 Hui Xu
Hui Xu Fengxia Zhang†
Fengxia Zhang†
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 15 August 2022
Sec. Breast Cancer
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.968306
This article is part of the Research Topic Reviews in Breast Cancer View all 20 articles
Breast cancer has a marked recurrence and metastatic trait and is one of the most prevalent malignancies affecting women’s health worldwide. Tumor initiation and progression begin after the cell goes from a quiescent to an activated state and requires different mechanisms to act in concert to regulate t a specific set of spectral genes for expression. Cancer stem cells (CSCs) have been proven to initiate and drive tumorigenesis due to their capability of self-renew and differentiate. In addition, CSCs are believed to be capable of causing resistance to anti-tumor drugs, recurrence and metastasis. Therefore, exploring the origin, regulatory mechanisms and ultimate fate decision of CSCs in breast cancer outcomes has far-reaching clinical implications for the development of breast cancer stem cell (BCSC)-targeted therapeutic strategies. In this review, we will highlight the contribution of BCSCs to breast cancer and explore the internal and external factors that regulate the fate of BCSCs.
Breast cancer is the second major risk of cancer death in women (1). At present, surgical resection is the preferred treatment for breast cancer, including breast-conserving surgery (BCS) and mastectomy, and a series of comprehensive treatment measures such as chemotherapy, radiao-therapy, hormone therapy and other novel therapies are combined according to clinical-pathology. Despite increasingly accessible systems for early diagnosis and therapy of breast cancer, it remains the most prevalent of female malignancies in terms of mortality. Recurrence and metastasis are the main reason for the increase in mortality (2–4). Most breast cancer patients express receptors for estrogen (ER) and progesterone (PR) and therefore respond to hormone therapy or aromatase inhibitors. However, triple negative breast cancer (TNBC) lacks the expression of ER, PR and human epidermal growth factor receptor-2 (HER-2) (5). Breast cancer contains a heterogeneous cell population and is divided into for major molecular subtypes according to genetic expression, including luminal A, luminal B, HER2-enriched, and triple-negative (6, 7). Certain subtypes are prone to drug resistance, resulting in limited treatment efficacy, which poses a significant challenge to clinical cure and survival of breast cancer patients.
BCSCs are a class of cells with the ability to continuously self-renew, proliferate indefinitely and differentiate in multiple directions, and possess multiple drug-resistant molecules that are the main cause of drug resistance in breast cancer (8–10). There are two hypotheses on the origin of BCSCs: one is that BCSCs originate from adult stem cells and can acquire malignant behaviors by changing their genetic characteristics; the other is that BCSCs are transformed by early progenitor cells that have acquired the ability to self-renew (11, 12). The concept of BCSCs has been further developed to be involved in mediating tumor heterogeneity, with the ability to clonally regenerate tumors after seemingly successful treatment, and is of profound importance in understanding and treating hierarchically organized breast cancer (13). Therefore, further understanding of the fate decisions of BCSCs, identifying significant roles in tumor recurrence, metastasis and drug resistance, and developing therapeutic strategies to target BCSCs are of great clinical significance for the treatment of breast cancer. Hence, we outline the hierarchy of BCSCs in the origin of breast cancer and their role in tumor heterogeneity, recurrence, metastasis and drug resistance, in conjunction with a discussion of the potential of BCSCs as therapeutic targets to provide clinicians with new strategies to improve breast cancer treatment.
A highly dynamic organ that produces and secretes milk to nourish offspring, the mammary gland undergoes multiple phases of remodeling throughout a female’s life and consists of two main parts: the parenchyma and surrounding stroma. The parenchyma contains mainly epithelial cells, glandular cells and myoepithelial cells: the epithelial cells are located in the inner layer of the milk ducts; the glandular cells form the alveoli, whose main function during lactation is to secrete milk; and the myoepithelial cells form the basement membrane, which usually surrounds or separates the epithelial cells from the glandular cells (14, 15). The proliferation and differentiation of the mammary gland is regulated by hormones and growth factors, for example estradiol, progesterone and prolactin. According to the characteristics of mammary gland development, it can be roughly divided into six developmental stages: embryonic stage, birth to early sexual maturity, sexual maturity, pregnancy, lactation and involution, as well as each estrous/menstrual cycle, both local and systemic stimuli can set off the mammary cell expansions and/or differentiation (16).
The mammary gland shows such obvious periodicity because a hierarchical array of mammary stem cells (MaSCs) and progenitor cells (PCs) are located in the organ, which maintain the homeostasis under physiological conditions (17). The differentiation of MaSCs is a two-step journey, consisting of cell lineage determination (from MaSCs to PCs of a specific lineage) and maturation (from PCs to particular cell types). These cells can yield all the mature cell types in the mammary gland, including ductal, alveolar and myoepithelial, and the primary outgrowths contain daughter cells that have the same regenerative capacity as the original stem cells (18). Thus, these cells have the dual hallmarks of stem cells, multidirectional differentiation and the ability to self-renew. Stem cell fate decisions, which begin after the cells differentiate from a quiescent to an activated state, require different mechanisms to coordinate and regulate the expression of a specific set of lineage genes. The presence of stem cells is necessary for the regeneration of the mammary gland and is important for studying the mechanisms of organogenesis and cell differentiation, but abnormal differentiation and proliferation of stem cells can lead to occurrence of tumors.
Unmasking the origins of breast cancer to be still a challenging and creative topic in the field of oncology research. The cellular origin of cancer continues to be an important scientific question. Two major models have been developed to describe the cellular origin of cancer. In the somatic mutation model, the stepwise accumulation of a series of independent mutations in differentiated cells promote the capability to gradually reprogram and obtain malignant genotypes (19, 20), while the second hypothesis involves mutations in stem or progenitor cells (21). It is inevitable that these two models will co-exist. About 5-10% of breast cancers are inherited susceptibility due to germline mutations, such as BRCA1 and BRCA2 (22–24). Using single-cell assays, in BRCA1 mutation breast cancer, basal-like breast cancer (ER-) and luminal breast cancer (ERhigh) respectively derive from luminal progenitors and mature luminal cells respectively (25). These discoveries indicate that breast cancers may be initiated by mutations in differentiated cells. Interestingly, it is evident that cancer cells and stem cells share many characteristics, including high proliferative capacity, longevity, pliancy and the activity of molecular pathways that regulate stem cells (26). Corinne A Boulanger1 and Gilbert H Smith’s inventive research in breast cancer was the first to demonstrate that the mammary epithelial stem cells were indeed responsible for the evolution of carcinogenesis in mature mammary gland and formed tumor stem cell populations (27). Increased expression of stemness-associated genes, such as pseudokinase Tribble 3 (TRIB3) (28), NOTCH1 (29) and SOX9 (30), is positively correlated with the development of breast cancer. Patients with TNBC tend to have a comparatively worse outcome than other subtypes, due to their inherently invasive trait and the lack of molecular targets for treatment (31). TNBC is often, but not always, a basal-like subtype and expresses basal like markers (K5, K14, ITGA6, P-cadherin and Id4) with the character of stemness (32, 33). Definitely, high expression of stem cell-related gene traits in the BCSC subpopulation were a potential predictor of worse prognosis (34, 35). Interestingly, it is hypothesized that the apparent heterogeneity within breast cancer tumors reflects the different mammary epithelial cells as the cellular origin and drivers of malignant transformation (36, 37). A comparison between specific molecular features of normal breast epithelial subpopulations and different breast cancer subtypes revealed that the tumor subtypes appeared to have similar differentiation characteristics to normal breast cells. The basal-like subtype expresses intracavitary progenitor cell markers. This appears to correlate with the basal MaSC molecular subtype and therefore intuitively indicates that MaSC are a potential cellular source of basal-like breast cancer. Correspondingly, the HER2, luminal A, and luminal B subtypes express luminal lineage markers (18) (Figure 1). Collectively, these discoveries provide several insights into the origin of breast cancer cells: 1. Is oncogene-induced transdifferentiation of mammary gland cell sufficient to explain the plasticity and heterogeneity observed in breast cancer? 2. Does the continuous differentiation of normal stem cells to replenish the pool of progenitor cells during the maintenance of mammary gland homeostasis contribute to tumorigenesis?
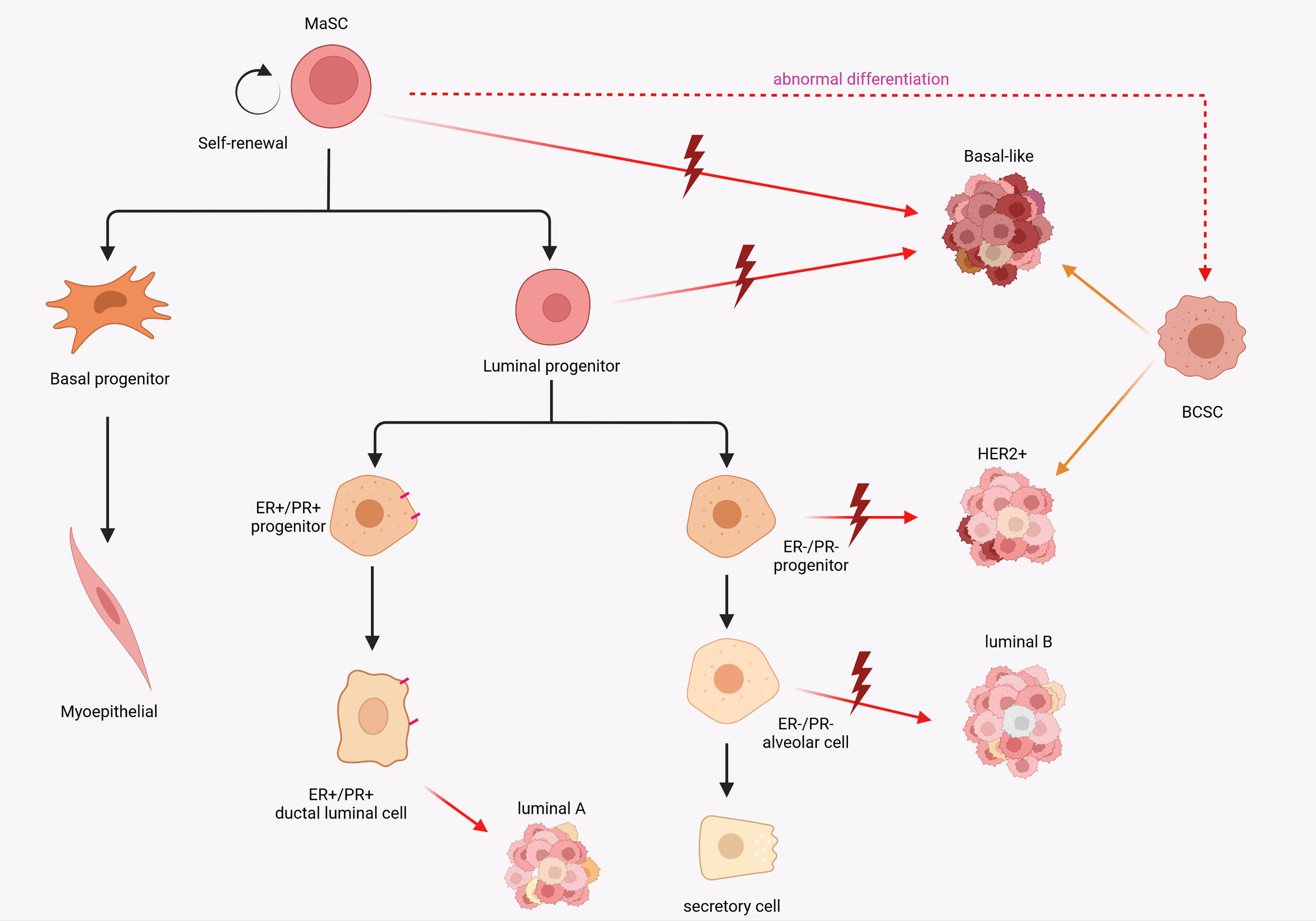
Figure 1 Schematic diagram of the potential relevance of breast epithelial cell hierarchy and breast cancer stem cell origin to breast cancer subtypes. MaSCs expose to mutations that cause abnormal differentiation and transformation into cancer stem cells. A comparison between specific molecular features of normal breast epithelial subpopulations and different breast cancer subtypes revealed that the tumor subtypes appeared to have similar differentiation characteristics to normal breast cells.
During the last few decades, numerous studies have demonstrated that both tissue stem cells and CSCs can survive for long periods of time and have a great proliferative capacity, which means not only that they can accumulate many mutations, but also that they share the same capacity for reversibly entering a quiescent cell-cell state, multidirectional differentiation, an overlapping immunophenotype and gene regulatory networks (26). Through xenotransplant experiments, Al-Hajj together with colleagues presented directly the first investigated evidence for the presence of so-called BCSCs (CD44+CD24-/lowLineage-), which are located at the top of breast cancer with a hierarchical structure (38). Subsequently, Christophe Ginestier and colleagues raveled out that aldehyde dehydrogenase (ALDH) can act as a potential marker for BCSCs, these cells with the widest lineage differentiation potential and the greatest capacity for growth (39). Surprisingly, BCSCs (CD44+CD24-) are predominantly quiescent and localized at the front of the tumor invasion, whereas epithelial-like BCSCs expressing ALDH are more proliferative and more centrally located (40). High-throughput sequencing technologies developed in recent decades have enabled us to accumulate a wealth of relevant data on BCSC hierarchies and fate decisions. At the single-cell level, BCSCs showed high tumorigenesis and expressed stemness and EMT-related genes, in particular ZEB2, SOX2, ID1 and TWIST1 (41). Along with the ground, acquiring the properties of EMT allows the cells to be reprogrammed to a more stem-like state (42). Importantly, BCSCs, located at the top of the cancer hierarchy, are considered to be in line with their healthy MaSCs, and they can exacerbate breast cancer. More studies have confirmed that BCSCs expressing relevant cell surface markers have biological properties similar to MaSCs, including ALDH1+ (39), CK5+ (43), CD49f+ (44), ITGA6+ (45). These discoveries provide insightful evidence not only of the clinical relevance of BCSCs in breast cancer, but also indicate that breast cancer should be uniquely therapeutic according to their gene profile (Figure 2).
