 Stefan S. Bielack
Stefan S. Bielack
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 08 September 2022
Sec. Head and Neck Cancer
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.966073
This article is part of the Research Topic Primary Bone Sarcomas of the Craniofacial Skeleton: Special Biology, Challenging Treatment and Reconstruction View all 5 articles
Introduction: Craniofacial bones may be the site of origin of various sarcomas. We review the various malignancies affecting this region of the body and attempt to put systemic treatment approaches into perspective.
Material and methods: Non-systematic literature review
Results: Conventional types of osteosarcoma, Ewing sarcoma, and chondrosarcoma are the most frequent bone sarcomas occurring in craniofacial region, but variants may occur. The tumors’ biologies and the resulting treatment strategies vary distinctly. As a general rule, local control remains paramount regardless of histology. The efficacy of antineoplastic chemotherapy varies by type of malignancy. It is clearly indicated in Ewing sarcoma and related tumors, potentially of benefit in high-grade osteosarcoma, undifferentiated pleomorphic sarcoma, dedifferentiated and mesenchymal chondrosarcoma, and of no proven benefit in the others.
Conclusions: Various histologies demand various and distinct treatment approaches, with local control remaining paramount in all. The efficacy of systemic treatments varies by type of tumor. Prospective trials would help in all of these to better define systemic treatment strategies.
Systemic therapy is an integral part of therapy for some of the most common malignant bone tumors. Successful approaches were, however, generally developed outside of the head and neck. They may not always apply to this region of the body without modifications. This review aims to focus on systemic therapies for the various histotypes of craniofacial bone sarcomas, especially where it deviates from other sites of the body.
Osteosarcoma is the most frequent primary bone cancer. It mainly affects teenagers and young adults and there the extremity bones (1, 2). Most osteosarcomas are high-grade, but there are some rare low-grade malignancies which carry a lower risk of systemic spread and are treated by surgery alone. The further text refers to high-grade tumors. These carry a very high risk of metastases, mainly to the lungs, rarer to distant bones or even other sites (1, 2). Micrometastases are likely even if the disease appears localized at diagnosis.
Accordingly, local therapy of the primary alone will rarely lead to cure. Only the introduction of chemotherapy into a multimodal treatment context resulted in frequent cure (3). Doxorubicin, high-dose methotrexate, cisplatin, and ifosfamide are considered the most effective agents. Local treatment, however, remains essential. Surgery must result in wide margins. The whole tumor, covered by an unviolated cuff of soft tissue, must be removed in one piece (4). With surgery and intensive chemotherapy, 60-70% of patients with seemingly localized extremity disease may be cured (5). The outlook is much worse for patients with primary metastases (6).
With below 5-10% of all primaries, the craniofacial bones rank among osteosarcoma´s rarer sites. The median age of affected patients is far older than in the more common extremity locations (7, 8). There is a preponderance of secondary malignancies at this location, particularly after prior radiotherapy (9). Radiotherapy was then often administered for other cancers. The most notable example is radiotherapy given for retinoblastoma (10, 11). Other cancers, however, may also have been present.
Local treatment principles appropriate for extremity osteosarcoma also apply to the head and neck region. There, local control poses much more of a challenge than in the limbs, as amputation cannot be an option even for the most extensive lesions. Owing to the reduced ability to achieve wide surgical margins (4), the local failure rate is much higher than usual. Radiotherapy can only be a substitute for surgery if administered at very high dosages, probably exceeding 60-70 Gy. Such doses are more likely to be reached with proton or heavy ion irradiation (12–16), which may offer an option for selected craniofacial osteosarcomas.
Craniofacial osteosarcomas may not metastasize quite as frequently as their extremity counterparts. This may be due to more of the tumors having a low-grade histology. Hence, the use of systemic chemotherapy is not universally accepted (17). A true multitude of usually small mono-institutional series has described outcomes with varying forms of treatment. In such analyses, none, some, or all craniofacial osteosarcomas may have been treated with chemotherapy, other therapies may have differed. It is nay impossible to make any clear deductions about the role of antineoplastic therapy from these reports.
At the end of the last millennium, a meta-analysis of the literature finally pointed to a potential beneficial effect of adjuvant treatment, although its magnitude remained a matter of debate (18). Another meta-analysis, published almost simultaneously, came to quite the opposite conclusion (19). A summary of these and other relevant publications investigating the matter is presented in Table 1. The reported outcomes are puzzling and any opinion will find a report to support it. This may be so because investigators may have been inclined to cure local therapeutic inadequacies by systemic chemotherapy. This, however, is bound to fail at any site.
