 Caitlin M. Tilsed
Caitlin M. Tilsed Scott A. Fisher
Scott A. Fisher Anna K. Nowak
Anna K. Nowak Richard A. Lake1,2
Richard A. Lake1,2 W. Joost Lesterhuis
W. Joost Lesterhuis- 1National Centre for Asbestos Related Diseases, Institute for Respiratory Health, Nedlands, WA, Australia
- 2School of Biomedical Sciences, University of Western Australia, Crawley, WA, Australia
- 3Medical School, University of Western Australia, Crawley, WA, Australia
- 4Department of Medical Oncology, Sir Charles Gairdner Hospital, Nedlands, WA, Australia
- 5Telethon Kids Institute, University of Western Australia, West Perth, WA, Australia
Chemotherapy has historically been the mainstay of cancer treatment, but our understanding of what drives a successful therapeutic response remains limited. The diverse response of cancer patients to chemotherapy has been attributed principally to differences in the proliferation rate of the tumor cells, but there is actually very little experimental data supporting this hypothesis. Instead, other mechanisms at the cellular level and the composition of the tumor microenvironment appear to drive chemotherapy sensitivity. In particular, the immune system is a critical determinant of chemotherapy response with the depletion or knock-out of key immune cell populations or immunological mediators completely abrogating the benefits of chemotherapy in pre-clinical models. In this perspective, we review the literature regarding the known mechanisms of action of cytotoxic chemotherapy agents and the determinants of response to chemotherapy from the level of individual cells to the composition of the tumor microenvironment. We then summarize current work toward the development of dynamic biomarkers for response and propose a model for a chemotherapy sensitive tumor microenvironment.
Introduction
The discovery and development of cytotoxic chemotherapies undoubtedly changed the landscape of cancer treatment. The first indication that a chemotherapy, or a cytotoxic chemical, could be a potential treatment for cancer was an incidental observation made in individuals exposed to a biochemical weapon, mustard gas. Soldiers exposed to mustard gas experienced severe leukopenia and marked depletion of the bone marrow and lymph nodes (1). This discovery was translated to one of the commonly studied cancers of that period, lymphoma, with the treatment resulting in significant, albeit temporary, regression. This stimulated the development and implementation of drug screening programs that tested multiple compounds in vitro for anti-cancer properties (2). Promising compounds were moved into animal cancer models, clinical trials in patients and developed into our current oncological treatment paradigm.
Despite the development of new treatment modalities, including oncogene-targeted therapies such as tyrosine kinase inhibitors, and immunotherapies such as immune checkpoint inhibitors, chemotherapy remains the first-line treatment for many cancers. In fact, despite the global search for new therapies that work synergistically with immune checkpoint blockade, combinations with classic chemotherapy so far have shown the best results (3). In many localized cancers, chemotherapy before or after surgery and/or combined with radiotherapy can provide durable, long-term survival benefits for many patients, such as chemoradiotherapy in esophageal cancer (4) or adjuvant chemotherapy in colon cancer (5). However, there are only a few scenarios in which chemotherapy results in robust and durable cures for metastatic solid cancers, with testicular cancer being the most important example (6–8). In almost all other metastatic cancers, clinical responses to systemic chemotherapy are partial at best, and then only in a subset of patients (Figure 1). This variability in chemo‐responsiveness occurs not only between patients with different tumor types, but also within groups of patients with the same tumor type. For example, in patients with esophageal cancer treated with carboplatin/paclitaxel in combination with radiotherapy, 30% have a histologically confirmed complete regression of their tumor, while 20% display no clinical response (4). Similarly, in early stage testicular cancer, adjuvant chemotherapy is curative and induces a robust clinical response in all but a small subset of patients (8). Also, in settings where chemotherapy rarely, if ever, results in complete regression, such as in mesothelioma, responses are still diverse, with approximately 40% of patients displaying an objective clinical response (12). This heterogeneity in response between patients with the same cancer type is not well understood. Given the frequent and sometimes severe toxicity of many chemotherapeutics, weighed against a beneficial response in only a subset of patients, there remains an urgent need for predictive biomarkers. However, despite many attempts, there are no robust and validated pre-treatment biomarkers that can guide clinical decision making.
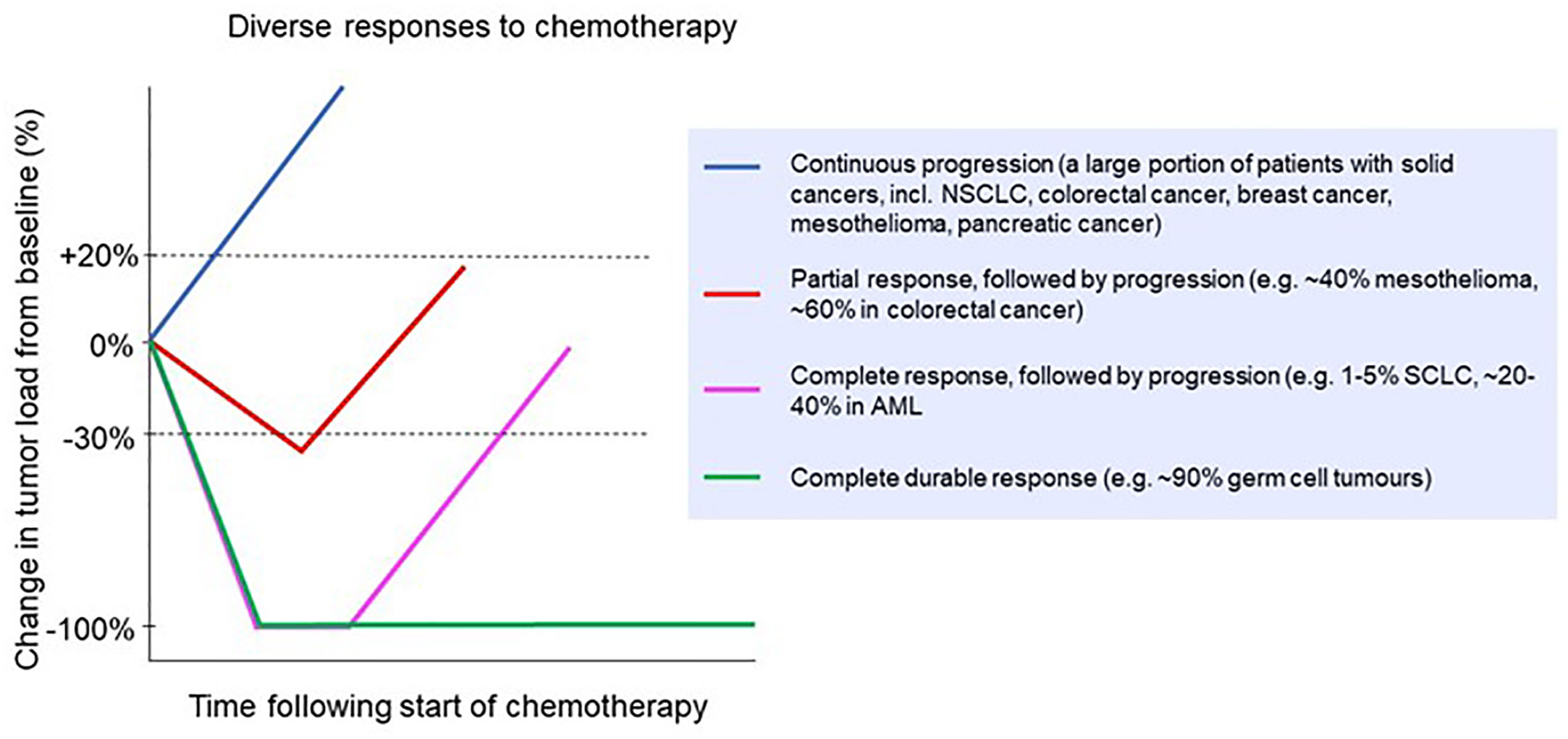
Figure 1 Clinical responses to chemotherapy in a range of cancer types. Patients might experience no response (continuous progression) or a partial response followed by progression [e.g. non-small cell lung cancer (9), colorectal cancer (10) breast cancer (11), mesothelioma (12) and pancreatic cancer (13)]; a complete response followed by progression [e.g. small cell lung cancer (14) and acute myeloid leukemia (15)]; or a complete durable response [e.g. germ cell tumors (16)].
In this review, we explore both the cell intrinsic (factors at the individual cell level) and cell extrinsic (factors within the tumor microenvironment) drivers of chemotherapy sensitivity or resistance. We then summarize the literature regarding the relationship between proliferation rate and chemotherapy sensitivity. Lastly, we describe the components of the tumor microenvironment and the roles they play in chemotherapy efficacy and propose a model of a chemo-sensitive tumor.
Mechanisms of inherent individual cell sensitivity and resistance to cytotoxic chemotherapy
Conventional chemotherapies are divided into several classes based on their primary or ostensible mechanism of action. They include alkylating agents and platinum analogues, which induce inter- or intra- strand DNA crosslinks that destabilize DNA and cause DNA breakage; antimetabolites that inhibit the synthesis of DNA, RNA or their components; topoisomerase inhibitors that block the DNA unwinding enzymes; and microtubular poisons that act on tubulin, impeding the mitotic spindle and stalling cell division (Table 1; Figure 2A). These drugs also have known secondary mechanisms of action, such as effects on mitochondrial biogenesis (22) or the production of reactive oxygen species (32), which contributes to their cytotoxicity (Figure 2B).

Table 1 Mechanism of action of classic chemotherapies.
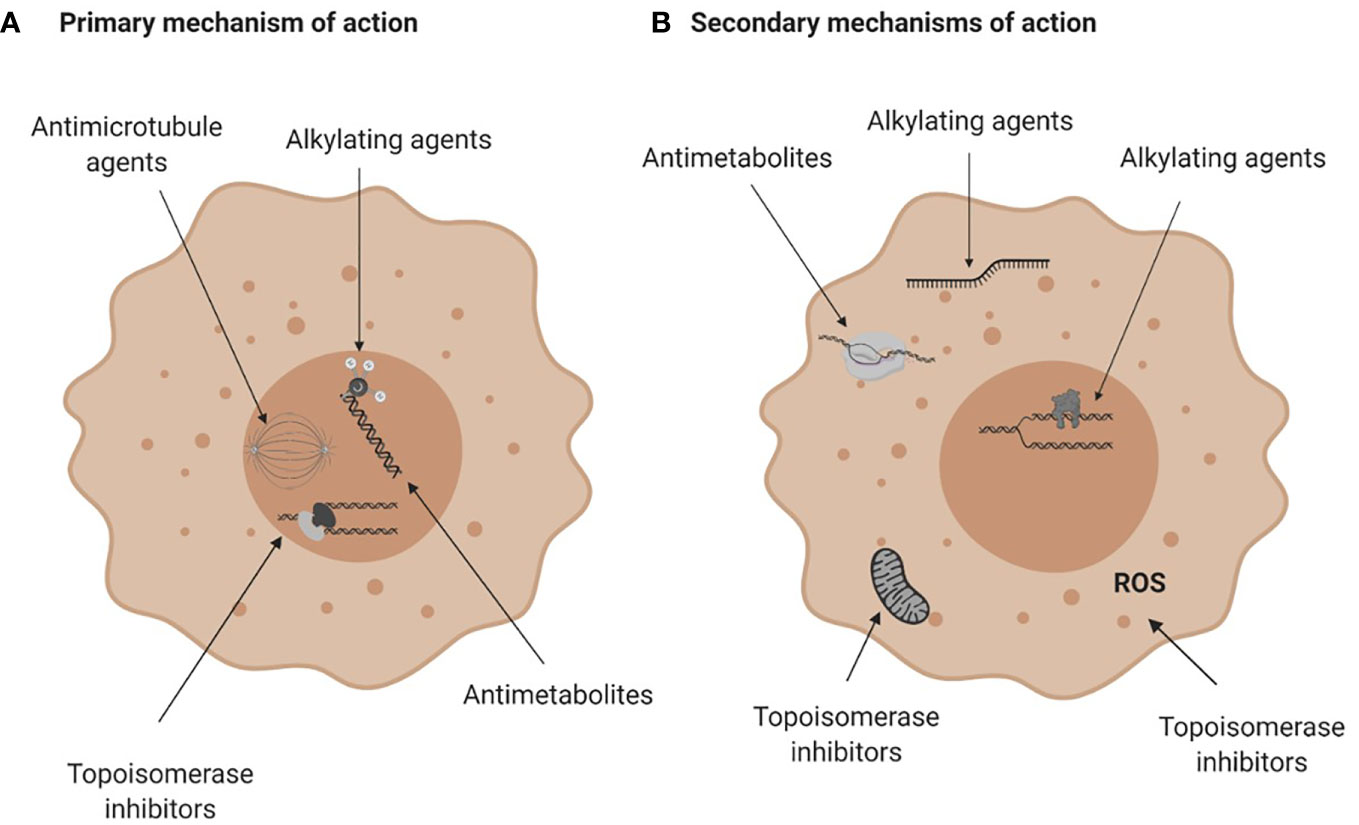
Figure 2 Mechanisms of action of conventional chemotherapies. (A) Primary mechanisms of action. Alkylating agents induce DNA breaks, anti-metabolites are incorporated into DNA or RNA and interfere with DNA and RNA synthesis, topoisomerase (Top) inhibitors damage the Top I or Top II enzymes halting DNA replication and anti-microtubule agents damage microtubules and affect mitosis. (B) Secondary mechanisms of action of chemotherapies. Alkylating agents can bind to RNA or induce protein-DNA crosslinks, antimetabolites can inhibit enzymes crucial for DNA or RNA synthesis and topoisomerase inhibitors can impair mitochondria biogenesis or generate reactive oxygen species. For more see Table 1. Figure created with BioRender.com.
