Abstract
Multiple myeloma (MM) is an incurable plasma cell malignancy with a heterogeneous genetic background. Each MM subtype may have its own therapeutic vulnerabilities, and tailored therapy could improve outcomes. However, the cumulative frequency of druggable targets across patients is very low, which has precluded the widespread adoption of precision therapy for patients with MM. Amplification of the long arm of chromosome 1 (Amp1q) is one of the most frequent genetic alterations observed in patients with MM, and its presence predicts inferior outcomes in the era of proteasome inhibitors and immunomodulatory agents. Therefore, establishing precision medicine for MM patients with Amp1q stands to benefit a large portion of patients who are otherwise at higher risk of relapse. In this article, we review the prevalence and clinical significance of Amp1q in patients with MM, its pathogenesis and therapeutic vulnerabilities, and discuss the opportunities and challenges for Amp1q-targeted therapy.
Background
Multiple myeloma (MM) is an incurable plasma cell malignancy that is usually confined to the bone marrow (1). It is preceded by two asymptomatic conditions, monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM) (2). These are delineated by the amount of tumor detected in the bone marrow or monoclonal protein detected in the blood, and thus have a different risk of progression to overt disease (2). Approximately 5% of people over the age of 40, and 17% of Black people over the age of 50 have MGUS (3, 4), but on average only 1% of those will develop MM every year (5). As such, despite the high prevalence of gammopathy in the general population, MM accounts for 1% of all cancers (1). Approximately 0.5% of people over the age of 40 years have SMM (6); their progression risk follows a logistic growth model with a yearly risk of 10% for the first 5 years and a gradual decrease in risk after that until an MGUS-like slope is reached (7). The mixed nature of this risk pattern indicates that, while useful, the clinical definitions of MGUS and SMM are broad and encompass multiple biological entities (8–13). Likewise, the clinical entity of MM is, in biological terms, a collection of different entities with similar cell morphology and clinical presentation (14). Cytogenetics-based and gene expression-based classifications of MM have been validated and delineate several subtypes of disease with variable outcomes (15–19). Since their underlying biology is different, it is reasonable to assume that each subtype may have its own therapeutic vulnerabilities and that tailored therapy or precision medicine may improve patient outcomes (20, 21). In practice, though, today the only subtype with an actionable, class-wide vulnerability is MM with translocation t(11;14), which is sensitive to BCL2 inhibition (20, 22, 23). Arguably, MM with t(11;14) is also the subtype with the most distinctive biological characteristics, including lymphoplasmacytic morphology, frequent expression of B-cell surface antigens, and a possible origin in bone marrow pro-B cells, as opposed to most other myelomas, which arise from post-germinal center B cells (24–26). Although the survival of patients with MM has improved significantly over the past decades (27), that has been thanks to effective one-size-fits-all regimens that are tailored to the cell type rather than the genetic alterations of the tumor (28). The limited success of precision medicine in MM is partly because class-defining alterations are either translocations that dysregulate undruggable targets, such as WHSC1 or MAF, or somatic copy number alterations (SCNAs), whose vulnerabilities are challenging to study and target. It is also due to the low cumulative frequency of actionable mutations, which precludes a broader adoption of precision therapy for patients with MM (20, 29, 30).
The amplification of the long arm of chromosome 1 (chr1q) is one of the most common SCNAs in patients with MM (31). Its presence is associated with poor outcomes (32). It follows that a large fraction of patients with MM stand to benefit from precision therapy approaches for MM with amplification of chr1q (Amp1q). In this review article, we summarize the evidence surrounding the prevalence and clinical significance of Amp1q in MM and its precursors, pathogenesis, and therapeutic vulnerabilities and discuss the opportunities and challenges in establishing precision medicine for MM patients with Amp1q.
Prevalence of Amp1q in patients with MM and its precursors
Increases in the copy number of chr1q are sometimes categorized as “gains” when the total number of copies is 3 (i.e., 1 extra copy), and “amplifications” when the total number of copies is larger (i.e., 2 or more extra copies) (33). In this review, the term “amplification” will be used to denote all increases in copy number, irrespective of the number of copies gained, as not all publications summarized here present data separately for gains and amplifications; it should be noted, however, that amplifications may confer more aggressive disease compared to gains (32, 33). The amplification of chr1q is the second most common arm-level SCNA in patients with MM, following Del13q (31). Patients with newly diagnosed MM have a high frequency of Amp1q by fluorescence in situ hybridization (FISH) [43%, n = 479, 43% of whom had 4 copies or more (34); 40.5%, n = 205, 22% of whom had 4 copies or more (35); 39%, n = 767 (36); 37%, n = 880, 25% of whom had 4 copies or more (37); 33%, n = 520 (38)], comparative genomic hybridization (CGH) [45%, n = 51 (39)], or Multiplex Ligation-dependent Probe Amplification (MLPA) [34%, n = 1,716 (37)]. In a whole-exome sequencing (WES) study of 1,074 patients with newly diagnosed MM, 29% had Amp1q, and 21% of those had 4 copies or more (29); whereas, in a whole-genome sequencing (WGS) study of 871 patients, 35% had Amp1q (40). Similar prevalence estimates have been reported in patients with SMM either by FISH [45%, n = 31 (34); 41%, n = 114 (41); 30%, n = 245 (42)] or next-generation sequencing (NGS) [25%, n = 77 (11); 28.5%, n = 214 (8); 24.4%, n = 90 (10)]. This is consistent with exome- or genome-wide analyses showing similar genomic profiles in patients with SMM and newly diagnosed MM (8, 10–13). However, in patients with MGUS, the prevalence of Amp1q has been reported to be lower by FISH [0%, n = 14 (34); 20%, n = 88 (37); 29%, n = 79 (43); 16.8%, n = 155 (44)] and NGS [4.3%, n = 23 (10)]; this is consistent with reports of lower prevalence for other secondary genomic alterations in MGUS, such as Del13q and Del17p (13, 43, 44). Lastly, the prevalence of Amp1q in patients with relapsed/refractory MM has been reported to be higher by FISH [72%, n = 45, 60% of whom had 4 copies or more (34); 44%, n = 81 (45); ~56%, n = 178 (46)]. Across all studies discussed here, Amp1q is present on average in approximately 14% (SE: 5.3%) of patients with MGUS, 32.3% of patients with SMM (SE: 3.5%), 37.3% of patients with MM (SE: 1.7%), and 57.3% (SE: 8.1%) of patients with RRMM (Figure 1A). Its increasing frequency along the stages of disease progression suggests a potential role for Amp1q in disease aggressiveness.
