 Hidenori Sahashi1
Hidenori Sahashi1 Akihisa Kato1*
Akihisa Kato1* Michihiro Yoshida1
Michihiro Yoshida1 Kazuki Hayashi1
Kazuki Hayashi1 Itaru Naitoh1
Itaru Naitoh1 Yasuki Hori1Makoto Natsume1Naruomi Jinno1Kenta Kachi1Go Asano1Tadashi Toyohara1Yusuke Kito1
Yasuki Hori1Makoto Natsume1Naruomi Jinno1Kenta Kachi1Go Asano1Tadashi Toyohara1Yusuke Kito1 Sudhakar Ammanamanchi2Hiromi Kataoka1
Sudhakar Ammanamanchi2Hiromi Kataoka1- 1Department of Gastroenterology and Metabolism, Graduate School of Medical Sciences, Nagoya City University, Nagoya, Japan
- 2Department of Internal Medicine, University of Arizona College of Medicine, Phoenix, AZ, United States
Urolithin A (UA; 3,8-dihydroxybenzo[c]chromen-6-one), a metabolite generated by intestinal bacteria during the biotransformation of ellagitannins, has gained considerable attention in treating several cancers. Cholangiocarcinoma (CCA) remains one of the most lethal cancers; it grows in a special environment constantly exposed to both blood and bile. Since UA is known to undergo enterohepatic recirculation, we hypothesized that UA might have significant antitumor effects in CCA. Here, we investigated the therapeutic potential of UA in CCA and aimed to elucidate its mechanisms, including autophagy. UA treatment inhibited cell proliferation and induced G2/M phase cell cycle arrest in CCA cells. UA also suppressed cell migration and invasion, but did not cause apoptosis. Furthermore, Western blotting and immunocytochemistry demonstrated increased LC3-II accumulation, while electron microscopy demonstrated induced autophagosomes after UA treatment, suggesting that UA upregulated autophagy in CCA cells. In xenograft mice treated with UA, tumor growth was inhibited with increased LC3-II levels. On the other hand, phospho-kinase array demonstrated downregulation of the AKT/WNK1 pathway. LC3-II expression was elevated in WNK1 knocked down cells, indicating that WNK1 is the key signal for regulating autophagy. Thus, UA exerted antitumor effects by suppressing the AKT/WNK1 signaling pathway and inducing autophagy. In conclusion, UA, a natural, well-tolerated compound, may be a promising therapeutic candidate for advanced CCA.
Introduction
Natural compounds have been extensively researched over the past several decades for their potential in cancer prevention and treatment (1). Ellagitannins (ETs) are naturally occurring polyphenolic compounds with a wide range of pharmacological effects, including antioxidant, anti-inflammatory, and antitumor effects (2, 3). ETs are hydrolyzed in the gut to release ellagic acid (EA), mainly present in pomegranates, strawberries, blueberries, nuts, and dried fruits (4). However, the absorption of EA is limited due to its hydrophobic nature (5).
Urolithins are metabolites of EA produced by the intestinal bacteria (6). Urolithins are much better absorbed than ETs and EA, and may provide various health benefits such as anti-obesity, antimicrobial, anti-inflammatory, anti-tumor effects (7–9). Various types of urolithins have been identified, including urolithin A (UA; 3,8-dihydroxybenzo[c]chromen-6-one), B (UB), C, and D (10, 11). Urolithins are produced in the gut from tetrahydroxy-urolithin by removal of one of the lactone rings of ellagic acid, and the subsequent removal of a hydroxyl group, resulting in the formation of UA and UB (11). Of these, UA is the major microbial metabolite observed in human, which possess anti-inflammatory and antioxidant properties (12, 13). UA has been found to induce mitophagy efficiently and improve mitochondrial function in the model organism, Caenorhabditis elegans (14). In addition, antitumor effects of UA on lung, prostate, colon, bladder, pancreatic, and neuroblastoma cancers have also been demonstrated (15–21). Several reports indicate that UA induces autophagy, but not mitophagy, in vitro and in vivo (18, 22, 23). Espín et al. reported the pharmacokinetics and tissue distribution of urolithins in Iberian pigs, which feed on oak acorns rich in ellagitannins (24). An analysis of urolithins in plasma, urine, bile, jejunum, colon, and feces revealed that UA undergoes enterohepatic recirculation and, therefore, persists in the body for long periods (24).
Cholangiocarcinoma (CCA) is the second most common primary hepatic malignancy, accounting for 10–20% of newly-diagnosed liver cancers with features of biliary tract differentiation (25, 26). Unfortunately, most CCAs are diagnosed at an advanced stage and have to be treated with systemic chemotherapy instead of surgery. However, effective chemotherapy for CCA is still limited, and the development of new therapies is required. Since CCA grows in a special environment that is constantly exposed to both blood and bile, we hypothesized that UA would have significant antitumor effects in CCA because of enterohepatic recirculation. Despite promising effects in other cancers, the antitumor effects of UA in CCA are currently unknown. We aimed to investigate the antitumor effects of UA in CCA and elucidate its mechanism, including autophagy.
Materials and methods
Cell cultures
Human intrahepatic cholangiocarcinoma cell lines, HuCCT-1 and SSP-25, were obtained from the RIKEN cell bank. All cell lines were cultured in RPMI-1640 medium (FUJIFILM Wako Pure Chemical Corp., Osaka, Japan), supplemented with 10% fetal bovine serum (FBS), in an incubator with 5% CO2 at 37°C.
Cell viability assays
Cell viability was measured using a Cell Counting Kit-8 assay (Dojindo, Kumamoto, Japan), and evaluated by the absorption of WST‐1. The cells were seeded at a density of 4.0 × 103 cells/well on 96-well plates. After overnight incubation, the cells were treated with or without different concentrations of UA (Cayman Chemical Co., Ann Arbor, MI, USA) for 48 h.
Wound-healing assay (scratch assay)
The cells were grown to confluence in 12-well plates, and then a straight wound was made using a sterile 200-μL pipette tip. UA (10 or 40 μmol/L) was then added to the cells. The straight wound was photographed and measured under a microscope at 0 and 12 h. These investigations were independently performed three times.
Transwell invasion assay
Transwell assay was performed using Corning®Matrigel™ Invasion Chamber with 8.0-μm pore membranes (top chamber) for the 24-well culture plate (Corning, NY, USA). The cells were seeded at a density of 2 × 105 (HuCCT-1) cells or 1 × 105 (SSP-25) cells with serum-free FBS in the top chamber of the 24-well plate, and treated with or without UA (10 or 40 μmol/L). Complete medium was added to the lower chamber. After incubation for 24 h, the invading cells were fixed with 10% formalin, stained with crystal violet, and microscopically counted.
Western blot analysis
The cells were lysed in lysis buffer, and 20 µL of protein lysate sample was fractionated on polyacrylamide gels (TGX™ FastCast™ Acrylamide Kit; Bio-Rad Laboratories, Hercules, CA, USA) and then electroblotted to nitrocellulose membranes. The membranes were blocked with 5% skim milk in phosphate buffered saline-Tween 20 (PBS-T). The membranes were incubated with primary and then secondary antibodies. They were then treated with enhanced chemiluminescence detection reagents (Amersham™; Cytiva, Marlborough, MA, USA), and chemiluminescent signals were visualized as bands using a LAS 4000 mini analyzer (Cytiva).
Antibodies against phospho-cdc2 (Try15), cyclin D1, cyclin B1, cleaved caspase-3, caspase-3, phospho-AKT (Ser473), AKT, phospho-WNK1 (Thr60), WNK1, phospho-GSK-3β (Ser9), GSK-3β, phospho-mTOR (Ser2448), and mTOR were purchased from Cell Signaling Technology (Beverly, MA, USA). Monoclonal beta-actin antibody (FUJIFILM Wako Pure Chemical Corp.) was used to probe an internal control.
Flow cytometry analysis
The cells were seeded in 60-mm dishes and cultured overnight, and then treated with or without UA (40 μmol/L) for 24, 48 and 72 h. After treatment, floating and attached cells were collected and stained, and flow cytometric analysis was performed using a flow cytometer (FACSCanto II, BD Biosciences; San Jose, CA, USA). Cell cycles were evaluated by PI staining (PI solution, Dojindo) and apoptosis was detected using the Annexin V Cell Apoptosis Detection Kit 1 (BD Biosciences) according to the manufacturer’s instructions. Camptothecin (Merck, Darmstadt, Germany) was used as a positive control for the apoptosis assay.
