
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Oncol. , 25 July 2022
Sec. Molecular and Cellular Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.955892
This article is part of the Research Topic Insights in Molecular and Cellular Oncology: 2022 View all 23 articles
 Om Saswat Sahoo1†
Om Saswat Sahoo1† Karthikeyan Pethusamy2†
Karthikeyan Pethusamy2† Tryambak P. Srivastava2†
Tryambak P. Srivastava2† Joyeeta Talukdar2†Mohammed S. Alqahtani3,4
Joyeeta Talukdar2†Mohammed S. Alqahtani3,4 Mohamed Abbas5,6Ruby Dhar2*†
Mohamed Abbas5,6Ruby Dhar2*† Subhradip Karmakar2*†
Subhradip Karmakar2*†Cancer stem cells (CSC) are the minor population of cancer originating cells that have the capacity of self-renewal, differentiation, and tumorigenicity (when transplanted into an immunocompromised animal). These low-copy number cell populations are believed to be resistant to conventional chemo and radiotherapy. It was reported that metabolic adaptation of these elusive cell populations is to a large extent responsible for their survival and distant metastasis. Warburg effect is a hallmark of most cancer in which the cancer cells prefer to metabolize glucose anaerobically, even under normoxic conditions. Warburg’s aerobic glycolysis produces ATP efficiently promoting cell proliferation by reprogramming metabolism to increase glucose uptake and stimulating lactate production. This metabolic adaptation also seems to contribute to chemoresistance and immune evasion, a prerequisite for cancer cell survival and proliferation. Though we know a lot about metabolic fine-tuning in cancer, what is still in shadow is the identity of upstream regulators that orchestrates this process. Epigenetic modification of key metabolic enzymes seems to play a decisive role in this. By altering the metabolic flux, cancer cells polarize the biochemical reactions to selectively generate “onco-metabolites” that provide an added advantage for cell proliferation and survival. In this review, we explored the metabolic-epigenetic circuity in relation to cancer growth and proliferation and establish the fact how cancer cells may be addicted to specific metabolic pathways to meet their needs. Interestingly, even the immune system is re-calibrated to adapt to this altered scenario. Knowing the details is crucial for selective targeting of cancer stem cells by choking the rate-limiting stems and crucial branch points, preventing the formation of onco-metabolites.
Cancer stem cells (CSCs), also known as tumor-initiating cells, constitute a rare subset of cells in cancerous tumors, characterized by an enhanced capacity for self-renewal, multipotency, tumor initiation, and tolerating foreign niches required for the growth of the tumor bulk. Located at the apex of the pyramid of tumor cells, they foster the very nature of malignancy and thus play a vital role in regenerating the heterogeneous cell population, unrestrained proliferation, metastatic dissemination, and sustaining therapy.
CSCs were first identified in AML, using cell surface markers, followed by other solid malignancies such as brain, breast, colon, pancreas, etc. Despite their heterogeneity amidst different tumor bulks, CSCs of almost all the subsets possess identical genetic backgrounds via the acquisition of stemness genes, expression of markers like CD44, CD133, ALDH, activation of specific signaling pathways, etc. As a result, the study of CSCs has not been restricted to just cell surface markers; instead, other complementary methods have been envisaged to measure the functional activation of CSCs on a molecular level. Accumulating shreds of evidence have also suggested striking parallels between metabolic phenotypes and CSCs in particular. Metabolic vulnerability is believed to be the hallmarks of these CSCs, thus providing another broad field of research. Yet another novel field of CSC interactions is how the epigenetic system harnesses tumorigenesis and stemness of the CSCs and cancer cells. However, another element of the tumor microenvironment is the immune system in tumorigenesis, which plays a supportive and inhibitive role in the development of CSCs an d cancer. Though these three domains of CSCs stay as integral elements of the tumor microenvironment, they participate quite inclusively in tumorigenesis. Metabolic addictions, epigenetic modifications and the pro/anti tumorigenic properties of the immune system play hand in hand to develop the framework of CSCs and the cancer cells, thus establishing cancer to be one of the leading causes of death globally.
Analogous to a hive’s queen bee, that the queen bees produce and nurture worker bees, killing the queen bee has always led to the hive’s demise. Similarly, destroying the CSCs has always been the appeal to eradicate tumor bulks. Though having an aberrant clinical behavior, several studies have helped determine the prognostic significance of CSCs and thus have suggested profound implications for cancer treatments. Combinational therapies of intra/inter phenotypic features of CSCs have been the demand for treating CSCs, because of its relapsing nature. In this perspective, the emerging fields of the CSC domains, viz CSC metabolism, CSC epigenetics, and CSC immunology, have been elucidated. Although domain-wise reviews are available in the literature, no collective review has been made to put forward into a single picture. In the present study, an effort has been made to congregate the epitranscriptional and immunogenic orchestration of genetic and metabolic programming and how they further impede the CSC microenvironment. The significance of biomarkers to detect CSC and cancer bulk populations has also been discussed. Further, the review will discuss the prospects of targeting all the above phenotypes for CSC therapy.
Since the discovery of CSCs, the role of metabolism in such cells has profoundly been studied. It has been conclusively demonstrated that the origins of these CSCs are substantial because of the upsurge of mutational events occurring in non-tumor cells, further characterized by elevated metabolic activities and plasticity. Characterized by high proliferation rates, their energy demand usually leads to increased biosynthesis of macromolecules and tight regulation of the cellular redox status (1). Thereby, they can be designated as metabolic omnivores, as they can sustain on a wide range of substrates to produce ATP molecules. Moreover, it has been observed that these CSCs have a noticeable ability to adapt to changes according to their physiological environment by selecting the most efficient substrate.
CSCs follow a different metabolic phenotype; however, their characterization is still contentious (2). With discrepancies in the biological function of normal differentiated tumor cells and CSCs, it has been observed that CSCs mimic the metabolic phenotype of the stem cell hierarchy. It was expected that CSC metabolism would conversely orient towards mitochondrial OXPHOS, unlike the widely accepted aerobic glycolysis (Warburg effect) (3). However, only a few studies support the fact that the primary metabolic phenotype of CSCs is mitochondrial OXPHOS. Results based on samples of glioma (4), PDAC (5) and leukemia stem cells (6) show a positive response; still, data obtained from breast CSC samples show discouraging responses in this regard (7).
Further, another misleading interpretation comes from the fact that cell cultures are based on non-physiological high glucose concentrations; thereby, there are possibilities that CSCs must be shifting their primary metabolic phenotype to adapt to the environmental changes. Factors such as biochemical reactions, paracrine cross-communications, and local ecological factors actively stimulate self-renewal pathways for maintaining a CSC niche (8). Though evidence for glucose-based and mitochondrial oxidation has been broadly accepted, it has also been observed that lipids and amino acids, like lysine and glutamine, may also act as alternative metabolic fuels for CSCs (9). The following section describes the above in detail.
CSCs usually exhibit an enhanced glucose metabolism pathway, with higher glucose uptake, lactate production, expression of glycolytic enzymes, and high ATP content. Several studies show that CSCs tend to follow a glycolytic pathway to meet their glucose demands. In certain tumors, it has also been observed that deprivation in glucose intake induces in vitro depletion of CSCs. The increase in the expression of proteins and other factors associated with glucose metabolism confers a longer lifespan for CSCs (10). This was further discussed by Cluntun et al., that CSCs rely on glutamine metabolism, too as glutamines are carbon and amino-nitrogen sources (11).
CSCs from CD133+ hepatic, breast, and nasopharyngeal carcinoma shows a preferential inclination towards aerobic glycolysis (12). Methylation of Fructose Biphosphate-1, FBP1 and other gluconeogenesis enzymes has been seen to help glycolysis retain stemness features in an aggressive form of breast cancer, BLBC (13). However, overexpression of FBP-1 and an increase of ROS have shown commendable results in the reduction of CD44high/CD24low/EpCAM+ in BLBC, thereby reducing their tumor-forming capability in vivo (13). This phenomenon was later amended by Peng et al. that BLBCs show profound levels of PDK 1, which is known to inhibit mitochondrial OXPHOS (14). A depletion of PDK1 drastically decreases the presence of ALDH-1 positive BLBCs, thereby decreasing tumor spheroid forming ability. A similar response was also noted when snail-mediated lowering of cytochrome C oxidase and FBP1 expression inhibited OXPHOS and activated glycolysis (15). Snail also plays a vital role in regulating glucose flux and PPP by inhibiting phosphofructokinase platelets (PFKPs), under conditions with nutrient deficiency (16). It has also been profoundly noticed that CSCs exhibit an upregulation in glycolytic enzymes and gene expressions like GLUT1, HK-1, HK-2, PDK-1, c-Myc, CD-44, and PGAM-1 along with downregulation of gluconeogenic enzymes like G6PC and PEPCK (17, 18). Hsp90, in coordination with other epigenetic factors like PKM2, also has been seen to mediate the metabolic signature of CSCs and promote glycolysis (19). PKM2 following Oct4 and β-catenin has been seen to express glycolytic gene expression, hence promoting stemness quality in cells (20–22). Many hypotheses have also confirmed the role of hypoxia as one of the characteristic features of the CSC niche. Glucose has a lower diffusion rate than oxygen; however, the solubility of glucose is higher than that of oxygen, which also delineates the credibility of a glucose-based metabolism in CSCs. Thereby, in a hypoxic niche, the cells compensate for their low oxygen conditions by inducing HIFs, which mediate the expression of a considerable number of genes involved in this regard. HIF-1α and the mTOR/Akt/beta-catenin stem cell regulatory pathway stimulate the transcription of the essential GLUTs and almost all the glycolytic enzymes, namely, LDH, GLUT, HK2, MCTs thereby endorsing that hypoxic conditions favor a glycolytic based metabolism (23–25).
In sum, the above observations help us postulate that activation of the glycolytic phenotype favors stemness in CSCs.
Despite the above-reported studies, an overwhelming amount of publications signify that CSCs use mitochondrial biogenesis and OXPHOS predominantly. In fact, Sato et al., in their experiment, demonstrated the reprogrammed metabolome analysis of the CSCs via the TCA cycle (26). Compared to non-CSCs, it was determined that activation of TCA cycles could be a characteristic feature of CSCs. A similar trend was observed in small lung cancer cells when CSCs were compared based on dependence upon OXPHOS and mitochondrial function (27). Convincing results were also reported concerning CD133+ glioblastoma (28), ROSlow quiescent leukemia stem cells (6), ovarian (29), and PDAC (30) to privilege OXPHOS as their metabolic phenotype. Furthermore, Janiszewska et al. showed that glioma-spheres were outlined by oncofetal insulin-like growth factor 2 mRNA-binding protein 2 (IMP2), a pivotal OXPHOS regulating factor (28). A study performed on CD133+ CD49f+ hepatocellular carcinoma cells showed that NANOG’s pluripotency factor contributes towards reprogramming mitochondrial metabolism to meet cellular demands (31). NANOG ChIP-seq analysis and metabolomic profiling determined that OXPHOS and FAO are NANOG-mediated oncogenic pathways. NANOG silencing leads to increased OXPHOS activity and downregulation of both transcripts and protein levels of FAO-associated genes.
Yet another study found out that transcription co-activator PGC-1α is a vital factor in stimulating mitochondrial biogenesis and OXPHOS, enhancing the cancer cells’ metastatic, migratory and invasive capability (32, 33). However, it has also been observed that inhibition of PGC-1α leads to a reduction of stemness properties in breast CSCs (32–35). Interestingly, overexpression of MYC genes curbed stemness properties via negatively controlling the expression of PGC-1α. It has been observed in breast tumor cells that this c-Myc oncoprotein has been shown to promote mitochondrial glutaminolysis as a part of its reprogramming of cellular metabolism. Being a global transcriptional regulator, MYC significantly induces changes in mitochondrial morphology and dynamics to promote the biosynthesis of metabolic precursors and cell development. MYC establishes a stereotypic gene expression in human tumors and has an inverse relationship with YAP/TAZ activity to drive clonogenic growth (36). Apart from these epigenetic factors, signaling pathways like Notch, PI3K/Akt, PTEN, NF-ĸB, Wnt/β-catenin, KRAS, and HIFs also influence CSC metabolism (37).
Studies also show that the metabolic profile of epithelial ovarian CSCs is characterized by high glucose intake and elevated PPP and OXPHOS. Elevated OXPHOS and reduced glycolysis activity were also deliberately expressed by high telomerase activity (hTERT-high) CSCs, isolated from lung and ovarian cancer models (38). The CD44+CD117+ cells of the ovary give rise to a substantial amount of mitochondrial reactive oxygen species (ROS) and die when the mitochondrial respiratory chain is blocked (29). However, research shows that CSCs show a significantly lower amount of ROS, reflecting that CSCs have enhanced ROS defenses, contributing to an increased expression of free radical scavenging system compared with their non-tumorigenic cells counterparts (39).
Several other mitochondrial carriers like SLC25A10 and NT5M were seen to be upregulated in varied cancer cell lines. Further analysis has shown that this SLC25A10 upregulation dysregulates a diverse amount of processes such as gluconeogenesis, urea cycle, FAO etc. However, overexpression of NT5M has been seen in maintaining dTTP levels of mitochondria, hence maintaining mitochondrial DNA synthesis and preserving its DNA copy number. Other genes associated with dTTP syntheses like RRM2, TYMS, and TK1 have also been seen to be overexpressed (as high as 20-folds) in cancer cell lines in vivo. This elevated dTTP pool plays a vital role in providing genetic stability, counteracting the imbalances occurring in the mitochondria thus protecting the mitochondrial genome (40).
Apart from generating ATP molecules for cancer cells, mitochondria communicate with the cytosol, release cytochrome C to commence cell death, and release ROS and other metabolites (41–43). They also appear to be responsible for regulating the stemness of CSCs irrespective of the metabolic phenotype of the CSC (27, 30, 39). Based on the above observations, it can be deduced that mitochondrial health plays a pivotal role in the maintenance of CSCs.
CSCs also happen to rely on metabolic phenotypes, other than glycolysis and OXPHOS to sustain self-renewal, maintain stemness features and avoid anoikis. As mentioned above, CSCs from hepatocellular carcinoma cells rely on FAO as one of the metabolic phenotypes (31). Pasto et al. also stated an overexpression of genes associated with PPP and FAO in ovarian CD44+CD117+ cells (29). Promoters of FAO genes like Acads (Acyl CoA dehydrogenase short chains) and ECHS1 (enoyl-CoA hydratase short chain 1) have been seen to bind with NANOG and trigger lipid biosynthesis to maintain stemness in CSCs (44). Upregulation of CD36 also plays a vital role in fatty acid import, thus promoting fatty acid synthesis. Recent advances in proteomics have indicated that fatty acid metabolisms play an essential role in redox homeostasis and determining the metabolic fate of CSCs. Mitochondrial FAO also plays a pivotal role in the epigenetic regulation of gene transcriptional factors. Key enzymes of lipogenesis like SREBP1 (45–47), FASN (48), CPT1 (49) have been overexpressed in many forms of human cancers. Upon activation of EGFR signaling, the interaction between SREBP1 and PKM2 potentiates SREBP genes to turn active, triggering lipid biosynthesis (44).
In addition, metabolic rewiring between PPP and glycolysis has also been noticed in glioblastoma CSCs. PPP enzymes were overexpressed in acute oxygenation regions and under-expressed in the hypoxia-mediated areas, unlike glycolysis-based enzymes, where the reverse occurs (50). Intriguingly, glutamine has been identified as an EMT-associated metabolite, demonstrating its role in CSCs from several tumors (26, 51). Recent studies have revealed that the c-Myc gene and Dlx2/GLS1/Glutamine axis enhances glutaminase expression (the enzyme responsible for catalyzing glutamine to glutamate), thereby suggesting the oncogenic signature of glutamine metabolism (52, 53). The glutamine uptake in CSCs has also been associated with upregulation of alanine, serine, cysteine-referring transporter 2 (ASCT2). Glutamate oxaloacetate transaminases (GOT1 and GOT2), enzymes that are responsible for converting glutamate-derived aspartate to oxaloacetate, also show an elevated expression in CSCs (54).
Epigenetic regulation governs a cell’s fate, followed by chromatin remodeling, leading to a normal somatic cell becoming its stem cell counterpart. A similar change in the epigenetic landscape in tumor-initiating cells can give rise to potential CSCs. Epigenetic modulation usually occurs via three mechanisms, DNA methylation, histone modification, and chromatin remodeling; however, several other methods have been identified in the recently (Figure 1). However, it has also been noticed that aberrations in these epigenetic/genetic factors and several cellular signaling pathways, demonstrating that these pathways also choreograph the metabostemness (as coined by Menendez), thus manipulating the metabolic programming of CSCs intrinsically. As further explained by Waddington’s buffering and canalization theory, metabostemness is driven by basically 2 methods; the first including the modification and methylation of DNA and histones, accounting to the reprogramming of metabolism and the second, comprising of the oncometabolite which decipher their presence in the form of chromatin modeling. This reprogramming also encompasses the impairment of the hallmark tumour microenvironment features thus remodeling both the epigenetic landscape and the dynamic metabotypes of CSCs (55). This epigenetic mediated switching of metabolic phenotypes also intervenes with the energy blockades enunciated during CSC treatment protocols, thereby promoting the flexible metabolic bonafide of CSCs and hence expanding their resilience of stemness. The further section describes the various reports of how epigenetic codes influenced the metabolic fates of CSCs and potentiated a positive feedback loop for signaling pathways in CSCs.
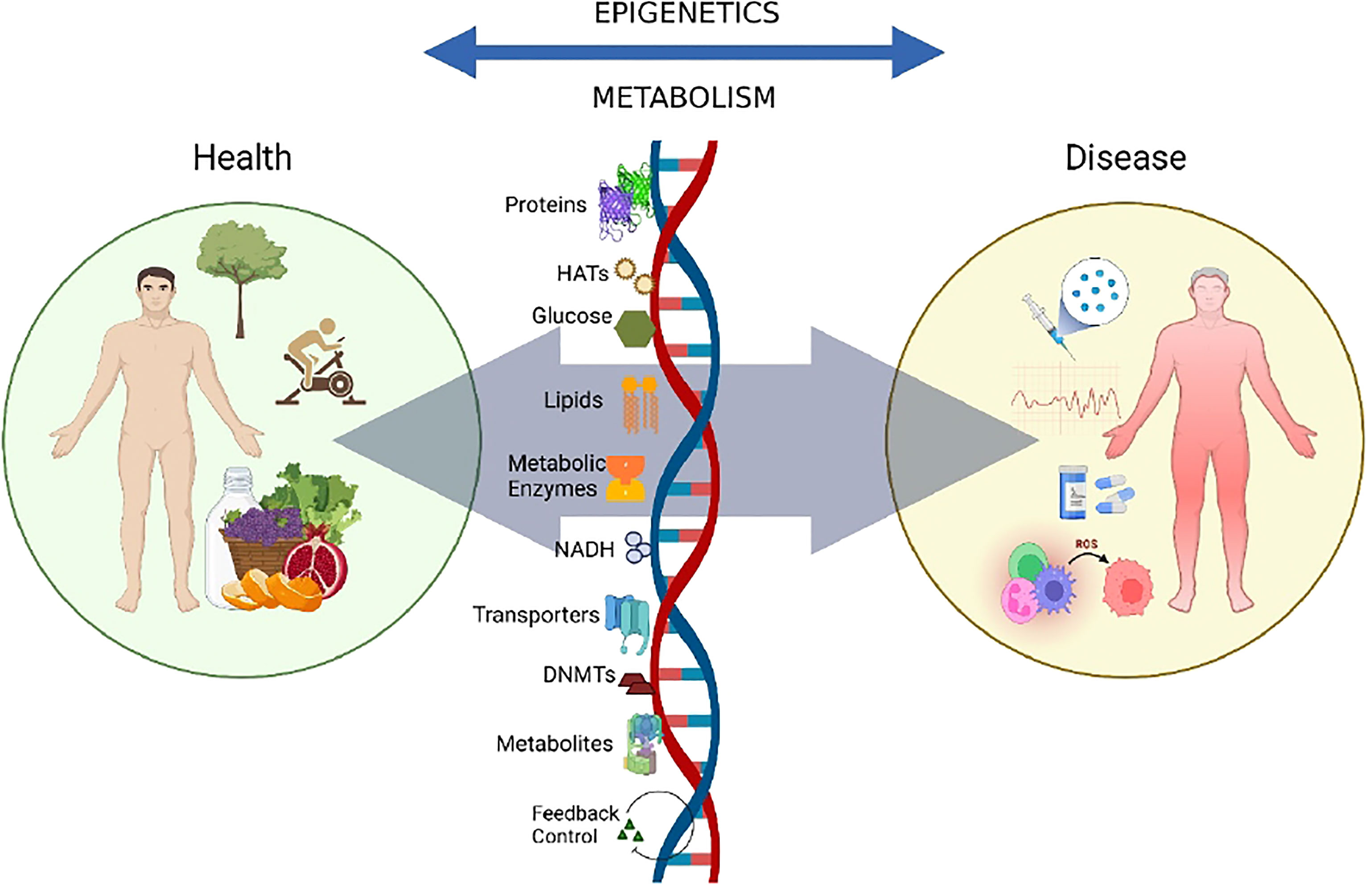
Figure 1 Crosstalk between epigenetics and metabolism.The crosstalk between epigenetics and metabolism is implicated in a variety of contexts related to disease progression. The availability of modifiers like metabolites, co-factors, chromatin-modifying enzymes, and other environmental factors like nutrition, exercise, and the gut microbiome, modulate the dynamics of the genome, thus contributing towards metabolic and epigenetic control and disease progression.
A complex array of metabolic pathways generating different metabolites not only carried the flux of matter but also bioactive molecules that can influence other metabolic pathways or signaling cascades (Figure 2).