Cancer cells experience different fates after drug exposure; some cells are killed while others escape cell death and survive (33, 34). Factors that contribute to the induction of apoptosis can act before DNA damage occurs (at the level of uptake of drug into the cell and the efflux of the drug out of the cell or the metabolism of drug to active metabolites); be associated with DNA damage (at the level of drug binding to target molecules, altered DNA repair enzymes or tolerance to DNA damage); or act after DNA damage has occurred (due to altered sensitivity to apoptosis, altered cell signaling or stochastic effects) and vary depending on the mechanism of action of the chemotherapy. Despite our increased understanding of these mechanisms of sensitivity and resistance this has not translated to the clinical implementation of a predictive biomarker of response to chemotherapy, nor the widespread use of combination therapies that exploit these pathways to improve drug effectiveness.
Uptake and efflux of chemotherapy drugs
While it is perhaps not surprising that cellular chemotherapy uptake varies between patients and tumor types, large differences in uptake are also observed in vitro between cells within the same clonal culture, resulting in differential therapeutic sensitivity (35, 36). Cellular features that can modulate intracellular levels of chemotherapy include efflux pumps which have been implicated in chemotherapy resistance (37–39). These drug efflux pumps, most notably p-glycoprotein, impede the transportation of chemotherapy into the cell. There have been numerous studies over the last few decades demonstrating that inhibition of these pumps improves chemotherapy uptake and tumor sensitivity in vitro (40–42) and in vivo (43–45).
Genetic variations
Cancer cells can harbor intrinsic mutations that render them less sensitive to chemotherapy. It is important to distinguish these inherent mutations that are present before treatment, from acquired mutations that the cells gain after treatment which provide a selective survival advantage (46, 47). For example, the dysregulation of components of the apoptotic pathway that enhance survival can increase drug resistance (48). Mutations in TP53, a key tumor suppressor, are associated with resistance to DNA damage induced by chemotherapy (49–52). Abnormalities in another tumor suppressor commonly dysregulated in cancer, retinoblastoma protein, is also associated with chemotherapy response in patients with lung cancer (53), breast cancer (54, 55), non-small cell lung cancer (56), and colorectal cancer (57) with the absence of retinoblastoma protein correlating with improved survival. Intrinsic mutations in the components of the apoptotic pathway are also associated with reduced sensitivity to chemotherapy. Lastly, genetic variations can occur in the proteins that some chemotherapies, primarily antimetabolites, target. For example, methotrexate binds to the enzyme DHFR to execute its anti-tumor effect and mutations in DHFR can alter chemo-sensitivity (58, 59).
Altered DNA damage repair pathways
As highlighted, a key mechanism of action of many chemotherapies is the induction of DNA damage which leads to the activation of cell death pathways. Acting against DNA damage are multiple repair pathways; base excision repair, mismatch repair, homologous recombination and non-homologous end-joining (60). Increased expression of nucleotide excision repair related genes correlates with resistance to platinum-based drugs. For example high expression of the excision repair cross-complementation group 1 factor is a known mechanism for cisplatin resistance in numerous cancers (61). Another example is ribonucleotide reductase subunit M1 which converts ribonucleotides into the deoxyribonucleotides required for DNA replication and DNA repair (62) and is inhibited by gemcitabine. Expression of RRM1 is inversely correlated with survival and sensitivity to platinum-based chemotherapy and gemcitabine (63, 64) in lung cancer or pancreatic cancer patients, though these findings vary with other studies finding no association with survival (65, 66).
Cell cycle
The cell cycle is intrinsically linked to chemotherapy efficacy because the primary mechanism of action of many drugs is to affect components crucial to cell division such as DNA replication or the formation of mitotic spindles. The cell cycle specificity of chemotherapies has been demonstrated in vitro. In the case of anti-mitotic chemotherapies, cytotoxicity is rarely induced until the cell enters mitosis, where it is most vulnerable (34). In vivo models to assess the effects of drugs on cell cycle often utilize the Fluorescent Ubiquitination-based Cell Cycle Indicator (FUCCI) system, in which cells express different fluorescent proteins as they progress throughout the cell cycle (67). In mice with orthotopic human gastric cancers, 68% of the cells were in S, G2 or M phase before treatment and 32% in G1 or G0 (68). After treatment with cisplatin or paclitaxel, more than 90% were in G1 or G0, indicating that the cells actively undergoing proliferation (those in S, G2 or M) were selectively targeted by the drugs. When cancer cells are treated with Salmonella Typhimurium AR-1 or recombinant methionine, trapping the cells in S/G2, they become more sensitive to subsequent treatment with cisplatin or paclitaxel, further demonstrating the cell cycle specificity of chemotherapy (69).
Cancer stem cells
Cancer stem cells (CSCs) are a small population of cancer cells with the capability of self-renewal and have high tumorigenic and metastatic potential (70). CSCs can be inherently resistant to chemotherapy due to a multitude of factors such as their slow proliferation rate and quiescent nature (71), active anti-apoptotic machinery (72, 73), efficient DNA repair systems (74, 75), effective modulation of reactive oxygen species (76) and their robust and stable expression of drug efflux pumps (77).
Chemotherapy induced senescence
A wide range of chemotherapies spanning most of the classes have been found to induce senescence both in vivo and in patient samples collected after treatment (78). These cells remain viable and metabolically active but are unable to proliferate. The induction of senescence by chemotherapy could be both beneficial and harmful to patient outcomes. As these cells do not divide and remain arrested in G1 or G2/M and can remain in this dormant state for an extended period of time, there is some degree of disease control (79, 80). However, the induction of senescence can be incomplete and is reversable, with the treatment resistant clones escaping cell cycle arrest and inducing disease relapse (81).
Stochastic differences affecting chemotherapy sensitivity
Lastly, the sensitivity of individual cancer cells to chemotherapy may differ due to stochastic differences. In vitro studies using genetically identical clonal cell lines, exposed to identical drug levels and corrected for cell cycle, found that there were significant differences in chemotherapy sensitivity between individual cells (33, 34, 82). This highlights that seemingly identical cancer cells differ in their chemo-sensitivity, even when all external factors are controlled for. Cancer cells exist in an equilibrium of pro- and anti- apoptotic proteins, where an additional stimulus can easily induce apoptosis. These cancer cells are termed ‘primed’ for apoptosis (83, 84) and are more chemo-sensitive than ‘un-primed’ cells (85). Heterogeneity in the chemotherapy response can be attributed to variability in the expression of key proteins, whereby some cells are primed for apoptosis and have a lower threshold of stimuli for the activation of cell death pathways due to the up- or down-regulation of specific pathways. For example, multiple myeloma is characterized by the overexpression of the anti-apoptotic proteins Bcl‐2 or Mcl-1 which favors cancer cell resistance to chemotherapy (86).
The influence of stochastic differences in chemotherapy sensitivity is further demonstrated in the observation that there is a moderate level of cell-cell variability in protein abundance in untreated cells and only 20% of this variability can be attributed to differences in cell cycle stage (33). When protein levels were measured before and after chemotherapy treatment, most of the profiles were similar before and after chemotherapy exposure in each individual cell. Interestingly, there was a small subset of proteins that displayed bimodal behavior, with increased levels in a subset of cells and decreased levels in others. Two of these proteins showed behavior that correlated with cell fate, indicating that the stochastic differences in protein expression between cells may contribute to the ability to escape chemotherapy induced cell death. These studies also demonstrated that the fate of individual sister cells can be independent from each other (34) and that individual subclones exhibit heterogeneity in the response to chemotherapy (34, 82), further highlighting the role of stochastically driven heterogeneity in the chemosensitivity of cancer cells.
Approaches to target cellular mechanisms of resistance to improve chemotherapy efficacy
Since the identification of the mechanisms of inherent cellular resistance to chemotherapy, novel drugs have been developed to target and inhibit drivers of resistance to improve chemotherapy efficacy. Targeting DNA repair pathways using poly(ADP-ribose) polymerases (PARP) inhibitors is one avenue that has shown promise. As PARP acts to recruit DNA repair proteins to promote repair of DNA breaks as well as homologous recombination, the inhibition of PARP limits DNA repair after damage which could augment the effects of chemotherapies that damage DNA (87). The addition of PARP inhibitors to chemotherapy have shown some clinical efficacy (Table 2) and work is ongoing to expand these findings to other cancers and chemotherapy combinations (107).
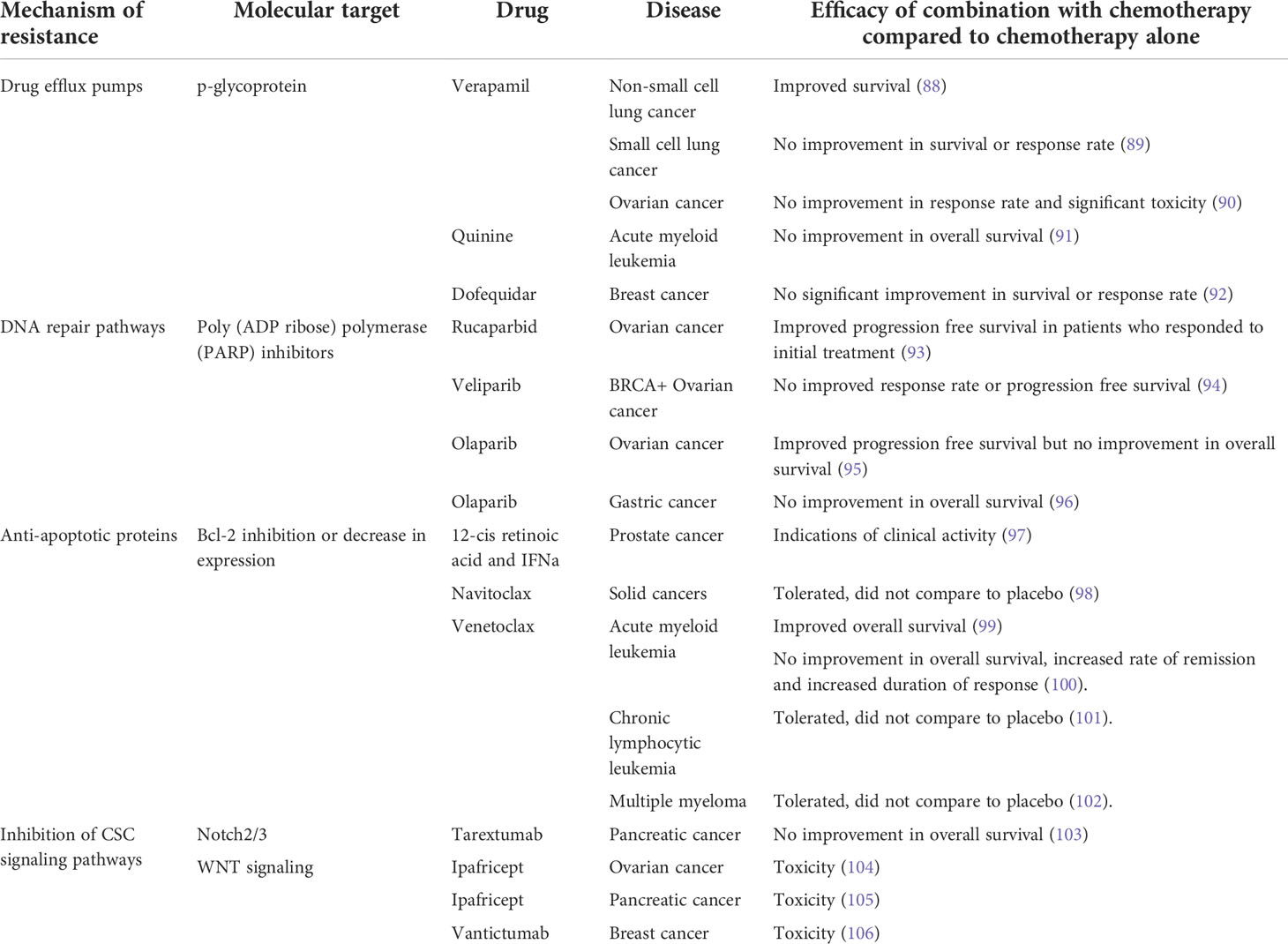
Table 2 Clinical studies that combine chemotherapy with agents that target cellular mechanisms of chemo-resistance. PARP, poly(ADP ribose) polymerase.
However, the expansion of this strategy to other pathways or drugs has proven difficult with mixed results from clinical trials and the discontinuation of studies due to excessive toxicity (Table 2). One example relates to drugs that target drug efflux pumps, particularly those mediated by p-glycoprotein. Clinical trials have not found a significant survival benefit using combination therapy with drug efflux pump inhibitors and chemotherapy (89–91) or only a slight improvement in a subset of patients (88), and development has been hindered by the levels of toxicity associated with the dose required for a clinical benefit to be achieved. Similarly, some drugs targeting the WNT signaling pathway which is important in both conventional stem cells and CSCs (108) have had to be discontinued due to toxicity, primarily in the bone marrow leading to increased incidence of fractures (105). Lastly, drugs that inhibit or decrease the expression of the anti-apoptotic protein Bcl-2 are well tolerated and induce substantial responses when used as a monotherapy or combined with dexamethasone in chronic lymphocytic leukemia or multiple myeloma (101, 102) or combined with azacytidine, decitabine or low-dose cytarabine in acute myeloid leukemia (99), the latter receiving FDA approval.
Proliferation and chemotherapy sensitivity-the proliferation rate hypothesis
An underlying commonality between the classes of chemotherapeutics discussed in this review is that their mechanism of action primarily affects biological processes associated with cell division, either by inducing DNA damage or by directly inhibiting mitotic progression. Since a key hallmark of cancer is rapid and uncontrolled cellular division, cancer cells are therefore thought to be more sensitive to chemotherapeutic drugs then normal tissues. This has led to the general acceptance within the biomedical community that the cancer specificity of chemotherapy comes from the preferential killing of rapidly proliferating cells (109). However, the experimental results that support this notion are relatively limited and mixed.