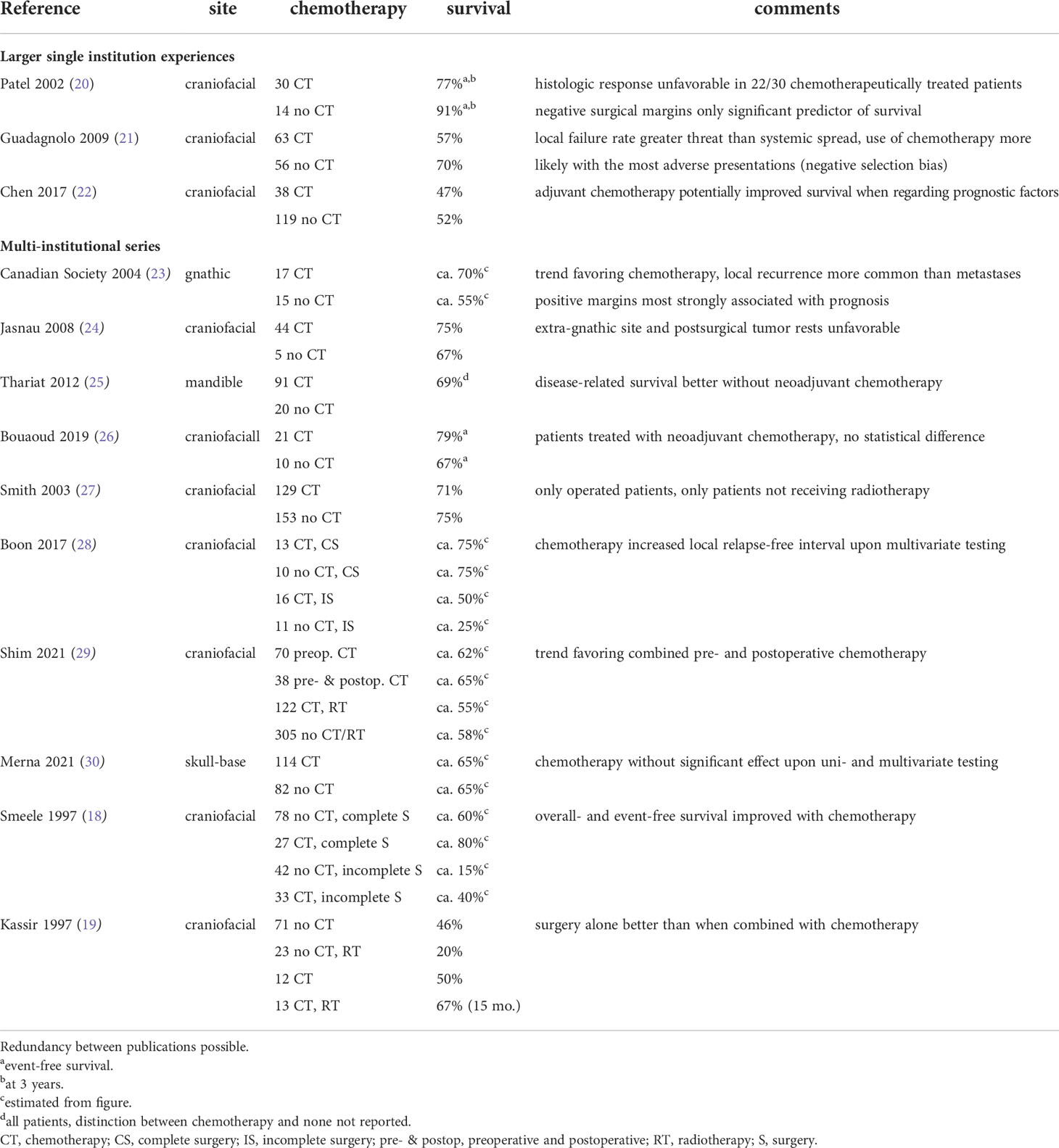
Table 1 Five-year survival of patients with craniofacial osteosarcoma receiving or not receiving chemotherapy.
With the data available, the general view has become that patients affected by craniofacial osteosarcomas benefit to some extent from systemic chemotherapy. Accordingly, the current European guidelines favor a multimodal - local plus systemic - approach (5). The choice of drugs then resembles that used against its extremity counterpart (2). It must be noted that older adults do often not tolerate high-dose methotrexate, an integral part of chemotherapy in the young (31). Ifosfamide may offer a reasonable substitute (32). Patients over the age of 65 generally tolerate chemotherapy very poorly and there is no evidence-base on which to decide if and which regimen to use. Single agent doxorubicin may be one option, as may be others.
The response of an osteosarcoma to preoperative chemotherapy has been shown to be a major prognostic factor (33). Predicting this response can be of value in helping to decide the next therapeutic steps. Various methods exist, none is close to perfect. An analysis specifically focusing on head and neck osteosarcomas was able to demonstrate that 18FDG PET/CT was more reliable than standard imaging in evaluating response to neo-adjuvant chemotherapy in this location (34).
The administration of targeted therapies, especially tyrosine kinase inhibitors, may prolong life for a few months in patients with unresectable extremity osteosarcomas (35–40). Such therapy may also be considered for craniofacial lesions. Unfortunately, targeted therapies alone will never be curative in any location.
Sometimes, tumors which would otherwise be classified as osteosarcoma do not produce any osteoid. These lesions are then characterized as undifferentiated pleomorphic sarcoma (UPS; formerly: malignant fibrous histiocytoma, MFH) (41). Their overall treatment strategy, prognostic factors and outcomes closely resemble that of its more frequent counterpart, osteosarcoma, even though the tumor response rate to chemotherapy seems to be lower (42, 43). Without relevant data about craniofacial primaries, it seems appropriate to treat such tumors just as one would osteosarcomas, with systemic chemotherapy.
Ewing sarcomas are fully malignant tumors which may arise in bone or, rarer, soft tissues. Primary sites can be in the extremities or axial skeleton. Tumors mostly affect young adolescents, but may occur at any age (44). Treatment can only be successful if a combination of local (surgery, radiation, or both) and systemic chemotherapy is administered. Current regimens include an anthracycline, generally doxorubicin, alkykators, vincristine, and etoposide (45). The so called augmented interval-compressed VDC/IE-scheme (vincristine, doxorubicin, cyclophosphamide/ifosfamide, etoposide) (46) may be more efficacious and less toxic than the VIDE-regimen (vincristine, ifosfamide, doxorubicin, etopside), which was previously used in many European counties (47). High-dose chemotherapy with stem-cell rescue has its role in some highly selected patients (47). If treated appropriately, some two thirds of affected individuals may become long-term survivors (44).
Ewing tumors primarily affecting craniofacial bones are detected in no more than five percent of patients (48). In the American Intergroup Ewing’s Sarcoma Study (IESS), for instance, it was reported that this osseous site was affected in approximately 4% of primaries (49). Craniofacial Ewing sarcomas, hence, may be considered rare even in the largest treatment centers. As an example, the University of Florida reported only eight such individuals observed over a 40-year period (50).
Ewing sarcomas of the craniofacial bones have been the focus of multiple publications. Mono-centric reports include, for example, a series of 14 patients who were subjected to tumor surgery, adjuvant chemotherapy, and radiotherapy. Eight patients survived five years or longer (51). The University of Florida reported no more than nine chemotherapeutically treated patients, of which one was clearly of extraosseous origin. Two thirds became 10-year survivors (50). A study from Toronto focused on the sino-nasal tract and maxillary bone and reported eight affected patients with follow-up data, of whom six survived for 1 – 158 months, 5 without evidence of disease (52). An Indian group reported an event-free survival of 59% at five years for 25 chemotherapeutically treated osseous primaries of the head and neck, three of which had metastases (53). The Mayo Clinic published 17 chemotherapeutically treated craniofacial Ewing sarcomas, five of these in the cervical spine, two with primary metastases. Five-year overall survival was given as 87% (54).