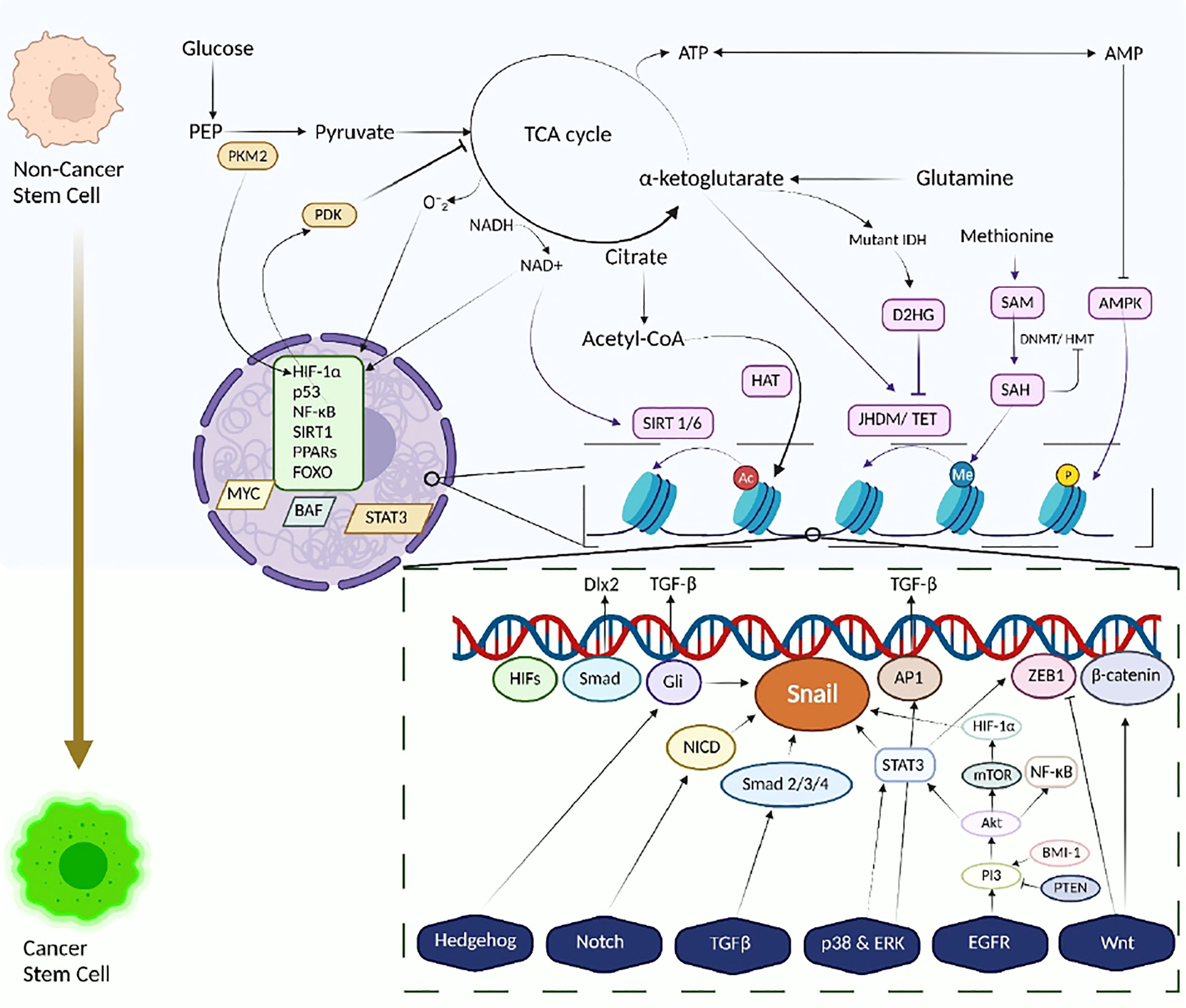
Figure 2 The metabolism-epigenetics axis of Cancer Stem Cells. Transformation of Stem cell to Cancer Stem cells along with metabolic reprogramming and Tumour metabolome modulates and links energy-generating biochemical reactions with several epigenetic pathways, thus integrating metabolism and a variety of signaling pathways with epigenetic modifications, histone changes. Signaling pathways involving HIF-1α and p53 dysregulate glucose and glutamine metabolism, contributing to enhanced production of acetyl Co-A and α-ketoglutarate. This enhanced production influences HAT activity, hence increasing the acetylation profile of cancer cells. A similar response is also observed in NADH/ NAD+ activity wherein the activity of Sirtuins changes. Other metabolites like D2HG and SAM, also play a vital role in epigenetic modifications, eliciting and inhibiting HMT/ DNMT activities. Energetic stress is also observed due to activation of AMPK leading to histone phosphorylation. The dysregulation of the metabolic homeostasis, in accordance with aberrant signaling pathways or mutations and epigenetic modifications, reprograms the stemness and pluripotency of the normal/ cancer cells towards cancer stem cells. Cancer stem cells exploit these altered metabolic pathways for their benefit and survival.
The Wnt signaling cascade is a mysterious complex encompassing 19 Wnt ligands and more than 15 receptors orchestrating many developmental processes like regulating cell homeostasis and maintaining adult stem cells in their pluripotent state (56). It encompasses three signaling pathways, canonical (mediated through β-catenin-T cell-specific transcription factor), non-canonical (independent of a β-catenin-T cell-specific transcription factor), and non-canonical Wnt-calcium pathway (regulates intracellular calcium levels) (57). The canonical pathway activates when Wnt ligands bind to Frizzled receptors, a G-protein-coupled receptor family, and a low-density lipoprotein related protein (LRP 5 and 6) on a neighboring cell (58). After the Wnt signaling gets activated, the β-catenin drives cellular responses via the transactivation of target genes and LEF/TCF transcription factor (59).
Aberrant activation of the Wnt/β-catenin pathway has also been linked with DNA methylation through subsequent silencing of various Wnt inhibitors like WIF-1, AXIN2, SFRP-1 and DKK1 in breast and CRCs (60–62). Methylation of Wnt negative regulators like DKK3 and NKD1 has also been correlated with gastric cancers (63). Apart from methylations, dysregulation of the pathway is also mediated by several histone modifications. Recruitment of SIRT1, EZH2 and PCR2 and decreased acetylation of H3K16 and increased H3K27 trimethylation has been eminent with deregulation of several Wnt antagonists (64). Wang et al. demonstrated the relevance of lncRNAs as epigenetic modulators of the Wnt signaling pathway, when they found lncRNAs of lncTCF7 induced TCF7 expression by recruiting SWI/SNF chromatin remodeling in liver CSCs, thus promoting self-renewal and tumorigenesis in the liver (65).
Experimental evidence suggests that Wnt signaling is activated in several malignancies, including CRC, breast, leukemia, lymphoma, medulloblastoma, and hepatocellular (66). In addition, Wnt activation has been seen in non-melanoma cutaneous tumor cells of murine models and potentially in humans (67). It also exerts influence over the glycolysis by upregulating Myc, MCTs and PDK. Distal-less homeobox -2 (Dlx-2), a protein-coding gene, also has been seen to trigger the glycolytic switch stimulated by Wnt/TGF-β and represses mitochondrial biogenesis by upregulating expression of snail and inhibiting mitochondrial complex IV, i.e. COX. Glutaminase-1, a predominant molecule expressed during glutamine metabolism has been seen to be induced by Wnt/TGF-β in a Dlx-2 dependent pathway (68).
The Hedgehog (Hh) signaling pathway is essential for developing embryos, tissue homeostasis, and EMT transition (69). The three HH homologs of the HH pathway – Sonic, Desert, and Indian mediate the Patched receptor’s (PTCH1) inhibitory effect on Smoothened (SMO), thereby allowing Gli transcription proteins to get isolated. When activated, Gli proteins (Gli-1 and Gli-2) are released to facilitate transcription of target genes, thereby mediating the HH signaling transduction. It has been observed that the HH pathway gets stimulated through up-regulation of Gli by several intracellular signals, which are conveyed by several other epigenetic factors and signaling pathways like KRAS-MAPK/RAF/MEK, mTOR, TGF-β, Snf5, and Phosphoinositide 3-kinase/Akt pathway (70–74). In addition, histone modifications and DNA methylations also play a major role in regulating the HH pathway. Enhanced expressions of the Shh ligands have been seen in breast and gastric cancers due to hypomethylation of the Shh promoter (75, 76). Similar activity with NF-ĸB led to the activation and transcription of Shh, resulting in the upregulation of the ligand, thus promoting self-renewal and invasiveness of varied cancers (77). The Gli proteins have also been associated with HDAC1 in virtue of a positive autoregulatory loop, thus dysregulating neural progenitor and tumor cells (78).
However, deregulations in this HH pathway implicate tumorigenesis and tumor growth. Several studies have suggested the role of HH signaling in various CSCs, including basal cell carcinoma (BCC), medulloblastoma, rhabdomyosarcoma, glioblastoma, colon cancer, and chronic myeloid leukemia (CMC) (79–81). In addition, HH signaling was also seen in glioma spheres of nude mice and humans (82). Concerning metabolism, the HH pathway has been associated with lncRNAs like breast cancer anti-estrogen resistance 4 (BCAR4), a downstream target of YAP. It enhances the expression of YAP-dependent glycolysis activators, viz. HK2 and PFKFB3, thus making YAP/BCAR4 signaling axis a potential target for breast cancer treatment (83). PKM2 also has been seen to be associated with transcriptional repressor TGF- β-induced factor homeobox 2 (TGIF2) to repress the transcription of E-cadherin by inducing HDAC3.
Like Wnt and HH pathway, Notch signaling occurs between neighboring cells via transmembrane protein ligands (Delta 1/3/4 and Jagged 1/2) (84, 85). The interaction initiates by binding the ligands, which cleaves the Notch intracellular- domain (NICD), by -secretase. NICD then translocates into the nucleus, interacts with recombination signal binding protein for immunoglobin kappa J region (RBPJ-ĸ) to finally activate transcriptional factors, like MYC and HES1 (86, 87). It is regarded as highly conserved, it helps regulate cell fate specification, stem cell differentiation, stem cell renewal and triggers multiple aspects of cancers. Aberrations in the Notch signaling pathway have been shown to regulate tumor progressions in leukemia, breast, colon (CRC), pancreas, glioblastoma, lungs, and multiple myeloma. Notch has also been seen to synergize with HIF-1α and induce metastasis, and also maintaining stemness characteristics. Neural and breast CSCs show overexpression of the notch genes, leading to increased formation of progenitor cells. On the contrary, several studies also support the role of Notch signaling as a tumor suppressor. They have been seen to impair epidermal differentiation and skin barrier integrity in skin cancers. In another study, Notch signaling has been shown to induce cell death by increasing p53 activity in cancers like HCC, cervical cancers, and Ewing’s sarcoma (88). Overexpression of Jagged2 was also associated with HDACs leading to subsequent activation of Notch signalling in multiple myeloma (89). Jin et al. also demonstrated the interaction of serine-threonine kinase receptor-associated protein (STRAP) with EZH2 and PRC2 complex, inhibiting histone methylation of H3K27 on HES1 and HES5 promoters, further leading to regulating stemness potential in CSCs (90).
In addition to modulating the epigenetic landscapes of CSCs, the role of Notch signaling has also been seen in regulating metabolites and metabolic enzymes both directly and indirectly. Notch signaling stimulated transcriptional factors like HIF-1α, MYC, p53, and many others show active participation in conferring advantages in virtue of metabolic pathways for survival and proliferation of both cancer bulk cells and CSCs. For instance, HIF-1α acts as a pivotal regulator of glycolytic enzymes, including GLUT1, GLUT3, HK, lactate dehydrogenase, and MCT, thereby promoting glycolysis (23, 91, 92). It also downregulates OXPHOS and oxygen intake by inducing PDK, thus destabilizing the TCA cycle (23, 92). HIF-1/PKM2 positive feedback loop also enhances the stimulation of glycolysis mediating proteins, thus enhancing the glycolytic switch of oncogenic cells (93–95). On the contrary, MYC has been seen to increase mitochondrial OXPHOS and ROS, enhancing resistance towards chemotherapy in TNBC (96). Intriguingly it has also been documented that MYC/PGC-1α balance in pancreatic cancers governs metabolophenotype and CSC plasticity (30). Besides, p53 also regulates metabolic changes by inducing several other transcriptional factors and metabolic enzymes. Metabolic enzymes like G6PD, GLUT1, GLUT3, GLUT4, GLS2, ME1, ME2 and PANK1 have been seen to be affected with the expression of p53 (68). It upregulates the expression of SCO2 and downregulates TIGAR, thereby enhancing mitochondrial biogenesis and suppressing glycolysis respectively. Stipulation of mitochondrial respiration by p53 is further influenced by GLS2 where the amino acid metabolism decreases ROS levels thus protecting cell DNA damage. Aerobic glycolysis is inhibited by the expression of TIGAR and repression of glucose transporters 1, 3, and 4, which altogether decrease glucose intake. It is also associated with AMPK and PTEN-mechanisms, further inhibiting the Warburg effect and enhancing gluconeogenesis. Several shreds of evidence have also stated the inhibition of NADPH and glutamine metabolism, lipogenesis and PPP, due to p53 (22, 68).
DNA methylation refers to the process of methylation of cytosine residues of the CpG dinucleotides present in mammalian DNA, using DNMTs – DNMT1, DNMT3A, DNMT 3B. DNMT1 is recognized as a maintenance methyltransferase, which ensures the accuracy of replication of inherited epigenetic patterns. DNMT3A and DNMT 3B are required for de novo DNA methylation at unmethylated CpG sites (97). Though DNMTs have distinct roles, they ally to safeguard DNA methylation around the genome loci, viz. CpG islands (CGI) loci are obstinate to de novo DNMT3A2 action, which results in DNA methylation (98). However, these DNMT-dependent workouts play a vital role in the regulation of maintaining cellular identity, stemness of a cell, and regulation of CSCs (99). Studies have demonstrated an association between PcG (Polycomb group) targeted methylation in cytogenetically-normal AML (100). A comparative study showed that hypomethylation was reported in a total of 68 differently methylated regions of breast CSCs, like that of non-breast CSCs (101). DMNT3A-knockout derived myeloid carcinomas led to the expansion of preleukemia stem cells, indicating that methylation aberrations induce the growth of CSCs (102). Notably, several pioneer epigenetic changes, including gene silencing and tumor progression, occur due to DNA demethylation. It is the process of removing methyl groups from cytosines of DNAs through the conversion of 5-methyl cytosine to thymine by ten-eleven translocation proteins (TET) and activation-induced deaminase (AID). TET enzymes, TET1 and TET2, promote gene expression of pluripotency genes Oct 4 and facilitate the formation of iPSCs (103).
Oncogenic dysfunction due to DNA methylation is also induced by endogenous metabolite levels and other environmental stress and vice versa (104). Recruitment of DNMTs at the FBP1 promoter was seen to be critical for the expression of FBP1 in BLBCs thus promoting glycolysis (13). Epigenetic inactivation of FBP1 has also observed in HCC, gastric, and colon cancers due to promoter hypermethylation (105, 106). Goel et al. also previewed the significance of methylation-demethylation in the expression of HK2 in tumour infected hepatocytes (107). The demethylation prospective of the Jumonji-C domain-containing Histone demethylases (JHDMs) and TETs also have been seen to be associated with α-ketoglutarate and succinate which further control the TCA cycle (22, 108). Yet another linkage of metabolism with methyl transfer is the usage of S-adenosylmethionine (SAM) as substrates. SAM is spawned from the coupled cycles of methionine and folate, often called one-carbon metabolism. This one-carbon metabolism involves the synthesis of micronutrients like zinc, methionine and the family members of vitamin B and hence is profoundly found active in cancers (109). However, the transfer of the methyl group from SAM to the substrate forms S-adenosyl homocysteine (SAH), which further builds up to the level of inhibiting DNMT and HMT (Histone methyltransferases) activity.
Histone modifications refer to the alteration of gene expressions via acetylation, methylation, ubiquitylation, or phosphorylation (110). These alterations in histone landscapes help establish cellular identity, signifying that they may be involved in acquiring stemness within the cells. In particular, repressive complexes Polycomb group complex 1 and 2 (PRC1 and PRC2) mediated overexpression of EZH2 have been shown to induce tumorigenesis and show relevant characteristics of CSCs in cancers like prostate, breast, PDACs, and different types of lymphomas (111). Several studies have demonstrated that BMI-1 reinforces bivalent histone domains in multipotent progenitor cells and is implicated in the maintenance and invasiveness of cancers like leukemia, GBM, human nasopharyngeal cancer, breast cancer, and endometrial cancer (112–115). Similarly, Brahma-related gene 1 (BRG1) has also shown indispensable aberrations in the Wnt pathway leading to intestinal adenoma, AML, non-small cell lung cancer, breast cancer, and pancreatic cancer (116–120).
Likewise, histone deacetylase (HDAC) 7 also promoted tumorigenesis of the lungs by inhibiting Stat3 (121). Higher levels of HDACs have been associated with advanced tumorigenesis and poor prognosis (122). HDAC7 protein level was overexpressed in human pancreatic cancers, breast CSCs, and CD-10 positive acute lymphoblastic leukemia (123–125). It was determined by Chang et al. that upregulation of HDAC7 was involved in regulating cell proliferation, cell death, differentiation, and metastatic properties, signifying the invasiveness of the protein. It suppresses transcriptional factor MMP10 by mediating myocyte enhancer factor-2 (MEF2), suggesting that HDAC7 regulates vascular permeability (126). HDAC1/7 was associated with CSC-suppressor miR-34a (127), a potential functional signature for the prognosis of glioma and osteosarcoma (128, 129).
Theoretically, active chromatin at promoters can change cellular identity, i.e., de-differentiation and, most importantly, switching to CSCs. A direct association between CSC generation and bivalency was shown in CD44+ breast cancers (130), where the response of ZEB1 with TGFβ led to an increase in transcription of ZEB1, which further led to switching of non-CSCs to CSCs. Expression of ZEB1 and ZEB2 have also been associated with glycolytic phosphoglucose isomerase (PGI) in several breast cancer cells (131). Similarly, other bivalent genes are involved in regulating embryonic stem cell differentiation. Polycomb-like 3 (Pcl3) mediates PRC2 in binding to target genes and promotes the expression of pluripotency in ESCs. Pcl3 was also found to be elevated in multiple primary tumor samples viz. cancers of the colon, epithelial, skin, uterus, cervix, and liver. Overexpression of such factors in such cancers can be potentially used for diagnostic purposes (132, 133).
Concerning metabolism, several shreds of evidence suggest the significance of histone modifications (acetylations, methylations, ubiquitylation etc.) in deciding the oncometabolite fate and vice versa. Oncometabolites like 2-hydroxyglutarate (2-HG), fumarate, and succinate accrete in virtue of the mutations in isocitrate dehydrogenases (IDHs), fumarate hydratase, and succinate dehydrogenases respectively, and further induce alterations in the histones and DNAs, which thereby illustrate a metabolic pseudohypoxia, reprogramming the metabolic fate (134, 135). These mutations in IDHs (IDH1 and IDH2) promote tumorigenesis by accumulating a rare oncometabolite D-2-hydroxyglutarate (D2HG). This D2HG further occupies the α-ketoglutarate binding sites, thus inhibiting JHDMs, a major family of enzymes responsible for the regulation of histone methylation. Some reports have also shown the intervention of D2HG with the stability of transcriptional factors HIF1 and HIF2, which further target the hypoxic metabolic pathway, however, the accuracy of the results remains contentious. Mutations in IDH enzymes have also been seen to upset the NADP/NADPH ratio and alter the forward-backward reactions of α-ketoglutarate to isocitrate while consuming NADPH, thus altering the metabolic microenvironment of tumors (136, 137).
The basic unit of DNA packaging is the nucleosome, consisting of DNA wrapping eight histone proteins, two each of H2A, H2B, H3, and H4. The presence of nucleosomes usually decides the sensitivity of gene expression in the chromatin, preventing access to the transcription machinery. Hence, nucleosome positioning also plays a vital role in the pluripotency of stem cells and the formation of CSCs (138). Four families of ATP-dependent complexes achieve positioning of a nucleosome, namely switch/sucrose non-fermenting (SWI2/SNF2), imitation switch (ISWI), inositol requiring 80 (INO80), and Mi-2/CHD (139, 140). Aberrant mutations in these complexes, however, have been linked with cancer. Mutations in SWI/SNF and hSNF5/INII have shown a distinct role in the aggression of tumors like malignant rhabdoid tumors, ovarian clear cell carcinoma, gastric cancer, pancreatic cancer, multiple myeloma, breast cancer, GBM, HCC etc (141–160). Likewise, research carried out by Lee et al. showed that INO80 subunits were frequently present in high copy numbers in the colon, suggesting that they promote colon cancer (161). Zhang et al., 2017 defined the role of INO80 in the enhancement of non-small cell lung cancer and revealed a potential therapeutic strategy for inhibiting cancer transcription networks using INO80 (162). Similar results were obtained by Zhou et al., where INO80 governs tumor growth in melanoma (163). It was also found that INO80/NANOG was overexpressed in cervical cancers, suggesting that INO80 can act as a potential therapeutic method for treating cancers (164).
Reprogramming of metabolic cofactors also possesses significant alterations in chromatin remodeling from the aspect of CSCs. Metabolic cofactors like NAD, FAD, PARP, SAM, LSD1, Coenzyme A have shown transformational changes in chromatin structure (54, 165). PGC-1α has been known to be associated with SIRT1 and AMPK to perturb the metabolic stature of cells. In addition, this SIRT1 has been seen to facilitate NAD+ dependent deacetylation of target molecules and orchestrate metabolic agitations by stimulating transcriptional factors like PPARs, p53, FOXO etc. NAD+/NADH ratio also helps in maintaining stemness features, mitochondrial quality, and active aerobic glycolysis, thus highlighting the significance of SIRT1 as a metabolic game-changer (54). Other factors like BAF/PBAF also regulate the energy metabolism pathways in cancer, thereby being a critical regulator of metabolic homeostasis (166).
miRNAs are small non-coding RNAs that regulate post-transcriptional gene expression by destabilizing the mRNA and translational silencing. These miRNAs have been seen playing a crucial role in maintaining stemness in both normal stem cells and CSCs. Deregulated expressions of miRNAs have been seen to play a critical role in tumor initiation and prognosis. Though genetic modifications in these miRNAs are crucial for initiating tumor suppressor genes, several theories have hypothesized the underlying oncogenic potential of miRNAs associated with the maintenance, growth, and function of CSCs (167). Some signal transduction pathways, such as Wnt/β-catenin, Notch, HH, JAK/STAT, NF-ĸB etc., are often distorted due to the dysregulated expression of miRNAs, further orchestrating carcinogenesis and regulation of CSCs. However, several miRNAs such as miR-134, mirR-296, and miR-470 suppressed the expression of pluripotency maintenance factors Oct4, Sox2 and NANOG. Similarly, let-7 and miR-200 inhibited the expression of Lin 28 and c-Myc, respectively, which were notable self-renewing factors. Further, recent studies about the regulatory functions of miRNAs on CSCs are summarized below.