Firstly, the relationship between proliferation rates and chemo-sensitivity in vitro is not straightforward. For example, Kondoh et al. analyzed the correlation between doubling time and sensitivity of anticancer drugs against the NCI-60 panel of cancer cell lines (110). Although the authors found that majority of anticancer drugs had higher efficacy in faster dividing cell lines, this was not the case for all chemotherapeutics and not for all cancer cell lines. These conflicting and varying results are also evident from other studies (41, 111), including studies in which the proliferation rate was modulated, through either pharmacological means or gene silencing, showing varying degrees of both decreased (110, 112) and increased sensitivity to chemotherapy (113–116).
Secondly, there is limited in vivo validation of the increased effect of chemotherapy in highly proliferating tumors. Many studies measured proliferation at either a fixed timepoint within the tumor or used in vitro rates of cell division and correlated this with in vivo response. For example, Nakasone et al. demonstrated that the in vivo difference in sensitivity between different tumor models could not be attributed to differences in in vitro proliferation rate (117). The development of intravital fluorescent imaging, utilizing the FUCCI system (67) has made it possible to assess proliferation over time within the tumor itself, overcoming the caveats of previous studies. Yano et al. utilized these methods to monitor the cell cycle progression in an orthotopic model of liver cancer during chemotherapy treatment (68). When tumors with most cells in S/G2/M phase (an actively cycling or proliferating tumor) cisplatin or paclitaxel treatment resulted in significant cancer cytotoxicity, while there was little anti-tumor effect when cells were mainly in G0/G1 (69). Although these data suggest a correlation between cell cycle stage (and by extension proliferation rate), clinical data to substantiate this hypothesis are limited and mixed; the use of proliferation rate as a biomarker for response to chemotherapy varies greatly between cancers and is limited in its predictive power. For example, there is a striking absence of any significant and reproducible correlation between high expression of Ki67 and chemotherapy response in many cancer types (Figure 3). A systematic review in breast cancer found a correlation between Ki67 expression before neoadjuvant chemotherapy and overall/progression-free survival in 10/20 and 17/33 studies, respectively (118). Similarly, one meta-analysis found that high (>10%) Ki67 positivity is associated with decreased survival (119), while another reported that high Ki67 could predict response and clinical benefit from neoadjuvant chemotherapy (120).
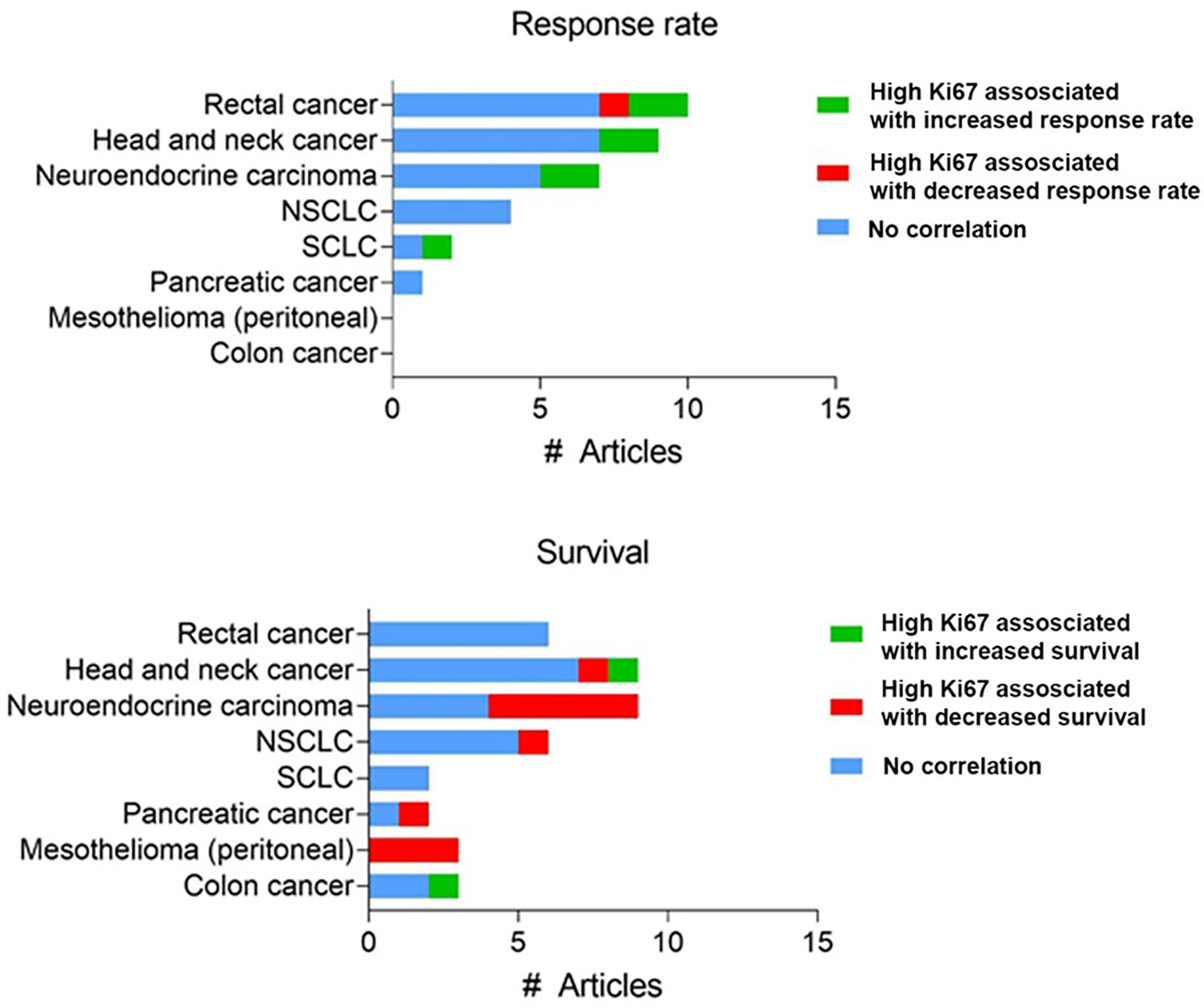
Figure 3 Reported clinical studies testing a correlation between Ki67 expression and chemotherapy response. Number of studies assessing Ki67 expression using immunohistochemistry with reported correlation between Ki67 expression and response rate or survival. Full dataset in Supplementary Table 1.
For these reasons, others have previously critically challenged the assumption that chemotherapy particularly targets cancer cells because they are rapidly proliferating (18, 109, 121). Regardless, the inability to consistently correlate cell proliferation rate with chemotherapy response in patients highlights that there are likely additional drivers of chemotherapy sensitivity.
The tumor microenvironment: the driver of chemotherapy sensitivity
Stroma and vasculature: a role in chemotherapy resistance
A tumor is a complex and dynamic environment of immune cells, extracellular matrix, fibroblasts and vasculature which make up the tumor stroma, which can all influence chemotherapy sensitivity (Figure 4).
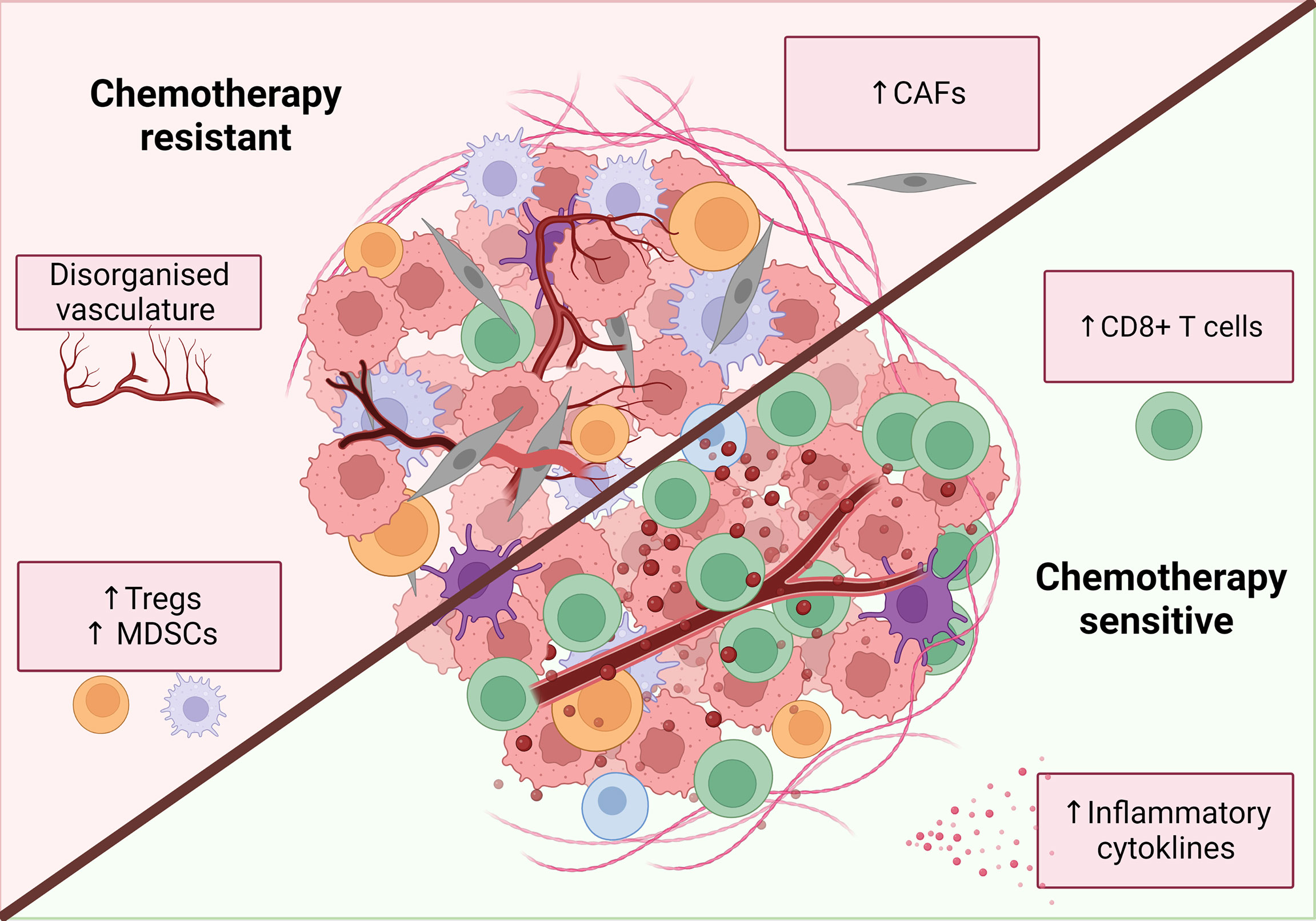
Figure 4 Characteristics of a chemotherapy sensitive TME. An inflammatory, immune infiltrated ‘hot’ tumor is associated with response to classical chemotherapies. These tumors are characterized by the infiltration of immune cells, particularly increased CD8+ T cells. release of inflammatory mediators such as IFNs and TNFα and decreased levels of immunosuppressive cells. Additionally, tumor vasculature can have both positive and negative effects on chemotherapy response while CAFs are primarily associated with chemotherapy resistance. Figure created with BioRender.com.
Cancer associated fibroblasts (CAFs) are one of the key components of the tumor stroma that have been implicated in tumor progression and resistance to chemotherapy. CAFs are phenotypically different from other fibroblasts and secrete cancer-promoting factors including TGF‐β, vascular endothelial growth factor, platelet-derived growth factor and fibroblast growth factor 2 (122). The secretion of these factors are associated with an enhancement in the invasive and metastatic ability of cancer cells (123, 124), stimulation of angiogenesis (125, 126),and expression of anti-apoptotic proteins (127) and promote an immunosuppressive tumor microenvironment (128) which modify the TME to support tumor cell survival and chemotherapy resistance (129). In vitro studies have confirmed that these soluble factors induce chemotherapy resistance (130–133) and targeting of CAFs in vivo enhances the anti-tumor effects of chemotherapy in animal models (134–136). CAFs also contribute to desmoplasia which is the formation of fibrotic tissue around tumor cells. This creates not only a physical barrier around the tumor, limiting the penetration of drugs, but also can increases interstitial pressure which compresses blood vessels, also decreasing drug availability within the TME (137, 138). Clinical studies found high levels of CAFs were associated with poor PFS following chemotherapy (139–142).
As chemotherapy is typically administered systemically, it is essential that the drug reaches the tumor at a sufficient concentration. Therefore, tumor vasculature is not only essential for cancer growth, but also for the distribution of chemotherapy to the tumor. Patients with tumors exhibiting a lower density of blood vessels indeed have a poorer response to chemotherapy (143, 144). However, tumor vasculature is often disorganized and characterized by neo-angiogenesis and the modification of existing vessels within the tissue stroma which can impede the distribution of chemotherapy. Strategies to ‘normalize’ tumor vasculature and reverse the dysfunctional structure have proven to be effective in murine studies (145–148). When drugs targeting tumor vasculature are combined with chemotherapy, both the chemotherapeutic dose reaching the tumor and subsequent anti-tumor response is more effective compared to chemotherapy alone (145, 146, 149). Clinical studies have demonstrated primarily positive results. For example, the addition of bevacizumab (a VEGF-A inhibitor) to chemotherapy regimens lead to an increase in both progression free and overall survival in mesothelioma (150) and ovarian cancer (151, 152). No clinical benefit was seen when combined with chemotherapy in early stage NSCLC (153) while in colon cancer bevacizumab improved survival in the metastatic but not adjuvant setting (154, 155).