A summary of multi-centric analyses is presented in Table 2. The Intergroup Ewing’s Sarcoma Study (IESS) reported that head and neck tumors comprised only 4% of all primaries, the gnathic bones being most commonly affected. Their prognosis was found to be significantly better than that of Ewing sarcoma in general (49). The Italian Association of Pediatric Hematology and Oncology reported a ten year actuarial survival of 64% in 21 multi-modally treated pediatric patients with localized Ewing sarcomas of the craniofacial bones (55). The German-Dutch Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH) reported 51 craniofacial Ewing sarcomas treated on the E.W.I.N.G.-99 trial. Their median age was 12 years. Approximately nine out of ten tumors were of osseous origin. Three-year event-free survival was 74% for 44 patients with localized disease (56). Forty-seven French patients were registered on the same Euro-E.W.I.N.G.-99 trial, 42 of those arose within bone. Primary metastases were observed in less than 10% of affected individuals. Three-year, event-free survival was reported as 79% (57).
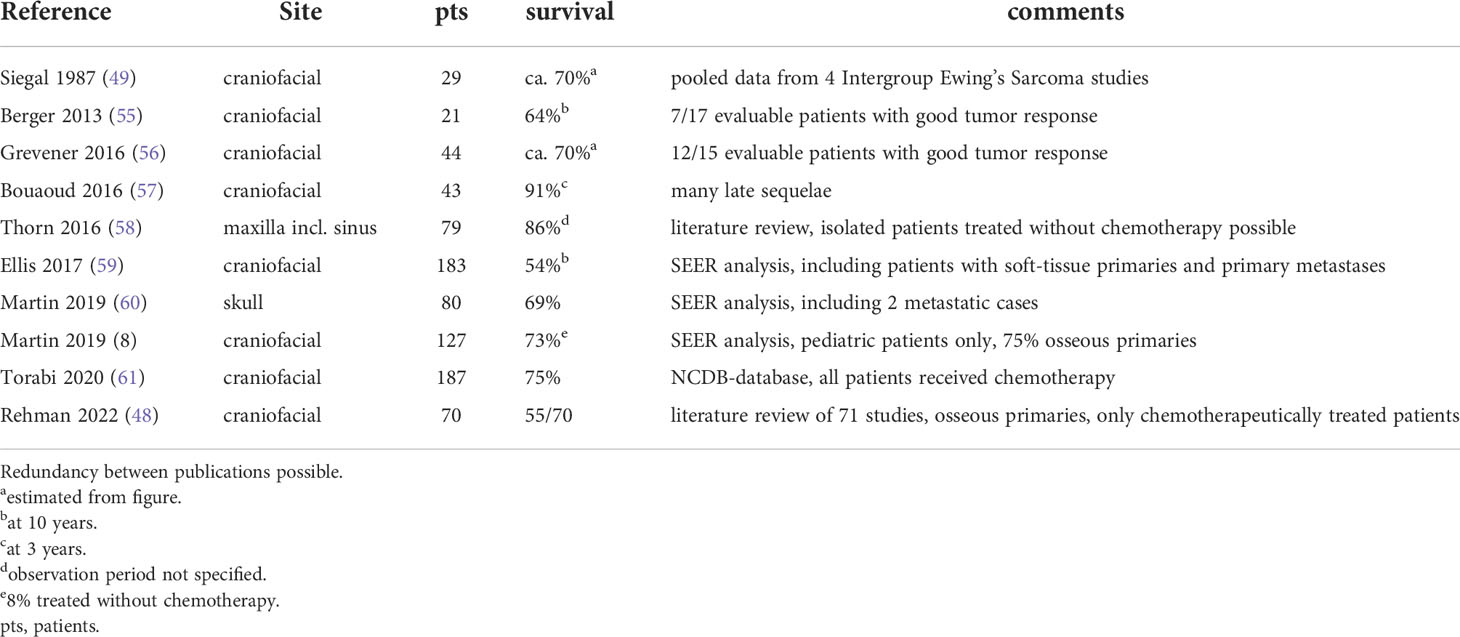
Table 2 Overview of multi-institutional analyses of chemotherapy in Ewing sarcoma of the head and neck.
As for reports about pooled data, Thorn et al. reviewed the published literature on Ewing tumors of the maxilla and maxillary sinus and found 93 cases. Over 90% of patients received combined local and systemic treatment, 70/79 individuals with any follow-up information remained alive, 68 of these disease-free (58). A Surveillance, Epidemiology, and End Results Program (SEER)-analysis of 183 craniofacial primaries found a lower tumor size and metastatic rate and a superior survival rate for craniofacial compared to other primaries (59). Another SEER-analysis identified 127 pediatric patients with Ewing sarcomas of the head and neck, of which some three quarters were of osseous origin. Five-year overall survival was reported as 72.9%, without any effect of age, sex, or irradiation (60). Focusing on 80 Ewing sarcomas of the skull, the same authors observed a five-year survival of 69% (8). A review of the National Cancer Database (NCDB) found 75% of chemotherapeutically treated patients with craniofacial Ewing sarcomas to survive beyond five years (61). Finally, a review of pertinent publications showed 55 of 70 chemotherapeutically treated individuals with craniofacial tumors to survive (48).
Recently, a rare malignant tumor characterized by the EWSR1:Friend leukemia integration 1 (FLI1)-translocation and complex epithelial differentiation was described and characterized as adamantinoma-like Ewing sarcoma (62). In even more uncommon instances, it seems to arise from osseous sites. Awareness of the morphologic and immunohistochemistry spectrum of this tumor is definitely required, but there is no justification for treating it differently from conventional Ewing sarcoma. It remains to be determined whether these tumors are only morphologically or also clinically distinct.