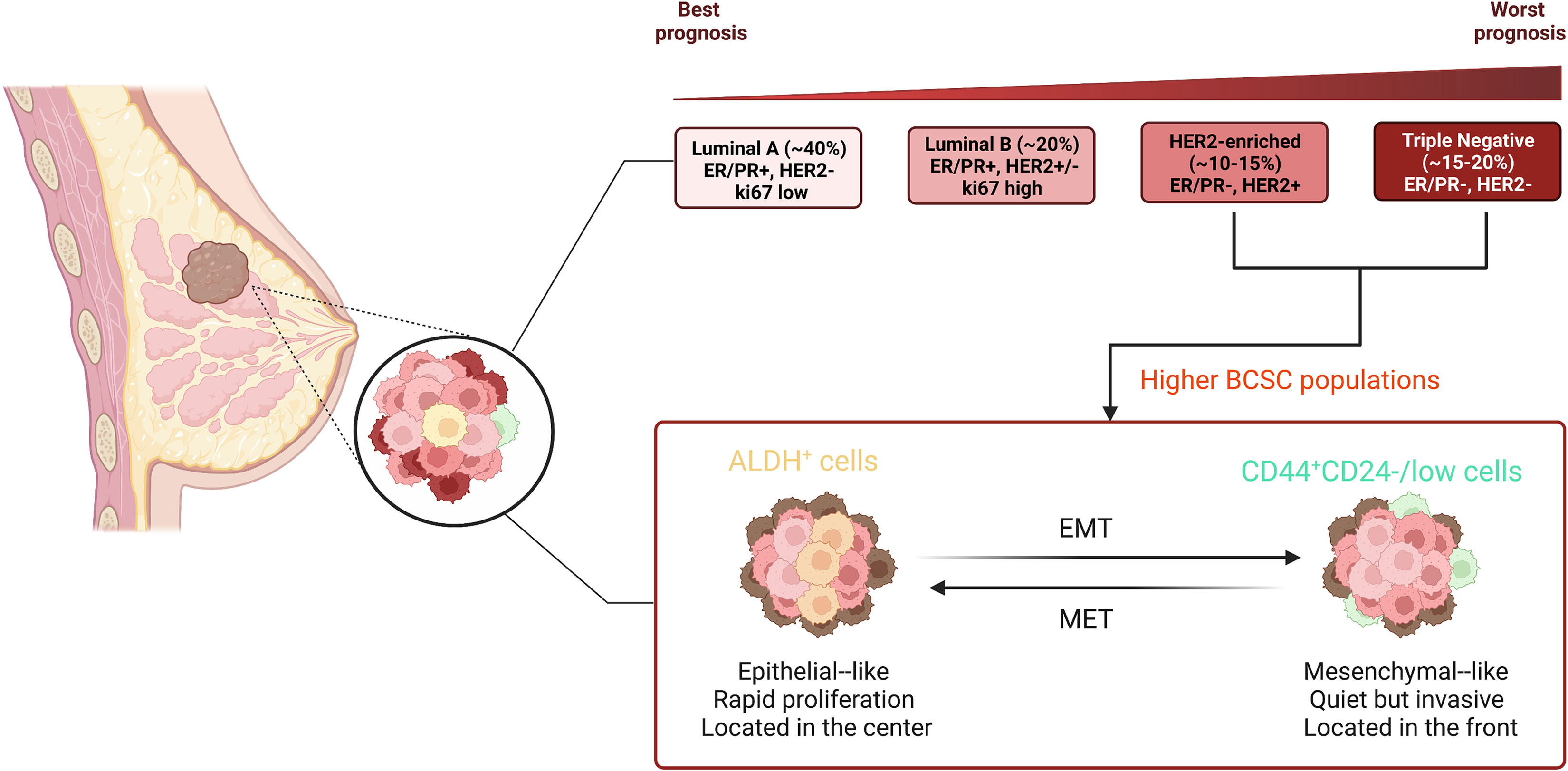
Figure 2 Different subtypes of breast cancer and distinct state of BCSCs. Breast cancer contains a heterogeneous cell population and is divided into four major molecular subtypes according to genetic expression, including luminal A, luminal B, HER2-enriched, and triple-negative. The typical molecular expression in each subtype is shown in the figure. There are two major distinct phenotypes of BCSCs: CD44+CD24-/low and ALDH+. BCSCs (CD44+CD24-) are mesenchymal-like and predominantly quiescent and localized at the front of the tumor invasion, whereas epithelial-like BCSCs expressing ALDH are more proliferative and more centrally located.
Noticeably, not all cancers are considered to be stringently hierarchical in organization. It is generally believed that the CSC lies at the top of the hierarchy, whereas in reality the CSC hypothesis is more complex than a simple linear model. Several discoveries have shown that some non-CSCs can lead to dedifferentiation through genetic mutation, and exhibit plasticity by reversibly transitioning between a stem and non-stem state (46). Importantly, the hierarchical heterogeneity is beyond genetic mutations and covers non-genetic characteristics with regard to epigenetic programs, immune characteristics, inflammatory states and microenvironmental composition. Lineage plasticity is important for the development of aggressive BLBC, transcription factor SOX9 can regulate cell phenotypic plasticity and breast cancer progression (30). Similarly, EMT-mediated phenotypic plasticity has important clinical implications for breast cancer progression and drug resistance (47). Similar to normal breast cells, BCSCs are able to respond to external or internal stimuli, debugging their phenotype and behavior via EMT, reversible quiescence and senescence or metabolic plasticity to counteract the stress of treatment. All of these characteristics contribute to the resistance to treatment of CSCs. For example, BCSCs have built-in mechanisms to promote phospholipid metabolism and the generation of free fatty acids, which activate the relevant signals and maintain stemness, thus contributing to chemo-resistance as cisplatin, doxorubicin, or tamoxifen. In this case, phospholipase A2 inhibitors, such as Giripladib, are required in combination to effectively eliminate BCSCs and inhibit tumorigenesis (48). Alexander Swarbrick and his colleagues demonstrated that stromal cues form CAFs, including FGF5 and fibrillar collagen, are capable of inducing and maintain a stem-like phenotype in TNBC cells by providing a supportive niche (49). Corporately, these evidences suggest that BCSCs are not in a specific-widespread phenotype but stay at a certain plasticity. In conclusion, the model of tumor origin and evolution is not limited to a single hierarchical level, but also needs to take fully into account the polyclonal heterogeneity that characterizes the successive interactions between different cell populations. Certainly, they are not mutually exclusive and there may be transitions between BCSC and BCSC-like states, and the concept of BCSC populations needs to be treated more dialectically, that is, there may be multiple BCSC populations of different subtypes.
Breast cancers show marked heterogeneity due to genomic, transcriptomic and microenvironmental differences, resulting in different phenotypes and variability in biological behavior (50). Due to inter-tumor heterogeneity, they can be classified into different types based on their morphology, molecular expression or genomic copy number patterns. There are currently two models that address the issue of heterogeneous origins: the clonal evolution model and the CSC model (37). These two hypotheses are not independent, but rather a coexisting, dynamic process that provides a theoretical basis for explaining inter-tumor heterogeneity and intrinsic differences in the regenerative capacity of breast cancer (51). The clonal evolutionary model assumes that any undifferentiated and differentiated cell is capable of accumulating mutations that lead to the creation of clonal populations of cells within a tumor, while individual tumor cells in a monoclonal clone share a degree of identical genetic variation, and different subpopulations of tumor cells have the ability to mutate individually during tumor evolution, thereby mediating the creation of tumor heterogeneity. The CSC model holds that a tumor actually consists of a cluster of stem cells, as well as cells that are unevenly differentiated, and can explain breast cancer tumorigenesis (52, 53). Typically, CSCs in tumors are genetically unstable, with multiple isoforms, and CSCs that survive adaptively in a clonal pool following altered microenvironmental niches and targeted therapeutic approaches, mediating the intar- and/or inter-tumor hierarchy and promoting malignant progression. In conclusion, further refinement and emphasis on the evolutionary and adaptive CSC dynamic concept is complementary to explain the possible causes of tumorigenesis, recurrence and metastasis. Therefore, eliminating the most diverse types of tumor cells, including BCSCs, is the most fundamental strategy for curing breast cancer.
The development of BCSC-specific biomarkers for breast cancer has expanded the understanding of heterogeneity and has been further validated in both in vivo and in vitro breast cancer models. These breast cancer stem cells represent only a small fraction of the cells within the tumor and are extracted by flow cytometry technique capable of identifying certain patterns of surface markers (54, 55). A growing number of studies have revealed and characterized BCSC markers, and these markers have been shown to identify different stem cell populations well. As mentioned above, CD44+CD24- and ALDH+ are common molecular markers for BCSCs. Equally important, due to the highly heterogeneous character of breast cancer, in which more different phenotypes of BCSCs may exist, the discovery and identification of their biological functions could achieve a substantially more constructive reaction to anti-cancer therapy in the design of new drugs targeting BCSCs (Table 1).
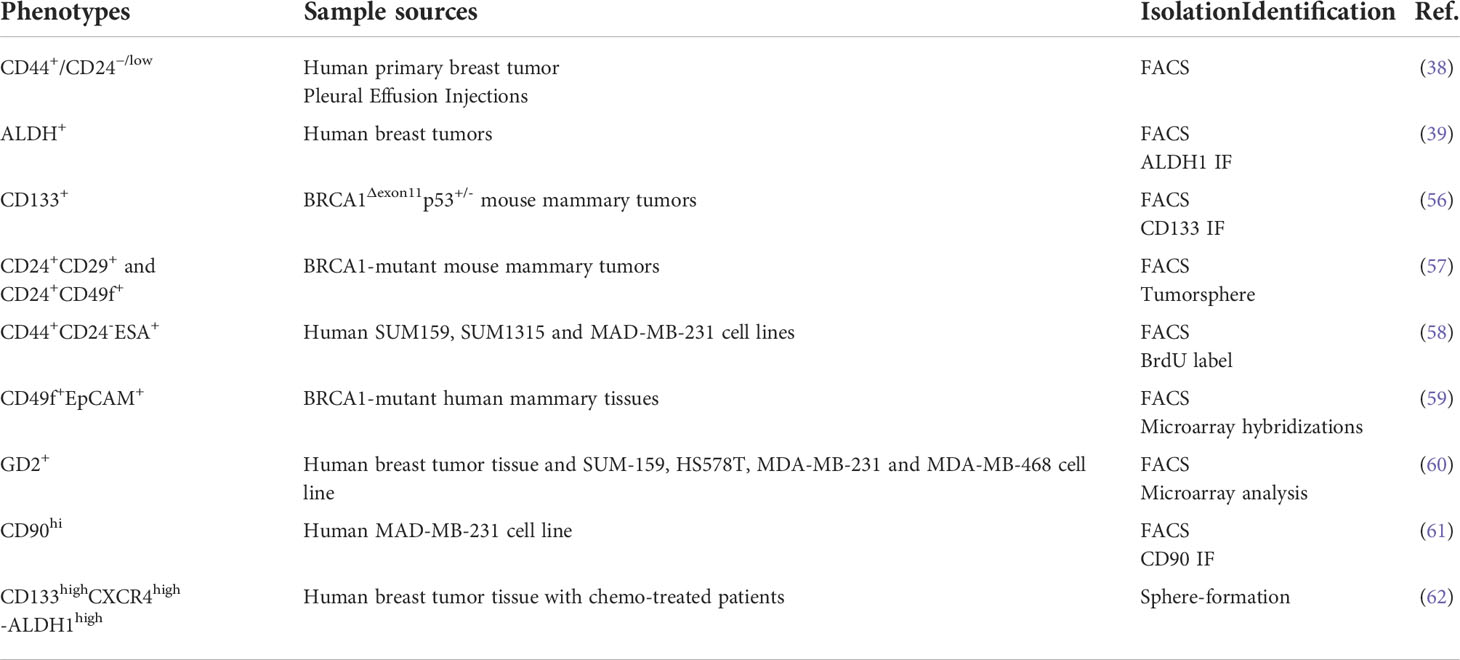
Table 1 Principal BCSC identity markers.
The establishment of the BCSC theory provides the theoretical basis for explaining the hierarchy and heterogeneity of breast cancer. These fickle BCSC populations initiate and fuel tumor growth and are intimately associated with intrinsic treatment-resistant. BCSCs possess significant stemness and plasticity, and their fate decisions that extensive and complex regulatory mechanisms are required to coordinate and regulate the expression of specific lineages of genes, starting after the cell differentiates from a quiescent to an activated state. Here we focus on the contribution of transcriptional regulation, signaling pathway, epigenetic regulation, and post-transcriptional modifications that occur during this process.
Transcription factors (TFs), also known as trans-acting factors, are functional protein molecules that specifically bind to DNA and regulate gene transcription. Most TFs bind to DNA before forming dimers or multimers through protein-protein interactions. In addition to TFs that bind DNA directly, there are regulatory proteins that do not bind DNA directly, but rather bind DNA indirectly through protein-protein interactions, regulating gene transcription and thus forming expression regulatory complexes. The gene expression that defines the phenotype is highly coordinated. As a result, regulatory programs meticulously curated by crucial TFs have been posited to have a central function in the determination of cell fate.
