 Eleonora Lauricella1
Eleonora Lauricella1 Barbara Mandriani1
Barbara Mandriani1 Federica Cavallo1
Federica Cavallo1 Gaetano Pezzicoli1
Gaetano Pezzicoli1 Nada Chaoul1Camillo Porta1,2†
Nada Chaoul1Camillo Porta1,2† Mauro Cives1,2*†
Mauro Cives1,2*†- 1Department of Interdisciplinary Medicine, University of Bari “Aldo Moro”, Bari, Italy
- 2Division of Medical Oncology, A.O.U. Consorziale Policlinico di Bari, Bari, Italy
Neuroendocrine neoplasms (NENs) are highly vascularized malignancies arising from cells of the diffuse neuroendocrine system. An intricated cross-talk exists between NEN cells and the tumor microenvironment, and three main molecular circuits (VEGF/VEGFR pathway, FGF-dependent signaling and PDGF/PDGFR axis) have been shown to regulate angiogenesis in these neoplasms. Multiple randomized trials have investigated antiangiogenic agents over the past two decades, and sunitinib is currently approved for the treatment of advanced, progressive, G1/G2 pancreatic NENs. In recent years, two phase III clinical trials have demonstrated the efficacy and safety of surufatinib, a multi-tyrosine kinase angioimmune inhibitor, in patients with well-differentiated pancreatic and extrapancreatic NENs, and two studies of this agent are currently underway in Europe and US. The HIF-2α inhibitor belzutifan has recently received regulatory approval for the treatment of tumors arising in the context of Von-Hippel Lindau syndrome including pancreatic NENs, and a study of this drug in patients with sporadic tumors is presently ongoing. Combinations of antiangiogenic agents with chemotherapeutics and targeted drugs have been tested, with accumulating toxicities being a matter of concern. The potential of antiangiogenic agents in fine-tuning the immune microenvironment of NENs to enhance the activity of immune checkpoint inhibitors has been only partially elucidated, and further research should be carried out at this regard. Here, we review the current understanding of the biology of angiogenesis in NENs and provide a summary of the latest clinical investigations on antiangiogenic drugs in this malignancy.
Introduction
Neuroendocrine neoplasms (NENs) are heterogeneous malignancies arising from cells of the diffuse neuroendocrine system. They are often characterized by an indolent behavior and the ability to secrete a variety of peptide hormones and biogenic amines (1). The incidence of NENs has steadily increased in the last four decades, and NENs currently constitute the second most prevalent cancer of the gastroenteropancreatic (GEP) tract (2). According to the 2019 WHO classification (3), GEP-NENs can be subdivided in well-differentiated neuroendocrine tumors (NETs) and poorly differentiated neuroendocrine carcinomas (NECs). Neuroendocrine tumors can be further subdivided in low-grade (G1), intermediate-grade (G2) and high-grade (G3) tumors according to their proliferative activity, and large series have proven the prognostic relevance of such a classification (4, 5).
Neuroendocrine tumors are highly vascularized malignancies, and their intratumor vessel density is estimated to be approximately 10-fold higher than in carcinomas (6, 7). This feature is not particularly surprising, as it recapitulates the microscopic architecture of normal endocrine glands which are characterized by a dense vascular network facilitating hormone secretion. In this context, evidence demonstrates that the aberrant activation of the hypoxia-inducible factor-1 (HIF-1) transcriptional program is a frequent event in NETs, driving the production of large amounts of proangiogenic molecules such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), semaphorins and angiopoietins (8).
Clinical strategies encompassing angiogenesis inhibition have a definite place in the therapeutic armamentarium against NETs. The oral tyrosine kinase inhibitor (TKI) sunitinib is currently approved for pancreatic NETs (panNETs) (9), and a variety of new antiangiogenic agents are presently under clinical scrutiny for both GEP and bronchopulmonary (BP) NETs (10). In this review, we provide an overview of the current understanding of the molecular events driving neoangiogenesis in NENs, also discussing present and future therapeutic applications of antiangiogenic agents in the clinical arena.
Angiogenesis in NETs
Tumor angiogenesis is a complex process through which a neoplasm creates its own vascularization, essential for obtaining the oxygen and nutrients necessary to grow beyond a certain, and well defined, volume. Moreover, this vascularization provides an access to the bloodstream that the tumor uses to metastasize. This is true for almost all malignancies, including NETs (7). Angiogenesis is tightly regulated by a complex balance between pro- and anti-angiogenic molecules, and a cross-talk exists between endothelial cells, pericytes and tumor cells. Indeed, while anti-apoptotic factors supporting the tumor growth are released by activated endothelial cells of the newly formed vessels, pro-angiogenic molecules are produced in turn by tumor cells, thereby sustaining the so called “angiogenic switch” and engaging neovascularization (11). In this context, pericytes can stimulate an autocrine VEGF-mediated prosurvival signaling in endothelial cells, further promoting neovascular sprouting and, indirectly, tumor growth (12). Influenced by the same family of molecular cues driving angiogenesis, tumor lymphangiogenesis has also a key role in metastasis formation, and possibly resistance to antiangiogenic therapy. In this context, VEGFs other than VEGF-A have been described to mediate the outgrowth of lymphatic vessels in NETs thereby leading to progression to stages of greater malignancy (13–15).
The vascular alterations observed in NETs are both quantitative and qualitative. Extensive neovascularization in the presence of low endothelial proliferation is indeed a hallmark of well-differentiated NETs, while a lower intratumor microvascular density is typically observed in poorly differentiated carcinomas (8, 16). Such a phenomenon, named as “neuroendocrine paradox”, is possibly related to the capability of well-differentiated NET cells to retain their normal precursors’ ability to stimulate the formation of a dense vascular network, with the angiogenesis of poorly differentiated neoplasms being instead primarily dependent on proliferation-induced hypoxia. The newly formed blood vessels are structurally and functionally aberrant in NETs (7). In particular, endothelial cells appear to contain multiple fenestrations (which are also typical of normal endocrine glands) and trans-endothelial channels while basement membranes are discontinuous and lack pericyte coverage, thus resulting in increased interstitial fluid pressure, vessel tortuosity and leakiness, as well as frequent hemorrhage.
As depicted in Figure 1, three main molecular circuits regulate angiogenesis in NETs: the VEGF/VEGFR pathway, the FGF-dependent signaling and the PDGF/PDGFR axis (17). Vascular endothelial growth factor is constitutively expressed by normal neuroendocrine cells. Its expression is retained in up to 80% of GEP-NETs, where it drives angiogenesis through interaction with VEGFR-1 and VEGFR-2. The expression of VEGF is higher in well-differentiated malignancies with respect to poorly differentiated NENs, and parallels the expression of its receptors on both tumor and endothelial cells (8, 18–20). Tumor and stromal cells are not the only sources of VEGF in NETs, as tumor-infiltrating neutrophils have been shown to mobilize latent VEGF from the extracellular matrix through the release of metalloproteinase 9 (MMP-9), at least in mice (21). Mechanistically, VEGF acts in an autocrine or paracrine fashion triggering both vascular endothelial mitogenesis and permeability via activation of the Notch signaling in endothelial cells (18, 20, 22, 23). Evidence from the RIP1-Tag2 transgenic mouse model demonstrates that VEGF exerts a critical role throughout the whole course of the multistage process of pancreatic endocrine tumorigenesis (19). In particular, the selective knockout of VEGF in β cells of RIP1-Tag2 mice dampens both angiogenic switch and neovasculature formation in dysplastic islets, thus preventing the growth of panNETs (24). As in other cancers, the production of VEGF by NET cells is primarily regulated by local oxygen availability through the sensing activity of HIF-1 (23, 25). In this context, evidence demonstrates that panNETs arising in patients with Von-Hippel Lindau disease, a condition characterized by uncoupled oxygen levels/HIF-1 activity, show a distinct proangiogenic molecular signature when compared with sporadic panNETs, thus suggesting that different evolutionary trajectories are followed by these two entities (26).