In summary, craniofacial location per se does not provide any reason to modify systemic Ewing sarcoma treatment, that is: All patients are to receive (neo-)adjuvant chemotherapy. If treated such, the prognosis of patients with craniofacial Ewing sarcomas may even be somewhat better than in other sites. It must be noted that, due to a limited amount of soft tissue able to hide tumor growth, craniofacial Ewing sarcomas are usually rather small when detected. As tumor size is an important prognostic factor in this disease (44, 45), this probably leads to a prognostic benefit for craniofacial primaries.
In recent years, several molecularly defined tumors which morphologically resemble Ewing sarcoma but carry distinct genetic translocations have been identified (63–65). Among these, protein capicua homolog (CIC)- and B-cell lymphoma 6 corepressor (BCOR)-rearranged sarcomas along with sarcomas carrying Ewing sarcoma breakpoint region 1 (EWSR1)-non-erythroblast transformation specific (ETS) fusions feature most prominently. A growing variety of others is also being described.
The tumor biology and the clinical course associated with these heterogeneous malignancies vary greatly. Tumors with CIC-rearrangements usually arise in adults and seem to behave even more aggressively than classical Ewing tumors. They are hence associated with a particularly poor outcome (66). Primaries often involve the soft tissues rather than bone and seem to affect craniofacial sites only very infrequently. The exact role of systemic therapy and the optimal regimen to be used are still open, even with tumors of the more frequent locations.
Tumors with BCOR-alterations, on the other hand, seem to have a more favorable outlook. Patients tend to be younger than those with CIC-fusions, with a male predominance (67). Many patients receive chemotherapy as for Ewing sarcoma. Again, specific information about craniofacial tumors is very hard to come by.
Even less can be said about other translocations. At present, all should be included in appropriate trials and registries to learn more about their respective behavior. Until that is achieved, there seems to be no reason to choose one particular systemic therapy regimen over another.
Conventional chondrosarcomas are mesenchymal cartilaginous tumors of, generally, older adults. A significant proportion have their origin not in bone, but in soft tissues. While many are not highly malignant initially, recurrences tend to be of a higher grade, pointing to the need for meticulous local therapy (5).
This tumor-type is considered largely resistant to systemic therapies. Treatment is therefore generally by surgery only, with radiotherapy and, particularly, cytotoxic chemotherapy reserved for truly desperate situations (5). Drugs employed then often resemble those used against osteosarcoma. A very recent review suggested that chondrosarcoma patients might benefit from antiangiogenic therapy and that tumors harboring isocitrate dehydrogenase 1 (IDH1)-mutations might benefit from treatment with IDH1-inhibitors. Mammalian target of rapamycin (mTOR)-inhibitors and tyrosine kinase inhibitors were suggested for relapsed disease (68).
Rarely, chondrosarcomas may also arise in craniofacial sites. It is challenging o delineate any therapeutic differences in relation to this location. A review of the literature on skull-base chondrosarcomas concluded that maximal safe resection followed by radiotherapy was the treatment of choice for Grade II and III lesions, while there was no current role for chemotherapy (69). A literature review of 161 sino-nasal chondrosarcomas suggested aggressive surgical resection as the most common treatment modality for this condition, with adjuvant radiotherapy being used for prevention of local recurrence after subtotal or total resection, but, again, did not elaborate on chemotherapy (70).
Larygeal primaries may form a distinct subgroup among head and neck chondrosacomas. They involve the cricoid, thyroid cartilage, epiglottis, or arytenoid cartilages and represent these structures’ most frequent malignancy. Treatment is again local, systemic chemotherapy being very rarely employed. Disease-specific survival at 10 years has been reported as 82% in a recent systematic review of 592 patients. Here, only.2% of patients received chemotherapy (71).
In summary, the role of chemotherapy for conventional craniofacial chondrosarcoma, even if high-grade, is close to zero.
Dedifferentiated chondrosarcoma is morphologically and clinically very distinct from its conventional counterpart. This malignancy arises from a conventional chondrosarcoma, which is often low-grade, by dedifferentiation. It may then resemble any type of spindle cell sarcoma. The tumor extremely often metastasizes and the prognosis is hence dismal (72).
The only collaborative, prospective trial of 57 eligible patients, 34 of those with primarily localized disease, recently suggested that a multi-drug chemotherapy regimen originally developed against osteosarcoma might have some efficacy in this condition. Median overall survival at five years was reported as 39%. However, the number of craniofacial primaries, if any, was not specified (73). Systemic treatment options may include immunotherapy in IDH1-mutant tumors. This is also still under discussion (68).
Data on systemic therapy for craniofacially located dedifferentiated chondrosarcomas is sparse to almost non-existent. It was suggested that chemotherapy improved survival in a series of only 6 such lesions of the skull-base (74), but the numbers were so small that it was difficult to draw any firm conclusions. However, there is also no evidence suggesting that these tumors would behave differently than in the rest of the body. So, if the evidence was considered sufficient, it should also be followed for craniofacial primaries.
Mesenchymal chondrosarcoma is a malignant tumor which may arise intra- or extraosseously. It is a small cell malignancy containing well-differentiated cartilage. Of note, it seems to be a fusion driven tumor, with Hes Related Family BHLH Transcription Factor With YRPW Motif 1: nuclear receptor coactivator 2 (HEY1-NCOA2) and interferon regulatory factor 2 binding protein 2: caudal type homeobox 1 (IRF2BP2-CDX1) fusions having recently been described. The disease may cause very late recurrences which manifest well over a decade after initial presentation. Surgery remains the mainstay of treatment (75).
Previously, from a limited experience, it was believed that chemotherapy was not effective (76). This may have to be questioned. In an intergroup series of 15 chemotherapeutically treated pediatric patients, 6/15 tumors were of craniofacial origin, only 4 of all tumors were located intraosseously. Actuarial 10-year overall and event-free survival were 67% and 53%, respectively (77). A European multicenter analysis of patients aged 11-80 years found 13% of 113 cases to arise in the head and neck, of whom 53/96 with localized disease received chemotherapy. The median progression free and overall survival for all 96 were 7 and 20 years, respectively. Primary site did not affect survival. Here, chemotherapy administration was definitely associated with reduced risks of recurrence and death (78).