Evidently, intratumoral hypoxia is a common manifestation in advanced cancers. In hypoxic breast cancer cells, HIFs activate the transcription of target genes that play important roles in tumor progression, metabolic reprogramming, motility and chemoresistance (63). Numerous studies have shown that the response of BCSCs to hypoxia requires HIFs to regulate and maintain the direct or indirect transcriptional regulation of BCSC stemness-related factors including NANOG, SOX2, and KLF4 (64, 65). In addition, HIF-1α maintains the onset of hypoxia-induced EMT and regulates the plasticity of BCSC (66, 67). Recently, researches showed that HIF-dependent ALKBH5 and S100A10 expression mediates the enrichment of BCSCs in the hypoxic tumor microenvironment (68, 69). Similarly, HIF-1 can directly activate calreticulin (CALR) transcription and facilitate breast cancer progression by promoting the BCSC phenotype in hypoxic (70). Collectively, these discoveries exhibit that hypoxia increases the percentage of BCSCs and governs their phenotypic transformation in a HIF-dependent manner.
Metastasis is the cause of up to 90% of cancer-related deaths, yet it continues to be the least known integral part of cancer pathogenesis. The most common sites of metastasis from breast cancer are bone, lung, brain and liver. Truncated glioma-associated oncogene homolog 1 (TGLI1) was found to transcriptionally activate the expression of CD44 and OCT4, contributing to BCSC renewal and thus promoting brain metastasis (71). Mechanistically, malignant progression in breast cancer is accompanied by an increase in the proportion of these BCSCs within the tumor and activation of the EMT (72, 73). EMT is a complex transdifferentiation program characterized by the loss of epithelial-specific features accompanied by the acquisition of mesenchymal phenotypes that fuels non-transformed cells and tumor cells to acquire stemness (74, 75). The loss of epithelial-specific features means that the tumor is more aggressive and has a poorer prognosis. Intrinsically, EMT-associated TFs (EMT-TFs) were crucial regulatory mechanism for tumor progression and metastasis including, Snail 2, Twist 1, Slug, SOX2/9 and Zeb1/2.
In contrast to well-known molecular signaling pathways, the involvement of non-coding RNAs (ncRNAs) in CSC lineage commitment has only just been discovered. Based on their biological functions, ncRNAs are divided into two major categories: housekeeping ncRNAs and regulatory ncRNAs. Regulatory ncRNAs can be divided into short chain ncRNAs and long chain ncRNAs according to the sequence length. ncRNAs with short chains include microRNA (miRNA), small interfering RNA (siRNA), piRNA and transcription initiation RNA (tiRNA) have the characteristics of small molecule and high sequence conservation. Whole-genome sequencing revealed that ncRNAs comprise 98% of the human gene transcriptome and consist mainly of miRNAs and LncRNAs that do not have protein-coding functions (76). A variety of miRNAs and LncRNAs are responsible for the modulation of BCSCs.
miRNAs are commonly expressed in organisms that are approximately 18-25 nucleotides in length and can complement the 3’-UTR of mRNA, leading to the degradation and/or translational repression of target genes (77). miRNAs act as regulators in stem cell proliferation, differentiation, apoptosis, and metabolism (78, 79). In this way, miRNAs act as a switch of gene networks, either as an oncogene or as a tumor suppressor gene, and these miRNAs have quickly become an important class of regulatory genes controlling developmental and disease processes. In contrast to transcription factors and molecular signaling pathways, miRNAs involved in stem cell lineage determination have only just started to be studied. An increasing number of miRNAs have been found to be implicated in BCSCs to regulate fate decisions.
Interestingly, miRNAs with micro size but macro function are known to have profound effects on maintaining and regulating the behavior of BCSCs by specifically targeting relevant TFs and oncogenic signaling pathways and play an important role in breast cancer initiation and prognosis. miRNAs serve as oncogenes as well as tumor suppressors. Based on the current findings, we will focus on describing the regulatory role and underlying mechanisms of miRNAs management of BSCS self-renewal, differentiation, metastasis, EMT, drug resistance and recurrence as potential links to breast cancer pathogenesis. Analysis of 11 surgically resected breast cancer patient samples revealed differentially expressed miRNAs in human BCSCs versus nontumorigenic cells (NTG cells) (80). Three clusters of miRNAs, including miRNA-200c-141, miR-200b-200a-429 and miR-183-96-182 cluster, were consistently downregulated in human BCSCs (80). The miR‐200 family maintains the stemness of BCSCs and is able to target the EMT-associated transcription factor ZEB1, thereby up-regulating E-cadherin, the expression of which is reduced and its inhibitory effect on EMT is diminished (80–83). Furthermore, other miRNAs, including let-7, miR-27b and miR-185-3p, were differentially expressed in BCSCs and NTG cells (84–86). It was recently shown that in BCSCs E2F1 binds to the Nanog gene to promote its transcription and that miR-185-3p can target E2F1 leading to a reduction in its expression, thereby inhibiting the stemness of BCSCs (86). Similarly, miR-378a-3p and miR-378d can activate the WNT and NOTCH pathways through targeted inhibition of DKK3 and NUMB, leading to doxorubicin (DOX) and paclitaxel (PTX) resistance (87). Taken together, these discoveries show that miRNAs are instrumental in determining the fate of BCSCs by targeting key coding TFs and related signaling pathways.
LncRNAs are ncRNAs with transcripts longer than 200 nucleotides and little or no protein-coding function. They regulate gene expression and are involved in biological processes such as apoptosis, metastasis, stemness maintenance, proliferation, differentiation, metabolism and drug resistance. LncRNAs can repress or activate gene expression through a variety of mechanisms and exhibit specific expression patterns in different cell and tissue types, respond to different stimuli, and regulate cell fate (88). In the last decade, researchers have shown great interest in the role of LncRNAs in CSC lineage Commitment and differentiation.
LncRNAs influence cell growth, apoptosis and tumor metastasis by participating in epigenetic, transcriptional or post-transcriptional gene regulation. Brown and colleagues summarized the LncRNAs in BCSCs and revealed that a series of BCSC-associated LncRNAs were enriched in TNBC (89). Notably, LncRNAs show differential expression in BCSCs versus non-BCSCs. LncRNA lnc030, which is highly expressed in BCSCs, is able to stabilize squalene epoxidase (SQLE) mRNA cooperating with poly(rC) binding protein 2 (PCBP2) and promote cholesterol synthesis, thereby activating PI3K/Akt to amplify the stemness properties of BCSCs (90). Likewise, high expression of LncRNA-ROPM can increase the stability of PLA2G16 mRNA, thereby promoting phospholipid metabolism and activating PI3K/AKT signaling (48). In addition, other LncRNAs that are upregulated in BCSCs, such as LncRNA-ROR, LncRNA-HOTAIR, LncRNA-HAL, LncRNA-Hh (91) and LncRNA-PVT1 (92), are able to induce EMT, consequently increasing the percentage of BCSC population and stemness. As LncRNAs research progresses, more and more LncRNAs will be demonstrated in the regulation of BCSCs. LncRNAs is a novel regulator of BCSCs by regulating mRNAs, miRNAs and other LncRNAs and will improve the understanding of new molecular regulation of BCSCs.
The tumor microenvironment (TME) plays a pivotal function in several steps of tumorigenesis and progression, including drug resistance, immune escape and distant metastasis. The microenvironment regulates the biological behavior of BCSCs through direct contact or ECM and paracrine factors (93). The microenvironment provides fuel and a proper niche for BCSCs, highly regulates their fate, protects them from genotoxicity and improves their tolerance to treatment. Reciprocally, BCSCs are able to influence the TME while adapting to changes in the TME. The TME mainly consists of surrounding normal tissue cells, tumor stroma and microvessels. For example, tumor cells can release immune inhibitory cytokines to evade detection by immune cells in TEM, resulting in immune escape (94). Concurrently, the TME provides the driving force for BCSC plasticity, inducing angiogenesis and recruitment of immune and stromal cells, which in turn accelerates tumor invasion and metastasis.
Stromal cells, such as cancer-associated fibroblasts (CAFs), are verified that affect BCSC activity through the cell-cell interactions, the secretion growth factors, cytokines, chemokines, and the remodeling of the ECM (95). These secreted factors are involved in a variety of regulatory roles for cells in TME and tumor cells. In particular, CAFs, a major component of the stroma, have been shown to support CSC function by secreting cytokines such as IL-6, IL-8 and IL-1β, activating signaling pathways, and promoting BCSC stemness and plasticity (96). The origin of CAFs is now thought to be multiple, including transference of resident fibroblasts (97), transdifferentiation of perivascular cells (97), differentiation of mesenchymal stem cells (MSCs) and EMT. CAFs haven been found to be able to secrete periostin, which in turn recruits Wnt ligands, activates intracellular Wnt signaling in BCSCs, remodels the ECM, establishes a nascent stromal niche and creates the conditions for metastatic colonization of BCSCs (98). Similarly, CAFs also secrete FGF5, which promotes fibronectin collagen formation and remodels the ECM, resulting in the induction of a reversible BCSC phenotype preferentially at the tumor-stromal interface (49).
In addition, CAFs are involved in regulating the biological behavior of BCSCs through their association with other signaling pathways. Activation of WNT/β-catenin and HGF/Met signaling in the mammary gland tumors accelerates the secretion of the Hedgehog ligand SHH in BCSCs, which regulates CAFs via a paracrine pathway, and in turn CAFs further secrete factors (99). Accordingly, the Hedgehog inhibitor vismodegib was able to reduce the activity of fibroblasts and breast cancer-forming cells, mechanistically indicating that Hedgehog signaling to CAFs is a potential mediator of CSC plasticity and an intriguing new therapeutic target in breast cancer (49). MSCs and CAFs express high levels of PEAK1 protein in a PEAK1-dependent manner, which activates p-AKT, enhancing tumorigenesis (100). In addition, when MSCs were co-cultured with breast cancer cells, they were able to induce aberrant expression of microRNAs, such as mir-199a upregulation, providing breast cancer cells with enhanced BCSC properties (101). Collectively, these findings identify a potential mechanism of crosstalk between stromal cells and BCSCs and aberrant signaling pathway perturbations, and therefore the development of targeted inhibitors may offer a novel therapy strategy for the management of breast cancer.
Macrophages are a group of plastic and heterogeneous cells that are involved in the innate immune response as another major component of the TME and are capable of regulating the formation and maintenance of BCSCs through the modulating the M1/M2 phenotype. It has been shown that tumor-associated macrophages (TAMs) can activate Src and NF-κB via EphA4, which in turn induce the secretion of a variety of cytokines such as IL-6, IL-8 and GM-CSF, thereby establishing a BCSC niche (102). Consistently, in breast cancer, the reduction of macrophages reduced the number of BCSC population (103). In addition, TME-derived endothelial cells provided Jag1 to neighboring BCSCs, increasing the upregulation of zeb1, which in turn increased VEGFA production by ectopic zeb1, inducing endothelial cells to express jag1 in a paracrine manner (29). Similarly, the cell-cell interaction of BCSCs with CD8+ T lymphocytes in TME can establish immune tolerance, mainly due to the ability of BCSCs highly expressing PD-L1 to bind to the PD-1 receptor on the surface of T cells, which in turn exerts an inhibitory effect and leads to T cell exhaustion (104). In addition, ECM, a major component of TME, is a niche that determines the behavior of BCSCs, such as hydroxylated collagen, hyaluronic acid, integrates the intra-/extra- cellular environment signals and activates multiple signaling pathways leading to BCSC metastatic growth (105, 106).
In a nutshell, the TME provides a niche for BCSCs and governs their biological behavior. Importantly, the TME varies markedly between patients, so an exhaustive understanding of the interactivity of the components of the TME on tumor progression is paramount. It has been revealed that TME is potentially of a complex character (107). In parallel, the heterogeneity of TME has been shown to be a potential prognostic factor in identifying different subtypes of breast cancer (108). Building on this, further refinement of breast cancer types and understanding of the specificity of BCSCs offers the potential to accurately predict tumor prognosis and develop new personalized treatment strategies (Figure 3).