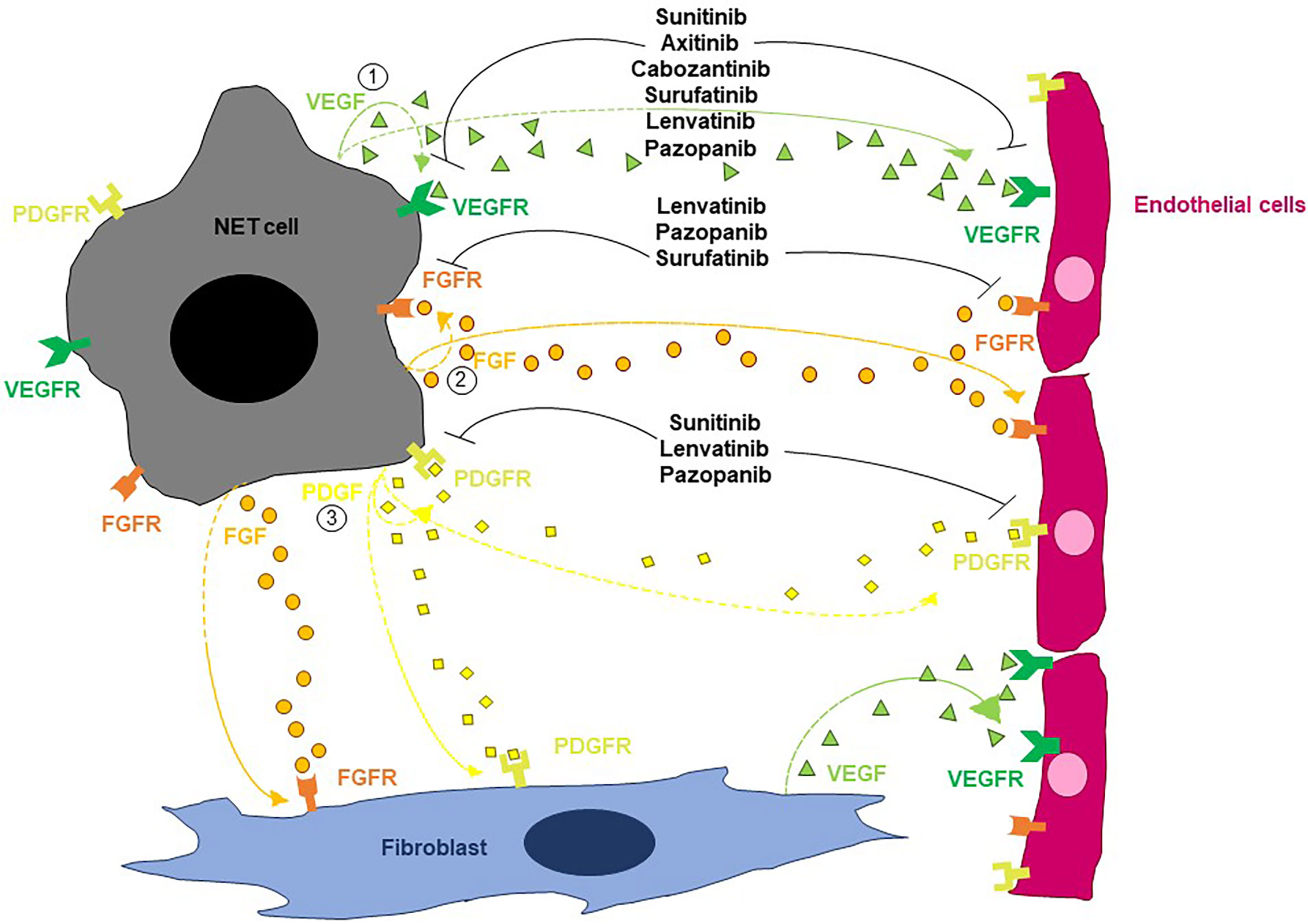
Figure 1 Schematic overview of the main pathways regulating angiogenesis in NETs. By acting on endothelial cells, VEGF stimulates both vascular endothelial mitogenesis and permeability. FGFs trigger endothelial cell migration, proliferation and differentiation as well as vessel formation. PDGF contributes to the angiogenic process by stimulating the recruitment of pericytes and the resulting vessel coverage. Multiple TKIs can interfere with the angiogenic process in NETs.
A second angiogenic pathway modulating the progression of NETs involves FGF and its cognate receptors. The family of FGF is known to comprise 23 members (although there are only 18 FGFR ligands) and exerts multiple functions through the activation of FGFRs (27). This pathway has both direct and indirect effects on angiogenesis. Indeed, while directly stimulating endothelial cell migration, proliferation and differentiation as well as vessel formation and maturation, FGF also acts as a key regulator of proangiogenic molecules including VEGF and angiopoietins (28, 29). Fibroblast growth factor-1 and fibroblast growth factor-2 are expressed in approximately 40% and 100% of GEP-NENs respectively, while fibroblast growth factor receptor (FGFR) 1-4 are expressed by the 68-88% of these malignancies (30, 31). Fibroblast growth factor has a key role in maintaining tumor angiogenesis after an initiation phase primarily guided by the VEGF signaling, and the inhibition of the FGF/FGFR axis suppresses neoangiogenesis and tumor growth in the RIP1-Tag2 transgenic mouse model (32). Evidence demonstrates that FGF is a critical driver of VEGF-independent revascularization of panNETs and can therefore mediate evasive resistance to antioangiogenic therapy (33, 34).
The PDGF/PDGFR axis is another crucial mediator of NET progression. Platelet-derived growth factor contributes to the angiogenic process by stimulating the recruitment of pericytes and the resulting vessel coverage (35). Expression of PDGFR-α and PDGFR-β has been described in approximately 75% and 60% of GEP-NETs respectively (36, 37). In particular, while PDGFR-α is predominantly expressed by tumor cells, PDGFR-β is mainly expressed by pericytes and stromal cells. A positive association between PDGFR-α expression and tumor grade as well as between PDGFR-β expression and tumor microvascular density has been documented (36, 37), and the paracrine secretion of PDGF-DD by endothelial cells has been shown to stimulate NET proliferation (38). In this context, experiments in the RIP1-Tag2-PDGFD knockout model demonstrate that the disruption of the PDGF-DD signaling significantly delays panNET growth (7).
Mounting evidence indicates that semaphorins and angiopoietins contribute to neoangiogenesis in NETs. Semaphorins have shown both pro- and anti-angiogenic effects in NETs, and their activity is the result of the interaction with neuropilin and plexin receptors (7). Neuropilin receptors have been found in both pancreatic, intestinal and pulmonary NETs (39–41), while data on the expression of plexin receptors in NETs are currently lacking. Experiments in RIP1-Tag2 mice have shown that the expression of semaphorin 3A (SEMA3A) is progressively lost during tumor progression and that the inhibition of SEMA3A during the angiogenic switch may enhance tumor formation. Of note, re-expression of SEMA3A by viral gene transfer during late stages of pancreatic endocrine tumorigenesis leads to normalization of the tumor vasculature, increased pericyte coverage and inhibition of tumor progression (42, 43). Similar antiangiogenic effects have been also documented for SEMA3F in ileal NETs (44). On the other hand, protumorigenic activities have been attributed to SEMA4D and SEMA5A. In particular, inhibition of SEMA4D has been recently associated with impaired tumor growth via pericyte coverage alteration and vascular function modification in RIP1-Tag2 mice (45). SEMA5A can elicit angiogenesis, tumor growth, invasion and metastasis by activating c-met downstream its interaction with Plexin-B3 (46). Angiopoietins and angiopoietins receptors are widely expressed in NETs (47, 48). The overexpression of Angiopoietin-2 (Ang-2) in orthothopic NET xenografts in nude mice drives increased microvascular density and enhanced metastatic spread through lymphatic vessels (49). The blockade of the interaction between Ang-2 and its cognate receptor TIE2 determines regression of the tumor vasculature and inhibition of tumor progression in the RIP1-Tag2 mouse model of pancreatic endocrine cancerogenesis (50).
Molecular mechanisms of resistance to antiangiogenic therapies in NETs
Inhibition of angiogenesis has revealed therapeutic efficacy in NET patients. Nevertheless, resistance to antiangiogenic therapies inevitably occurs, and the biological events leading to such a phenomenon have been only partly elucidated. While primary resistance refers to an intrinsic unresponsiveness to antiangiogenic treatments, secondary (or acquired) resistance stems from tumor adaption to therapy, mostly as result of the activation of alternative proangiogenic circuits (51). Figure 2 depicts the main biological events driving resistance to antiangiogenic therapies in NETs.
