in the eµ-TCL1 mouse model of chronic lymphocytic leukemia. Cancers (Basel). (2021) 13(18):4518. doi: 10.3390/cancers13184518
111. Velasco-Hernandez T, Hyrenius-Wittsten A, Rehn M, Bryder D, Cammenga J. HIF-1α can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood (2014) 124(24):3597–607. doi: 10.1182/blood-2014-04-567065
112. Velasco-Hernandez T, Soneji S, Hidalgo I, Erlandsson E, Cammenga J, Bryder D. Hif-1α deletion may lead to adverse treatment effect in a mouse model of MLL-AF9-Driven AML. Stem Cell Rep (2019) 12(1):112–21. doi: 10.1016/j.stemcr.2018.11.023
Keywords: hypoxic stress, bone marrow, HIF - 1α, HIF - 2, αhypoxia, inducible-factor, leukemia
Citation: Magliulo D and Bernardi R (2022) Hypoxic stress and hypoxia-inducible factors in leukemias. Front. Oncol. 12:973978. doi: 10.3389/fonc.2022.973978
Received: 20 June 2022; Accepted: 21 July 2022;
Published: 18 August 2022.
Edited by:
Francesco Piazza, University of Padua, ItalyReviewed by:
Persio Dello Sbarba, University of Florence, ItalyCopyright © 2022 Magliulo and Bernardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa Bernardi, YmVybmFyZGkucm9zYUBoc3IuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.