Immunity, inflammation and chemotherapy efficacy
Spurred on by the emergence and success of immunotherapy, the role of the immune system within the tumor stroma is now a key focus of research into chemotherapy efficacy. However, because chemotherapy is leukodepleting, it was historically thought to be predominantly immunosuppressive. The important role of immune cells in the efficacy of chemotherapy has been highlighted in recent years, first demonstrated by Schwartz in 1973 (156) and more recently driven by the work of Nowak (157, 158) and Zitvogel and Kroemer (159, 160). The latter group compared the response to chemotherapy in immunodeficient (predominantly either Nu/Nu or Rag1-/- mice which lack functional T cells and functional T and B cells respectively) and immunocompetent mice (159, 161). Chemotherapy was significantly less effective in mice lacking an intact immune system, while these drugs were extremely effective in the wildtype counterparts. The requirement of an intact immune system for chemotherapy to induce an effective anti-tumor response has been tested using numerous cancer models and chemotherapies (Table 3). Genetic mouse models did not provide the same convincing results as transplantable models, which tend to be more immunogenic (165).
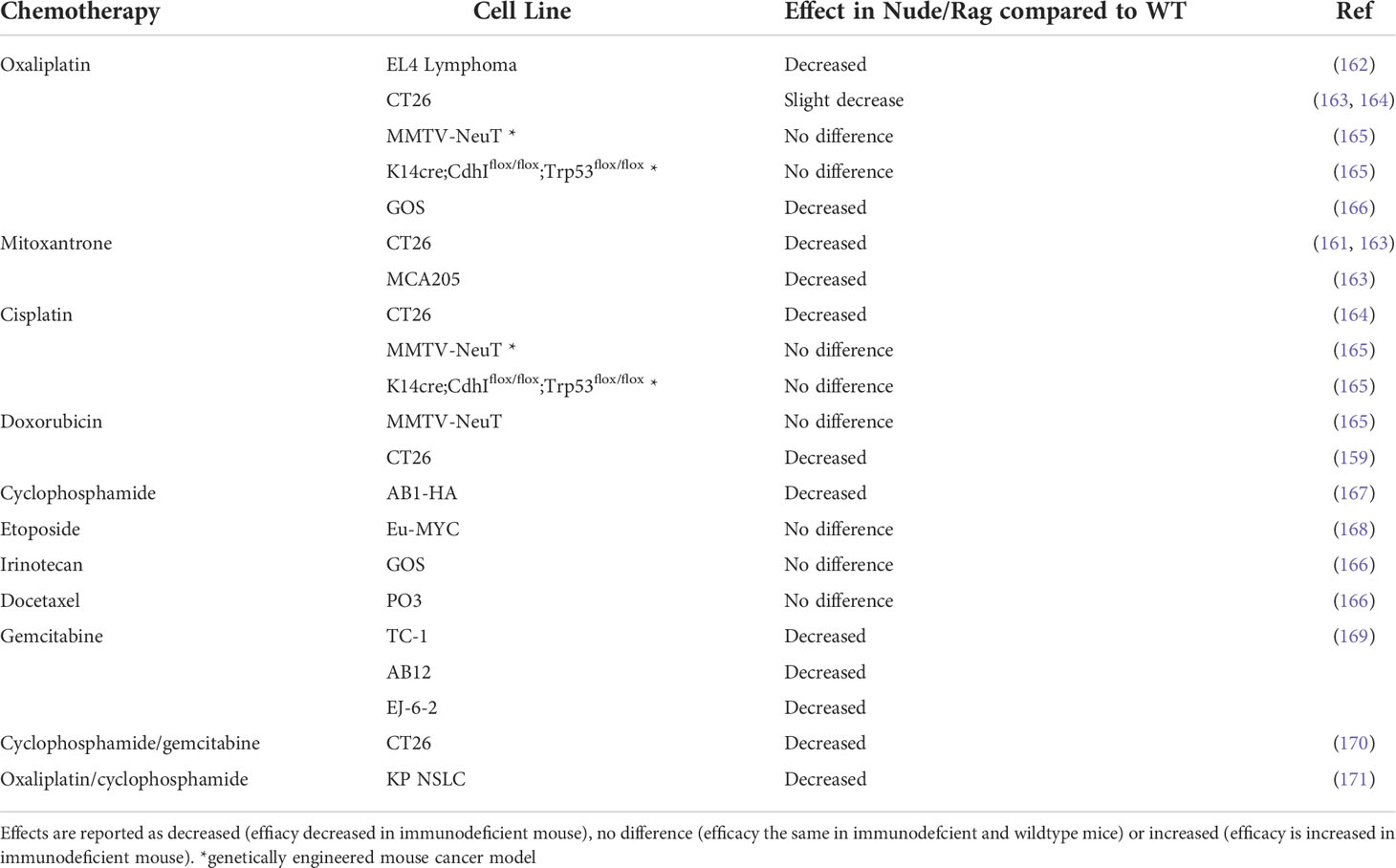
Table 3 Effect of chemotherapy in immunodeficient Nude/Rag mice compared to immunocompetent wildtype (WT) mice.
Studies using depleting antibodies against specific immune cells and cytokines or the knockout of specific genes or cellular receptors have helped to define the cell types and pathways that play an important role in the chemotherapy response (Tables 4, 5). Experiments where IFNγ was neutralized using knock‐out mouse models found that the absence of signaling decreased chemotherapy efficacy in vivo (162, 167, 173, 174). The idea of an immunostimulatory ‘hot’ TME has been popularized in the context of immunotherapy: highly immune infiltrated, inflammatory tumors with increased expression of IFN-related genes are associated with improved response to immune checkpoint therapy (179–181). Indeed, several studies suggest an inflammatory, immune infiltrated TME is associated with chemotherapy sensitivity, with complete responders to chemotherapy having increased immune infiltration (178, 182–184). In particular, responders to chemotherapy have increased levels of CD8+ T cells (182, 185, 186) and the upregulation of IFN related genes (178, 182). The presence of immunosuppressive cells like regulatory T cells (Treg) or myeloid suppressor cells (MDSC) are associated with a decreased response to chemotherapy, presumably due to the promotion of an immunosuppressive TME (187–191). The contribution of other immune cells to chemotherapy sensitivity is less well characterized and may vary depending on drug or tumor model (Table 3) and further studies are required to fully elucidate their role in chemotherapy‐driven anti-tumor immunity.
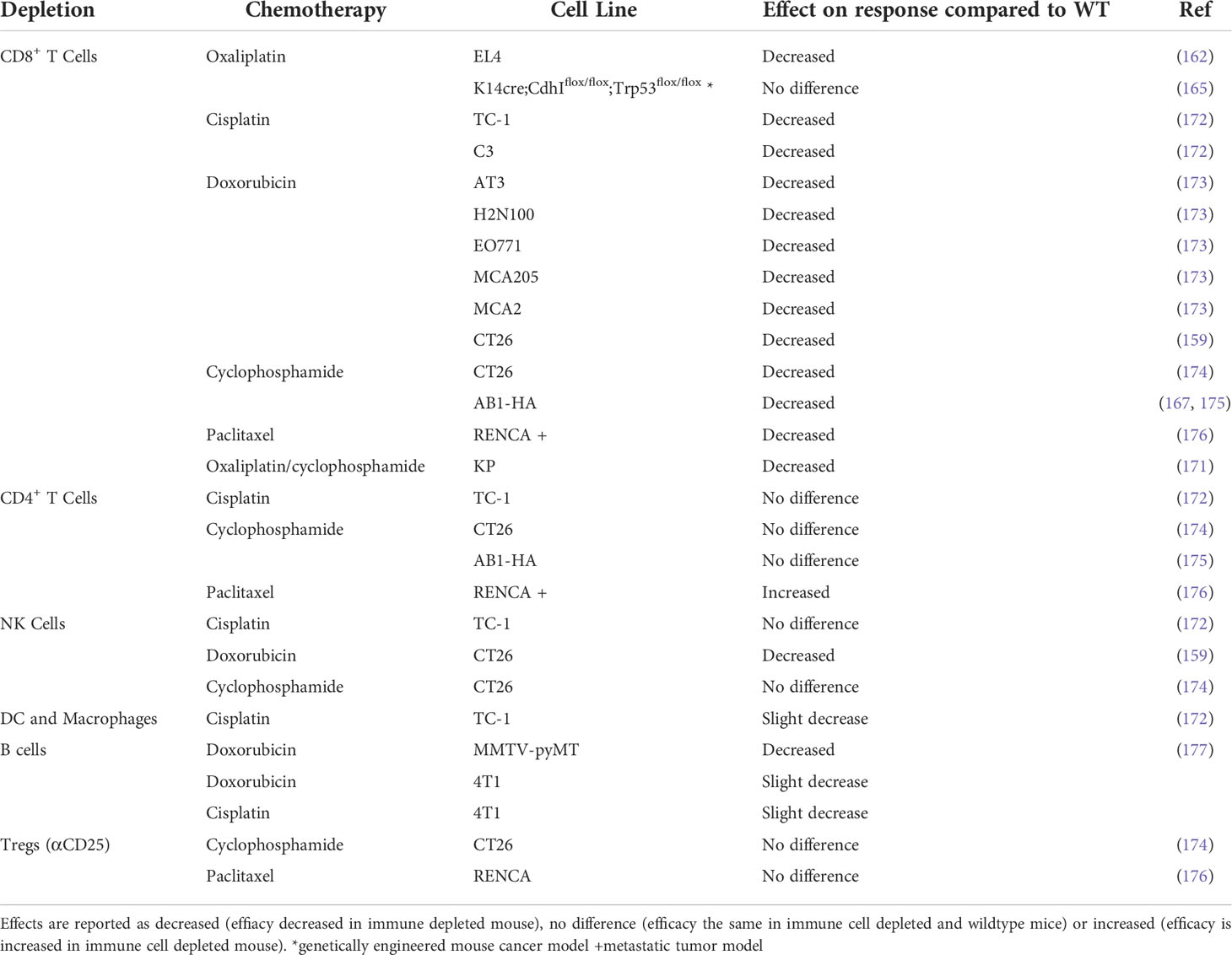
Table 4 Effect of chemotherapy in immune cell depleted mice compared to immunocompetent wildtype mice.
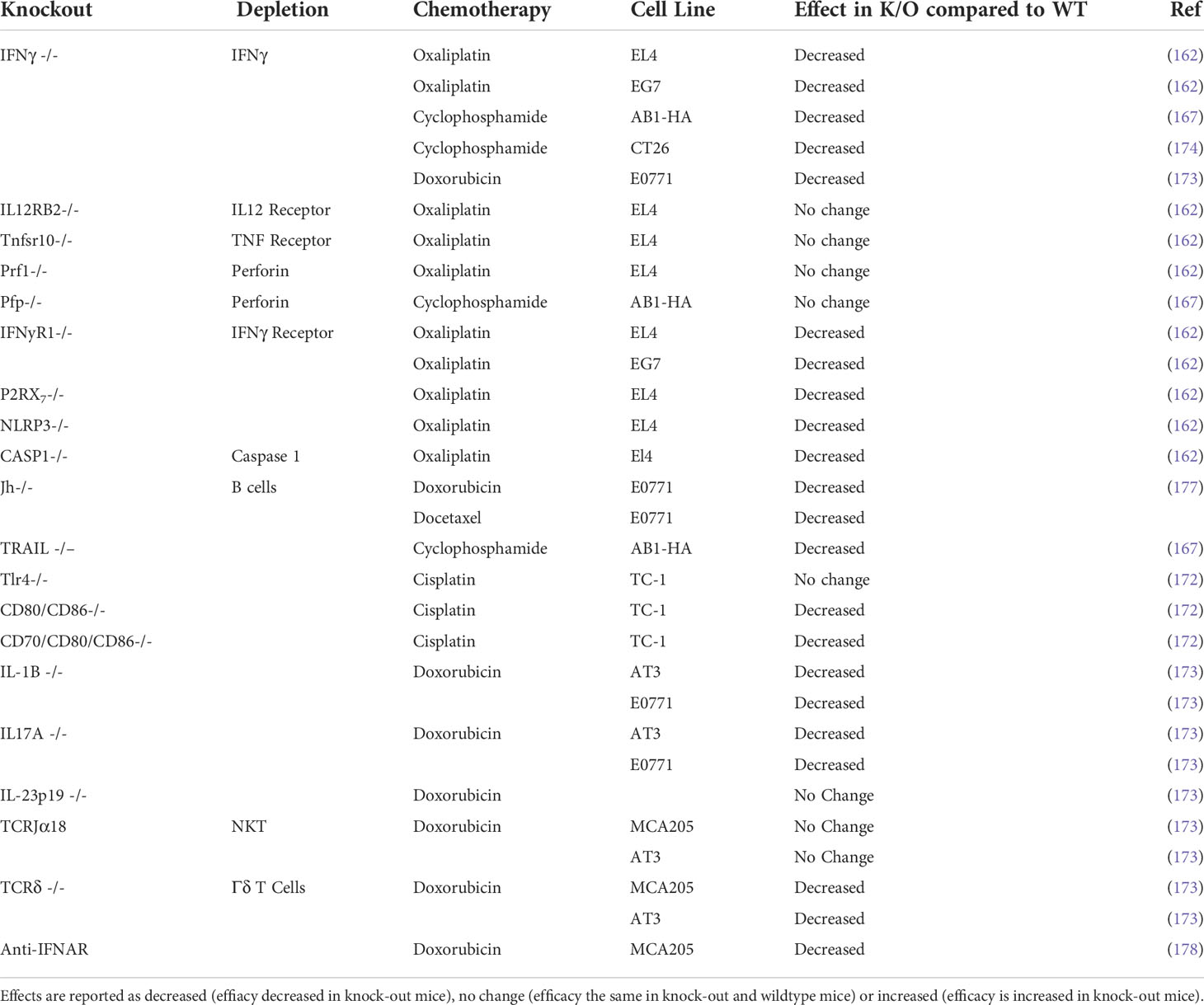
Table 5 Effect of chemotherapy in knock out mice compared to immunocompetent wildtype mice.