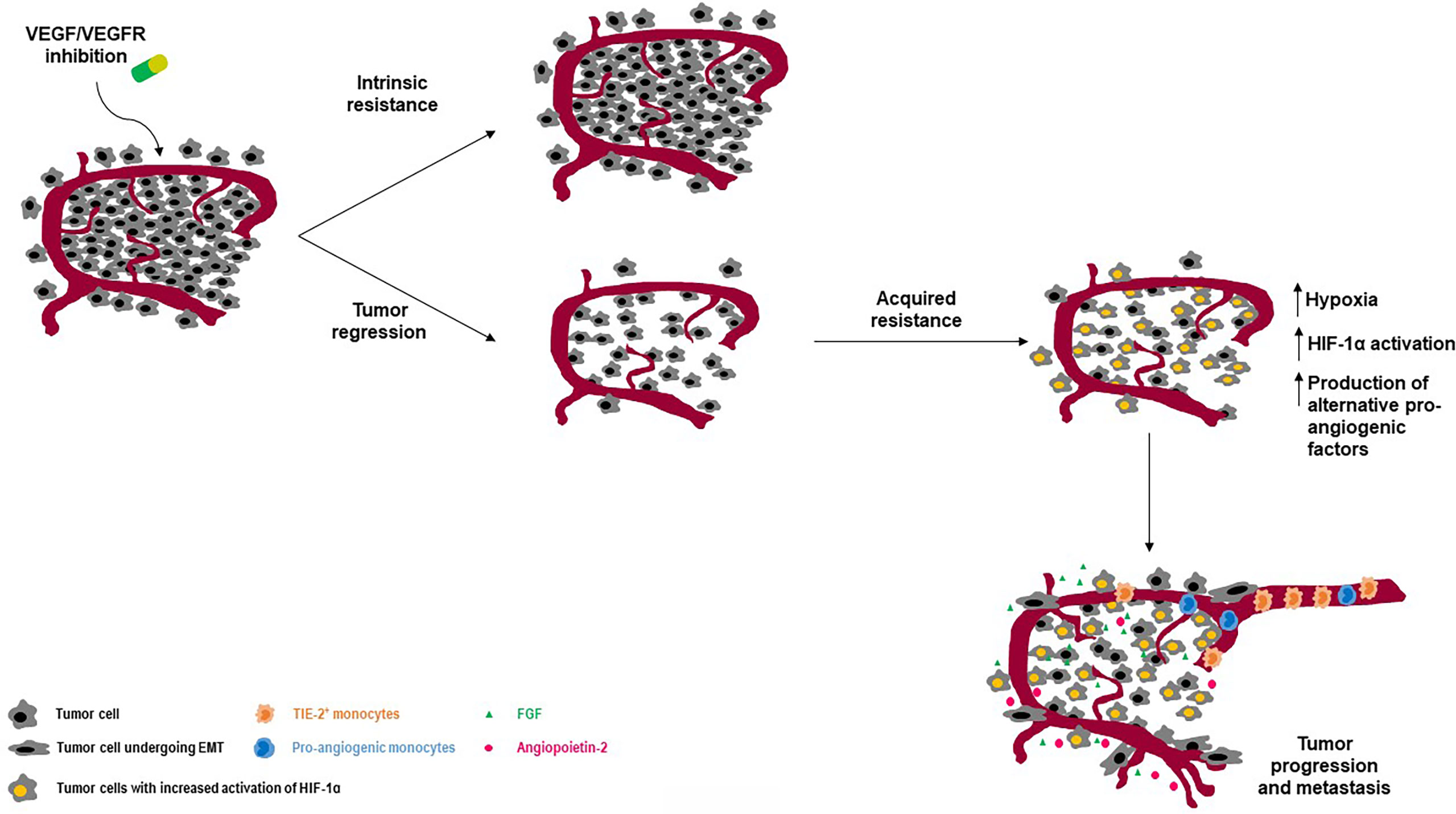
Figure 2 Intrinsic and acquired resistance to angiogenesis blockade: an overview on molecular determinants. While some tumors may show primary resistance to antiangiogenic agents, other may develop resistance upon blockade of the VEGF/VEGFR pathway. One of the main mechanisms leading to secondary resistance is the activation of HIF-1 as result of antiangiogenesis-induced hypoxia. Among other important events, there are the recruitment and/or activation of pro-angiogenic cells including TIE-2 expressing macrophages and the activation of the epithelial-to-mesenchymal transcriptional program in NEN cells.
An established cause of resistance to antiangiogenic agents primarily acting through VEGF suppression is tumor hypoxia. Tumor hypoxia can stimulate HIF-1 activation, thus triggering neoangiogenesis through VEGF-independent mechanisms involving FGF, angiopoietins, and ephrins (33, 52). Evidence from the RIP1-Tag2 model demonstrates that inhibition of both VEGF and FGF signaling at the time of VEGF-independent tumor revascularization attenuates both revascularization and tumor growth (33). In this context, brivanib, a first-in-class, dual FGF-VEGF inhibitor has shown superior preclinical antitumor activity against panNETs when compared with single VEGF suppression or single FGF inhibition (34). A marked upregulation of Ang-2 and TIE2 has been observed in tumors from late-stage RIP1-Tag2 mice resistant to VEGFR blockade (53). In this context, dual Ang-2/VEGFR inhibition was shown to suppress tumor revascularization and progression, suggesting that the adaptive enforcement of Ang2-TIE2 signaling plays a key role in the establishment of evasive tumor resistance to anti-VEGF therapy. The upregulation of c-Met is another consequence of the chronic HIF-1 activation induced by tumor hypoxia. In RIP1-Tag2 mice, VEGFR blockade results in c-Met overexpression, leading to increased tumor growth, proliferation and invasion (54). Concurrent inhibition of VEGF and c-Met signaling is able to revert such effects in vivo (55).
The recruitment of bone marrow derived cells such as endothelial progenitor cells, pro-angiogenic monocytic cells or TIE2-expressing macrophages has been described in panNETs as a result of the hypoxic environment generated by VEGFR inhibition (13, 53). These cells promote the sprouting of new vessels, maintaining the high-demanded blood supply of tumor cells, while concurring to the generation of new premetastatic niches. A progressive increase in the number of tumor-associated macrophages has been described during the sequential progression from hyperplastic islets to angiogenic islets and ultimately invasive tumors in the RIP1-Tag2 model (56). A possible involvement of these cells in the establishment of adaptive resistance to VEGFR blockade through the exploitation of alternative proangiogenic pathways has been inferred.
An increased pericyte coverage has been detected in murine panNETs resistant to VEGFR2 inhibition as compared to those responsive to antiangiogenic therapy (57). Such a phenomenon has been related to a non-angiogenic mechanism of tumor vascularization named vascular co-option. When vascular co-option is activated, cancer cells grow around normal vessels pre-existing in the adjacent “normal” tissue, without the need of generating new vessels. Evidence demonstrates that this process contributes to the emergence of resistance to VEGFR inhibition in the RIP1-Tag2 model (33). Another non-angiogenic mechanism driving resistance to antiangiogenic therapy in panNETs is named “vascular mimicry”, and consists of cancer cells forming vascular channels to autonomously sustain their growth (58). An increased expression of Snail, vimentin and N-cadherin as well as a concurrent downregulation of E-cadherin has been observed in tumors treated with sunitinib, and the hypoxia-driven epithelial-to-mesenchymal transition can be therefore listed as an additional mechanism of resistance to angiogenesis blockade and tumor aggressiveness (43, 59).
Ion trapping and degradation of hydrophobic TKIs within the acidic lysosomal compartment is another mechanism leading to resistance to antiangiogenic therapy. Chloroquine, an agent able to permeabilize the lysosomal membrane, has been shown to enhance the antitumor activity of sunitinib in murine models of pancreatic endocrine carcinogenesis by stimulating the release of the TKI in the cytoplasm (60).
Targeting angiogenesis in NETs: established and investigational agents
Proangiogenic pathways can be blocked at different levels in NETs. Both direct suppression of proangiogenic molecules such as VEGF and inhibition of receptors tyrosine kinase including VEGFR and FGFR through TKIs (i.e., sunitinib) or mAb (i.e., ramucirumab) have been exploited in clinical trials. Table 1 provides an overview of the clinical investigations of antiangiogenic agents in patients with NETs. Being the targets of TKIs usually multiple, it is currently difficult to precisely determine to what extent their therapeutic effects are related to anti-angiogenesis, antiproliferative activity against tumor cells per se, or interference in the mechanisms of cross-talk between tumor cells and their microenvironment.
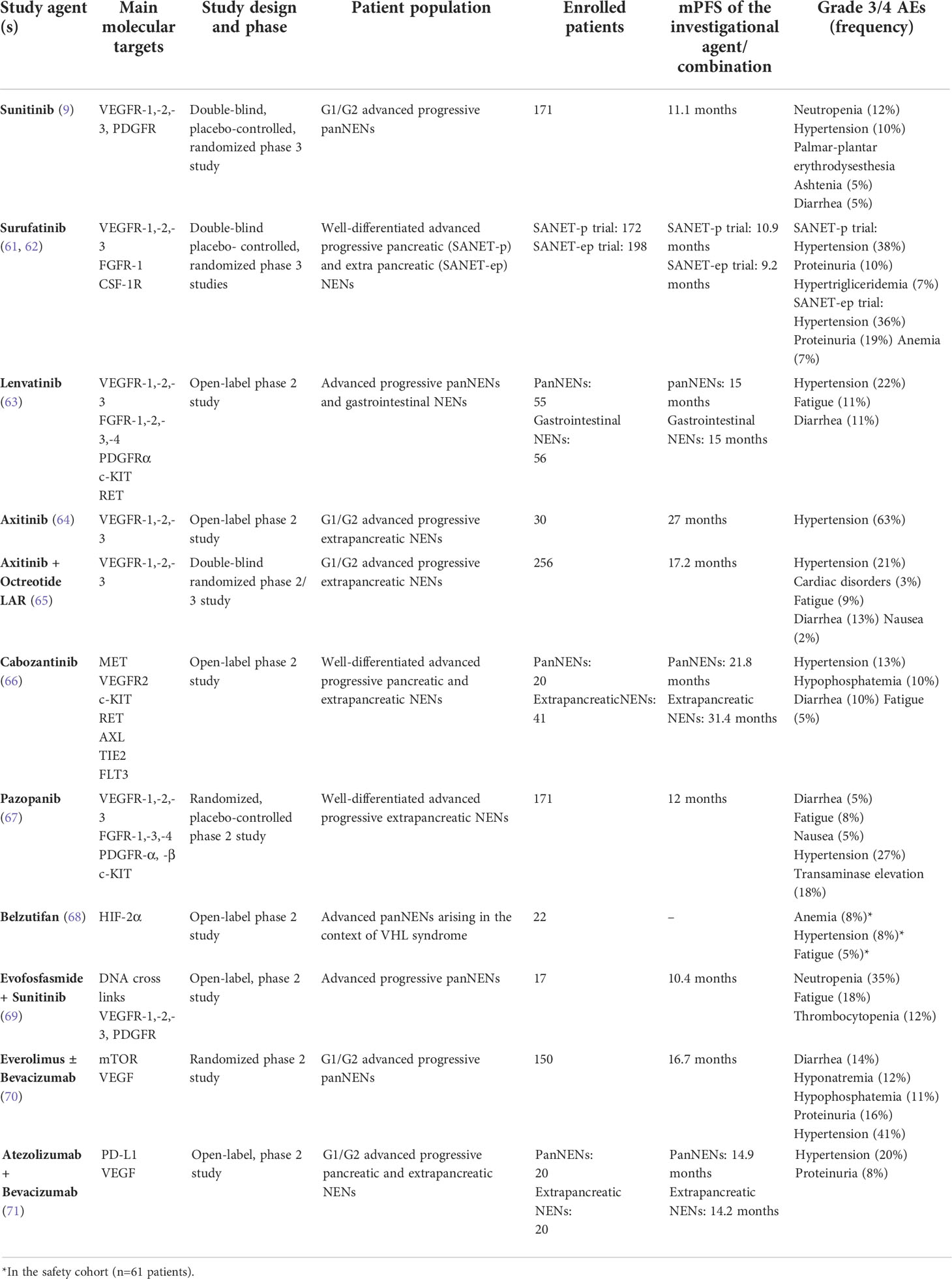
Table 1 An overview of completed studies with antiangiogenic agents in patients with NENs.