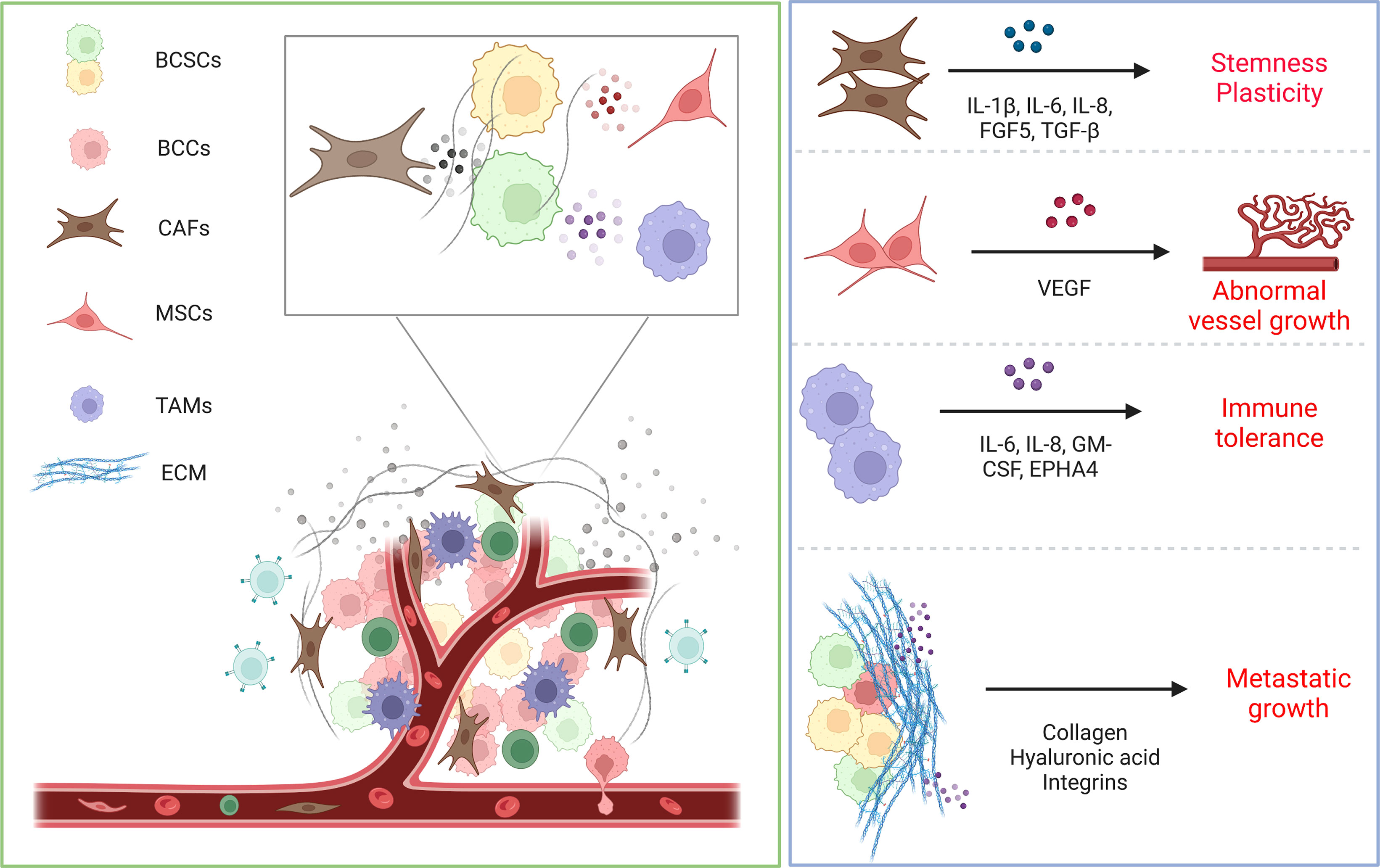
Figure 3 Schematic representation of interactions between TME and BCSCs. The microenvironment regulates the biological behavior of BCSCs through direct contact or ECM and paracrine factors. CAFs secrete cytokines such as IL-6, IL-8 and IL-1β to promote BCSC stemness and plasticity. MSCs secrete VEGF to feed BCSCs, leading to abnormal vessel growth. Macrophages likewise secrete various cytokines that establish the BCSC niche and lead to immune tolerance. ECM offers protection to BCSCs from treatment pressure and safeguards their metastatic growth.
BCSCs are usually quiescent and are able to transforming their phenotype through EMT, metabolic plasticity, and microenvironment, resulting in limited specific markers. Therefore, more researches have focused on defining the mechanisms of relevant signaling pathways regulating the tumor initiating ability of CSCs. Exhilaratingly, insightful investigations have verified that many key signaling pathways are implicated in modulating the lineage commitment and biological processes of BCSCs.
The Wnt family consists of a large number of secreted glycoproteins with both paracrine and autocrine functions (109). Wnt is participating in many important biological processes (109–111). Wnt ligands bind to the seven transmembrane structural domains of the frizzled receptor, FZD) and LRP5/6 co-receptors and stabilizes β-catenin by preventing its phosphorylation (112). The Wnt pathway plays a key role in BCSC fate.
Abnormal WNT/β-catenin signals are more prevalent in breast cancer, and clinical evidence indicates that increased WNT/β-catenin signals are correlated with higher tumor grade and poorer prognosis (98, 113). In BCSCs, WNT/β-catenin is relevant to stemness, plasticity and microsphere formation (114). Canonical and noncanonical Wnt signaling pathways by promoting the expression CD44 and ALDH1, which in turn increase the stemness of BCSCs. Canonical Wnt signaling through β-catenin stabilization and subsequent nuclear translocation leads to transcriptional activation of β-catenin-TCF/LEF target genes. Inhibition of β-catenin reduces BCSC population, tumor size and resistance to doxorubicin (Dox) in TNBC cells (115). Non-canonical WNT/Ca2+ signaling regulates the biological behavior of BCSCs through the activation of RTKs such as ROR1/2 and PI3K/AKT. Just as, Wnt plays an important role in BCSC, so targeting the canonical and/or noncanonical Wnt signaling pathway may be an effective marker for eliminating BCSCs. Recent studies showed that DKK1 inhibited lung metastasis by inhibiting PTGS2-induced macrophage and neutrophil recruitment and thereby antagonizing non-classical WNT/PCP-RAC1-JNK signaling. Conversely, DKK1 promotes bone metastasis by regulating canonical Wnt signaling of osteoblasts (114). These results reveal that amplified Wnt signaling is instrumental in the self-renewal, apoptosis inhibition and metastasis of BCSCs, and therefore inhibition of wnt is essential for the elimination of BCSCs (Table 2). A growing number of preclinical researches are treating breast cancer by targeting inhibition of Wnt signaling in BCSCs including OMP-18R5 (Vantictumab) (123), NSC668036 (124) and Pyrvinium pamoate (PP) (122).

Table 2 Antagonist of WNT signaling and their effects on BCSCs.
The Notch signaling pathway enables ligand-receptor interactions through direct cell-to-cell contact. In mammals, the Notch signaling pathway involves the Notch receptor (Notch1-4) and Notch ligand, which is divided into two classes (Jagged1-2 and Delta-like 1,3,4) that differ due to the presence of a cysteine-rich structural domain in the Jagged ligand (125). Notch receptors are activated by ligands on their neighboring cells, which trigger signals regulating various cellular differentiation processes.
Notch signaling plays a variety of roles in cancer, including oncogenesis, carcinogenesis or both. Concurrently, notch pathway is associated with many aspects of cancer biology, including metabolism, metastasis, drug resistance and the maintenance of CSCs. Multiple discoveries have confirmed that Notch signaling is associated with CSC activity in various forms of breast cancer. A meta-analysis of tumor molecular landscapes and several pathological studies have shown that Notch1 activity is associated with the risk of recurrence in ER+ breast cancer (126). Endocrine resistant BCSCs, most of which are Notch4-dependent, are a major factor in tumor recurrence and death (127). Interestingly, unlike Notch4, which is predominantly located in the basal cell population, Notch1 is predominantly expressed in the luminal cells of normal breast epithelium, indirectly suggesting that both may play this specific role in different subpopulations of BCSCs (127, 128). In patients with trastuzumab-resistant and HER2+ breast cancer, Notch1 expression was associated with poorer prognosis (129). Under the circumstances, abrogation of Notch1 expression resulted in a significant reduction of cancer proliferation in vivo (130). In particular, Notch3 was capable to act as a mediator of PD-L1 overexpression in BCSCs, activating mTOR and maintaining the self-renewal and invasive capacity of BCSCs (131). What’s more, it has been reported that Notch3 does effectively downregulate Notch1 signaling by repressing the expression of the downstream genes Hes1 and Hes5 (132). Interestingly, In ER- human breast cancer samples, survival advantage of Notch2High over Notch2Low patients in primary and bone metastatic breast cancer (133). Taken together, these observations suggest a common theme: deciphering the variation in the expression of Notch family members in different breast cancer types is necessary to develop effective treatments for the eradication of BCSCs.
Eph receptors are the largest family of RTKs in mammals and are activated by membrane-linked Ephrin ligands (134–136). The Eph receptor and its Ephrin ligand have been implicated as cell-cell communication complexes that influence the behavior of epithelial cells (137). The function of the Eph/Ephrin in the initiation of breast cancer has been analyzed in detail. In the Eph/ephrin system, chromosomal abnormalities, gene methylation, and alterations in transcription regulators induce dysregulation of the Eph/ephrin expression and tumorigenesis (136). It was demonstrated that EPHB6, an intrinsically catalytically inactive member of the Eph group, partially inhibits EMT, synergistically activates RAS-ERK signaling and promotes the expression of OCT4 in BCSCs, thus exhibiting higher stemness (138). PF-06647263 was a humanized monoclonal antibody that selected Ephrin-A4 as a pharmacological target to inhibit the activity of Ephrin-A4, which was highly expressed in BCSCs, in order to alleviate the clinical symptoms of TNBC (139). Importantly, understanding the complexity of the Eph/Ephrin system will help to elucidate the mechanisms of breast cancer.
Hedgehogs signaling includes SHH, IHH and DHH. The precursors can be cleaved to produce an active 19kd N-terminal fragment which binds to the membrane protein Patched gene (Ptc) and Smoothened gene (Smo). As Hedgehog genes are linked, Smo is released, leading to the activation of transcription factors (Gli1-3). In BCSCs, tetraspanin-8 (TSPAN8) was significantly upregulated, recruiting the deubiquitinating enzyme ATXN3 to inhibit the degradation of the SHH/PTCH1 complex, leading to SMO translocation to cilia, causing resistance to chemotherapeutic agents in CSCs and enhancing tumorigenesis in mice (140). Dehydrocholesterol reductase (DHCR24), a key enzyme in cholesterol synthesis, could promote breast cancer development by enhancing the Hedgehog and BCSC populations (141). While, Neuropilin-2 (NRP2) had the ability to activate Gli-1 and α6β1 integrins to induce BCSC initiation (142). Further in depth, Gli-1 and α6β1 integrins mediated the self-renewal and progression of BCSC by promoting angiogenesis and triggering focal adhesion kinase (FAK) signaling, respectively (143, 144). Consequently, targeting the SHH, α6β1, TSPAN8, and FAK can represent an attractive strategy for breast cancer treatment. Curcumin, a polyphenolic compound from the rhizome of Curcuma longa, has been reported to inhibit the proliferation and metastasis of TNBC cells, EMT and BCSC characteristics via the Hedgehog/Gli1 pathway (145). Similarly, genistein reduced the population of BCSCs by inhibiting Hedgehog (146). In summary, the search for integrated interventions in Hedgehog signaling and targeted inhibition of BCSC biological behavior could provide a new direction for breast cancer treatment.
PI3K is an intracellular phosphatidylinositol kinase (147). AKT is composed of three main isoforms (AKT1-3), which are key effectors of PI3K and can be directly activated by PI3K (148). PI3K/AKT is involved in regulating BCSC self-renewal, EMT and invasion (149, 150). PI3K/AKT also induced the of activation WNT signaling, which in turn increased the stemness and metastasis of BCSCs. HER2 dysregulation leads to aberrant activation of (PI3K)-Akt and/or WNT signaling and enhanced activity of the BCSC population, resulting in trastuzumab treatment resistance (151). Reciprocally, the role of the HER2 signaling in BCSCs can be enhanced by the PI3K/Akt pathway (152). Therefore, an open-label phase II study demonstrated that trastuzumab and lapatinib, which targeted HER2, inhibited the expression of FOXO, STAT5 and PI3K/AKT and suppress BCSC subpopulations (153).
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase consisting mainly of two distinct protein complexes, mTORC1 and mTORC2, which are key target genes downstream of AKT (154). Activation of PI3K promotes activation of mTORC1 and mTORC2, while the mTOR activity is frequently upregulated in human cancers (155). What’s more, the mTOR pathway is generally considered to be over-activated in CSCs. The inhibitory effect of some mTOR inhibitors on CSCs has been demonstrated (156). Rapamycin, everolimus and PF-04691502 inhibit tamoxifen-induced activation of BCSCs (157). Inhibition of mTOR restores AKT/mTOR-induced resistance to radiotherapy in BCSCs (158). Although mTOR has a role in suppressing BCSCs, a study showed that treatment of TNBC cells with mTOR inhibitors upregulated FGF1-FGFR-Notch1 signaling, leading to an increase in BCSC population (159). In this case, combined blockade of FGFR or Notch1 may prevent resistance to mTORC1/2 inhibitors by eliminating BCSCs (160). Mechanistically, adaptation or resistance to mTOR inhibition in BCSCs is manifested mainly by transcriptional reprogramming of the EVI1 and SOX9 to upregulate REHB and RAPTOR and metastasis-associated mediators (FSCN1 and SPARC) (161). Corporately, a link between PI3K-Akt-mTOR and BCSCs is evident.
As described previously, these intricate signal transduction pathways are not linear. The crosstalk among multiple pathways is also common in breast cancer, for instance, a discovery has revealed that Syndecan-1 promoted the activation of IL-6/STAT3 and EGFR via Notch to regulate inflammation and phenotype of BCSCs (162). The Hippo transducer TAZ confers BCSC-related features, including self-renew and tumor-initiation capacities, through MET (42). FAK can regulate YAP/TAZ activation (163). Aberrant regulation of signaling pathways, such as ERα, Notch and Hedgehog, can lead to abnormal activation of Hippo, resulting in BCSC fate perturbations (164–166). The cumulative effect of aberrant regulation of these pathways in breast cancer maintains and enhances the characteristics of BCSCs, ultimately culminating in malignant tumor progression. Consequently, a thorough insight into the perturbations of different pathways in individual patients is necessary to optimize personalized therapeutic strategies. Importantly, fully assessing the characteristics and subpopulation distribution of BCSCs and developing novel vehicles to eliminate them (Figure 4).