The aberrant overexpression of miR-1301-3p promotes prostate CSC by inhibiting SFRP1 and GSK3β (168). Further, miR-424 and mir-7 were found upregulated in prostate tumors and featured enhanced cell migration and invasion, increased stemness characteristics, and formation of prostatospheres (169, 170). However, several miRNAs like miR-34a (via CD44), miR-7 (via KLF4/PI3K/Akt/p21 pathway), and miR-320 (via Wnt/β-catenin signaling) inhibit prostate CSC growth reduce stemness and metastatic features. Class III HDACs like SIRT1, is also overexpressed in prostate tumors, where they inactivate other tumor suppressors like HIC1 and activate tumor-promoting genes like p53, N-Myc, cortactin etc. Other histone marks like H3K27 methylation, modification of H4K16, H3K4, H3K56, H3K9, K14, H4K5, H4K12, H3K18, substrates of SIRT1, LSD1, H3R2, H3R42 and EZH2 and mutations in KAT7 and KAT2A are also noted in prostate cancers (170–173).
Overexpression of miR-6875-3p, miR-106b-5p (via PTEN/PI3K/Akt pathway), miR-55 (via NF-ĸB), miR-191 (via HIF-2α), miR217 (via DKK1), miR-500a-3p (via negative regulators of SOCS2/SOCS4/PTPN11 of STAT3) and miR-1246 (via Wnt/β-catenin signaling) in HCCs promotes tumorigenesis, stemness and metastasis. Overexpression of miR-137 also induces drug resistance by degrading ANT2 in HCCs. Enhanced stemness markers such as CD90, EpCAM, and Oct4 due to exposure to arsenic also led to the spheroid formation in liver CSCs (174). Negative regulation by miR-612 and CD-133 reduced stemness and metastatic characteristics in HCCs and subsequently inhibited the activity of Sp1 and NANOG (174–183). PAD4 of the partitioning and anchoring domain family, which is known to citrullinate several histones like H2A, H3, H4, and H1R54 was also found to be overexpressed in HCCs, thus contributing to tumorigenesis. Besides, like prostate cancer, modification of H4K16, H3K4, H3K56, substrates of SIRT7 and KMT5A was correlated with HCCs (173, 184, 185).
Breast CSCs show up-regulation of miR-29a (basic fibroblast growth factor-induced), miR-137 (via β3/Wnt pathway), and miR-221/222 (via PTEN signaling), signifying that the listed miRNAs are required for the maintenance of the CSCs. However, unlike other cancers, up-regulation of other miRNAs inhibited cell proliferation and induced apoptosis in triple-negative breast cancer and breast CSCs. MiRNAs like miR-1287-5p, miR-27a, miR-34a, miR-628, miR-142-3p, miR Let-7, and others were found to reduce tumor growth, potentiate the effectiveness of therapeutic therapies and modulate signaling pathways, thereby inhibiting the renewal of CSCs (186–194). In fact, class I HDAC1 like H4K16 and H3K56, HDAC2, class III HDACs like SIRT1, SIRT2, SIRT3 and SIRT7, HDAC5 (p53), HDAC6 (HSP90 and cortactin), HDAC7 and HDAC11 overexpress themselves in several breast carcinomas. Modification levels of H3K9, H3K18, H4K12, H4K16, H3K4, H4K20, and H4R3 also were related to breast tumorigenesis (185). Histone marks like KAT2A, KAT4, KAT7, KAT6A, LSD1, JMJD2B, JMJD2C, p300, SETD2, SMYD3, and substrates of EZH2 and PAD4 were also seen to be mutated in breast cancer (173).
Poor regulation of miRNAs plays a vital role in cellular apoptosis, proliferation, inflammatory response, maintenance of stemness, and metastasis development of the lungs. Associated with self-renewal of lung CSCs and tumor differentiation, micro RNAs like miR-5100, miR-494-3p, miR-19a/19b, and miR-1246 showed upregulation (195–198). Shi et al. showed that miR-34a demonstrated negative regulation of tumor properties in CD44hi lung CSCs and non-small cell lung cancer (NSCLC) (199). Aberrant regulation of miR-218 overexpressed stemness features and IL-6/JAK-STAT3 signaling in ALDH-positive lungs (200). Downregulation of tumor suppressor miR-128 showed tumor differentiation and metastasis by targeting ERK/AKT and p38 signaling pathways (201). Alike HCCs and breast cancer, lung cancers (esp NSCLCs) show upregulated expression of PAD4, compared to their normal or benign hyperplastic tissues (184). Other than that, mutations in KAT2A, KAT2B, KAT4, KAT6A, KAT6B, KAT7, and MYST1are quite prevalent in lung cancers. Though HDAC1 Class I and HDAC 2 display an overexpressed state in lung tumors, lower expression of HDAC9 and HDAC10 are linked to poor prognosis in the same. Histone targets like H3K9, H3K36, and H4K20 have also been seen to be associated with several lines of lung cancer (173).
Several reports revealed the role of miRNAs associated with the maintenance of stemness, cancer invasiveness, and metastasis of pancreatic CSCs and PDAC is quite crucial. Overexpression of miR-30a, -30b, and -30c in CD133+ pancreatic CSCs show high metastatic features with increased mesenchymal phenotype markers (202). Dysregulated expressions of miR-744 are often related to Wnt/β-catenin signaling, one of the main pathways of pancreatic CSCs. Upregulation of miR-744 and downregulation of miR-200 potentiates stemness features and drug resistance of pancreatic CSCs (203, 204). HDAC1 substrates like H4K16, H3K56, and HDAC7 substrates also show an overpowered expression in pancreatic cancers, which further leads to poor survival. Low levels of SIRT7 substrate H3K18ac also display a poor prognosis in PDACs, thus contributing to the tumorigenesis of pancreatic cancers (173).
CSCs are endowed with various biological characteristics, which reflect the heterogeneity of these stem cells. However, to promote and maintain tumorigenesis, many a time, these characteristics also play an interdisciplinary role within each other. The immune system’s role which has been primarily attributed towards eliminating CSCs, plays a dual role in both promoting and eliminating cancer progression, suggesting CSC’s enhanced immunoediting mechanisms. This enhanced immunoediting mechanism is primarily regulated by several metabolic and epigenetic factors which decide the fate of the immune responses, thereby promoting cancer progression. Playing as major hallmarks of immune cell phenotype, both metabolism and epigenetics have displayed several pieces of evidence in reinstating tumorigenesis and metastasis in CSCs. In virtue of these factors, tumor cells induce changes in the immune microenvironment by modulating innate immune cells like macrophages, MDSCs, and Tregs and transforming the environment into an immunosuppressive one. In this part of the review, interactions of CSCs, the oncometabolite, and the epigenetic factors with the immune system will be discussed, as well as we will highlight the therapeutic potential of targeting immune cell interactions to invade CSCs. Possible interactions have been summarized below and in Table 1.
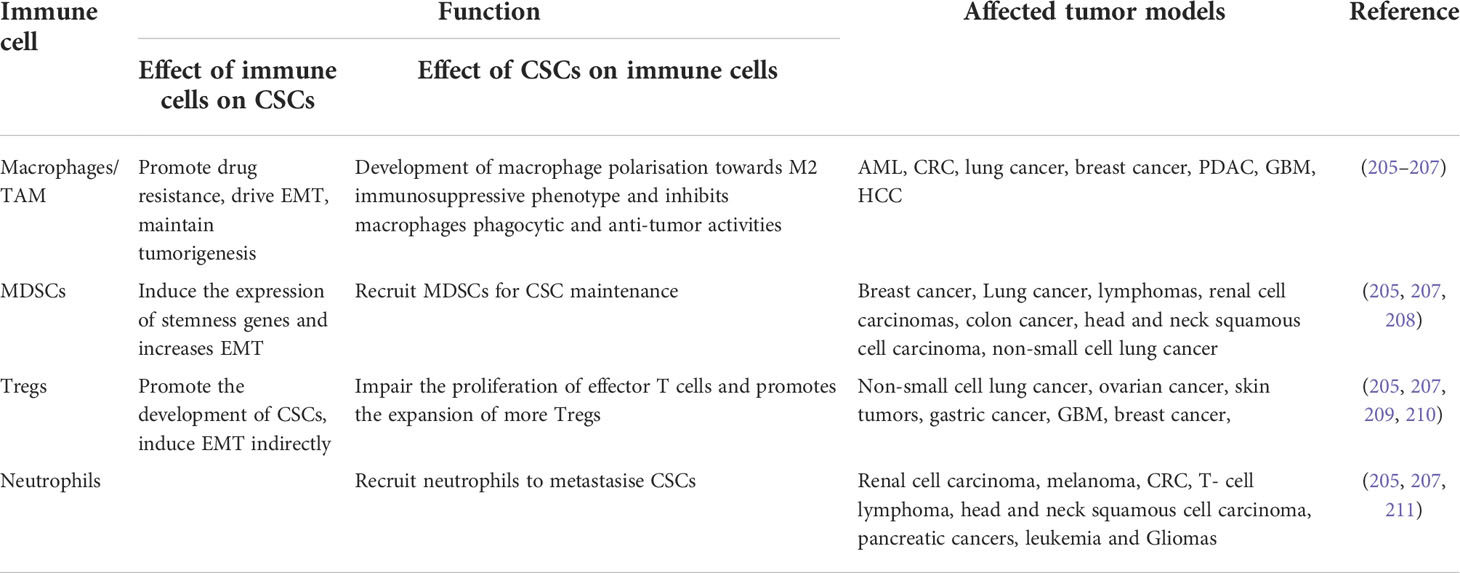
Table 1 A summarized interaction between CSCs and the immune system.
Macrophages are notable leukocytes that have roles in tissue homeostasis and immunity. Depending upon the tumor microenvironment (TME), these macrophages, also referred to as TAMs, exhibit pro (M1 macrophage) and anti-tumor (M2 macrophage) characteristics (212–214). Several studies have reported the significance of TAMs in mediating immune-stimulatory agents and the destruction of tumor bulks. On the contrary, there is also evidence where they have been seen to participate in tumorigenesis, promoting cancer motility and metastasis of CSCs (215–217). The diversity of such functions is governed in response to local signals (chemokines and cytokines). Though TAMs mostly show an M2- like phenotype, a high M1/M2 ratio is tried to maintain so as to suppress cancer entities. These functional changes also are dependent upon the underlying metabolic changes, thus specifying the complex relationships of metabolic and functional reprogramming of macrophages.
In the early stages of tumors, TAMs produce cytokines like milk fat globule epidermal growth factor (MFG-E8) and IL-6, which activate several signaling molecules and pathways in CSCs, like STAT3 and Hedgehog, thereby mediating DNA damage, cancer-related inflammations, and drug resistance (218). They further modulate CSC plasticity by producing factors like TNF-α, TGF-β1, which is supported by other stromal collagen fibers and stemness factors like IL-8 and CXCL12 (8). Hypoxic conditions also favor CSC angiogenesis and metastasis, as hypoxic factors like HIF-1α and HIF-2α promote VEGF-A production, a major proangiogenic cytokine (219).
However, the M1 macrophages try to restrain tumor growth by releasing type 1 pro-inflammatory cytokines. By upregulating IFN-γ, they characterize the secretion of several ROS and nitrogen intermediates, major histocompatibility complex class II molecules, and other immune-stimulatory cytokines such as IL-12, IL-23, CXCL9, and CXCL10 in CSC niches. These, in turn, drive the recruitment of TH1 cells, thereby amplifying a type 1 response. This is further supplemented by a shift in iron homeostasis, which contributes to bacteriostatic effects (220). In relation to metabolism, Liu et al. in their experiment comprehended a similar analysis on tumor-extract stimulated bone-marrow-derived macrophages (TES-TAMs), and found an upregulated aerobic glycolysis. It exhibited a blend of M1/M2 phenotype with upregulation of HK2 and other cytokines like ARG1, IL4Rα, and PLIN2 (221). An upregulation of cytokine production and aerobic glycolysis was also discovered by Arts et al. using immunohistochemistry, showing enhanced expressions of PFKFB3 and PKM2 in TAMs of thyroid carcinomas (222). It was also further supported by Penny et al. when they inhibited HK2 with 2DG and showed disruption in prometastatic phenotype in PDAC cell lines (223). REDD1/mTOR axis was also seen to be upregulated in hypoxic TAMs leading to suppression of glycolysis and increased angiogenic responses (224), thus potentiating the fact that the general preference of TAMs is aerobic glycolysis and further metastasis is promoted and regulated by the expression of HIF-1α/mTOR. This regulated expression also further induces an elevated ARG1 and VEGF expression, which also plays a pivotal role in tumor progression using the ARG1-dependent polyamine synthesis pathway, thus enhancing the polarization of the macrophages towards an M2-like phenotype. Several shreds of evidence have also stated the significance of FA metabolism in influencing the tumoricidal and immunogenic functions of TAMs, viz. overexpressed prostaglandin production, activation of PPARβ/γ via IL-4/STAT6 pathway, fatty acid accumulation, and redistribution (225–228).
Concerning the epigenetic modifications relevant to TAMs, several reports have reported the switch of the M2 phenotype to M1 due to these changes, thereby re-polarizing an anti-tumor activity. DNMT3B, TET2, H3K4 methyltransferase, SMYD3, HDAC9 are some of the speculated factors known to be associated with M2 polarization. In fact, this TAM polarization is also associated with TLR4 ligands and LPS, which result in a shift towards glycolysis and impaired mitochondrial biogenesis. The glycolytic shift is accompanied by an increase in lactate which inhibits class II-HDACs. As mentioned above, TET and JHDM, the two crucial oncometabolite also serve as vital metabolic switches for M1/M2 macrophages. STAT6/PPARγ/PGC-1α and the mTORC2-IRF4 signaling axes also play a pivotal role in the regulation of OXPHOS and mitochondrial biogenesis, thus modulating the polarization of TAMs (229). Besides, dysregulation of NF-ĸB signaling in ovarian cancers, overexpression of miR155 in Lewis lung carcinomas, and CSF-1R inhibition in gliomas have shown robust regression of tumor bulks, thereby potentiating epigenetics as a therapeutic opportunity in treating cancers (230–232).
MDSCs are a subset of immature granulocytes which exert an immune-suppressive phenotype in the CSC niche. They have been seen to suppress NK cells, natural killer T cells, and other T-cell responses, thereby modulating the innate immune system and releasing reactive nitrogen and oxygen species (233–235). Besides, CSCs produce VEGF and SDF-1 to recruit MDSCs into the CSC niche. Tumor-induced chemokines like CCL2, CCL15, CXCL5, and CXCL12 also mediate MDSC recruitment, which further suppresses the immune function, thereby facilitating CSC maintenance (205).
Several evidence has been reported indicating that MDSCs induce stemness genes and drive the EMT in CSCs. They have also been seen to express a dynamic metabolic flux, with increased carbon metabolism, i.e. enhanced glycolysis, PPP and TCA cycle. Owing to the high glycolysis and glutaminolysis rates, several pathways like the PI3K/AKT/mTOR, LXR, PPARγ, AMPK, STAT, PGE2 pathways support cellular proliferation for the synthesis of glucose, fatty acids, proteins, and nucleic acids. Under hypoxic conditions, the mTOR pathway activates HIF-1α (236, 237), which further stimulates enhanced glucose, lactate transporters, glycolytic enzymes, and a repressed OXPHOS, thus mediating the switch from OXPHOS to glycolysis in MDSCs. Besides, carbon metabolism, MDSCs also show an upregulated expression of FA transport proteins, FA translocase CD36, CPT1 and CPT3, thus suggesting FAO as one of the generic fuels for MDSCs (238, 239). G-MDSCs in certain tumors express higher levels of Arg1, whereas M-MDSCs express an overexpressed iNOS to catabolize Arg1 to inhibit MDSC functions, thus associating amino acids with MDSC metabolism (240).
Epigenetic modifications also have been seen to result in a heritable regulation of MDSCs leading to the reframing of the tumor microenvironment of several cancer species. In addition, to support tumor growth and progression, they have been seen to stimulate factors like VEGF, β-EGF, MMP9, and other angiogenesis factors (241–243). Higher expressions of Arg1 and STAT3 have led to the accumulation of MDSCs in cancer models, thus suggesting their tumor-suppressive functions. In a study by Sahakian et al., HDAC11 was noted to be a regulator of MDSC maturation, thus indicating its role as a gatekeeper in myeloid differentiation (244). Concerning microRNA interference, miR-210 enhances the expression of CXCL12, IL-16, Arg-1 in MDSCs, thereby establishing a link between microRNAs and MDSC mediated immune suppression. The expression of miR-101 in ovarian cancer cells and prostaglandin E2 in cervical cancer cells is mediated by MDSCs, thus demonstrating their role in tumorigenesis and metastasis (245, 246). Other microRNAs like miR-9, miR-690, miR-155, miR-21, miR-223, miR-34a, miR-146a, miR-424, miR-181b, miR-17-5p, and miR-20a has been associated with MDSC development and differentiation, and if regulated can promote overall tumor immunity (244). In addition, MDSCs have been seen to foster STAT3 signaling in breast CSCs and PDACs by inducing stemness factors like IL-6 and nitric oxide (NO), which subsequently increase ALDH1+ CSCs (247, 248).
T regulatory cells (Tregs) orchestrate the function and activation of other immune cells and play a crucial role in controlling T cell-mediated autoimmunity (249). In regard to cancer, CSCs alter the immune landscape and promote the expansion of immunosuppressive and pro-tumorigenic Tregs. Several studies have shown that CSCs from glioblastoma, breast cancer, prostate cancer, mesothelioma cell lines, and head and neck squamous cell carcinoma impairs the production of effector T cells and stimulates Tregs production (250–253). CSCs also produce TGF-β pathway members, which act in concert with the regulation of Tregs. With increased tumor formation, this TGF-β inhibits the accumulation of CD8+ T cells and secrete inflammatory cytokines such as IFN-γ, TNF, IL-6, and CCL2, thereby maintaining stemness in CSCs (254).
On the contrary, Tregs also produce TGF-β, which promotes invasion, angiogenesis and drives the epithelial-to-mesenchymal transition (EMT) process (255). Furthermore, Tregs also influence the expression of programmed cell death 1 on CD8+ T cells and enhance VEGF levels in the presence of hypoxia, which promotes angiogenesis, thereby affecting the stemness and progression of CSCs (256, 257). In addition, recent studies have shown that FOXP3+ Tregs induce IL-17, which under a hypoxic condition, potentiates CSC development in CRCs. Further results showed that IL-17 acts in a STAT3 dependent manner (258).
Expression of FOXP3 is also mediated by notable metabolic factors like PPARγ, HIF-1α, and mTOR and PD-L1, which are maintained by the balance between glycolysis and FAO in Tregs, thus caveating the control of metabolism over differentiation of Tregs (228, 259–261). Depending upon the tumor microenvironment, Tregs avail alternate substrates for their metabolic sustenance. In steady states, they are known to display an enhanced glycolytic rate and lipid biosynthesis with the hyperactivation of mTOR. During proliferation, they increase the expression of GLUT1, activate mTORC1, and display a comparatively higher glycolytic activity, thereby suggesting that Tregs are the most enthusiastically proliferating compartment in vivo (262–265). However, deletion of PGC-1α and SIRT3 abrogated Tregs suppressive function both in vivo and in vitro, hence suggesting the role of OXPHOS in the proliferation of Tregs (266, 267). Yet, another interesting theory also suggests the engagement of epigenetics in the differentiation program of Tregs. The role of Enolase/MYC Binding Protein-1α also has been associated with the expression of glycolysis and FOXP3 splicing variants (268). Suppression of EZH2 in the tumor microenvironment also has shown sustained generation of Tregs from naïve precursors, thus potentiating tumor growth (269). Evidence has also stated that enhanced expression of tryptophan metabolizing enzyme IDO also leads to increased Treg cell infiltration in several cancers like CRC, HCC, and cervical cancers, and also has been seen to increase the frequency of metastasis in these cancer lines (267).
Neutrophils account for the maximum amount of leukocytes in the human blood. Thus, apart from eliminating pathogens and maintaining a circuit of adaptive immunity, neutrophils constitute a vital portion of the immune cells invading the CSC niche. Several studies have demonstrated the role of TANs in cancer progression and metastasis. However, similar to TAMs, TANs also show a cytokine-driven polarisation wherein they can be modulated into a pro- (N2) or an anti- (N1) tumorigenic phenotype.
The extent of neutrophil infiltration was suggested by Hira et al. when they examined glioma stem-like cells in high-grade glioma patients (270). A similar prognosis was also obtained in cases of aggressive forms of renal cell carcinoma, melanoma, CRC, head and neck cancer, pancreatic neoplasia, and micro-papillary carcinomas (271, 272). TANs have also been shown to facilitate metastasis by suppressing NK-cell mediated clearance of tumor cells and recruiting TAMs at inflammatory sites through the secretion of IL-8, TNF-α and myeloperoxidase (273, 274). However, in the presence of type-1 IFNs, TANs display an antitumor N1 phenotype with increased tumor cytotoxicity and escalated levels of ICAM1, TNF-α, and NET (neutrophil extracellular trap) (275).. In addition, there were upregulations of pro-inflammatory cytokines and chemokines like IL-12, GM-CSF, CXCL10, CCL7, CCL2, and CCL3 in the N1 phenotype. This cytokine expression promotes CD8+ T cell recruitment and activation, thereby potentiating the antitumor effect, thence suppressing tumor growth (211).