VEGF/VEGFR-targeting agents
Bevacizumab is a mAb against VEGF and preliminary evidence suggested the antitumor activity of the drug in patients with NETs (69, 72, 73). However, no benefit in PFS was recorded in a randomized phase 3 trial comparing bevacizumab plus octreotide versus interferon plus octreotide in 427 patients who had high-risk NETs (74). In recent years, bevacizumab has been investigated in combination with chemotherapy, targeted agents or immunotherapy. In 2 separate phase 2 studies, bevacizumab has been tested in association with the capecitabine-oxaliplatin or FOLFOX regimens (75) in 40 and 36 patients with progressive NETs or NECs. Neither study met its primary endpoint, leading to objective responses in 18% and 25% of patients respectively. Another phase 2 trial investigated bevacizumab with 5-FU and streptozocin in 34 patients with progressive, well-differentiated panNETs. A median PFS of 23.7 months was observed, in the presence of an ORR of 56% (76). In a phase 2 trial, 150 patients with advanced panNETs were randomized to receive the mTOR inhibitor everolimus plus octreotide with or without bevacizumab (70). Of note, preclinical evidence suggests that the antitumor activity of mTOR inhibition in NETs results from a combination of antiproliferative and antiangiogenic effects (77). The treatment arm containing the antiangiogenic agent resulted in improved PFS compared to the control arm (16.7 months compared to 14 months; hazard ratio, 0.8; p=0.10), with objective responses seen in 31% and 12% of patients treated with or without bevacizumab respectively (p=0.005). Despite the encouraging efficacy outcomes, the higher rate of treatment-related toxicities observed in the investigational arm may limit further investigations on this combination. Similar results were achieved in a phase II study evaluating the combination of bevacizumab and sorafenib in 44 patients with advanced NETs (78). Despite a median PFS of 12.4 months, grade 3/4 toxicities were described in the 63% of the enrolled patients. Based on the evidence that anti-angiogenic agents such as bevacizumab may modulate the tumor immune microenvironment and decrease the expression of regulatory checkpoints on tumor-infiltrating lymphocytes (79), a single-arm, open-label, nonrandomized study has recently evaluated the association of bevacizumab with the anti-PD-L1 Ab atezolizumab in patients with advanced, progressive, well-differentiated NETs (71). Overall, 20 patients with panNETs and 20 patients with extra-pancreatic NETs have been enrolled, and an ORR of 20% and 15% has been recorded in the two cohorts. A median PFS of 14.9 months and 14.2 months has been reported in the pancreatic and extra-pancreatic cohort, suggesting the potential efficacy of this combination. Hypertension and proteinuria have been described as the most common treatment-emergent toxicities. Bevacizumab is currently being investigated in combination with chemotherapy or chemo-immunotherapy in multiple trials of patients with extrapulmonary NECs (80).
Ramucirumab, a humanized mAb targeting VEGFR2, has demonstrated preliminary evidence of efficacy when used in combination with chemotherapy in patients with gastric NEC (81). A prospective, multicenter, single-arm study is currently investigating ramucirumab plus dacarbazine in patients with advanced, progressive, well-differentiated panNETs (82).
The VEGF trap aflibercept has been investigated in a phase II, single-arm trial of 21 patients with advanced panNETs (83). An ORR of 9% has been reported, a finding consistent with other antiangiogenic agents in panNETs.
Sunitinib
Sunitinib is the only antiangiogenic agent currently approved for the treatment of NETs. Sunitinib is an oral TKI targeting, among a number of different kinases, VEGFR-1, -2, -3 and PDFGR, and has demonstrated efficacy in the treatment of advanced, progressive panNETs. A double-blind, placebo-controlled, phase 3 study evaluated sunitinib 37.5 mg daily in 171 patients with low-to-intermediate grade, progressive panNETs (9). The trial demonstrated a statistically significant improvement in median PFS from 5.5 months on the placebo arm to 11.1 months on the sunitinib arm (hazard ratio, 0.42; p<0.001). A nonsignificant overall survival (OS) improvement of approximately 10 months was observed in the sunitinib arm compared with the placebo arm (84). Nausea, diarrhea, fatigue, cytopenia, hypertension and palmar-plantar erythrodydesthesia were the main treatment-related toxicities. A similar toxicity profile has been described in a recent phase IV study (85).
Other multikinase inhibitors
Dual inhibitors of the VEGF/FGF signaling carry the promise of overcoming the mechanisms leading to adaptive resistance to sunitinib, and recent clinical research has focused on agents including surufatinib, lenvatinib, axitinib, cabozantinib and pazopanib. Surufatinib is an oral, selective inhibitor of VEGFR-1, -2, -3, FGFR-1 and colony stimulating factor-1 receptor (CSF-1R). The TKI has been tested at a dosage of 300 mg daily in a single-arm, multicenter, phase 1b/2 trial of 81 patients with low-to-intermediate grade advanced NETs (86). A median PFS of 21.2 months and 13.4 months was reported in 42 patients with panNETs and 39 patients with extrapancreatic NETs respectively. Two randomized, double-blind, placebo-controlled, phase 3 studies have recently investigated the safety and efficacy of surufatinib in Chinese patients with well-differentiated, progressive, advanced pancreatic (SANET-p trial) and extrapancreatic NETs (SANET-ep trial). The SANET-p trial (61) randomized 172 patients with panNETs to receive surufatinib or placebo in a 2:1 ratio. The investigator-assessed median PFS was 10.9 months for surufatinib versus 3.7 months for placebo (hazard ratio: 0.49; p=0.001), with an investigator-assessed overall response rate (ORR) of 19% in the investigational arm. The SANET-ep trial (62) randomized 198 patients with extrapancreatic NETs to receive surufatinib or placebo in a 2:1 ratio. The investigator-assessed median PFS was 9.2 and 3.8 months in the surufatinib and placebo arms respectively (hazard ratio: 0.33; p<0.0001). The ORR was 15%, and the majority of enrolled patients (84%) had G2 tumors. Overall, hypertension, proteinuria, hypertriglyceridemia and diarrhea were reported as the most frequent treatment-related grade 3 or worse adverse events. The occurrence of treatment-related adverse events including hypertension, proteinuria and hemorrhage in the first 4 weeks of treatment has been recently described to predict the antitumor efficacy of surufatinib (87). The efficacy and safety of surufatinib are being currently evaluated in two ongoing trials in the US (NCT02549937) and Europe (NCT04579679), and their results might lead to the approval of the drug in Western countries. It remains currently unclear whether surufatinib may be active in patients progressing to prior antiangiogenic therapy, and current investigations exclude from enrollment patients who have received prior VEGF/VEGFR targeted therapy.