Detection of autophagy
Autophagic cells were detected with LC3 using autophagy watch (Medical & Biological Laboratories, Aichi, Japan), according to the manufacturer’s instructions. For Western blot analysis, HuCCT-1 and SSP-25 cells were treated with UA (40 μmol/L) and/or Chloroquine (CQ; 20 μmol/L) for 24 h, and the analysis was performed with 20 µL of protein lysate sample using anti-LC3 monoclonal antibody-HRP-DirecT (Autophagy watch). For immunocytochemistry, the cells were evaluated using anti-LC3 monoclonal antibody (Autophagy watch), with Alexa Fluor 488-conjugated goat anti-rabbit IgG (H + L; Thermo Fisher Scientific, Waltham, MA, USA) as the secondary antibody. All sections were counterstained using 4′,6-diamidino-2-phenylindole (DAPI; Fluoromount-G; Southern Biotech, Birmingham, AL, USA). HuCCT-1 cells were seeded in 4-well glass slides (Lab-Tek® Chamber Slide™ system; Thermo Fisher Scientific) and incubated for 24 h, and then treated under the respective conditions for 24 h. Images were obtained using a confocal laser scanning fluorescence microscope (FV3000; Olympus, Tokyo, Japan).
Transmission electron microscopy
HuCCT-1 cells were seeded at a density of 1.5 × 105 cells/well on 6-well plates. After overnight incubation, the cells were treated with UA (40 μmol/L) and/or CQ (20 μmol/L) for 24 h, and the samples were pre-fixed with 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) at 4°C. After fixation, the specimens were post-fixed with 2% osmium tetroxide in 0.1 M phosphate buffer (pH 7.4) for 45 min. They were subsequently dehydrated in a graded series of ethanol and embedded in epoxy resin. Ultra-thin sections were cut using an Ultracut-UCT (LEICA, Wetzlar, Germany) with a diamond knife, and stained with 2% uranyl acetate in distilled water for 15 min followed by a lead staining solution for 5 min. Sections were examined with a JEM-1400 plus (JEOL, Tokyo, Japan) electron microscope.
Human Phospho-kinase array
Phosphorylated proteins were analyzed using the Human Phospho-Kinase Array Kit (ARY003C; R&D Systems, Minneapolis, MN, USA). HuCCT-1 and SSP-25 cells were treated with or without UA (40 μmol/L) for 3 h and according to the manufacturer’s instructions. Signals were detected using chemiluminescence detection reagents (Amersham™, Cytiva), and array images were analyzed using the ImageJ software.
Transfection
Small interfering RNA (siRNA) transfection was performed using Lipofectamine RNAi-MAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. HuCCT-1 cells were transfected with the desired siRNA using siGENOME non-targeting siRNA (siNT) control pool and siGENOME human WNK1 siRNA SMART pool (Dharmacon, Lafayette, CO, USA). Two days after transfection, the cells were treated with each condition for 3 or 24 h.
In vivo experiments
The protocols for all animal studies were approved by Nagoya City University Center for Experimental Animal Science, and the mice were housed according to the guidelines of Nagoya City University for Animal Experiments. Female nude mice (BALB/c Slc-nu/nu), aged 7 weeks, were obtained from Japan SLC Inc. The mice were acclimatized for 2 weeks before the experiments, and were kept in individual cages with unrestricted access to food and water. All mice were maintained under specific pathogen-free conditions with a 12-h light/dark cycle. To prepare the xenograft models, HuCCT-1 cells were injected into the mouse flanks with 5 × 106 cells in 100 μL of media. One day after implantation, the mice were randomly allocated into two groups. Two weeks after subcutaneous tumor transplantation, UA (20 mg/kg, 3 times a week) or dimethyl sulfoxide (DMSO; control) was administered by oral gavage, as in a previous study (20). The maximum tumor diameter (L) and the diameter at right angles to that axis (W) were measured using calipers twice a week, and the tumor volume was calculated according to the formula: (L×W2)/2. The weights of the mice were also recorded twice a week. The mice were sacrificed 35 days after the start of medication, and the transplanted tumors were excised and fixed in formalin or frozen in liquid nitrogen for protein lysate.
In vivo immunohistochemistry
The tumors were excised, and immediately fixed in formalin and embedded in paraffin blocks. Then, the block specimens were sectioned (4 µm) and stained using Ki-67 antibodies (Cell Signaling Technology). High spot areas were captured under a microscope, and the positive areas were counted visually. Data were expressed as means ± SD (Standard Deviation) of five independent experiments.
Statistical analysis
The data were analyzed using Student’s t test and Mann-Whitney U test. Differences were considered statistically significant at P <.05. Data were expressed as means ± SD.
Results
UA treatment inhibited cell proliferation and induced G2/M phase cell cycle arrest in cholangiocarcinoma cell lines
The chemical structure of UA is shown in Figure 1A. To assess sensitivity for UA, a cell viability assay was performed with HuCCT-1 and SSP-25 cells. We found that the viabilities of the two cell lines treated with UA for 48 h were reduced in a dose-dependent manner (Figure 1B). We further explored the effect of UA on the cell cycle using flow cytometry (FACS). HuCCT-1 and SSP-25 cells treated with 40 μmol/L UA for 48 h showed accumulation of cells in the G2/M phase compared to the controls (control vs. 40 μmol/L UA in HuCCT-1 cells: 21.3± 1.9% vs. 31.5 ± 3.7%; and in SSP-25 cells: 40.9 ± 3.9% vs. 48.5 ± 1.3%, respectively, P < 0.05) (Figure 1C). As shown in Supplementary Figure 1, the G2/M phase cell accumulation was also observed under the conditions treated with UA for 24 or 72 h. Then, to confirm major cell cycle regulators of the G2/M phase, we examined the changes in phospho-cdc2 (Try15), cyclin B1, and cyclin D1 using Western blot analysis. HuCCT-1 cells treated with UA for 48 h upregulated the expression of phospho-CDC2 (Tyr15) and cyclin B1 without influencing cyclin D1 levels, consistent with the observed G2/M cell cycle arrest (Figure 1D).