Metabolically, TANs like other immune cells strongly rely on aerobic glycolysis and PPP as their dominant form of metabolism. Mitochondrial action also progressively diminishes as neutrophils mature, as they lose their c-kit expressions; however immature neutrophils and glucose-restricted tumor microenvironment rely on mitochondrial biogenesis, thereby maintaining a local immune suppression and ROS production (276). This comes in line with the fact that NADPH, an important cofactor for NADPH oxidase is produced by the PPP pathway, also stimulates neutrophil microbicidal functions. Yet, another function of neutrophils, the formation of neutrophil extracellular traps (NETs), a mixture of several epigenetic elements, i.e. DNA, histones, antimicrobial-peptides; also works in virtue of the metabolic shift towards glycolysis, glucose uptake, and PPP (277, 278). NETs also have been associated with several metastatic factors and other proinflammatory adhesion molecules, which further contribute to cancer-stimulated organ failures, thus demonstrating the characterization of NETs towards tumor progression (228, 279).
Classically, the metabolism signature of CSCs is quite different from that of regular cancer cells, which offers novel opportunities to target CSCs specifically. Several approaches have been made targeting glycolysis, mitochondrial biogenesis, OXPHOS, PPP, and FAO.
Drugs have been developed targeting glycolysis to suppress GLUT1 and regulate pyruvate metabolism. Glycolysis blockers along with competitive inhibition of phosphoglucoisomerase induced cell death in human CD44+/CD24low breast CSCs (only, not their non-CSC equivalent) (280). Similar approaches were tried with prostate CSCs, which resulted in induced cell death, reduced tumor spheroid formation, and higher dependency on glutamine, which can further be treated with glutaminase blockers (281). GLUT1 inhibition in vivo, by administration of WZB117, inhibited tumor initiation by CD133+ affiliated CSCs (glioma, pancreatic and ovarian CSCs) (282). Experiments by Li et al., and Isayev et al., extended the knowledge about anti-cancerous properties of 3-bromopyruvate (3BrP) (51, 283). 3BrP is known to suppress the cell viability of clonogenic RPMI8226 myeloma cells, KG1 leukemia cells, HepG2 hepatoma cells, and DU145 prostate cancer cell line by inhibiting ATP-dependent efflux pumps (284). Yet, another promising candidate in this field turned out to be metformin, an anti-diabetic drug. Compelling evidence showed that metformin induces an energy crisis by inhibiting mitochondrial complex I and impairing the ability to switch to glycolysis, subjecting to cell death (30, 285).
Suppression of mitochondrial OXPHOS by blocking ETC complexes also helps in modulating CSC populations (30). Treatment with oligomycin targets mitochondrial H+-ATP synthase and has shown commendable results in treating pancreatic, CD44+/CD117+ ovarian, and CD87+ lung CSCs and their non-tumor bulk variant too (29, 281, 286). SLC25A10 inhibition in combination with radiation therapy has shown also significant improvements in treating cancers, especially solid tumors with hypoxic cell fractions (40).
Considering the importance of lipid metabolism in CSCs, drugs intervening in different aspects of fatty acid metabolism have yielded positive results. Inhibition of FASN has showed suppressed tumor growth in brain CSCs, CD24-/CD44+/ESA+ ovarian CSCs, and breast CSCs (287, 288).
Intriguingly, mTORC1 activation leads to glutamine anaplerosis by activating glutamate dehydrogenase. On the other hand, mTORC1 downregulates SIRT4, which is a critical negative regulator for glutamine metabolism. However, mTOR inhibitors like rapamycin targets show efficacy in deregulating the mTOR pathway, thereby destabilizing the glutamine metabolism of the CSCs (289). These mTOR inhibitors have shown potency in reducing angiomyolipomas’ tumor growth in clinical trials of tuberous sclerosis (290).
Recent studies have also shown that combined therapy of chemotherapeutic drugs and Vitamin C induces an effective treatment mechanism. Vit. C interferes with the metabolic interests of the CSCs and also disturbs the epigenome regulation in them (291). Further research should focus on using Vit. C as a viable source to facilitate cancer treatment. Furthermore, evaluating a combined drug-controlled process aiming to destroy metabolic adaptability would condemn CSC survival.
As a whole, several characteristic metabolic, genetic, and epigenetic signatures can act as biomarkers for detecting CSCs. Over the past couple of decades, numerous methods have been envisaged regarding the dysregulation of cell signaling or cell surface proteins to treat CSCs.
DNMT inhibitors were among the first epigenetic therapeutic molecules which were incorporated for cancer treatment. Low doses of these inhibitors show profound efficacy in decreasing DNA methylation, thereby reducing tumorigenesis. Transient exposure to such low-dosed inhibitors induced antitumor response and decreased stemness properties in primary leukemia and epithelial tumor cells. DNMT inhibitors like decitabine and azacitidine have been broadly used to treat cancers of the breast, colon, and lungs. They have also shown significant downregulation of stemness factors like NANOG and Oct4 in prostate cancer (292, 293). Knockdown of DNMT1 decreased IL-6 mediated lung tumorigenesis and cancer stemness (294). Administration of SGI-110, along with other chemotherapeutic drugs, has repressed the stemness properties of ALDH+ ovarian cancer cells and inhibited tumorigenesis (295). Clinical trials with SGI-110 are still ongoing to treat various other CSCs like Liver, Leukemia, Ovarian, etc.
Yet, another procedure for treating CSCs is targeting histone modifications, like HDAC, LSD1 inhibitors etc. LSD1 or Lysine-specific demethylase 1A mediates transcriptional activation/repression, thereby enhancing proliferation, invasiveness, and cell motility. However, effective LSD1inhibitors and their corresponding analogs have shown significant antitumor activities across solid tumors and CSCs (296). HDACs have also been known to remove additional acetyl groups, thereby stimulating gene expressions. HDAC inhibitors like vorinostat and romidepsin have been approved to treat cutaneous T-cell lymphomas. HDAC inhibitors have also been shown to repress stemness in certain CSCs by reprogramming cancer cells. Gottlicher et al. demonstrated that valproic acid, a potent HDAC inhibitor known to be an antiepileptic drug, relieves HDAC mediated transcriptional repression, which further induces apoptosis of carcinoma cells, hematopoietic stem cells, and leukemia blasts from AML patients. This was further amended by Travaglini et al., that valproic acid reprograms the malignant mammary epithelial cells to a more physiologic phenotype and improves the sensitivity towards chemotherapic treatment (297, 298). Yet, another inhibitor of class I HDACs, entinostat, has been shown to decrease tumor-initiating cells from triple-negative breast cancer, hence reducing expression of several stemness factors like BMI-1, NANOG, and Oct-4. They have also led to prevent tumor formation at primary sites and distant metastasis (299).
Similarly, miRNAs also play a crucial role in regulating CSCs, which can become therapeutic tools when exploited. MiR-34a has been associated with CSCs of the prostate and pancreas, which, when supplemented with inhibitors of DNMTs and HDACs, lead to inhibition of stem cell proliferation and metastasis (172, 300). Suppression of miR-200c and miR-9 were found to be associated with the treatment of breast CSCs and inhibiting tumor formation in the breast (301, 302). MiR-126, when inhibited, led to the eradication of leukemic cells in AML (303). Combinations of anti-miRNA nucleotides and chemotherapeutic drug Sunitinib have shown anticancer effect in PDAC, indicating that miRNA inhibition therapies, when complemented with chemotherapeutic drugs, suppress stemness markers and stem cell properties of the tumor (304).
As discussed above, it can be postulated that CSCs can shape the immune system to harness their own needs. Vice versa, the infiltrating immune cells interact with CSCs change their immunogenic signature, and promote stemness, tumorigenesis, and metastasis of CSCs. However, the review also mentions the anti-tumorigenic potential of M1-TAMs and N1-TANs, and if appropriately modulated, might improve the clinical outcome of the patients. However, as mentioned earlier, immune cell-CSC interactions, including M2-TAMs, N2-TAMs, Tregs, and MDSCs, can be fruitful therapeutic structures if reversed using signaling techniques. Possible therapeutic strategies have been summarized in Table 2.
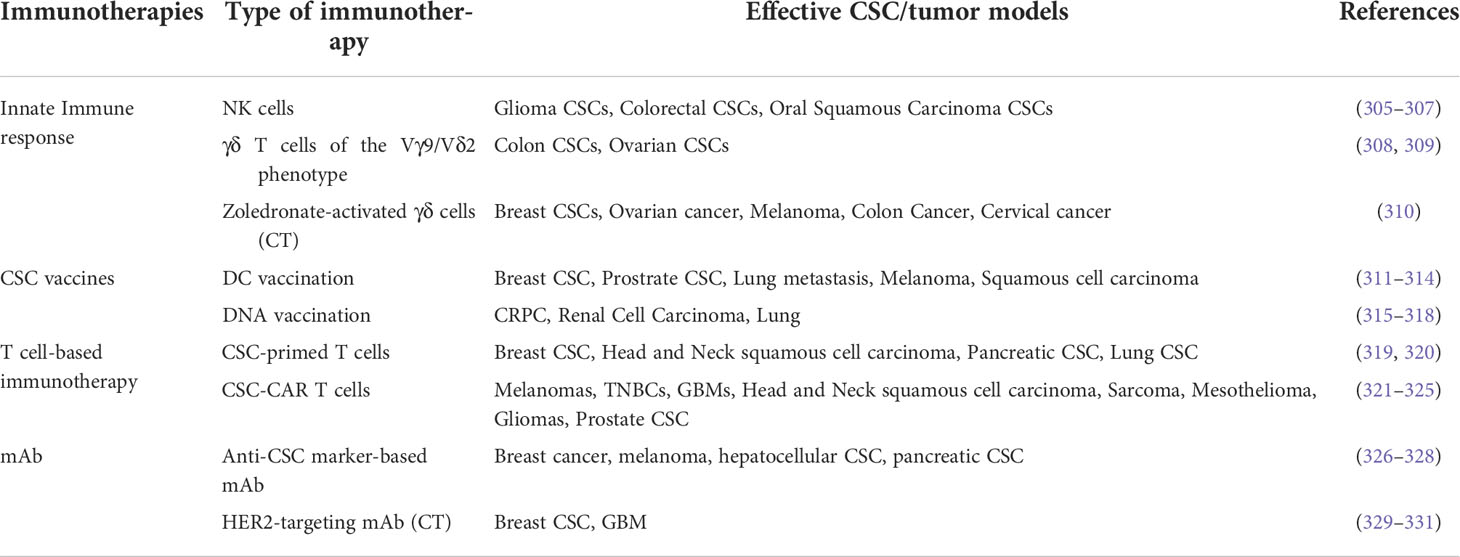
Table 2 A summarized form of immunotherapies (*CT = Combinational therapy).
As frontline workers of the immune system, the natural killer (NK) cells constitute the primary lymphocytes responsible for invading tumor cells. However, regarding CSCs, the role of NK cells remains quite contentious. For example, Wu et al. suggested that CD133+ and CD133- cells in brain CSCs adopt a mechanism to escape immune response mediated by MHC1 or NK cell-activating ligands, thereby making them obstinate to the innate or adaptive immune monitoring. Further, treatment of CD133+ cells with IFN-γ renders them sensitive to NK cells in vitro (332). Similar observations were reported in the case of breast CSCs, where CSCs dodge NK cell-mediated immunity due to downregulation of MICA and MICB (MHC I-related chain A and B), the two ligands for the stimulatory NK cell receptor NKG2D due to aberrant expression of miR-20a (333).
However, conversely, certain CSCs like glioma, colorectal and oral squamous carcinoma have expressed a significant amount of NK cell receptors, thereby mediating NK cell toxicity (305–307). These conflicting results thereby help us determine that CSCs in different tumor systems foster different sensitivities.
Yet another mode of therapy is done by γδ T cells of the Vγ9/Vδ2 phenotype, a part of the innate immune family. Human Vγ9/Vδ2 cells have been seen to be effective against colon CSCs, ovarian CSCs, and neuroblastoma. Zoledronate-activated γ cells showed positive responses against breast CSCs, solid tumors like ovarian, melanoma, colon, and cervical and enhanced the potential of CD8+ T cells via the IFN-γ driven overexpression of MHC I and ICAM 1 (308–310).
T-cell-based immunotherapy entirely relies on the production of effector T cells, accompanied by the adoptive transfer of CD8+ T cells back into patients. For CSC-primed T cells, CSCs derived from carcinoma cell lines like breast, head, and neck, and pancreas could induce a CD8+ T cell response. Adoptive therapy with ALDH-specific CD8+ T cells could be used to kill ALDHhi CSCs in vitro, thereby suppressing tumor growth and metastasis (319). Similar techniques were adopted by Luo et al. when they used CSC lysate-pulsed dendritic cells to stimulate CD8+ T cells to inhibit lung CSCs. Their study showed that ALDHhi-CD8+ T cells mediated antitumor effects, thereby suppressing tumorigenesis and metastasis (320).
Nevertheless, CAR T cells also symbolized to be successful immunotherapy. Using ex vivo genetic crafting, T cells can be genetically engineered to manifest a T cell receptor or CAR to recognize TAAs and inhibit tumor growth. CSC antigens like CSPG4 in melanomas, TNBCs, GBMs, head and neck squamous cell carcinoma, sarcoma and mesothelioma (321), CD 133 in GBMs (322), EGFRvIII and IL13Rα2 in gliomas (323, 324), and EpCAM in prostate carcinomas (325), are used as targets for CAR T-cell based immunotherapies. Neoantigens, which can be identified efficiently by T cells, also show promising CAR T cell therapy candidacy.
mAb-based immunotherapy has been considered a successful cancer therapy in the 21st century. Immunomodulatory drugs, demonstrated by anti-PD-1, anti-PD-L1, and immune checkpoints targeting anti-CTLA-4 antibodies, have shown profound success in clinical trials against cancer. The expression levels of CSC markers also act as attractive targets for antibody-based immunotherapy. For, e.g., anti-CD44 mAb inhibited tumorigenesis of murine breast tumors and showed regression in human melanoma metastasis (326, 327). CIK cells bound with anti-CD3/anti-CD133 bispecific antibodies were used to inhibit the growth of CD133hi CSCs of the liver and pancreas (328).
HER2-targeting antibodies, like trastuzumab and pertuzumab, also dramatically reduced CSC populations in GBMs and Breast CSCs. However, as CSC markers are not genuinely defined for specific subsets of CSCs, single mAb therapy may cause side effects to neighboring normal cells. Moreover, combinational therapy using mAb cocktails can efficiently eradicate CSC populations while reducing the side effects to other normal cells (329–331).
Immunotherapeutic strategies are based on the endogenous activation of T-cell responses to malignant cells via administering tumor-associated antigens (TAAs) to patients, which is successfully performed using vaccines (Dendritic cell, peptide, whole-cell or genetic). Specifically, the DC-mediated tumor immune response is stimulated by CSCs, which act as antigen sources (334, 335). However, it has been observed that enriched immunogenic CSCs are more effective than their non-stem counterpart. Administration of dendritic ell vaccines to syngeneic tumor models of immunocompetent mice has led to a substantial decrease in lung metastasis, squamous carcinoma cells, prostate CSCs, and regression in tumor models of melanoma D5 and squamous cancer SCC7 (311–313). Breast CSC-Dendritic Cell vaccines also showed a CTL-antitumor response by stimulating CD8+ and CD45+ T cells (314).
On the contrary, DNA vaccinations have been used as a new strategy to target cancers. They involve injecting genetically engineered plasmids for direct production of antigens, leading to a culminating immune response, thereby preventing cancer. Albeit in clinical trials, several DNA-based vaccines have successfully elicited antigen-specific T cell immune responses. In human castration-resistant prostate cancer, both PAP (prostate acid phosphatase) and PSA (prostate-specific antigen) is administered using DNA-based vaccinations (315, 316). Immunization with CSC-specific DNAJB8 expression plasmids also showed commendable anti-tumor response compared to immunization with TAA survivin (expressed in renal cell carcinomas) (317). Polakova et al., in their experiment, also used DNA vaccinations to induce antitumor effect against mouse oncogenic Sox2-expressing lung TC-1/B7 tumor cells (318). In sum, it can be said that CSC-DNA vaccines hold great potential to serve as a tool for immunotherapy, and they will turn out to become a great asset in cancer therapy in the years to come.
CSCs are regulated via three perspectives, namely metabolism, epigenetics, and immunology. It has been identified that CSCs acknowledge various phenotypes of metabolic reprogramming, the glycolytic pathway, oxidative phosphorylation, mitochondrial biogenesis, glutamine metabolism, Pentose Phosphate Pathway etc. Epigenetic dysregulation of CSC-related signaling pathways, i.e. Wnt, Notch, and Hedgehog leads to self-renewal ability and therapeutic resistance. Immune cell-CSC interactions have also been seen to alter the functional and genetic makeup of both the immune cell subsets and CSCs, leading to impaired CSC recognition and elimination by the immune system. With several processes in hand, CSCs evolve with varied niches and render their lifestyle procedures efficiently. (HK= Hexokinase, LDH= Lactate dehydrogenase, PK= Pyruvate kinase, PDK= Pyruvate dehydrogenase kinase, G6PDH= Glucose-6-phosphate dehydrogenase, SCD= Stearoyl-CoA, FASN= Fatty Acid synthase, GDH= Glutamate dehydrogenase, GLS= Glutaminase, VEGF= Vascular endothelial growth factor) (Created with BioRender.com)
Manifesting properties like self-renewal and stemness, metabolic alterations, epigenetic modifications, and immunogenic rewiring, cancer cells and their stem cell analogous can be considered different from naïve cells. With varied such properties, they characterize themselves with several markers, which are also known as biomarkers. Facilitating with the molecular definition of tumor, these biomarkers are subject to dynamic modulation and are expected to enhance our understanding of tumor metabolism, epigenetic and immunogenic influence. This would further provide insights into new therapeutic approaches for eradicating CSCs and their tumor bulks.
Enhanced carbon metabolism also plays a prominent and fundamental change in many cancers irrespective of their nature of origin or subsequent histological changes, thus entailing a great potential as cancer biomarkers. Enhanced glucose uptake using GLUTs and HK2 levels (336) has been observed to be efficient candidates for conferring the glycolysis metabolism inside tumors. Mutations in IDH enzymes also implicate metabolic rewiring, thus providing references to tumors (337). Positron emission tomography (PET) metabolism biomarkers like 18F-fluorodeoxyglucose, 18F-fluoroethyltyrosine, 18F-dihydroxyphenylalanine, 18F-fluorothymidine, and other radiotracers like 13N-ammonia, 15O-water, 82Rb-chloride, 11C-palmitate, 11C-glucose, 11C-acetate, etc. are also associated with perfusion and metabolic index of specific cancers (338).
Cancer, also being a cluster of alterations, confers a broad spectrum of genetic/epigenetic alterations, mutations, gene amplification, accompanied by varied methylation and acetylation changes. With DNA hypo- and hyper-methylation being the apparent epigenetic events responsible for promoting cancer, several studies have also shown the involvement of DNMTs in developing abnormalities in naïve cells. Blood/serum gene panels have shown promoter hypermethylation of several genes like GSTP1/TIG1/PTGS2/RPRM for prostate carcinomas (339), APC, RARβ2, RASSF1A, and p16/INK4a for lung cancers (340, 341), ITIH5, RASSF1A, and DKK3 for breast cancers (342), MLH1, VIM, TFPI2/HPP1 and APC for CRCs (343–345), MGMT, RASSF1A, p15/INK4b and p14/ARF for gliomas (346), etc. Concerning histone modifications, H3K4, H3K9, H3 and H4 in prostate cancer, H4K20 in preneoplastic lesions, H2A in breast cancer, H3K27 in midline gliomas are some of the notable expressions associated with early cancer detections (347). MicroRNAs and lncRNAs also play a critical role in detecting tumors and have already been described in the review. Immuno-specific antigen-based biomarkers like prostate-specific antigens, alpha-foetoprotein, cancer antigens, carcinoembryonic antigen, human chorionic gonadotrophin (hCG), HSPs, TGF-β, VEGF, EGFR, YKL, and MMPs are also seen to be expressed in many malignant tumors (339, 348).
However, to date, identifying markers to specific CSCs remains a challenge, as most of the CSC biomarkers label a heterogeneous stem cell population. However, combinations of extracellular and intracellular markers give prominence towards the isolation and characterization of specific CSCs. A short summary of some prominent CSC markers showing prominent prognostic approaches towards specific CSC models is also pointed out (also in Table 3).

Table 3 A summarized form of certain CSC markers expressed on their target CSCs.
With advances in technology, our understanding of CSCs and cancer have strengthened dramatically. The crosstalk among metabolic, epigenetic, and immunological aspects involved in CSC generation and recurrence substantially enhance tumorigenesis and metastasis. Several shreds of evidence have indicated that it is not just a specific domain that orchestrates tumor formation; instead, each and every domain has its role in developing the CSC niche (Figure 3). Though the origin of CSC has always been a contentious matter, it can be postulated that CSCs show stemness features and are resistant to chemo/radiotherapy alone. Thus, targeting the CSC population would give better insights into anti-tumor treatment, thereby eradicating cancer. Identifying the CSC with specific cell surface markers as mentioned before, forms fundamentally the first step in targeting the CSC. There are several universal CSC markers, with a few been extensively used in multiple studies.