Lenvatinib is an oral TKI targeting VEGFR-1, -2, -3, FGFR-1, -2, -3, -4, platelet-derived growth factor receptor α (PDGFRα), KIT and RET. The drug has been recently investigated in the phase 2 TALENT study at a dosage of 24 mg daily (63). A total of 55 patients with advanced panNETs and 56 patients with advanced gastrointestinal NETs have been enrolled. All patients had progressive disease according to RECIST criteria, and prior therapy with targeted agents was mandatory for inclusion in the panNET cohort. By central radiology review, the ORR was 44% and 16% for panNETs and gastrointestinal NETs respectively, and the median duration of response was 20 and 34 months in the two cohorts respectively. After a median follow-up of 23 months, the median PFS was 15 months for either panNETs and gastrointestinal NETs. Hypertension, fatigue and diarrhea were the most frequent G3/4 treatment-emergent adverse events. Dose reductions or interruptions were required in the 94% of patients. Although the ORR observed in the TALENT study is the highest reported to date with a TKI in advanced NETs, further clinical investigations of this agent in NETs have not been planned so far.
Axitinib is a TKI that selectively targets VEGFR-1, -2 and -3. In an open-label phase 2 study, axitinib 5 mg twice daily was investigated in 30 patients with progressive, advanced, low-to-intermediate grade NETs of extra-pancreatic origin (64). After a median follow-up of 29 months, a median PFS of 27 months was observed. Grade 3/4 hypertension was recorded in the 63% of the cohort, leading to treatment discontinuation in one fifth of enrolled patients. The double-blind, phase 2/3 AXINET trial has recently randomized 256 patients with advanced, low-to-intermediate grade, progressive, extra-pancreatic NETs to receive axitinib plus octreotide LAR or placebo plus octreotide LAR (65). Per blinded independent central review, the median PFS was 16.6 and 9.9 months in the axitinib and placebo arms respectively (HR: 0.69; p=0.01). An ORR of 13% and 3% has been reported in the investigational and control group respectively (p=0.004). Grade 3 or worse adverse events occurred in the 52% of the enrolled patients and included hypertension, cardiac disorders, fatigue, diarrhea and nausea/vomiting. One treatment-emergent death was reported in the axitinib arm. Overall, axitinib appears a promising candidate for future regulatory approval in patients with NETs.
Cabozantinib is an oral, potent inhibitor of MET, VEGFR2, KIT, RET, AXL, TIE2 and FLT3. The TKI has been tested at 60 mg daily in a two-cohort, phase 2 study enrolling 20 patients with panNETs and 41 patients with extra-pancreatic NETs (66). All patients had well-differentiated tumors and progressive disease according to RECIST 1.1 criteria. The ORR was 15% in either cohort, while a median PFS of 21.8 and 31.4 months was recorded in patients with pancreatic and extra-pancreatic neoplasms respectively. Hypertension, hypophosphatemia, diarrhea and fatigue were among the most common grade 3/4 adverse events. Dose reductions were required in the 80% of patients. The phase 3 CABINET trial (NCT03375320) is currently randomizing patients with well-differentiated, advanced, progressive, pancreatic or extra-pancreatic NET to receive cabozantinib 60 mg daily or placebo. Combinations of cabozantinib plus temozolomide (NCT04893785), lanreotide (NCT04427787) or 177Lu-DOTATATE (NCT05249114) are presently under scrutiny in phase 2 studies.
Pazopanib is an oral TKI targeting VEGFR -1, -2, -3, FGFR-1, -3, -4, PDGFR-α and -β and c-KIT. The drug has been investigated in the open-label, phase 2 PAZONET trial (88). In 44 patients with advanced, progressive, well-differentiated NETs, the TKI was associated with a median PFS of 9 months. The most common grade 3/4 toxicities of pazopanib included diarrhea, fatigue and hypertension, and drug dosage reductions were required in approximately one fifth of enrolled patients. More recently, pazopanib has been tested at 800 mg daily in the phase 2 Alliance A021202 study (67). The trial randomized 171 patients with well-differentiated, progressive, extrapancreatic NETs to receive pazopanib or placebo. After a median follow-up of 31 months, a median PFS of 12 and 8 months was recorded in patients treated with pazopanib or placebo respectively (HR: 0.53; p=0.0005). Pazopanib was associated with an ORR of only 2%. Among pazopanib-treated patients, treatment-related grade 3/4 adverse events were reported in 61% of cases, and hypertension, fatigue, nausea, diarrhea and transaminases elevation were the most common toxicities. Pazopanib has also demonstrated clinical activity against panNETs arising in the context of von Hippel-Lindau syndrome. In a single-arm study enrolling 31 patients with this inherited syndrome, the drug induced objective responses in 53% of 17 pancreatic lesions (89).
Nintedanib is an oral inhibitor of FGFR1-3, VEGFR1-3 and PDGFR. The drug has been tested in a phase II study of 32 patients with extra-pancreatic NETs on a stable dose of somatostatin analog (90). A median PFS of 11 months has been observed, and toxicities were manageable.
HIF inhibitors and hypoxia-activated prodrugs
Novel antiangiogenic compounds investigated in patients with NETs comprise the HIF-2α inhibitor belzutifan and the hypoxia-activated prodrug evofosfamide. Belzutifan has been recently tested at 120 mg daily in an open-label, phase 2 trial of 61 patients with von Hippel-Lindau syndrome. Among 22 patients harboring a panNET, objective responses were seen in 90% of cases, with complete responses in 14% of the cohort (68). Anemia and fatigue were the most common adverse events, being reported in 90% and 66% of patients respectively. On this basis, belzutifan has received regulatory approval for the treatment of tumors arising in the context of Von-Hippel Lindau syndrome. An international phase 2 study of belzutifan in patients with sporadic panNETs is currently ongoing (NCT04924075). Evofosfamide is a prodrug of the alkylating agent bromoisophosphoramide mustard. The release of the active drug occurs exclusively under hypoxic conditions, and results in intra- and inter-strand DNA cross links in tumor cells. Given the well-known ability of sunitinib in inducing intratumor hypoxia, evofosfamide has been recently investigated in combination with sunitinib in the open-label, Simon’s two-stage design, phase II SUNEVO trial (69). The study enrolled 17 patients with advanced panNETs, and only prior therapy with somatostatin analogs was permitted. After a median follow-up of 16 months, three objective responses were recorded, in the presence of a median PFS of 10.4 months. Grade 3 or worse treatment-related adverse events were reported in the 65% of the cohort, the most frequent being neutropenia, fatigue and thrombocytopenia. Overall, treatment discontinuation due to toxicity was required in 88% of the patients. In light of the unfavorable safety profile and the modest efficacy shown by sunitinib and evofosfamide in this study, further clinical investigations of this combination have not been planned.
Future directions for angiogenesis blockade in NETs
Developing new antiangiogenic agents, testing new combinations of antiangiogenic agents with targeted drugs or immunotherapy and defining the correct positioning of antiangiogenic therapies in the context of treatment sequences are among the main priorities for future research on angiogenesis blockade in NETs.
Angiogenesis is a complex process involving distinct biological mechanisms. Mechanistically, endothelial cell proliferation, vessel guidance, vessel maturation, stabilization and quiescence are driven by different families of molecular cues, and angiogenic processes can be thereby inhibited at different levels. There is a need to identify and characterize additional molecular regulators of angiogenesis in NETs in order to develop the next generation of antiangiogenic drugs to be tested (alone or in combination) in clinical trials. Moreover, since evidence demonstrates that different angiogenic molecules may be expressed differently during tumor progression, a precise understanding of the molecular events driving neoangiogenesis during NET evolution might be instrumental to provide molecular-level guidance on the correct positioning of angiogenesis blockade throughout the treatment journey of NET patients. Combinatorial strategies aimed at concurrently disrupting key pathways operating in NET progression (i.e., concurrent inhibition of angiogenesis and mTOR signaling) have been only partially investigated. In a phase II study of bevacizumab and temsirolimus, an ORR of 41% and a median PFS of 13.2 months were observed among 58 patients with panNETs, in the presence of toxicities leading to treatment discontinuation in approximately one third of the cohort (91). While the efficacy/toxicity ratio of combinatorial treatments should be always carefully scrutinized in relatively indolent tumors such as NETs, clinical trials should explore the impact of targeted agent combinations in disease settings where tumor shrinkage is the goal of treatment (i.e., neoadjuvant setting). Accumulating evidence indicates that a tight link exists between aberrant tumor angiogenesis and the immune microenvironment, and antiangiogenic agents have been shown to synergize with immune checkpoint inhibitors in malignancies including renal cell carcinoma, endometrial cancer and hepatocellular carcinoma (92). Future studies should assess the potential of antiangiogenic agents in tuning the microenvironment of NETs from an immune-suppressive to an immune-supportive one, thus enhancing the efficacy of immunotherapy.
Conclusions
The concept of angiogenesis inhibition as a potential weapon against cancer was first proposed by Folkman in the 70s (93). After the initial skepticism of the scientific community, multiple lines of evidence have demonstrated that antiangiogenic agents can be clinically effective in controlling tumor growth. Sunitinib is the only antiangiogenic drug approved for the treatment of NETs, and its use is restricted to pancreatic primaries. Newer TKIs including surufatinib, cabozantinib, axitinib and lenvatinib seem to possess more potent antitumor activity, probably as result of their multi-targeting potential, and might be utilized as monotherapy or as backbone for combinatorial treatment of both pancreatic and extra-pancreatic NETs in the near future. While it is currently unclear whether combinations of antiangiogenic agents with chemotherapeutics, targeted agents or immunotherapy are more effective than the monotherapy, the results of the first study comparing the efficacy of sunitinib versus PRRT in patients with progressive panNETs are awaited soon (NCT02230176). In the absence of predictors of response and given the lack of high-level evidence on optimal treatment sequences, clinical wisdom continues to be critical in defining the timing of antiangiogenic therapy in patients with NETs.