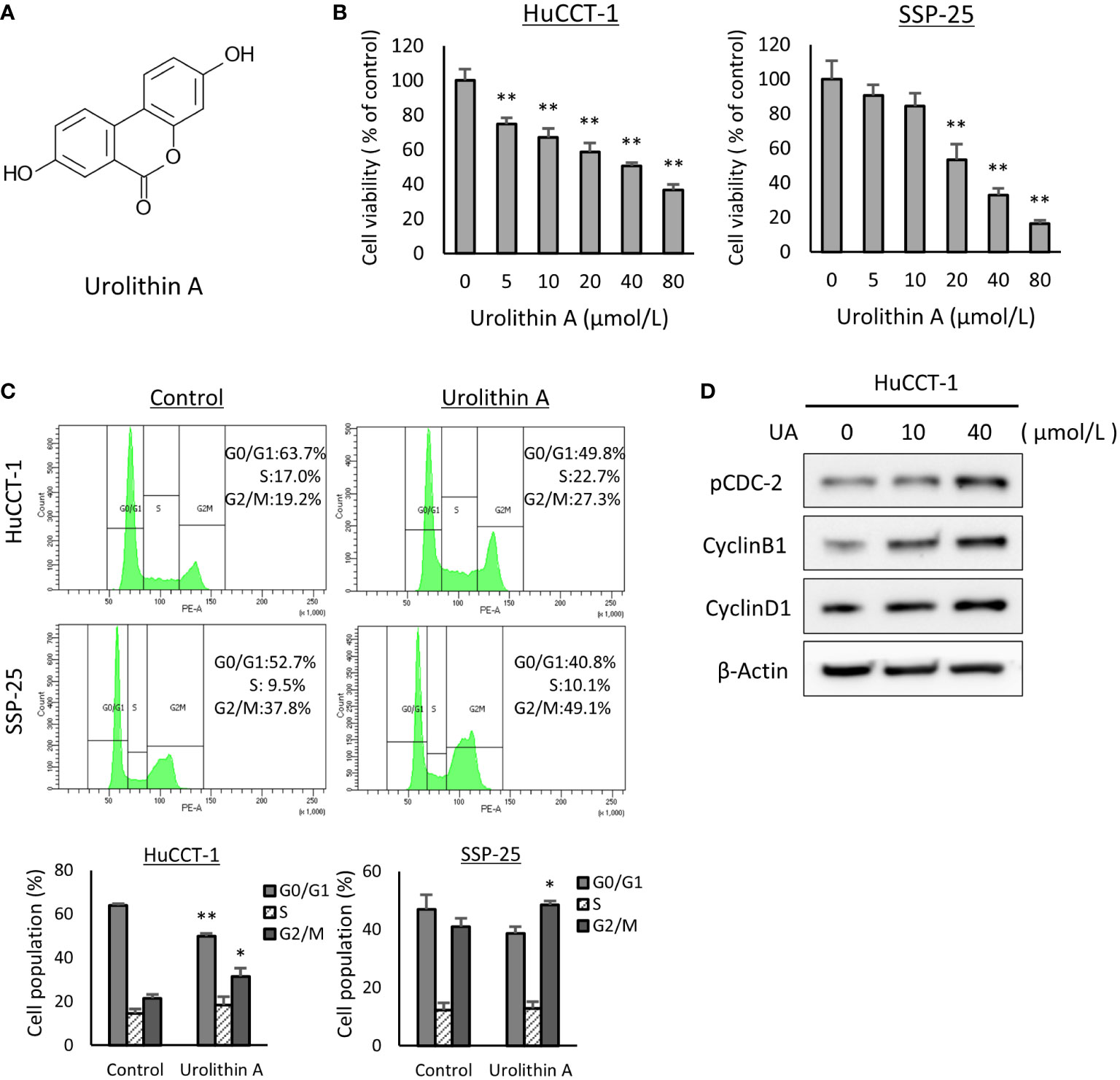
Figure 1 UA treatment inhibits cell proliferation and induces G2/M phase cell cycle arrest in cholangiocarcinoma cell lines. (A) Chemical structures of UA. (B) HuCCT-1 and SSP-25 cells were treated with UA at 0–80 μmol/L for 48 h Cell viability was measured using the Cell Counting Kit-8 assay. Data represent the means of three independent experiments. Bars, standard deviation; **P < 0.01. (C) HuCCT-1 and SSP-25 cells were treated with 0 or 40 μmol/L UA for 48 h Cell cycles were determined using flow cytometry. Data represent the means of three independent experiments. Bars, standard deviation; *P < 0.05; **P < 0.01. (D) HuCCT-1 were treated with 0, 10, or 40 μmol/L UA for 48 h Expression of cell cycle regulators was analyzed by Western blotting for phospho (p)-cdc2 (Try15), cyclin B1, and Cyclin D1. β-actin was used as internal loading control.
Effects of UA on cell migration, invasion, and apoptosis progression in cholangiocarcinoma cell lines
To evaluate the effects of UA on cell migration, we conducted a wound-healing assay. UA treatment (40 μmol/L) significantly suppressed cell migration in both HuCCT-1 (0, 10, and 40 μmol/L UA: 81.2 ± 9.0%, 74.6 ± 15.5%, and 38.1 ± 9.3%, respectively, P < 0.01) and SSP-25 (0, 10, and 40 μmol/L UA: 74.1 ± 7.1%, 64.8 ± 1.9%, and 36.6 ± 3.0%, respectively, P < 0.01) cells (Figure 2A). We also performed the transwell assay to evaluate the effects of UA on cell invasion. UA significantly inhibited cell invasion at 40 μmol/L in both HuCCT-1 (0, 10 and 40 μmol/L UA: 1.0 ± 0.097, 0.91 ± 0.094, and 0.43 ± 0.106, respectively, P < 0.01) and SSP-25 (0, 10, and 40 μmol/L UA: 1.0 ± 0.119, 0.90 ± 0.091, and 0.63 ± 0.143, respectively, P < 0.01) cells (Figure 2B).

Figure 2 Effects of UA on cell migration, invasion, and apoptosis progression in cholangiocarcinoma cell lines. (A) Representative images obtained at 12 h after a scratch wound was made in confluent monolayers of HuCCT-1 and SSP-25 cells. After the scratch, 0, 10, or 40 μmol/L of UA were added. The migration rates were quantified by measuring the area of the injured region. Data represent the means of three independent experiments. Bars, standard deviation; **P < 0.01. (B) Representative transwell-membrane images stained with crystal violet show invasion cells after 12 h of treatment with 0, 10, or 40 μmol/L UA in HuCCT-1 and SSP-25 cells. Quantitative analysis of the invasion cells was expressed as fold change relative to untreated controls. Data represent the means of three independent experiments. Bars, standard deviation; **P < 0.01. (C) HuCCT-1 cells were treated with 0, 10, or 40 μmol/L UA for 24 h, and then stained with annexin-V FITC and PI. Apoptosis cells were evaluated using flow cytometry. (D) HuCCT-1 cells were treated with 40 μmol/L UA for 0, 1, 3, 6, or 24 h Expression of apoptosis-related factors was analyzed by Western blotting for cleaved caspase-3 and caspase-3. β-actin was used as an internal loading control.
To investigate the effects of UA on apoptosis, we used the AnnexinV-FITC/PI staining method with flow cytometry. As shown in Supplementary Figure 2, 30 μmol/L Camptothecin for 24 h was used as a positive control for the apoptosis assay. Interestingly, there was no difference between control and UA treatment in the percentage of apoptotic cells in HuCCT-1 cells, treated with or without UA for 24h (0, 10, and 40 μmol/L UA: 10.6%, 10.3% and 8.4%) (Figure 2C). And, as shown in Supplementary Figure 3, there was also no difference between them under the conditions treated with UA for 48 or 72 h. We also examined the effects of UA on apoptosis-related factors, total and cleaved caspase-3, using Western blot analysis. There were no apparent changes in the total and cleaved caspase-3 in HuCCT-1 cells treated with 40 μmol/L UA for 0, 1, 3, 6, or 24 h (Figure 2D).
UA-mediated upregulation of autophagy in cholangiocarcinoma cells
Increased LC3-II levels and the formation of LC3 puncta were used to determine whether UA treatment induced autophagy in cholangiocarcinoma cells. To confirm the contribution of UA treatment to autophagy, we performed autophagy flux assay with CQ, which blocks the fusion of autophagosomes with lysosomes and inhibits late-stage autophagy. We first examined the effects of UA on autophagy using Western blot analysis. It was found that UA treatment for 24 h caused an increase in LC3-II levels in HuCCT-1 and SSP-25 cells. CQ induced LC3-II expression, and addition of UA led to further accumulation of LC3-II in HuCCT-1 cells (Figure 3A). In addition, immunofluorescent staining revealed that UA, CQ, and their combination significantly increased LC3 puncta accumulation in the cytoplasm of cells compared to control (Figure 3B). Furthermore, transmission electron microscopy (TEM) demonstrated that there were more autophagosomes and autolysosomes in HuCCT-1 cells treated with UA for 24 h. After combined treatment with UA and CQ, autophagosomes that had stopped prematurely were clearly observed in the cytoplasm (Figure 3C).