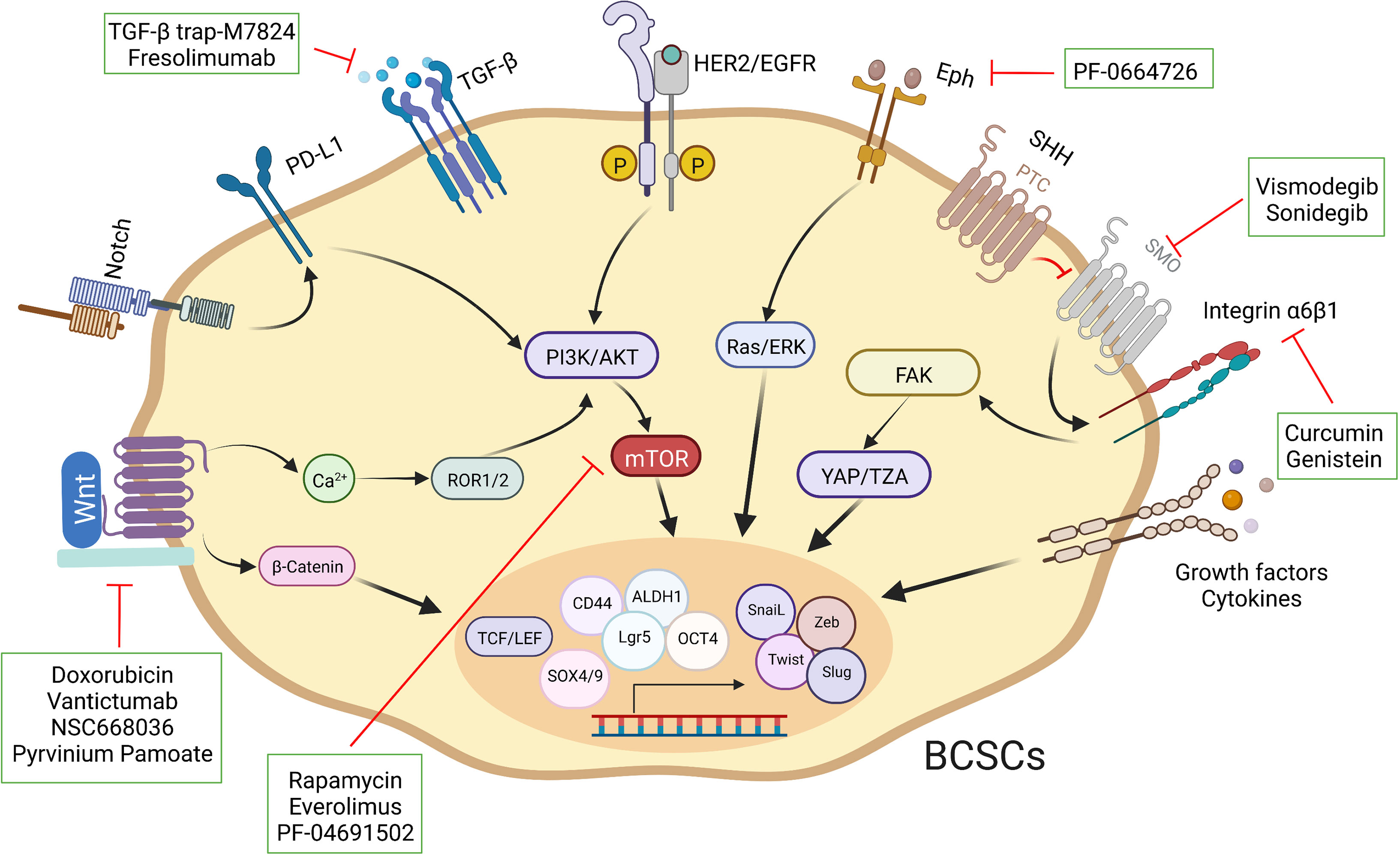
Figure 4 Schematic representation of different strategies used to target BCSCs. Specific pathways have been implicated in the fate of BCSCs. A select set of inhibitors have been developed to inhibit specific pathways.
From a clinical perspective, deciphering the relevance of BCSCs in therapy resistance, including chemotherapy, radiotherapy, immunotherapy and endocrinotherapy, is one of the major challenges in the clinical translation of anti-CSC therapies. Actually, CSCs are involved in tumor recurrence, metastasis and drug resistance, therefore targeting CSCs may be helpful and complementary to the treatment of breast cancer, combating concerns about safety and treatment failure. Excitingly, researchers have recently explored targeted therapeutic strategies for BCSC quiescence, maintenance pathways and specific markers.
The availability of BCSC-specific markers has facilitated researchers to effectively identify them, and commonly markers used to isolate BCSC include CD44+CD24-, CD133 and ALDH1. The high expression of these phenotypically and functionally significant markers in BCSCs compared to normal tissue could allow novel drugs to identify and block relevant BCSC signaling pathways, making them more susceptible to elimination by therapeutic strategies. CD44 is a cell surface receptor that binds to its ligand hyaluronic acid (HA) and that activates a variety of intracellular signals, and the interaction between them is used as a drug target. A study has demonstrated that lapatinib nanocrystals coated with HA have better therapeutic efficacy than uncoated HA in TNBC (167). Comparably, CD133, a membrane glycoprotein, has a demonstrated association with tumor resistance and recurrence. Polymeric nanoparticles loaded with paclitaxel targeting CD133 can markedly reinforce CD133+ cell internalization while significantly suppressing tumor regrowth in a xenograft model (168). Going further, conjugation anti-CD133 mAbs with saporin causes CD133+ BCSC proliferation arrest followed by cell death (169). However, unlike the traditional membrane proteins, ALDH1 is an enzyme with an activity that is intimately associated with the ability of BCSCs to self-renewal. Therefore, targeting ALDH1 is an effective therapeutic agent to eliminate BCSCs.
In fact, the surface phenotype of BCSCs is constantly in flux during cancer progression, differentiating or evolving into different cancer cells and thus obtaining distinct phenotypic recurrences. As a consequence, this will be the most prominent challenge in the design of targeted BCSC therapeutic interventions. Mechanistically, BCSCs undergo cell fate shifts in response to therapeutic pressure or metastasis, leading to malignant progression, which is mainly driven by their inherent genomic and epigenetic instability. Consequently, the strategy applied in clinical trials should take adequate consideration of the comprehensive range of elements leading to selective cell fate decisions, including the tumor microenvironment, intratumor heterogeneous cells and signaling cascades. The TME supports the self-renewal and differentiation of BCSCs, providing a niche to regulate their cell fate in the form of secreted factors and intercellular communication. The xenograft NOD/SCID mice model demonstrated that by affecting the expression of IL-6 in the local microenvironment of BCSCs, it was possible to regain ER expression and subsequently CD133hi cells were able to respond to hormone therapy (170). Inevitably, the damage to a single local microenvironment established through these animal models alone cannot fully replicate the reality of human breast cancer progression, but these adventurous research methods provide an important theoretical and temporal basis for extending preclinical studies. With technological advances, methods such as primary cell culture, organoid culture and microfluidic 3D biomimetic model allow for an improved mimicking of the normal tissue microenvironment, thus providing a new voyage to target the variable traits of BCSCs (171, 172).
Of vital note, signaling pathways are one of the key factors regulating the maintenance and evolution of BCSCs, and therefore targeting these key signals has proven to be an invaluable vehicle for the elimination of BCSCs. The major signaling pathways include Wnt, Notch, Eph, Hedgehogs and PI3K/AKT, which often interact with together in breast cancer stem cells during the development of breast cancer. Equally excitingly, with intensive research into cellular immunity, an additional option for oncology treatments has been developed with novel anti-BCSC immunotherapies such as immunologic checkpoint blocking or CAR-T cell therapies. PD-L1 is detected in 20% of TNBC (173). Deletion of RBMS1 expression by specific shRNA activates PD-L1 immune checkpoint receptor blockade to promote anti-tumor immunity in TNBC (174). In a phase I clinical study of 54 TNBC patients, Atezolizumab showed an objective response rate of 19% as an inhibitor of PD-L1 (175). For CAR-T cell therapy, TEM8 (176) and NKG2D (177) have been used for BCSC-targeted immunotherapy. Collaboratively, these discoveries shed new perspective on the preparation of clinically feasible therapeutic strategies for targeting BCSCs (Table 3).
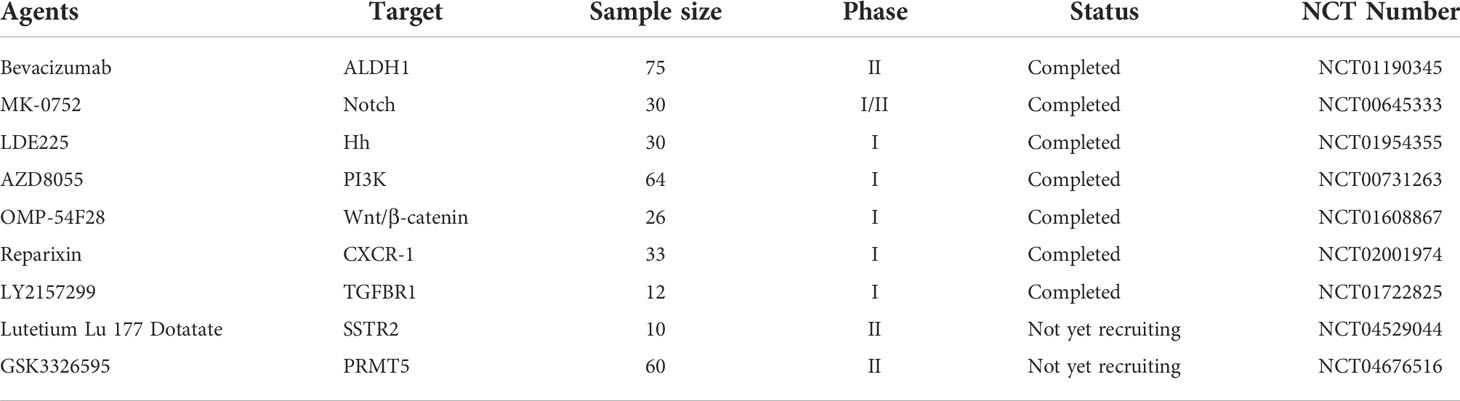
Table 3 Targeting BCSCs with different agents in clinical trials.
To date, we recognize that BCSCs are a small population of cancer cells with self-renewal and differentiation potential that are involved in mediating tumor heterogeneity, recurrence, metastasis and treatment resistance. There is current research indicating that BCSCs are an attractive target for tackling resistance and recurrence in the clinical therapy of breast cancer. Fortunately, BCSCs have the expression of their own specific markers that can provide post-therapeutic local biopsies with timely information on treatable targets for the remaining tumor tissue on the basis of variable biomarkers, thus allowing the selection of targets for the use of personalized and precise second-line therapy (178, 179). Especially, it is the introduction of the breast cancer stem cell concept, which focuses on biomarkers of BCSCs in the post-treatment period, that offers a new alternative to combating tumor recurrence. However, further attention needs to be given to the fact that normal stem cells in the tissue may also express the overlapping biomarkers and signaling pathways as BCSCs. This therefore requires that the possible side effects of targeting BCSCs for the treatment of breast cancer be fully considered, which in turn requires the rigorous elaboration of identity markers and signaling patterns that are specific or even unique to the targeted BCSCs. Meanwhile, BCSCs tend to have quiescent properties during response therapy, so therapeutic strategies to inhibit tumor progression do not fully prove to be due to the efficacy of targeted inhibition of BCSCs. In addition, BCSCs exist in a specific niche surrounded by heterogeneous cells such as TAMs, MSCs and CAFs that maintain their long-term survival. However, most current researches deficient a microenvironment have used isolated BCSCs and the relationship between BCSCs and their niches is currently ambiguous. Finally, it is undeniable that the immunodeficient animal models lacking adaptive immunity used in the current studies on BCSCs are not capable of recapitulating the biological complexity of tumors in the clinic (180). Collectively, there are still many obstacles to cross in achieving efficient and safe elimination of BCSCs.
In conclusion, the discovery of BCSCs has well revealed that individual cancer cells from the same tumor exhibit essential heterogeneity in terms of mutations, transcriptional programs, immune characteristics and functional properties. Indeed, BCSCs exist in a dynamic state, with multiple pools in individual tumors, so combining multiple treatment strategies to eradicate the pools of therapy-resistant BCSCs on the top of the heterogeneity is clinically important for preventing cancer recurrence. Deeply, cellular plasticity that mediates stemness, fueling cancer heterogeneity and responding to therapeutic pressure, further leading to the limitations of anti-CSC therapeutic strategies. Importantly, the BCSC concept not only has broad and profound implications for our understanding of cancer origins and progression, but also has significant clinical value for the design of more effective and personalized treatment options in the future. Therefore, a combination of conventional cytotoxic drugs, immunotherapy agents, endocrine therapy and eradication of BCSC therapy is a future direction of great significance for improving the clinical prognosis of breast cancer.
HX and LZ contributed to the conception of the study. HX and FZ were responsible for the collection and assembly of data. XG and QZ were responsible for literature search. All authors were involved in the writing and final approval of the manuscript.