Author contributions
All authors contributed to literature review and to the writing the manuscript. All authors approved the final version of the manuscript.
Funding
This work was supported by the Associazione Italiana per la Ricerca sul Cancro [MFAG #23583] and Associazione per la Ricerca Biomolecolare Onlus, Acquaviva, Italy [2020].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cives M, Strosberg JR. Gastroenteropancreatic neuroendocrine tumors. CA Cancer J Clin (2018) 68(6):471–87. doi: 10.3322/caac.21493
2. Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y, et al. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the united states. JAMA Oncol (2017) 3(10):1335–42. doi: 10.1001/jamaoncol.2017.0589
3. Klimstra DS, Kloppel G, La Rosa S, Rindi G. WHO classification of tumors of the digestive system. 5th ed Vol. Volume 1. . Lyon, France: IARC Press (2019).
4. Strosberg JR, Cheema A, Weber J, Han G, Coppola D, Kvols LK. Prognostic validity of a novel American joint committee on cancer staging classification for pancreatic neuroendocrine tumors. J Clin Oncol (2011) 29(22):3044–9. doi: 10.1200/JCO.2011.35.1817
5. Strosberg JR, Weber JM, Feldman M, Coppola D, Meredith K, Kvols LK. Prognostic validity of the American joint committee on cancer staging classification for midgut neuroendocrine tumors. J Clin Oncol (2013) 31(4):420–5. doi: 10.1200/JCO.2012.44.5924
6. Marion-Audibert AM, Barel C, Gouysse G, Dumortier J, Pilleul F, Pourreyron C, et al. Low microvessel density is an unfavorable histoprognostic factor in pancreatic endocrine tumors. Gastroenterology (2003) 125(4):1094–104. doi: 10.1016/S0016-5085(03)01198-3
7. Carrasco P, Zuazo-Gaztelu I, Casanovas O. Sprouting strategies and dead ends in antiangiogenic targeting of NETs. J Mol Endocrinol (2017) 59(1):R77–91. doi: 10.1530/JME-17-0029
8. Couvelard A, O’ Toole D, Turley H, Leek R, Sauvanet A, Degott C, et al. Microvascular density and hypoxia-inducible factor pathway in pancreatic endocrine tumours: negative correlation of microvascular density and VEGF expression with tumour progression. Br J Cancer (2005) 92(1):94–101. doi: 10.1038/sj.bjc.6602245
9. Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med (2011) 364(6):501–13. doi: 10.1056/NEJMoa1003825
10. Cives M, Pelle’ E, Strosberg J. Emerging treatment options for gastroenteropancreatic neuroendocrine tumors. J Clin Med (2020) 9(11):3655. doi: 10.3390/jcm9113655
11. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell (1996) 86(3):353–64. doi: 10.1016/S0092-8674(00)80108-7
12. Franco M, Roswall P, Cortez E, Hanahan D, Pietras K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-a signaling and bcl-w expression. Blood (2011) 118(10):2906–17. doi: 10.1182/blood-2011-01-331694
13. Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell (2009) 15(3):220–31. doi: 10.1016/j.ccr.2009.01.027
14. Sennino B, Ishiguro-Oonuma T, Schriver BJ, Christensen JG, McDonald DM. Inhibition of c-met reduces lymphatic metastasis in RIP-Tag2 transgenic mice. Cancer Res (2013) 73(12):3692–703. doi: 10.1158/0008-5472.CAN-12-2160
15. Chang TM, Chu PY, Hung WC, Shan YS, Lin HY, Huang KW, et al. C-myc promotes lymphatic metastasis of pancreatic neuroendocrine tumor through VEGFC upregulation. Cancer Sci (2021) 112(1):243–53. doi: 10.1111/cas.14717
16. Scoazec JY. Angiogenesis in neuroendocrine tumors: therapeutic applications. Neuroendocrinology (2013) 97(1):45–56. doi: 10.1159/000338371
17. Cives M, Pelle' E, Quaresmini D, Rizzo FM, Tucci M, Silvestris F. The tumor microenvironment in neuroendocrine tumors: Biology and therapeutic implications. Neuroendocrinology (2019) 109(2):83–99. doi: 10.1159/000497355
18. Terris B, Scoazec JY, Rubbia L, Bregeaud L, Pepper MS, Ruszniewski P. Et al: Expression of vascular endothelial growth factor in digestive neuroendocrine tumours. Histopathology (1998) 32(2):133–8. doi: 10.1046/j.1365-2559.1998.00321.x
19. Besig S, Voland P, Baur DM, Perren A, Prinz C. Vascular endothelial growth factors, angiogenesis, and survival in human ileal enterochromaffin cell carcinoids. Neuroendocrinology (2009) 90(4):402–15. doi: 10.1159/000245900
20. La Rosa S, Uccella S, Finzi G, Albarello L, Sessa F, Capella C. Localization of vascular endothelial growth factor and its receptors in digestive endocrine tumors: correlation with microvessel density and clinicopathologic features. Hum Pathol (2003) 34(1):18–27. doi: 10.1053/hupa.2003.56
21. Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci USA (2006) 103(33):12493–8. doi: 10.1073/pnas.0601807103
22. Kuroda M, Oka T, Oka Y, Yamochi T, Ohtsubo K, Mori S, et al. Colocalization of vascular endothelial growth factor (vascular permeability factor) and insulin in pancreatic islet cells. J Clin Endocrinol Metab (1995) 80(11):3196–200. doi: 10.1210/jcem.80.11.7593426
23. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: Beyond discovery and development. Cell (2019) 176(6):1248–64. doi: 10.1016/j.cell.2019.01.021
24. Inoue M, Hager JH, Ferrara N, Gerber HP, Hanahan D. VEGF-a has a critical, nonredundant role in angiogenic switching and pancreatic beta cell cancinogenesis. Cancer Cell (2002) 1(2):193–202. doi: 10.1016/S1535-6108(02)00031-4
25. Haugen M, Dammen R, Svejda B, Gustafsson BI, Pfragner R, Modlin I, et al. Differential signal pathway activation and 5-HT function: the role of enterochromaffin cells as oxygen sensors. Am J Physiol Gastrointest Liver Physiol (2012) 303(10):G1164–73. doi: 10.1152/ajpgi.00027.2012
26. Speisky D, Duces A, Bièche I, Rebours V, Hammel P, Sauvanet A, et al. Couvelard a for the GTE group (Groupe d'Etude des tumeurs endocrines): Molecular profiling of pancreatic neuroendocrine tumors in sporadic and Von hippel-lindau patients. Clin Cancer Res (2012) 18(10):2838–49. doi: 10.1158/1078-0432.CCR-11-2759
27. Yun YR, Won JE, Jeon E, Lee S, Kang W, Jo H, et al. Fibroblast growth factors: biology, function, and application for tissue regeneration. J Tissue Eng (2010) 2010:218142. doi: 10.4061/2010/218142
28. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discovery (2009) 8:235–53. doi: 10.1038/nrd2792
29. Vitale G, Cozzolino A, Malandrino P, Minotta R, Puliani G, Saronni D, et al. Role of FGF system in neuroendocrine neoplasms: Potential therapeutic applications. Front Endocrinol (Lausanne) (2021) 12:665631. doi: 10.3389/fendo.2021.665631
30. Chaudhry A, Funa K, Oberg K. Expression of growth factor peptides and their receptors in neuroendocrine tumors of the digestive system. Acta Oncol (1993) 32:107–14. doi: 10.3109/02841869309083898
31. La Rosa S, Uccella S, Erba S, Capella C, Sessa F. Immunohistochemical detection of fibroblast growth factor receptors in normal endocrine cells and related tumors of the digestive system. Appl Immunohistochem Mol Morphol (2001) 9:319–28. doi: 10.1097/00129039-200112000-00006
32. Compagni A, Wilgenbus P, Impagnatiello MA, Cotten M, Christofori G. Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res (2000) 60(24):7163–9. PMID: 11156426
33. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell (2005) 8(4):299–309. doi: 10.1016/j.ccr.2005.09.005
34. Allen E, Walters IB, Hanahan D. Brivanib, a dual FGF/VEGF inhibitor, is active both first and second line against mouse pancreatic neuroendocrine tumors developing adaptive/evasive resistance to VEGF inhibition. Clin Cancer Res (2011) 17(16):5299–310. doi: 10.1158/1078-0432.CCR-10-2847
35. Allt G, Lawrenson JG. Pericytes: cell biology and pathology. Cells Tissues Organs (2001) 169:1–11. doi: 10.1159/000047855