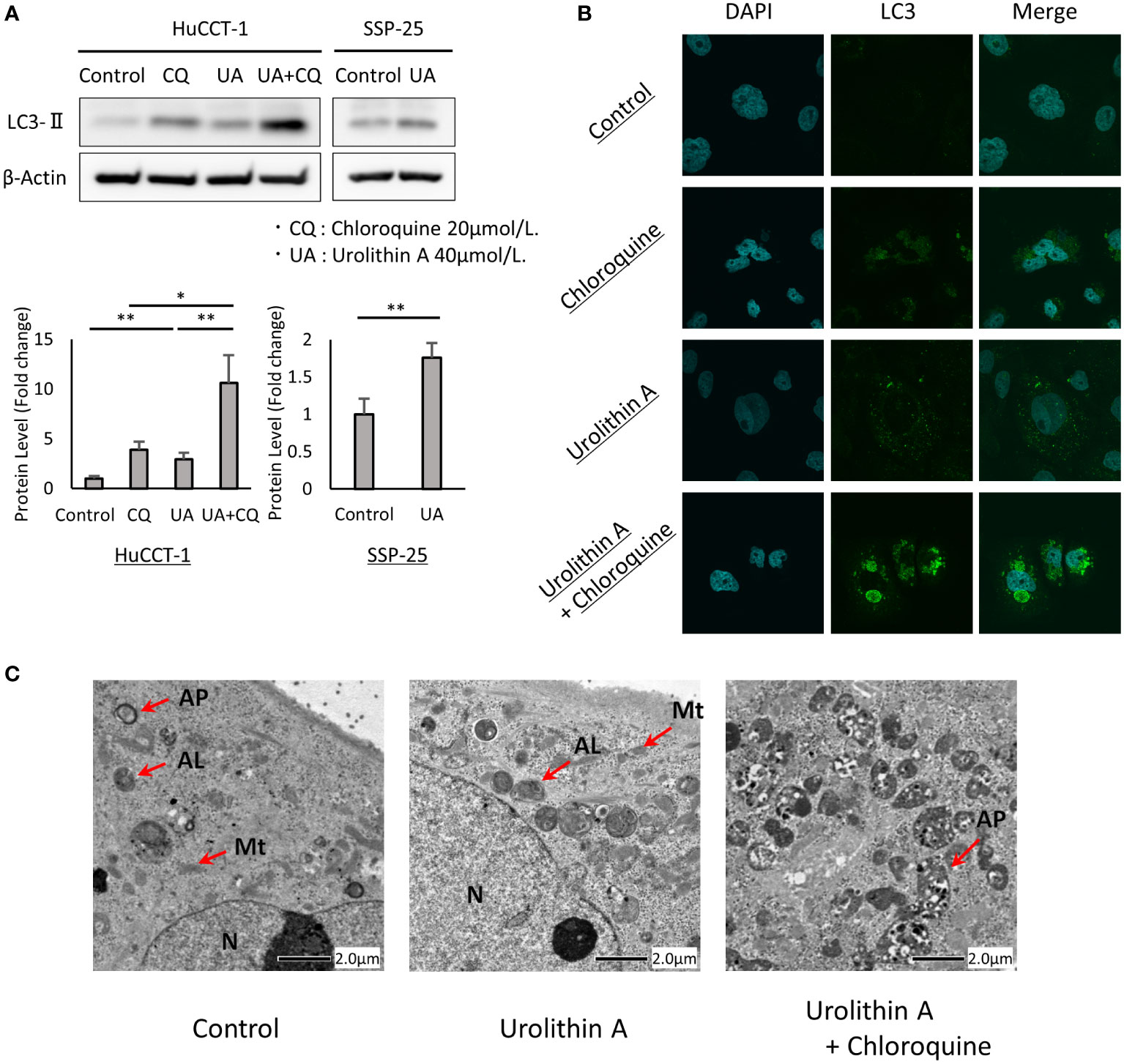
Figure 3 UA-mediated upregulation of autophagy in cholangiocarcinoma cells. (A) HuCCT-1 and SSP-25 cells were treated with 20 μmol/L CQ and 40 μmol/L UA for 24 h Autophagy was detected by Western blotting for LC3-II. β-actin was used as an internal loading control. LC3-II levels were normalized against β-Actin and represented the means of three independent experiments. Bars, standard deviation; *P < 0.05; **P < 0.01. (B) Immunofluorescence for LC3 (green) was performed after the same treatment as shown in (A). Blue staining denotes DAPI-labeled nuclei. (C) Electron microscopy after the same treatment as shown in (A). N, nucleus; Mt, mitochondrion; AP, autophagosome; AL, autolysosome.
UA inhibited xenograft tumor growth in vivo
The above-mentioned results demonstrated the efficacy of UA in cholangiocarcinoma cells. To verify these effects in vivo, we subcutaneously injected HuCCT-1 cells into the flank of nude mice as xenograft models. UA (20 mg/kg, 3 times a week) or DMSO (control) was administered by oral gavage for 35 days (Figure 4A). There was no body weight loss in the treatment group compared to the control group during the treatment (data not shown), which suggested that the volume of UA used was not harmful to the mice. Tumor volume and weight significantly reduced in the UA-treated mice compared to controls (Figures 4B, C). The proliferative potential of mice tumor samples were analyzed by Ki-67 immunostaining. The number of Ki-67 positive cells in the high spot area was significantly suppressed in the UA treatment group compared to the control group (Figure 4D). Western blot analysis revealed that the UA treatment group had significantly higher LC3-II levels than the control group (Figure 4E). These results suggested that UA could suppress tumor growth and might induce autophagy in cholangiocarcinoma.
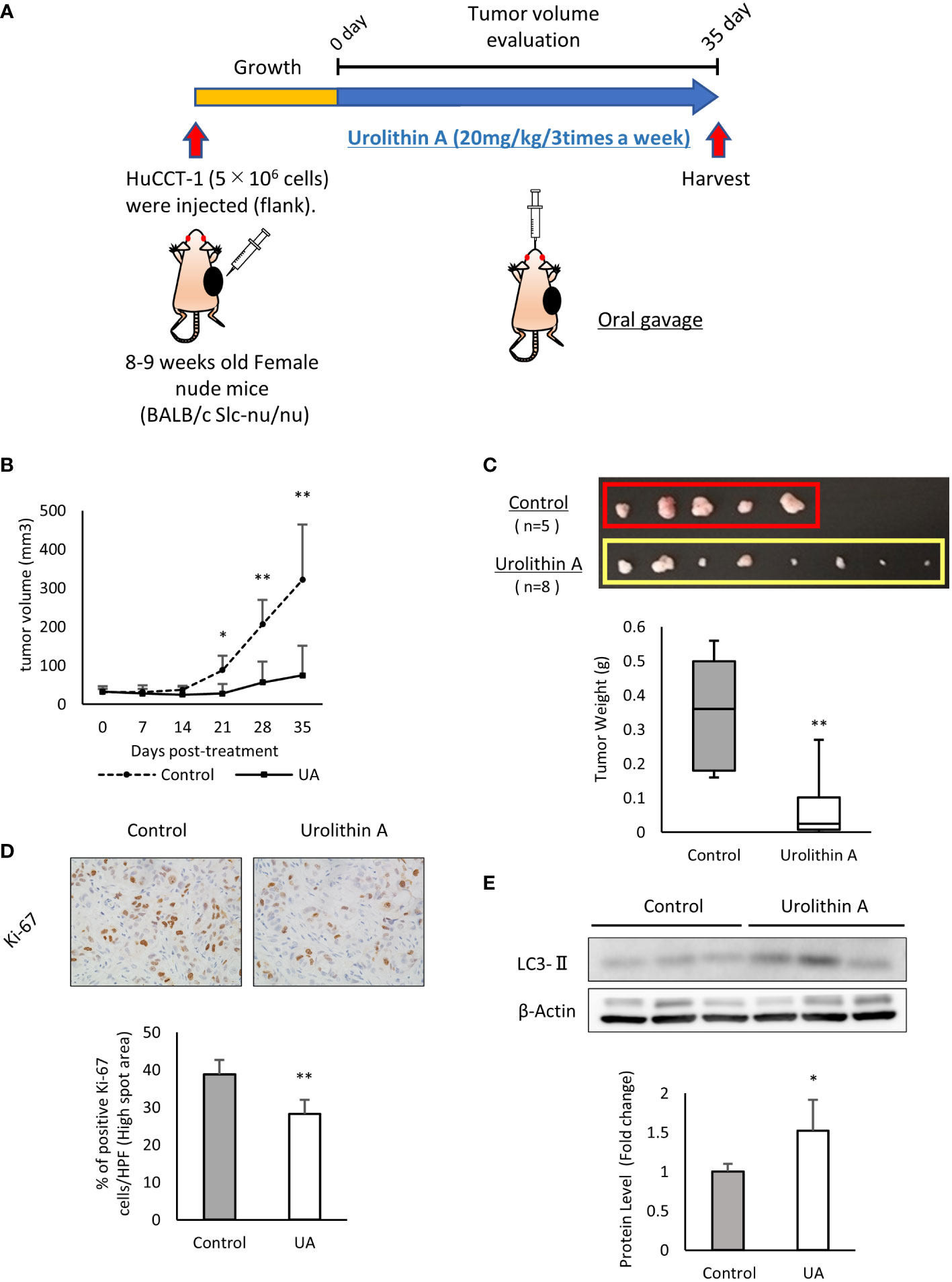
Figure 4 UA inhibits xenograft tumor growth in vivo. (A) Experimental design for UA treatment in the xenograft model. HuCCT-1 cells were injected into the flank of nude mice. UA (20 mg/kg, 3 times a week) or DMSO (control) were administered by oral gavage for 35 days. (B) The volume of subcutaneous tumors in the xenograft model was measured twice a week. Data represent the means of the control group (n = 5) or the UA group (n = 8). Bars, standard deviation; *P < 0.05; **P < 0.01. (C) Representative macroscopic images of tumors in nude mice obtained at day 35 after the start of the treatment. Data represent the means of the control group (n = 5) or the UA group (n = 8). Bars, standard deviation; **P < 0.01. (D) Representative Ki67-stained immunohistochemical images of the two groups. The positive rates of Ki67 staining were quantified by measuring five high spot areas from each tumor. Data represent the means of the control group (n = 4) or the UA group (n = 4). Bars, standard deviation; **P < 0.01. (E) Autophagy was detected by Western blotting for LC3-II in the two groups. β-actin was used as an internal loading control. LC3-II levels were normalized against β-Actin and represented the means of three independent experiments. Bars, standard deviation; *P < 0.05.