It is evident from the above studies in mouse models and patients that the composition of the TME plays a critical role in chemotherapy efficacy. It must also be noted that chemotherapy has numerous positive and negative effects on immune cells, which have been reviewed in detail previously (160, 192). The fact that chemotherapy is one of the most efficacious combinatorial treatments with immune checkpoint therapy, suggests at the very least that chemotherapy treatment is not an immunological null-event, and that its beneficial immunological effects can be exploited therapeutically (3, 193, 194).
Tracking dynamic changes in the tumor microenvironment correlating with treatment outcome
Increased understanding of the role of the TME in the response to chemotherapy has led to a large body of work on identifying predictive biomarkers from the TME. A detailed understanding of the effects of chemotherapy on the various components of the TME would help with the selection of cell populations, genes or proteins for use as a predictive biomarker. While there has been a significant amount of work in this field, there has been little progress of integrating the use of pre-treatment biomarkers into routine use within the clinic that could predict response. It must be noted that whether a patient achieves a pathological complete response when chemotherapy is used in a neoadjuvant setting is a robust indicator of clinical outcome, as for most cancers, these patients have increased disease-free survival and overall survival compared to patients with residual disease at surgery (195).
One of the limitations of the most common approach to biomarker discovery is that tumor or blood samples are only collected at a single timepoint, usually before treatment. This only gives a ‘snapshot’ of the tumor microenvironment, or the systemic environment in the case of blood sample. Taking serial samples would allow the effects of chemotherapy to be monitored throughout therapy. Whether a patient is responding or not would be able to be determined earlier, allowing physicians to make a more informed decision on whether to continue with the current treatment or not. Moreover, it would allow a deeper understanding of the biological mechanisms that are responsible for an effective chemotherapy response, allowing the development of novel rational combination therapies. Measuring the changes within the tumor that are induced by chemotherapy is hindered by the inability to obtain serial tumor samples from patients throughout the course of their therapy, primarily due to the location of the cancer and invasive procedures required to retrieve a biopsy. Often clinical studies use peripheral blood (196, 197) or effusions as a surrogate for the tumor microenvironment, however it is not clear whether these samples provide a meaningful representation of the events occurring within the tumor itself (198). Studies that do examine the changes within the TME during therapy differ in the parameters measured, often only measuring a selection of markers, making it difficult to compare between studies, and likely resulting in an incomplete representation of what happens throughout the whole TME.
The few studies that investigated changes in the TME during chemotherapy and correlated these changes with clinical response highlight the importance of serial measurements instead of pre- or post- treatment snapshots. Many studies found no difference between responders and non-responders when baseline levels of their chosen markers were compared (Supplementary Table 2). However, when the change in expression from pre-treatment to post-treatment was interrogated, the differences between responders and non-responders became apparent (199–202). Molecules involved in chemotherapy resistance, for example GSTP1 (an enzyme associated with decreased sensitivity to cytotoxic agents including anthracyclines (203)) or ALDH1 (an enzyme involved in detoxifying aldehydes into weaker metabolites), decrease in patients whose tumors respond to chemotherapy (200, 204). Decreased GSTP1 expression after chemotherapy, is more prominent in tumors of patients with breast cancer that respond to doxorubicin and cyclophosphamide chemotherapy and is associated with improved progression free survival (204). Similarly, decreased tumor expression of biomarkers associated with tumorigenesis (COX-2) or immune evasion (PD-L1) is noted throughout treatment, with the decrease more prominent in responders (199, 205). The primary limitation of these studies is that they only assessed a small number of markers or cell populations, using immunohistochemistry or flow cytometry. This makes it difficult to capture the complexity of the TME. The increasing ability to obtain high dimensional biological data using for example single cell RNAseq or spatial transcriptomics provides an avenue for a deeper characterization of the TME during chemotherapy treatment.
Summary and key outstanding questions
Based on the available data, an incomplete picture emerges of a chemotherapy-sensitive TME, which includes CD8 T cell infiltration, activation of inflammatory pathways such as IFNs, low levels of CAFs and a normalized vasculature (Figure 4).
An added complexity is the wide range of chemotherapeutics used in the clinic, spanning different classes with vastly different mechanisms of action and immune effects. Uncovering what drives chemotherapy efficacy opens the door to the development of predictive biomarkers and novel combination treatments. While immune checkpoint therapy has shown promise in a multitude of cancer types, like chemotherapy, it is not effective in all patients. Having a predictive biomarker for a robust response to chemotherapy, either on its own or in combination with immunotherapy, would significantly improve the potential of clinical decision making, allowing patients to stratified based on their likelihood of a beneficial response to either treatment.
An additional question is whether the TME can be modulated and transformed from chemotherapy-resistant to chemotherapy-sensitive. Pre-treating a patient to induce a sensitive TME phenotype has improved the response to immunotherapy in preclinical models (206, 207). The dependence of chemotherapy efficacy on the immune system and early indications of synergy between chemotherapy and immune checkpoint therapy (208–210) highlights the opportunity to alter the tumor immune milieu to improve the anti-tumor immune response generated by chemotherapeutics.
Author contributions
CT wrote the article and generated the figures and tables. SF, RL, AN, and WL critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
WL was supported by fellowships from the Simon Lee Foundation, NHMRC (grant names APP1126076 and APP1196605) and Cancer Council WA. The National Centre for Asbestos Related Diseases received funding through the NHMRC Centre of Research Excellence scheme (grant number APP1197652).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.960317/full#supplementary-material
References
1. Krumbhaar EB, Krumbhaar HD. The blood and bone marrow in yelloe cross gas (Mustard gas) poisoning: Changes produced in the bone marrow of fatal cases. J Med Res (1919) 40(3):497–508.
2. DeVita VT, Chu E. A history of cancer chemotherapy. Cancer Res (2008) 68(21):8643–53. doi: 10.1158/0008-5472.CAN-07-6611
3. Schmidt EV, Chisamore MJ, Chaney MF, Maradeo ME, Anderson J, Baltus GA, et al. Assessment of clinical activity of PD-1 checkpoint inhibitor combination therapies reported in clinical trials. JAMA Netw Open (2020) 3(2):e1920833. doi: 10.1001/jamanetworkopen.2019.20833
4. van Hagen P, Hulshof MCCM, van Lanschot JJB, Steyerberg EW, Henegouwen MI van B, Wijnhoven BPL, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. New Engl J Med (2012) 366(22):2074–84. doi: 10.1056/NEJMoa1112088
5. Achilli P, Crippa J, Grass F, Mathis KL, D’Angelo ALD, Aziz MAAE, et al. Survival impact of adjuvant chemotherapy in patients with stage IIA colon cancer: Analysis of the national cancer database. Int J Cancer (2021) 148(1):161–9. doi: 10.1002/ijc.33203
6. Tandstad T, Ståhl O, Håkansson U, Dahl O, Haugnes HS, Klepp OH, et al. One course of adjuvant BEP in clinical stage I nonseminoma mature and expanded results from the SWENOTECA group. Ann Oncol (2014) 25(11):2167–72. doi: 10.1093/annonc/mdu375
7. Chau C, Cathomas R, Wheater M, Klingbiel D, Fehr M, Bennett J, et al. Treatment outcome and patterns of relapse following adjuvant carboplatin for stage I testicular seminomatous germ-cell tumour: results from a 17-year UK experience. Ann Oncol (2015) 26(9):1865–70. doi: 10.1093/annonc/mdv254
8. Dieckmann KP, Dralle-Filiz I, Matthies C, Heinzelbecker J, Bedke J, Ellinger J, et al. Testicular seminoma clinical stage 1: treatment outcome on a routine care level. J Cancer Res Clin Oncol (2016) 142(7):1599–607. doi: 10.1007/s00432-016-2162-z
9. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non–Small-Cell lung cancer. New Engl J Med (2015) 373(2):123–35. doi: 10.1056/NEJMoa1504627
10. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med (2004) 350(23):2335–42. doi: 10.1056/NEJMoa032691
11. Bear HD, Tang G, Rastogi P, Geyer CE, Robidoux A, Atkins JN, et al. Bevacizumab added to neoadjuvant chemotherapy for breast cancer. N Engl J Med (2012) 366(4):310–20. doi: 10.1056/NEJMoa1111097
12. Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol (2003) 21(14):2636–44. doi: 10.1200/JCO.2003.11.136
13. Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. New Engl J Med (2018) 379(25):2395–406. doi: 10.1056/NEJMoa1809775
14. Cheng S, Evans WK, Stys-Norman D, Shepherd FA. Chemotherapy for relapsed small cell lung cancer: A systematic review and practice guideline. J Thorac Oncol (2007) 2(4):348–54. doi: 10.1097/01.JTO.0000263720.15062.51
15. Löwenberg B, Ossenkoppele GJ, van Putten W, Schouten HC, Graux C, Ferrant A, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med (2009) 361(13):1235–48. doi: 10.1056/NEJMoa0901409
16. Rajpert-De Meyts E, McGlynn KA, Okamoto K, Jewett MAS, Bokemeyer C. Testicular germ cell tumours. Lancet (2016) 387(10029):1762–74. doi: 10.1016/S0140-6736(15)00991-5
17. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by taxol. Nature (1979) 277(5698):665–7. doi: 10.1038/277665a0
18. Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T. Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol (2011) 8(4):244–50. doi: 10.1038/nrclinonc.2010.228
19. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer (2004) 4(4):253–65. doi: 10.1038/nrc1317
20. Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science (1984) 226(4673):466–8. doi: 10.1126/science.6093249
21. Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. i. anthracycline radical formation by NADH dehydrogenase. J Biol Chem (1986) 261(7):3060–7. doi: 10.1016/S0021-9258(17)35746-0
22. Henriksen PA. Anthracycline cardiotoxicity: an update on mechanisms, monitoring and prevention. Heart (2018) 104(12):971–7. doi: 10.1136/heartjnl-2017-312103
23. DeVita VT, Lawrence TS, Rosenberg SA. DeVita, hellman, and rosenberg’s cancer: Principles & practice of oncology Vol. 1748 p. Lippincott Williams & Wilkins (Wolters Kluwer Health) (2008).
24. Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer (2012) 12(2):104–20. doi: 10.1038/nrc3185
25. Mizutani H, Tada-Oikawa S, Hiraku Y, Kojima M, Kawanishi S. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci (2005) 76(13):1439–53. doi: 10.1016/j.lfs.2004.05.040
26. Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R, et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat Commun (2013) 4(1):1908. doi: 10.1038/ncomms2921
27. Gewirtz D. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol (1999) 57(7):727–41. doi: 10.1016/S0006-2952(98)00307-4
28. Basourakos SP, Li L, Aparicio AM, Corn PG, Kim J, Thompson TC. Combination platinum-based and DNA damage response-targeting cancer therapy: Evolution and future directions. Curr Med Chem (2017) 24(15):1586–606. doi: 10.2174/0929867323666161214114948
29. Hato SV, Khong A, de Vries IJM, Lesterhuis WJ. Molecular pathways: the immunogenic effects of platinum-based chemotherapeutics. Clin Cancer Res (2014) 20(11):2831–7. doi: 10.1158/1078-0432.CCR-13-3141
30. Mikuła-Pietrasik J, Witucka A, Pakuła M, Uruski P, Begier-Krasińska B, Niklas A, et al. Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell Mol Life Sci (2019) 76(4):681–97. doi: 10.1007/s00018-018-2954-1
31. Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol (2006) 17 Suppl 5:v7–12. doi: 10.1093/annonc/mdj941
32. Yang H, Villani RM, Wang H, Simpson MJ, Roberts MS, Tang M, et al. The role of cellular reactive oxygen species in cancer chemotherapy. J Exp Clin Cancer Res (2018) 37(1):266. doi: 10.1186/s13046-018-0909-x
33. Cohen AA, Geva-Zatorsky N, Eden E, Frenkel-Morgenstern M, Issaeva I, Sigal A, et al. Dynamic proteomics of individual cancer cells in response to a drug. Science (2008) 322(5907):1511–6. doi: 10.1126/science.1160165
34. Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell (2008) 14(2):111–22. doi: 10.1016/j.ccr.2008.07.002
35. Durand RE, Olive PL. Flow cytometry studies of intracellular adriamycin in single cells in vitro. Cancer Res (1981) 41(9 Pt 1):3489–94.
36. Ganapathi R, Gulick P, Miller R, Grabowski D, Turinic R, Valeunzuela R, et al. Analysis of heterogeneity in daunorubicin uptake by human leukemia cells using laser flow cytometry. Invest New Drugs (1985) 3(3):273–7. doi: 10.1007/BF00179431
37. Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Targets (2013) 13(3):326–46. doi: 10.2174/15680096113139990076
38. Ughachukwu P, Unekwe P. Efflux pump-mediated resistance in chemotherapy. Ann Med Health Sci Res (2012) 2(2):191–8. doi: 10.4103/2141-9248.105671
39. Muley H, Fadó R, Rodríguez-Rodríguez R, Casals N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem Pharmacol (2020) 177:113959. doi: 10.1016/j.bcp.2020.113959
40. Gagliano T, Gentilin E, Benfini K, Di Pasquale C, Tassinari M, Falletta S, et al. Mitotane enhances doxorubicin cytotoxic activity by inhibiting p-gp in human adrenocortical carcinoma cells. Endocrine (2014) 47(3):943–51. doi: 10.1007/s12020-014-0374-z
41. Kibria G, Hatakeyama H, Akiyama K, Hida K, Harashima H. Comparative study of the sensitivities of cancer cells to doxorubicin, and relationships between the effect of the drug-efflux pump p-gp. Biol Pharm Bull (2014) 37(12):1926–35. doi: 10.1248/bpb.b14-00529
42. Fultang N, Illendula A, Lin J, Pandey MK, Klase Z, Peethambaran B. ROR1 regulates chemoresistance in breast cancer via modulation of drug efflux pump ABCB1. Sci Rep (2020) 10(1):1821. doi: 10.1038/s41598-020-58864-0
43. Mistry P, Stewart AJ, Dangerfield W, Okiji S, Liddle C, Bootle D, et al. In vitro and in vivo reversal of p-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res (2001) 61(2):749–58.