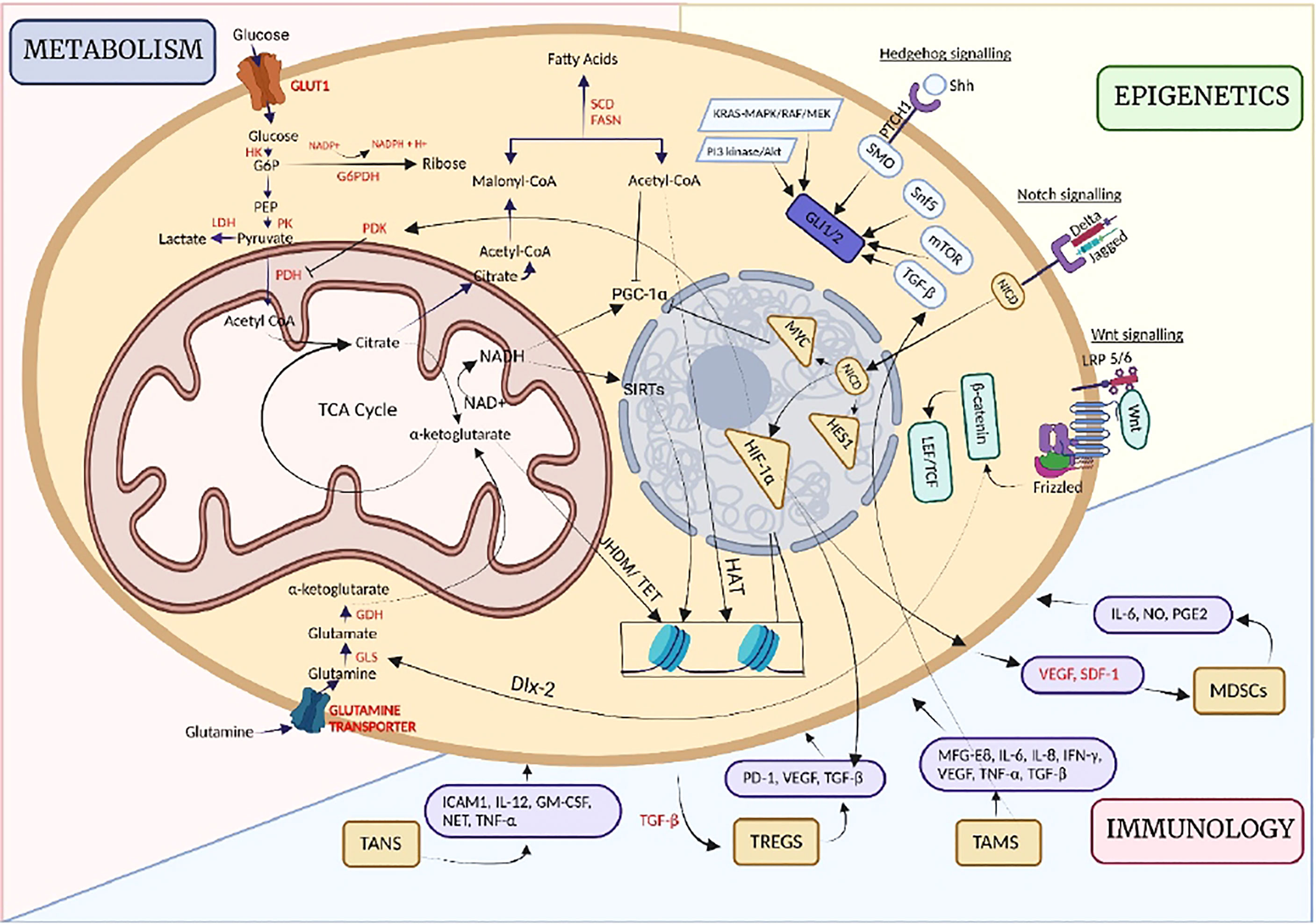
Figure 3 Pictorial representation of the different functional domains of Cancer Stem Cells. Epigenetic mechanisms have a profound influence on regulating the activities of key metabolic enzymes. Several of the enzymatic reactions, in turn, produce metabolites and onco-metabolites that in turn orchestrate key signaling pathways. TET ( Ten Eleven translocases) are one such mediator. Epigenetic mechanisms in turn also regulate mitogenic signals and alter the way cell interacts with their surroundings.
ALDH1A1 and ALDH1A2 are used to isolate CSC populations, with their expression related to stemness. They seems to be associated with different cancers in a context specific manner (Figure 4). While it is downregulated in few cancers, it is upregulated in few others indicating a complex interplay between cells and environment.

Figure 4 ALDH1A1 is differentially expressed in a cancer specific manner with few cancers showing downregulation(LAML, BRCA, A, B), upregulation (KIRP, C) and no changes (PAAD, D). TCGA data was analyzed using GEPIA tools.
A coordinated effort has been made to present all the interdisciplinary domains of CSCs under the purview of a single review in an elegant manner. The various dimensions of CSC, viz metabolic, epigenetic, and immunogenic interactions of the CSC population with the host organism, have been elucidated. Further discussion has also been made on treating these CSC subsets and cancer populations using novel therapeutic methods. We believe that the combined therapeutic trials’ success will definitely provide a promising path to pursue in future. However, the authors feel that there are still areas that require further investigation and research. In particular:
● To obtain a better understanding of the mechanisms involved in the driving of CSC initiation from normal naïve cells.
● To be able to differentiate the mechanisms followed by CSCs, naïve cancer cells, and normal stem cells.
● To ascertain the relative inter-contribution of the metabolic, epigenetic, and immunogenic domains, to CSC generation and determine if any particular domain can eradicate the transformed state of cancer.
● A combined approach to study the intra/inter CSC domains to target CSC populations.
● Delineating the different metaboepigenetic and other immunogenic approaches to circumvent the limitations caused due to less research in the field.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
OS drafted the manuscript and made the images with assistance from TS, JT, MAl, MAb and KP. MAl and MAb provided critical scientific and technological inputs and assistance. RD and SK conceptualized and oversaw the entire work. All authors contributed to the article and approved the submitted version.
SK acknowledge Department of Health Research (DHR)–Grant in aid (2020-9644) for funding. TS acknowledge Council of Scientific and Industrial Research (CSIR) for fellowship (564/(CSIR-UGC NET JUNE 2019). JT acknowledge Indian Council of Medical Research (ICMR) for financial support (2021-11320/F1). MSA and MA extend their appreciation to the Deanship of Scientific Research at King Khalid University (KKU) for funding this work through the Research Group Program Under the Grant Number:(R.G.P.2/248/43).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Authors (RD and SK) thank the Department of Health Research (DHR) and Indian Council of Medical Research (ICMR) for providing partial funding to carry on this work. Authors (MAl, MAb) extend their appreciation to the Deanship of Scientific Research at King Khalid University (KKU) for funding this work through the Research Group Program Under the Grant Number:(R.G.P.2/248/43).”
CSC, Cancer Stem Cell; ALDH, Aldehyde dehydrogenase; AML, Acute myeloid leukemia; BLBC, Basal,like breast cancer; CRC, Colorectal Cancer; CXCL, Chemokine (C,X,C motif) ligand; DNMT, DNA methyltransferase; Dlx,2, Distal-less homobox-2; FAO, Fatty Acid Oxidation; GBM, Glioblastoma multiforme; HCC, Hepatocellular Carcinoma; HH, Hedgehog; HIF, Hypoxia-inducible factor; IFN-γ, Interferon-gamma; JHDM, Jumonji-C domain-containing Histone demethylase; mAB, Monoclonal antibody; MDSC, Myeloid,derived suppressor cells; MCT, Monocarboxylate transporters; mTORC, Mammalian Target of Rapamycin Complex; NET, Neutrophil Extracellular Trap; OXPHOS, Oxidative Phosphorylation; PDAC, Pancreatic Ductal Adenocarcinoma; PDK, Pyruvate Dehydrogenase kinase; PGC-1α, Peroxisome proliferator-activated receptor gamma co-activator 1α; PKM2 – Pyruvate kinase M2; PPP, Pentose Phosphate Pathway; ROS, Reactive Oxygen Species; TAM, Tumor,associated macrophages; TAN, Tumor,associated neutrophils; TCA, Tricarboxylic acid; TET, Ten,eleven translocation proteins; TGF β, Transforming growth factor β; TNBC, Triple,negative breast cancer; TNF α, Tumor necrosis factor α; Tregs, Regulatory T cells.
1. Heiden MGV, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Sci (80- ) (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
2. Dando I, Dalla Pozza E, Biondani G, Cordani M, Palmieri M, Donadelli M. The metabolic landscape of cancer stem cells. IUBMB Life (2015) 67(9):687–93. doi: 10.1002/iub.1426
3. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
4. Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A (2011) 108(38):16062–7. doi: 10.1073/pnas.1106704108
5. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature (2014) 514(7524):628–32. doi: 10.1038/nature13611
6. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell (2013) 12(3):329–41. doi: 10.1016/j.stem.2012.12.013
7. Vlashi E, Lagadec C, Vergnes L, Reue K, Frohnen P, Chan M, et al. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res Treat (2014) 146(3):525–34. doi: 10.1007/s10549-014-3051-2
8. Plaks V, Kong N, Werb Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell (2015) 16(3):225–38. doi: 10.1016/j.stem.2015.02.015
9. Liu J, Qin X, Pan D, Zhang B, Jin F. Amino acid-mediated metabolism: A new power to influence properties of stem cells. Stem Cells Int (2019). doi: 10.1155/2019/6919463
10. Liu PP, Liao J, Tang ZJ, Wu WJ, Yang J, Zeng ZL, et al. Metabolic regulation of cancer cell side population by glucose through activation of the akt pathway. Cell Death Differ (2014) 21(1):124–35. doi: 10.1038/cdd.2013.131
11. Cluntun AA, Lukey MJ, Cerione Richard A, Jason L. Glutamine metabolism in cancer: Understanding the heterogeneity. Trends CANCER (2017) 3(3):169–80. doi: 10.1016/j.trecan.2017.01.005
12. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer (2016) 114(12):1305–12. doi: 10.1038/bjc.2016.152
13. Dong C, Yuan T, Wu Y, Wang Y, Fan TWM, Miriyala S, et al. Loss of FBP1 by snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell (2013) 23(3):316–31. doi: 10.1016/j.ccr.2013.01.022
14. Peng F, Wang JH, Fan WJ, Meng YT, Li MM, Li TT, et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene (2018) 37(8):1062–74. doi: 10.1038/onc.2017.368
15. Dong C, Yuan T, Wu Y, Wang Y, Fan TWM, Miriyala S, et al. Loss of FBP1 by snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell (2013) 23(3):316–31. doi: 10.1016/j.ccr.2013.01.022
16. Kim NH, Cha YH, Lee J, Lee SH, Yang JH, Yun JS, et al. Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat Commun [Internet] (2017) 8(1):1–12. doi: 10.1038/ncomms14374
17. Penkert J, Ripperger T, Schieck M, Schlegelberger B, Steinemann D, Illig T. On metabolic reprogramming and tumor biology: A comprehensive survey of metabolism in breast cancer. Oncotarget (2016) 7(41):67626–49. doi: 10.18632/oncotarget.11759
18. Tamada M, Nagano O, Tateyama S, Ohmura M, Yae T, Ishimoto T, et al. Modulation of glucose metabolism by CD44 contributes to antioxidant status and drug resistance in cancer cells. Cancer Res (2012) 72(6):1438–48. doi: 10.1158/0008-5472.CAN-11-3024
19. Xu Q, Tu J, Dou C, Zhang J, Yang L, Liu X, et al. HSP90 promotes cell glycolysis, proliferation and inhibits apoptosis by regulating PKM2 abundance via thr-328 phosphorylation in hepatocellular carcinoma. Mol Cancer (2017) 16(1):1–16. doi: 10.1186/s12943-017-0748-y
20. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. Corrigendum: Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature (2017) 480(7375):118–22. doi: 10.1038/nature10598
21. Lee J, Kim HK, Han YM, Kim J. Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4 in regulating transcription. Int J Biochem Cell Biol (2008) 40(5):1043–54. doi: 10.1016/j.biocel.2007.11.009
22. Kinnaird A, Zhao S, Wellen KE, Michelakis ED. Metabolic control of epigenetics in cancer. Nat Rev Cancer (2016) 16(11):694–707. doi: 10.1038/nrc.2016.82
23. Meijer TWH, Kaanders JHAM, Span PN, Bussink J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin Cancer Res (2012) 18(20):5585–94. doi: 10.1158/1078-0432.CCR-12-0858
24. Semenza GL. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J (2017) 36(3):252–9. doi: 10.15252/embj.201695204
25. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A. (2012) 109(8):2784–9. doi: 10.1073/pnas.1018866109
26. Sato M, Kawana K, Adachi K, Fujimoto A, Yoshida M, Nakamura H, et al. Spheroid cancer stem cells display reprogrammed metabolism and obtain energy by actively running the tricarboxylic acid (TCA) cycle. Oncotarget (2016) 7(22):33297–305. doi: 10.18632/oncotarget.8947
27. Lamb R, Bonuccelli G, Ozsvári B, Peiris-Pagès M, Fiorillo M, Smith DL, et al. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget (2015) 6(31):30453–71. doi: 10.18632/oncotarget.5852
28. Janiszewska M, Suvà ML, Riggi N, Houtkooper RH, Auwerx J, Clément-Schatlo V, et al. Imp2 controls oxidative phosphorylation and is crucial for preservin glioblastoma cancer stem cells. Genes Dev (2012) 26(17):1926–44. doi: 10.1101/gad.188292.112
29. Pastò A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G, et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget (2014) 5(12):4305–19. doi: 10.18632/oncotarget.2010
30. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab (2015) 22(4):590–605. doi: 10.1016/j.cmet.2015.08.015
31. Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, et al. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab (2016) 23(1):206–19. doi: 10.1016/j.cmet.2015.12.004
32. Cassim S, Raymond VA, Lacoste B, Lapierre P, Bilodeau M. Metabolite profiling identifies a signature of tumorigenicity in hepatocellular carcinoma. Oncotarget (2018) 9(42):26868–83. doi: 10.18632/oncotarget.25525
33. Piccinin E, Villani G, Moschetta A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: the role of PGC1 coactivators. Nat Rev Gastroenterol Hepatol (2019) 16(3):160–74. doi: 10.1038/s41575-018-0089-3
34. De Luca A, Fiorillo M, Peiris-Pagès M, Ozsvari B, Smith DL, Sanchez-Alvarez R, et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget (2015) 6(17):14777–95. doi: 10.18632/oncotarget.4401
35. Lebleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol (2014) 16(10):992–1003. doi: 10.1038/ncb3039
36. von Eyss B, Jaenicke LA, Kortlever RM, Royla N, Wiese KE, Letschert S, et al. A MYC-driven change in mitochondrial dynamics limits YAP/TAZ function in mammary epithelial cells and breast cancer. Cancer Cell (2015) 28(6):743–57. doi: 10.1016/j.ccell.2015.10.013
37. Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res (2016) 18(1):1–10. doi: 10.1186/s13058-016-0712-6
38. Bonuccelli G, Peiris-Pages M, Ozsvari B, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Targeting cancer stem cell propagation with palbociclib, a CDK4/6 inhibitor: Telomerase drives tumor cell heterogeneity. Oncotarget (2017) 8(6):9868–84. doi: 10.18632/oncotarget.14196
39. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. (2009) 458(7239):780–3. doi: 10.1038/nature07733
40. Rohatgi N, Ghoshdastider U, Baruah P, Kulshrestha T, Skanderup AJ. A pan-cancer metabolic atlas of the tumor microenvironment. bioRxiv (2020) 39(6):110800. doi: 10.1101/2020.10.16.342519
41. Chandel NS. Mitochondria as signaling organelles. BMC Biol (2014) 12:34. doi: 10.1186/1741-7007-12-34
42. Martínez-Reyes I, Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer (2021) 21(10):669–80. doi: 10.1038/s41568-021-00378-6
43. Cassim S, Vučetić M, Ždralević M, Pouyssegur J. Warburg and beyond: The power of mitochondrial metabolism to collaborate or replace fermentative glycolysis in cancer. Cancers (Basel) (2020) 12(5):13–5. doi: 10.3390/cancers12051119
44. Yi M, Li J, Chen S, Cai J, Ban Y, Peng Q, et al. Emerging role of lipid metabolism alterations in cancer stem cells. J Exp Clin Cancer Res (2018) 37(1):1–18. doi: 10.1186/s13046-018-0784-5
45. Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab (2013) 1(1):3. doi: 10.1186/2049-3002-1-3
46. Li W, Tai Y, Zhou J, Gu W, Bai Z, Zhou T, et al. Repression of endometrial tumor growth by targeting SREBP1 and lipogenesis. Cell Cycle (2012) 11(12):2348–58. doi: 10.4161/cc.20811
47. Luo D, Xiao H, Dong J, Li Y, Feng G, Cui M, et al. B7-H3 regulates lipid metabolism of lung cancer through SREBP1-mediated expression of FASN. Biochem Biophys Res Commun (2017) 482(4):1246–51. doi: 10.1016/j.bbrc.2016.12.021
48. Yasumoto Y, Miyazaki H, Vaidyan LK, Kagawa Y, Ebrahimi M, Yamamoto Y, et al. Inhibition of fatty acid synthase decreases expression of stemness markers in glioma stem cells. PloS One (2016) 11(1):e0147717. doi: 10.1371/journal.pone.0147717
49. Flaig TW, Salzmann-Sullivan M, Su LJ, Zhang Z, Joshi M, Gijón MA, et al. Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget (2017) 8(34):56051–65. doi: 10.18632/oncotarget.17359
50. Kathagen A, Schulte A, Balcke G, Phillips HS, Martens T, Matschke J, et al. Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol (2013) 126(5):763–80. doi: 10.1007/s00401-013-1173-y
51. Li D, Fu Z, Chen R, Zhao X, Zhou Y, Zeng B, et al. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget (2015) 6(31):31151–63. doi: 10.18632/oncotarget.5150
52. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. C-myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. (2009) 458(7239):762–5. doi: 10.1038/nature07823
53. Lee SY, Jeon HM, Ju MK, Jeong EK, Kim CH, Park HG, et al. Dlx-2 and glutaminase upregulate epithelial-mesenchymal transition and glycolytic switch. Oncotarget (2016) 7(7):7925–39. doi: 10.18632/oncotarget.6879
54. Naik PP, Panigrahi S, Parida R, Praharaj PP, Bhol CS, Patil S, et al. Metabostemness in cancer: Linking metaboloepigenetics and mitophagy in remodeling cancer stem cells. Stem Cell Rev Rep (2021) 18(1):198–213. doi: 10.1007/s12015-021-10216-9
55. Huang S. The molecular and mathematical basis of waddington’s epigenetic landscape: A framework for post-Darwinian biology? BioEssays (2012) 34(2):149–57. doi: 10.1002/bies.201100031
56. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discovery (2014) 13(7):513–32. doi: 10.1038/nrd4233
57. Chien AJ, Conrad WH, Moon RT. A wnt survival guide: From flies to human disease. J Invest Dermatol (2009) 129(7):1614–27. doi: 10.1038/jid.2008.445
58. Clevers H, Loh KM, Nusse R. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Sci (80- ). (2014) 346(6205):1248012. doi: 10.1126/science.1248012
59. Arce L, Yokoyama NN, Waterman ML. Diversity of LEF/TCF action in development and disease. Oncogene. (2006) 25(57):7492–504. doi: 10.1038/sj.onc.1210056
60. Klarmann GJ, Decker A, Farrar WL. Epigenetic gene silencing in the wnt pathway in breast cancer. Epigenetics (2008) 3(2):59–63. doi: 10.4161/epi.3.2.5899
61. Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet (2004) 36(4):417–22. doi: 10.1038/ng1330
62. Koinuma K, Yamashita Y, Liu W, Hatanaka H, Kurashina K, Wada T, et al. Epigenetic silencing of AXIN2 in colorectal carcinoma with microsatellite instability. Oncogene. (2006) 25(1):139–46. doi: 10.1038/sj.onc.1209009
63. Yoda Y, Takeshima H, Niwa T, Kim JG, Ando T, Kushima R, et al. Integrated analysis of cancer-related pathways affected by genetic and epigenetic alterations in gastric cancer. Gastric Cancer (2015) 18(1):65–76. doi: 10.1007/s10120-014-0348-0
64. Hussain M, Rao M, Humphries AE, Hong JA, Liu F, Yang M, et al. Tobacco smoke induces polyeomb-mediated repression of dickkopf-1 in lung cancer cells. Cancer Res (2009) 69(8):3570–8. doi: 10.1158/0008-5472.CAN-08-2807
65. Wang Y, He L, Du Y, Zhu P, Huang G, Luo J, et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of wnt signaling. Cell Stem Cell (2015) 16(4):413–25. doi: 10.1016/j.stem.2015.03.003
66. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. (2017) 36(11):1461–73. doi: 10.1038/onc.2016.304
67. Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, et al. Cutaneous cancer stem cell maintenance is dependent on β-catenin signalling. Nature. (2008) 452(7187):650–3. doi: 10.1038/nature06835
68. Lee SY, Ju MK, Jeon HM, Lee YJ, Kim CH, Park HG, et al. Oncogenic metabolism acts as a prerequisite step for induction of cancer metastasis and cancer stem cell phenotype. Oxid Med Cell Longev (2018), 2018:1027453. doi: 10.1155/2018/1027453
69. Beachy PA, Hymowitz SG, Lazarus RA, Leahy DJ, Siebold C. Interactions between hedgehog proteins and their binding partners come into view. Genes Dev (2010) 24(18):2001–12. doi: 10.1101/gad.1951710
70. Riobó NA, Lu K, Ai X, Haines GM, Emerson CP. Phosphoinositide 3-kinase and akt are essential for sonic hedgehog signaling. Proc Natl Acad Sci U S A. (2006) 103(12):4505–10. doi: 10.1073/pnas.0504337103
71. Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem (2007) 282(19):14048–55. doi: 10.1074/jbc.M611089200
72. Larsen LJ, Møller LB. Crosstalk of hedgehog and mTORC1 pathways. Cells. (2020) 9(10):1–21. doi: 10.3390/cells9102316
73. Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the hedgehog-gli pathway. Nat Med (2010) 16(12):1429–34. doi: 10.1038/nm.2251
74. Dennler S, André J, Alexaki I, Li A, Magnaldo T, Ten Dijke P, et al. Induction of sonic hedgehog mediators by transforming growth factor-β: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res (2007) 67(14):6981–6. doi: 10.1158/0008-5472.CAN-07-0491
75. Cui W, Wang LH, Wen YY, Song M, Li BL, Chen XL, et al. Expression and regulation mechanisms of sonic hedgehog in breast cancer. Cancer Sci (2010) 101(4):927–33. doi: 10.1111/j.1349-7006.2010.01495.x
76. Wang LH, Choi YL, Hua XY, Shin YK, Song YJ, Youn SJ, et al. Increased expression of sonic hedgehog and altered methylation of its promoter region in gastric cancer and its related lesions. Mod Pathol (2006) 19(5):675–83. doi: 10.1038/modpathol.3800573
77. Duan ZH, Wang HC, Zhao DM, Ji XX, Song M, Yang XJ, et al. Cooperatively transcriptional and epigenetic regulation of sonic hedgehog overexpression drives malignant potential of breast cancer. Cancer Sci (2015) 106(8):1084–91. doi: 10.1111/cas.12697
78. Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante P, et al. Histone deacetylase and Cullin3-REN KCTD11 ubiquitin ligase interplay regulates hedgehog signalling through gli acetylation. Nat Cell Biol (2010) 12(2):132–42. doi: 10.1038/ncb2013
79. Merchant AA, Matsui W. Targeting hedgehog - a cancer stem cell pathway. Clin Cancer Res (2010) 16(12):3130–40. doi: 10.1158/1078-0432.CCR-09-2846
80. Hutchin ME, Kariapper MST, Grachtchouk M, Wang A, Wei L, Cummings D, et al. Sustained hedgehog signaling is required for basal cell carcinoma proliferation and survival: Conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev (2005) 19(2):214–23. doi: 10.1101/gad.1258705
81. Ng JMY, Curran T. The hedgehog’s tale: Developing strategies for targeting cancer. Nat Rev Cancer (2011) 11(7):493–501. doi; 10.1038/nrc3079
82. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol (2007) 17(2):165–72. doi: 10.1016/j.cub.2006.11.033.