UA treatment downregulated AKT and WNK1 pathways, and induced autophagy in cholangiocarcinoma cells
To clarify the key regulatory pathways of UA treatment, we utilized the human Phospho-kinase array. UA treatment downregulated the expressions of phospho-WNK1, phospho-AKT, and phospho-GSK-3β in HuCCT-1 and SSP-25 cells (Figure 5A). The significant changes of phosphorylation for WNK1 and AKT were also confirmed in the two cell lines using Western blot analysis, but were not seen in GSK-3β (Figure 5B). Therefore, we hypothesized that UA treatment might induce autophagy via the AKT/WNK1 pathway. To verify our hypothesis, we analyzed LC3-II expression in WNK1 knocked down HuCCT-1 cells. Western blot analysis for LC3-II revealed that the targeted knockdown of WNK1 elevated LC3-II protein without UA treatment, suggesting the importance of WNK1 in the activation of autophagy. Furthermore, UA treatment indicated similar up-regulation of LC3-II, regardless of the knockdown of WNK1. These results suggested that UA induced autophagy mainly via the AKT/WNK1 pathway (Figure 5C). In addition, Western blotting with IGF-1, known as the AKT-WNK1 signal activator, demonstrated that UA downregulated phospho-WNK1 levels even in HuCCT-1 cells treated with IGF-1. We further assessed the mTOR activity, which is another pathway for induction of autophagy, by measuring phosphorylation of mTOR, and found that the mTOR pathway was not affected by UA treatment and WNK1 knockdown (Figure 5D).
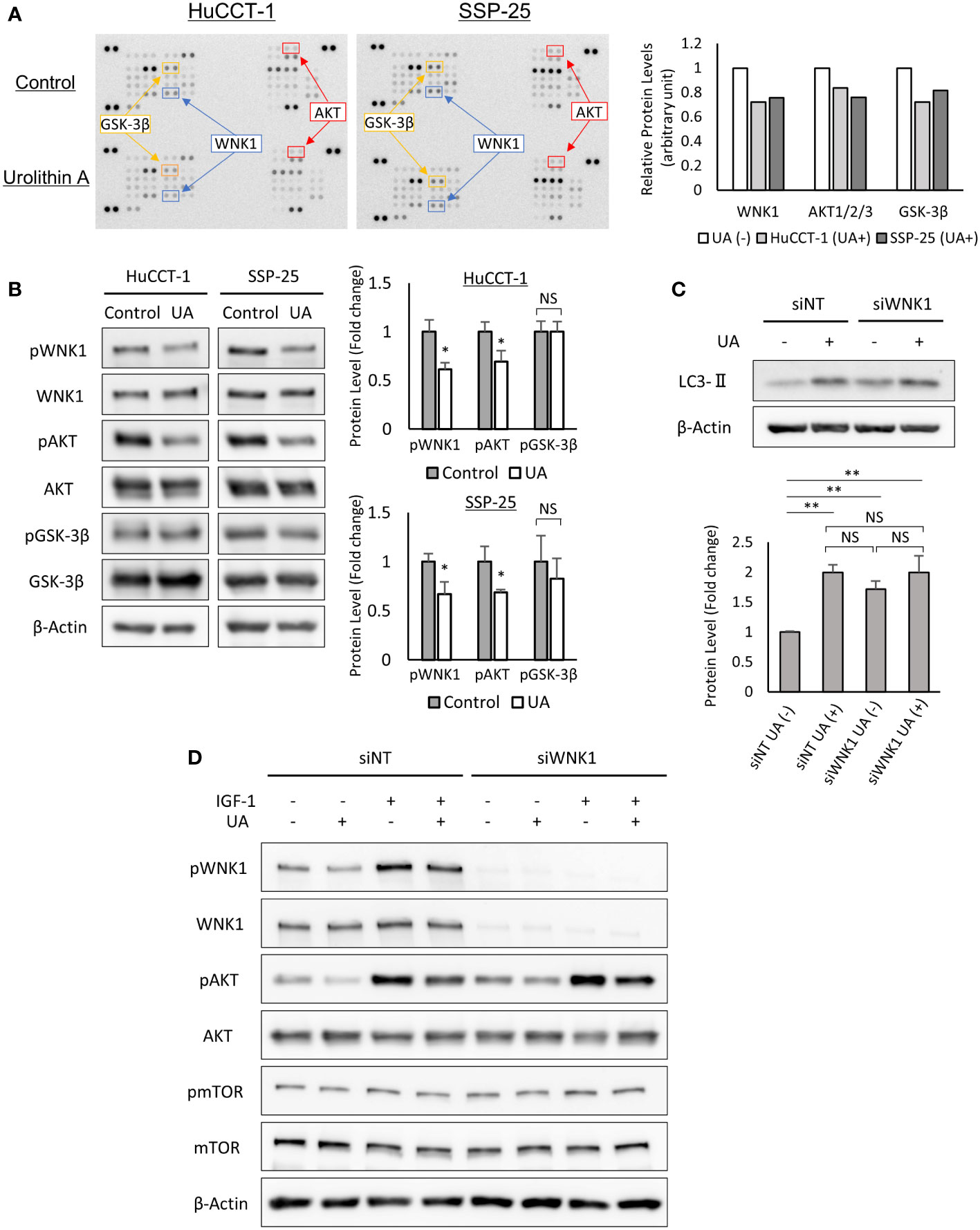
Figure 5 UA treatment downregulated AKT and WNK1 pathways, and induced autophagy in cholangiocarcinoma cells. (A) HuCCT-1 and SSP-25 cells were treated with 40 μmol/L UA for 3 h and analyzed using the human Phospho-Kinase array. Relative levels of protein phosphorylation (normalized intensity for each antibody) were quantified as a ratio of the UA-treated sample to the untreated one. (B) Results of the human Phospho-Kinase array were verified by Western blotting. β-actin was used as an internal loading control. Protein phosphorylation levels were normalized against β-Actin and represented the means of three independent experiments. Bars, standard deviation; NS, not significant; *P < 0.05. (C) Western blotting for LC3-II in WNK1 knocked down HuCCT-1 cells. Cells were treated with 40 μmol/L UA for 24 h β-actin was used as an internal loading control. LC3-II levels were normalized against β-Actin and represented the means of three independent experiments. Bars, standard deviation; NS, not significant; **P < 0.01. (D) Western blotting for WNK1 (Thr60 and total), mTOR (Ser2448 and total), and AKT (Ser473 and total) in HuCCT-1 cells transfected with control (siNT) or WNK1-specific (siWNK1) small interfering RNAs. Cells were treated with 40 μmol/L UA and 50 ng/mL IGF-1 for 3 h.
Discussion
Systemic chemotherapy with a combination of gemcitabine and is globally considered the standard first-line therapy for advanced CCA (27). However, effective chemotherapy for CCA is still limited, and the development of new therapies has not proressed sufficiently. Many targeted therapies for CCA, targeting FGFR2 fusions (28), IDH mutations (29, 30), major downstream pathways (31), and growth factor receptors (32), have been reported. However, clinical trials on therapies that appeared promising on basic research have not led to clinical breakthroughs due to various challenges (33).
