 Chien-shan Cheng
Chien-shan Cheng Pei-wen Yang1,2†
Pei-wen Yang1,2† Yun Sun
Yun Sun Shao-li Song
Shao-li Song Zhen Chen
Zhen Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 03 October 2022
Sec. Molecular and Cellular Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.969731
This article is part of the Research TopicNew Insights into Fibrotic Signaling in CancerView all 11 articles
Fibroblast activation protein-α (FAP) is a type II transmembrane serine protease that has specific endopeptidase activity. Given its well-established selective expression in the activated stromal fibroblasts of epithelial cancers, although not in quiescent fibroblasts, FAP has received substantial research attention as a diagnostic marker and therapeutic target. Pancreatic cancer is characterized by an abundant fibrotic or desmoplastic stroma, leading to rapid progression, therapeutic resistance, and poor clinical outcomes. Numerous studies have revealed that the abundant expression of FAP in cancer cells, circulating tumor cells, stromal cells, and cancer-associated fibroblasts (CAFs) of pancreatic adenocarcinoma is implicated in diverse cancer-related signaling pathways, contributing to cancer progression, invasion, migration, metastasis, immunosuppression, and resistance to treatment. In this article, we aim to systematically review the recent advances in research on FAP in pancreatic adenocarcinoma, including its utility as a diagnostic marker, therapeutic potential, and correlation with prognosis. We also describe the functional role of FAP-overexpressing stromal cells, particulary CAFs, in tumor immuno- and metabolic microenvironments, and summarize the mechanisms underlying the contribution of FAP-overexpressing CAFs in pancreatic cancer progression and treatment resistance. Furthermore, we discuss whether targeting FAP-overexpressing CAFs could represent a potential therapeutic strategy and describe the development of FAP-targeted probes for diagnostic imaging. Finally, we assess the emerging basic and clinical studies regarding the bench-to-bedside translation of FAP in pancreatic cancer.
Pancreatic adenocarcinoma (PAAD) is a highly aggressive and lethal cancer requiring novel diagnostic and therapeutic approaches. Characterized by an abundant fibrotic and extensive desmoplastic stromal response, cancer-associated fibroblasts (CAFs) are the most abundant component of the PAAD stroma. CAFs accumulate in pancreatic tumor tissue, wherein they enhance collagen family protein and fibronectin expression in a paracrine manner, thereby maintaining and remodeling the extracellular matrix structure. They also generate multiple factors, including exosomes, growth factors, immune- and metabolism-associated metabolites in an autocrine manner to communicate with cancer cells. Moreover, they contribute to promoting the desmoplastic response and mediate the early invasion, high recurrence rate, and treatment resistance of PAAD (1). Owing to abundant extracellular matrix deposition, vasculature, and fibroblasts, the pancreatic cancer stroma acts as a firm haven protecting cancer cells from different interventions, thereby contributing to treatment resistance. Thus, rather than playing the roles of a mere tumorigenic onlooker, the desmolastic stroma functions as an active participant in pancreatic cancer progression, invasion, migration, metastasis, immunosuppression, and resistance to treatment. Therefore, CAFs have become an attractive therapeutic target in the stroma of pancreatic cancers (Figure 1).
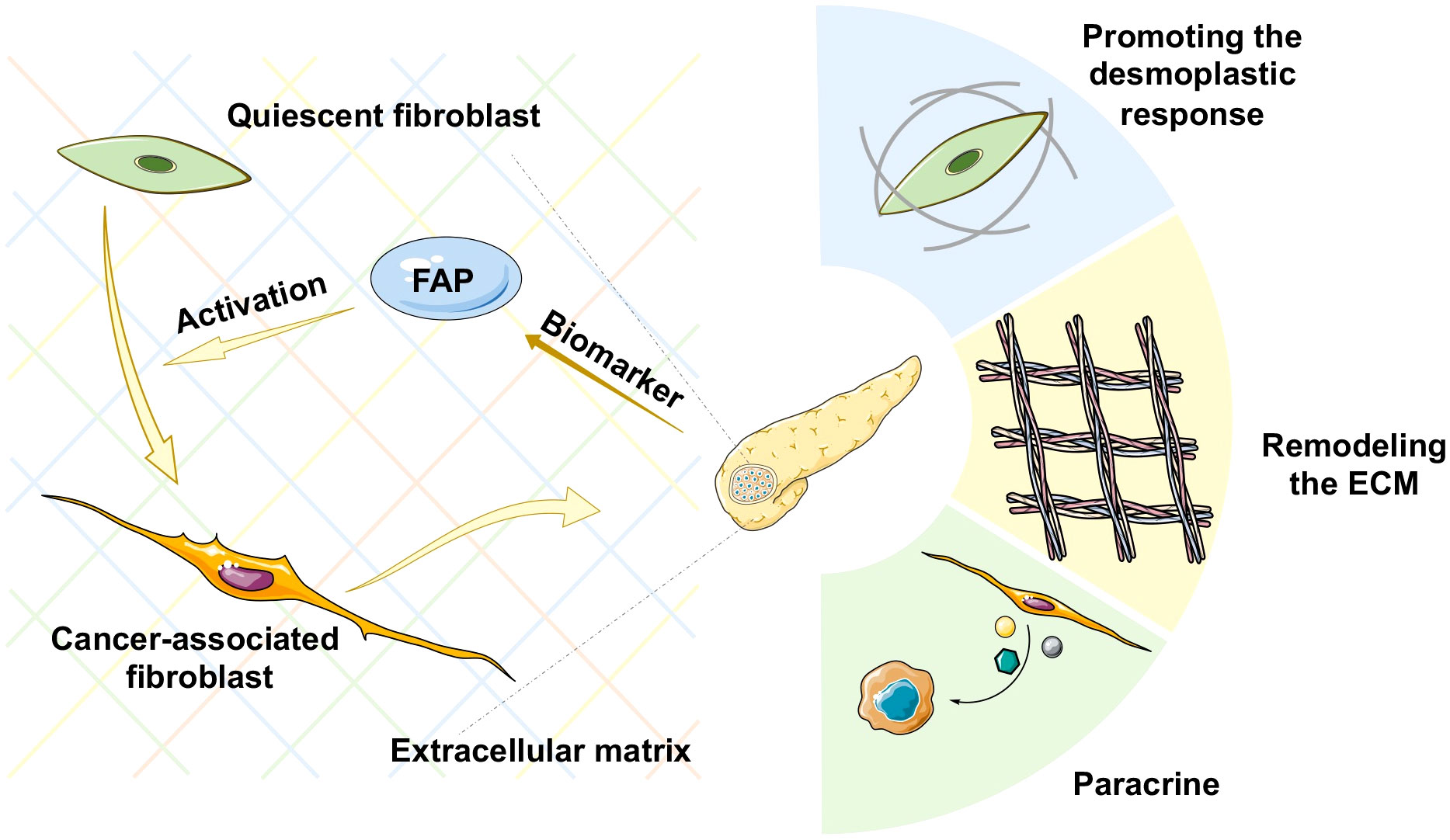
Figure 1 Schematic representation of various mechanisms of cancer-associated fibroblast (CAF) activation in pancreatic cancer. ECM, extracellular matrix.
Fibroblast activation protein-α (FAP), named after its activating role, is a type II transmembrane serine protease with specific endopeptidase activity that is upregulated in different cancer types. Given its well-established selective expression in the activated stromal fibroblasts of epithelial cancers, although not in quiescent fibroblasts, FAP has received considerable research attention as a diagnostic marker and therapeutic target. FAP was first identified in 1986 by Wolfgang Rettig based on its role in activating fibroblasts (2). Initial research conducted on FAP focused on its role as a cell surface antigen. Subsequent studies, however, revealed that FAP could be shed from the cell membrane, thereby undergoing conversion to a soluble derivative referred to as circulating antiplasmin-cleaving enzyme (APCE) (3). In normal tissues and plasma, FAP is typically expressed at low levels and appears to serve as a redundant or non-essential protease in developmental processes. However, FAP was subsequently established to be involved in diverse pathological processes, including wound healing; inflammation, such as arthritis, atherosclerosis, and fibrosis; and epithelial cancers. The abundant expression of FAP in cancer and stromal cells, and particularly in CAFs, has been implicated in different cancer-related signaling pathways, thereby contributing to cancer progression, invasion, migration, metastasis, immunosuppression, and resistance to treatment. Consequently, FAP+ CAFs have emerged as important regulators and potential treatment targets.
The high levels of FAP expression in pancreatic cancer stroma have also been correlated with the poor prognosis of patients and desmoplasia (4, 5), and FAP has been identified as an independent predictor of pancreatic cancer survival (6, 7). The extensive fibrotic matrix of PAAD serves as a physical barrier that contributes to resistance to chemoradiotherapy or immunotherapy. Treatment paradigms targeting cancer cells are unable to overcome this obstacle, and consequently, considerable research effort has been devoted to characterizing the mechanisms underlying the treatment resistance of PAAD and identifying novel therapeutic strategies that can target the associated stroma and fibroblasts. In this review article, we summarize the promising theragnostic role of FAP in PAAD, with the aim of gaining further insights regarding the clinical implications.