The available data now argues for adjuvant chemotherapy in this type of tumor, with little reliable data on craniofacial lesions in particular. The optimal drug combination to be employed has not been defined. Mostly, patients will be offered Ewing based or, rarer, osteosarcoma based approaches.
Clear cell chondrosarcoma is another uncommon, slowly growing variant of chondrosarcoma which usually affects extracranial sites. There, the tumor is treated by local therapy only (79). There is no reason to do so differently for craniofacial primaries.
In addition to those tumors discussed above, a variety of ultra-rare semi-malignant and malignant sarcomas may affect craniofacial bones. These are not further discussed here.
The role of chemotherapy for craniofacial sarcomas of bone differs by histology. It is clearly indicated in Ewing sarcoma and its variants. It may be useful in high-grade osteosarcoma, undifferentiated pleomorphic sarcoma (UPS), dedifferentiated and mesenchymal chondrosarcoma. It is as of completely unproven value in conventional, myxoid, and clear cell chondrosarcoma.
The author confirms being the sole contributor of this work and has approved it for publication.
This study was supported by Förderkreis krebskranke Kinder Stuttgart e. V.
SB reports having received personal funds from Ipsen, Hoffmann-La Roche, Bayer Healthcare, Boehringer-Ingelheim, EISAI, MAP Biopharma, all outside the submitted work.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Meyers PA. Osteosarcoma. In: Pappo A, editor. Pediatric bone and soft tissue sarcomas. Berlin Heidelberg NewYork: Springer (2006), ISBN: ISBN-13 978-3-540-40843-7. p. 219–34.
2. Bielack S, Cable MG, Gorlick R, Hecker-Nolting S, Kager L, Marina N, et al. Osteosarcoma - approach to therapy. In: Arndt CAS, editor. Sarcomas of bone and soft tissues in children and adolescents (Cham, Switzerland:Springer Nature Switzerland AG) (2021). Available at: 10.1007/978-3-030-51160-9_8. Pediatric Oncology.
3. Rosen G, Tan C, Sanmaneechai A, Beattie EJ Jr, Marcove R, Murphy ML. The rationale for multiple drug chemotherapy in the treatment of osteogenic sarcoma. Cancer (1975) 35(3 suppl):936–45. doi: 10.1002/1097-0142(197503)35:3+<936::aid-cncr2820350714>3.0.co;2-b
4. Enneking WF, Spanier SS, Goodman MA. A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop Relat Res (1980) 153:106–20. doi: 10.1097/00003086-198011000-00013
5. Strauss SJ, Frezza AM, Abecassis N, Bajpai J, Bauer S, Biagini R, et al. Bone sarcomas: ESMO-EURACAN-GENTURIS-ERN PaedCan clinical practice guideline for diagnosis, treatment and follow-up. Ann Oncol (2021) 32(12):1520–36. doi: 10.1016/j.annonc.2021.08.1995
6. Kager L, Zoubek A, Pötschger U, Kastner U, Flege S, Kempf-Bielack B, et al. Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol (2003) 21(10):2011–8. doi: 10.1200/JCO.2003.08.132
7. Duchman KR, Gao Y, Miller BJ. Prognostic factors for survival in patients with high-grade osteosarcoma using the surveillance, epidemiology, and end results (SEER) program database. Cancer Epidemiol (2015) 39(4):593–9. doi: 10.1016/j.canep.2015.05.001
8. Martin E, Senders JT, Ter Wengel PV, Smith TR, Broekman MLD. Treatment and survival of osteosarcoma and Ewing sarcoma of the skull: a SEER database analysis. Acta Neurochir (Wien) (2019) 161(2):317–25. doi: 10.1007/s00701-018-3754-y
9. Koshy M, Paulino AC, Mai WY, Teh BS. Radiation-induced osteosarcomas in the pediatric population. Int J Radiat Oncol Biol Phys (2005) 63(4):1169–74. doi: 10.1016/j.ijrobp.2005.04.008
10. Temming P, Arendt M, Viehmann A, Eisele L, Le Guin CH, Schündeln MM, et al. Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatr Blood Cancer (2017) 64(1):71–80. doi: 10.1002/pbc.26193
11. Yamanaka R, Hayano A. Secondary craniofacial sarcomas following retinoblastoma: A systematic review. World Neurosurg (2017) 101:722–730.e4. doi: 10.1016/j.wneu.2017.02.031
12. Ciernik IF, Niemierko A, Harmon DC, Kobayashi W, Chen YL, Yock TI, et al. Proton-based radiotherapy for unresectable or incompletely resected osteosarcoma. Cancer (2011) 117(19):4522–30. doi: 10.1002/cncr.26037
13. Leroy R, Benahmed N, Hulstaert F, Van Damme N, De Ruysscher D. Proton therapy in children: A systematic review of clinical effectiveness in 15 pediatric cancers. Int J Radiat Oncol Biol Phys (2016) 95(1):267–78. doi: 10.1016/j.ijrobp.2015.10.025
14. Demizu Y, Mizumoto M, Onoe T, Nakamura N, Kikuchi Y, Shibata T, et al. Proton beam therapy for bone sarcomas of the skull base and spine: A retrospective nationwide multicenter study in Japan. Cancer Sci (2017) 108(5):972–7. doi: 10.1111/cas.13192
15. Vogel J, Both S, Kirk M, Chao HH, Bagatell R, Li Y, et al. Proton therapy for pediatric head and neck malignancies. Pediatr Blood Cancer (2018) 65:(2). doi: 10.1002/pbc.26858
16. Seidensaal K, Mattke M, Haufe S, Rathke H, Haberkorn U, Bougatf N, et al. The role of combined ion-beam radiotherapy (CIBRT) with protons and carbon ions in a multimodal treatment strategy of inoperable osteosarcoma. Radiother Oncol (2021) 159:8–16. doi: 10.1016/j.radonc.2021.01.029
17. Baumhoer D, Brunner P, Eppenberger-Castori S, Smida J, Nathrath M, Jundt G. Osteosarcomas of the jaws differ from their peripheral counterparts and require a distinct treatment approach. experiences from the DOESAK registry. Oral Oncol (2014) 50(2):147–53. doi: 10.1016/j.oraloncology.2013.10.017