Figure 1
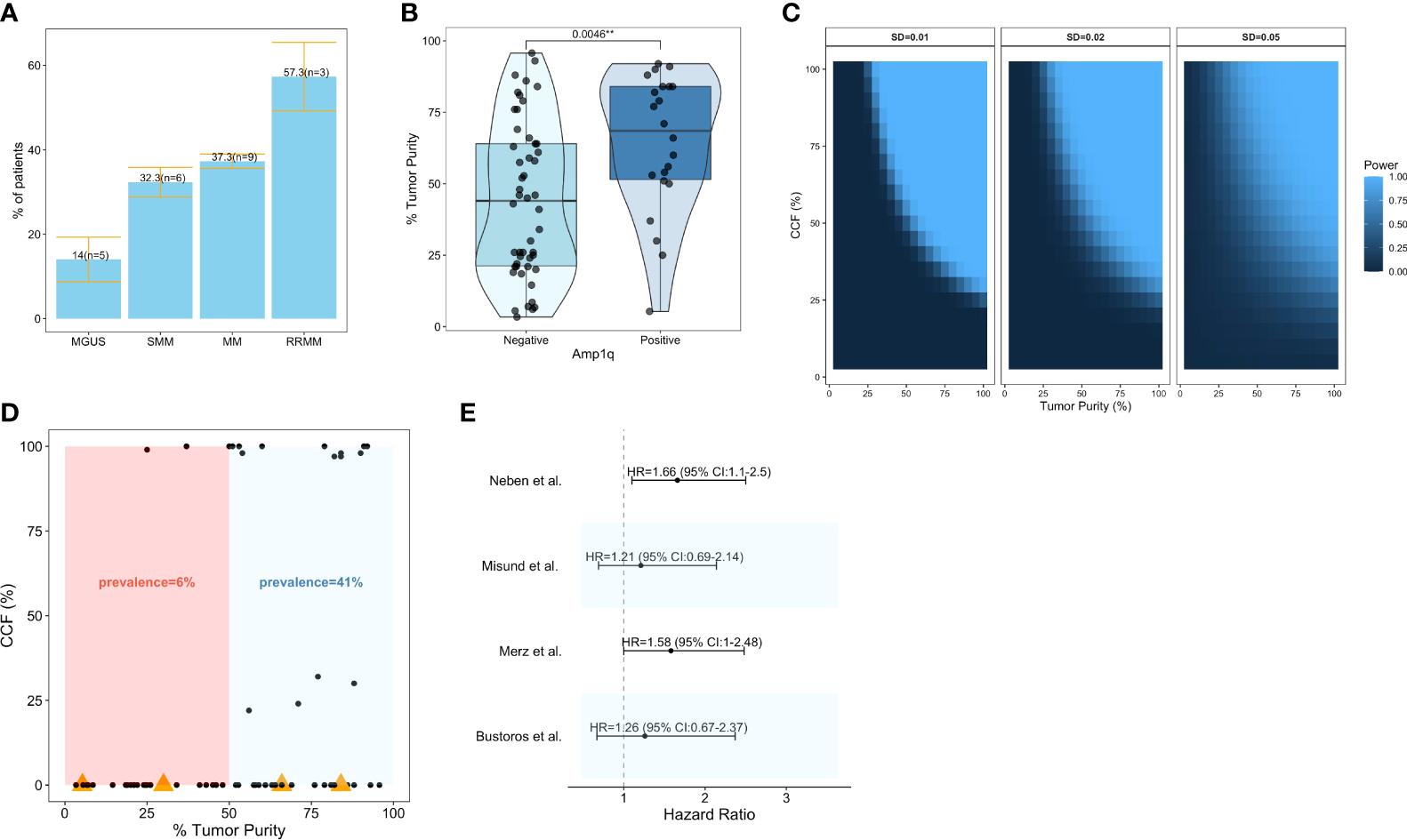
Prevalence and clinical significance of Amp1q in patients with MM and its precursors. (A) Barplots visualizing the mean ± standard error of the prevalence of Amp1q per disease stage. The number of studies (n) considered in those estimates is also visualized. (B) Boxplots comparing the tumor purity of samples in which Amp1q was detected to those in which it was not detected in Bustoros et al. (8). (C) Power analysis for the detection of arm-level copy number variants in sequencing experiments as a function of tumor purity (x-axis) and the abnormality’s cancer cell fraction (CCF). Each panel corresponds to a different standard deviation (SD). (D) Scatterplot of tumor purity (x-axis) and CCF (y-axis) in patients from Bustoros et al. (8). Samples in which Amp1q was not detected by whole-exome sequencing are shown with a CCF of 0. Samples in which Amp1q was not detected by whole-exome sequencing but was detected by fluorescence in situ hybridization (FISH) are shown with an orange triangle. (E) Forest plot summarizing the impact of Amp1q on the risk of progression from smoldering multiple myeloma to overt disease in four studies. Its effect is visualized as a hazard ratio with a 95% confidence interval.
It is important to note that the true prevalence of Amp1q may be underestimated in early-stage disease where the tumor purity of the samples (i.e., the fraction of malignant cells in the sample) can be low, and secondary alterations like Amp1q are often subclonal with low/intermediate cancer cell fraction (CCF) (i.e., the fraction of malignant cells with the abnormality). In Bustoros et al. (8), a study of WES in patients with SMM, the average tumor purity was 47%, and Amp1q was detected in 28% (n = 214) of patients; indeed, the detection of Amp1q was significantly associated with higher tumor purity (Wilcoxon, p = 0.004), suggesting that the true frequency of this abnormality may be higher in this cohort (Figure 1B). To estimate our power to detect Amp1q in a sequencing experiment of good quality, we can assume that chr1q copy ratios are normally distributed with relatively small standard deviations (SD) and that they are a function of purity and ploidy as outlined in Carter et al. (47). By requiring a copy ratio of at least 1.1 to detect the abnormality, then in a sample with 45% tumor purity and SD of 0.01 we would be well powered (power >0.8) to detect Amp1q provided its CCF is at least 65% (Figure 1C), which we argue may not be the case in patients with early-stage disease. Indeed, in Bustoros et al. (8), the majority of samples in which Amp1q was detected had a tumor purity of at least 50%, and if we calculated the prevalence of Amp1q based on samples with at least 50% tumor purity, it would be closer to that reported by FISH (41%) (Figure 1D). Furthermore, different studies may have different amounts of noise in the data, necessitating different thresholds for calling the abnormality in a sample. This problem is not unique to NGS studies. For example, when FISH is used to detect Amp1q, a CCF threshold is used to report the detection of the abnormality, which is usually around 10–30% (i.e., the abnormality needs to be present in at least this fraction of cancer cells studied for it to be called), and which can differ from study to study. Other sources of this bias in FISH studies include the type of cells tested (e.g., mononuclear cells or magnetically sorted CD138+ plasma cells), the number of nuclei tested (typically 100–200 nuclei), and whether cytoplasmic kappa/lambda staining is performed to enrich for cells expressing the light chain of the clone, all of which can affect the probability of detecting a subclonal Amp1q (48). This is important to keep in mind as we consider estimates of the clinical significance of Amp1q since different studies may have different proportions of patients with low-CCF Amp1q subclones, which may impact survival as much as clonal abnormalities (49).
Clinical significance of Amp1q in patients with MM and its precursors
In patients with SMM, the detection of Amp1q by FISH conferred a ~60% higher risk of progression to overt MM in two different studies (42, 50). However, Amp1q was not a significant predictor of progression in two other studies that used WES and custom panels to detect it (Figure 1E) (8, 10). While multiple studies have shown an excellent correlation between FISH and NGS in patients with MM (51–53), it is possible that FISH (which studies single nuclei) can detect subclonal Amp1q in more patients than NGS (which studies DNA fragments in bulk). This could explain the dampening of the survival impact of Amp1q in NGS studies of patients with SMM. In Bustoros et al. (8), FISH detected Amp1q in four more patients than WES, and for two of those, the tumor purity was above 65%, suggesting that the abnormalities missed were subclonal (Figure 1D). Nevertheless, including these 4 patients in the Amp1q group did not change its effect on the risk of progression, suggesting that while detection sensitivity is an issue, it does not fully explain the differences observed in hazard ratios.
As patients with newly diagnosed MM receive treatment, the effect of Amp1q detection on their overall survival (OS) is also a function of the type of treatment administered, and therefore can change as the standard of care improves. In patients with newly diagnosed MM who were treated with vincristine, adriamycin, and dexamethasone (VAD) followed by melphalan and autologous stem cell transplantation (ASCT) in the IFM 99-02, IFM 99-04, and CMG2002 trials, the detection of Amp1q by FISH or SNP array was associated with significantly shorter OS in multivariate analyses (38, 54, 55). Furthermore, the detection of Amp1q was associated with significantly shorter OS in patients with newly diagnosed MM who were treated on the Total Therapy 2 (TT2), Myeloma IX, and the GMMG-HD-3/HD-4 trials, which started incorporating thalidomide and bortezomib into chemotherapy induction regimens (34, 56, 57). However, except in the TT2 study, Amp1q was not a significant predictor in the multivariate setting, raising questions about its usefulness in risk models, given that it frequently co-occurs with other high-risk abnormalities (33, 34, 56, 57). This was also the case in an analysis that looked at patients from the Mayo Clinic who were treated with high-dose therapy and transplantation, as well as an analysis of a subset of patients from the TT2 trial (58). Nevertheless, recent data from patients who were treated with proteasome inhibitors and immunomodulatory agents indicate that Amp1q is a significant independent risk factor for patients with newly diagnosed MM in this era (31, 59–64).
In summary, in the era of proteasome inhibition and immunomodulatory agents, Amp1q is a significant predictor of poor outcomes in patients with newly diagnosed MM. In patients with SMM, Amp1q may be a risk factor for progression, although more and larger studies are needed to confirm this and prove that its effect is independent of other high-risk abnormalities, such as Myc translocations. Therefore, establishing precision therapy for patients with MM and Amp1q stands to benefit a large fraction of patients who are still at higher risk of poor outcomes, despite improvements in plasma cell-targeting therapies.
