
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 02 August 2022
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.954879
This article is part of the Research Topic Improving our Understanding of the Management and Pathogenesis of Rare and Neglected Tumors of the Central and Peripheral Nervous System View all 23 articles
 Mythili Merchant1Margarita Raygada1
Mythili Merchant1Margarita Raygada1 Ying Pang1Martha Quezado2
Ying Pang1Martha Quezado2 Mark Raffeld2
Mark Raffeld2 Liqiang Xi2Jung Kim2
Liqiang Xi2Jung Kim2 Manoj Tyagi2Zied Abdullaev2
Manoj Tyagi2Zied Abdullaev2 Olga Kim1Zach Sergi1Tina Pillai1
Olga Kim1Zach Sergi1Tina Pillai1 Byram Ozer1
Byram Ozer1 Kareem Zaghloul3
Kareem Zaghloul3 John D. Heiss3
John D. Heiss3 Terri S. Armstrong1
Terri S. Armstrong1 Mark R. Gilbert1
Mark R. Gilbert1 Kenneth Aldape2
Kenneth Aldape2 Jing Wu1*
Jing Wu1*Most tumors, including brain tumors, are sporadic. However, a small subset of CNS tumors are associated with hereditary cancer conditions like Lynch Syndrome (LS). Here, we present a case of an oligodendroglioma, IDH-mutant and 1p/19q-codeleted, and LS with a germline pathogenic PMS2 mutation. To our knowledge, this has only been reported in a few cases in the literature. While the family history is less typical of LS, previous studies have indicated the absence of a significant family history in patient cohorts with PMS2 mutations due to its low penetrance. Notably, only a handful of studies have worked on characterizing PMS2 mutations in LS, and even fewer have looked at these mutations in the context of brain tumor development. This report aims to add to the limited literature on germline PMS2 mutations and oligodendrogliomas. It highlights the importance of genetic testing in neuro-oncology.
Oligodendroglioma, IDH-mutant and 1p/19q-codeleted, is a subset of diffuse gliomas that primarily develop sporadically. Very few patients with oligodendroglioma have been associated with a hereditary cancer predisposition syndrome. According to the most recent CBTRUS Statistical Report, these tumors have an adjusted annual incidence rate estimated at 0.11 cases per 100,000 population and account for 0.4% of all primary brain tumors (1). The most recent WHO classification of Central Nervous System (CNS) Tumors defines oligodendrogliomas as a diffusely infiltrating glioma with an isocitrate dehydrogenase (IDH) mutation and codeletion of chromosomes 1p and 19q (CNS WHO grade 2 or 3) (2).
Brain tumors have been associated with several hereditary syndromes including Lynch Syndrome (LS), which has an autosomal dominant inheritance pattern (3–5). In a study conducted to compare the incidence of brain tumors in families with LS versus that of the general population, it was found that the brain tumor incidence in LS families was 3.35% by the age of 85, compared to 0.47% in the general population (6). LS is caused by germline mutations in DNA mismatch repair (MMR) genes like MLH1, MSH2, MSH6, PMS1, and PMS2. Individuals that carry mutations in these genes have an increased susceptibility to cancers of the colon, endometrium, stomach, ovary, urinary tract, brain, skin, and many more (7–9). PMS2 accounts for approximately 5% of the pathogenic variants involved in LS and has a lower penetrance than the other MMR genes (9–11). There is limited validated data on germline monoallelic PMS2 mutations and, therefore, a lack of clinical and biological phenotype associations (10).
The patient is a 65-year-old gentleman diagnosed with a recurrent oligodendroglioma, IDH-mutant and 1p/19q-codeleted, CNS WHO grade 3. He was initially found to have a non-enhancing lesion in the left posterior temporal lobe at the age of 30. As illustrated in Figure 1, he received a craniotomy with tumor resection about thirty-six years ago. Pathological exam revealed an oligodendroglioma, grade 2. Following three years of clinical observation, he underwent a second tumor resection and received a course of radiation therapy following the surgery. His tumor remained stable until the disease progressed again twenty-three years after the initial diagnosis. At this time, the tumor was confirmed to be a recurrent oligodendroglioma, IDH-mutant and 1p/19q-codeleted, CNS WHO grade 3 after a tumor biopsy. He then received his first chemotherapy regimen, which involved 18 cycles of temozolomide. Approximately ten years later, the MRI findings were suggestive of disease progression (Figure 2A). He underwent a left temporoparietal craniotomy with resection of the tumor. Pathology demonstrated oligodendroglioma, IDH-mutant and 1p/19q-codeleted, CNS WHO grade 3 (Figure 2B). DNA methylation profiling was consistent with the diagnosis of oligodendroglioma, IDH-mutant and 1p/19q-codeleted (Figure 2C). He received his second regimen of chemotherapy with 6 cycles of temozolomide. The patient has been under clinical monitoring and has remained clinically stable since he completed the second round of chemotherapy.