18. Smeele LE, Snow GB, van der Waal I. Osteosarcoma of the head and neck: meta-analysis of the nonrandomized studies. Laryngoscope (1998) 108(6):946. doi: 10.1097/00005537-199806000-00030
19. Kassir RR, Rassekh CH, Kinsella JB, Segas J, Carrau RL, Hokanson JA. Osteosarcoma of the head and neck: meta-analysis of nonrandomized studies. Laryngoscope (1997) 107(1):56–61. doi: 10.1097/00005537-199701000-00013
20. Patel SG, Meyers P, Huvos AG, Wolden S, Singh B, Shaha AR, et al. Improved outcomes in patients with osteogenic sarcoma of the head and neck. Cancer (2002) 95(7):1495–503. doi: 10.1002/cncr.10849
21. Guadagnolo BA, Zagars GK, Raymond AK, Benjamin RS, Sturgis EM. Osteosarcoma of the jaw/craniofacial region: outcomes after multimodality treatment. Cancer (2009) 115(14):3262–70. doi: 10.1002/cncr.24297
22. Chen Y, Gokavarapu S, Shen Q, Liu F, Cao W, Ling Y, et al. Chemotherapy in head and neck osteosarcoma: Adjuvant chemotherapy improves overall survival. Oral Oncol (2017) 73:124–31. doi: 10.1016/j.oraloncology.2017.08.017
23. Canadian Society of Otolaryngology-Head and Neck Surgery Oncology Study Group. Osteogenic sarcoma of the mandible and maxilla: a Canadian review (1980-2000). J Otolaryngol (2004) 33(3):139–44. doi: 10.2310/7070.2004.03013
24. Jasnau S, Meyer U, Potratz J, Jundt G, Kevric M, Joos UK, et al. Craniofacial osteosarcoma experience of the cooperative German-Austrian-Swiss osteosarcoma study group. Oral Oncol (2008) 44(3):286–94. doi: 10.1016/j.oraloncology.2007.03.001
25. Thariat J, Julieron M, Brouchet A, Italiano A, Schouman T, Marcy PY, et al. Osteosarcomas of the mandible: are they different from other tumor sites? Crit Rev Oncol Hematol (2012) 82(3):280–95. doi: 10.1016/j.critrevonc.2011.07.001
26. Bouaoud J, Beinse G, Epaillard N, Amor-Sehlil M, Bidault F, Brocheriou I, et al. Lack of efficacy of neoadjuvant chemotherapy in adult patients with maxillo-facial high-grade osteosarcomas: A French experience in two reference centers. Oral Oncol (2019) 95:79–86. doi: 10.1016/j.oraloncology.2019.06.011
27. Smith RB, Apostolakis LW, Karnell LH, Koch BB, Robinson RA, Zhen W, et al. National cancer data base report on osteosarcoma of the head and neck. Cancer (2003) 98(8):1670–80. doi: 10.1002/cncr.11716
28. Boon E, van der Graaf WT, Gelderblom H, Tesselaar ME, van Es RJ, Oosting SF, et al. Impact of chemotherapy on the outcome of osteosarcoma of the head and neck in adults. Head Neck (2017) 39(1):140–6. doi: 10.1002/hed.24556
29. Shim T, Chillakuru Y, Darwish C, Chalif E, Strum D, Benito DA, et al. Head and neck osteosarcomas: Analysis of treatment trends and survival outcomes in the united states (2004-2016). Head Neck (2021) 43(11):3294–305. doi: 10.1002/hed.26817
30. Merna C, Lehrich BM, Diaz-Aguilar LD, Goshtasbi K, Sahyouni R, Hsu FPK, et al. Determinants of survival in skull base osteosarcoma: A national cancer database study. World Neurosurg (2021) 151:e828–38. doi: 10.1016/j.wneu.2021.04.135
31. Widemann BC, Adamson PC. Understanding and managing methotrexate nephrotoxicity. Oncologist (2006) 11(6):694–703. doi: 10.1634/theoncologist.11-6-694
32. Ferrari S, Bielack SS, Smeland S, Longhi A, Egerer G, Sundby Hall K, et al. EURO-B.O.S.S.: A European study on chemotherapy in bone-sarcoma patients aged over 40: Outcome in primary high-grade osteosarcoma. Tumori (2018) 104(1):30–6. doi: 10.5301/tj.5000696
33. Bielack SS, Kempf-Bielack B, Delling G, Exner GU, Flege S, Helmke K, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol (2002) 20(3):776–90. doi: 10.1200/JCO.2002.20.3.776
34. Frezza AM, Beale T, Bomanji J, Jay A, Kalavrezos N, Dileo P, et al. Is [F-18]-fluorodeoxy-D-glucose positron emission tomography of value in the management of patients with craniofacial bone sarcomas undergoing neo-adjuvant treatment? BMC. Cancer (2014) 14:23. doi: 10.1186/1471-2407-14-23
35. Grignani G, Palmerini E, Dileo P, Asaftei SD, D’Ambrosio L, Pignochino Y, et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: an Italian sarcoma group study. Ann Oncol (2012) 23:508–16. doi: 10.1093/annonc/mdr151
36. Grignani G, Palmerini E, Ferraresi V, D’Ambrosio L, Bertulli R, Asaftei SD, et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. Lancet Oncol (2015) 16):98–107. doi: 10.1016/S1470-2045(14)71136-2
37. Duffaud F, Mir O, Boudou-Rouquette P, Piperno-Neumann S, Penel N, Bompas E, et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: a non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol (2019) 20:120–33. doi: 10.1016/S1470-2045(18)30742-3
38. Davis LE, Bolejack V, Ryan CW, Ganjoo KN, Loggers ET, Chawla S, et al. Randomized double-blind phase II study of regorafenib in patients with metastatic osteosarcoma. J Clin Oncol (2019) 37:1424–31. doi: 10.1200/JCO.18.02374
39. Italiano A, Mir O, Mathoulin-Pelissier S, Penel N, Piperno-Neumann S, Bompas E, et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol (2020) 21:446–55. doi: 10.1016/S1470-2045(19)30825-3
40. Gaspar N, Campbell-Hewson Q, Gallego Melcon S, Locatelli F, Venkatramani R, Hecker-Nolting S, et al. Phase I/II study of single-agent lenvatinib in children and adolescents with refractory or relapsed solid malignancies and young adults with osteosarcoma (ITCC-050). ESMO Open (2021) 6(5):100250. doi: 10.1016/j.esmoop.2021.100250
41. WHO Classification of Tumours Editorial Board. Soft tissue and bone tumours. In: WHO classification of tumours, 5th edition, vol. 3. Geneva, Switzerland: WHO Press, World Health Organization (2020).