According to the National Cancer Institute Clinical Proteomic Tumor Analysis Consortium (CPTAC) dataset, FAP protein expression is significantly higher in primary PAAD tumor tissues than in normal tissues. Similarly, stromal FAP expression has been detected in approximately 98% of specimens in a sample of 48 patients with surgically resected PAAD (7). Consistently, the findings of a further immunohistochemical study involving 134 patients with PAAD revealed that FAP was expressed in both stromal fibroblasts (98/134, 73.1%) and cancer cells (102/134, 76.1%), with expression being significantly associated with patient age (p < 0.001), tumor size (p < 0.001), fibrotic foci (p = 0.003), and perineural invasion (p = 0.009) (4), thereby highlighting the importance of FAP in the clinicopathological characterization of PAAD.
In humans and some mammals, plasma contains small amounts of FAP, at concentrations of approximately 100 ng/mL or 0.6 nmol/L (8). FAP is also detectable in peripheral blood, the levels of which have been used for disease characterizations. For example, FAP has been used as a molecular marker in the identification of subpopulations of circulating tumor cell (9). Moreover, the measurement of FAP-specific substrates in body fluids has also been reported (10). However, as to whether the levels of FAP enzyme activity can serve as an informative diagnostic tool and in the assessment of disease progression in the clinical setting has yet to be sufficiently ascertained. Although the potential utility of FAP immunohistochemistry and plasma FAP as a biomarker in renal tumors for the differential diagnosis at an early stage have previously been reported (11), there have to date been no comparable studies with respect to pancreatic cancer.
The overexpression of FAP has also been implicated in the poor prognosis of PAAD patients (shown in Figure 2). Survival analyses have revealed that high FAP expression is associated with lower overall survival (12). A KPC mice model in which FAP had been deleted was characterized by delayed primary tumor onset and a more prolonged survival (13). Studies have also shown that higher CD8 expression and lower FAP expression in stromal cells can independently predict the prognosis in PAAD patients, and that blockade of FAP may improve the prognosis of patients with PAAD. It may increase CD8 cell levels (6). Furthermore, a comprehensive transcriptome analysis (14) identified two four-hub gene modules (including FAP) as specific predictive features for PAAD diagnosis and prognosis, thereby providing a new perspective with respect to clinical trials. Moreover, by applying weighted gene co-expression network analysis, Wang et al. (15) established that FAP is involved in a core module associated with PAAD type. Collectively, the findings of these studies highlight the potential utility of FAP expression patterns as a prognostic marker and an immune checkpoint target in PAAD.
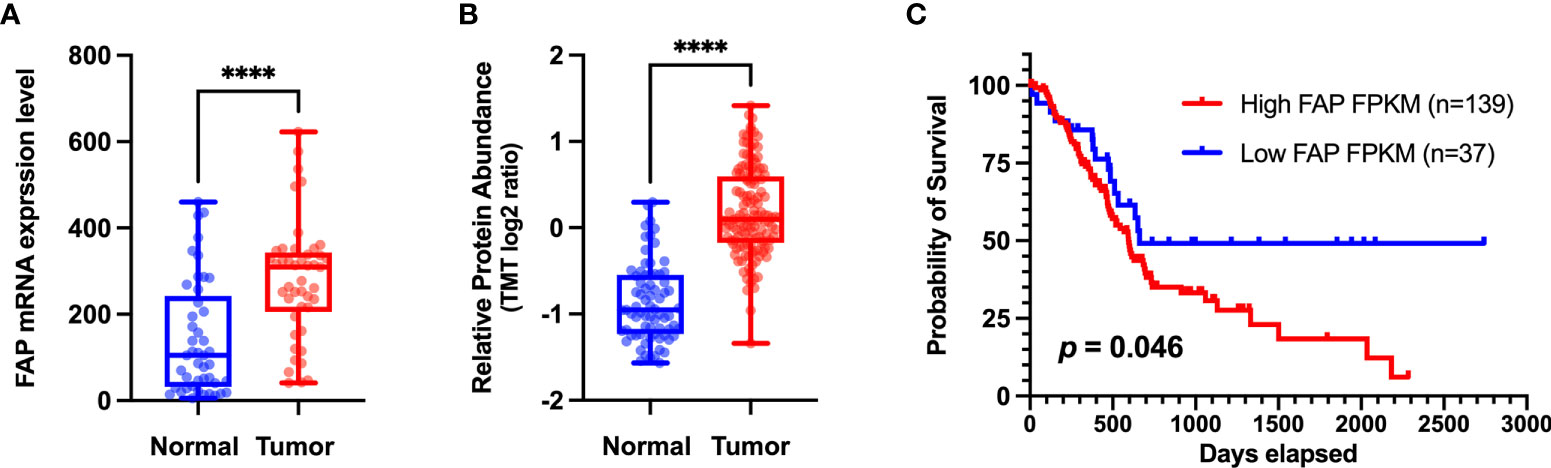
Figure 2 (A) Different levels of FAP mRNA expression in normal and tumor tissues. Transcriptome data of pancreatic adenocarcinoma (PAAD) and matched normal tissue were obtained from the Gene Expression Omnibus dataset (http://www.ncbi.nlm.nih.gov/geo) (GSE28735) for validation at the transcriptomic level. (B) Different levels of FAP protein expression in normal and tumor tissues. Proteome data were obtained from the CPTAC dataset (12) for validation at the protein level. (C) Overall survival analysis of two groups of patients with PAAD from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). The Kaplan–Meier method was used to compare the survival of different FAP expression groups (the expression cutoff = 2.8), and the log-rank method was used for statistical analysis. GraphPad 9.0 software was used for data visualization. ****p < 0.0001.
Previous diagnostic and therapeutic paradigms have tended to focus almost exclusively on tumor cells, and have failed to make any significant breakthroughs in PAAD therapy during the past decade. In this context, it has been established that FAP is expressed predominantly in stromal cells, whereas only very low levels of expression are detected in cancer cells in resected pancreatic ductal adenocarcinoma (PDAC) tissues (7). At the cellular level, pancreatic cancer cell lines have been shown to express FAP protein to a greater or lesser extent (4), and mRNA at different levels (Figure 3). Furthermore, the expression of FAP has been shown to alter the expression of stromal extracellular matrix proteins, including tenascin C, collagen I, fibronectin, and α-SMA, and also to enhance fibronectin fiber patterned orientation, thereby resulting in the rapid disease progression and increasing resistance to treatment (16). PAAD cells communicate with stromal cells, recruit fibroblasts, and activate these to yield cancer-associated fibroblasts. The finding of numerous studies have established the link between stromal-expressed FAP, particularly FAP+ CAFs, and pancreatic cancer, thereby indicating the potential ultility of FAP as a target in PAAD treatment (Figure 4). Moreover, it has been found that FAP+ stroma promotes cancer immune escape (17). In addition, findings relating to the enzymatic role, expressional patterns, and stroma modulation role of FAP in PAAD have provided insights for further examination of its pro-tumorigenic role and provided evidence to indicate that FAP could serve as both a novel marker in PAAD diagnosis and as therapeutic target (Figure 5).
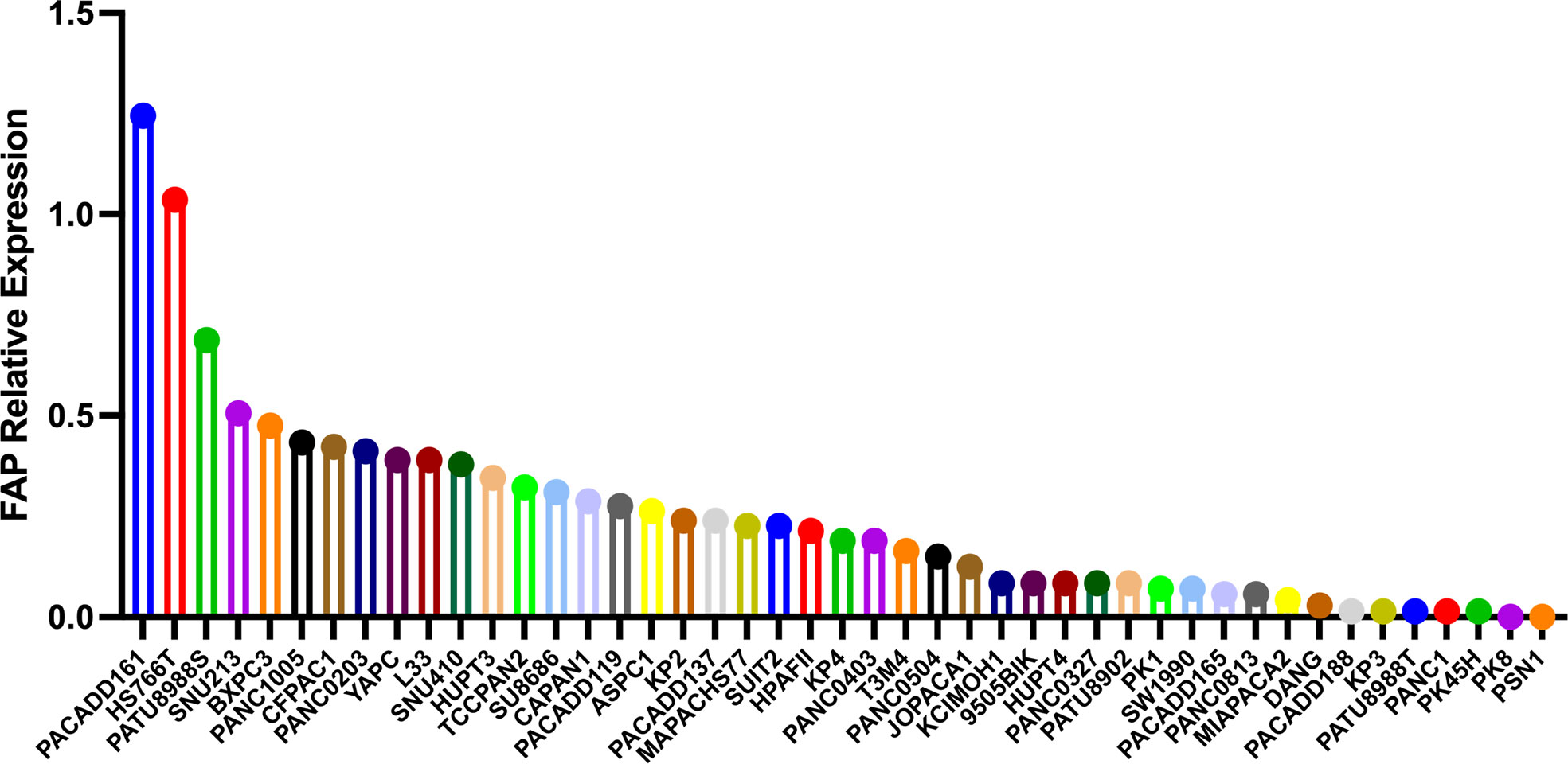
Figure 3 The different mRNA levels of FAP protein expression in pancreatic cancer cell lines using data obtained from the Motivations for the Cancer Cell Line Encyclopedia database (https://portals.broadinstitute.org/ccle).