Pathogenesis of MM with Amp1q
Chromosome 1q can be amplified as part of a trisomy (where an extra copy of the entire chr1 is generated, perhaps due to missegregation during mitosis), or whole-genome doubling, but arm-level events (Amp1q) are observed in more than 75% of patients (65). Patients with MM and Amp1q show a preponderance of breakpoints in the pericentromeric heterochromatin region of cytoband 1q12 (66). Chromosome 1 has the longest pericentromeric heterochromatin region, which is rich in satellite 2 and 3 DNA repeats and is one of the most frequent breakpoint locations in cancer (67, 68). The pericentromeric chromatin of Chromosome 1 is maintained in a repressed state (heterochromatin) through epigenetic regulation, involving the tri-methylation of the 9th lysine residue (H3K9m3) of histone 3 by Suv39h methyltransferases and the subsequent binding of these methylated residues by heterochromatin protein 1 (HP1) (67, 69, 70). Suv39h methyltransferases accumulate around centromeres during mitosis, and together with HP1, orchestrate the methylation of pericentromeric satellite repeats by DNA methyltransferases 3A and 3B (DNMT3A, DNMT3B) (71, 72). These interactions play a key role in chromosome segregation and stability by preserving pericentromeric chromatin in its more condensed form. Loss of Suv39h leads to hypomethylation of the pericentromeric heterochromatin, chromosomal instability, and malignant transformation in vivo (70). Germline mutations in DNMT3B in patients with immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome lead to hypomethylation and decondensation of pericentromeric heterochromatin in chr1; this results in elongated, thread-like pericentromeric chromatin and multiradial chromosomal formations comprising multiple copies of whole arms, which lead to missegregation, chromosomal translocations, and copy number gains in chr1 (73, 74). Similar chromosomal anomalies can be induced in human lymphocytes with drug-induced pericentromeric hypomethylation, using, for example, the methyltransferase inhibitor 5-azacytidine (75–77).
Multiple myeloma is characterized by global hypomethylation with significant interpatient variability, although MM with Amp1q does not appear to have a characteristic methylation trace compared with other cytogenetic abnormalities (78, 79). In patients with MM, metaphase cytogenetics shows active decondensation of pericentromeric heterochromatin with multiradial chr1 formations, which leads to jumping chr1q translocations (whereby the same donor fragment is translocated onto two or more chromosomes, in different cells), isochromosome 1q (whereby chr1p is deleted, and chr1q is duplicated), segmental duplications (whereby certain segments of chr1q are duplicated, sometimes repeatedly so), and breakage–fusion–bridge cycles (Figure 2) (80–83). Importantly, these alterations can continue to evolve, producing subclones with more copies of chr1q and more complex karyotypes. While such abnormalities can be reproduced in vitro by treating peripheral blood mononuclear cells from patients with MM with the hypomethylating agent 5-azacytidine (77), no drug has, thus far, been able to reverse or halt this state of hypomethylation and chromosomal instability in MM cells. More studies on the pathogenesis of Amp1q in patients with MM may reveal novel class-wide vulnerabilities and improve patient outcomes.
Figure 2
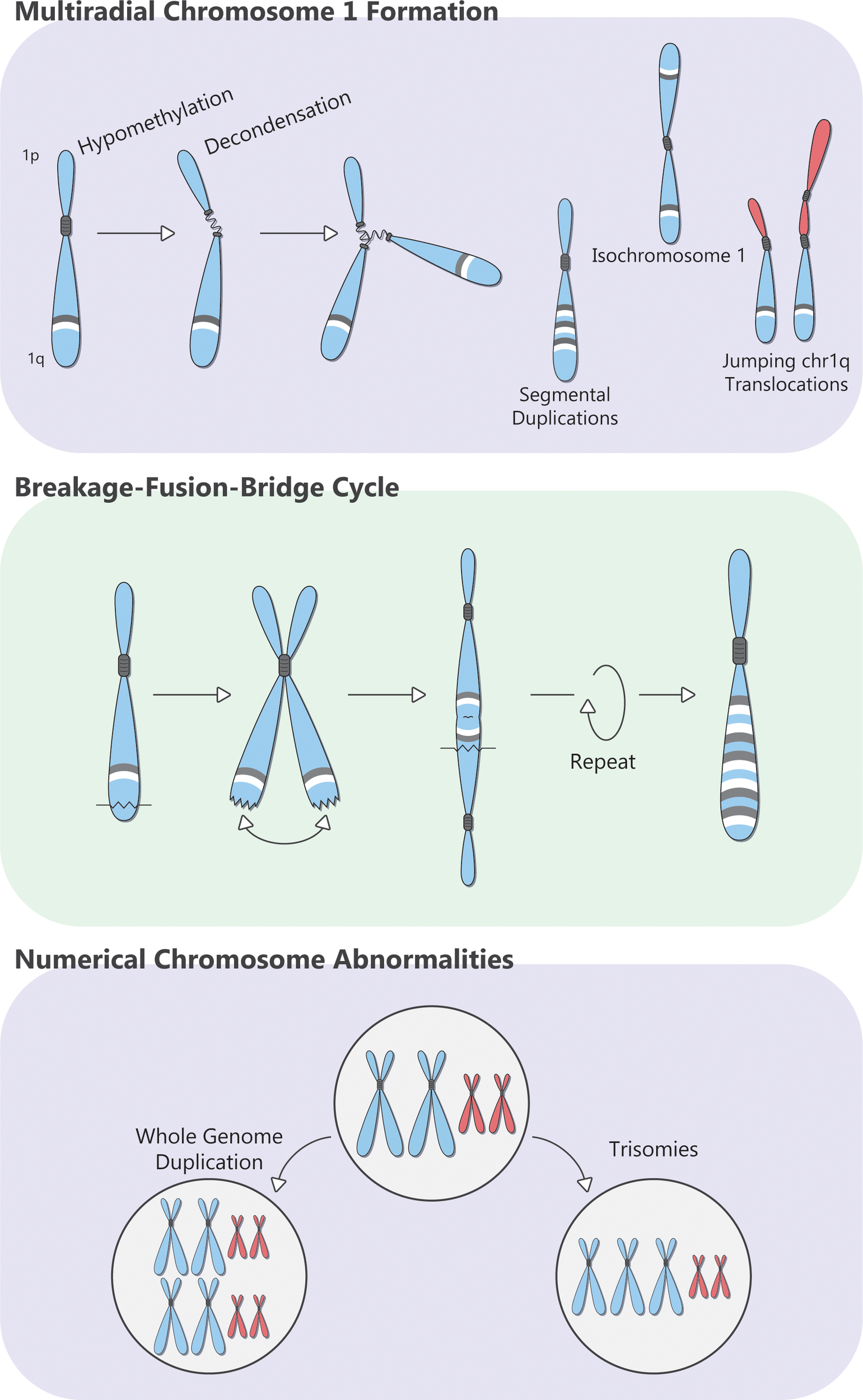
Overview of the pathogenesis of Amp1q in patients with MM.
Therapeutic vulnerabilities of MM with Amp1q
The identification of therapeutic vulnerabilities for MM with Amp1q has focused on genes located in cytoband 1q21 (Figure 3). Despite Amp1q typically being an arm-level abnormality, minimal common regions have been identified in cytobands 1q21–1q22 and 1q43–1q44 (84). Such regions are presumed to contain genes critical for tumor cell proliferation and survival; in this case, genes such as MCL1, BCL9, CKS1B, and ILF2, and members of the ubiquitin-proteasome pathway are found in the affected regions (84). CDC28 Protein Kinase Regulatory Subunit 1B (CKS1B) promotes the ubiquitination and degradation of p27, a cell cycle inhibitor encoded by CDKN1B (85, 86), and activates STAT3 and the MEK/ERK pathway (87). Inhibition of STAT3 and MEK1, or inhibition of p27 degradation via a NEDD8 inhibitor, may be particularly effective in tumors with high levels of CKS1B, such as MM with Amp1q (86, 88). Myeloid Cell Factor 1 (MCL1) encodes an antiapoptotic protein that most MM tumors depend on for survival through both tumor-intrinsic and extrinsic mechanisms (89–94). Multiple myeloma with Amp1q is particularly dependent on MCL1, which results in pronounced sensitivity to MCL1 inhibitors (95–97). B-cell lymphoma 9 (BCL9) acts as a co-activator of β-catenin, and promotes cell proliferation, metastasis, and angiogenesis in MM (98). Disruption of its protein–protein interaction with β-catenin, which may be particularly relevant for MM with Amp1q, can suppress tumor growth in vivo. However, therapeutically targeting this signaling pathway has proved challenging (99, 100). Interleukin Enhancer Binding Factor 2 (ILF2) encodes NF45, a subunit of the transcription factor NFAT, and regulates genomic stability in MM with Amp1q; knockdown of ILF2 in vivo leads to significantly prolonged OS, although currently no drug has been shown to degrade this transcription factor in patients with MM and Amp1q (101). In recent years, technological advances have enabled the systematic interrogation of all of chr1q using large-scale screens and multi-omics approaches, which have nominated new actionable targets, including the PI3K pathway (96), the kinase CLK2 (102), and the transcription factor PBX1 (103).