44. Slater LM, Murray SL, Wetzel MW, Wisdom RM, DuVall EM. Verapamil restoration of daunorubicin responsiveness in daunorubicin-resistant Ehrlich ascites carcinoma. J Clin Invest (1982) 70(5):1131–4. doi: 10.1172/JCI110702
45. Wang F, Zhou F, Kruh GD, Gallo JM. Influence of blood-brain barrier efflux pumps on the distribution of vincristine in brain and brain tumors. Neuro-oncology (2010) 12(10):1043–9. doi: 10.1093/neuonc/noq056
46. Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell (2018) 173(4):879–93. doi: 10.1016/j.cell.2018.03.041
47. Faltas BM, Prandi D, Tagawa ST, Molina AM, Nanus DM, Sternberg C, et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat Genet (2016) 48(12):1490–9. doi: 10.1038/ng.3692
48. Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell (2002) 108(2):153–64. doi: 10.1016/S0092-8674(02)00625-6
49. Darb-Esfahani S, Denkert C, Stenzinger A, Salat C, Sinn B, Schem C, et al. Role of TP53 mutations in triple negative and HER2-positive breast cancer treated with neoadjuvant anthracycline/taxane-based chemotherapy. Oncotarget (2016) 7(42):67686–98. doi: 10.18632/oncotarget.11891
50. Robl B, Pauli C, Botter SM, Bode-Lesniewska B, Fuchs B. Prognostic value of tumor suppressors in osteosarcoma before and after neoadjuvant chemotherapy. BMC Cancer (2015) 15:379. doi: 10.1186/s12885-015-1397-4
51. Zenz T, Eichhorst B, Busch R, Denzel T, Häbe S, Winkler D, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol (2010) 28(29):4473–9. doi: 10.1200/JCO.2009.27.8762
52. Zhang SS, Huang QY, Yang H, Xie X, Luo KJ, Wen J, et al. Correlation of p53 status with the response to chemotherapy-based treatment in esophageal cancer: a meta-analysis. Ann Surg Oncol (2013) 20(7):2419–27. doi: 10.1245/s10434-012-2859-4
53. Cecchini MJ, Ishak CA, Passos DT, Warner A, Palma DA, Howlett CJ, et al. Loss of the retinoblastoma tumor suppressor correlates with improved outcome in patients with lung adenocarcinoma treated with surgery and chemotherapy. Hum Pathol (2015) 46(12):1922–34. doi: 10.1016/j.humpath.2015.08.010
54. Treré D, Brighenti E, Donati G, Ceccarelli C, Santini D, Taffurelli M, et al. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Ann Oncol (2009) 20(11):1818–23. doi: 10.1093/annonc/mdp209
55. Witkiewicz AK, Ertel A, McFalls J, Valsecchi ME, Schwartz G, Knudsen ES. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res (2012) 18(18):5110–22. doi: 10.1158/1078-0432.CCR-12-0903
56. Ludovini V, Gregorc V, Pistola L, Mihaylova Z, Floriani I, Darwish S, et al. Vascular endothelial growth factor, p53, Rb, bcl-2 expression and response to chemotherapy in advanced non-small cell lung cancer. Lung Cancer (2004) 46(1):77–85. doi: 10.1016/j.lungcan.2004.03.018
57. Ikai A, Watanabe M, Sowa Y, Kishimoto M, Yanagisawa A, Fujiwara H, et al. Phosphorylated retinoblastoma protein is a potential predictive marker of irinotecan efficacy for colorectal cancer. Int J Oncol (2016) 48(3):1297–304. doi: 10.3892/ijo.2016.3332
58. Evenson DA, Adams J, McIvor RS, Wagner CR. Methotrexate resistance of mouse dihydrofolate reductase: effect of substitution of phenylalanine-31 by serine or tryptophan. J Med Chem (1996) 39(9):1763–6. doi: 10.1021/jm950793d
59. Volpato JP, Fossati E, Pelletier JN. Increasing methotrexate resistance by combination of active-site mutations in human dihydrofolate reductase. J Mol Biol (2007) 373(3):599–611. doi: 10.1016/j.jmb.2007.07.076
60. Hoeijmakers JHJ. Genome maintenance mechanisms for preventing cancer. Nature (2001) 411(6835):366–74. doi: 10.1038/35077232
61. Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res (2016) 106:27–36. doi: 10.1016/j.phrs.2016.01.001
62. Besse B, Olaussen KA, Soria JC. ERCC1 and RRM1: Ready for prime time? JCO (2013) 31(8):1050–60. doi: 10.1200/JCO.2012.43.0900
63. Han QL, Zhou YH, Lyu Y, Yan H, Dai GH. Effect of ribonucleotide reductase M1 expression on overall survival in patients with pancreatic cancer receiving gemcitabine chemotherapy: A literature-based meta-analysis. J Clin Pharm Ther (2018) 43(2):163–9. doi: 10.1111/jcpt.12655
64. Rosell R, Danenberg KD, Alberola V, Bepler G, Sanchez JJ, Camps C, et al. Ribonucleotide reductase messenger RNA expression and survival in Gemcitabine/Cisplatin-treated advanced non-small cell lung cancer patients. Clin Cancer Res (2004) 10(4):1318–25. doi: 10.1158/1078-0432.CCR-03-0156
65. Zhu CM, Lian XY, Bi YH, Hu CC, Liang YW, Li QS. Prognostic value of ribonucleotide reductase subunit M1 (RRM1) in non-small cell lung cancer: A meta-analysis. Clin Chim Acta (2018) 485:67–73. doi: 10.1016/j.cca.2018.05.042
66. Jordheim LP, Sève P, Trédan O, Dumontet C. The ribonucleotide reductase large subunit (RRM1) as a predictive factor in patients with cancer. Lancet Oncol (2011) 12(7):693–702. doi: 10.1016/S1470-2045(10)70244-8
67. Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell (2008) 132(3):487–98. doi: 10.1016/j.cell.2007.12.033
68. Yano S, Zhang Y, Miwa S, Tome Y, Hiroshima Y, Uehara F, et al. Spatial–temporal FUCCI imaging of each cell in a tumor demonstrates locational dependence of cell cycle dynamics and chemoresponsiveness. Cell Cycle (2014) 13(13):2110–9. doi: 10.4161/cc.29156
69. Yano S, Takehara K, Zhao M, Tan Y, Han Q, Li S, et al. Tumor-specific cell-cycle decoy by salmonella typhimurium A1-r combined with tumor-selective cell-cycle trap by methioninase overcome tumor intrinsic chemoresistance as visualized by FUCCI imaging. Cell Cycle (2016) 15(13):1715–23. doi: 10.1080/15384101.2016.1181240
70. Najafi M, Mortezaee K, Majidpoor J. Cancer stem cell (CSC) resistance drivers. Life Sci (2019) 234:116781. doi: 10.1016/j.lfs.2019.116781
71. Davis JE, Kirk J, Ji Y, Tang DG. Tumor dormancy and slow-cycling cancer cells. Adv Exp Med Biol (2019) 1164:199–206. doi: 10.1007/978-3-030-22254-3_15
72. Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene (2008) 27(12):1749–58. doi: 10.1038/sj.onc.1210811
73. Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell (2007) 1(4):389–402. doi: 10.1016/j.stem.2007.08.001
74. Yuan M, Eberhart CG, Kai M. RNA Binding protein RBM14 promotes radio-resistance in glioblastoma by regulating DNA repair and cell differentiation. Oncotarget (2014) 5(9):2820–6. doi: 10.18632/oncotarget.1924
75. Lim YC, Roberts TL, Day BW, Harding A, Kozlov S, Kijas AW, et al. A role for homologous recombination and abnormal cell-cycle progression in radioresistance of glioma-initiating cells. Mol Cancer Ther (2012) 11(9):1863–72. doi: 10.1158/1535-7163.MCT-11-1044
76. Park HK, Hong JH, Oh YT, Kim SS, Yin J, Lee AJ, et al. Interplay between TRAP1 and sirtuin-3 modulates mitochondrial respiration and oxidative stress to maintain stemness of glioma stem cells. Cancer Res (2019) 79(7):1369–82. doi: 10.1158/0008-5472.CAN-18-2558
77. Boesch M, Zeimet AG, Rumpold H, Gastl G, Sopper S, Wolf D. Drug transporter-mediated protection of cancer stem cells from ionophore antibiotics. Stem Cells Trans Med (2015) 4(9):1028–32. doi: 10.5966/sctm.2015-0054
78. Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst (2010) 102(20):1536–46. doi: 10.1093/jnci/djq364
79. Ewald J, Desotelle J, Almassi N, Jarrard D. Drug-induced senescence bystander proliferation in prostate cancer cells in vitro and in vivo. Br J Cancer (2008) 98(7):1244–9. doi: 10.1038/sj.bjc.6604288
80. Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst CMAM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature (2005) 436(7051):720–4. doi: 10.1038/nature03890
81. Duy C, Li M, Teater M, Meydan C, Garrett-Bakelman FE, Lee TC, et al. Chemotherapy induces senescence-like resilient cells capable of initiating AML recurrence. Cancer Discovery (2021) 11(6):1542–61. doi: 10.1158/2159-8290.CD-20-1375
82. Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, Brown AMK, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science (2013) 339(6119):543–8. doi: 10.1126/science.1227670
83. Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell (2006) 9(5):351–65. doi: 10.1016/j.ccr.2006.03.027
84. Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer (2008) 8(2):121–32. doi: 10.1038/nrc2297
85. Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell (2007) 12(2):171–85. doi: 10.1016/j.ccr.2007.07.001
86. Touzeau C, Maciag P, Amiot M, Moreau P. Targeting bcl-2 for the treatment of multiple myeloma. Leukemia (2018) 32(9):1899–907. doi: 10.1038/s41375-018-0223-9
87. Zhu H, Wei M, Xu J, Hua J, Liang C, Meng Q, et al. PARP inhibitors in pancreatic cancer: molecular mechanisms and clinical applications. Mol Cancer (2020) 19(1):49. doi: 10.1186/s12943-020-01167-9
88. Millward MJ, Cantwell BM, Munro NC, Robinson A, Corris PA, Harris AL. Oral verapamil with chemotherapy for advanced non-small cell lung cancer: a randomised study. Br J Cancer (1993) 67(5):1031–5. doi: 10.1038/bjc.1993.189
89. Milroy R. A randomised clinical study of verapamil in addition to combination chemotherapy in small cell lung cancer. West of Scotland lung cancer research group, and the Aberdeen oncology group. Br J Cancer (1993) 68(4):813–8. doi: 10.1038/bjc.1993.433
90. Ozols RF, Cunnion RE, Klecker RW, Hamilton TC, Ostchega Y, Parrillo JE, et al. Verapamil and adriamycin in the treatment of drug-resistant ovarian cancer patients. J Clin Oncol (1987) 5(4):641–7. doi: 10.1200/JCO.1987.5.4.641
91. Solary E, Drenou B, Campos L, de Crémoux P, Mugneret F, Moreau P, et al. Quinine as a multidrug resistance inhibitor: a phase 3 multicentric randomized study in adult de novo acute myelogenous leukemia. Blood (2003) 102(4):1202–10. doi: 10.1182/blood-2002-11-3419
92. Saeki T, Nomizu T, Toi M, Ito Y, Noguchi S, Kobayashi T, et al. Dofequidar fumarate (MS-209) in combination with cyclophosphamide, doxorubicin, and fluorouracil for patients with advanced or recurrent breast cancer. J Clin Oncol (2007) 25(4):411–7. doi: 10.1200/JCO.2006.08.1646
93. Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2017) 390(10106):1949–61. doi: 10.1016/S0140-6736(17)32440-6
94. Kummar S, Oza AM, Fleming GF, Sullivan DM, Gandara DR, Naughton MJ, et al. Randomized trial of oral cyclophosphamide and veliparib in high-grade serous ovarian, primary peritoneal, or fallopian tube cancers, or BRCA-mutant ovarian cancer. Clin Cancer Res (2015) 21(7):1574–82. doi: 10.1158/1078-0432.CCR-14-2565
95. Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RHJ, Sonke GS, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol (2015) 16(1):87–97. doi: 10.1016/S1470-2045(14)71135-0
96. Bang YJ, Xu RH, Chin K, Lee KW, Park SH, Rha SY, et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol (2017) 18(12):1637–51. doi: 10.1016/S1470-2045(17)30682-4
97. DiPaola RS, Chen YH, Stein M, Vaughn D, Patrick-Miller L, Carducci M, et al. A randomized phase II trial of mitoxantrone, estramustine and vinorelbine or bcl-2 modulation with 13-cis retinoic acid, interferon and paclitaxel in patients with metastatic castrate-resistant prostate cancer: ECOG 3899. J Trans Med (2010) 8(1):20. doi: 10.1186/1479-5876-8-20
98. Cleary JM, Lima CMSR, Hurwitz HI, Montero AJ, Franklin C, Yang J, et al. A phase I clinical trial of navitoclax, a targeted high-affinity bcl-2 family inhibitor, in combination with gemcitabine in patients with solid tumors. Invest New Drugs (2014) 32(5):937–45. doi: 10.1007/s10637-014-0110-9
99. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med (2020) 383(7):617–29. doi: 10.1056/NEJMoa2012971
100. Wei AH, Panayiotidis P, Montesinos P, Laribi K, Ivanov V, Kim I, et al. 6-month follow-up of VIALE-c demonstrates improved and durable efficacy in patients with untreated AML ineligible for intensive chemotherapy. Blood Cancer J (2021) 11(10):1–8. doi: 10.1038/s41408-021-00555-8
101. Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med (2016) 374(4):311–22. doi: 10.1056/NEJMoa1513257
102. Kaufman JL, Gasparetto C, Schjesvold FH, Moreau P, Touzeau C, Facon T, et al. Targeting BCL-2 with venetoclax and dexamethasone in patients with relapsed/refractory t (11;14) Multiple myeloma. Am J Hematol (2021) 96(4):418–27. doi: 10.1002/ajh.26083
103. O’Reilly EM, Sahai V, Bendell JC, Bullock AJ, LoConte NK, Hatoum H, et al. Results of a randomized phase II trial of an anti-notch 2/3, tarextumab (OMP-59R5, TRXT, anti-Notch2/3), in combination with nab-paclitaxel and gemcitabine (Nab-P+Gem) in patients (pts) with untreated metastatic pancreatic cancer (mPC). JCO (2017) 35(4_suppl):279–9. doi: 10.1200/JCO.2017.35.4_suppl.279