We are grateful to the BioRender.com platform for providing online drawing tools (Agreement number of Figures 1-4: SG24652NWG, AP241A6DG7, JE241A6DHK, KU241A6DJM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
2. Liang Y, Zhang H, Song X, Yang Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin Cancer Biol (2020) 60:14–27. doi: 10.1016/j.semcancer.2019.08.012
3. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature (2012) 490(7418):61–70. doi: 10.1038/nature11412
4. Early Breast Cancer Trialists' Collaborative G. Long-term outcomes for neoadjuvant versus adjuvant chemotherapy in early breast cancer: Meta-analysis of individual patient data from ten randomised trials. Lancet Oncol (2018) 19(1):27–39. doi: 10.1016/S1470-2045(17)30777-5
5. Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers (2019) 5(1):66. doi: 10.1038/s41572-019-0111-2
6. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature (2000) 406(6797):747–52. doi: 10.1038/35021093
7. Sorlie T, Wang Y, Xiao C, Johnsen H, Naume B, Samaha RR, et al. Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: Gene expression analyses across three different platforms. BMC Genomics (2006) 7:127. doi: 10.1186/1471-2164-7-127
8. Bai X, Ni J, Beretov J, Graham P, Li Y. Cancer stem cell in breast cancer therapeutic resistance. Cancer Treat Rev (2018) 69:152–63. doi: 10.1016/j.ctrv.2018.07.004
9. Dandawate PR, Subramaniam D, Jensen RA, Anant S. Targeting cancer stem cells and signaling pathways by phytochemicals: Novel approach for breast cancer therapy. Semin Cancer Biol (2016) 40-41:192–208. doi: 10.1016/j.semcancer.2016.09.001
10. Carnero A, Garcia-Mayea Y, Mir C, Lorente J, Rubio IT, ME LL. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat Rev (2016) 49:25–36. doi: 10.1016/j.ctrv.2016.07.001
11. Polyak K. Breast cancer: Origins and evolution. J Clin Invest (2007) 117(11):3155–63. doi: 10.1172/JCI33295
12. Skibinski A, Kuperwasser C. The origin of breast tumor heterogeneity. Oncogene (2015) 34(42):5309–16. doi: 10.1038/onc.2014.475
13. Trumpp A, Haas S. Cancer stem cells: The adventurous journey from hematopoietic to leukemic stem cells. Cell (2022) 185(8):1266–70. doi: 10.1016/j.cell.2022.03.025
14. Inman JL, Robertson C, Mott JD, Bissell MJ. Mammary gland development: Cell fate specification, stem cells and the microenvironment. Development (2015) 142(6):1028–42. doi: 10.1242/dev.087643
15. Sakakura T, Suzuki Y, Shiurba R. Mammary stroma in development and carcinogenesis. J Mammary Gland Biol Neoplasia (2013) 18(2):189–97. doi: 10.1007/s10911-013-9281-9
16. Srivastava V, Huycke TR, Phong KT, Gartner ZJ. Organoid models for mammary gland dynamics and breast cancer. Curr Opin Cell Biol (2020) 66:51–8. doi: 10.1016/j.ceb.2020.05.003
17. Rios AC, Fu NY, Lindeman GJ, Visvader JE. In situ identification of bipotent stem cells in the mammary gland. Nature (2014) 506(7488):322–7. doi: 10.1038/nature12948
18. Fu NY, Nolan E, Lindeman GJ, Visvader JE. Stem cells and the differentiation hierarchy in mammary gland development. Physiol Rev (2020) 100(2):489–523. doi: 10.1152/physrev.00040.2018
19. Puisieux A, Pommier RM, Morel AP, Lavial F. Cellular pliancy and the multistep process of tumorigenesis. Cancer Cell (2018) 33(2):164–72. doi: 10.1016/j.ccell.2018.01.007
20. Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, et al. Universal patterns of selection in cancer and somatic tissues. Cell (2017) 171(5):1029–41.e21. doi: 10.1016/j.cell.2017.09.042
21. Fu N, Lindeman GJ, Visvader JE. The mammary stem cell hierarchy. Curr Top Dev Biol (2014) 107:133–60. doi: 10.1016/B978-0-12-416022-4.00005-6
22. Ripperger T, Gadzicki D, Meindl A, Schlegelberger B. Breast cancer susceptibility: Current knowledge and implications for genetic counselling. Eur J Hum Genet (2009) 17(6):722–31. doi: 10.1038/ejhg.2008.212
23. Fackenthal JD, Olopade OI. Breast cancer risk associated with Brca1 and Brca2 in diverse populations. Nat Rev Cancer (2007) 7(12):937–48. doi: 10.1038/nrc2054
24. Couch FJ, Nathanson KL, Offit K. Two decades after brca: Setting paradigms in personalized cancer care and prevention. Science (2014) 343(6178):1466–70. doi: 10.1126/science.1251827
25. Hu L, Su L, Cheng H, Mo C, Ouyang T, Li J, et al. Single-cell rna sequencing reveals the cellular origin and evolution of breast cancer in Brca1 mutation carriers. Cancer Res (2021) 81(10):2600–11. doi: 10.1158/0008-5472.CAN-20-2123
26. Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell (2012) 148(5):1015–28. doi: 10.1016/j.cell.2012.02.008
27. Boulanger CA, Smith GH. Reducing mammary cancer risk through premature stem cell senescence. Oncogene (2001) 20(18):2264–72. doi: 10.1038/sj.onc.1204312
28. Yu JM, Sun W, Wang ZH, Liang X, Hua F, Li K, et al. Trib3 supports breast cancer stemness by suppressing Foxo1 degradation and enhancing Sox2 transcription. Nat Commun (2019) 10(1):5720. doi: 10.1038/s41467-019-13700-6
29. Jiang H, Zhou C, Zhang Z, Wang Q, Wei H, Shi W, et al. Jagged1-Notch1-Deployed tumor perivascular niche promotes breast cancer stem cell phenotype through Zeb1. Nat Commun (2020) 11(1):5129. doi: 10.1038/s41467-020-18860-4
30. Christin JR, Wang C, Chung CY, Liu Y, Dravis C, Tang W, et al. Stem cell determinant Sox9 promotes lineage plasticity and progression in basal-like breast cancer. Cell Rep (2020) 31(10):107742. doi: 10.1016/j.celrep.2020.107742
31. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med (2010) 363(20):1938–48. doi: 10.1056/NEJMra1001389
32. Vieira AF, Ricardo S, Ablett MP, Dionisio MR, Mendes N, Albergaria A, et al. P-cadherin is coexpressed with Cd44 and Cd49f and mediates stem cell properties in basal-like breast cancer. Stem Cells (2012) 30(5):854–64. doi: 10.1002/stem.1075
33. Prat A, Adamo B, Cheang MC, Anders CK, Carey LA, Perou CM. Molecular characterization of basal-like and non-Basal-Like triple-negative breast cancer. Oncologist (2013) 18(2):123–33. doi: 10.1634/theoncologist.2012-0397
34. Vijay GV, Zhao N, Den Hollander P, Toneff MJ, Joseph R, Pietila M, et al. Gsk3beta regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res (2019) 21(1):37. doi: 10.1186/s13058-019-1125-0
35. Wang X, Sun Y, Wong J, Conklin DS. Ppargamma maintains Erbb2-positive breast cancer stem cells. Oncogene (2013) 32(49):5512–21. doi: 10.1038/onc.2013.217
36. Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, et al. Control of mammary stem cell function by steroid hormone signalling. Nature (2010) 465(7299):798–802. doi: 10.1038/nature09027
37. Visvader JE. Cells of origin in cancer. Nature (2011) 469(7330):314–22. doi: 10.1038/nature09781
38. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci United States America (2003) 100(7):3983–8. doi: 10.1073/pnas.0530291100
39. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. Aldh1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell (2007) 1(5):555–67. doi: 10.1016/j.stem.2007.08.014
40. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep (2014) 2(1):78–91. doi: 10.1016/j.stemcr.2013.11.009
41. Colacino JA, Azizi E, Brooks MD, Harouaka R, Fouladdel S, McDermott SP, et al. Heterogeneity of human breast stem and progenitor cells as revealed by transcriptional profiling. Stem Cell Rep (2018) 10(5):1596–609. doi: 10.1016/j.stemcr.2018.03.001
42. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, et al. The hippo transducer taz confers cancer stem cell-related traits on breast cancer cells. Cell (2011) 147(4):759–72. doi: 10.1016/j.cell.2011.09.048
43. Kabos P, Haughian JM, Wang X, Dye WW, Finlayson C, Elias A, et al. Cytokeratin 5 positive cells represent a steroid receptor negative and therapy resistant subpopulation in luminal breast cancers. Breast Cancer Res Treat (2011) 128(1):45–55. doi: 10.1007/s10549-010-1078-6
44. Ye F, Zhong X, Qiu Y, Yang L, Wei B, Zhang Z, et al. Cd49f can act as a biomarker for local or distant recurrence in breast cancer. J Breast Cancer (2017) 20(2):142–9. doi: 10.4048/jbc.2017.20.2.142
45. Cariati M, Naderi A, Brown JP, Smalley MJ, Pinder SE, Caldas C, et al. Alpha-6 integrin is necessary for the tumourigenicity of a stem cell-like subpopulation within the Mcf7 breast cancer cell line. Int J Cancer (2008) 122(2):298–304. doi: 10.1002/ijc.23103
46. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci United States America (2011) 108(19):7950–5. doi: 10.1073/pnas.1102454108
47. Navas T, Kinders RJ, Lawrence SM, Ferry-Galow KV, Borgel S, Hollingshead MG, et al. Clinical evolution of epithelial-mesenchymal transition in human carcinomas. Cancer Res (2020) 80(2):304–18. doi: 10.1158/0008-5472.CAN-18-3539
48. Liu S, Sun Y, Hou Y, Yang L, Wan X, Qin Y, et al. A novel lncrna ropm-mediated lipid metabolism governs breast cancer stem cell properties. J Hematol Oncol (2021) 14(1):178. doi: 10.1186/s13045-021-01194-z
49. Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun (2018) 9(1):2897. doi: 10.1038/s41467-018-05220-6
50. Russnes HG, Navin N, Hicks J, Borresen-Dale AL. Insight into the heterogeneity of breast cancer through next-generation sequencing. J Clin Invest (2011) 121(10):3810–8. doi: 10.1172/JCI57088
51. Kai K, Arima Y, Kamiya T, Saya H. Breast cancer stem cells. Breast Cancer (2010) 17(2):80–5. doi: 10.1007/s12282-009-0176-y
52. Pontier SM, Muller WJ. Integrins in mammary-Stem-Cell biology and breast-cancer progression–a role in cancer stem cells? J Cell Sci (2009) 122(Pt 2):207–14. doi: 10.1242/jcs.040394
53. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature (2001) 414(6859):105–11. doi: 10.1038/35102167
54. Boman BM, Wicha MS. Cancer stem cells: A step toward the cure. J Clin Oncol (2008) 26(17):2795–9. doi: 10.1200/JCO.2008.17.7436
55. Velasco-Velazquez MA, Popov VM, Lisanti MP, Pestell RG. The role of breast cancer stem cells in metastasis and therapeutic implications. Am J Pathol (2011) 179(1):2–11. doi: 10.1016/j.ajpath.2011.03.005
56. Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct Cd44+/Cd24- and Cd133+ cells with cancer stem cell characteristics. Breast Cancer Res (2008) 10(1):R10. doi: 10.1186/bcr1855
57. Vassilopoulos A, Wang RH, Petrovas C, Ambrozak D, Koup R, Deng CX. Identification and characterization of cancer initiating cells from Brca1 related mammary tumors using markers for normal mammary stem cells. Int J Biol Sci (2008) 4(3):133–42. doi: 10.7150/ijbs.4.133
58. Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res (2008) 10(2):R25. doi: 10.1186/bcr1982
59. Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in Brca1 mutation carriers. Nat Med (2009) 15(8):907–13. doi: 10.1038/nm.2000
60. Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside Gd2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest (2012) 122(6):2066–78. doi: 10.1172/JCI59735
61. Lu H, Clauser KR, Tam WL, Frose J, Ye X, Eaton EN, et al. Addendum: A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol (2015) 17(12):1607. doi: 10.1038/ncb3281
62. Saha SK, Choi HY, Kim BW, Dayem AA, Yang GM, Kim KS, et al. Krt19 directly interacts with beta-Catenin/Rac1 complex to regulate numb-dependent notch signaling pathway and breast cancer properties. Oncogene (2017) 36(3):332–49. doi: 10.1038/onc.2016.221
63. Semenza GL. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim Biophys Acta (2016) 1863(3):382–91. doi: 10.1016/j.bbamcr.2015.05.036
64. Zhang H, Lu H, Xiang L, Bullen JW, Zhang C, Samanta D, et al. Hif-1 regulates Cd47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc Natl Acad Sci United States America (2015) 112(45):E6215–23. doi: 10.1073/pnas.1520032112
65. Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci United States America (2014) 111(50):E5429–38. doi: 10.1073/pnas.1421438111
66. Yoo YG, Christensen J, Gu J, Huang LE. Hif-1alpha mediates tumor hypoxia to confer a perpetual mesenchymal phenotype for malignant progression. Sci Signal (2011) 4(178):pt4. doi: 10.1126/scisignal.2002072
67. Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell (2011) 145(6):926–40. doi: 10.1016/j.cell.2011.04.029
68. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by hif-dependent and Alkbh5-mediated M(6)a-demethylation of nanog mrna. Proc Natl Acad Sci United States America (2016) 113(14):E2047–56. doi: 10.1073/pnas.1602883113
69. Lu H, Xie Y, Tran L, Lan J, Yang Y, Murugan NL, et al. Chemotherapy-induced S100a10 recruits Kdm6a to facilitate Oct4-mediated breast cancer stemness. J Clin Invest (2020) 130(9):4607–23. doi: 10.1172/JCI138577
70. Liu X, Xie P, Hao N, Zhang M, Liu Y, Liu P, et al. Hif-1-Regulated expression of calreticulin promotes breast tumorigenesis and progression through Wnt/Beta-catenin pathway activation. Proc Natl Acad Sci USA (2021) 118(44):e2109144118. doi: 10.1073/pnas.2109144118
71. Sirkisoon SR, Carpenter RL, Rimkus T, Doheny D, Zhu D, Aguayo NR, et al. Tgli1 transcription factor mediates breast cancer brain metastasis Via activating metastasis-initiating cancer stem cells and astrocytes in the tumor microenvironment. Oncogene (2020) 39(1):64–78. doi: 10.1038/s41388-019-0959-3
72. Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell (2010) 140(1):62–73. doi: 10.1016/j.cell.2009.12.007
73. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science (2011) 331(6024):1559–64. doi: 10.1126/science.1203543
74. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139(5):871–90. doi: 10.1016/j.cell.2009.11.007
75. Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, et al. Distinct emt programs control normal mammary stem cells and tumour-initiating cells. Nature (2015) 525(7568):256–60. doi: 10.1038/nature14897
76. Rinn J, Guttman M. Rna function. rna and dynamic nuclear organization. Science (2014) 345(6202):1240–1. doi: 10.1126/science.1252966
77. Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking mirna-1-2. Cell (2007) 129(2):303–17. doi: 10.1016/j.cell.2007.03.030
78. Rottiers V, Naar AM. Micrornas in metabolism and metabolic disorders. Nat Rev Mol Cell Biol (2012) 13(4):239–50. doi: 10.1038/nrm3313
79. Singh SK, Pal Bhadra M, Girschick HJ, Bhadra U. Micrornas–micro in size but macro in function. FEBS J (2008) 275(20):4929–44. doi: 10.1111/j.1742-4658.2008.06624.x
80. Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, et al. Downregulation of mirna-200c links breast cancer stem cells with normal stem cells. Cell (2009) 138(3):592–603. doi: 10.1016/j.cell.2009.07.011
81. Christoffersen NR, Silahtaroglu A, Orom UA, Kauppinen S, Lund AH. Mir-200b mediates post-transcriptional repression of Zfhx1b. RNA (2007) 13(8):1172–8. doi: 10.1261/rna.586807
82. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The mir-200 family and mir-205 regulate epithelial to mesenchymal transition by targeting Zeb1 and Sip1. Nat Cell Biol (2008) 10(5):593–601. doi: 10.1038/ncb1722
83. Park SM, Gaur AB, Lengyel E, Peter ME. The mir-200 family determines the epithelial phenotype of cancer cells by targeting the e-cadherin repressors Zeb1 and Zeb2. Genes Dev (2008) 22(7):894–907. doi: 10.1101/gad.1640608
84. Liu C, Tang DG. Microrna regulation of cancer stem cells. Cancer Res (2011) 71(18):5950–4. doi: 10.1158/0008-5472.CAN-11-1035
85. Takahashi RU, Miyazaki H, Takeshita F, Yamamoto Y, Minoura K, Ono M, et al. Loss of microrna-27b contributes to breast cancer stem cell generation by activating Enpp1. Nat Commun (2015) 6:7318. doi: 10.1038/ncomms8318
86. Lu G, Li Y, Ma Y, Lu J, Chen Y, Jiang Q, et al. Long noncoding rna Linc00511 contributes to breast cancer tumourigenesis and stemness by inducing the mir-185-3p/E2f1/Nanog axis. J Exp Clin Cancer Res (2018) 37(1):289. doi: 10.1186/s13046-018-0945-6
87. Yang Q, Zhao S, Shi Z, Cao L, Liu J, Pan T, et al. Chemotherapy-elicited exosomal mir-378a-3p and mir-378d promote breast cancer stemness and chemoresistance Via the activation of Ezh2/Stat3 signaling. J Exp Clin Cancer Res (2021) 40(1):120. doi: 10.1186/s13046-021-01901-1
88. Conte I, Banfi S, Bovolenta P. Non-coding rnas in the development of sensory organs and related diseases. Cell Mol Life Sci (2013) 70(21):4141–55. doi: 10.1007/s00018-013-1335-z
89. Brown JM, Wasson MD, Marcato P. The missing lnc: The potential of targeting triple-negative breast cancer and cancer stem cells by inhibiting long non-coding rnas. Cells (2020) 9(3):763. doi: 10.3390/cells9030763
90. Qin Y, Hou Y, Liu S, Zhu P, Wan X, Zhao M, et al. A novel long non-coding rna Lnc030 maintains breast cancer stem cell stemness by stabilizing sqle mrna and increasing cholesterol synthesis. Adv Sci (Weinh) (2021) 8(2):2002232. doi: 10.1002/advs.202002232
91. Zhou M, Hou Y, Yang G, Zhang H, Tu G, Du YE, et al. Lncrna-hh strengthen cancer stem cells generation in twist-positive breast cancer Via activation of hedgehog signaling pathway. Stem Cells (2016) 34(1):55–66. doi: 10.1002/stem.2219
92. Cho SW, Xu J, Sun R, Mumbach MR, Carter AC, Chen YG, et al. Promoter of lncrna gene Pvt1 is a tumor-suppressor DNA boundary element. Cell (2018) 173(6):1398–412.e22. doi: 10.1016/j.cell.2018.03.068
93. Santamaria-Martinez A, Huelsken J. The niche under siege: Novel targets for metastasis therapy. J Intern Med (2013) 274(2):127–36. doi: 10.1111/joim.12024
94. Joyce JA, Fearon DT. T Cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348(6230):74–80. doi: 10.1126/science.aaa6204
95. Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214(3):579–96. doi: 10.1084/jem.20162024
96. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. Cd10(+)Gpr77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell (2018) 172(4):841–56.e16. doi: 10.1016/j.cell.2018.01.009
97. Bartoschek M, Oskolkov N, Bocci M, Lovrot J, Larsson C, Sommarin M, et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell rna sequencing. Nat Commun (2018) 9(1):5150. doi: 10.1038/s41467-018-07582-3
98. Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature (2011) 481(7379):85–9. doi: 10.1038/nature10694
99. Valenti G, Quinn HM, Heynen G, Lan L, Holland JD, Vogel R, et al. Cancer stem cells regulate cancer-associated fibroblasts Via activation of hedgehog signaling in mammary gland tumors. Cancer Res (2017) 77(8):2134–47. doi: 10.1158/0008-5472.CAN-15-3490
100. Hamalian S, Guth R, Runa F, Sanchez F, Vickers E, Agajanian M, et al. A Snai2-Peak1-Inhba stromal axis drives progression and lapatinib resistance in Her2-positive breast cancer by supporting subpopulations of tumor cells positive for antiapoptotic and stress signaling markers. Oncogene (2021) 40(33):5224–35. doi: 10.1038/s41388-021-01906-2
101. Cuiffo BG, Campagne A, Bell GW, Lembo A, Orso F, Lien EC, et al. Msc-regulated micrornas converge on the transcription factor Foxp2 and promote breast cancer metastasis. Cell Stem Cell (2014) 15(6):762–74. doi: 10.1016/j.stem.2014.10.001
102. Lu H, Clauser KR, Tam WL, Frose J, Ye X, Eaton EN, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol (2014) 16(11):1105–17. doi: 10.1038/ncb3041
103. Yang J, Liao D, Chen C, Liu Y, Chuang TH, Xiang R, et al. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine Egfr/Stat3/Sox-2 signaling pathway. Stem Cells (2013) 31(2):248–58. doi: 10.1002/stem.1281
104. Boyle ST, Kochetkova M. Breast cancer stem cells and the immune system: Promotion, evasion and therapy. J Mammary Gland Biol Neoplasia (2014) 19(2):203–11. doi: 10.1007/s10911-014-9323-y
105. Elia I, Rossi M, Stegen S, Broekaert D, Doglioni G, van Gorsel M, et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature (2019) 568(7750):117–21. doi: 10.1038/s41586-019-0977-x
106. Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, et al. Cd44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest (2011) 121(3):1064–74. doi: 10.1172/JCI44540
107. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell (2018) 174(5):1293–308 e36. doi: 10.1016/j.cell.2018.05.060
108. Tekpli X, Lien T, Rossevold AH, Nebdal D, Borgen E, Ohnstad HO, et al. An independent poor-prognosis subtype of breast cancer defined by a distinct tumor immune microenvironment. Nat Commun (2019) 10(1):5499. doi: 10.1038/s41467-019-13329-5
109. Clevers H, Loh KM, Nusse R. Stem cell signaling. an integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science (2014) 346(6205):1248012. doi: 10.1126/science.1248012
110. Sherwood V. Wnt signaling: An emerging mediator of cancer cell metabolism? Mol Cell Biol (2015) 35(1):2–10. doi: 10.1128/MCB.00992-14
111. Kahn M. Wnt signaling in stem cells and cancer stem cells: A tale of two coactivators. Prog Mol Biol Transl Sci (2018) 153:209–44. doi: 10.1016/bs.pmbts.2017.11.007
112. Muruganandan S, Roman AA, Sinal CJ. Adipocyte differentiation of bone marrow-derived mesenchymal stem cells: Cross talk with the osteoblastogenic program. Cell Mol Life Sci (2009) 66(2):236–53. doi: 10.1007/s00018-008-8429-z
113. Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic latency and immune evasion through autocrine inhibition of wnt. Cell (2016) 165(1):45–60. doi: 10.1016/j.cell.2016.02.025
114. Zhuang X, Zhang H, Li X, Li X, Cong M, Peng F, et al. Differential effects on lung and bone metastasis of breast cancer by wnt signalling inhibitor Dkk1. Nat Cell Biol (2017) 19(10):1274–85. doi: 10.1038/ncb3613
115. Miller-Kleinhenz J, Guo X, Qian W, Zhou H, Bozeman EN, Zhu L, et al. Dual-targeting wnt and upa receptors using peptide conjugated ultra-small nanoparticle drug carriers inhibited cancer stem-cell phenotype in chemo-resistant breast cancer. Biomaterials (2018) 152:47–62. doi: 10.1016/j.biomaterials.2017.10.035
116. Katoh M. Antibody-drug conjugate targeting protein tyrosine kinase 7, a receptor tyrosine kinase-like molecule involved in wnt and vascular endothelial growth factor signaling: Effects on cancer stem cells, tumor microenvironment and whole-body homeostasis. Ann Transl Med (2017) 5(23):462. doi: 10.21037/atm.2017.09.11
117. Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, et al. Wnt pathway inhibition Via the targeting of frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci United States America (2012) 109(29):11717–22. doi: 10.1073/pnas.1120068109
118. Shetti D, Zhang B, Fan C, Mo C, Lee BH, Wei K. Low dose of paclitaxel combined with Xav939 attenuates metastasis, angiogenesis and growth in breast cancer by suppressing wnt signaling. Cells (2019) 8(8):892. doi: 10.3390/cells8080892
119. Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, et al. Targeting wnt-driven cancer through the inhibition of porcupine by Lgk974. Proc Natl Acad Sci United States America (2013) 110(50):20224–9. doi: 10.1073/pnas.1314239110
120. Huang C, Chen Y, Liu H, Yang J, Song X, Zhao J, et al. Celecoxib targets breast cancer stem cells by inhibiting the synthesis of prostaglandin E2 and down-regulating the wnt pathway activity. Oncotarget (2017) 8(70):115254–69. doi: 10.18632/oncotarget.23250
121. Li Y, Zhang T, Korkaya H, Liu S, Lee HF, Newman B, et al. Sulforaphane, a dietary component of Broccoli/Broccoli sprouts, inhibits breast cancer stem cells. Clin Cancer Res (2010) 16(9):2580–90. doi: 10.1158/1078-0432.CCR-09-2937
122. Xu L, Zhang L, Hu C, Liang S, Fei X, Yan N, et al. Wnt pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Int J Oncol (2016) 48(3):1175–86. doi: 10.3892/ijo.2016.3337
123. Zhang Y, Wang X. Targeting the Wnt/Beta-catenin signaling pathway in cancer. J Hematol Oncol (2020) 13(1):165. doi: 10.1186/s13045-020-00990-3
124. Abetov D, Mustapova Z, Saliev T, Bulanin D, Batyrbekov K, Gilman CP. Novel small molecule inhibitors of cancer stem cell signaling pathways. Stem Cell Rev Rep (2015) 11(6):909–18. doi: 10.1007/s12015-015-9612-x
125. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: Cell fate control and signal integration in development. Science (1999) 284(5415):770–6. doi: 10.1126/science.284.5415.770
126. Mollen EWJ, Ient J, Tjan-Heijnen VCG, Boersma LJ, Miele L, Smidt ML, et al. Moving breast cancer therapy up a notch. Front Oncol (2018) 8:518. doi: 10.3389/fonc.2018.00518
127. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res (2010) 70(2):709–18. doi: 10.1158/0008-5472.CAN-09-1681