83. Zheng X, Han H, Liu G, Ma Y, Pan R, Sang L, et al. Lnc RNA wires up hippo and hedgehog signaling to reprogramme glucose metabolism. EMBO J (2017) 36(22):3325–35. doi: 10.15252/embj.201797609.
84. Andersson ER, Lendahl U. Therapeutic modulation of notch signalling-are we there yet? Nat Rev Drug Discovery (2014) 13(5):357–78. doi: 10.1038/nrd4252.
86. Andersson ER, Sandberg R, Lendahl U. Notch signaling: Simplicity in design, versatility in function. Development. (2011) 138(17):3593–612. doi: 10.1242/dev.063610
87. Toh TB, Lim JJ, Chow EKH. Epigenetics in cancer stem cells. Mol Cancer (2017) 16(1):1–21. doi: 10.1186/s12943-017-0596-9
88. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat Rev Cancer (2011) 11(5):338–51. doi: 10.1038/nrc3035
89. Ghoshal P, Nganga AJ, Moran-Giuati J, Szafranek A, Johnson TR, Bigelow AJ, et al. Loss of the SMRT/NCoR2 corepressor correlates with JAG2 overexpression in multiple myeloma. Cancer Res (2009) 69(10):4380–7. doi: 10.1158/0008-5472.CAN-08-3467
90. Jin L, Vu T, Yuan G, Datta PK. STRAP promotes stemness of human colorectal cancer via epigenetic regulation of the NOTCH pathwaya. Cancer Res (2017) 77(20):5464–78. doi: 10.1158/0008-5472.CAN-17-0286
91. El Hout M, Cosialls E, Mehrpour M, Hamaï A. Crosstalk between autophagy and metabolic regulation of cancer stem cells. Mol Cancer (2020) 19(1):1–17. doi: 10.1186/s12943-019-1126-8.
92. Singh D, Arora R, Kaur P, Singh B, Mannan R, Arora S. Overexpression of hypoxia-inducible factor and metabolic pathways: Possible targets of cancer. Cell Biosci (2017) 7(1):1–9. doi: 10.1186/s13578-017-0190-2
94. Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell (2011) 145(5):732–44. doi: 10.1016/j.cell.2011.03.054
95. Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget (2011) 2(7):551–6. doi: 10.18632/oncotarget.299
96. Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab (2017) 26(4):633–647.e7. doi: 10.1016/j.cmet.2017.09.009
97. Okano M, Bell DW, Haber DA, Li E. DNA Methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. (1999) 99(3):247–57. doi: 10.1016/S0092-8674(00)81656-6
98. Noh KM, Wang H, Kim HR, Wenderski W, Fang F, Li CH, et al. Engineering of a histone-recognition domain in Dnmt3a alters the epigenetic landscape and phenotypic features of mouse ESCs. Mol Cell (2015) 59(1):89–103. doi: 10.1016/j.molcel.2015.05.017
99. Pathania R, Ramachandran S, Elangovan S, Padia R, Yang P, Cinghu S, et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat Commun (2015) 6(1):1–11. doi: 10.1038/ncomms7910
100. Deneberg S, Guardiola P, Lennartsson A, Qu Y, Gaidzik V, Blanchet O, et al. Prognostic DNA methylation patterns in cytogenetically normal acute myeloid leukemia are predefined by stem cell chromatin marks. Blood. (2011) 118(20):5573–82. doi: 10.1182/blood-2011-01-332353
101. El Helou R, Wicinski J, Guille A, Adélaïde J, Finetti P, Bertucci F, et al. Brief reports: A distinct DNA methylation signature defines breast cancer stem cells and predicts cancer outcome. Stem Cells (2014) 32(11):3031–6. doi: 10.1002/stem.1792
102. Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, et al. Hematopoiesis and stem cells: Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood (2015) 125(4):629–38. doi: 10.1182/blood-2014-08-594648
103. Gao Y, Chen J, Li K, Wu T, Huang B, Liu W, et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell (2013) 12(4):453–69. doi: 10.1016/j.stem.2013.02.005
104. Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, et al. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect (2015) 122(12):1271–8. doi: 10.1289/ehp.1408418
105. Chen M, Zhang J, Li N, Qian Z, Zhu M, Li Q, et al. Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PloS One (2011) 6(10):e25564. doi: 10.1371/journal.pone.0025564
106. Liu X, Wang X, Zhang J, Lam EKY, Shin VY, Cheng ASL, et al. Warburg effect revisited: An epigenetic link between glycolysis and gastric carcinogenesis. Oncogene. (2010) 29(3):442–50. doi: 10.1038/onc.2009.332
107. Goel A, Mathupala SP, Pedersen PL. Glucose metabolism in cancer: Evidence that demethylation events play a role in activating type II hexokinase gene expression. J Biol Chem (2003) 278(17):15333–40. doi: 10.1074/jbc.M300608200
108. Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab (2012) 16(1):9–17. doi: 10.1016/j.cmet.2012.06.001
109. Nowicki S, Gottlieb E. Oncometabolites: Tailoring our genes. FEBS J (2015) 282(15):2796–805. doi: 10.1111/febs.13295
110. Kouzarides T. Chromatin modifications and their function. Cell. (2007) 128(4):693–705. doi: 10.1016/j.cell.2007.02.005
111. Venkatesan N, Wong JF, Tan KP, Chung HH, Yau YH, Cukuroglu E, et al. EZH2 promotes neoplastic transformation through VAV interaction-dependent extranuclear mechanisms. Oncogene. (2018) 37(4):461–77. doi: 10.1038/onc.2017.309
112. Guo BH, Feng Y, Zhang R, Xu LH, Li MZ, Kung HF, et al. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol Cancer (2011) 10:1–23. doi: 10.1186/1476-4598-10-10
113. Dong P, Kaneuchi M, Watari H, Hamada J, Sudo S, Ju J, et al. MicroRNA-194 inhibits epithelial to mesenchymal transition of endometrial cancer cells by targeting oncogene BMI-1. Mol Cancer (2011) 10:1–9. doi: 10.1186/1476-4598-10-99
114. Abdouh M, Facchino S, Chatoo W, Balasingam V, Ferreira J, Bernier G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci (2009) 29(28):8884–96. doi: 10.1523/JNEUROSCI.0968-09.2009
115. Yuan J, Takeuchi M, Negishi M, Oguro H, Ichikawa H, Iwama A. Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia. (2011) 25(8):1335–43. doi: 10.1038/leu.2011.85
116. Bai J, Mei P, Zhang C, Chen F, Li C, Pan Z, et al. BRG1 is a prognostic marker and potential therapeutic target in human breast cancer. PloS One (2013) 8(3):1–9. doi: 10.1371/journal.pone.0059772
117. Holik AZ, Young M, Krzystyniak J, Williams GT, Metzger D, Shorning BY, et al. Brg1 loss attenuates aberrant wnt-signalling and prevents wnt-dependent tumourigenesis in the murine small intestine. PloS Genet (2014) 10(7):e1004453. doi: 10.1371/journal.pgen.1004453
118. Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat (2008) 29(5):617–22. doi: 10.1002/humu.20730
119. Roy N, Malik S, Villanueva KE, Urano A, Lu X, Von Figura G, et al. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev (2015) 29(6):658–71. doi: 10.1101/gad.256628.114
120. Buscarlet M, Krasteva V, Ho L, Simon C, Hébert J, Wilhelm B, et al. Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood. (2014) 123(11):1720–8. doi: 10.1182/blood-2013-02-483495
121. Lei Y, Liu L, Zhang S, Guo S, Li X, Wang J, et al. Hdac7 promotes lung tumorigenesis by inhibiting Stat3 activation. Mol Cancer (2017) 16(1):1–13. doi: 10.1186/s12943-017-0736-2
122. Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett (2009) 277(1):8–21. doi: 10.1016/j.canlet.2008.08.016
123. Ouaïssi M, Sielezneff I, Silvestre R, Sastre B, Bernard JP, Lafontaine JS, et al. High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann Surg Oncol (2008) 15(8):2318–28. doi: 10.1245/s10434-008-9940-z
124. Moreno DA, Scrideli CA, Cortez MAA, De Paula Queiroz R, Valera ET, Da Silva Silveira V, et al. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia: Research paper. Br J Haematol (2010) 150(6):665–73. doi: 10.1111/j.1365-2141.2010.08301.x
125. Witt AE, Lee CW, Lee TI, Azzam DJ, Wang B, Caslini C, et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene (2017) 36(12):1707–20. doi: 10.1038/onc.2016.337
126. Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. (2006) 126(2):321–34. doi: 10.1016/j.cell.2006.05.040
127. Wu MY, Fu J, Xiao X, Wu J, Wu RC. MiR-34a regulates therapy resistance by targeting HDAC1 and HDAC7 in breast cancer. Cancer Lett (2014) 354(2):311–9. doi: 10.1016/j.canlet.2014.08.031
128. Gao H, Zhao H, Xiang W. Expression level of human miR-34a correlates with glioma grade and prognosis. J Neurooncol (2013) 113(2):221–8. doi: 10.1007/s11060-013-1119-1
129. Wang Y, Jia LS, Yuan W, Wu Z, Wang HB, Xu T, et al. Low miR-34a and miR-192 are associated with unfavorable prognosis in patients suffering from osteosarcoma. Am J Transl Res (2015) 7(1):111–9.
130. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, et al. XPoised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell (2013) 154(1):61. doi: 10.1016/j.cell.2013.06.005
131. Ahmad A, Aboukameel A, Kong D, Wang Z, Sethi S, Chen W, et al. Phosphoglucose isomerase/autocrine motility factor mediates epithelial-mesenchymal transition regulated by miR-200 in breast cancer cells. Cancer Res (2011) 71(9):3400–9. doi: 10.1158/0008-5472.CAN-10-0965
132. Hunkapiller J, Shen Y, Diaz A, Cagney G, McCleary D, Ramalho-Santos M, et al. Polycomb-like 3 promotes polycomb repressive complex 2 binding to CpG islands and embryonic stem cell self-renewal. PloS Genet (2012) 8(3):e1002576. doi: 10.1371/journal.pgen.1002576
133. Wang S, Robertson GP, Zhu J. A novel human homologue of drosophila polycomblike gene is up-regulated in multiple cancers. Gene. (2004) 343(1):69–78. doi: 10.1016/j.gene.2004.09.006
134. Menendez JA, Alarcón T, Joven J. Gerometabolites: The pseudohypoxic aging side of cancer oncometabolites. Cell Cycle (2014) 13(5):699–709. doi: 10.4161/cc.28079
135. Yang M, Soga T, Pollard PJ, Adam J. The emerging role of fumarate as an oncometabolite. Front Oncol (2012) 2:85. doi: 10.3389/fonc.2012.00085
136. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov (2013) 3(7):730–41. doi: 10.1158/2159-8290.CD-13-0083
137. Frezza C, Pollard PJ, Gottlieb E. Inborn and acquired metabolic defects in cancer. J Mol Med (2011) 89(3):213–20. doi: 10.1007/s00109-011-0728-4
138. French R, Pauklin S. Epigenetic regulation of cancer stem cell formation and maintenance. Int J Cancer (2020) 148(12):2884–97. doi: 10.1002/ijc.33398
139. Varga-Weisz P. ATP-dependent chromatin remodeling factors: Nucleosome shufflers with many missions. Oncogene. (2001) 20(24):3076–85. doi: 10.1038/sj.onc.1204332
140. Chen L, Cai Y, Jin G, Florens L, Swanson SK, Washburn MP, et al. Subunit organization of the human INO80 chromatin remodeling complex: An evolutionarily conserved core complex catalyzes ATP-dependent nucleosome remodeling. J Biol Chem (2011) 286(13):11283–9. doi: 10.1074/jbc.M111.222505
141. Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med (2011) 365(26):2497–506. doi: 10.1056/NEJMoa1109016
142. Shain AH, Giacomini CP, Matsukuma K, Karikari CA, Bashyam MD, Hidalgo M, et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci U S A. (2012) 109(5):E252-9. doi: 10.1073/pnas.1114817109
143. Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large b-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. (2012) 109(10):3879–84. doi: 10.1073/pnas.1121343109
144. DeCristofaro MF, Betz BL, Wang W, Weissman BE. Alteration of hSNF5/INI1/BAF47 detected in rhabdoid cell lines and primary rhabdomyosarcomas but not wilms’ tumors. Oncogene. (1999) 18(52):7559–65. doi: 10.1038/sj.onc.1203168
145. Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Sci (80- ). (2007) 318(5853):1108–13. doi: 10.1126/science.1145720
146. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med (2010) 363(16):1532–43. doi: 10.1056/NEJMoa1008433
147. Misawa A, Hosoi H, Imoto I, Iehara T, Sugimoto T, Inazawa J. Translocation (1;22)(p36;q11.2) with concurrent del(22)(q11.2) resulted in homozygous deletion of SNF5/INI1 in a newly established cell line derived from extrarenal rhabdoid tumor. J Hum Genet (2004) 49(10):586–9. doi: 10.1007/s10038-004-0191-y
148. Parsons DW, Jones S, Zhang X, Lin JCH, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Sci (80- ). (2008) 321(5897):1807–12. doi: 10.1126/science.1164382
149. Worley MJ, Welch WR, Berkowitz RS, Ng SW. Endometriosis-associated ovarian cancer: A review of pathogenesis. Int J Mol Sci (2013) 14(3):5367–79. doi: 10.3390/ijms14035367
150. Biegel JA, Fogelgren B, Zhou JY, James CD, Janss AJ, Allen JC, et al. Mutations of the INI1 rhabdoid tumor suppressor gene in medulloblastomas and primitive neuroectodermal tumors of the central nervous system. Clin Cancer Res (2000) 6(7):2759–63.
151. Oike T, Ogiwara H, Nakano T, Yokota J, Kohno T. Inactivating mutations in SWI/SNF chromatin remodeling genes in human cancer. Jpn J Clin Oncol (2013) 43(9):849–55. doi: 10.1093/jjco/hyt101
152. Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PloS One (2013) 8(1):e55119. doi: 10.1371/journal.pone.0055119
153. Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large b-cell lymphoma. Nat Genet (2011) 43(9):830–7. doi: 10.1038/ng.892
154. Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet (2012) 44(1):47–52. doi: 10.1038/ng.1032
155. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. (2011) 471(7339):467–72. doi: 10.1038/nature09837
156. Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet (2011) 43(9):828–9. doi: 10.1038/ng.903
157. Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet (2012) 44(2):133–9. doi: 10.1038/ng.1026
158. Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. (2011) 469(7331):539–42. doi: 10.1038/nature09639
159. Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet (2011) 43(12):1219–23. doi: 10.1038/ng.982
160. Jones S, Wang TL, Shih IM, Mao TL, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Sci (80- ). (2010) 330(6001):228–31. doi: 10.1126/science.1196333
161. Lee SA, Lee HS, Hur SK, Kang SW, Oh GT, Lee D, et al. INO80 haploinsufficiency inhibits colon cancer tumorigenesis via replication stress-induced apoptosis. Oncotarget (2017) 8(70):115041–53. doi: 10.18632/oncotarget.22984
162. Zhang S, Zhou B, Wang L, Li P, Bennett BD, Snyder R, et al. INO80 is required for oncogenic transcription and tumor growth in non-small cell lung cancer. Oncogene. (2017) 36(10):1430–9. doi: 10.1038/onc.2016.311
163. Zhou B, Wang L, Zhang S, Bennett BD, He F, Zhang Y, et al. INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev (2016) 30(12):1440–53. doi: 10.1101/gad.277178.115
164. Hu J, Liu J, Chen A, Lyu J, Ai G, Zeng Q, et al. Ino80 promotes cervical cancer tumorigenesis by activating nanog expression. Oncotarget (2016) 7(44):72250–62. doi: 10.18632/oncotarget.12667
165. Menendez JA, Alarcón T. Metabostemness: A new cancer hallmark. Front Oncol (2014) 4:262. doi: 10.3389/fonc.2014.00262
166. Morrison AJ. Chromatin-remodeling links metabolic signaling to gene expression. Mol Metab (2020) 38(March):100973. doi: 10.1016/j.molmet.2020.100973
167. Takahashi RU, Miyazaki H, Ochiya T. The role of microRNAs in the regulation of cancer stem cells. Front Genet (2013) 4:295. doi: 10.3389/fgene.2013.00295.