UA is a metabolite generated by intestinal bacteria after ingestion of EA- and ET-rich foods and health supplements (34). UA is reported to have antitumor effects in many cancers, such as lung, prostate, colon, bladder, pancreatic, and neuroblastoma cancers (15–21). Given that UA undergoes enterohepatic recirculation (24), we speculated that UA might have significant antitumor effects in CCA, which grows in a special environment that is constantly exposed to both blood and bile. In this study, we demonstrated that UA showed antitumor effects by inhibiting cell viability, migration, and invasion in HuCCT-1 and SSP-25 cells (Figure 1). In addition, UA administration dramatically reduced tumor growth in a xenograft mice model (Figure 4).
The mechanism of the antitumor effects of UA is characterized by various factors that regulate intracellular molecule targets, ultimately influencing cancer cell survival. Our data suggested that UA showed antitumor effects mainly via autophagy in cholangiocarcinoma cells (Figure 3). Autophagy is a self-degradative process required to maintain cellular homeostasis, development, differentiation, survival, and death (35). In cancer, suppression or induction of autophagy can exert antitumor effects through promotion of cell death or survival, which are two main therapeutic targets (36). Thus, it is essential to identify key autophagy targets for new therapeutic agents. Previous studies have reported a cross-talk relationship between autophagy and apoptosis in anti-tumor therapy (37), but in our study, UA treatment did not lead to apoptosis (Figure 2). We suggest that UA significantly affects cancer cell survival by inducing autophagy in cholangiocarcinoma.
Autophagy is mainly mediated through the PI3K/Akt/mTOR and AMPK/mTOR signaling pathways, the molecular mechanisms by which mTOR kinase induces autophagy (36). We examined the change in mTOR phosphorylation after UA treatment and found that UA did not cause any change in mTOR phosphorylation (Figure 5D). Kankanamalage et al. reported that reduced WNK1 expression accelerates autophagy independently of the mTOR signaling pathway (38). In concordance with that report, we found that WNK1 knockdown induced autophagy regardless of the mTOR signaling pathway in HuCCT-1 cells (Figures 5C, D).
WNKs (With-no-lysine kinases) are a family of four serine-threonine protein kinases, WNK1–4, with an atypical placement of the catalytic lysine (39). Initial attention was focused on these enzymes as regulators of blood pressure because mutations of two family members, WNK1 and WNK4, caused pseudohypoaldosteronism type II, a heritable form of hypertension (39). WNK1 was also reported to be involved in PI3K-AKT pathway activation in several cancers (40). Likewise, our study indicated that IGF-1 stimulation upregulated AKT phosphorylation in WNK1 knockdown cells, indicating that WNK1 was downstream of AKT (Figure 5D). From these results, we proposed a schematic representation of the signaling pathway involved in the inhibition of cancer growth by UA-modulation of the AKT/WNK1 axis (Figure 6).
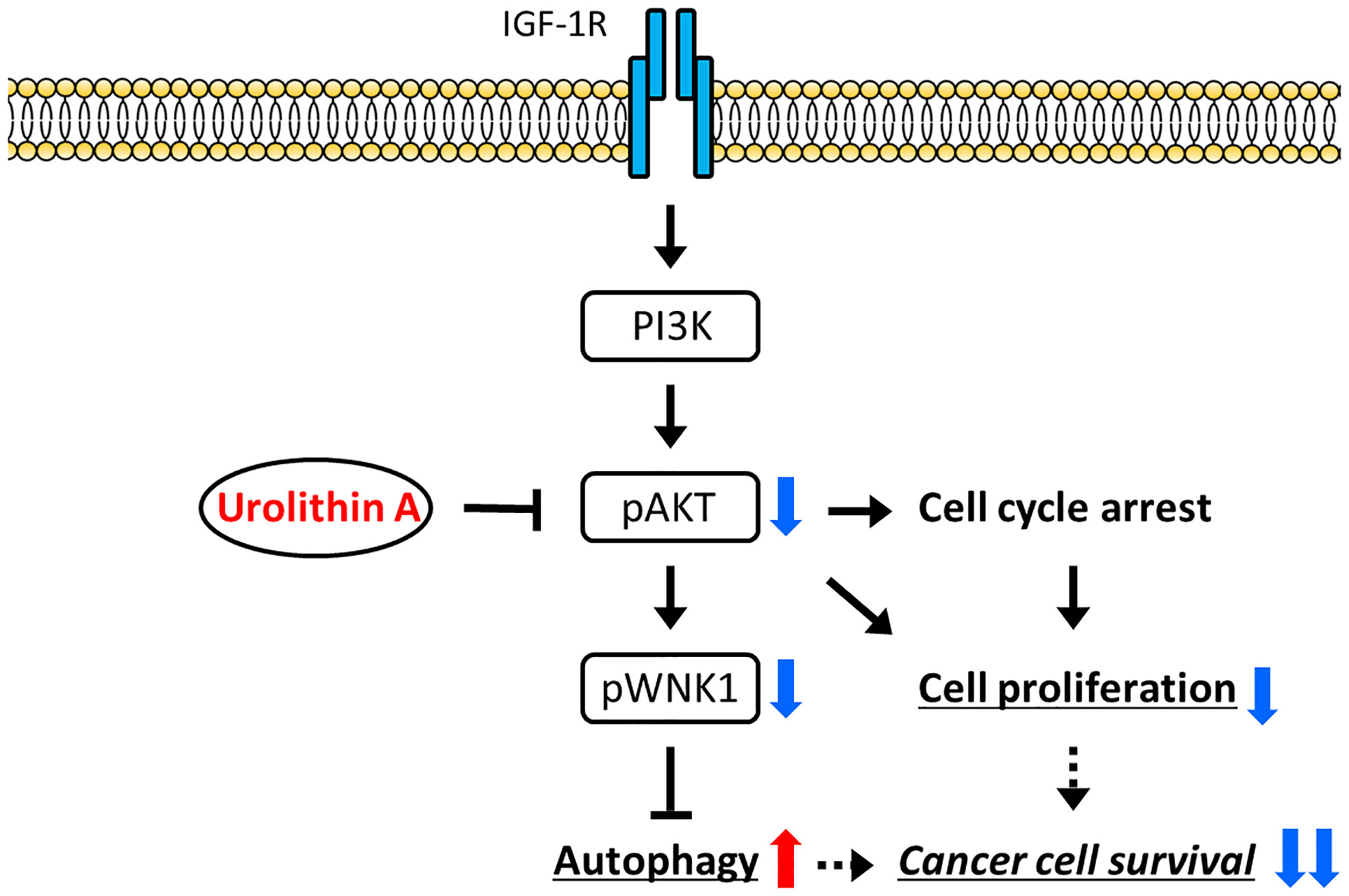
Figure 6 A proposed model of the mechanism. UA treatment reduces cell proliferation by inhibiting the activation of AKT, and inducing autophagy via the WNK1 pathway. As a result, cancer cell survival is suppressed by UA in cholangiocarcinoma cells.
According to recent pharmacokinetic studies, UA is reported to undergo phase-II metabolism, to be mainly glucuronides, after absorption (41). Several in vitro studies showed that UA phase-II metabolites have lower bioactivity than deconjugated UA, including anti-tumor effects and inflammation (42–44). However, in the present study, UA oral administration exerts significant anti-tumor effects in xenograft model. Some reports indicated UA glucuronides are susceptible to β-Glucuronidase, which is known to present at high concentration in the microenvironment of most solid cancers (45, 46). On the basis of these findings, we speculated that β-Glucuronidase might be related to deconjugation of UA in in vivo study. Further investigation is needed.
In terms of safety of UA supplementation, a human clinical study revealed that UA was biologically safe and improved mitochondrial function in older adults (47). A recent randomized, double-blind, placebo-controlled clinical study demonstrated that daily 1000-mg UA supplementation in healthy older adults for 4 months was biologically safe, and improved muscle endurance and mitochondrial health (48). In our study, the UA dose used in mice (20 mg/kg) was convertible to a human equivalent dose (HED) of approximately 1.62 mg/kg for adults (49), and is expected to be safe. The potential clinical application of UA appears promising on the basis of its safety and benefits.
Collectively, our in vitro and in vivo data revealed that UA exerted antitumor effects by suppressing the AKT/WNK1 signaling pathway and inducing autophagy. Thus, UA, a natural, well-tolerated compound, may be a promising therapeutic candidate for advanced CCA.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by Nagoya City University Center for Experimental Animal Science.