42. Picci P, Bacci G, Ferrari S, Mercuri M. Neoadjuvant chemotherapy in malignant fibrous histiocytoma of bone and in osteosarcoma located in the extremities: analogies and differences between the two tumors. Ann Oncol (1997) 8(11):1107–15. doi: 10.1023/a:1008283516969
43. Bielack SS, Schroeders A, Fuchs N, Bacci G, Bauer HC, Mapeli S, et al. Malignant fibrous histiocytoma of bone: a retrospective EMSOS study of 125 cases. European musculo-skeletal oncology society. Acta Orthop Scand (1999) 70(4):353–60. doi: 10.3109/17453679908997824
44. Riggi N, Suvà ML, Stamenkovic I. Ewing’s sarcoma. N Engl J Med (2021) 384(2):154–64. doi: 10.1056/NEJMra2028910
45. Pappo AS, Dirksen U. Rhabdomyosarcoma, Ewing sarcoma, and other round cell sarcomas. J Clin Oncol (2018) 36(2):168–79. doi: 10.1200/JCO.2017.74.7402
46. Womer RB, West DC, Krailo MD, Dickman PS, Pawel BR, Grier HE, et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the children’s oncology group. J Clin Oncol (2012) 30(33):4148–54. doi: 10.1200/JCO.2011.41.5703
47. Whelan J, Le Deley MC, Dirksen U, Le Teuff G, Brennan B, Gaspar N, et al. High-dose chemotherapy and blood autologous stem-cell rescue compared with standard chemotherapy in localized high-risk Ewing sarcoma: Results of Euro-E.W.I.N.G.99 and Ewing-2008. J Clin Oncol (2018) 36(31):JCO2018782516. doi: 10.1200/JCO.2018.78.2516
48. Rehman R, Osto M, Parry N, Awada N, Agemy J, Arianpour K, et al. Ewing Sarcoma of the craniofacial bones: A qualitative systematic review. Otolaryngol Head Neck Surg (2021) 166(4):608–14. doi: 10.1177/01945998211022228
49. Siegal GP, Oliver WR, Reinus WR, Gilula LA, Foulkes MA, Kissane JM, et al. Primary ewing’s sarcoma involving the bones of the head and neck. Cancer (1987) 60(11):2829–40. doi: 10.1002/1097-0142(19871201)60:11<2829::aid-cncr2820601139>3.0.co;2-s
50. Whaley JT, Indelicato DJ, Morris CG, Hinerman RW, Amdur RJ, Mendenhall WM, et al. Ewing tumors of the head and neck. Am J Clin Oncol (2010) 33(4):321–6. doi: 10.1097/COC.0b013e3181aaca71
51. Desai KI, Nadkarni TD, Goel A, Muzumdar DP, Naresh KN, Nair CN. Primary ewing’s sarcoma of the cranium. Neurosurgery (2000) 46(1):62–8; discussion 68-9.
52. Hafezi S, Seethala RR, Stelow EB, Mills SE, Leong IT, MacDuff E, et al. Ewing’s family of tumors of the sinonasal tract and maxillary bone. Head Neck Pathol (2011) 5(1):8–16. doi: 10.1007/s12105-010-0227-x
53. Biswas B, Thakar A, Mohanti BK, Vishnubhatla S, Bakhshi S. Prognostic factors in head and neck Ewing sarcoma family of tumors. Laryngoscope (2015) 125(3):E112–7. doi: 10.1002/lary.24985
54. Olson MD, Van Abel KM, Wehrs RN, Garcia JJ, Moore EJ. Ewing Sarcoma of the head and neck: The Mayo clinic experience. Head Neck (2018) 40(9):1999–2006. doi: 10.1002/hed.25191
55. Berger M, Fagioli F, Abate M, Riccardi R, Prete A, Cozza R, et al. Unusual sites of Ewing sarcoma (ES): a retrospective multicenter 30-year experience of the Italian association of pediatric hematology and oncology (AIEOP) and Italian sarcoma group (ISG). Eur J Cancer (2013) 49(17):3658–65. doi: 10.1016/j.ejca.2013.06.045
56. Grevener K, Haveman LM, Ranft A, van den Berg H, Jung S, Ladenstein R, et al. Management and outcome of Ewing sarcoma of the head and neck. Pediatr Blood Cancer (2016) 63(4):604–10. doi: 10.1002/pbc.25830
57. Bouaoud J, Temam S, Cozic N, Galmiche-Rolland L, Belhous K, Kolb F, et al. Ewing’s sarcoma of the head and neck: Margins are not just for surgeons. Cancer Med (2018) 7(12):5879–88. doi: 10.1002/cam4.1801
58. Thorn D, Mamot C, Krasniqi F, Metternich F, Prestin S. Multimodality treatment in ewing’s sarcoma family tumors of the maxilla and maxillary sinus: Review of the literature. Sarcoma (2016) 2016:3872768. doi: 10.1155/2016/3872768
59. Ellis MA, Gerry DR, Neskey DM, Lentsch EJ. Ewing Sarcoma of the head and neck. Ann Otol Rhinol Laryngol (2017) 126(3):179–84. doi: 10.1177/0003489416681322
60. Martin E, Radomski S, Harley EH. Pediatric Ewing sarcoma of the head and neck: A retrospective survival analysis. Int J Pediatr Otorhinolaryngol (2019) 117:138–42. doi: 10.1016/j.ijporl.2018.11.026
61. Torabi SJ, Izreig S, Kasle DA, Benchetrit L, Salehi PP, Judson BL. Clinical characteristics and treatment-associated survival of head and neck Ewing sarcoma. Laryngoscope (2020) 130(10):2385–92. doi: 10.1002/lary.28412