128. Raouf A, Zhao Y, To K, Stingl J, Delaney A, Barbara M, et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell (2008) 3(1):109–18. doi: 10.1016/j.stem.2008.05.018
129. Shah D, Wyatt D, Baker AT, Simms P, Peiffer DS, Fernandez M, et al. Inhibition of Her2 increases Jagged1-dependent breast cancer stem cells: Role for membrane Jagged1. Clin Cancer Res (2018) 24(18):4566–78. doi: 10.1158/1078-0432.CCR-17-1952
130. Baker A, Wyatt D, Bocchetta M, Li J, Filipovic A, Green A, et al. Notch-1-Pten-Erk1/2 signaling axis promotes Her2+ breast cancer cell proliferation and stem cell survival. Oncogene (2018) 37(33):4489–504. doi: 10.1038/s41388-018-0251-y
131. Mansour FA, Al-Mazrou A, Al-Mohanna F, Al-Alwan M, Ghebeh H. Pd-L1 is overexpressed on breast cancer stem cells through Notch3/Mtor axis. Oncoimmunology (2020) 9(1):1729299. doi: 10.1080/2162402X.2020.1729299
132. Zhou W, Wang G, Guo S. Regulation of angiogenesis Via notch signaling in breast cancer and cancer stem cells. Biochim Biophys Acta (2013) 1836(2):304–20. doi: 10.1016/j.bbcan.2013.10.003
133. Capulli M, Hristova D, Valbret Z, Carys K, Arjan R, Maurizi A, et al. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br J Cancer (2019) 121(2):157–71. doi: 10.1038/s41416-019-0501-y
134. Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol (2005) 6(6):462–75. doi: 10.1038/nrm1662
135. Lackmann M, Boyd AW. Eph, a protein family coming of age: More confusion, insight, or complexity? Sci Signal (2008) 1(15):re2. doi: 10.1126/stke.115re2
136. Pasquale EB. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat Rev Cancer (2010) 10(3):165–80. doi: 10.1038/nrc2806
137. Perez White BE, Getsios S. Eph receptor and ephrin function in breast, gut, and skin epithelia. Cell Adh Migr (2014) 8(4):327–38. doi: 10.4161/19336918.2014.970012
138. Toosi BM, El Zawily A, Truitt L, Shannon M, Allonby O, Babu M, et al. Ephb6 augments both development and drug sensitivity of triple-negative breast cancer tumours. Oncogene (2018) 37(30):4073–93. doi: 10.1038/s41388-018-0228-x
139. Damelin M, Bankovich A, Park A, Aguilar J, Anderson W, Santaguida M, et al. Anti-Efna4 calicheamicin conjugates effectively target triple-negative breast and ovarian tumor-initiating cells to result in sustained tumor regressions. Clin Cancer Res (2015) 21(18):4165–73. doi: 10.1158/1078-0432.CCR-15-0695
140. Zhu R, Gires O, Zhu L, Liu J, Li J, Yang H, et al. Tspan8 promotes cancer cell stemness Via activation of sonic hedgehog signaling. Nat Commun (2019) 10(1):2863. doi: 10.1038/s41467-019-10739-3
141. Qiu T, Cao J, Chen W, Wang J, Wang Y, Zhao L, et al. 24-dehydrocholesterol reductase promotes the growth of breast cancer stem-like cells through the hedgehog pathway. Cancer Sci (2020) 111(10):3653–64. doi: 10.1111/cas.14587
142. Goel HL, Pursell B, Chang C, Shaw LM, Mao J, Simin K, et al. Gli1 regulates a novel neuropilin-2/Alpha6beta1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol Med (2013) 5(4):488–508. doi: 10.1002/emmm.201202078
143. Ge X, Lyu P, Gu Y, Li L, Li J, Wang Y, et al. Sonic hedgehog stimulates glycolysis and proliferation of breast cancer cells: Modulation of Pfkfb3 activation. Biochem Biophys Res Commun (2015) 464(3):862–8. doi: 10.1016/j.bbrc.2015.07.052
144. Habib JG, O'Shaughnessy JA. The hedgehog pathway in triple-negative breast cancer. Cancer Med (2016) 5(10):2989–3006. doi: 10.1002/cam4.833
145. Li M, Guo T, Lin J, Huang X, Ke Q, Wu Y, et al. Curcumin inhibits the invasion and metastasis of triple negative breast cancer Via Hedgehog/Gli1 signaling pathway. J Ethnopharmacol (2022) 283:114689. doi: 10.1016/j.jep.2021.114689
146. Fan P, Fan S, Wang H, Mao J, Shi Y, Ibrahim MM, et al. Genistein decreases the breast cancer stem-like cell population through hedgehog pathway. Stem Cell Res Ther (2013) 4(6):146. doi: 10.1186/scrt357
147. Tasian SK, Teachey DT, Rheingold SR. Targeting the Pi3k/Mtor pathway in pediatric hematologic malignancies. Front Oncol (2014) 4:108. doi: 10.3389/fonc.2014.00108
148. Wang Q, Chen X, Hay N. Akt as a target for cancer therapy: More is not always better (Lessons from studies in mice). Br J Cancer (2017) 117(2):159–63. doi: 10.1038/bjc.2017.153
149. Gao X, Qin T, Mao J, Zhang J, Fan S, Lu Y, et al. Ptenp1/Mir-20a/Pten axis contributes to breast cancer progression by regulating pten Via Pi3k/Akt pathway. J Exp Clin Cancer Res (2019) 38(1):256. doi: 10.1186/s13046-019-1260-6
150. Bai J, Chen WB, Zhang XY, Kang XN, Jin LJ, Zhang H, et al. Hif-2alpha regulates Cd44 to promote cancer stem cell activation in triple-negative breast cancer Via Pi3k/Akt/Mtor signaling. World J Stem Cells (2020) 12(1):87–99. doi: 10.4252/wjsc.v12.i1.87
151. Choi HJ, Jin S, Cho H, Won HY, An HW, Jeong GY, et al. Cdk12 drives breast tumor initiation and trastuzumab resistance Via wnt and Irs1-Erbb-Pi3k signaling. EMBO Rep (2019) 20(10):e48058. doi: 10.15252/embr.201948058
152. Alanazi IO, Khan Z. Understanding egfr signaling in breast cancer and breast cancer stem cells: Overexpression and therapeutic implications. Asian Pac J Cancer Prev (2016) 17(2):445–53. doi: 10.7314/apjcp.2016.17.2.445
153. Holmes FA, Espina V, Liotta LA, Nagarwala YM, Danso M, McIntyre KJ, et al. Pathologic complete response after preoperative anti-Her2 therapy correlates with alterations in pten, foxo, phosphorylated Stat5, and autophagy protein signaling. BMC Res Notes (2013) 6:507. doi: 10.1186/1756-0500-6-507
154. Alzahrani AS. Pi3k/Akt/Mtor inhibitors in cancer: At the bench and bedside. Semin Cancer Biol (2019) 59:125–32. doi: 10.1016/j.semcancer.2019.07.009
155. Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mtor for cancer therapy. J Hematol Oncol (2019) 12(1):71. doi: 10.1186/s13045-019-0754-1
156. Francipane MG, Lagasse E. Therapeutic potential of mtor inhibitors for targeting cancer stem cells. Br J Clin Pharmacol (2016) 82(5):1180–8. doi: 10.1111/bcp.12844
157. Karthik GM, Ma R, Lovrot J, Kis LL, Lindh C, Blomquist L, et al. Mtor inhibitors counteract tamoxifen-induced activation of breast cancer stem cells. Cancer Lett (2015) 367(1):76–87. doi: 10.1016/j.canlet.2015.07.017
158. Lai Y, Yu X, Lin X, He S. Inhibition of mtor sensitizes breast cancer stem cells to radiation-induced repression of self-renewal through the regulation of mnsod and akt. Int J Mol Med (2016) 37(2):369–77. doi: 10.3892/ijmm.2015.2441
159. Bhola NE, Jansen VM, Koch JP, Li H, Formisano L, Williams JA, et al. Treatment of triple-negative breast cancer with Torc1/2 inhibitors sustains a drug-resistant and notch-dependent cancer stem cell population. Cancer Res (2016) 76(2):440–52. doi: 10.1158/0008-5472.CAN-15-1640-T
160. Hoxhaj G, Hughes-Hallett J, Timson RC, Ilagan E, Yuan M, Asara JM, et al. The Mtorc1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep (2017) 21(5):1331–46. doi: 10.1016/j.celrep.2017.10.029
161. Mateo F, Arenas EJ, Aguilar H, Serra-Musach J, de Garibay GR, Boni J, et al. Stem cell-like transcriptional reprogramming mediates metastatic resistance to mtor inhibition. Oncogene (2017) 36(19):2737–49. doi: 10.1038/onc.2016.427
162. Ibrahim SA, Gadalla R, El-Ghonaimy EA, Samir O, Mohamed HT, Hassan H, et al. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype Via the il-6/Stat3, notch and egfr signaling pathways. Mol Cancer (2017) 16(1):57. doi: 10.1186/s12943-017-0621-z
163. Wang S, Englund E, Kjellman P, Li Z, Ahnlide JK, Rodriguez-Cupello C, et al. Ccm3 is a gatekeeper in focal adhesions regulating mechanotransduction and Yap/Taz signalling. Nat Cell Biol (2021) 23(7):758–70. doi: 10.1038/s41556-021-00702-0
164. Britschgi A, Duss S, Kim S, Couto JP, Brinkhaus H, Koren S, et al. The hippo kinases Lats1 and 2 control human breast cell fate Via crosstalk with eralpha. Nature (2017) 541(7638):541–5. doi: 10.1038/nature20829
165. Lim SK, Lu SY, Kang SA, Tan HJ, Li Z, Adrian Wee ZN, et al. Wnt signaling promotes breast cancer by blocking itch-mediated degradation of Yap/Taz transcriptional coactivator Wbp2. Cancer Res (2016) 76(21):6278–89. doi: 10.1158/0008-5472.CAN-15-3537
166. Ko YC, Choi HS, Liu R, Lee DS. Physalin a, 13,14-Seco-16, 24-Cyclo-Steroid, inhibits stemness of breast cancer cells by regulation of hedgehog signaling pathway and yes-associated protein 1 (Yap1). Int J Mol Sci (2021) 22(16):8718. doi: 10.3390/ijms22168718
167. Agrawal S, Dwivedi M, Ahmad H, Chadchan SB, Arya A, Sikandar R, et al. Cd44 targeting hyaluronic acid coated lapatinib nanocrystals foster the efficacy against triple-negative breast cancer. Nanomedicine (2018) 14(2):327–37. doi: 10.1016/j.nano.2017.10.010
168. Swaminathan SK, Roger E, Toti U, Niu L, Ohlfest JR, Panyam J. Cd133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J Control Release (2013) 171(3):280–7. doi: 10.1016/j.jconrel.2013.07.014
169. Bostad M, Olsen CE, Peng Q, Berg K, Hogset A, Selbo PK. Light-controlled endosomal escape of the novel Cd133-targeting immunotoxin Ac133-saporin by photochemical internalization - a minimally invasive cancer stem cell-targeting strategy. J Control Release (2015) 206:37–48. doi: 10.1016/j.jconrel.2015.03.008
170. Sansone P, Ceccarelli C, Berishaj M, Chang Q, Rajasekhar VK, Perna F, et al. Self-renewal of Cd133(Hi) cells by Il6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat Commun (2016) 7:10442. doi: 10.1038/ncomms10442
171. Dekkers JF, van Vliet EJ, Sachs N, Rosenbluth JM, Kopper O, Rebel HG, et al. Long-term culture, genetic manipulation and xenotransplantation of human normal and breast cancer organoids. Nat Protoc (2021) 16(4):1936–65. doi: 10.1038/s41596-020-00474-1
172. Berger Fridman I, Kostas J, Gregus M, Ray S, Sullivan MR, Ivanov AR, et al. High-throughput microfluidic 3d biomimetic model enabling quantitative description of the human breast tumor microenvironment. Acta Biomater (2021) 132:473–88. doi: 10.1016/j.actbio.2021.06.025
173. Sabatier R, Finetti P, Mamessier E, Adelaide J, Chaffanet M, Ali HR, et al. Prognostic and predictive value of Pdl1 expression in breast cancer. Oncotarget (2015) 6(7):5449–64. doi: 10.18632/oncotarget.3216
174. Zhang J, Zhang G, Zhang W, Bai L, Wang L, Li T, et al. Loss of Rbms1 promotes anti-tumor immunity through enabling pd-L1 checkpoint blockade in triple-negative breast cancer. Cell Death Differ (2022) 9(24):19. doi: 10.1038/s41418-022-01012-0
175. Gibson J. Anti-Pd-L1 for metastatic triple-negative breast cancer. Lancet Oncol (2015) 16(6):e264. doi: 10.1016/S1470-2045(15)70208-1
176. Byrd TT, Fousek K, Pignata A, Szot C, Samaha H, Seaman S, et al. Tem8/Antxr1-specific car T cells as a targeted therapy for triple-negative breast cancer. Cancer Res (2018) 78(2):489–500. doi: 10.1158/0008-5472.CAN-16-1911
177. Han Y, Xie W, Song DG, Powell DJ Jr. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated Nkg2d car T cells. J Hematol Oncol (2018) 11(1):92. doi: 10.1186/s13045-018-0635-z
178. Phi LTH, Sari IN, Yang YG, Lee SH, Jun N, Kim KS, et al. Cancer stem cells (Cscs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int (2018) 2018:5416923. doi: 10.1155/2018/5416923
179. Shibue T, Weinberg RA. Emt, cscs, and drug resistance: The mechanistic link and clinical implications. Nat Rev Clin Oncol (2017) 14(10):611–29. doi: 10.1038/nrclinonc.2017.44
Keywords: breast cancer stem cells, breast cancer, heterogeneity, tumor microenvironment, transcription factors, non-coding RNAs
Citation: Xu H, Zhang F, Gao X, Zhou Q and Zhu L (2022) Fate decisions of breast cancer stem cells in cancer progression. Front. Oncol. 12:968306. doi: 10.3389/fonc.2022.968306
Received: 13 June 2022; Accepted: 26 July 2022;
Published: 15 August 2022.
Edited by:
Maria Rosaria De Miglio, University of Sassari, ItalyReviewed by:
Arunasalam Dharmarajan, Sri Ramachandra Institute of Higher Education and Research, IndiaCopyright © 2022 Xu, Zhang, Gao, Zhou and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linhai Zhu, eXp6bGhhaUAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.