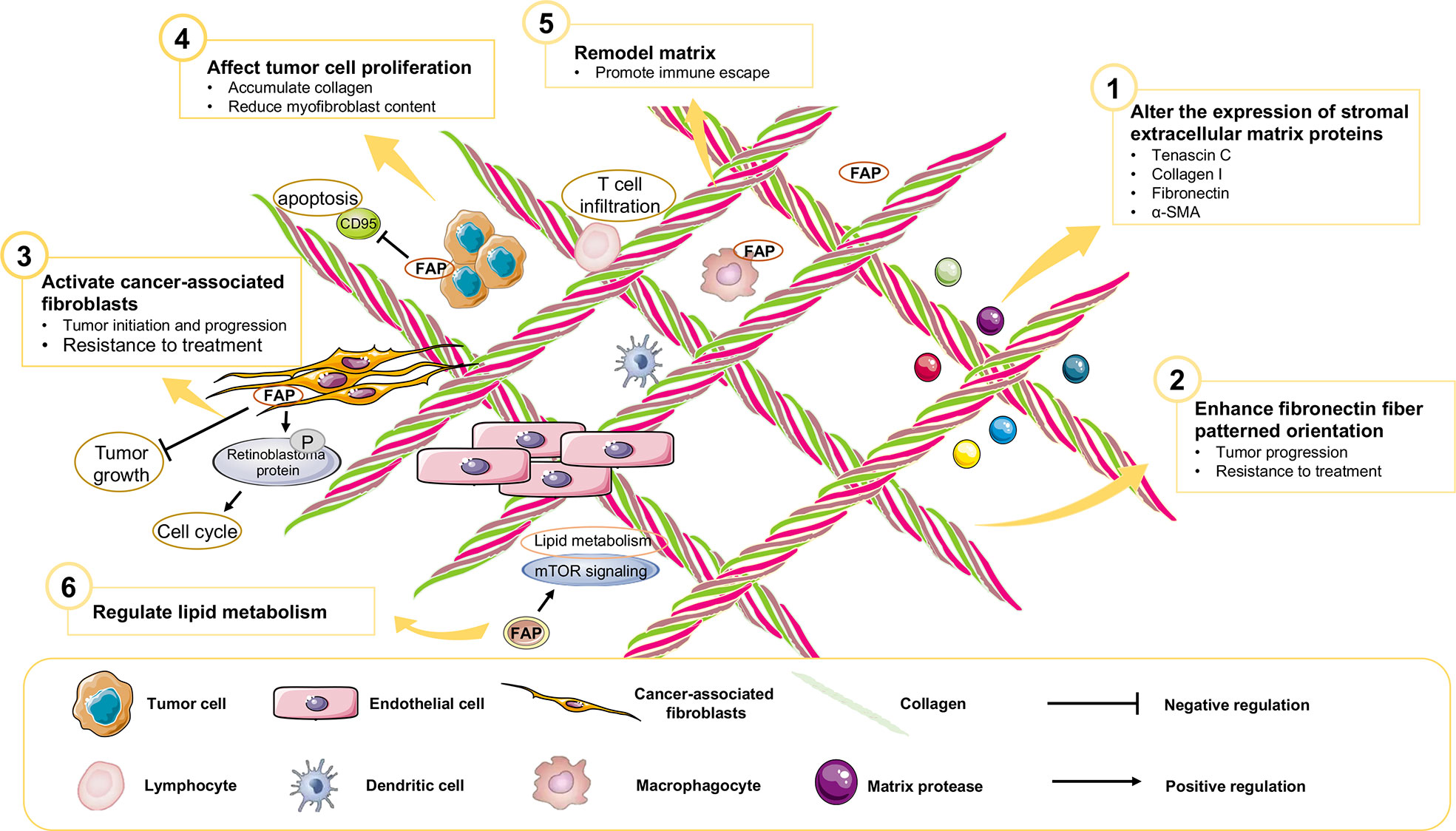
Figure 4 The role of FAP in the PAAD tumor microenvironment.
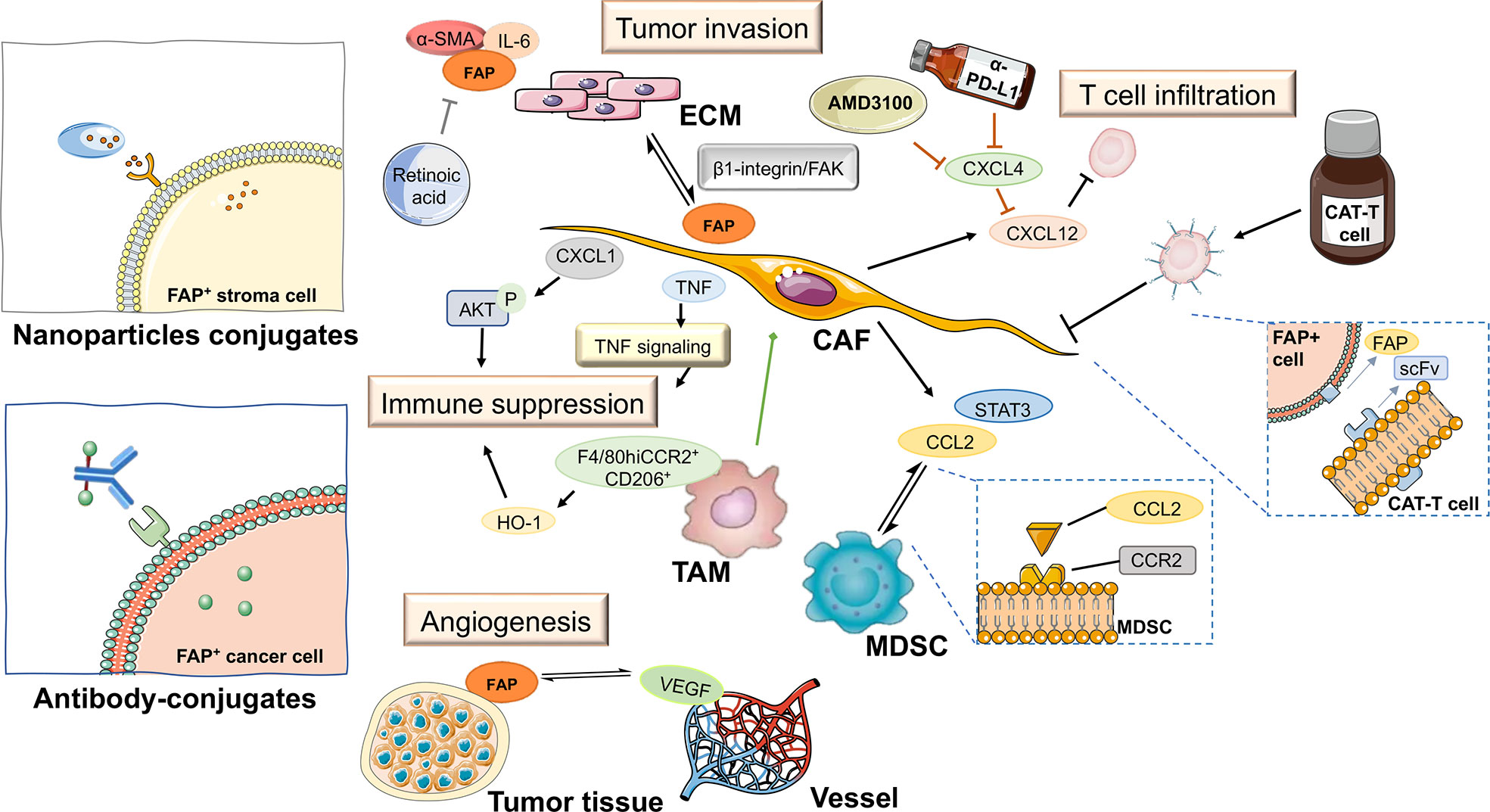
Figure 5 FAP-targeted therapies and their expected impact on pancreatic tumor tissue. ECM, extracellular matrix; TAM, tumor-associated macrophage; CAT-T cell, chimeric antigen receptor T cell; MDSC, myeloid-derived suppressor cell; CAF, cancer-associated fibroblast; scFv, fragments of single-chain antibody.