Author contributions
AK and HS, MY designed the study, and drafted the paper. HS mainly contributed in vitro and in vivo data on cholangiocarcinoma cells. MN and NJ, KK, GA, TT performed in vivo experiments. MY and IN, YH, YK analyzed the data. SA and KH, HK finalized and revised the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by a JSPS KAKENHI grant number 22K15973 and Pancreas Research Foundation of Japan (to AK), and a JSPS KAKENHI grant number 20K08291 (to MY), and a JSPS KAKENHI grant numbert 17K09479 (to IN).
Acknowledgments
We acknowledge the assistance of the Research Equipment Sharing Center at the Nagoya City University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.963314/full#supplementary-material
Supplementary Figure 1 | UA treatment inhibits cell proliferation and induces G2/M phase cell cycle arrest in cholangiocarcinoma cell lines. HuCCT-1 and SSP-25 cells were treated with 0 or 40 μmol/L UA for 24 and 72 h. Cell cycles were determined using flow cytometry. Data represent the means of three independent experiments. Bars, standard deviation; **P < 0.01.
Supplementary Figure 2 | Positive control for the apoptosis assay. HuCCT-1 cells were treated with 30 μmol/L Camptothecin for 24 h, and then stained with annexin-V FITC and PI. Apoptosis cells were evaluated using flow cytometry.
Supplementary Figure 3 | Effects of UA on apoptosis progression. HuCCT-1 cells were treated with 0 or 40 μmol/L UA for 48 or 72 h, and then stained with annexin-V FITC and PI. Apoptosis cells were evaluated using flow cytometry.
References
1. Newman DJ, Cragg GM. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Natural Prod (2020) 83(3):770–803. doi: 10.1021/acs.jnatprod.9b01285
2. Ríos JL, Giner RM, Marín M, Recio MC. A pharmacological update of ellagic acid. Planta Med (2018) 84(15):1068–93. doi: 10.1055/a-0633-9492
3. Ismail T, Calcabrini C, Diaz AR, Fimognari C, Turrini E, Catanzaro E, et al. Ellagitannins in cancer chemoprevention and therapy. Toxins (2016) 8(5):151. doi: 10.3390/toxins8050151
4. Ceci C, Lacal PM, Tentori L, De Martino MG, Miano R, Graziani G. Experimental evidence of the antitumor, antimetastatic and antiangiogenic activity of ellagic acid. Nutrients (2018) 10(11):1756. doi: 10.3390/nu10111756
5. Muku GE, Murray IA, Espín JC, Perdew GH. Urolithin a is a dietary microbiota-derived human aryl hydrocarbon receptor antagonist. Metabolites (2018) 8(4):86. doi: 10.3390/metabo8040086
6. Kujawska M, Jodynis-Liebert J. Potential of the ellagic acid-derived gut microbiota metabolite - urolithin a in gastrointestinal protection. World J Gastroenterol (2020) 26(23):3170–81. doi: 10.3748/wjg.v26.i23.3170
7. Abdulrahman AO, Kuerban A, Alshehri ZA, Abdulaal WH, Khan JA, Khan MI. Urolithins attenuate multiple symptoms of obesity in rats fed on a high-fat diet. Diabetes Metab Syndr Obesity: Targets Ther (2020) 13:3337–48. doi: 10.2147/dmso.s268146
8. Abdelazeem KNM, Kalo MZ, Beer-Hammer S, Lang F. The gut microbiota metabolite urolithin a inhibits nf-κb activation in lps stimulated bmdms. Sci Rep (2021) 11(1):7117. doi: 10.1038/s41598-021-86514-6
9. Al-Harbi SA, Abdulrahman AO, Zamzami MA, Khan MI. Urolithins: The gut based polyphenol metabolites of ellagitannins in cancer prevention, a review. Front Nutr (2021) 8:647582. doi: 10.3389/fnut.2021.647582
10. Tomás-Barberán FA, González-Sarrías A, García-Villalba R, Núñez-Sánchez MA, Selma MV, García-Conesa MT, et al. Urolithins, the rescue of “Old” metabolites to understand a “New” concept: Metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status. Mol Nutr Food Res (2017) 61(1):1500901. doi: 10.1002/mnfr.201500901
11. Espín JC, Larrosa M, García-Conesa MT, Tomás-Barberán F. Biological significance of urolithins, the gut microbial ellagic acid-derived metabolites: The evidence so far. Evidence-Based Complement Altern Med: eCAM (2013) 2013:270418. doi: 10.1155/2013/270418
12. Tomás-Barberán FA, García-Villalba R, González-Sarrías A, Selma MV, Espín JC. Ellagic acid metabolism by human gut microbiota: Consistent observation of three urolithin phenotypes in intervention trials, independent of food source, age, and health status. J Agric Food Chem (2014) 62(28):6535–8. doi: 10.1021/jf5024615
13. Ishimoto H, Shibata M, Myojin Y, Ito H, Sugimoto Y, Tai A, et al. In vivo anti-inflammatory and antioxidant properties of ellagitannin metabolite urolithin A. Bioorg. Med Chem Lett (2011) 21(19):5901–4. doi: 10.1016/j.bmcl.2011.07.086
14. Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Félix AA, et al. Urolithin a induces mitophagy and prolongs lifespan in c. Elegans and increases muscle function in rodents. Nat Med (2016) 22(8):879–88. doi: 10.1038/nm.4132
15. Cheng F, Dou J, Zhang Y, Wang X, Wei H, Zhang Z, et al. Urolithin a inhibits epithelial-mesenchymal transition in lung cancer cells via P53-Mdm2-Snail pathway. OncoTargets Ther (2021) 14:3199–208. doi: 10.2147/ott.s305595
16. Mohammed Saleem YI, Albassam H, Selim M. Urolithin a induces prostate cancer cell death in P53-dependent and in P53-independent manner. Eur J Nutr (2020) 59(4):1607–18. doi: 10.1007/s00394-019-02016-2
17. Norden E, Heiss EH. Urolithin a gains in antiproliferative capacity by reducing the glycolytic potential via the P53/Tigar axis in colon cancer cells. Carcinogenesis (2019) 40(1):93–101. doi: 10.1093/carcin/bgy158
18. Zhao W, Shi F, Guo Z, Zhao J, Song X, Yang H. Metabolite of ellagitannins, urolithin a induces autophagy and inhibits metastasis in human Sw620 colorectal cancer cells. Mol carcinogenesis (2018) 57(2):193–200. doi: 10.1002/mc.22746
19. Liberal J, Carmo A, Gomes C, Cruz MT, Batista MT. Urolithins impair cell proliferation, arrest the cell cycle and induce apoptosis in Umuc3 bladder cancer cells. Invest New Drugs (2017) 35(6):671–81. doi: 10.1007/s10637-017-0483-7
20. Totiger TM, Srinivasan S, Jala VR, Lamichhane P, Dosch AR, Gaidarski AA 3rd, et al. Urolithin a, a Novel natural compound to target Pi3k/Akt/Mtor Pathway in pancreatic cancer. Mol Cancer Ther (2019) 18(2):301–11. doi: 10.1158/1535-7163.MCT-18-0464
21. Liu CL, Zhao D, Li JJ, Liu S, An JJ, Wang D, et al. Inhibition of glioblastoma progression by urolithin a in vitro and in vivo by regulating Sirt1-Foxo1 axis via Erk/Akt signaling pathways. Neoplasma (2021) 69(1):80–94. doi: 10.4149/neo_2021_210623N834
22. Zhang Y, Zhang Y, Halemahebai G, Tian L, Dong H, Aisker G. Urolithin a, a pomegranate metabolite, protects pancreatic β cells from apoptosis by activating autophagy. J Ethnopharmacol (2021) 272:113628. doi: 10.1016/j.jep.2020.113628
23. Ahsan A, Zheng YR, Wu XL, Tang WD, Liu MR, Ma SJ, et al. Urolithin a-activated autophagy but not mitophagy protects against ischemic neuronal injury by inhibiting er stress in vitro and in vivo. CNS Neurosci Ther (2019) 25(9):976–86. doi: 10.1111/cns.13136