Figure 3
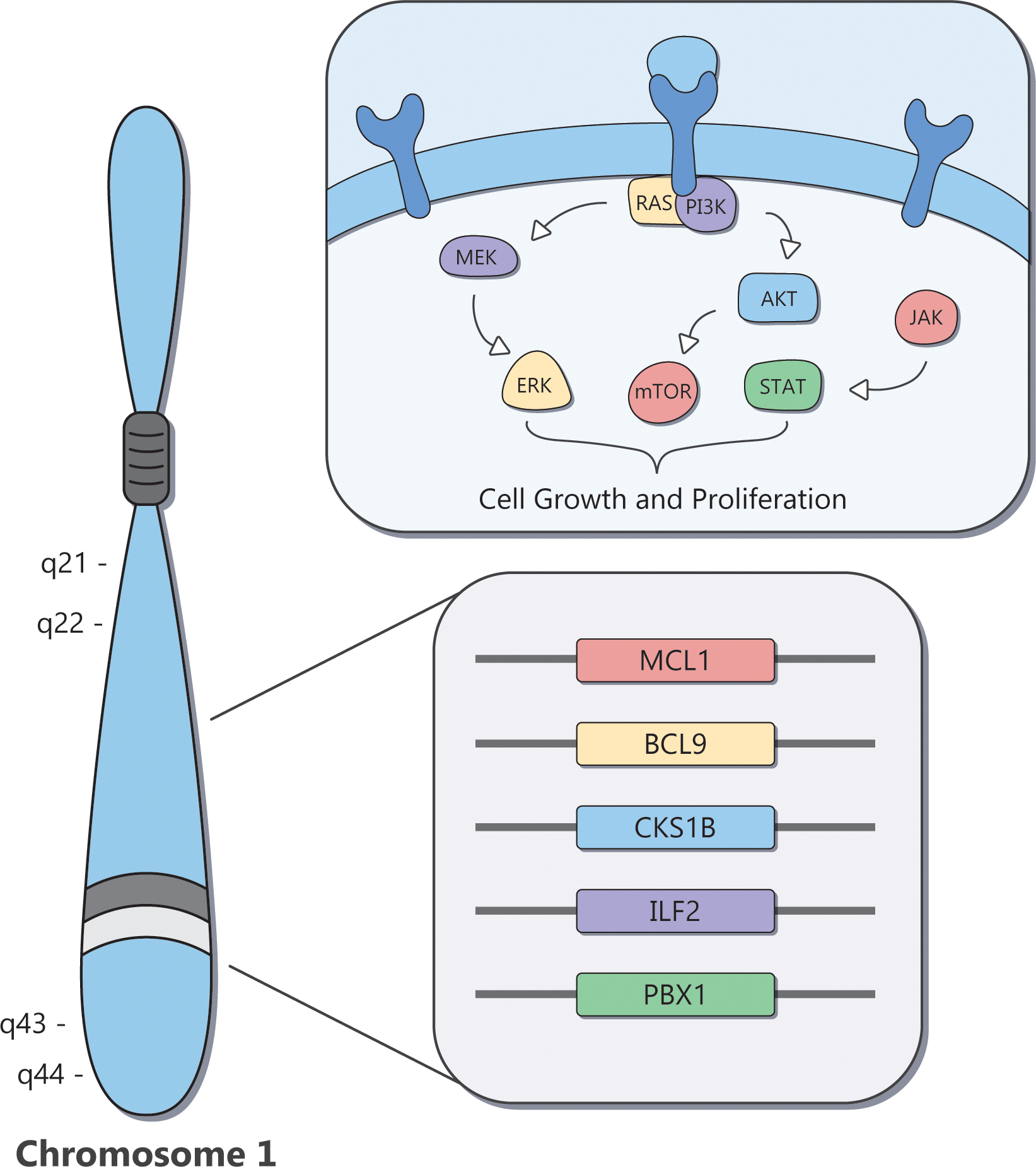
Overview of the therapeutic vulnerabilities of MM with Amp1q.
In summary, the presence of Amp1q in a MM tumor confers specific therapeutic vulnerabilities, some of which may already be actionable today. However, these discoveries have not been translated into clinical practice, as candidate drugs have not been tested in patients with Amp1q yet and have been hampered by suboptimal efficacy and toxicity profiles. Furthermore, it is unclear how these vulnerabilities may be modified by the presence of other genomic abnormalities and whether further patient stratification will be necessary to achieve optimal responses to targeted therapy.
Opportunity and challenges in targeting MM with Amp1q
Because of its frequency and its impact on patient outcomes, multiple myeloma with Amp1q represents an opportunity for precision medicine to improve the lives of many patients who have not benefitted as much from the therapeutic developments of recent years. Furthermore, the discovery of vulnerabilities that are either specific to Amp1q or enhanced by the presence of Amp1q indicates that precision approaches can indeed be developed for patients with this abnormality. A major limitation in developing precision medicine for MM with Amp1q is the lack of genome engineering methods that enable the generation of arm-level amplifications in human cell line models. Methods exist for the generation of single-chromosome trisomies in human cells. However, those are not frequently seen in patients with MM and Amp1q, possibly due to the existence of potent tumor suppressors on chr1p, which is frequently deleted in patients with MM (104–106). In the absence of syngeneic models of MM with Amp1q, we are forced to draw conclusions from suboptimal comparisons of cell lines that have Amp1q to cell lines that do not, which differ by more than just the presence or absence of Amp1q. Moreover, approximately 91% of MM cell lines have Amp1q, which restricts the pool of negative control cell lines available (34). It is also unclear whether all amplifications of chr1q generate similar vulnerabilities; perhaps certain targets are dose-dependent (i.e., a certain number of chr1q copies is required), or depend upon the type of the amplification (e.g., segmental duplication, translocation, isochromosome), the borders of the amplification (i.e., is the target contained within the borders or not), the size of the amplification (e.g., focal or arm-level), or the co-occurrence of Amp1q with another genomic abnormality, which may modify its effect on cell fitness. Furthermore, many of the dependencies discovered may not be directly actionable (84, 96). Considering the vulnerabilities that are actionable today, none have been tested in patients with Amp1q yet. MEK inhibitors may be effective in combination with BCL2 and/or PD-L1 inhibitors in patients with MM (20), but PI3K inhibitors can be quite toxic and often lead to lackluster responses (20, 107), and MCL1 inhibitors may be effective but may not be tolerated well, generating concerns of cardiotoxicity, for example (20, 108). Furthermore, resistance mechanisms have been described, including MEK/ERK activation following PI3K inhibition and vice versa (109), and BCL2 or BCLXL activation following MCL1 inhibition (95, 110), suggesting that combinatorial regimens may be necessary for successful treatment of these tumors.
Conclusion
The survival of patients with MM has improved drastically over the last couple of decades. However, patients with high-risk genomic features, such as Amp1q, have not benefited as much. The variety of methods used to detect Amp1q complicates the interpretation of changes in its prevalence over stages of disease progression and its clinical significance in newly diagnosed patients. However, multiple studies confirm that its prevalence is high and that it is an independent predictor of poor outcomes in multivariate analyses even in the era of proteasome inhibitors and immunomodulatory agents. In patients with SMM, larger studies are needed to assess the clinical significance of Amp1q in analyses that account for other genomic and/or clinical variables. Tailoring treatment to the underlying abnormality could improve outcomes for patients with MM and Amp1q. However, developing precision approaches has proved challenging. Despite the role of hypomethylation in the generation and continuous evolution of Amp1q, no epigenetic therapeutic approaches have been developed to target it. Instead, precision approaches for MM with Amp1q typically target genes that are located on 1q21, the most commonly amplified region of chr1q, such as MCL1. When actionable, these approaches may be effective. However, they are often toxic and may require a combination with other inhibitors to preempt escape mechanisms and drug resistance. Until now, none of these approaches has been specifically tested in patients with Amp1q, where the therapeutic window may be different, and it is unclear how the presence of other abnormalities may modify the effect of these treatments. Establishing precision therapy for patients with MM and Amp1q will require clinical trials to systematically enroll patients with Amp1q and report results for this subgroup of patients.