FAP is a member of the dipeptidyl peptidase (DPP) 4 family of proteins that hydrolyzes a prolyl bond at sites two amino acids from the N-terminus of a protein. The cyclic character of proline residue makes targeting the specific catalytic ability of FAP attractive. However, the finding that mice in which FAP is not expressed show no an aberrant phenotype would tend to indicate that under physiological conditions, FAP is a redundant or non-essential protease (18).Moreover, in preclinical or clinical research, it has been demonstrated that inhibitors of FAP fail to have significant anti-cancer effect in preclinical or clinical research (19–21). These observations could be ascribed to one of several possible factors. Firstly, FAP could act indirectly on tumor cells, as has been indicated by the finding that the depletion of FAP indirectly inhibits tumor cell proliferation via an enhancement of collagen accumulation and the impediment of myofibroblast content (19). Mechanistically, it has been found that the regulation of FAP-1 enzymatic activity negatively regulates CD95 (Fas, APO-1)-mediated apoptosis in pancreatic cancer cells (22). Additionally, as a known collagen-degrading enzyme, FAP mediates matrix remodeling and in part regulates lipid metabolism via mTOR signaling (23). Secondly, the development of talabostat as an anti-cancer agent has focused mainly on its immune-mediated activity, and to date, there have been no studies that have specifically sought to evaluate the efficacy of talabostat in FAP-expressing tumors, or its use in combination with other targeted therapies. Collectively, these findings provide evidence to indicate a potential regulatory role of FAP under pathological conditions, and that this FAP-mediated regulation of lipid metabolism and remodeling of the extracellular matrix (ECM) could serve as a potential novel therapeutic target.
Despite efforts designed to suppress the expression and enzymatic activity of FAP as anti-cancer treatments, studies, such as those involving the development of a murine anti-FAP mAb F19 and its humanized version unconjugated sibrotuzumab (BIBH 1) (24–26), have failed to demonstrate promising clinical efficacy. On the basis of the studies conducted to date, there is no evidence to indicate that direct targeting of FAP would be either safe or effective. Consequently, given the overexpression of FAP in a wide variety of stromal cells during tumorigenesis, this would tend to identify the stroma as a target for anti-cancer drugs. Indeed, it has been found that depletion of FAP+ cells has the effect of delaying subcutaneous tumor growth and contributes to the development of a more immunogenic environment, thereby indicating that FAP-expressing stromal cells could play important roles as immunomodulators (27).
Malignant cells drive the generation of desmoplastic and immunosuppressive tumor microenvironments, and emerging evidence indicates that FAP is aberrantly expressed in CAFs, chimeric antigen receptor T (CAR-T) cells, and a subset of M2 macrophages. Furthermore, FAP-expressing cells have also been associated with the degradation of extracellular matrix proteins and glycosaminoglycans, as well as a reduction in tumor vessel density, which jointly contributes to the continual remodeling of the tumor microenvironment during the progression of PAAD. Furthermore, the emerging development of nanoparticle conjugates and antibody-conjugates is offering a potentially wide range of therapeutic options from the perspective of targeting FAP in cancer.
CAFs are the main component of the PAAD stroma, thereby indicating that targeting CAFs could represent a viable therapeutic strategy. CAFs play a pleiotropic role in PAAD initiation and progression, and in line with expectations, it has been demonstrated that depletion of CAFs can accelerate tumor progression (28, 29). In this context, attention has focused on targeting precise subpopulations of CAFs that are known to be oncogenic and pro-progression, particularly FAP-positive CAFs. These FAP-expressing CAFs have been proposed to accelerate proliferation, invasion, and therapeutic resistance in ovarian cancer cells (30), and have also been suggested to reprogram tumor inflammatory environments in human intrahepatic cholangiocarcinoma via FAP-STAT3-CCL2 signaling (31). However, in-depth research on PAAD cells is still lacking. By promoting retinoblastoma protein phosphorylation, FAP+ CAFs have been suggested to alter the cell cycle of co-cultured PAAD cells (7). Furthermore, the depleting of FAP-expressing cells (including all FAP+ CAFs), has been found to delay tumor growth in a subcutaneous model of PAAD (27).
It is well established that PAAD is highly resistant to immunotherapy, and it has been demonstrated that FAP-positive stromal cells (including almost all the CAFs) mediated local immunosuppression and immune escape, thereby compromising the efficacy of immunotherapy, and in particular, immune checkpoint blockade treatment (27, 32). Studies have indicated that the production of chemokine (C-X-C motif) ligand 12 (CXCL12) by FAP+ CAFs within the cancer cell-rich region of a tumor impedes T cell infiltration leading to immunosuppression (32). To counteract this suppressive effect, a synergized treatment approach has been proposed, which entails inhibition of the CXCL12 receptor by the suppression of chemokine (C-X-C motif) receptor 4 (CXCR4) using α-PD-L1, a checkpoint antagonist. This was demonstrated to promote T-cell accumulation and contribute to tumor regression (32). Recently, a CXCR4 small-molecule inhibitor, AMD3100 (plerixafor), was assessed in a phase I clinical trial (NCT02179970) that aimed to evaluate its safety in PAAD patients. The findings of the comparative transcriptional analysis revealed an unusually rapid enhancement of intra-tumoral B and T cell responses within 7 days of treatment, thereby indicating the possible induction of an integrated anti-cancer immune response. Given these promising findings, further clinical trials are warranted that evaluate responses in a larger number of patients who have undergone repeated cycles of combination therapy with CXCR4 inhibitor and immune checkpoint antagonist, along with additional studies examining tolerability, pharmacokinetics, and pharmacodynamics to further assess the therapeutic activity.
However, the fact that several well-established biomarkers of CAFs are also typically expressed on the surface of other cell types, raises the likelihood that CAF-targeted therapy might have certain unwanted side effects. As an alternative strategy for the treatment of PAAD, researchers are examining the effects of blocking the interactions between CAFs and their neighboring cells. Determining the mechanisms underlying the accumulation of CAFs in cancer and developing methods that can be used to identify different CAF subsets in the tumor microenvironment will contribute to establishing therapeutic strategies that could convert the tumor-promoting microenvironment into a tumor-suppressing microenvironment. Considerable evidence has accumulated to indicate that FAP plays an important role in this alteration, and thus further research on the synergetic roles of FAP+ CAFs in PAAD immunotherapy is warranted.
The infiltration of immature myeloid cells, including the myeloid-derived suppressor cells (MDSCs) and macrophages, is one of the well-defined hallmarks of cancer (33). In this regard, Yang et al. (31) have demonstrated that FAP+ CAFs can enhance the recruitment of MDSCs via STAT3-CCL2 signaling and thereby promote tumor progression in a murine liver tumor model. FAP+ CAFs are characterized by a distinct inflammatory phenotype of STAT3 activation and pro-inflammatory cytokine CCL2 upregulation. As the primary source of CCL2, FAP+ CAFs undergo crosstalk with circulating MDSCs expressing the CCL2 receptor (CCR2), thereby forming a CCL2–CCR2 axis that plays a role in mediating tumor promotion. In line with expectations, it has been observed that the tumor progression promoting and MDSC recruiting effects of FAP+ CAFs are abolished in Ccr2-deficient mice. However, current evidence indicates that the activity of tumoral FAP+ stromal cells contributes to immune suppression, the function of FAP+ CAFs in the recruitment of MDSCs in PAAD remains largely undetermined, and accordingly warrants further investigations.
Recently, a direct mechanism has been proposed for the generation of CAFs, which indicates that tumor-associated macrophage (TAMs) expressing CAF markers could be a potential source of CAFs (34, 35). The findings of research have indicated that these FAP-positive cells comprise two subpopulations, namely, a CD45− mesenchymal subset and a CD45+ hematopoietic subset (36). In this regard, a previous study on immunogenic Lewis lung carcinoma cells identified FAP+CD45+ cells as a subset of F4/80hiCCR2+CD206+ M2 macrophages and the main tumoral source of the immune inhibitory enzyme heme oxygenase-1 (HO-1). The depletion of FAP+CD45+ cells or administration of the HO-1 inhibitor Sn mesoporphyrin has been shown to promote the immune-dependent arrest of subcutaneous tumor growth (36). Moreover, FAP+/F4/80+/HO-1+ stromal cells have been identified in a PAAD subcutaneous tumor model. Sn mesoporphyrin has been established to induce an immune response to a currently unidentified antigen(s) and inhibits tumor growth in a tumor-bearing mouse model of PAAD (37). In PAAD, TAMs in the tumor stroma also secretes FAP, shifting their phenotype to an immunosuppressive M2 macrophage type, suppressing the adaptive immune response, and promoting a positive feedback loop in PAAD progression (37).
PAAD is histologically characterized by a large stroma volume, and previous studies have consistently reported the suppression of intra-tumoral effector T-cells by the FAP-expressing stromal cells among different cancer types (27, 32, 38). In PAAD, FAP+ CAF-abundant areas have been shown to be characterized by limited CD8+ lymphocytes infiltration, thereby indicating the contribution to the spatial exclusion of intra-tumoral CD8+ T cells (39), along with Tregs and neutrophil accumulation and cancer-associated pathways modulation (40). Given that heterogeneous and differential FAP+ CAFs in PAAD tumors may be involved to varying degrees in stromal variation, immunosuppression, and differential responses to potential immunotherapy, further experimental studies are necessary to clarify the potential causal inference.