104. Moore KN, Gunderson CC, Sabbatini P, McMeekin DS, Mantia-Smaldone G, Burger RA, et al. A phase 1b dose escalation study of ipafricept (OMP54F28) in combination with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer. Gynecol Oncol (2019) 154(2):294–301. doi: 10.1016/j.ygyno.2019.04.001
105. Weekes C, Berlin J, Lenz HJ, O’Neil B, Messersmith W, Cohen S, et al. Phase 1b study of WNT inhibitor ipafricept (IPA, decoy receptor for WNT ligands) with nab-paclitaxel (Nab-p) and gemcitabine (G) in patients (pts) with previously untreated stage IV pancreatic cancer (PC). Ann Oncol (2016) 27:vi117. doi: 10.1093/annonc/mdw368.10
106. Diamond JR, Becerra C, Richards D, Mita A, Osborne C, O’Shaughnessy J, et al. Phase ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res Treat (2020) 184(1):53–62. doi: 10.1007/s10549-020-05817-w
107. Matulonis UA, Monk BJ. PARP inhibitor and chemotherapy combination trials for the treatment of advanced malignancies: does a development pathway forward exist? Ann Oncol (2017) 28(3):443–7. doi: 10.1093/annonc/mdw697
108. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature (2005) 434(7035):843–50. doi: 10.1038/nature03319
109. Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell (2012) 23(1):1–6. doi: 10.1091/mbc.e10-04-0335
110. Kondoh E, Mori S, Yamaguchi K, Baba T, Matsumura N, Barnett JC, et al. Targeting slow-proliferating ovarian cancer cells. Int J Cancer (2010) 126(10):2448–56. doi: 10.1002/ijc.24919
111. Campiglio M, Somenzi G, Olgiati C, Beretta G, Balsari A, Zaffaroni N, et al. Role of proliferation in HER2 status predicted response to doxorubicin. Int J Cancer (2003) 105(4):568–73. doi: 10.1002/ijc.11113
112. Vanas V, Haigl B, Stockhammer V, Sutterlüty-Fall H. MicroRNA-21 increases proliferation and cisplatin sensitivity of osteosarcoma-derived cells. PloS One (2016) 11(8). doi: 10.1371/journal.pone.0161023
113. Liu H, Song X, Liu C, Xie L, Wei L, Sun R. Knockdown of astrocyte elevated gene-1 inhibits proliferation and enhancing chemo-sensitivity to cisplatin or doxorubicin in neuroblastoma cells. J Exp Clin Cancer Res (2009) 28(1):19. doi: 10.1186/1756-9966-28-19
114. Chen X, Xiong D, Ye L, Yang H, Mei S, Wu J, et al. SPP1 inhibition improves the cisplatin chemo-sensitivity of cervical cancer cell lines. Cancer Chemother Pharmacol (2019) 83(4):603–13. doi: 10.1007/s00280-018-3759-5
115. Bialk P, Wang Y, Banas K, Kmiec EB. Functional gene knockout of NRF2 increases chemosensitivity of human lung cancer A549 cells in vitro and in a xenograft mouse model. Mol Ther Oncolytics (2018) 11:75–89. doi: 10.1016/j.omto.2018.10.002
116. Gobin B, Huin MB, Lamoureux F, Ory B, Charrier C, Lanel R, et al. BYL719, a new α-specific PI3K inhibitor: Single administration and in combination with conventional chemotherapy for the treatment of osteosarcoma. Int J Cancer (2015) 136(4):784–96. doi: 10.1002/ijc.29040
117. Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell (2012) 21(4):488–503. doi: 10.1016/j.ccr.2012.02.017
118. Li L, Han D, Wang X, Wang Q, Tian J, Yao J, et al. Prognostic values of ki-67 in neoadjuvant setting for breast cancer: a systematic review and meta-analysis. Future Oncol (2017) 13(11):1021–34. doi: 10.2217/fon-2016-0428
119. Petrelli F, Viale G, Cabiddu M, Barni S. Prognostic value of different cut-off levels of ki-67 in breast cancer: a systematic review and meta-analysis of 64,196 patients. Breast Cancer Res Treat (2015) 153(3):477–91. doi: 10.1007/s10549-015-3559-0
120. Chen X, He C, Han D, Zhou M, Wang Q, Tian J, et al. The predictive value of ki-67 before neoadjuvant chemotherapy for breast cancer: a systematic review and meta-analysis. Future Oncol (2017) 13(9):843–57. doi: 10.2217/fon-2016-0420
121. Chan KS, Koh CG, Li HY. Mitosis-targeted anti-cancer therapies: where they stand. Cell Death Dis (2012) 3(10):e411. doi: 10.1038/cddis.2012.148
122. Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol Lett (2017) 14(3):2611–20. doi: 10.3892/ol.2017.6497
123. Peña C, Céspedes MV, Lindh MB, Kiflemariam S, Mezheyeuski A, Edqvist PH, et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res (2013) 73(4):1287–97. doi: 10.1158/0008-5472.CAN-12-1875
124. Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer (2014) 110(3):724–32. doi: 10.1038/bjc.2013.768
125. Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, et al. PDGF-c mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell (2009) 15(1):21–34. doi: 10.1016/j.ccr.2008.12.004
126. Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, Takeyama H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br J Cancer (2014) 110(2):469–78. doi: 10.1038/bjc.2013.748
127. Kharaziha P, Rodriguez P, Li Q, Rundqvist H, Björklund AC, Augsten M, et al. Targeting of distinct signaling cascades and cancer-associated fibroblasts define the efficacy of sorafenib against prostate cancer cells. Cell Death Dis (2012) 3:e262. doi: 10.1038/cddis.2012.1
128. Zhao Q, Huang L, Qin G, Qiao Y, Ren F, Shen C, et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett (2021) 518:35–48. doi: 10.1016/j.canlet.2021.06.009
129. Harper J, Sainson RCA. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin Cancer Biol (2014) 25:69–77. doi: 10.1016/j.semcancer.2013.12.005
130. Gonçalves-Ribeiro S, Díaz-Maroto NG, Berdiel-Acer M, Soriano A, Guardiola J, Martínez-Villacampa M, et al. Carcinoma-associated fibroblasts affect sensitivity to oxaliplatin and 5FU in colorectal cancer cells. Oncotarget (2016) 7(37):59766–80. doi: 10.18632/oncotarget.11121
131. Steinbichler TB, Metzler V, Pritz C, Riechelmann H, Dudas J. Tumor-associated fibroblast-conditioned medium induces CDDP resistance in HNSCC cells. Oncotarget (2016) 7(3):2508–18. doi: 10.18632/oncotarget.6210
132. Zhang H, Xie C, Yue J, Jiang Z, Zhou R, Xie R, et al. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFβ1 signaling loop in esophageal squamous cell carcinoma. Mol Carcinog (2017) 56(3):1150–63. doi: 10.1002/mc.22581
133. Díaz-Maroto NG, Garcia-Vicién G, Polcaro G, Bañuls M, Albert N, Villanueva A, et al. The blockade of tumoral IL1β-mediated signaling in normal colonic fibroblasts sensitizes tumor cells to chemotherapy and prevents inflammatory CAF activation. Int J Mol Sci (2021) 22(9):4960. doi: 10.3390/ijms22094960
134. Sharma M, Turaga RC, Yuan Y, Satyanarayana G, Mishra F, Bian Z, et al. Simultaneously targeting cancer-associated fibroblasts and angiogenic vessel as a treatment for TNBC. J Exp Med (2021) 218(4):e20200712. doi: 10.1084/jem.20200712
135. Peran I, Dakshanamurthy S, McCoy MD, Mavropoulos A, Allo B, Sebastian A, et al. Cadherin 11 promotes immunosuppression and extracellular matrix deposition to support growth of pancreatic tumors and resistance to gemcitabine in mice. Gastroenterology (2021) 160(4):1359–1372.e13. doi: 10.1053/j.gastro.2020.11.044
136. Li M, Li M, Yin T, Shi H, Wen Y, Zhang B, et al. Targeting of cancer−associated fibroblasts enhances the efficacy of cancer chemotherapy by regulating the tumor microenvironment. Mol Med Rep (2016) 13(3):2476–84. doi: 10.3892/mmr.2016.4868
137. Ebelt ND, Zamloot V, Manuel ER. Targeting desmoplasia in pancreatic cancer as an essential first step to effective therapy. Oncotarget (2020) 11(38):3486–8. doi: 10.18632/oncotarget.27745
138. Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, et al. Stromal biology and therapy in pancreatic cancer. Gut (2011) 60(6):861–8. doi: 10.1136/gut.2010.226092
139. Hasebe T, Iwasaki M, Akashi-Tanaka S, Hojo T, Shimizu C, Andoh M, et al. Atypical tumor-stromal fibroblasts in invasive ductal carcinomas of the breast treated with neoadjuvant therapy. Hum Pathol (2011) 42(7):998–1006. doi: 10.1016/j.humpath.2010.10.020
140. Koriyama H, Ishii G, Yoh K, Neri S, Morise M, Umemura S, et al. Presence of podoplanin-positive cancer-associated fibroblasts in surgically resected primary lung adenocarcinoma predicts a shorter progression-free survival period in patients with recurrences who received platinum-based chemotherapy. J Cancer Res Clin Oncol (2015) 141(7):1163–70. doi: 10.1007/s00432-014-1891-0
141. Tao L, Huang G, Wang R, Pan Y, He Z, Chu X, et al. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci Rep (2016) 6:38408. doi: 10.1038/srep38408
142. Zheng H, Liu H, Ge Y, Wang X. Integrated single-cell and bulk RNA sequencing analysis identifies a cancer associated fibroblast-related signature for predicting prognosis and therapeutic responses in colorectal cancer. Cancer Cell Int (2021) 21(1):552. doi: 10.1186/s12935-021-02252-9
143. Gadducci A, Ferrero A, Cosio S, Zola P, Viacava P, Dompé D, et al. Intratumoral microvessel density in advanced epithelial ovarian cancer and its use as a prognostic variable. . Anticancer Res (2006) 26(5B):3925–32.
144. Hansen TF, Nielsen BS, Jakobsen A, Sørensen FB. Visualising and quantifying angiogenesis in metastatic colorectal cancer : A comparison of methods and their predictive value for chemotherapy response. Cell Oncol (Dordr) (2013) 36(4):341–50. doi: 10.1007/s13402-013-0139-3
145. Kim SJ, Jung KH, Son MK, Park JH, Yan HH, Fang Z, et al. Tumor vessel normalization by the PI3K inhibitor HS-173 enhances drug delivery. Cancer Lett (2017) 10(403):339–53. doi: 10.1016/j.canlet.2017.06.035
146. Dickson PV, Hamner JB, Sims TL, Fraga CH, Ng CYC, Rajasekeran S, et al. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin Cancer Res (2007) 13(13):3942–50. doi: 10.1158/1078-0432.CCR-07-0278
147. Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res (2004) 64(11):3731–6. doi: 10.1158/0008-5472.CAN-04-0074
148. Sun Y, Chen W, Torphy RJ, Yao S, Zhu G, Lin R, et al. Blockade of the CD93 pathway normalizes tumor vasculature to facilitate drug delivery and immunotherapy. Sci Transl Med (2021) 13(604):eabc8922. doi: 10.1126/scitranslmed.abc8922
149. Zhou F, Hu J, Shao JH, Zou SB, Shen SL, Luo ZQ. Metronomic chemotherapy in combination with antiangiogenic treatment induces mosaic vascular reduction and tumor growth inhibition in hepatocellular carcinoma xenografts. J Cancer Res Clin Oncol (2012) 138(11):1879–90. doi: 10.1007/s00432-012-1270-7
150. Zalcman G, Mazieres J, Margery J, Greillier L, Audigier-Valette C, Moro-Sibilot D, et al. Bevacizumab for newly diagnosed pleural mesothelioma in the mesothelioma avastin cisplatin pemetrexed study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet (2016) 387(10026):1405–14. doi: 10.1016/S0140-6736(15)01238-6
151. Coleman RL, Brady MF, Herzog TJ, Sabbatini P, Armstrong DK, Walker JL, et al. Bevacizumab and paclitaxel–carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic oncology group study GOG-0213): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol (2017) 18(6):779–91. doi: 10.1016/S1470-2045(17)30279-6
152. Pignata S, Lorusso D, Joly F, Gallo C, Colombo N, Sessa C, et al. Carboplatin-based doublet plus bevacizumab beyond progression versus carboplatin-based doublet alone in patients with platinum-sensitive ovarian cancer: a randomised, phase 3 trial. Lancet Oncol (2021) 22(2):267–76. doi: 10.1016/S1470-2045(20)30637-9
153. Wakelee HA, Dahlberg SE, Keller SM, Tester WJ, Gandara DR, Graziano SL, et al. Adjuvant chemotherapy with or without bevacizumab in patients with resected non-small-cell lung cancer (E1505): an open-label, multicentre, randomised, phase 3 trial. Lancet Oncol (2017) 18(12):1610–23. doi: 10.1016/S1470-2045(17)30691-5
154. André T, Vernerey D, Im SA, Bodoky G, Buzzoni R, Reingold S, et al. Bevacizumab as adjuvant treatment of colon cancer: updated results from the s-AVANT phase III study by the GERCOR group. Ann Oncol (2020) 31(2):246–56. doi: 10.1016/j.annonc.2019.12.006
155. Kabbinavar FF, Schulz J, McCleod M, Patel T, Hamm JT, Hecht JR, et al. Addition of bevacizumab to bolus fluorouracil and leucovorin in first-line metastatic colorectal cancer: Results of a randomized phase II trial. JCO (2005) 23(16):3697–705. doi: 10.1200/JCO.2005.05.112
156. Schwartz HS, Grindey GB. Adriamycin and daunorubicin: a comparison of antitumor activities and tissue uptake in mice following immunosuppression. Cancer Res (1973) 33(8):1837–44.