Funding
The authors would like to thank Anna V. Justis, PhD, for her editing services, Sarah Nersersian, MSc, for illustration support, and Nicholas J. Haradhvala, PhD, for his suggestions regarding the power analysis for the detection of SCNAs in sequencing experiments. RS-P is supported by the Multiple Myeloma Research Foundation Fellowship Award, the International Waldenstrom’s Macroglobulinemia Foundation’s Robert A. Kyle Award, and the Claudia Adams-Barr Award for Innovative Basic Cancer Research. GG is partially supported by the Paul C. Zamecnik Chair in Oncology at Massachusetts General Hospital Cancer Center. IMG and GG would like to acknowledge funding from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and the Multiple Myeloma Research Foundation (MMRF) and Dana-Farber Prevention Program. This research was also supported by a Stand Up To Cancer Dream Team Research Grant (Grant Number: SU2C-AACR-DT-28-18). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of Stand Up To Cancer. Opinions, interpretations, conclusions, and recommendations are those of the author(s) and are not necessarily endorsed by Stand Up To Cancer, the Entertainment Industry Foundation, or the American Association for Cancer Research.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
RS-P conducted the literature review and analysis, and wrote the manuscript. GG, MP, and IG reviewed, edited and approved the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
GG receives research funds from IBM and Pharmacyclics, and is also an inventor on patent applications filed by the Broad Institute related to MSMuTect, MSMutSig, POLYSOLVER, SignatureAnalyzer-GPU, and MSIDetect. He is also a founder, consultant, and holds privately held equity in Scorpion Therapeutics. IG has a consulting or advisory role with AbbVie, Adaptive, Amgen, Aptitude Health, Bristol Myers Squibb, GlaxoSmithKline, Huron Consulting, Janssen, Menarini Silicon Biosystems, Oncopeptides, Pfizer, Sanofi, Sognef, Takeda, The Binding Site, and Window Therapeutics; has received speaker fees from Vor Biopharma and Veeva Systems, Inc.; and her spouse is the CMO and equity holder of Disc Medicine.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Rajkumar SV . Multiple Myeloma: 2020 Update on Diagnosis, Risk-Stratification and Management. Am J Hematol (2020) 95(5):548–67. doi: 10.1002/ajh.25791
2
Kyle RA Durie BGM Rajkumar SV Landgren O Blade J Merlini G et al . Monoclonal Gammopathy of Undetermined Significance (MGUS) and Smoldering (Asymptomatic) Multiple Myeloma: IMWG Consensus Perspectives Risk Factors for Progression and Guidelines for Monitoring and Management. Leukemia (2010) 24(6):1121–7. doi: 10.1038/leu.2010.60
3
Kristinsson SY Rognvaldsson S Thorsteinsdottir S Reed ER Oskarsson JTT Petursdottir I et al . Screening for Monoclonal Gammopathy of Undetermined Significance: A Population-Based Randomized Clinical Trial. First Results From the Iceland Screens, Treats, or Prevents Multiple Myeloma (Istopmm) Study. Am Soc Hematol Annu Meeting (2021) . doi: 10.1182/blood-2021-152333
4
El-Khoury H Lee D Alberge J Redd R Cea-Curry C Perry J et al . Prevalence of Monoclonal Gammopathies and Clinical Outcomes in a High-Risk US Population Screened by Mass Spectrometry: A Multicentre Cohort Study. Lancet Haematol (2022) 9(5):e340–9. doi: 10.1016/S2352-3026(22)00069-2
5
Kyle R Larson D Therneau T Dispenzieri A Kumar S Cerhan J et al . Long-Term Follow-Up of Monoclonal Gammopathy of Undetermined Significance. N Engl J Med (2018) 378(3):241–9. doi: 10.1056/NEJMoa1709974
6
Thorsteinsdottir S Aspelund T Rögnvaldsson S Oskarsson JT Petursdottir I Sigurdsson JK et al . Prevalence of Smoldering Multiple Myeloma: Results From the Iceland Screens, Treats, or Prevents Multiple Myeloma (Istopmm) Study. Am Soc Hematol Annu Meeting (2021). doi: 10.1182/blood-2021-148617
7
Kyle R Remstein E Therneau T Dispenzieri A Kurtin P Hodnefield J et al . Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma. N Engl J Med (2007) 356(25):2582–90. doi: 10.1056/NEJMoa070389
8
Bustoros M Sklavenitis-Pistofidis R Park J Redd R Zhitomirsky B Dunford A et al . Genomic Profiling of Smoldering Multiple Myeloma Identifies Patients at a High Risk of Disease Progression. J Clin Oncol (2020) 38(21):2380–9. doi: 10.1200/JCO.20.00437
9
Bustoros M Kastritis E Sklavenitis-Pistofidis R Liu C Hornburg K Kanellias N et al . Bone Marrow Biopsy in Low-Risk Monoclonal Gammopathy of Undetermined Significance Reveals a Novel Smoldering Multiple Myeloma Risk Group. Am J Hematol (2019) 94(5):E146–9. doi: 10.1002/ajh.25441
10
Misund K Keane N Stein C Asmann Y Day G Welsh S et al . MYC Dysregulation in the Progression of Multiple Myeloma. Leukemia (2019) 34(1):322–6.
11
Boyle E Deshpande S Tytarenko R Ashby C Wang Y Bauer M et al . The Molecular Make Up of Smoldering Myeloma Highlights the Evolutionary Pathways Leading to Multiple Myeloma. Nat Commun (2021) 12(1):293. doi: 10.1038/s41467-020-20524-2
12
Bolli N Maura F Minvielle S Gloznik D Szalat R Fullam A et al . Genomic Patterns of Progression in Smoldering Multiple Myeloma. Nat Commun (2018) 9(1):3363. doi: 10.1038/s41467-018-05058-y
13
Oben B Froyen G Maclachlan K Leongamornlert D Abascal F Zheng-Lin B et al . Whole-Genome Sequencing Reveals Progressive Versus Stable Myeloma Precursor Conditions as Two Distinct Entities. Nat Commun (2021) 12(1):1861. doi: 10.1038/s41467-021-22140-0
14
Kumar S Rajkumar S . The Multiple Myelomas — Current Concepts in Cytogenetic Classification and Therapy. Nat Rev Clin Oncol (2018) 15(7):409–21. doi: 10.1038/s41571-018-0018-y
15
Palumbo A Avet-Loiseau H Oliva S Lokhorst H Goldschmidt H Rosinol L et al . Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol (2015) 33(26):2863–9. doi: 10.1200/JCO.2015.61.2267
16
Fonseca R . Many and Multiple Myeloma(s). Leukemia (2003) 17(10):1943–4. doi: 10.1038/sj.leu.2403090
17
Kumar S Rajkumar V Kyle R van Duin M Sonneveld P Mateos M et al . Multiple Myeloma. Nat Rev Dis Primers (2017) 3(1):17046. doi: 10.1038/nrdp.2017.46
18
Zhan F Huang Y Colla S Stewart J Hanamura I Gupta S et al . The Molecular Classification of Multiple Myeloma. Blood (2006) 108(6):2020–8. doi: 10.1182/blood-2005-11-013458
19
Barwick B Neri P Bahlis N Nooka A Dhodapkar M Jaye D et al . Multiple Myeloma Immunoglobulin Lambda Translocations Portend Poor Prognosis. Nat Commun (2019) 10(1):1911. doi: 10.1038/s41467-019-09555-6
20
Pan D Richter J . Where We Stand With Precision Therapeutics in Myeloma: Prosperity, Promises, and Pipedreams. Front Oncol (2022) 11. doi: 10.3389/fonc.2021.819127
21
Harding T Baughn L Kumar S Van Ness B . The Future of Myeloma Precision Medicine: Integrating the Compendium of Known Drug Resistance Mechanisms With Emerging Tumor Profiling Technologies. Leukemia (2019) 33(4):863–83. doi: 10.1038/s41375-018-0362-z
22
Ahmed Z Nabeel S Farooqui A Imtiaz H Ashraf A Farooq U et al . Advances in Precision Therapy With Venetoclax in Multiple Myeloma With T(11;14) and High BCL2 Expression: A Systematic Review. J Clin Oncol (2020) 38(15_suppl):e20535. doi: 10.1200/JCO.2020.38.15_suppl.e20535
23
Gupta V Barwick B Matulis S Shirasaki R Jaye D Keats J et al . Venetoclax Sensitivity in Multiple Myeloma Is Associated With B-Cell Gene Expression. Blood (2021) 137(26):3604–15. doi: 10.1182/blood.2020007899
24
Walker B Wardell C Johnson D Kaiser M Begum D Dahir N et al . Characterization of IGH Locus Breakpoints in Multiple Myeloma Indicates a Subset of Translocations Appear to Occur in Pregerminal Center B Cells. Blood (2013) 121(17):3413–9. doi: 10.1182/blood-2012-12-471888
25
Bazarbachi A Avet-Loiseau H Szalat R Samur A Hunter Z Shammas M et al . IgM-MM is Predominantly a Pre–Germinal Center Disorder and has a Distinct Genomic and Transcriptomic Signature From WM. Blood (2021) 138(20):1980–5. doi: 10.1182/blood.2021011452
26
Fonseca R Blood E Oken M Kyle R Dewald G Bailey R et al . Myeloma and the T(11;14)(Q13;Q32); Evidence for a Biologically Defined Unique Subset of Patients. Blood (2002) 99(10):3735–41. doi: 10.1182/blood.V99.10.3735
27
Kumar S Dispenzieri A Lacy M Gertz M Buadi F Pandey S et al . Continued Improvement in Survival in Multiple Myeloma: Changes in Early Mortality and Outcomes in Older Patients. Leukemia (2013) 28(5):1122–8.