In addition to cellular constituents, important components of the tumor microenvironment include the ECM, blood vessels, and cytokines (41). FAP activity has been found to direct stromal ECM organization via β1-integrin-dependent cell matrix interactions, in which suppression of FAP activity limits β1-integrin/FAK-mediated invasive capacity in PAAD cells (16). Moreover, the findings of a further study have indicated that retinoic acid, a vitamin A derivative, plays a role in transforming FAP-activated CAFs into static fibroblasts via the down-regulated expression of α-SMA, FAP, and IL-6 and the inhibition of the ECM production (42). These findings provide important insights regarding the selective targeting of the main source of ECM proteins and highlight the potential utility of FAP-mediated disruption of the ECM as a novel therapeutic approach for PAAD.
Levels of vascular endothelial growth factor (VEGF), a promotor of angiogenesis and biomarker of vessel density, are found to be raised in PDAC tissues and are linked to liver metastases and poor prognosis. It has previously been demonstrated that in the cancerous tissues of PAAD patients, the expression of FAP is positively correlated with VEGF (41). This significant positive correlation between FAP and VEGF indicates that the two factors conjunctly influence PAAD survival, and that contributing to the complexity of these interactions are pericytes, inflammatory cells, and fibroblastic cells, which together with PAAD cells, shape the tumor vasculature.
Cytokines and chemokines, including tumor necrosis factor (TNF), and CXCL1, secreted by FAP+ CAFs contribute to the further suppression of adaptive immunity in PAAD (38, 43) and are currently attracting considerable attention among researchers. Moreover, with an increasing number of studies focusing on TNF, its roles in fibroblasts within the tumor microenvironment have gradually become more precisely elucidated, and it has accordingly been established that its functions are not confined to merely driving fibroblast activation. However, the tumor-promoting and immunosuppressive activity of FAP+ fibroblasts have been demonstrated to suppress TNF signaling (27, 44, 45), thereby contributing to cancer–stroma–cancer interaction loop that promotes tumor progression (46). For example, the pancreatic stellate cells (PSCs), the FAP+ resident cells of the pancreas, have been implicated in cancer-related fibrosis, induction of PAAD migration, and invasion via the activation of CXCL1-mediated AKT phosphorylation. Furthermore, the secretion of transforming growth factor-β1 (TGF-β1) PAAD cells has been observed to induce the expression of FAP in hitherto quiescent PSCs, thereby promoting a further releasing CXCL1, promoting tyrosine kinase receptors phosphorylation, and forming a positive feed-forward loop promoting PAAD progression (47). Collectively, the multifarious aspects of the tumor microenvironment provide a pool of secreted cytokines that stimulates or suppresses different components of this microenvironment, thereby contributing to the progression of PAAD.
As previously mentioned, the FAP+ stroma and CAFs play pivotal roles in mediating the immunosuppressive characteristics of PAAD (32) and are responsible for the ineffective activity of known T-cell checkpoint antagonists. Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) or its ligand PD-L1 function as important checkpoint receptors on T cells, and accordingly present ideal targets for inhibition by antagonistic antibodies. However, given the ineffectual nature of immunotherapy and therapeutic approaches. In this regard, targeting tumoral FAP+ stromal cells is seen as a novel alternative therapeutic option. Emerging evidence indicates that the malignant behavior of PAAD, such as resistance to treatment, immune escape, and metastasis, is associated with the complex interaction between stromal and tumor cells. However, the current therapeutic paradigm focuses on tumor-stroma interactions in PAAD rather than the expression of FAP protein. It is believed that gaining a better understanding of the role of this protein in the FAP-mediated interaction between stroma and tumor cells could contribute to optimizing the FAP-inhibition strategy.
Chimeric antigen receptor T cells (CAT-T cells) are genetically engineered T cells that are becoming increasingly widely used as components in novel types of anti-cancer immunotherapy. T cells contain fragments of a single-chain antibody that can recognize predefined surface antigens, thus enabling T cells to effectively identify and target cancer cells, and thereby affording a considerable range of opportunities to modify and improve CAR T cell therapy. Traditionally, predefined surface antigens on cancer cells have been the primary targets in anti-cancer therapy. However, the findings of recent studies have identified a number of antigens, which, although expressed at very low levels on normal cells, are highly expressed on the surface of tumor stromal cells, thereby indicating their potential value as novel targets for CAR T cell therapy that can minimize on-target/off-tumor toxicity. This has thus stimulated an increasing interest in the development of CAR tools with FAP specificity for T cell modification.
Studies using mouse models have shown that FAP-CAR T cells can exhaust FAP-expressing stromal cells and inhibit tumor growth by producing immunostimulatory cytokines, promoting tumor cytolysis (48), and enhancing the anti-tumor response of endogenous CD8+ T cells (49). Moreover, synchronously targeting both malignant cells and FAP+ stroma has been established to produce a robust anti-tumor effect. However, the findings of a further study have indicated that the presence of highly reactive FAP-specific CAR-modified T cells can promote severe cachexia and dose-limiting bone toxicity without significantly affecting tumor suppression among different syngeneic tumor implantation models in mice. These observations tend to indicate that FAP-expressing stroma derived from different sources might contribute to contrasting outcomes. Hence, this emphasizes the necessity to take into consideration the location of FAP expression in stromal cells during PAAD initiation and progression and the requirement for a more precise biomarker of FAP+ CAFs. These results warrant further assessment for optimizing FAP-specific CAR in preclinical and clinical trials of human PAAD.
The findings of studies that have used murine pancreatic cancer models have tended to indicate that the adoptive transfer of FAP-CAR T can also inhibit tumor growth in an immune-independent manner (50–52). Furthermore, the adoptive transfer of FAP-targeted CAT-T cells has been demonstrated to reduce tumor vascularity, ECM proteins, and glycosaminoglycans (48, 53, 54). Moreover, FAP-specific CAR T transfer has been found to selectively recognizes and depletes the FAP+ subsets of cancer stem cells, which play a vital role in maintaining the tumor stroma and eliminating FAP+ CAFs. Consequently, the adoptive transfer of FAP-CAR T has provided insights that will contribute to the further development of FAP+-specific stromal cell-targeted therapies for treating PAAD by targeting FAP+ cells in the tumor microenvironment.
Investigations that have focused on the depletion of FAP+ cells in the PAAD tumor microenvironment have revealed an anti-tumor effect associated with α-CTLA-4 and α-PD-L1 treatment, thereby providing further evidence in support of the immune-suppressive role of FAP+ stromal cells in the poor responsiveness to T-cell checkpoints antagonists. As previously mentioned, CXCL12 produced by FAP+ CAFs may direct tumor immune evasion in a model of human PAAD and synergized treatment of CXCL12 receptor inhibition with α-PD-L1 (32), thereby remodeling the immunosuppressive microenvironment. In this context, Ji et al. (55) designed a cleavable amphiphilic peptide (CAP) marker to be specifically responsive to FAP stroma, in which, at the site of FAP+ cell abundance, the CAP was cleaved, and nanoparticles rapidly disassembled and unloaded the drug into a solid tumor. In addition, an amphiphilic bifunctional PD-1/PD-L1 peptide antagonist has been developed to deliver doxorubicin and R848 after being cleaved by FAP, which in combination with PD-1 blockade therapy was observed to trigger a stronger immune response on activating immunogenic cell death and reprogramming tumor-associated macrophages (56). Furthermore, it is worth noting that stromal factors such as TGF-β and FAP, which are commonly characterized by altered expression in PAAD, were found to be associated with resistance to neoadjuvant atezolizumab therapy (targeting PD-1, PD-L1) in operable urothelial carcinoma in the ABACUS trial (NCT02662309) (57). The findings of these studies thus indicate the utility of FAP as a clinical biomarker in T-cell checkpoint antagonist treatments, and consequently, have potential implications for comparable treatment in PAAD warrants further studies.
Currently, several clinical trials targeting FAP, especially RO6874281, are ongoing across cancers. An immunocytokine RO6874281 is composed of an interleukin-2 variant (IL-2v) targeting FAP and pembrolizumab (anti-PD-1), leading to the blockade of T cells migration. RO6874281 is applied to immunotherapy of several cancers, including renal cell carcinoma (NCT03063762), metastatic melanoma (NCT03875079), solid tumor, breast cancer, and cancer of head and neck (NCT02627274), and metastatic PAAD(NCT03193190). These trials are active or recruiting, without results yet, but their preclinical studies are promising. Therefore, it is valuable to keep a watchful eye on the results.
As a protease, FAP has also been exploited in drug innovation studies, and several methods have been adopted that target FAP-expressing cells based on FAP enzyme activity. For example, nanomaterial‐based drug delivery systems are a common platform used to deliver drugs to tumor sites. In this regard, the stroma can act as a barrier that impedes the passage of drugs targeting tumor sites. However, the aberrant expression of FAP provides a strategy for overcoming this impediment, namely, the development of smart nanomaterials that respond to FAP-positive CAFs. Nanomaterial‐based drug delivery systems have shown considerable opportunity to optimize the drug specificity, biocompatibility, and pharmacokinetic features of these materials.
Sum et al. (58) have assessed a bispecific FAP-CD40 antibody that triggers a potent FAP-dependent stimulation of CD40, thereby enhancing tumor-specific T-cell priming and inducing tumor growth inhibition in vivo. By specifically inducing CD40 stimulation in the presence of FAP, this novel bispecific antibody was demonstrated to overcome the systemic toxicity associated with the CD40 agonist, thereby providing a promising approach for cancer immunotherapy. A further antibody-conjugate targeting FAP, FAP5-DM1, has also been shown to have an inhibitory effect on tumor progression, inducing complete regression in a PAAD xenograft model with good efficacy and tolerability (59). These studies have accordingly demonstrated the potential utility of combined targeting antibody conjugates as novel drug candidates for PAAD and stimulated further clinical translational studies toward clinical applications.