157. Nowak AK, Lake RA, Marzo AL, Scott B, Heath WR, Collins EJ, et al. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol (2003) 170(10):4905–13. doi: 10.4049/jimmunol.170.10.4905
158. Nowak AK, Robinson BWS, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res (2003) 63(15):4490–6.
159. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med (2005) 202(12):1691–701. doi: 10.1084/jem.20050915
160. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol (2008) 8(1):59–73. doi: 10.1038/nri2216
161. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13(1):54–61. doi: 10.1038/nm1523
162. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med (2009) 15(10):1170–8. doi: 10.1038/nm.2028
163. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (2011) 334(6062):1573–7. doi: 10.1126/science.1208347
164. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene (2010) 29(4):482–91. doi: 10.1038/onc.2009.356
165. Ciampricotti M, Hau CS, Doornebal CW, Jonkers J, de Visser KE. Chemotherapy response of spontaneous mammary tumors is independent of the adaptive immune system. Nat Med (2012) 18(3):344–6; author reply 346. doi: 10.1038/nm.2652
166. Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev (2007) 220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x
167. van der Most RG, Currie AJ, Cleaver AL, Salmons J, Nowak AK, Mahendran S, et al. Cyclophosphamide chemotherapy sensitizes tumor cells to TRAIL-dependent CD8 T cell-mediated immune attack resulting in suppression of tumor growth. PloS One (2009) 4(9):e6982. doi: 10.1371/journal.pone.0006982
168. West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, et al. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res (2013) 73(24):7265–76. doi: 10.1158/0008-5472.CAN-13-0890
169. Suzuki E, Sun J, Kapoor V, Jassar AS, Albelda SM. Gemcitabine has significant immunomodulatory activity in murine tumor models independent of its cytotoxic effects. Cancer Biol Ther (2007) 6(6):880–5. doi: 10.4161/cbt.6.6.4090
170. Tongu M, Harashima N, Monma H, Inao T, Yamada T, Kawauchi H, et al. Metronomic chemotherapy with low-dose cyclophosphamide plus gemcitabine can induce anti-tumor T cell immunity in vivo. Cancer Immunol Immunother (2013) 62(2):383–91. doi: 10.1007/s00262-012-1343-0
171. Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity (2016) 44(2):343–54. doi: 10.1016/j.immuni.2015.11.024
172. Beyranvand Nejad E, van der Sluis TC, van Duikeren S, Yagita H, Janssen GM, van Veelen PA, et al. Tumor eradication by cisplatin is sustained by CD80/86-mediated costimulation of CD8+ T cells. Cancer Res (2016) 76(20):6017–29. doi: 10.1158/0008-5472.CAN-16-0881
173. Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res (2011) 71(14):4809–20. doi: 10.1158/0008-5472.CAN-11-0753
174. Radojcic V, Bezak KB, Skarica M, Pletneva MA, Yoshimura K, Schulick RD, et al. Cyclophosphamide resets dendritic cell homeostasis and enhances antitumor immunity through effects that extend beyond regulatory T cell elimination. Cancer Immunol Immunother (2010) 59(1):137–48. doi: 10.1007/s00262-009-0734-3
175. van der Most RG, Currie AJ, Mahendran S, Prosser A, Darabi A, Robinson BWS, et al. Tumor eradication after cyclophosphamide depends on concurrent depletion of regulatory T cells: a role for cycling TNFR2-expressing effector-suppressor T cells in limiting effective chemotherapy. Cancer Immunol Immunother (2009) 58(8):1219–28. doi: 10.1007/s00262-008-0628-9
176. Vicari AP, Luu R, Zhang N, Patel S, Makinen SR, Hanson DC, et al. Paclitaxel reduces regulatory T cell numbers and inhibitory function and enhances the anti-tumor effects of the TLR9 agonist PF-3512676 in the mouse. Cancer Immunol Immunother (2009) 58(4):615–28. doi: 10.1007/s00262-008-0586-2
177. Lu Y, Zhao Q, Liao JY, Song E, Xia Q, Pan J, et al. Complement signals determine opposite effects of b cells in chemotherapy-induced immunity. Cell (2020) 180(6):1081–1097.e24. doi: 10.1016/j.cell.2020.02.015
178. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med (2014) 20(11):1301–9. doi: 10.1038/nm.3708
179. Newman JH, Chesson CB, Herzog NL, Bommareddy PK, Aspromonte SM, Pepe R, et al. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc Natl Acad Sci U S A. (2020) 117(2):1119–28. doi: 10.1073/pnas.1904022116
180. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science (2018) 362(6411). doi: 10.1126/science.aar3593
181. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest (2017) 127(8):2930–40. doi: 10.1172/JCI91190
182. Brockwell NK, Rautela J, Owen KL, Gearing LJ, Deb S, Harvey K, et al. Tumor inherent interferon regulators as biomarkers of long-term chemotherapeutic response in TNBC. NPJ Precis Oncol (2019) 3:21. doi: 10.1038/s41698-019-0093-2
183. Loi S, Sirtaine N, Piette F, Salgado R, Viale G, Van Eenoo F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol (2013) 31(7):860–7. doi: 10.1200/JCO.2011.41.0902
184. Tekpli X, Lien T, Røssevold AH, Nebdal D, Borgen E, Ohnstad HO, et al. An independent poor-prognosis subtype of breast cancer defined by a distinct tumor immune microenvironment. Nat Commun (2019) 10(1):5499. doi: 10.1038/s41467-019-13329-5
185. West NR, Milne K, Truong PT, Macpherson N, Nelson BH, Watson PH. Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Res (2011) 13(6):R126. doi: 10.1186/bcr3072
186. Mahmoud SMA, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AHS, et al. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol (2011) 29(15):1949–55. doi: 10.1200/JCO.2010.30.5037
187. Montero AJ, Diaz-Montero CM, Deutsch YE, Hurley J, Koniaris LG, Rumboldt T, et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II-IIIc breast cancer. Breast Cancer Res Treat (2012) 132(1):215–23. doi: 10.1007/s10549-011-1889-0
188. Li F, Zhao Y, Wei L, Li S, Liu J. Tumor-infiltrating treg, MDSC, and IDO expression associated with outcomes of neoadjuvant chemotherapy of breast cancer. Cancer Biol Ther (2018) 19(8):695–705. doi: 10.1080/15384047.2018.1450116
189. Mizukoshi E, Yamashita T, Arai K, Terashima T, Kitahara M, Nakagawa H, et al. Myeloid-derived suppressor cells correlate with patient outcomes in hepatic arterial infusion chemotherapy for hepatocellular carcinoma. Cancer Immunol Immunother (2016) 65(6):715–25. doi: 10.1007/s00262-016-1837-2
190. Xu H, Mao Y, Dai Y, Wang Q, Zhang X. CD4CD25+ regulatory T cells in patients with advanced gastrointestinal cancer treated with chemotherapy. Onkologie (2009) 32(5):246–52. doi: 10.1159/000210023
191. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol (2006) 24(34):5373–80. doi: 10.1200/JCO.2006.05.9584
192. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell (2015) 28(6):690–714. doi: 10.1016/j.ccell.2015.10.012
193. Cook AM, Lesterhuis WJ, Nowak AK, Lake RA. Chemotherapy and immunotherapy: mapping the road ahead. Curr Opin Immunol (2016) 39:23–9. doi: 10.1016/j.coi.2015.12.003
194. Lesterhuis WJ, Salmons J, Nowak AK, Rozali EN, Khong A, Dick IM, et al. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PloS One (2013) 8(4):e61895. doi: 10.1371/journal.pone.0061895
195. Spring LM, Fell G, Arfe A, Sharma C, Greenup R, Reynolds KL, et al. Pathologic complete response after neoadjuvant chemotherapy and impact on breast cancer recurrence and survival: A comprehensive meta-analysis. Clin Cancer Res (2020) 26(12):2838–48. doi: 10.1158/1078-0432.CCR-19-3492
196. Mignot G, Hervieu A, Vabres P, Dalac S, Jeudy G, Bel B, et al. Prospective study of the evolution of blood lymphoid immune parameters during dacarbazine chemotherapy in metastatic and locally advanced melanoma patients. PloS One (2014) 9(8):e105907. doi: 10.1371/journal.pone.0105907
197. Young RJ, Tin AW, Brown NJ, Jitlal M, Lee SM, Woll PJ. Analysis of circulating angiogenic biomarkers from patients in two phase III trials in lung cancer of chemotherapy alone or chemotherapy and thalidomide. Br J Cancer (2012) 106(6):1153–9. doi: 10.1038/bjc.2012.50
198. Principe N, Kidman J, Goh S, Tilsed CM, Fisher SA, Fear VS, et al. Tumor infiltrating effector memory antigen-specific CD8+ T cells predict response to immune checkpoint therapy. Front Immunol (2020) 11:584423/full#h3. doi: 10.3389/fimmu.2020.584423/full#h3
199. Chuah BYS, Putti T, Salto-Tellez M, Charlton A, Iau P, Buhari SA, et al. Serial changes in the expression of breast cancer-related proteins in response to neoadjuvant chemotherapy. Ann Oncol (2011) 22(8):1748–54. doi: 10.1093/annonc/mdq755
200. Alamgeer M, Ganju V, Kumar B, Fox J, Hart S, White M, et al. Changes in aldehyde dehydrogenase-1 expression during neoadjuvant chemotherapy predict outcome in locally advanced breast cancer. Breast Cancer Res (2014) 16(2):R44. doi: 10.1186/bcr3648
201. Bottini A, Berruti A, Bersiga A, Brizzi MP, Bruzzi P, Aguggini S, et al. Relationship between tumour shrinkage and reduction in Ki67 expression after primary chemotherapy in human breast cancer. Br J Cancer (2001) 85(8):1106–12. doi: 10.1054/bjoc.2001.2048
202. Tokuda E, Horimoto Y, Arakawa A, Himuro T, Senuma K, Nakai K, et al. Differences in Ki67 expressions between pre- and post-neoadjuvant chemotherapy specimens might predict early recurrence of breast cancer. Hum Pathol (2017) 63:40–5. doi: 10.1016/j.humpath.2017.02.005
203. Strange RC, Spiteri MA, Ramachandran S, Fryer AA. Glutathione-s-transferase family of enzymes. Mutat Res (2001) 482(1–2):21–6. doi: 10.1016/S0027-5107(01)00206-8
204. Eralp Y, Keskin S, Akışık E, Akışık E, İğci A, Müslümanoğlu M, et al. Predictive role of midtreatment changes in survivin, GSTP1, and topoisomerase 2α expressions for pathologic complete response to neoadjuvant chemotherapy in patients with locally advanced breast cancer. Am J Clin Oncol (2013) 36(3):215–23. doi: 10.1097/COC.0b013e318243913f
205. Sheng J, Fang W, Yu J, Chen N, Zhan J, Ma Y, et al. Expression of programmed death ligand-1 on tumor cells varies pre and post chemotherapy in non-small cell lung cancer. Sci Rep (2016) 6:20090. doi: 10.1038/srep20090
206. Zemek R, de Jong E, Chin M, Schuster I, Fear V, Casey T, et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci Trans Med (2019) 11(501):eaav7816. doi: 10.1126/scitranslmed.aav7816
207. Zemek RM, Chin WL, Nowak AK, Millward MJ, Lake RA, Lesterhuis WJ. Sensitizing the tumor microenvironment to immune checkpoint therapy. Front Immunol (2020) 11:223/full. doi: 10.3389/fimmu.2020.00223/full
208. Nowak AK, Lesterhuis WJ, Kok PS, Brown C, Hughes BG, Karikios DJ, et al. Durvalumab with first-line chemotherapy in previously untreated malignant pleural mesothelioma (DREAM): a multicentre, single-arm, phase 2 trial with a safety run-in. Lancet Oncol (2020) 21(9):1213–23. doi: 10.1016/S1470-2045(20)30462-9
209. Paz-Ares L, Ciuleanu TE, Cobo M, Schenker M, Zurawski B, Menezes J, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol (2021) 22(2):198–211. doi: 10.1016/S1470-2045(20)30641-0
210. Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet (2019) 394(10212):1929–39. doi: 10.1016/S0140-6736(19)32222-6
Keywords: chemotherapy, tumor microenvironment, cell death, immune response, mechanism of action, cancer chemotherapy
Citation: Tilsed CM, Fisher SA, Nowak AK, Lake RA and Lesterhuis WJ (2022) Cancer chemotherapy: insights into cellular and tumor microenvironmental mechanisms of action. Front. Oncol. 12:960317. doi: 10.3389/fonc.2022.960317
Received: 02 June 2022; Accepted: 01 July 2022;
Published: 29 July 2022.
Edited by:
Gautam Sethi, National University of Singapore, SingaporeReviewed by:
Chakrabhavi Mohan, University of Mysore, IndiaMilad Ashrafizadeh, Sabancı University, Turkey
Manoj Garg, Amity University, India
Copyright © 2022 Tilsed, Fisher, Nowak, Lake and Lesterhuis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: W. Joost Lesterhuis, Sm9vc3QuTGVzdGVyaHVpc0B0ZWxldGhvbmtpZHMub3JnLmF1