168. Song XL, Huang B, Zhou BW, Wang C, Liao ZW, Yu Y, et al. miR-1301-3p promotes prostate cancer stem cell expansion by targeting SFRP1 and GSK3β. BioMed Pharmacother (2018) 99:369–74. doi: 10.1016/j.biopha.2018.01.086
169. Dallavalle C, Albino D, Civenni G, Merulla J, Ostano P, Mello-Grand M, et al. MicroRNA-424 impairs ubiquitination to activate STAT3 and promote prostate tumor progression. J Clin Invest (2016) 126(12):4585–602. doi: 10.1172/JCI86505
170. Chang YL, Zhou PJ, Wei L, Li W, Ji Z, Fang YX, et al. MicroRNA-7 inhibits the stemness of prostate cancer stem-like cells and tumorigenesis by repressing KLF4/PI3K/Akt/p21 pathway. Oncotarget (2015) 6(27):24017–31. doi: 10.18632/oncotarget.4447
171. Hsieh IS, Chang KC, Tsai YT, Ke JY, Lu PJ, Lee KH, et al. MicroRNA-320 suppresses the stem cell-like characteristics of prostate cancer cells by downregulating the wnt/beta-catenin signaling pathway. Carcinogenesis. (2013) 34(3):530–8. doi: 10.1093/carcin/bgs371
172. Li J, Lam M, Iorns E, Gunn W, Tan F, Lomax J, et al. Registered report: The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med (2015) 17(2):211–5. doi: 10.7554/eLife.06434.003
173. Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol (2016) 8(4):a019521. doi: 10.1101/cshperspect.a019521
174. Cassim S, Raymond VA, Dehbidi-Assadzadeh L, Lapierre P, Bilodeau M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle (2018) 17(7):903–16. doi: 10.1080/15384101.2018.1460023
175. Jiang C, Yu M, Xie X, Huang G, Peng Y, Ren D, et al. MiR-217 targeting DKK1 promotes cancer stem cell properties via activation of the wnt signaling pathway in hepatocellular carcinoma. Oncol Rep (2017) 38(4):2351–9. doi: 10.3892/or.2017.5924
176. Lu AQ, Lv B, Qiu F, Wang XY, Cao XH. Upregulation of miR-137 reverses sorafenib resistance and cancer-initiating cell phenotypes by degrading ANT2 in hepatocellular carcinoma. Oncol Rep (2017) 37(4):2071–8. doi: 10.3892/or.2017.5498
177. Chai S, Ng KY, Tong M, Lau EY, Lee TK, Chan KW, et al. Octamer 4/microRNA-1246 signaling axis drives wnt/β-catenin activation in liver cancer stem cells. Hepatology. (2016) 64(6):2062–76. doi: 10.1002/hep.28821
178. Chen C, Yang Q, Wang D, Luo F, Liu X, Xue J, et al. MicroRNA-191, regulated by HIF-2α is involved in EMT and acquisition of a stem cell-like phenotype in arsenite-transformed human liver epithelial cells. Toxicol Vitr (2018) 48:128–36. doi: 10.1016/j.tiv.2017.12.016
179. Chen C, Luo F, Yang Q, Wang D, Yang P, Xue J, et al. NF-κB-regulated miR-155, via repression of QKI, contributes to the acquisition of CSC-like phenotype during the neoplastic transformation of hepatic cells induced by arsenite. Mol Carcinog (2018) 57(4):483–93. doi: 10.1002/mc.22772
180. Yu LX, Zhang BL, Yang MY, Liu H, Xiao CH, Zhang SG, et al. MicroRNA-106b-5p promotes hepatocellular carcinoma development via modulating FOG2. Onco Targets Ther (2019) 12:5639–47. doi: 10.2147/OTT.S203382
181. Jiang C, Long J, Liu B, Xu M, Wang W, Xie X, et al. MiR-500a-3p promotes cancer stem cells properties via STAT3 pathway in human hepatocellular carcinoma. J Exp Clin Cancer Res (2017) 36(1):1–13. doi: 10.1186/s13046-017-0568-3
182. Liu Y, Liu DL, Dong LL, Wen D, Shi DM, Zhou J, et al. MIR-612 suppresses stem cell-like property of hepatocellular carcinoma cells by modulating Sp1/ nanog signaling. Cell Death Dis (2016) 7(9):e2377–10. doi: 10.1038/cddis.2016.282
183. Xie Y, Du J, Liu Z, Zhang D, Yao X, Yang Y. MiR-6875-3p promotes the proliferation, invasion and metastasis of hepatocellular carcinoma via BTG2/FAK/Akt pathway. J Exp Clin Cancer Res (2019) 38(1):1–14. doi: 10.1186/s13046-018-1020-z
184. Chang X, Han J, Pang L, Zhao Y, Yang Y, Shen Z. Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer (2009) 9:1–11. doi: 10.1186/1471-2407-9-40
185. Zhu P, Fan Z. Cancer stem cells and tumorigenesis. Biophys Rep (2018) 4(4):178–88. doi: 10.1007/s41048-018-0062-2
186. Ren YQ, Fu F, Han J. MiR-27a modulates radiosensitivity of triple- negative breast cancer (TNBC) cells by targeting CDC27. Med Sci Monit (2015) 21:1297–303. doi: 10.12659/MSM.893974
187. Chen F, Luo N, Hu Y, Li X, Zhang K. MiR-137 suppresses triple-negative breast cancer stemness and tumorigenesis by perturbing BCL11A-DNMT1 interaction. Cell Physiol Biochem (2018) 47(5):2147–58. doi: 10.1159/000491526
188. Sun X, Xu C, Xiao G, Meng J, Wang J, Tang SC, et al. Breast cancer stem-like cells are sensitized to tamoxifen induction of self-renewal inhibition with enforced let-7c dependent on wnt blocking. Int J Mol Med (2018) 41(4):1967–75. doi: 10.3892/ijmm.2018.3388
189. Li B, Lu Y, Yu L, Han X, Wang H, Mao J, et al. miR-221/222 promote cancer stem-like cell properties and tumor growth of breast cancer via targeting PTEN and sustained Akt/NF-κB/COX-2 activation. Chem Biol Interact (2017) 277:33–42. doi: 10.1016/j.cbi.2017.08.014
190. Troschel FM, Böhly N, Borrmann K, Braun T, Schwickert A, Kiesel L, et al. miR-142-3p attenuates breast cancer stem cell characteristics and decreases radioresistance in vitro. Tumor Biol (2018) 40(8):1–10. doi: 10.1177/1010428318791887
191. Bonetti P, Climent M, Panebianco F, Tordonato C, Santoro A, Marzi MJ, et al. Dual role for miR-34a in the control of early progenitor proliferation and commitment in the mammary gland and in breast cancer. Oncogene (2019) 38(3):360–74. doi: 10.1038/s41388-018-0445-3
192. Lin C, Gao B, Yan X, Lei Z, Chen K, Li Y, et al. MicroRNA 628 suppresses migration and invasion of breast cancer stem cells through targeting SOS1. Onco Targets Ther (2018) 11:5419–28. doi: 10.2147/OTT.S164575
193. Schwarzenbacher D, Klec C, Pasculli B, Cerk S, Rinner B, Karbiener M, et al. MiR-1287-5p inhibits triple negative breast cancer growth by interaction with phosphoinositide 3-kinase CB, thereby sensitizing cells for PI3Kinase inhibitors. Breast Cancer Res (2019) 21(1):1–16. doi: 10.1186/s13058-019-1104-5
194. Wu Y, Shi W, Tang T, Wang Y, Yin X, Chen Y, et al. miR-29a contributes to breast cancer cells epithelial–mesenchymal transition, migration, and invasion via down-regulating histone H4K20 trimethylation through directly targeting SUV420H2. Cell Death Dis (2019) 10(3):176. doi: 10.1038/s41419-019-1437-0
195. Zhu J, Wang S, Chen Y, Li X, Jiang Y, Yang X, et al. miR-19 targeting of GSK3β mediates sulforaphane suppression of lung cancer stem cells. J Nutr Biochem (2017) 44:80–91. doi: 10.1016/j.jnutbio.2017.02.020
196. Zhang WC, Chin TM, Yang H, Nga ME, Lunny DP, Lim EKH, et al. Tumour-initiating cell-specific MIR-1246 and MIR-1290 expression converge to promote non-small cell lung cancer progression. Nat Commun (2016) 7(1):1–16. doi: 10.1038/ncomms11702
197. Li QQ, Xie YK, Wu Y, Li LL, Liu Y, Miao XB, et al. Sulforaphane inhibits cancer stem-like cell properties and cisplatin resistance through miR-214-mediated downregulation of c-MYC in non-small cell lung cancer. Oncotarget (2017) 8(7):12067–80. doi: 10.18632/oncotarget.14512
198. Faversani A, Amatori S, Augello C, Colombo F, Porretti L, Fanelli M, et al. miR-494-3p is a novel tumor driver of lung carcinogenesis. Oncotarget (2017) 8(5):7231–47. doi: 10.18632/oncotarget.13933
199. Shi Y, Liu C, Liu X, Tang DG, Wang J. The microRNA miR-34a inhibits non-small cell lung cancer (NSCLC) growth and the CD44hi stem-like NSCLC cells. PloS One (2014) 9(3):1–8. doi: 10.1371/journal.pone.0090022
200. Yang Y, Ding L, Hu Q, Xia J, Sun J, Wang X, et al. MicroRNA-218 functions as a tumor suppressor in lung cancer by targeting IL-6/STAT3 and negatively correlates with poor prognosis. Mol Cancer (2017) 16(1):1–13. doi: 10.1186/s12943-017-0710-z
201. Hu J, Cheng Y, Li Y, Jin Z, Pan Y, Liu G, et al. MicroRNA-128 plays a critical role in human non-small cell lung cancer tumourigenesis, angiogenesis and lymphangiogenesis by directly targeting vascular endothelial growth factor-c. Eur J Cancer (2014) 50(13):2336–50. doi: 10.1016/j.ejca.2014.06.005
202. Tsukasa K, Ding Q, Miyazaki Y, Matsubara S, Natsugoe S, Takao S. miR-30 family promotes migratory and invasive abilities in CD133+ pancreatic cancer stem-like cells. Hum Cell (2016) 29(3):130–7. doi: 10.1007/s13577-016-0137-7
203. Zhou W, Li Y, Gou S, Xiong J, Wu H, Wang C, et al. MiR-744 increases tumorigenicity of pancreatic cancer by activating wnt/β-catenin pathway. Oncotarget (2015) 6(35):37557–69. doi: 10.18632/oncotarget.5317
204. Ma C, Huang T, Ding YC, Yu W, Wang Q, Meng B, et al. MicroRNA-200c overexpression inhibits chemoresistance, invasion and colony formation of human pancreatic cancer stem cells. Int J Clin Exp Pathol (2015) 8(6):6533–9.
205. Vahidian F, Duijf PHG, Safarzadeh E, Derakhshani A, Baghbanzadeh A, Baradaran B. Interactions between cancer stem cells, immune system and some environmental components: Friends or foes? Immunol Lett [Internet] (2019) 208:19–29. doi: 10.1016/j.imlet.2019.03.004
206. Raggi C, Mousa HS, Correnti M, Sica A, Invernizzi P. Cancer stem cells and tumor-associated macrophages: A roadmap for multitargeting strategies. Oncogene. (2016) 35(6):671–82. doi: 10.1038/onc.2015.132
207. Sultan M, Coyle KM, Vidovic D, Thomas ML, Gujar S, Marcato P. Hide-and-seek: The interplay between cancer stem cells and the immune system. Carcinogenesis. (2017) 38(2):107–18. doi: 10.1093/carcin/bgw115
208. Liu Y, Wei G, Cheng WA, Dong Z, Sun H, Lee VY, et al. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol Immunother (2018) 67(8):1181–95. doi: 10.1007/s00262-018-2175-3
209. Rezalotfi A, Ahmadian E, Aazami H, Solgi G, Ebrahimi M. Gastric cancer stem cells effect on Th17/Treg balance; a bench to beside perspective. Front Oncol (2019) 9:226. doi: 10.3389/fonc.2019.00226
210. Yu X, Li H, Ren X. Interaction between regulatory T cells and cancer stem cells. Int J Cancer (2012) 131(7):1491–8. doi: 10.1002/ijc.27634
211. Shaul ME, Fridlender ZG. Neutrophils as active regulators of the immune system in the tumor microenvironment. J Leukoc Biol (2017) 102(2):343–9. doi: 10.1189/jlb.5MR1216-508R
212. Cassim S, Pouyssegur J. Tumor microenvironment: A metabolic player that shapes the immune response. Int J Mol Sci (2019) 21(1):157. doi: 10.3390/ijms21010157
213. Singer K, Cheng WC, Kreutz M, Ho PC, Siska PJ. Immunometabolism in cancer at a glance. DMM Dis Model Mech (2018) 11(8):1–10. doi: 10.1242/dmm.034272
214. Porporato PE, Filigheddu N, Pedro JMBS, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res (2018) 28(3):265–80. doi: 10.1038/cr.2017.155
215. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol (2013) 229(2):176–85. doi: 10.1002/path.4133
216. Mantovani A, Allavena P, Sica A. Tumour-associated macrophages as a prototypic type II polarised phagocyte population: Role in tumour progression. Eur J Cancer (2004) 40(11):1660–7. doi: 10.1016/j.ejca.2004.03.016
217. Noy R, Pollard JW. Tumor-associated macrophages: From mechanisms to therapy. Immunity (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
218. Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A. (2011) 108(30):12425–30. doi: 10.1073/pnas.1106645108
219. Joshi S, Singh AR, Zulcic M, Durden DL. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1α and HIF2α stability and tumor growth, angiogenesis, and metastasis. Mol Cancer Res (2014) 12(10):1520–31. doi: 10.1158/1541-7786.MCR-13-0682
220. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat Immunol (2010) 11(10):889–96. doi: 10.1038/ni.1937
221. Liu D, Chang C, Lu N, Wang X, Lu Q, Ren X, et al. Comprehensive proteomics analysis reveals metabolic reprogramming of tumor-associated macrophages stimulated by the tumor microenvironment. J Proteome Res (2017) 16(1):288–97. doi: 10.1021/acs.jproteome.6b00604
222. Arts RJW, Plantinga TS, Tuit S, Ulas T, Heinhuis B, Tesselaar M, et al. Transcriptional and metabolic reprogramming induce an inflammatory phenotype in non-medullary thyroid carcinoma-induced macrophages. Oncoimmunology (2016) 5(12):1–13. doi: 10.1080/2162402X.2016.1229725
223. Penny HL, Sieow JL, Adriani G, Yeap WH, See Chi Ee P, San Luis B, et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology (2016) 5(8):1–15. doi: 10.1080/2162402X.2016.1191731
224. Wenes M, Shang M, Di Matteo M, Goveia J, Martín-Pérez R, Serneels J, et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab (2016) 24(5):701–15. doi: 10.1016/j.cmet.2016.09.008
225. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. (2008) 452(7184):230–3. doi: 10.1038/nature06734
226. Na YR, Je S, Seok SH. Metabolic features of macrophages in inflammatory diseases and cancer. Cancer Lett (2018) 413:46–58. doi: 10.1016/j.canlet.2017.10.044
227. Rabold K, Netea MG, Adema GJ, Netea-Maier RT. Cellular metabolism of tumor-associated macrophages – functional impact and consequences. FEBS Lett (2017) 591(19):3022–41. doi: 10.1002/1873-3468.12771
228. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immun [Internet] (2015) 43(3):435–49. doi: 10.1016/j.immuni.2015.09.001
229. Puthenveetil A, Dubey S. Metabolic reprograming of tumor-associated macrophages. Ann Transl Med (2020) 8(16):1030–0. doi: 10.21037/atm-20-2037
230. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-κB. J Exp Med (2008) 205(6):1261–8. doi: 10.1084/jem.20080108
231. Cai X, Yin Y, Li N, Zhu D, Zhang J, Zhang CY, et al. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J Mol Cell Biol (2012) 4(5):341–3. doi: 10.1093/jmcb/mjs044
232. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med (2013) 19(10):1264–72. doi: 10.1038/nm.3337
233. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol [Internet] (2012) 22(4):275–81. doi: 10.1016/j.semcancer.2012.01.011
234. Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: The blessing and the curse of morphological and functional heterogeneity. Eur J Immunol (2010) 40(11):2969–75. doi: 10.1002/eji.201040895
235. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. (2013) 138(2):105–15. doi: 10.1111/imm.12036
236. Yu Q, Dong L, Li Y, Liu G. SIRT1 and HIF1α signaling in metabolism and immune responses. Cancer Lett [Internet] (2018) 418:20–6. doi: 10.1016/j.canlet.2017.12.035
237. Yan D, Adeshakin AO, Xu M, Afolabi LO, Zhang G, Chen YH, et al. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front Immunol (2019) 10:1399. doi: 10.3389/fimmu.2019.01399
238. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res (2015) 3(11):1236–47. doi: 10.1158/2326-6066.CIR-15-0036
239. Al-Khami AA, Rodriguez PC, Ochoa AC. Metabolic reprogramming of myeloid-derived suppressor cells (MDSC) in cancer. Oncoimmunol [Internet] (2016) 5(8):1–2. doi: 10.1080/2162402X.2016.1200771
240. Hu C, Pang B, Lin G, Zhen Y, Yi H. Energy metabolism manipulates the fate and function of tumour myeloid-derived suppressor cells. Br J Cancer [Internet] (2020) 122(1):23–9. doi: 10.1038/s41416-019-0644-x
241. Tartour E, Pere H, Maillere B, Terme M, Merillon N, Taieb J, et al. Angiogenesis and immunity: A bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer Metastasis Rev (2011) 30(1):83–95. doi: 10.1007/s10555-011-9281-4
242. Ortiz ML, Lu L, Ramachandran I, Gabrilovich DI. Myeloid-derived suppressor cells in the development of lung cancer. Cancer Immunol Res (2014) 2(1):50–8. doi: 10.1158/2326-6066.CIR-13-0129
243. Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. (2009) 106(16):6742–7. doi: 10.1073/pnas.0902280106.
244. Zhang C, Wang S, Liu Y, Yang C. Epigenetics in myeloid derived suppressor cells: A sheathed sword towards cancer. Oncotarget (2016) 7(35):57452–63. doi: 10.18632/oncotarget.10767
245. Kuroda H, Mabuchi S, Yokoi E, Komura N, Kozasa K, Matsumoto Y, et al. Prostaglandin E2 produced by myeloid-derived suppressive cells induces cancer stem cells in uterine cervical cancer. Oncotarget (2018) 9(91):36317–30. doi: 10.18632/oncotarget.26347
246. Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CTBP2. Immunity. (2013) 39(3):611–21. doi: 10.1016/j.immuni.2013.08.025
247. Panni RZ, Sanford DE, Belt BA, Mitchem JB, Worley LA, Goetz BD, et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother (2014) 63(5):513–28. doi: 10.1007/s00262-014-1527-x
248. Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signaling. Cancer Res (2016) 76(11):3156–65. doi: 10.1158/0008-5472.CAN-15-2528
249. Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res (2003) 9(2):606–12.
250. Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther (2010) 9(1):67–78. doi: 10.1158/1535-7163.MCT-09-0734
251. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother (2014) 63(7):721–35. doi: 10.1007/s00262-014-1549-4
252. Stapelberg M, Zobalova R, Nguyen MN, Walker T, Stantic M, Goodwin J, et al. Indoleamine-2,3-dioxygenase elevated in tumor-initiating cells is suppressed by mitocans. Free Radic Biol Med [Internet] (2014) 67:41–50. doi: 10.1016/j.freeradbiomed.2013.10.003
253. Müller L, Tunger A, Plesca I, Wehner R, Temme A, Westphal D, et al. Bidirectional crosstalk between cancer stem cells and immune cell subsets. Front Immunol (2020) 11:140. doi: 10.3389/fimmu.2020.00140
254. Yu P, Lee Y, Liu W, Krausz T, Chong A, Schreiber H, et al. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J Exp Med (2005) 201(5):779–91. doi: 10.1084/jem.20041684
255. Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell (2007) 11(3):259–73. doi: 10.1016/j.ccr.2007.01.013
256. Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res (2006) 66(16):7843–8. doi: 10.1158/0008-5472.CAN-06-1010
257. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-a modulates expression of inhibitory checkpoints on CD8++ T cells in tumors. J Exp Med (2015) 212(2):139–48. doi: 10.1084/jem.20140559
258. Yang S, Wang B, Guan C, Wu B, Cai C, Wang M, et al. Foxp3 + IL-17 + T cells promote development of cancer-initiating cells in colorectal cancer. J Leukoc Biol (2011) 89(1):85–91. doi: 10.1189/jlb.0910506
259. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and treg cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
260. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20(11):1327–33. doi: 10.1038/nm.3704
261. Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, et al. The nuclear receptor PPARγ selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med (2009) 206(10):2079–89. doi: 10.1084/jem.20082771
262. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity (2016) 44(2):406–21. doi: 10.1016/j.immuni.2016.01.028
263. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. MTORC1 couples immune signals and metabolic programming to establish T reg-cell function. Nature. (2013) 499(7459):485–90. doi: 10.1038/nature12297
264. Procaccini C, De Rosa V, Galgani M, Abanni L, Calì G, Porcellini A, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. (2010) 33(6):929–41. doi: 10.1016/j.immuni.2010.11.024
265. Vukmanovic-Stejic M, Agius E, Booth N, Dunne PJ, Lacy KE, Reed JR, et al. The kinetics of CD4+Foxp3+ T cell accumulation during a human cutaneous antigen-specific memory response in vivo. J Clin Invest (2008) 118(11):3639–50. doi: 10.1172/JCI35834
266. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. (2010) 464(7285):121–5. doi: 10.1038/nature08778
267. De Rosa V, Di Rella F, Di Giacomo A, Matarese G. Regulatory T cells as suppressors of anti-tumor immunity: Role of metabolism. Cytokine Growth Factor Rev (2017) 35:15–25. doi: 10.1016/j.cytogfr.2017.04.001
268. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol (2015) 16(11):1174–84. doi: 10.1038/ni.3269
269. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol (2016) 17(1):95–103. doi: 10.1038/ni.3313
270. Hira VVV, Ploegmakers KJ, Grevers F, Verbovšek U, Silvestre-Roig C, Aronica E, et al. CD133+ and nestin+ glioma stem-like cells reside around CD31+ arterioles in niches that express SDF-1α, CXCR4, osteopontin and cathepsin K. J Histochem Cytochem (2015) 63(7):481–93. doi: 10.1369/0022155415581689
271. Reid MD, Basturk O, Thirabanjasak D, Hruban RH, Klimstra DS, Bagci P, et al. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod Pathol (2011) 24(12):1612–9. doi: 10.1038/modpathol.2011.113
272. Shen M, Hu P, Donskov F, Wang G, Liu Q, Du J. Tumor-associated neutrophils as a new prognostic factor in cancer: A systematic review and meta-analysis. PloS One (2014) 9(6):1–10. doi: 10.1371/journal.pone.0098259
273. Lefkowitz DL, Lefkowitz SS. Macrophage-neutrophil interaction: A paradigm for chronic inflammation revisited. Immunol Cell Biol (2001) 79(5):502–6. doi: 10.1046/j.1440-1711.2001.01020.x
274. Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, et al. Neutrophils suppress intraluminal NK cell-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discovery (2016) 6(6):630–49. doi: 10.1158/2159-8290.CD-15-1157
275. Andzinski L, Kasnitz N, Stahnke S, Wu CF, Gereke M, Von Köckritz-Blickwede M, et al. Type i IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer (2016) 138(8):1982–93. doi: 10.1002/ijc.29945
276. Rice CM, Davies LC, Subleski JJ, Maio N, Gonzalez-Cotto M, Andrews C, et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat Commun (2018) 9(1):5099. doi: 10.1038/s41467-018-07505-2
277. Azevedo EP, Rochael NC, Guimarães-Costa AB, De Souza-Vieira TS, Ganilho J, Saraiva EM, et al. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-acetate-induced neutrophil extracellular trap (NET) formation. J Biol Chem (2015) 290(36):22174–83. doi: 10.1074/jbc.M115.640094
278. Rodríguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MMB, López-Villegas EO, Sánchez-García FJ. Metabolic requirements for neutrophil extracellular traps formation. Immunology. (2015) 145(2):213–24. doi: 10.1111/imm.12437
279. Cedervall J, Zhang Y, Huang H, Zhang L, Femel J, Dimberg A, et al. Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor-bearing animals. Cancer Res (2015) 75(13):2653–62. doi: 10.1158/0008-5472.CAN-14-3299
280. Gordon N, Skinner AM, Pommier RF, Schillace RV, O’Neill S, Peckham JL, et al. Gene expression signatures of breast cancer stem and progenitor cells do not exhibit features of warburg metabolism. Stem Cell Res Ther (2015) 6(1):157. doi: 10.1186/s13287-015-0153-7
281. Aguilar E, Marin De Mas I, Zodda E, Marin S, Morrish F, Selivanov V, et al. Metabolic reprogramming and dependencies associated with epithelial cancer stem cells independent of the epithelial-mesenchymal transition program. Stem Cells (2016) 34(5):1163–76. doi: 10.1002/stem.2286
282. Shibuya K, Okada M, Suzuki S, Seino M, Seino S, Takeda H, et al. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget (2015) 6(2):651–61. doi: 10.18632/oncotarget.2892
283. Isayev O, Rausch V, Bauer N, Liu L, Fan P, Zhang Y, et al. Inhibition of glucose turnover by 3-bromopyruvate counteracts pancreatic cancer stem cell features and sensitizes cells to gemcitabine. Oncotarget (2014) 5(13):5177–89. doi: 10.18632/oncotarget.2120
284. Nakano A, Tsuji D, Miki H, Cui Q, El Sayed SM, Ikegame A, et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PloS One (2011) 6(11):e27222. doi: 10.1371/journal.pone.0027222
285. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife (2014) 3:e02242. doi: 10.7554/eLife.02242
286. Gao C, Shen Y, Jin F, Miao Y, Qiu X. Cancer stem cells in small cell lung cancer cell line H446: Higher dependency on oxidative phosphorylation and mitochondrial substrate-level phosphorylation than non-stem cancer cells. PloS One (2016) 11(5):1–16. doi: 10.1371/journal.pone.0154576
287. Pandey PR, Xing F, Sharma S, Watabe M, Pai SK, Iiizumi-Gairani M, et al. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene. (2013) 32(42):5111–22. doi: 10.1038/onc.2012.519
288. Pandey PR, Okuda H, Watabe M, Pai SK, Liu W, Kobayashi A, et al. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res Treat (2011) 130(2):387–98. doi: 10.1007/s10549-010-1300-6
289. Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. (2013) 153(4):840–54. doi: 10.1016/j.cell.2013.04.023
290. Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med (2008) 358(2):140–51. doi: 10.1056/NEJMoa063564
291. Satheesh NJ, Samuel SM, Büsselberg D. Combination therapy with vitamin c could eradicate cancer stem cells. Biomolecules. (2020) 10(1):79. doi: 10.3390/biom10010079
292. Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell (2012) 21(3):430–46. doi: 10.1016/j.ccr.2011.12.029
293. Tian J, Lee SO, Liang L, Luo J, Huang CK, Li L, et al. Targeting the unique methylation pattern of androgen receptor (AR) promoter in prostate stem/progenitor cells with 5-aza-2′-deoxycytidine (5-AZA) leads to suppressed prostate tumorigenesis. J Biol Chem (2012) 287(47):39954–66. doi: 10.1074/jbc.M112.395574
294. Liu CC, Lin JH, Hsu TW, Su K, Li AFY, Hsu HS, et al. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int J Cancer (2015) 136(3):547–59. doi: 10.1002/ijc.29033.