Although immune checkpoint blockade has shown promising results in the treatment of various cancers, the extensive fibrotic matrix and immunosuppressive effect of the PAAD tumor microenvironment have tended to hinder its wider application. In this respect, Yu et al. have proposed the use of novel thermo- and fibrotic matrix-sensitive liposomes encapsulating small-sized albumin nanoparticles loaded with an immune checkpoint inhibitor (BMS-202). Exposure to FAP and near-infrared laser radiation, with a mild elevation in localized temperature, induces the release of this agent at localized sites, thereby promoting the recovery of T lymphocyte activities within the immunosuppressive tumor microenvironment (60). By enhancing the accumulation of immune checkpoint inhibitors at the tumor site, these novel nanoparticles can promote the localized secretion of cytokines such as TNF-α and INF-γ, and potentiates the anti-tumor immune response in PAAD.
Despite significant advances in research on FAP as an anti-cancer target candidate with broad clinical application prospects in conjunction with checkpoint blockade immunotherapy of solid tumors, the potentiality of lethal adverse effects should be taken into consideration when targeting FAP for immunotherapeutic purposes. The findings of previous studies have indicated that targeting FAP triggers the recognition of multipotent bone marrow stromal cells, whereas cachexic mouse and human pluripotent bone marrow mesenchymal stem cells (BMSCs) are recognized by FAP-reactive T cells (61). Fatal ototoxicity and cachexia observed following cell-based immunotherapy against FAP accordingly cautions against its use as a generic target. Furthermore, as FAP expression by pluripotent BMSCs may point to the cellular origin of tumor stromal fibroblasts and at least in part result from the universal recognition of FAP-reactive T cells on multipotent BMSCs, lethal bone marrow hypocellularity and necrosis, and cachexia have been observed subsequently to FAP-targeting immunotherapy (61). Additionally, long non-coding RNAs (lncRNAs), which are expressed in cell-type and disease-specific manners, have been demonstrated to contribute to the development and activation of CAFs (62, 63). In turn, these activated CAFs can promote tumor development by affecting the gene expression and secretory properties of cells, altering the tumor microenvironment, and enhancing malignant biological processes in cancer cells via lncRNAs. These observations provide evidence to indicate that FAP+CAFs-specific lncRNAs could potentially be targeted for PAAD therapy without affecting the FAP expression of normal cells. In addition to these plausible obstacles, other challenges remain and should be carefully assessed to avert the potential risks of systemic toxicity when targeting FAP. Accordingly, prior to translational research evaluating the utility of FAP-related therapy, a comprehensive assessment of the safety and toxicity of these treatments using different animal models is of the utmost necessity.
In preclinical and clinical trials, FAP-targeting molecular imaging radiotracers have shown promising results with respect to tumor diagnosis. At present, the clinical assessment of FAP-targeting radiotracers is primarily performed in small cohorts of patients in single-center studies. However, small sample populations and certain disparities among studies have led to difficulties in drawing definitive conclusions. Given the widely reported potential for oncological diagnosis, application of the [68Ga]Ga-FAPI-04 probe, for example, has been reported in clinical trials (NCT04554719 and NCT04605939) with the uptake of [68Ga]Ga-FAPI-04 in benign pancreatic lesions being demonstrated (64). Nevertheless, in low [18F]F-FDG-avid tumors, such as in PAAD, [68Ga]Ga-FAPI PET/CT shows high sensitivity in detecting primary pancreatic tumors, involved lymph nodes, and metastases, and is superior in terms of TNM staging (65), thereby indicating the potential utility of [68Ga]Ga-FAPI-PET/CT as an effective imaging tool. Consequently, upon diagnosis, it is essential to undertake a comprehensive pathological assessment.
Furthermore, when applied in combination with magnetic resonance imaging (MRI), the [68Ga]Ga-FAPI-04 PET/MR probe may enhance diagnostic sensitivity and prevent the misdiagnosis of certain pancreatic lesions. Although compared with the current gold standard contrast-enhanced CT, FAPI-PET/CT appears to be a superior imaging modality for pancreatic cancer (66), prospective trials with larger patient populations are needed to evaluate whether [68Ga]Ga-FAPI PET/CT can elicit treatment modification in PAAD when compared with other imaging methodologies. Similarly, a preliminary study has reported the diagnostic potential of 18F-FAPI-74 in FAP-expressing PAAD xenografts with higher tumoral uptake than [68Ga]Ga-FAPI-04, thereby highlighting the advantageous properties of 18F, notably the higher rate of detection and wide availability [https://jnm.snmjournals.org/content/62/supplement_1/1492/tab-article-info].
In pancreatic cancer, FAP-targeted radiotracers have been proposed as a diagnostic and therapeutic tool. For example, [64Cu]FAPI-04 and [225Ac]FAPI-04 have shown utility in theranostics for the treatment of FAP-expressing PAAD (67), whereas application of the albumin binder-conjugated FAPI radiotracers [177Lu]TEFAPI-06 and [177Lu]TEFAPI-07 has been found to promote substantial growth inhibition in patient-derived PAAD xenografts with negligible side effects (68). Other radiotracers, such as [177Lu]FAPI-46 and [225Ac]FAPI-4, have both demonstrated rapid renal clearance, relatively high intra-tumoral accumulation, and tumor-suppressive effects in PAAD xenografts, with a slight reduction in body weight (69). Furthermore, FAP-targeted radioligand therapy with 90Y-FAPI-46 has also been assessed in three patients with advanced PAAD (70). The administration of low radiation doses to at-risk organs suggests the feasibility of repeat cycles of 90Y-FAPI-46 with well-tolerated treatment and a low rate of attributable adverse events. However, although evidence of tumor response was observed following 90Y-FAPI-46 treatment, further studies are warranted to determine efficacy and the toxicity profile in a larger cohort (71).
Given that the FAP is generally expressed at low levels on non-malignant cells, numerous studies have focused on targeting FAP+ CAF to achieve precise imaging of solid tumors and assess potential treatments (72). Since the development of [89Zr]Zr-B12 IgG as a selective imaging probe for FAP-expressing tumors, a series of compounds with the general structure of EB-FAPI-Bn have been synthesized based on FAP inhibitor (FAPI) variants (73), including 99mTc-labeled FAPI tracers for SPECT imaging and 188Re therapy (74). However, the rapid clearance of these molecules and their inadequate tumor retention have hindered them from further clinical translation into cancer therapeutics.
Examination of the complex and highly heterogeneous characteristics of tumors and the tumor microenvironment may lead to the development of novel treatments that can contribute to reducing suffering and enhance the overall prognosis of PAAD patients. Studies have shown that the desmoplastic response in PAAD is not only a “bystander” but also a source of dynamic cellular and non-cytokinetic factors that promote tumor progression, immunosuppression, and metastasis. To date, the findings of numerous studies have confirmed the potential value of FAP expression patterns as prognostic markers and immune checkpoint targets in PAAD. They have also unraveled the role of this enzyme in crosstalk with other cellular components of the tumor microenvironment, as well as non-cellular components, such as cytokines and chemokines, and have thereby provided insights on angiogenesis, matrix remodeling, and immunosuppression.
As an oncogenic subset of CAFs, FAP+ CAFs mediate local immunosuppression and immune escape. FAP+ CAFs have emerged as a therapeutic paradigm for new PAAD drug candidates with the development of novel nanoparticle conjugates and antibody-conjugates, which will contribute to overcoming matrix barriers to drug delivery, thereby facilitating the rapid accumulation of immune checkpoint inhibitors at tumor sites. Moreover, FAP-specific modification of CAR T cells can contribute to the depletion of FAP-expressing stromal cells and inhibit tumor growth by promoting the production of immunostimulatory cytokines, inducing tumor cell lysis, enhancing the anti-tumor response of endogenous CD8+ T cells, and favoring the anti-tumor effects of α-CTLA-4 and α-PD-L1 therapy.
Nevertheless, deciphering the complex interactive processes between tumor and stroma cells in PAAD warrants considerable further study. Although investigations that focus on the target-specific elimination of cancerous cells or pro-tumorigenesis components of the tumor microenvironment will undoubtedly contribute to identifying specific treatment options, future research should not be limited to singular components of PAAD. Studies on multiple compartments of the tumor microenvironment that evaluate combinations of drugs that target both tumors and stroma, as well as the inhibition of cancer-promoting signaling pathways and checkpoints, will aid in developing therapeutic targets and agents that are cytotoxic to cancerous cells and activate anti-tumor immune responses. In PAAD in particular, approaches designed to overcome the current therapeutic dilemma by suppressing desmoplastic responses, overcoming immunosuppression, and inhibiting tumor-promoting signaling pathways, may provide novel therapeutics that will comtribute to enhancing the overall treatment outcomes of PAAD patients.