28
Boise L Kaufman J Bahlis N Lonial S Lee K . The Tao of Myeloma. Blood (2014) 124(12):1873–9. doi: 10.1182/blood-2014-05-578732
29
Walker B Mavrommatis K Wardell C Ashby T Bauer M Davies F et al . Identification of Novel Mutational Drivers Reveals Oncogene Dependencies in Multiple Myeloma. Blood (2018) 132(6):587–97. doi: 10.1182/blood-2018-03-840132
30
Maura F Bolli N Angelopoulos N Dawson K Leongamornlert D Martincorena I et al . Genomic Landscape and Chronological Reconstruction of Driver Events in Multiple Myeloma. Nat Commun (2019) 10(1):3835. doi: 10.1038/s41467-019-11680-1
31
Walker B Mavrommatis K Wardell C Ashby T Bauer M Davies F et al . A High-Risk, Double-Hit, Group of Newly Diagnosed Myeloma Identified by Genomic Analysis. Leukemia (2018) 33(1):159–70.
32
Schmidt T Fonseca R Usmani S . Chromosome 1q21 Abnormalities in Multiple Myeloma. Blood Cancer J (2021) 11(4):83. doi: 10.1038/s41408-021-00474-8
33
Hanamura I . Gain/Amplification of Chromosome Arm 1q21 in Multiple Myeloma. Cancers (2021) 13(2):256. doi: 10.3390/cancers13020256
34
Hanamura I Stewart J Huang Y Zhan F Santra M Sawyer J et al . Frequent Gain of Chromosome Band 1q21 in Plasma-Cell Dyscrasias Detected by Fluorescence in Situ Hybridization: Incidence Increases From MGUS to Relapsed Myeloma and Is Related to Prognosis and Disease Progression Following Tandem Stem-Cell Transplantation. Blood (2006) 108(5):1724–32. doi: 10.1182/blood-2006-03-009910
35
Liebisch P Wendl C Wellmann A Kröber A Schilling G Goldschmidt H et al . High Incidence of Trisomies 1q, 9q, and 11q in Multiple Myeloma: Results From a Comprehensive Molecular Cytogenetic Analysis. Leukemia (2003) 17(12):2535–7. doi: 10.1038/sj.leu.2403153
36
Chiecchio L Dagrada G Cabanas E Protheroe R Stockley D Konn Z et al . Gain of 1q21 Does Not Predict for Immediate Progression in MGUS. Blood (2009) 114(22):123–3. doi: 10.1182/blood.V114.22.123.123
37
Weinhold N Salwender H Cairns D Raab M Waldron G Blau I et al . Chromosome 1q21 Abnormalities Refine Outcome Prediction in Patients With Multiple Myeloma - A Meta-Analysis of 2,596 Trial Patients. Haematologica (2021) 106(10):2754–8. doi: 10.3324/haematol.2021.278888
38
Avet-Loiseau H Attal M Campion L Caillot D Hulin C Marit G et al . Long-Term Analysis of the IFM 99 Trials for Myeloma: Cytogenetic Abnormalities [T(4;14), Del(17p), 1q Gains] Play a Major Role in Defining Long-Term Survival. J Clin Oncol (2012) 30(16):1949–52. doi: 10.1200/JCO.2011.36.5726
39
Gutieírrez N Garciía J Hernaíndez J Lumbreras E Castellanos M Rasillo A et al . Prognostic and Biologic Significance of Chromosomal Imbalances Assessed by Comparative Genomic Hybridization in Multiple Myeloma. Blood (2004) 104(9):2661–6. doi: 10.1182/blood-2004-04-1319
40
Skerget S Penaherrera D Chari A Jagannath S Siegel D Vij R et al . Genomic Basis of Multiple Myeloma Subtypes From the MMRF CoMMpass Study. (2021). doi: 10.1101/2021.08.02.21261211
41
Lopez-Corral L Mateos M Corchete L Sarasquete M de la Rubia J de Arriba F et al . Genomic Analysis of High-Risk Smoldering Multiple Myeloma. Haematologica (2012) 97(9):1439–43. doi: 10.3324/haematol.2011.060780
42
Neben K Jauch A Hielscher T Hillengass J Lehners N Seckinger A et al . Progression in Smoldering Myeloma Is Independently Determined by the Chromosomal Abnormalities Del(17p), T(4;14), Gain 1q, Hyperdiploidy, and Tumor Load. J Clin Oncol (2013) 31(34):4325–32. doi: 10.1200/JCO.2012.48.4923
43
López-Corral L Gutiérrez N Vidriales M Mateos M Rasillo A García-Sanz R et al . The Progression From MGUS to Smoldering Myeloma and Eventually to Multiple Myeloma Involves a Clonal Expansion of Genetically Abnormal Plasma Cells. Clin Cancer Res (2011) 17(7):1692–700. doi: 10.1158/1078-0432.CCR-10-1066
44
Merz M Hielscher T Hoffmann K Seckinger A Hose D Raab M et al . Cytogenetic Abnormalities in Monoclonal Gammopathy of Undetermined Significance. Leukemia (2018) 32(12):2717–9. doi: 10.1038/s41375-018-0202-1
45
Mohan M Weinhold N Schinke C Thanedrarajan S Rasche L Sawyer J et al . Daratumumab in High-Risk Relapsed/Refractory Multiple Myeloma Patients: Adverse Effect of Chromosome 1q21 Gain/Amplification and GEP70 Status on Outcome. Br J Haematol (2019) 189(1):67–71.
46
Croft J Ellis S Sherborne A Sharp K Price A Jenner M et al . Copy Number Evolution and its Relationship With Patient Outcome—An Analysis of 178 Matched Presentation-Relapse Tumor Pairs From the Myeloma XI Trial. Leukemia (2020) 35(7):2043–53.
47
Carter S Cibulskis K Helman E McKenna A Shen H Zack T et al . Absolute Quantification of Somatic DNA Alterations in Human Cancer. Nat Biotechnol (2012) 30(5):413–21. doi: 10.1038/nbt.2203
48
Ross F Avet-Loiseau H Ameye G Gutierrez N Liebisch P O'Connor S et al . Report From the European Myeloma Network on Interphase FISH in Multiple Myeloma and Related Disorders. Haematologica (2012) 97(8):1272–7. doi: 10.3324/haematol.2011.056176
49
An G Li Z Tai Y Acharya C Li Q Qin X et al . The Impact of Clone Size on the Prognostic Value of Chromosome Aberrations by Fluorescence In Situ Hybridization in Multiple Myeloma. Clin Cancer Res (2015) 21(9):2148–56. doi: 10.1158/1078-0432.CCR-14-2576
50
Merz M Hielscher T Schult D Mai E Raab M Hillengass J et al . Cytogenetic Subclone Formation and Evolution in Progressive Smoldering Multiple Myeloma. Leukemia (2019) 34(4):1192–6.
51
Cuenca I Medina A Rosinol L Vazquez I Sanchez R Gutierrez N et al . Clinical Validation of a NGS Capture Panel to Identify Mutations, Copy Number Variations and Translocations in Patients With Multiple Myeloma. Blood (2020) 136(Supplement 1):13–4. doi: 10.1182/blood-2020-140711
52
Sudha P Ahsan A Khera A Kazeroun M Kausar T Wang L et al . A Clinically Validated Targeted Capture Panel to Identify Translocations, Copy Number Abnormalities, and Mutations in Multiple Myeloma. Blood (2021) 138(Supplement 1):2676–6. doi: 10.1182/blood-2021-148692
53
Lomas O Gooding S Ramasamy K Hamblin A Cabes M Dreau H et al . Clinical-Grade Whole Genome Sequencing Reproduces FISH Cytogenetics and Provides Actionable Data in Newly Diagnosed Myeloma - A Pilot Study From the UK 100,000 Genomes Project. Blood (2019) 134(Supplement_1):3062–2. doi: 10.1182/blood-2019-125145
54
Avet-Loiseau H Li C Magrangeas F Gouraud W Charbonnel C Harousseau J et al . Prognostic Significance of Copy-Number Alterations in Multiple Myeloma. J Clin Oncol (2009) 27(27):4585–90. doi: 10.1200/JCO.2008.20.6136
55
Nemec P Zemanova Z Greslikova H Michalova K Filkova H Tajtlova J et al . Gain of 1q21 Is an Unfavorable Genetic Prognostic Factor for Multiple Myeloma Patients Treated With High-Dose Chemotherapy. Biol Blood Marrow Transplant (2010) 16(4):548–54. doi: 10.1016/j.bbmt.2009.11.025
56
Boyd K Ross F Chiecchio L Dagrada G Konn Z Tapper W et al . A Novel Prognostic Model in Myeloma Based on Co-Segregating Adverse FISH Lesions and the ISS: Analysis of Patients Treated in the MRC Myeloma IX Trial. Leukemia (2011) 26(2):349–55.