295. Wang Y, Cardenas H, Fang F, Condello S, Taverna P, Segar M, et al. Epigenetic targeting of ovarian cancer stem cells. Cancer Res (2014) 74(17):4922–36. doi: 10.1158/0008-5472.CAN-14-1022
296. French R, Pauklin S. Epigenetic regulation of cancer stem cell formation and maintenance. Int J Cancer (2021) 148(12):2884–97. doi: 10.1002/ijc.33398
297. Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J (2001) 20(24):6969–78. doi: 10.1093/emboj/20.24.6969
298. Travaglini L, Vian L, Billi M, Grignani F, Nervi C. Epigenetic reprogramming of breast cancer cells by valproic acid occurs regardless of estrogen receptor status. Int J Biochem Cell Biol (2009) 41(1):225–34. doi: 10.1016/j.biocel.2008.08.019
299. Schech A, Kazi A, Yu S, Shah P, Sabnis G. Histone deacetylase inhibitor entinostat inhibits tumor-initiating cells in triple-negative breast cancer cells. Mol Cancer Ther (2015) 14(8):1848–57. doi: 10.1158/1535-7163.MCT-14-0778
300. Nalls D, Tang SN, Rodova M, Srivastava RK, Shankar S. Targeting epigenetic regulation of mir-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PloS One (2011) 6(8):e24099. doi: 10.1371/journal.pone.0024099
301. Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell (2009) 138(3):592–603. doi: 10.1016/j.cell.2009.07.011
302. Gwak JM, Kim HJ, Kim EJ, Chung YR, Yun S, Seo AN, et al. MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast cancer. Breast Cancer Res Treat (2014) 147(1):39–49. doi: 10.1007/s10549-014-3069-5
303. De Leeuw DC, Denkers F, Olthof MC, Rutten AP, Pouwels W, Schuurhuis GJ, et al. Attenuation of microRNA-126 expression that drives CD34+38 - stem/progenitor cells in acute myeloid leukemia leads to tumor eradication. Cancer Res (2014) 74(7):2094–105. doi: 10.1158/0008-5472.CAN-13-1733
304. Miranda Furtado CL, Dos Santos Luciano MC, Da Silva Santos R, Furtado GP, Moraes MO, Pessoa C. Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics (2019) 14(12):1164–76. doi: 10.1080/15592294.2019.1640546
305. Tallerico R, Todaro M, Di Franco S, Maccalli C, Garofalo C, Sottile R, et al. Human NK cells selective targeting of colon cancer–initiating cells: A role for natural cytotoxicity receptors and MHC class I molecules. J Immunol (2013) 190(5):2381–90. doi: 10.4049/jimmunol.1201542
306. Tseng HC, Arasteh A, Paranjpe A, Teruel A, Yang W, Behel A, et al. Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or reprogramming activates NK cells. PloS One (2013) 190(5):2381–90. doi: 10.4049/jimmunol.1201542
307. Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol (2009) 182(6):3530–9. doi: 10.4049/jimmunol.0802845
308. Lai D, Wang F, Chen Y, Wang C, Liu S, Lu B, et al. Human ovarian cancer stem-like cells can be efficiently killed by γδ T lymphocytes. Cancer Immunol Immunother (2012) 61(7):979–89. doi: 10.1007/s00262-011-1166-4
309. Todaro M, D’Asaro M, Caccamo N, Iovino F, Francipane MG, Meraviglia S, et al. Efficient killing of human colon cancer stem cells by γδ T lymphocytes. J Immunol (2009) 182(11):7287–96. doi: 10.4049/jimmunol.0804288
310. Zhang D, Tang DG, Rycaj K. Cancer stem cells: Regulation programs, immunological properties and immunotherapy. Semin Cancer Biol (2018) 52:94–106. doi: 10.1016/j.semcancer.2018.05.001
311. Lu L, Tao H, Chang AE, Hu Y, Shu G, Chen Q, et al. Cancer stem cell vaccine inhibits metastases of primary tumors and induces humoral immune responses against cancer stem cells. Oncoimmunology. (2015) 4(3):1–11. doi: 10.4161/2162402X.2014.990767
312. Ning N, Pan Q, Zheng F, Teitz-Tennenbaum S, Egenti M, Yet J, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res (2012) 72(7):1853–64. doi: 10.1158/0008-5472.CAN-11-1400
313. Jachetti E, Mazzoleni S, Grioni M, Ricupito A, Brambillasca C, Generoso L, et al. Prostate cancer stem cells are targets of both innate and adaptive immunity and elicit tumor-specific immune responses. Oncoimmunology. (2013) 2(5):1–12. doi: 10.4161/onci.24520
314. Van Phuc P, Hou CJ, Thi N, Nguyet M, Thuy DT, Van Dong L, et al. Effects of breast cancer stem cell extract primed dendritic cell transplantation on breast cancer tumor murine models. Annu Rev Res Biol (2011) 1(1):1–13.
315. McNeel DG, Becker JT, Eickhoff JC, Johnson LE, Bradley E, Pohlkamp I, et al. Real-time immune monitoring to guide plasmid DNA vaccination schedule targeting prostatic acid phosphatase in patients with castration-resistant prostate cancer. Clin Cancer Res (2014) 20(14):3692–704. doi: 10.1158/1078-0432.CCR-14-0169
316. Pavlenko M, Roos AK, Lundqvist A, Palmborg A, Miller AM, Ozenci V, et al. A phase I trial of DNA vaccination with a plasmid expressing prostate-specific antigen in patients with hormone-refractory prostate cancer. Br J Cancer (2004) 91(4):688–94. doi: 10.1038/sj.bjc.6602019
317. Nishizawa S, Hirohashi Y, Torigoe T, Takahashi A, Tamura Y, Mori T, et al. HSP DNAJB8 controls tumor-initiating ability in renal cancer stem-like cells. Cancer Res (2012) 72(11):2844–54. doi: 10.1158/0008-5472.CAN-11-3062
318. Polakova I, Duskova M, Smahel M. Antitumor DNA vaccination against the Sox2 transcription factor. Int J Oncol (2014) 45(1):139–46. doi: 10.3892/ijo.2014.2402
319. Visus C, Wang Y, Lozano-Leon A, Ferris RL, Silver S, Szczepanski MJ, et al. Targeting ALDH bright human carcinoma-initiating cells with ALDH1A1-specific CD8 + T cells. Clin Cancer Res (2011) 17(19):6174–84. doi: 10.1158/1078-0432.CCR-11-1111
320. Luo H, Zeng C, Fang C, Seeruttun SR, Lv L, Wang W. A new strategy using ALDHhigh-CD8+T cells to inhibit tumorigenesis. PloS One (2014) 9(8):1–7. doi: 10.1371/journal.pone.0103193
321. Beard RE, Zheng Z, Lagisetty KH, Burns WR, Tran E, Hewitt SM, et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer (2014) 2(1):1–11. doi: 10.1186/2051-1426-2-25
322. Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget (2015) 6(1):171–84. doi: 10.18632/oncotarget.2767
323. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther (2012) 23(10):1043–53. doi: 10.1089/hum.2012.041
324. Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, et al. Stem-like tumor-initiating cells isolated from IL13Rα2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T cells. Clin Cancer Res (2012) 18(8):2199–209. doi: 10.1158/1078-0432.CCR-11-1669
325. Deng Z, Wu Y, Ma W, Zhang S, Zhang YQ. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol (2015) 16(1):1–9. doi: 10.1186/s12865-014-0064-x
326. Ghatak S, Misra S, Toole BP. Hyaluronan oligosaccharides inhibit anchorage-independent growth of tumor cells by suppressing the phosphoinositide 3-kinase/Akt cell survival pathway. J Biol Chem (2002) 277(41):38013–20. doi: 10.1074/jbc.M202404200
327. Narula J, Bigby M, Che X, Sy MS. Inhibition of human melanoma growth and metastasis in vivo by anti-CD44 monoclonal antibody. Cancer Res (1994) 54(6):1561–5.
328. Huang J, Li C, Wang Y, Lv H, Guo Y, Dai H, et al. Cytokine-induced killer (CIK) cells bound with anti-CD3/anti-CD133 bispecific antibodies target CD133high cancer stem cells in vitro and in vivo. Clin Immunol (2013) 149(1):156–68. doi: 10.1016/j.clim.2013.07.006
329. Swain SM, Kim SB, Cortés J, Ro J, Semiglazov V, Campone M, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): Overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol (2013) 14(6):461–71. doi: 10.1016/S1470-2045(13)70130-X
330. Diéras V, Miles D, Verma S, Pegram M, Welslau M, Baselga J, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol (2017) 8(6):732–42. doi: 10.1016/S1470-2045(17)30312-1
331. Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res (2010) 16(2):474–85. doi: 10.1158/1078-0432.CCR-09-1322
332. Wu A, Wiesner S, Xiao J, Ericson K, Chen W, Hall WA, et al. Expression of MHC I and NK ligands on human CD133+ glioma cells: possible targets of immunotherapy. J Neurooncol (2007) 83(2):121–31. doi: 10.1007/s11060-006-9265-3
333. Wang B, Wang Q, Wang Z, Jiang J, Yu SC, Ping YF, et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res (2014) 74(20):5746–57. doi: 10.1158/0008-5472.CAN-13-2563
334. Zhou L, Lu L, Wicha MS, Chang AE, Xia JC, Ren X, et al. Promise of cancer stem cell vaccine. Hum Vaccines Immunother (2015) 11(12):2796–9. doi: 10.1080/21645515.2015.1083661
335. Xu Q, Liu G, Yuan X, Xu M, Wang H, Ji J, et al. Antigen-specific T-cell response from dendritic cell vaccination using cancer stem-like cell-associated antigens. Stem Cells (2009) 27(8):1734–40. doi: 10.1002/stem.102
336. Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. (2006) 25(34):4777–86. doi: 10.1038/sj.onc.1209603
337. Yen KE, Bittinger MA, Su SM, Fantin VR. Cancer-associated IDH mutations: Biomarker and therapeutic opportunities. Oncogene. (2010) 29(49):6409–17. doi: 10.1038/onc.2010.444
338. Croteau E, Renaud JM, Richard MA, Ruddy TD, Bénard F, deKemp RA. PET metabolic biomarkers for cancer. biomark Cancer (2016) 8(Suppl 2):61–9. doi: 10.4137/BIC.S27483
339. Bhatt AN, Mathur R, Farooque A, Verma A, Dwarakanath BS. Cancer biomarkers - current perspectives. Indian J Med Res (2010) 132(8):129–49.
340. Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E, et al. Aberrant methylation of p16INK4a is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci U S A. (1998) 95(20):11891–6. doi: 10.1073/pnas.95.20.11891
341. Hoque MO, Begum S, Topaloglu O, Jeronimo C, Mambo E, Westra WH, et al. Detection of promoter hypermethylation of multiple genes in the tumor and bronchoalveolar lavage of patients with lung cancer. Cancer Res (2004) 10:2284–8. doi: 10.1158/1078-0432.ccr-1111-3.
342. Kloten V, Becker B, Winner K, Schrauder MG, Fasching PA, Anzeneder T, et al. Promoter hypermethylation of the tumor-suppressor genes ITIH5, DKK3, and RASSF1A as novel biomarkers for blood-based breast cancer screening. Breast Cancer Res (2013) 15(1):R4. doi: 10.1186/bcr3375
343. Kim MS, Lee J, Sidransky D. DNA Methylation markers in colorectal cancer. Cancer Metastasis Rev (2010) 29(1):181–206. doi: 10.1007/s10555-010-9207-6
344. Jung G, Hernández-Illán E, Moreira L, Balaguer F, Goel A. Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat Rev Gastroenterol Hepatol (2020) 17(2):111–30. doi: 10.1038/s41575-019-0230-y
345. Coppedè F, Lopomo A, Spisni R, Migliore L. Genetic and epigenetic biomarkers for diagnosis, prognosis and treatment of colorectal cancer. World J Gastroenterol (2014) 20(4):943–56. doi: 10.3748/wjg.v20.i4.943
346. Zachariah MA, Oliveira-Costa JP, Carter BS, Stott SL, Nahed BV. Blood-based biomarkers for the diagnosis and monitoring of gliomas. Neuro Oncol (2018) 20(9):1155–61. doi: 10.1093/neuonc/noy074
347. Costa-pinheiro P, Montezuma D. Diagnostic and prognostic epigenetic biomarkers in cancer. Epigenomics. (2015) 7(6):1003–15. doi: 10.2217/epi.15.56
348. Holdhoff M, Yovino SG, Boadu O, Grossman SA. Blood-based biomarkers for malignant gliomas. J Neurooncol (2013) 113(3):345–52. doi: 10.1007/s11060-013-1144-0
349. Lugli A, Iezzi G, Hostettler I, Muraro MG, Mele V, Tornillo L, et al. Prognostic impact of the expression of putative cancer stem cell markers CD133, CD166, CD44s, EpCAM, and ALDH1 in colorectal cancer. Br J Cancer (2010) 103(3):382–90. doi: 10.1038/sj.bjc.6605762
350. Su Y, Qiu Q, Zhang X, Jiang Z, Leng Q, Liu Z, et al. Aldehyde dehydrogenase 1 A1-positive cell population is enriched in tumor-initiating cells and associated with progression of bladder cancer. Cancer Epidemiol Biomarkers Prev (2010) 19(2):327–37. doi: 10.1158/1055-9965.EPI-09-0865
351. Ludwig K, Kornblum HI. Molecular markers in glioma. J Neurooncol (2017) 134(3):505–12. doi: 10.1007/s11060-017-2379-y
352. Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauß A, et al. Cancer stem cells–origins and biomarkers: Perspectives for targeted personalized therapies. Front Immunol (2020) 11(August):1–33. doi: 10.3389/fimmu.2020.01280
353. Wang K, Chen X, Zhan Y, Jiang W, Liu X, Wang X, et al. Increased expression of ALDH1A1 protein is associated with poor prognosis in clear cell renal cell carcinoma. Med Oncol (2013) 30(2):574. doi: 10.1007/s12032-013-0574-z
354. Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res (2009) 69(8):3382–9. doi: 10.1158/0008-5472.CAN-08-4418
355. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell (2007) 1(5):555–67. doi: 10.1016/j.stem.2007.08.014
356. Li T, Su Y, Mei Y, Leng Q, Leng B, Liu Z, et al. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients outcome. Lab Investig (2010) 90(2):234–44. doi: 10.1038/labinvest.2009.127
357. Islam F, Gopalan V, Wahab R, Smith RA, Lam AKY. Cancer stem cells in oesophageal squamous cell carcinoma: Identification, prognostic and treatment perspectives. Crit Rev Oncol Hematol (2015) 96(1):9–19. doi: 10.1016/j.critrevonc.2015.04.007
358. Brungs D, Aghmesheh M, Vine KL, Becker TM, Carolan MG, Ranson M. Gastric cancer stem cells: evidence, potential markers, and clinical implications. J Gastroenterol (2016) 51(4):313–26. doi: 10.1007/s00535-015-1125-5
359. Lingala S, Cui YY, Chen X, Ruebner BH, Qian XF, Zern MA, et al. Immunohistochemical staining of cancer stem cell markers in hepatocellular carcinoma. Exp Mol Pathol (2010) 89(1):27–35. doi: 10.1016/j.yexmp.2010.05.005
360. Rasheed ZA, Yang J, Wang Q, Kowalski J, Freed I, Murter C, et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J Natl Cancer Inst (2010) 102(5):340–51. doi: 10.1093/jnci/djp535
361. O’Flaherty JD, Barr M, Fennell D, Richard D, Reynolds J, O’Leary J, et al. The cancer stem-cell hypothesis: Its emerging role in lung cancer biology and its relevance for future therapy. J Thorac Oncol (2012) 7(12):1880–90. doi: 10.1097/JTO.0b013e31826bfbc6
362. Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer (2014) 135(12):2741–8. doi: 10.1002/ijc.28690
363. Jeter CR, Badeaux M, Choy G, Chandra D, Patrawala L, Liu C, et al. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells (2009) 27(5):993–1005. doi: 10.1002/stem.29
364. Niu CS, Li DX, Liu YH, Fu XM, Tang SF, Li J. Expression of NANOG in human gliomas and its relationship with undifferentiated glioma cells. Oncol Rep (2011) 26(3):593–601. doi: 10.3892/or.2011.1308
365. Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res (2008) 68(11):4311–20. doi: 10.1158/0008-5472.CAN-08-0364
366. Yu CC, Chen YW, Chiou GY, Tsai LL, Huang PI, Chang CY, et al. MicroRNA let-7a represses chemoresistance and tumourigenicity in head and neck cancer via stem-like properties ablation. Oral Oncol (2011) 47(3):202–10. doi: 10.1016/j.oraloncology.2010.12.001
367. Golestaneh AF, Atashi A, Langroudi L, Shafiee A, Ghaemi N, Soleimani M. miRNAs expressed differently in cancer stem cells and cancer cells of human gastric cancer cell line MKN-45. Cell Biochem Funct (2012) 30(5):411–8. doi: 10.1002/cbf.2815
368. Sun C, Sun L, Jiang K, Gao DM, Kang XN, Wang C, et al. NANOG promotes liver cancer cell invasion by inducing epithelial- mesenchymal transition through NODAL/SMAD3 signaling pathway. Int J Biochem Cell Biol (2013) 45(6):1099–108. doi: 10.1016/j.biocel.2013.02.017
369. Zhang J, Espinoza LA, Kinders RJ, Lawrence SM, Pfister TD, Zhou M, et al. NANOG modulates stemness in human colorectal cancer. Oncogene (2013) 32(37):4397–405. doi: 10.1038/onc.2012.461
370. Lagadec C, Vlashi E, Della Donna L, Dekmezian C, Pajonk F. Radiation-induced reprogramming of breast cancer cells. Stem Cells (2012) 30(5):833–44. doi: 10.1002/stem.1058
371. Hsu HS, Huang PI, Chang YL, Tzao C, Chen YW, Shih HC, et al. Cucurbitacin i inhibits tumorigenic ability and enhances radiochemosensitivity in nonsmall cell lung cancer-derived CD133-positive cells. Cancer. (2011) 117(13):2970–85. doi: 10.1002/cncr.25869
372. Kaufhold S, Garbán H, Bonavida B. Yin yang 1 is associated with cancer stem cell transcription factors (SOX2, OCT4, BMI1) and clinical implication. J Exp Clin Cancer Res (2016) 35(1):1–14. doi: 10.1186/s13046-016-0359-2
373. Guo Y, Liu S, Wang P, Zhao S, Wang F, Bing L, et al. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and nanog in human gliomas. Histopathology. (2011) 59(4):763–75. doi: 10.1111/j.1365-2559.2011.03993.x
374. Mohiuddin IS, Wei SJ, Kang MH. Role of OCT4 in cancer stem-like cells and chemotherapy resistance. Biochim Biophys Acta - Mol Basis Dis (2020) 1866(4):165432. doi: 10.1016/j.bbadis.2019.03.005
375. Van Schaijik B, Davis PF, Wickremesekera AC, Tan ST, Itinteang T. Subcellular localisation of the stem cell markers OCT4, SOX2, NANOG, KLF4 and c-MYC in cancer: A review. J Clin Pathol (2018) 71(1):88–91. doi: 10.1136/jclinpath-2017-204815
376. Zhang Q, Han Z, Zhu Y, Chen J, Li W. The role and specific mechanism of OCT4 in cancer stem cells: A review. Int J Stem Cells (2020) 13(3):312–25. doi: 10.15283/ijsc20097
377. Gzil A, Zarębska I, Bursiewicz W, Antosik P, Grzanka D, Szylberg Ł. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol Biol Rep (2019) 46(6):6629–45. doi: 10.1007/s11033-019-05058-1
378. Klonisch T, Wiechec E, Hombach-Klonisch S, Ande SR, Wesselborg S, Schulze-Osthoff K, et al. Cancer stem cell markers in common cancers - therapeutic implications. Trends Mol Med (2008) 14(10):450–60. doi: 10.1016/j.molmed.2008.08.003
Keywords: cancer stem cells, metabolism, epigenetics, cross talks, oncometabolites
Citation: Sahoo OS, Pethusamy K, Srivastava TP, Talukdar J, Alqahtani MS, Abbas M, Dhar R and Karmakar S (2022) The metabolic addiction of cancer stem cells. Front. Oncol. 12:955892. doi: 10.3389/fonc.2022.955892
Received: 29 May 2022; Accepted: 28 June 2022;
Published: 25 July 2022.
Edited by:
Aamir Ahmad, PhD University of Alabama at Birmingham, United StatesReviewed by:
Shamir Cassim, Addmedica, FranceCopyright © 2022 Sahoo, Pethusamy, Srivastava, Talukdar, Alqahtani, Abbas, Dhar and Karmakar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Subhradip Karmakar, c3ViaHJhZGlwLmtAYWlpbXMuZWR1; Ruby Dhar, cnVieWRoYXJAZ21haWwuY29t
†ORCID: Om Saswat Sahoo, orcid.org/0000-0003-4114-9461
Karthikeyan Pethusamy, orcid.org/0000-0001-7155-2257
Tryambak Pratap Srivastava, orcid.org/0000-0002-7903-5876
Joyeeta Talukdar, orcid.org/0000-0002-4922-2283
Ruby Dhar, orcid.org/0000-0003-3600-6554
Subhradip Karmakar, orcid.org/0000-0002-4757-8729
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.