24. Espín JC, González-Barrio R, Cerdá B, López-Bote C, Rey AI, Tomás-Barberán FA. Iberian Pig as a model to clarify obscure points in the bioavailability and metabolism of ellagitannins in humans. J Agric Food Chem (2007) 55(25):10476–85. doi: 10.1021/jf0723864
25. Massarweh NN, El-Serag HB. Epidemiology of hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Cancer Control: J Moffitt Cancer Center (2017) 24(3):1073274817729245. doi: 10.1177/1073274817729245
26. Banales JM, Cardinale V, Carpino G, Marzioni M, Andersen JB, Invernizzi P, et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European network for the study of cholangiocarcinoma (Ens-cca). Nat Rev Gastroenterol Hepatol (2016) 13(5):261–80. doi: 10.1038/nrgastro.2016.51
27. Sasaki T, Takeda T, Okamoto T, Ozaka M, Sasahira N. Chemotherapy for biliary tract cancer in 2021. J Clin Med (2021) 10(14):3108. doi: 10.3390/jcm10143108
28. Rizvi S, Yamada D, Hirsova P, Bronk SF, Werneburg NW, Krishnan A, et al. A hippo and fibroblast growth factor receptor autocrine pathway in cholangiocarcinoma. J Biol Chem (2016) 291(15):8031–47. doi: 10.1074/jbc.M115.698472
29. Salati M, Caputo F, Baldessari C, Galassi B, Grossi F, Dominici M, et al. Idh signalling pathway in cholangiocarcinoma: From biological rationale to therapeutic targeting. Cancers (Basel) (2020) 12(11):3310. doi: 10.3390/cancers12113310
30. Wu MJ, Shi L, Dubrot J, Merritt J, Vijay V, Wei TY, et al. Mutant idh inhibits ifnγ-Tet2 signaling to promote immunoevasion and tumor maintenance in cholangiocarcinoma. Cancer Discov (2021) 12(3):812–35. doi: 10.1158/2159-8290.Cd-21-1077
31. Loilome W, Juntana S, Namwat N, Bhudhisawasdi V, Puapairoj A, Sripa B, et al. Prkar1a is overexpressed and represents a possible therapeutic target in human cholangiocarcinoma. Int J Cancer (2011) 129(1):34–44. doi: 10.1002/ijc.25646
32. Sirica AE. Role of erbb family receptor tyrosine kinases in intrahepatic cholangiocarcinoma. World J Gastroenterol (2008) 14(46):7033–58. doi: 10.3748/wjg.14.7033
33. Tella SH, Kommalapati A, Borad MJ, Mahipal A. Second-line therapies in advanced biliary tract cancers. Lancet Oncol (2020) 21(1):e29–41. doi: 10.1016/s1470-2045(19)30733-8
34. Qiu Z, Zhou J, Zhang C, Cheng Y, Hu J, Zheng G. Antiproliferative effect of urolithin a, the ellagic acid-derived colonic metabolite, on hepatocellular carcinoma Hepg2.2.15 cells by targeting Lin28a/Let-7a axis. Braz J Med Biol Res = Rev Bras Pesqui Med e Biol (2018) 51(7):e7220. doi: 10.1590/1414-431x20187220
35. Yang L, Wang Q, Zhao Q, Yang F, Liu T, Huang X, et al. Deglycosylated epcam regulates proliferation by enhancing autophagy of breast cancer cells via Pi3k/Akt/Mtor pathway. Aging (Albany NY) (2022) 14(1):316–29. doi: 10.18632/aging.203795
36. Al-Bari MAA, Ito Y, Ahmed S, Radwan N, Ahmed HS, Eid N. Targeting autophagy with natural products as a potential therapeutic approach for cancer. Int J Mol Sci (2021) 22(18):9807. doi: 10.3390/ijms22189807
37. Xie Q, Liu Y, Li X. The interaction mechanism between autophagy and apoptosis in colon cancer. Transl Oncol (2020) 13(12):100871. doi: 10.1016/j.tranon.2020.100871
38. Gallolu Kankanamalage S, Lee AY, Wichaidit C, Lorente-Rodriguez A, Shah AM, Stippec S, et al. Multistep regulation of autophagy by Wnk1. Proc Natl Acad Sci U.S.A. (2016) 113(50):14342–7. doi: 10.1073/pnas.1617649113
39. Murthy M, Kurz T, O’Shaughnessy KM. Wnk signalling pathways in blood pressure regulation. Cell Mol Life Sci: CMLS (2017) 74(7):1261–80. doi: 10.1007/s00018-016-2402-z
40. Gallolu Kankanamalage S, Karra AS, Cobb MH. Wnk pathways in cancer signaling networks. Cell Commun Signal (2018) 16(1):72. doi: 10.1186/s12964-018-0287-1
41. García-Villalba R, Giménez-Bastida JA, Cortés-Martín A, Ávila-Gálvez M, Tomás-Barberán FA, Selma MV, et al. Urolithins: A comprehensive update on their metabolism, bioactivity, and associated gut microbiota. Mol Nutr Food Res (2022):e2101019. doi: 10.1002/mnfr.202101019
42. Ávila-Gálvez M, Espín JC, González-Sarrías A. Physiological relevance of the antiproliferative and estrogenic effects of dietary polyphenol aglycones versus their phase-ii metabolites on breast cancer cells: A call of caution. J Agric Food Chem (2018) 66(32):8547–55. doi: 10.1021/acs.jafc.8b03100
43. González-Sarrías A, Giménez-Bastida JA, Núñez-Sánchez M, Larrosa M, García-Conesa MT, Tomás-Barberán FA, et al. Phase-ii metabolism limits the antiproliferative activity of urolithins in human colon cancer cells. Eur J Nutr (2014) 53(3):853–64. doi: 10.1007/s00394-013-0589-4
44. Ávila-Gálvez MA, Giménez-Bastida JA, González-Sarrías A, Espín JC. Tissue deconjugation of urolithin a glucuronide to free urolithin a in systemic inflammation. Food Funct (2019) 10(6):3135–41. doi: 10.1039/c9fo00298g
45. Piwowarski JP, Stanisławska I, Granica S, Stefańska J, Kiss AK. Phase ii conjugates of urolithins isolated from human urine and potential role of β-glucuronidases in their disposition. Drug Metab Dispos (2017) 45(6):657–65. doi: 10.1124/dmd.117.075200
46. Tranoy-Opalinski I, Legigan T, Barat R, Clarhaut J, Thomas M, Renoux B, et al. β-Glucuronidase-Responsive prodrugs for selective cancer chemotherapy: An update. Eur J Med Chem (2014) 74:302–13. doi: 10.1016/j.ejmech.2013.12.045
47. Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, et al. The mitophagy activator urolithin a is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab (2019) 1(6):595–603. doi: 10.1038/s42255-019-0073-4
48. Liu S, D’Amico D, Shankland E, Bhayana S, Garcia JM, Aebischer P, et al. Effect of urolithin a supplementation on muscle endurance and mitochondrial health in older adults: A randomized clinical trial. JAMA Netw Open (2022) 5(1):e2144279. doi: 10.1001/jamanetworkopen.2021.44279
Keywords: Urolithin A, UA, cholangiocarcinoma, autophagy, WNK1
Citation: Sahashi H, Kato A, Yoshida M, Hayashi K, Naitoh I, Hori Y, Natsume M, Jinno N, Kachi K, Asano G, Toyohara T, Kito Y, Ammanamanchi S and Kataoka H (2022) Urolithin A targets the AKT/WNK1 axis to induce autophagy and exert anti-tumor effects in cholangiocarcinoma. Front. Oncol. 12:963314. doi: 10.3389/fonc.2022.963314
Received: 07 June 2022; Accepted: 05 September 2022;
Published: 23 September 2022.
Edited by:
Divya P. Kumar, JSS Academy of Higher Education and Research, IndiaReviewed by:
Hayrettin Ozan Gulcan, Eastern Mediterranean University, TurkeyElke Heiss, University of Vienna, Austria
Jakub P. Piwowarski, Medical University of Warsaw, Poland
Copyright © 2022 Sahashi, Kato, Yoshida, Hayashi, Naitoh, Hori, Natsume, Jinno, Kachi, Asano, Toyohara, Kito, Ammanamanchi and Kataoka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akihisa Kato, YWtpaGlzYUBtZWQubmFnb3lhLWN1LmFjLmpw