57
Neben K Jauch A Bertsch U Heiss C Hielscher T Seckinger A et al . Combining Information Regarding Chromosomal Aberrations T(4;14) and Del(17p13) With the International Staging System Classification Allows Stratification of Myeloma Patients Undergoing Autologous Stem Cell Transplantation. Haematologica (2010) 95(7):1150–7. doi: 10.3324/haematol.2009.016436
58
Fonseca R Van Wier S Chng W Ketterling R Lacy M Dispenzieri A et al . Prognostic Value of Chromosome 1q21 Gain by Fluorescent In Situ Hybridization and Increase CKS1B Expression in Myeloma. Leukemia (2006) 20(11):2034–40. doi: 10.1038/sj.leu.2404403
59
Schmidt T Barwick B Joseph N Heffner L Hofmeister C Bernal L et al . Gain of Chromosome 1q Is Associated With Early Progression in Multiple Myeloma Patients Treated With Lenalidomide, Bortezomib, and Dexamethasone. Blood Cancer J (2019) 9(12):94.
60
Abdallah N Greipp P Kapoor P Gertz M Dispenzieri A Baughn L et al . Clinical Characteristics and Treatment Outcomes of Newly Diagnosed Multiple Myeloma With Chromosome 1q Abnormalities. Blood Adv (2020) 4(15):3509–19. doi: 10.1182/bloodadvances.2020002218
61
Giri S Huntington S Wang R Zeidan A Podoltsev N Gore S et al . Chromosome 1 Abnormalities and Survival of Patients With Multiple Myeloma in the Era of Novel Agents. Blood Adv (2020) 4(10):2245–53. doi: 10.1182/bloodadvances.2019001425
62
Locher M Steurer M Jukic E Keller M Fresser F Ruepp C et al . The Prognostic Value of Additional Copies of 1q21 in Multiple Myeloma Depends on the Primary Genetic Event. Am J Hematol (2020) 95(12):1562–71. doi: 10.1002/ajh.25994
63
Shah V Sherborne A Walker B Johnson D Boyle E Ellis S et al . Prediction of Outcome in Newly Diagnosed Myeloma: A Meta-Analysis of the Molecular Profiles of 1905 Trial Patients. Leukemia (2017) 32(1):102–10.
64
D'Agostino M Ruggeri M Aquino S Giuliani N Arigoni M Gentile M et al . Impact of Gain and Amplification of 1q in Newly Diagnosed Multiple Myeloma Patients Receiving Carfilzomib-Based Treatment in the Forte Trial. Blood (2020) 136(Supplement 1):38–40. doi: 10.1182/blood-2020-137060
65
Rustad E Yellapantula V Glodzik D Maclachlan K Diamond B Boyle E et al . Revealing the Impact of Structural Variants in Multiple Myeloma. Blood Cancer Discov (2020) 1(3):258–73. doi: 10.1158/2643-3230.BCD-20-0132
66
Le Baccon P Leroux D Dascalescu C Duley S Marais D Esmenjaud E et al . Novel Evidence of a Role for Chromosome 1 Pericentric Heterochromatin in the Pathogenesis of B-Cell Lymphoma and Multiple Myeloma. Genes Chromosomes Cancer (2001) 32(3):250–64. doi: 10.1002/gcc.1189
67
Gjerstorff M . Novel Insights Into Epigenetic Reprogramming and Destabilization of Pericentromeric Heterochromatin in Cancer. Front Oncol (2020) 10. doi: 10.3389/fonc.2020.594163
68
Beroukhim R Mermel C Porter D Wei G Raychaudhuri S Donovan J et al . The Landscape of Somatic Copy-Number Alteration Across Human Cancers. Nature (2010) 463(7283):899–905. doi: 10.1038/nature08822
69
Fournier A Florin A Lefebvre C Solly F Leroux D Callanan M . Genetics and Epigenetics of 1q Rearrangements in Hematological Malignancies. Cytogenet Genome Res (2007) 118(2-4):320–7. doi: 10.1159/000108316
70
Peters A O'Carroll D Scherthan H Mechtler K Sauer S Schöfer C et al . Loss of the Suv39h Histone Methyltransferases Impairs Mammalian Heterochromatin and Genome Stability. Cell (2001) 107(3):323–37. doi: 10.1016/S0092-8674(01)00542-6
71
Lehnertz B Ueda Y Derijck A Braunschweig U Perez-Burgos L Kubicek S et al . Suv39h-Mediated Histone H3 Lysine 9 Methylation Directs DNA Methylation to Major Satellite Repeats at Pericentric Heterochromatin. Curr Biol (2003) 13(14):1192–200. doi: 10.1016/S0960-9822(03)00432-9
72
Fuks F . The DNA Methyltransferases Associate With HP1 and the SUV39H1 Histone Methyltransferase. Nucleic Acids Res (2003) 31(9):2305–12. doi: 10.1093/nar/gkg332
73
Tuck-Muller C Narayan A Tsien F Smeets D Sawyer J Fiala E et al . DNA Hypomethylation and Unusual Chromosome Instability in Cell Lines From ICF Syndrome Patients. Cytogenet Genome Res (2000) 89(1-2):121–8. doi: 10.1159/000015590
74
Xu G Bestor T Bourc'his D Hsieh C Tommerup N Bugge M et al . Chromosome Instability and Immunodeficiency Syndrome Caused by Mutations in a DNA Methyltransferase Gene. Nature (1999) 402(6758):187–91. doi: 10.1038/46052
75
Hernandez R Frady A Zhang X Varela M Ehrlich M . Preferential Induction of Chromosome 1 Multibranched Figures and Whole-Arm Deletions in a Human Pro-B Cell Line Treated With 5-Azacytidine or 5-Azadeoxycytidine. Cytogenet Genome Res (1997) 76(3-4):196–201. doi: 10.1159/000134548
76
Prada D González R Sánchez L Castro C Fabián E Herrera L . Satellite 2 Demethylation Induced by 5-Azacytidine is Associated With Missegregation of Chromosomes 1 and 16 in Human Somatic Cells. Mutat Res Fundam Mol Mech Mutagenesis (2012) 729(1-2):100–5. doi: 10.1016/j.mrfmmm.2011.10.007
77
Sawyer J Tian E Heuck C Johann D Epstein J Swanson C et al . Evidence of an Epigenetic Origin for High-Risk 1q21 Copy Number Aberrations in Multiple Myeloma. Blood (2015) 125(24):3756–9. doi: 10.1182/blood-2015-03-632075
78
Walker B Wardell C Chiecchio L Smith E Boyd K Neri A et al . Aberrant Global Methylation Patterns Affect the Molecular Pathogenesis and Prognosis of Multiple Myeloma. Blood (2011) 117(2):553–62. doi: 10.1182/blood-2010-04-279539
79
Heuck C Mehta J Bhagat T Gundabolu K Yu Y Khan S et al . Myeloma Is Characterized by Stage-Specific Alterations in DNA Methylation That Occur Early During Myelomagenesis. J Immunol (2013) 190(6):2966–75. doi: 10.4049/jimmunol.1202493
80
Sawyer J Tricot G Mattox S Jagannath S Barlogie B . Jumping Translocations of Chromosome 1q in Multiple Myeloma: Evidence for a Mechanism Involving Decondensation of Pericentromeric Heterochromatin. Blood (1998) 91(5):1732–41. doi: 10.1182/blood.V91.5.1732
81
Sawyer J Tricot G Lukacs J Binz R Tian E Barlogie B et al . Genomic Instability in Multiple Myeloma: Evidence for Jumping Segmental Duplications of Chromosome Arm 1q. Genes Chromosomes Cancer (2004) 42(1):95–106.