62. Rooper LM, Bishop JA. Soft tissue special issue: Adamantinoma-like Ewing sarcoma of the head and neck: A practical review of a challenging emerging entity. Head Neck Pathol (2020) 14(1):59–69. doi: 10.1007/s12105-019-01098-y
63. Sbaraglia M, Righi A, Gambarotti M, Dei Tos AP. Ewing Sarcoma and Ewing-like tumors. Virchows Arch (2020) 476(1):109–19. doi: 10.1007/s00428-019-02720-8
64. Davis JL, Rudzinski ER. Small round blue cell sarcoma other than Ewing sarcoma: What should an oncologist know? Curr Treat Options Oncol (2020) 21(11):90. doi: 10.1007/s11864-020-00785-1
65. Kallen ME, Hornick JL. From the ashes of “Ewing-like” sarcoma: A contemporary update of the classification, immunohistochemistry, and molecular genetics of round cell sarcomas. Semin Diagn Pathol (2022) 39(1):29–37. doi: 10.1053/j.semdp.2021.10.002
66. Antonescu CR, Owosho AA, Zhang L, Chen S, Deniz K, Huryn JM, et al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: A clinicopathologic and molecular study of 115 cases. Am J Surg Pathol (2017) 41(7):941–9. doi: 10.1097/PAS.0000000000000846
67. Kao YC, Owosho AA, Sung YS, Zhang L, Fujisawa Y, Lee JC, et al. BCOR-CCNB3 fusion positive sarcomas: A clinicopathologic and molecular analysis of 36 cases with comparison to morphologic spectrum and clinical behavior of other round cell sarcomas. Am J Surg Pathol (2018) 42(5):604–15. doi: 10.1097/PAS.0000000000000965
68. Rock A, Ali S, Chow WA. Systemic therapy for chondrosarcoma. Curr Treat Options Oncol (2022) 23(2):199–209. doi: 10.1007/s11864-022-00951-7
69. Awad M, Gogos AJ, Kaye AH. Skull base chondrosarcoma. J Clin Neurosci (2016) 24:1–5. doi: 10.1016/j.jocn.2015.10.029
70. Khan MN, Husain Q, Kanumuri VV, Boghani Z, Patel CR, Liu JK, et al. Management of sinonasal chondrosarcoma: a systematic review of 161 patients. Int Forum Allergy Rhinol (2013) 3(8):670–7. doi: 10.1002/alr.21162
71. Chin OY, Dubal PM, Sheikh AB, Unsal AA, Park RC, Baredes S, et al. Laryngeal chondrosarcoma: A systematic review of 592 cases. Laryngoscope (2017) 127(2):430–9. doi: 10.1002/lary.26068
72. Gelderblom H, Hogendoorn PC, Dijkstra SD, van Rijswijk CS, Krol AD, Taminiau AH, et al. The clinical approach towards chondrosarcoma. Oncologist (2008) 13(3):320–9. doi: 10.1634/theoncologist.2007-0237
73. Hompland I, Ferrari S, Bielack S, Palmerini E, Hall KS, Picci P, et al. Outcome in dedifferentiated chondrosarcoma for patients treated with multimodal therapy: Results from the EUROpean bone over 40 sarcoma study. Eur J Cancer (2021) 151:150–8. doi: 10.1016/j.ejca.2021.04.017
74. Raza SM, Gidley PW, Meis JM, Grosshans DR, Bell D, De Monte F. Multimodality treatment of skull base chondrosarcomas: The role of histology specific treatment protocols. Neurosurgery (2017) 81(3):520–30. doi: 10.1093/neuros/nyx042
75. El Beaino M, Roszik J, Livingston JA, Wang WL, Lazar AJ, Amini B, et al. Mesenchymal chondrosarcoma: a review with emphasis on its fusion-driven biology. Curr Oncol Rep (2018) 20(5):37. doi: 10.1007/s11912-018-0668-z
76. Pellitteri PK, Ferlito A, Fagan JJ, Suárez C, Devaney KO, Rinaldo A. Mesenchymal chondrosarcoma of the head and neck. Oral Oncol (2007) 43(10):970–5. doi: 10.1016/j.oraloncology.2007.04.007
77. Dantonello TM, Int-Veen C, Leuschner I, Schuck A, Furtwaengler R, Claviez A, et al. Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: experiences of the CWS and COSS study groups. Cancer (2008) 112(11):2424–31. doi: 10.1002/cncr.23457
78. Frezza AM, Cesari M, Baumhoer D, Biau D, Bielack S, Campanacci DA, et al. Mesenchymal chondrosarcoma: prognostic factors and outcome in 113 patients. a European musculoskeletal oncology society study. Eur J Cancer (2015) 51(3):374–81. doi: 10.1016/j.ejca.2014.11.007
Keywords: osteosarcoma, Ewing sarcoma, chondrosacoma, craniofacial, chemotherapy, cancer
Citation: Bielack SS (2022) Systemic treatment for primary malignant sarcomas arising in craniofacial bones. Front. Oncol. 12:966073. doi: 10.3389/fonc.2022.966073
Received: 10 June 2022; Accepted: 22 August 2022;
Published: 08 September 2022.
Edited by:
Rainer Lutz, University of Erlangen Nuremberg, GermanyReviewed by:
Alberto Righi, Rizzoli Orthopedic Institute (IRCCS), ItalyCopyright © 2022 Bielack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan S. Bielack, Y29zc0BrbGluaWt1bS1zdHV0dGdhcnQuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.