ZC, C, S-lS, and YS contributed to the conception and design of the review. C-sC and P-wY wrote the first draft of the manuscript. YS and S-lS wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81930115), Shanghai Science and Technology Commission (22YF1425800), and the Natural Science Foundation of Shanghai (21ZR1481800).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
FAP, fibroblast activation protein-α; APCE, antiplasmin-cleaving enzyme; CAFs, cancer-associated fibroblasts; PAAD, pancreatic adenocarcinoma; CPTAC, national cancer institute clinical proteomic tumor analysis consortium; PDAC, pancreatic ductal adenocarcinoma; DPP, dipeptidyl peptidase; ECM, extracellular matrix; BIBH 1, humanized version unconjugated sibrotuzumab; CAR-T, chimeric antigen receptor T; CXCL12, chemokine (C-X-C motif) ligand 12; CXCR4, chemokine (C-X-C motif) receptor 4; MDSCs, myeloid-derived suppressor cells; CCR2, CCL2 receptor; TAM, tumor-associated macrophage; HO-1, heme oxygenase-1; VEGF, vascular endothelial growth factor; TNF, tumor necrosis factor; PSCs, pancreatic stellate cells; TGFβ1, transforming growth factor-β; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; PD-1, programmed cell death protein 1; PD-L1, programmed cell death protein ligand 1; CAT-T cells, chimeric antigen receptor T cells; CAP, cleavable amphiphilic peptide; BMSCs, bone marrow stromal cells; lncRNAs, long non-coding RNAs.
1. Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol (2020) 17(8):487–505. doi: 10.1038/s41575-020-0300-1
2. Rettig WJ, Chesa PG, Beresford HR, Feickert HJ, Jennings MT, Cohen J, et al. Differential expression of cell surface antigens and glial fibrillary acidic protein in human astrocytoma subsets. Cancer Res (1986) 46(12 Pt 1):6406–12.
3. Lee KN, Jackson KW, Christiansen VJ, Lee CS, Chun JG, McKee PA, et al. Antiplasmin-cleaving enzyme is a soluble form of fibroblast activation protein. Blood (2006) 107(4):1397–404. doi: 10.1182/blood-2005-08-3452
4. Shi M, Yu DH, Chen Y, Zhao CY, Zhang J, Liu QH, et al. Expression of fibroblast activation protein in human pancreatic adenocarcinoma and its clinicopathological significance. World J Gastroenterol (2012) 18(8):840–6. doi: 10.3748/wjg.v18.i8.840
5. Cohen SJ, Alpaugh RK, Palazzo I, Meropol NJ, Rogatko A, Xu Z, et al. Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas (2008) 37(2):154–8. doi: 10.1097/MPA.0b013e31816618ce
6. MacNeil T, Vathiotis IA, Shafi S, Aung TN, Zugazagoitia J, Gruver AM, et al. Multiplex quantitative analysis of tumor-infiltrating lymphocytes, cancer-associated fibroblasts, and CD200 in pancreatic cancer. Cancers (Basel) (2021) 13(21):5501. doi: 10.3390/cancers13215501
7. Kawase T, Yasui Y, Nishina S, Hara Y, Yanatori I, Tomiyama Y, et al. Fibroblast activation protein-α-expressing fibroblasts promote the progression of pancreatic ductal adenocarcinoma. BMC Gastroenterol (2015) 15:109. doi: 10.1186/s12876-015-0340-0
8. Busek P, Mateu R, Zubal M, Kotackova L, Sedo A. Targeting fibroblast activation protein in cancer - prospects and caveats. Front Biosci (Landmark Ed) (2018) 23(10):1933–68. doi: 10.2741/4682
9. Witek MA, Aufforth RD, Wang H, Kamande JW, Jackson JM, Pullagurla SR, et al. Discrete microfluidics for the isolation of circulating tumor cell subpopulations targeting fibroblast activation protein alpha and epithelial cell adhesion molecule. NPJ Precis Oncol (2017) 1:24. doi: 10.1038/s41698-017-0028-8
10. Keane FM, Yao TW, Seelk S, Gall MG, Chowdhury S, Poplawski SE, et al. Quantitation of fibroblast activation protein (FAP)-specific protease activity in mouse, baboon and human fluids and organs. FEBS Open Bio (2013) 4:43–54. doi: 10.1016/j.fob.2013.12.001
11. Solano-Iturri JD, Errarte P, Etxezarraga MC, Echevarria E, Angulo J, López JI, et al. Altered tissue and plasma levels of fibroblast activation protein-α (FAP) inxrenal tumours. Cancers (Basel) (2020) 12(11):3393. doi: 10.3390/cancers12113393
12. Wei L, Jin Z, Yang S, Xu Y, Zhu Y, Ji Y. TCGA-assembler 2: software pipeline for retrieval and processing of TCGA/CPTAC data. Bioinformatics (2018) 34(9):1615–7. doi: 10.1093/bioinformatics/btx812
13. Lo A, Li CP, Buza EL, Blomberg R, Govindaraju P, Avery D, et al. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight (2017) 2(19): e92232. doi: 10.1172/jci.insight.92232
14. Zhou YY, Chen LP, Zhang Y, Hu SK, Dong ZJ, Wu M, et al. Integrated transcriptomic analysis reveals hub genes involved in diagnosis and prognosis of pancreatic cancer. Mol Med (2019) 25(1):47. doi: 10.1186/s10020-019-0113-2
15. Wang W, Xing H, Huang C, Pan H, Li D. Identification of pancreatic cancer type related factors by weighted gene Co-expression network analysis. Med Oncol (2020) 37(4):33. doi: 10.1007/s12032-020-1339-0
16. Lee HO, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E, Cheng JD. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer (2011) 11:245. doi: 10.1186/1471-2407-11-245
17. Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med (2013) 210(6):1137–51. doi: 10.1084/jem.20122344
18. Niedermeyer J, Kriz M, Hilberg F, Garin-Chesa P, Bamberger U, Lenter MC, et al. Targeted disruption of mouse fibroblast activation protein. Mol Cell Biol (2000) 20(3):1089–94. doi: 10.1128/MCB.20.3.1089-1094.2000
19. Santos AM, Jung J, Aziz N, Kissil JL, Puré E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest (2009) 119(12):3613–25. doi: 10.1172/JCI38988
20. Eager RM, Cunningham CC, Senzer N, Richards DA, Raju RN, Jones Bc. Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer. Clin Oncol (R Coll Radiol) (2009) 21(6):464–72. doi: 10.1016/j.clon.2009.04.007
21. Eager RM, Cunningham CC, Senzer NN, Stephenson Jr Anthony J SP, O'Day SJ, et al. Phase II assessment of talabostat and cisplatin in second-line stage IV melanoma. BMC Cancer (2009) 9:263. doi: 10.1186/1471-2407-9-263
22. Ungefroren H, Kruse ML, Trauzold A, Roeschmann S, Roeder C, Arlt A, et al. FAP-1 in pancreatic cancer cells: functional and mechanistic studies on its inhibitory role in CD95-mediated apoptosis. J Cell Sci (2001) 114(Pt 15):2735–46. doi: 10.1242/jcs.114.15.2735
23. Blomberg R, Beiting DP, Wabitsch M, Puré Ec, et al. Fibroblast activation protein restrains adipogenic differentiation and regulates matrix-mediated mTOR signaling. Matrix Biol (2019) 83:60–76. doi: 10.1016/j.matbio.2019.07.007
24. Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jäger D, Renner C, et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie (2003) 26(1):44–8. doi: 10.1159/000069863
25. Welt S, Divgi CR, Scott AM, Garin-Chesa P, Finn RD, Graham M, et al. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J Clin Oncol (1994) 12(6):1193–203. doi: 10.1200/JCO.1994.12.6.1193
26. Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res (2003) 9(5):1639–47.
27. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science (2010) 330(6005):827–30. doi: 10.1126/science.1195300
28. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
29. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell (2005) 7(5):469–83. doi: 10.1016/j.ccr.2005.04.023
30. Hussain A, Voisin V, Poon S, Karamboulas C, Bui NHB, Meens J, et al. Distinct fibroblast functional states drive clinical outcomes in ovarian cancer and are regulated by TCF21. J Exp Med (2020) 217(8):e20191094. doi: 10.1084/jem.20191094
31. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res (2016) 76(14):4124–35. doi: 10.1158/0008-5472.CAN-15-2973
32. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U.S.A. (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
33. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
34. Muliaditan T, Caron J, Okesola M, Opzoomer JW, Kosti P, Georgouli M, et al. Macrophages are exploited from an innate wound healing response to facilitate cancer metastasis. Nat Commun (2018) 9(1):2951. doi: 10.1038/s41467-018-05346-7
35. Tang PC, Chung JY, Xue VW, Xiao J, Meng XM, Huang XR, et al. Smad3 promotes cancer-associated fibroblasts generation via macrophage-myofibroblast transition. Adv Sci (Weinh) (2022) 9(1):e2101235. doi: 10.1002/advs.202101235
36. Arnold JN, Magiera L, Kraman M, Fearon DT, et al. Tumoral immune suppression by macrophages expressing fibroblast activation protein-α and heme oxygenase-1. Cancer Immunol Res (2014) 2(2):121–6. doi: 10.1158/2326-6066.CIR-13-0150
37. Mu W, Wang Z, Zöller M. Ping-Pong-Tumor and host in pancreatic cancer progression. Front Oncol (2019) 9:1359. doi: 10.3389/fonc.2019.01359
38. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer (2016) 16(9):582–98. doi: 10.1038/nrc.2016.73
39. Ogawa Y, Masugi Y, Abe T, Yamazaki K, Ueno A, Fujii-Nishimura Y, et al. Three distinct stroma types in human pancreatic cancer identified by image analysis of fibroblast subpopulations and collagen. Clin Cancer Res (2021) 27(1):107–19. doi: 10.1158/1078-0432.CCR-20-2298
40. McAndrews KM, Chen Y, Darpolor JK, Zheng X, Yang S, Carstens JL, et al. Identification of functional heterogeneity of carcinoma-associated fibroblasts with distinct IL6-mediated therapy resistance in pancreatic cancer. Cancer Discovery (2022) 12(6):1580–97. doi: 10.1158/2159-8290.CD-20-1484
41. Patsouras D, Papaxoinis K, Kostakis A, Safioleas MC, Lazaris AC, Nicolopoulou-Stamati P, et al. Fibroblast activation protein and its prognostic significance in correlation with vascular endothelial growth factor in pancreatic adenocarcinoma. Mol Med Rep (2015) 11(6):4585–90. doi: 10.3892/mmr.2015.3259
42. Guan J, Zhang H, Wen Z, Gu Y, Cheng Y, Sun Y, et al. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett (2014) 345(1):132–9. doi: 10.1016/j.canlet.2013.12.006
43. Whittle MC, Hingorani SR. Fibroblasts in pancreatic ductal adenocarcinoma: Biological mechanisms and therapeutic targets. Gastroenterology (2019) 156(7):2085–96. doi: 10.1053/j.gastro.2018.12.044
44. Chaudhry SI, Hooper S, Nye E, Williamson P, Harrington K, Sahai E. Autocrine IL-1β-TRAF6 signalling promotes squamous cell carcinoma invasion through paracrine TNFα signalling to carcinoma-associated fibroblasts. Oncogene (2013) 32(6):747–58. doi: 10.1038/onc.2012.91
45. Lau TS, Chan LK, Wong EC, Hui CW, Sneddon K, Cheung TH, et al. A loop of cancer-stroma-cancer interaction promotes peritoneal metastasis of ovarian cancer via TNFα-TGFα-EGFR. Oncogene (2017) 36(25):3576–87. doi: 10.1038/onc.2016.509
46. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20(3):174–86. doi: 10.1038/s41568-019-0238-1
47. Wen Z, Liu Q, Wu J, Xu B, Wang J, Liang Lc, et al. Fibroblast activation protein α-positive pancreatic stellate cells promote the migration and invasion of pancreatic cancer by CXCL1-mediated akt phosphorylation. Ann Transl Med (2019) 7(20):532. doi: 10.21037/atm.2019.09.164
48. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther (2013) 21(8):1611–20. doi: 10.1038/mt.2013.110
49. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2014) 2(2):154–66. doi: 10.1158/2326-6066.CIR-13-0027
50. Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell (2016) 167(2):419–432.e16. doi: 10.1016/j.cell.2016.09.011
51. Bansal R, Reshef R. Revving the CAR - combination strategies to enhance CAR T cell effectiveness. Blood Rev (2021) 45:100695. doi: 10.1016/j.blre.2020.100695
52. D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis (2018) 9(3):282. doi: 10.1038/s41419-018-0278-6
53. Lo A, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res (2015) 75(14):2800–10. doi: 10.1158/0008-5472.CAN-14-3041
54. Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med (2013) 11:187. doi: 10.1186/1479-5876-11-187
55. Ji T, Zhao Y, Ding Y, Wang J, Zhao R, Lang J, et al. Transformable peptide nanocarriers for expeditious drug release and effective cancer therapy via cancer-associated fibroblast activation. Angew Chem Int Ed Engl (2016) 55(3):1050–5. doi: 10.1002/anie.201506262
56. Sun M, Yao S, Fan L, Fang Z, Miao W, Hu Z, et al. Fibroblast activation protein-α responsive peptide assembling prodrug nanoparticles for remodeling the immunosuppressive microenvironment and boosting cancer immunotherapy. Small (2022) 18(9):e2106296. doi: 10.1002/smll.202106296
57. Powles T, Kockx M, Rodriguez-Vida A, Duran I, Crabb SJ, Van Der Heijden MS, et al. Clinical efficacy and biomarker analysis of neoadjuvant atezolizumab in operable urothelial carcinoma in the ABACUS trial. Nat Med (2019) 25(11):1706–14. doi: 10.1038/s41591-019-0628-7
58. Sum E, Rapp M, Fröbel P, Le Clech M, Dürr H, Giusti AM, et al. Fibroblast activation protein α-targeted CD40 agonism abrogates systemic toxicity and enables administration of high doses to induce effective antitumor immunity. Clin Cancer Res (2021) 27(14):4036–53. doi: 10.1158/1078-0432.CCR-20-4001
59. Ostermann E, Garin-Chesa P, Heider KH, Kalat M, Lamche H, Puri C, et al. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin Cancer Res (2008) 14(14):4584–92. doi: 10.1158/1078-0432.CCR-07-5211
60. Yu Q, Tang X, Zhao W, Qiu Y, He J, Wan D, et al. Mild hyperthermia promotes immune checkpoint blockade-based immunotherapy against metastatic pancreatic cancer using size-adjustable nanoparticles. Acta Biomater (2021) 133:244–56. doi: 10.1016/j.actbio.2021.05.002
61. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med (2013) 210(6):1125–35. doi: 10.1084/jem.20130110
62. Tang PM, Tang PC, Chung JY, Lan HY. TGF-β1 signaling in kidney disease: From smads to long non-coding RNAs. Noncoding RNA Res (2017) 2(1):68–73. doi: 10.1016/j.ncrna.2017.04.001
63. Fang Z, Xu J, Zhang B, Wang W, Liu J, Liang C, et al. The promising role of noncoding RNAs in cancer-associated fibroblasts: an overview of current status and future perspectives. J Hematol Oncol (2020) 13(1):154. doi: 10.1186/s13045-020-00988-x
64. Zhang X, Song W, Qin C, Liu F, Lan X, et al. Non-malignant findings of focal (68)Ga-FAPI-04 uptake in pancreas. Eur J Nucl Med Mol Imaging (2021) 48(8):2635–41. doi: 10.1007/s00259-021-05194-6
65. Dendl K, Finck R, Giesel FL, Kratochwil C, Lindner T, Mier W, et al. FAP imaging in rare cancer entities-first clinical experience in a broad spectrum of malignancies. Eur J Nucl Med Mol Imaging (2022) 49(2):721–31. doi: 10.1007/s00259-021-05488-9
66. Liermann J, Syed M, Ben-Josef E, Schubert K, Schlampp I, Sprengel SD, et al. Impact of FAPI-PET/CT on target volume definition in radiation therapy of locally recurrent pancreatic cancer. Cancers (Basel) (2021) 13(4):796. doi: 10.3390/cancers13040796
67. Watabe T, Liu Y, Kaneda-Nakashima K, Shirakami Y, Lindner T, Ooe K, et al. Theranostics targeting fibroblast activation protein in the tumor stroma: (64)Cu- and (225)Ac-labeled FAPI-04 in pancreatic cancer xenograft mouse models. J Nucl Med (2020) 61(4):563–9. doi: 10.2967/jnumed.119.233122
68. Xu M, Zhang P, Ding J, Chen J, Huo L, Liu Z. Albumin binder-conjugated fibroblast activation protein inhibitor radiopharmaceuticals for cancer therapy. J Nucl Med (2022) 63(6):952–8. doi: 10.2967/jnumed.121.262533
69. Liu Y, Watabe T, Kaneda-Nakashima K, Shirakami Y, Naka S, Ooe K. Fibroblast activation protein targeted therapy using [(177)Lu]FAPI-46 compared with [(225)Ac]FAPI-46 in a pancreatic cancer model. Eur J Nucl Med Mol Imaging (2022) 49(3):871–80. doi: 10.1007/s00259-021-05554-2
70. Ferdinandus J, Costa PF, Kessler L, Weber M, Hirmas N, Kostbade K, et al. Initial clinical experience with (90)Y-FAPI-46 radioligand therapy for advanced-stage solid tumors: A case series of 9 patients. J Nucl Med (2022) 63(5):727–34. doi: 10.2967/jnumed.121.262468
71. Gilardi L, Airò Farulla LS, Demirci E, Clerici I, Omodeo Salè E, Ceci F. Imaging cancer-associated fibroblasts (CAFs) with FAPi PET. Biomedicines (2022) 10(3):523. doi: 10.3390/biomedicines10030523
72. Roy J, Hettiarachchi SU, Kaake M, Mukkamala R, Low PS. Design and validation of fibroblast activation protein alpha targeted imaging and therapeutic agents. Theranostics (2020) 10(13):5778–89. doi: 10.7150/thno.41409
73. Wen X, Xu P, Shi M, Liu J, Zeng X, Zhang Y, et al. Evans Blue-modified radiolabeled fibroblast activation protein inhibitor as long-acting cancer therapeutics. Theranostics (2022) 12(1):422–33. doi: 10.7150/thno.68182
Keywords: pancreatic adenocarcinoma, fibroblast activation protein-α, cancer-associated fibroblasts, FAPI imaging, cancer theranostics
Citation: Cheng C-s, Yang P-w, Sun Y, Song S-l and Chen Z (2022) Fibroblast activation protein-based theranostics in pancreatic cancer. Front. Oncol. 12:969731. doi: 10.3389/fonc.2022.969731
Received: 15 June 2022; Accepted: 29 August 2022;
Published: 03 October 2022.
Edited by:
Patrick Ming-Kuen Tang, The Chinese University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Chiu Tsun Philip Tang, The Chinese University of Hong Kong, Hong Kong SAR, ChinaCopyright © 2022 Cheng, Yang, Sun, Song and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhen Chen, emNoZW56bEBmdWRhbi5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.