82
Sawyer J Tian E Thomas E Koller M Stangeby C Sammartino G et al . Evidence for a Novel Mechanism for Gene Amplification in Multiple Myeloma: 1q12 Pericentromeric Heterochromatin Mediates Breakage-Fusion-Bridge Cycles of a 1q12∼23 Amplicon. Br J Haematol (2009) 147(4):484–94. doi: 10.1111/j.1365-2141.2009.07869.x
83
Sawyer J Tian E Walker B Wardell C Lukacs J Sammartino G et al . An Acquired High-Risk Chromosome Instability Phenotype in Multiple Myeloma: Jumping 1q Syndrome. Blood Cancer J (2019) 9(8):62. doi: 10.1038/s41408-019-0226-4
84
Carrasco D Tonon G Huang Y Zhang Y Sinha R Feng B et al . High-Resolution Genomic Profiles Define Distinct Clinico-Pathogenetic Subgroups of Multiple Myeloma Patients. Cancer Cell (2006) 9(4):313–25. doi: 10.1016/j.ccr.2006.03.019
85
Shaughnessy J . Amplification and Overexpression of CKS1B at Chromosome Band 1q21 Is Associated With Reduced Levels of P27 Kip1 and an Aggressive Clinical Course in Multiple Myeloma. Hematology (2005) 10(sup1):117–26. doi: 10.1080/10245330512331390140
86
Spruck C Strohmaier H Watson M Smith A Ryan A Krek W et al . A CDK-Independent Function of Mammalian Cks1. Mol Cell (2001) 7(3):639–50. doi: 10.1016/S1097-2765(01)00210-6
87
Shi L Wang S Zangari M Xu H Cao T Xu C et al . Over-Expression of CKS1B Activates Both MEK/ERK and JAK/STAT3 Signaling Pathways and Promotes Myeloma Cell Drug-Resistance. Oncotarget (2010) 1(1):22–33. doi: 10.18632/oncotarget.105
88
Huang J Zhou Y Thomas G Gu Z Yang Y Xu H et al . NEDD8 Inhibition Overcomes CKS1B-Induced Drug Resistance by Upregulation of P21 in Multiple Myeloma. Clin Cancer Res (2015) 21(24):5532–42. doi: 10.1158/1078-0432.CCR-15-0254
89
Zhang B Gojo I Fenton R . Myeloid Cell Factor–1 is a Critical Survival Factor for Multiple Myeloma. Blood (2002) 99(6):1885–93. doi: 10.1182/blood.V99.6.1885
90
Peperzak V Vikström I Walker J Glaser S LePage M Coquery C et al . Mcl-1 Is Essential for the Survival of Plasma Cells. Nat Immunol (2013) 14(3):290–7. doi: 10.1038/ni.2527
91
Derenne S Monia B Dean N Taylor J Rapp M Harousseau J et al . Antisense Strategy Shows That Mcl-1 Rather Than Bcl-2 or Bcl-xL Is an Essential Survival Protein of Human Myeloma Cells. Blood (2002) 100(1):194–9. doi: 10.1182/blood.V100.1.194
92
Puthier D Derenne S Barillé S Moreau P Harousseau J Bataille R et al . Mcl-1 and Bcl-xL Are Co-Regulated by IL-6 in Human Myeloma Cells. Br J Haematol (1999) 107(2):392–5. doi: 10.1046/j.1365-2141.1999.01705.x
93
Jourdan M Veyrune J Vos J Redal N Couderc G Klein B . A Major Role for Mcl-1 Antiapoptotic Protein in the IL-6-Induced Survival of Human Myeloma Cells. Oncogene (2003) 22(19):2950–9. doi: 10.1038/sj.onc.1206423
94
Gupta V Matulis S Conage-Pough J Nooka A Kaufman J Lonial S et al . Bone Marrow Microenvironment–Derived Signals Induce Mcl-1 Dependence in Multiple Myeloma. Blood (2017) 129(14):1969–79. doi: 10.1182/blood-2016-10-745059
95
Slomp A Moesbergen L Gong J Cuenca M von dem Borne P Sonneveld P et al . Multiple Myeloma With 1q21 Amplification Is Highly Sensitive to MCL-1 Targeting. Blood Adv (2019) 3(24):4202–14. doi: 10.1182/bloodadvances.2019000702
96
Sklavenitis-Pistofidis R Reidy M Huynh D Zavidij O Berrios B Park J et al . Founding Precision Therapy for 1q-Amplified Multiple Myeloma. Clin Lymphoma Myeloma Leuk (2019) 19(10):e50. doi: 10.1016/j.clml.2019.09.076
97
Matulis S Gupta V Brown I Keats J Secrist P Cidado J et al . Preclinical Activity of Novel MCL1 Inhibitor AZD5991 in Multiple Myeloma. Blood (2018) 132(Supplement 1):952–2. doi: 10.1182/blood-2018-99-113504
98
Mani M Carrasco D Zhang Y Takada K Gatt M Dutta-Simmons J et al . BCL9 Promotes Tumor Progression by Conferring Enhanced Proliferative, Metastatic, and Angiogenic Properties to Cancer Cells. Cancer Res (2009) 69(19):7577–86. doi: 10.1158/0008-5472.CAN-09-0773
99
Takada K Zhu D Bird G Sukhdeo K Zhao J Mani M et al . Targeted Disruption of the BCL9/β-Catenin Complex Inhibits Oncogenic Wnt Signaling. Sci Trans Med (2012) 4(148):148ra117. doi: 10.1126/scitranslmed.3003808
100
Lyou Y Habowski A Chen G Waterman M . Inhibition of Nuclear Wnt Signalling: Challenges of an Elusive Target for Cancer Therapy. Br J Pharmacol (2017) 174(24):4589–99. doi: 10.1111/bph.13963
101
Marchesini M Ogoti Y Fiorini E Aktas Samur A Nezi L D'Anca M et al . ILF2 Is a Regulator of RNA Splicing and DNA Damage Response in 1q21-Amplified Multiple Myeloma. Cancer Cell (2017) 32(1):88–100.e6. doi: 10.1016/j.ccell.2017.05.011
102
Sklavenitis-Pistofidis R Reidy M Huynh D Salem K Park J Glavey S et al . Founding Precision Therapy in 1q-Amplified Multiple Myeloma. Blood (2018) 132(Supplement 1):1007–7. doi: 10.1182/blood-2018-99-112673
103
Trasanidis N Katsarou A Ponnusamy K Shen Y Kostopoulos I Bergonia B et al . Systems Medicine Dissection of Chr1q-Amp Reveals a Novel PBX1-FOXM1 Axis for Targeted Therapy in Multiple Myeloma. Blood (2022) 139(13):1939–53. doi: 10.1182/blood.2021014391
104
Taylor A Shih J Ha G Gao G Zhang X Berger A et al . Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell (2018) 33(4):676–689.e3.
105
Williams B Prabhu V Hunter K Glazier C Whittaker C Housman D et al . Aneuploidy Affects Proliferation and Spontaneous Immortalization in Mammalian Cells. Science (2008) 322(5902):703–9. doi: 10.1126/science.1160058
106
Stingele S Stoehr G Peplowska K Cox J Mann M Storchova Z . Global Analysis of Genome, Transcriptome and Proteome Reveals the Response to Aneuploidy in Human Cells. Mol Syst Biol (2012) 8(1):608. doi: 10.1038/msb.2012.40
107
Phillips T Michot J Ribrag V . Can Next-Generation PI3K Inhibitors Unlock the Full Potential of the Class in Patients With B-Cell Lymphoma? Clin Lymphoma Myeloma Leuk (2021) 21(1):8–20.e3. doi: 10.1016/j.clml.2020.08.022
108
Wang H Guo M Wei H Chen Y . Targeting MCL-1 in Cancer: Current Status and Perspectives. J Hematol Oncol (2021) 14(1):67. doi: 10.1186/s13045-021-01079-1
109
Haagensen E Kyle S Beale G Maxwell R Newell D . The Synergistic Interaction of MEK and PI3K Inhibitors Is Modulated by mTOR Inhibition. Br J Cancer (2012) 106(8):1386–94. doi: 10.1038/bjc.2012.70
110
Bolomsky A Vogler M Köse M Heckman C Ehx G Ludwig H et al . MCL-1 Inhibitors, Fast-Lane Development of a New Class of Anti-Cancer Agents. J Hematol Oncol (2020) 13(1):1386–94. doi: 10.1186/s13045-020-01007-9
Summary
Keywords
multiple myeloma, chr1q, amplification, precision medicine, therapy
Citation
Sklavenitis-Pistofidis R, Getz G, Ghobrial I and Papaioannou M (2022) Multiple Myeloma With Amplification of Chr1q: Therapeutic Opportunity and Challenges. Front. Oncol. 12:961421. doi: 10.3389/fonc.2022.961421
Received
04 June 2022
Accepted
20 June 2022
Published
14 July 2022
Volume
12 - 2022
Edited by
Alessandro Gozzetti, University of Siena, Italy
Reviewed by
Arleigh Robertson McCurdy, Ottawa Hospital, Canada; Carolina Terragna, Sant’Orsola-Malpighi Polyclinic, Italy
Updates
Copyright
© 2022 Sklavenitis-Pistofidis, Getz, Ghobrial and Papaioannou.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Papaioannou, papaioam@auth.gr; Irene Ghobrial, irene_ghobrial@dfci.harvard.edu
†These authors have contributed equally to this work and share senior authorship
This article was submitted to Hematologic Malignancies, a section of the journal Frontiers in Oncology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.