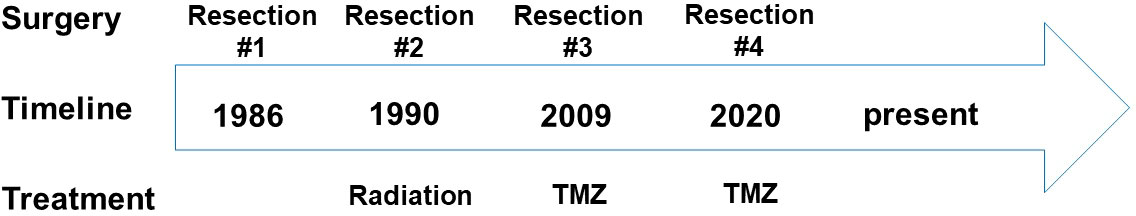
Figure 1 Timeline of the disease history and management. TMZ, Temozolomide.
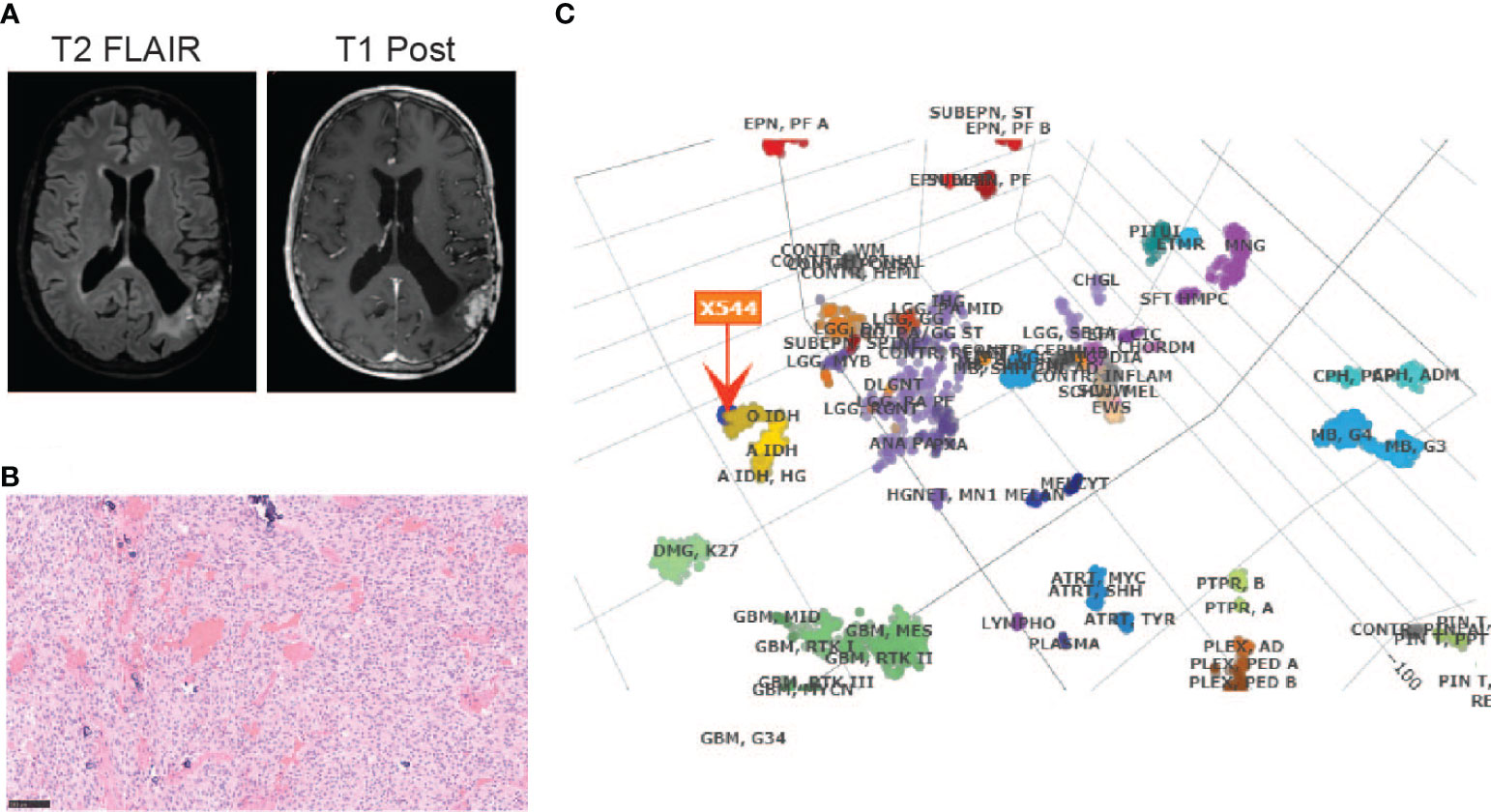
Figure 2 Diagnosis of an oligodendroglioma, IDH-mutant and 1p/19q co-deleted, CNS WHO grade 3 by (A) MRI before the most recent tumor resection; (B) H&E staining (20X) and (C) DNA methylation profiling of the tumor sample from the most recent tumor resection.
After his most recent tumor recurrence, matched Tumor/Normal Whole Exome Sequencing (T/N WES; NCI-COMPASS-NIH) revealed that he carries a pathogenic germline mutation in PMS2 (c.251-2A>T splice variant) (Figure 3A). This splice alteration affects a canonical splice acceptor at nucleotide position -2, upstream of coding exon 4 in