36. Funa K, Papanicolaou V, Juhlin C, Rastad J, Akerström G, Heldin CH, et al. Expression of platelet-derived growth factor beta-receptors on stromal tissue cells in human carcinoid tumors. Cancer Res (1990) 50(3):748–53. PMID: 2153446
37. Cavalcanti E, Ignazzi A, De Michele F, Caruso ML. PDGFRα expression as a novel therapeutic marker in well-differentiated neuroendocrine tumors. Cancer Biol Ther (2019) 20(4):423–30. doi: 10.1080/15384047.2018.1529114
38. Cortez E, Gladh H, Braun S, Bocci M, Cordero E, Bj.rkstr.m NK, et al. Functional malignant cell heterogeneity in pancreatic neuroendocrine tumors revealed by targeting of PDGF-DD. Proc Natl Acad Sci USA (2016) 113(7):E864–73. doi: 10.1073/pnas.1509384113
39. Bollard J, Patte C, Radkova K, Massoma P, Chardon L, Valantin J, et al. Neuropilin-2 contributes to tumor progression in preclinical models of small intestinal neuroendocrine tumors. J Pathol (2019) 249(3):343–55. doi: 10.1002/path.5321
40. Cohen T, Herzog Y, Brodzky A, Greenson JK, Eldar S, Gluzman-Poltorak Z. Et al: Neuropilin-2 is a novel marker expressed in pancreatic islet cells and endocrine pancreatic tumours. J Pathol (2002) 198(1):77–82. doi: 10.1002/path.1179
41. Lantuéjoul S, Constantin B, Drabkin H, Brambilla C, Roche J, Brambilla E. Expression of VEGF, semaphorin SEMA3F, and their common receptors neuropilins NP1 and NP2 in preinvasive bronchial lesions, lung tumours, and cell lines. J Pathol (2003) 200(3):336–47. doi: 10.1002/path.1367
42. Maione F, Molla F, Meda C, Latini R, Zentilin L, Giacca M, et al. Semaphorin 3A is an endogenous angiogenesis inhibitor that blocks tumor growth and normalizes tumor vasculature in transgenic mouse models. J Clin Invest (2009) 119(11):3356–72. doi: 10.1172/JCI36308
43. Maione F, Capano S, Regano D, Zentilin L, Giacca M, Casanovas O, et al. Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J Clin Invest (2012) 122(5):1832–48. doi: 10.1172/JCI58976
44. Bollard J, Massoma P, Vercherat C, Blanc M, Lepinasse F, Gadot N, et al. The axon guidance molecule semaphorin 3F is a negative regulator of tumor progression and proliferation in ileal neuroendocrine tumors. Oncotarget (2015) 6(34):36731–45. doi: 10.18632/oncotarget.5481
45. Zuazo-Gaztelu I, Pàez-Ribes M, Carrasco P, Martín L, Soler A, Martínez-Lozano M, et al. Antitumor effects of anti-semaphorin 4D antibody unravel a novel proinvasive mechanism of vascular-targeting agents. Cancer Res (2019) 79(20):5328–41. doi: 10.1158/0008-5472.CAN-18-3436
46. Saxena S, Hayashi Y, Wu L, Awaji M, Atri P, Varney ML, et al. Pathological and functional significance of semaphorin-5A in pancreatic cancer progression and metastasis. Oncotarget (2017) 9(5):5931–43. doi: 10.18632/oncotarget.23644
47. Figueroa-Vega N, Díaz A, Adrados M, Alvarez-Escolá C, Paniagua A, Aragonés J, et al. The association of the angiopoietin/Tie-2 system with the development of metastasis and leukocyte migration in neuroendocrine tumors. Endocr Relat Cancer (2010) 17(4):897–908. doi: 10.1677/ERC-10-0020
48. Puliani G, Sesti F, Anastasi E, Verrico M, Tarsitano MG, Feola T, et al. Angiogenic factors as prognostic markers in neuroendocrine neoplasms. Endocrine (2022) 76(1):208–2017. doi: 10.1007/s12020-021-02942-4
49. Detjen KM, Rieke S, Deters A, Schulz P, Rexin A, Vollmer S, et al. Angiopoietin-2 promotes disease progression of neuroendocrine tumors. Clin Cancer Res (2010) 16(2):420–9. doi: 10.1158/1078-0432.CCR-09-1924
50. Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell (2011) 19(4):512–26. doi: 10.1016/j.ccr.2011.02.005
51. Jimenez-Valerio G, Casanovas O. Antiangiogenic resistance: novel angiogenesis axes uncovered by antiangiogenic therapies research. Curr Drug Targets (2016) 17:1728–34. doi: 10.2174/1389450117666160301101425
52. Tijeras-Raballand A, Neuzillet C, Couvelard A, Serova M, de Gramont A, Hammel P, et al. Resistance to targeted therapies in pancreatic neuroendocrine tumors (PNETs): Molecular basis, preclinical data, and counteracting strategies. Target Oncol (2012) 7(3):173–81. doi: 10.1007/s11523-012-0229-6
53. Rigamonti N, Kadioglu E, Keklikoglou I, Wyser Rmili C, Leow CC, De Palma M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep (2014) 8(3):696–706. doi: 10.1016/j.celrep.2014.06.059
54. Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discovery (2012) 2(3):270–87. doi: 10.1158/2159-8290.CD-11-0240
55. You WK, Sennino B, Williamson CW, Falcón B, Hashizume H, Yao LC, et al. VEGF and c-met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res (2011) 71(14):4758–68. doi: 10.1158/0008-5472.CAN-10-2527
56. Krug S, Abbassi R, Griesmann H, Sipos B, Wiese D, Rexin P, et al. Therapeutic targeting of tumor-associated macrophages in pancreatic neuroendocrine tumors. Int J Cancer (2018) 143(7):1806–16. doi: 10.1002/ijc.31562
57. Franco M, Pàez-Ribes M, Cortez E, Casanovas O, Pietras K. Use of a mouse model of pancreatic neuroendocrine tumors to find pericyte biomarkers of resistance to anti-angiogenic therapy. Horm Metab Res (2011) 43(12):884–9. doi: 10.1055/s-0031-1284381
58. Chu X, Gao X, Jansson L, Quach M, Skogseid B, Barbu A. Multiple microvascular alterations in pancreatic islets and neuroendocrine tumors of a Men1 mouse model. Am J Pathol (2013) 182(6):2355–67. doi: 10.1016/j.ajpath.2013.02.023
59. Cives M, Rizzo F, Simone V, Bisceglia F, Stucci S, Seeber A, et al. Reviewing the osteotropism in neuroendocrine tumors: The role of epithelial-mesenchymal transition. Neuroendocrinology (2016) 103(3-4):321–34. doi: 10.1159/000438902
60. Wiedmer T, Blank A, Pantasis S, Normand L, Bill R, Krebs P, et al. Autophagy inhibition improves sunitinib efficacy in pancreatic neuroendocrine tumors via a lysosome-dependent mechanism. Mol Cancer Ther (2017) 16:2502–15. doi: 10.1158/1535-7163.MCT-17-0136
61. Xu J, Shen L, Bai C, Wang W, Li J, Yu X, et al. Surufatinib in advanced pancreatic neuroendocrine tumours (SANET-p): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol (2020) 21(11):1489–99. doi: 10.1016/S1470-2045(20)30493-9
62. Xu J, Shen L, Zhou Z, Li J, Bai C, Chi Y, et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol (2020) 21(11):1500–12. doi: 10.1016/S1470-2045(20)30496-4
63. Capdevila J, Fazio N, Lopez C, Teulé A, Valle JW, Tafuto S, et al. Lenvatinib in patients with advanced grade 1/2 pancreatic and gastrointestinal neuroendocrine tumors: Results of the phase II TALENT trial (GETNE1509). J Clin Oncol (2021) 39(20):2304–12. doi: 10.1200/JCO.20.03368
64. Strosberg JR, Cives M, Hwang J, Weber T, Nickerson M, Atreya CE, et al. A phase II study of axitinib in advanced neuroendocrine tumors. Endocr Relat Cancer (2016) 23(5):411–8. doi: 10.1530/ERC-16-0008
65. Garcia-Carbonero R, Benavent M, Jiménez Fonseca P, Castellano D, Alonso-Gordoa T, Teulé T, et al. The AXINET trial (GETNE1107): Axitinib plus octreotide LAR improves PFS by blinded central radiological assessment vs placebo plus octreotide LAR in G1-2 extrapancreatic NETs. Ann Oncol (2021) 32(suppl_5):S906–920. doi: 10.1016/j.annonc.2021.08.179
66. Chan JA, Faris JE, Murphy JE, Blaszkowsky LS, Kwak EL, McCleary NJ, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol (2017) 35:228. doi: 10.1200/JCO.2017.35.4_suppl.228
67. Bergsland EK, Mahoney MR, Asmis TR, Hall N, Kumthekar P, Maitland ML, et al. Et al: Prospective randomized phase II trial of pazopanib versus placebo in patients with progressive carcinoid tumors (CARC) (Alliance A021202). J Clin Oncol (2019) 37:4005. doi: 10.1200/JCO.2019.37.15_suppl.4005
68. Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, et al. Belzutifan for renal cell carcinoma in von hippel-lindau disease. N Engl J Med (2021) 385(22):2036–46. doi: 10.1056/NEJMoa2103425
69. Grande E, Rodriguez-Antona C, López C, Alonso-Gordoa T, Benavent M, Capdevila J, et al. Sunitinib and evofosfamide (TH-302) in systemic treatment-naïve patients with grade 1/2 metastatic pancreatic neuroendocrine tumors: The GETNE-1408 trial. Oncologist (2021) 26(11):941–9. doi: 10.1002/onco.13885
70. Kulke MH, Ou FS, Niedzwiecki D, Heubner L, Kunz P, Kennecke HF, et al. Everolimus with or without bevacizumab in advanced pNET: CALGB 80701 (Alliance). Endocr Relat Cancer (2022) 29(6):335–44. doi: 10.1530/ERC-21-0239
71. Halperin DM, Liu S, Dasari A, Fogelman D, Bhosale P, Mahvash A, et al. Assessment of clinical response following atezolizumab and bevacizumab treatment in patients with neuroendocrine tumors: A nonrandomized clinical trial. JAMA Oncol (2022) 8(6):904–9. doi: 10.1001/jamaoncol.2022.0212
72. Yao JC, Phan A, Hoff PM, Chen HX, Charnsangavej C, Yeung SC, et al. Targeting vascular endothelial growth factor in advanced carcinoid tumor: a random assignment phase II study of depot octreotide with bevacizumab and pegylated interferon alpha-2b. J Clin Oncol (2008) 26:1316–23. doi: 10.1200/JCO.2007.13.6374
73. Zhu M, Costello BA, Yin J, Pettinger AM, Strosberg JR, Erlichman C, et al. Phase II trial of bevacizumab monotherapy in pancreatic neuroendocrine tumors. Pancreas (2021) 50(10):1435–9. doi: 10.1097/MPA.0000000000001950
74. Yao JC, Guthrie KA, Moran C, Strosberg JR, Kulke MH, Chan JA, et al. Phase III prospective randomized comparison trial of depot octreotide plus interferon alfa-2b versus depot octreotide plus bevacizumab in patients with advanced carcinoid tumors: SWOGS0518. J Clin Oncol (2017) 35:1695–703. doi: 10.1200/JCO.2016.70.4072
75. Kunz PL, Balise RR, Fehrenbacher L, Pan M, Venook AP, Fisher GA, et al. Oxaliplatin-fluoropyrimidine chemotherapy plus bevacizumab in advanced neuroendocrine tumors: An analysis of 2 phase II trials. Pancreas (2016) 45(10):1394–400. doi: 10.1097/MPA.0000000000000659
76. Ducreux M, Dahan L, Smith D, O'Toole D, Lepère C, Dromain C, et al. Bevacizumab combined with 5-FU/streptozocin in patients with progressive metastatic well-differentiated pancreatic endocrine tumours (BETTER trial)–a phase II non-randomised trial. Eur J Cancer (2014) 50(18):3098–106. doi: 10.1016/j.ejca.2014.10.002
77. Couderc C, Poncet G, Villaume K, Blanc M, Gadot N, Walter T, et al. Targeting the PI3K/mTOR pathway in murine endocrine cell lines: in vitro and in vivo effects on tumor cell growth. Am J Pathol (2011) 178(1):336–44. doi: 10.1016/j.ajpath.2010.11.023
78. Castellano D, Capdevila J, Sastre J, Alonso V, Llanos M, García-Carbonero R, et al. Sorafenib and bevacizumab combination targeted therapy in advanced neuroendocrine tumour: a phase II study of Spanish neuroendocrine tumour group (GETNE0801). Eur J Cancer (2013) 49:3780–7. doi: 10.1016/j.ejca.2013.06.042
79. Yi M, Jiao D, Qin S, Chu Q, Wu K, Li A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer (2019) 18(1):60. doi: 10.1186/s12943-019-0974-6
80. Frizziero M, Kilgour E, Simpson KL, Rothwell DG, Moore DA, Frese KK, et al. Expanding therapeutic opportunities for extrapulmonary neuroendocrine carcinoma. Clin Cancer Res (2022) 28(10):1999–2019. doi: 10.1158/1078-0432.CCR-21-3058
81. Mishima S, Kawazoe A, Matsumoto H, Kuboki Y, Bando H, Kojima T, et al. Efficacy and safety of ramucirumab-containing chemotherapy in patients with pretreated metastatic gastric neuroendocrine carcinoma. ESMO Open (2018) 3(7):e000443. doi: 10.1136/esmoopen-2018-000443
82. Krug S, Kegel T, Gress TM, Rinke A, Apostolidis L, Jann H, et al. Ramucirumab in combination with dacarbazine in patients with progressive well-differentiated metastatic pancreatic neuroendocrine tumors (RamuNET): study protocol for a multicenter single-arm trial. BMC Cancer (2021) 21(1):1206. doi: 10.1186/s12885-021-08900-7
83. Halperin MD, Lee JJ, Ng CS, Strosberg JR, Estrella JS, Dagohoy CG, et al. A phase II trial of ziv-afliberecept in patients with advanced pancreatic neuroendocrine tumors. Pancreas (2019) 48(3):381–6. doi: 10.1097/MPA.0000000000001258
84. Faivre S, Niccoli P, Castellano D, Valle JW, Hammel P, Raoul JL, et al. Sunitinib in pancreatic neuroendocrine tumors: updated progression-free survival and final overall survival from a phase III randomized study. Ann Oncol (2017) 28(2):339–43. doi: 10.1093/annonc/mdw561
85. Fazio N, Kulke M, Rosbrook B, Fernandez K, Raymond E. Updated efficacy and safety outcomes for patients with well-differentiated pancreatic neuroendocrine tumors treated with sunitinib. Target Oncol (2021) 16(1):27–35. doi: 10.1007/s11523-020-00784-0
86. Xu J, Li J, Bai C, Xu N, Zhou Z, Li Z, et al. Surufatinib in advanced well-differentiated neuroendocrine tumors: A multicenter, single-arm, open-label, phase Ib/II trial. Clin Cancer Res (2019) 25:3486–94. doi: 10.1158/1078-0432.CCR-18-2994
87. Li J, Cheng Y, Bai C, Xu J, Shen L, Li J, et al. Treatment-related adverse events as predictive biomarkers of efficacy in patients with advanced neuroendocrine tumors treated with surufatinib: results from two phase III studies. ESMO Open (2022) 7(2):100453. doi: 10.1016/j.esmoop.2022.100453
88. Grande E, Capdevila J, Castellano D, Teulé A, Durán I, Fuster J, et al. Pazopanib in pretreated advanced neuroendocrine tumors: A phase II, open-label trial of the Spanish task force group for neuroendocrine tumors (GETNE). Ann Oncol (2015) 26:1987–93. doi: 10.1093/annonc/mdv252
89. Jonasch E, McCutcheon IE, Gombos DS, Ahrar K, Perrier ND, Liu D, et al. Pazopanib in patients with von hippel-lindau disease: a single-arm, single-centre, phase 2 trial. Lancet Oncol (2018) 19(10):1351–9. doi: 10.1016/S1470-2045(18)30487-X
90. Iyer RV, Konda B, Fountzilas C, Mukherjee S, Owen D, Attwood K, et al. Multicenter phase 2 trial of nintedanib in advanced nonpancreatic neuroendocrine tumors. Cancer (2020) 126(16):3689–97. doi: 10.1002/cncr.32994
91. Hobday TJ, Qin R, Reidy-Lagunes D, Moore MJ, Strosberg J, Kaubisch A, et al. Multicenter phase II trial of temsirolimus and bevacizumab in pancreatic neuroendocrine tumors. J Clin Oncol (2015) 33(14):1551–6. doi: 10.1200/JCO.2014.56.2082
92. Chambers A, Kundranda M, Rao S, Mahmoud F, Niu J. Anti-angiogenesis revisited: Combination with immunotherapy in solid tumors. Curr Oncol Rep (2021) 23(9):100. doi: 10.1007/s11912-021-01099-7
Keywords: cabozantinib, lenvatinib, pazopanib, carcinoid tumor, TKIs (tyrosine kinase inhibitors)
Citation: Lauricella E, Mandriani B, Cavallo F, Pezzicoli G, Chaoul N, Porta C and Cives M (2022) Angiogenesis in NENs, with a focus on gastroenteropancreatic NENs: from biology to current and future therapeutic implications. Front. Oncol. 12:957068. doi: 10.3389/fonc.2022.957068
Received: 01 June 2022; Accepted: 28 July 2022;
Published: 17 August 2022.
Edited by:
Cornelis F. M. Sier, Leiden University, NetherlandsReviewed by:
Francesca Spada, European Institute of Oncology (IEO), ItalyJean-Yves Scoazec, Institut Gustave Roussy, France
Copyright © 2022 Lauricella, Mandriani, Cavallo, Pezzicoli, Chaoul, Porta and Cives. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauro Cives, bWF1cm8uY2l2ZXNAdW5pYmEuaXQ=
†These authors have contributed equally to this work