PMS2. Despite the germline PMS2 mutation, it did not cause a complete loss of PMS2 protein expression, and all MMR proteins were positively expressed (Figure 3B). While his family history is less characteristic of LS, he has a brother with colon cancer (Figure 4). The patient, however, is unable to recall further details regarding the diagnosis, including age of the diagnosis. His father also had multiple myeloma, but there are no other known cancers or hereditary syndromes in the family.
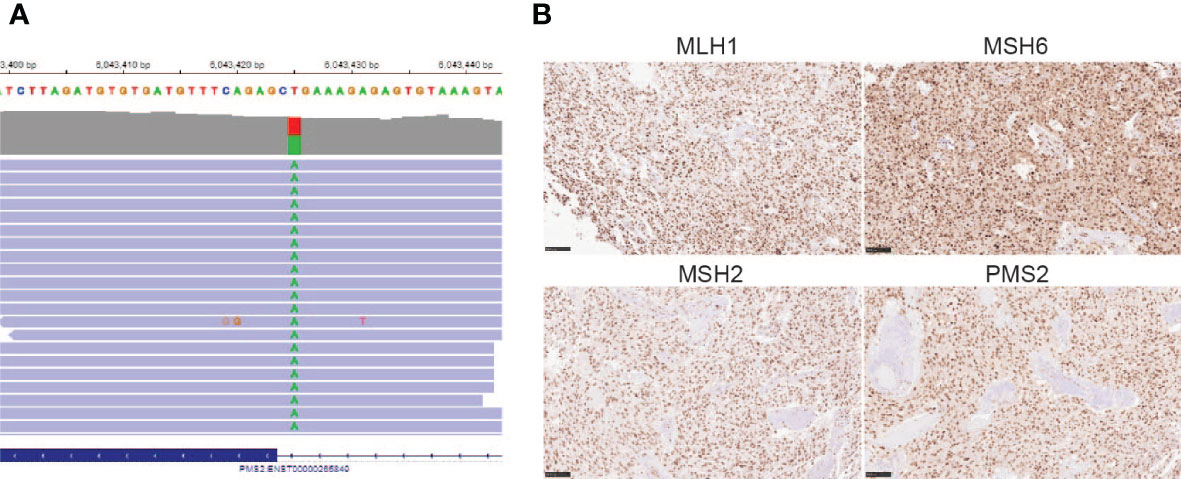
Figure 3 (A) PMS2 mutation found in the proband (c.251-2A>T); (B) Immunohistochemistry staining of MMR proteins, including MLH1, MSH6, MSH2, and PMS2 (20X) using the tumor sample from the most recent tumor resection.
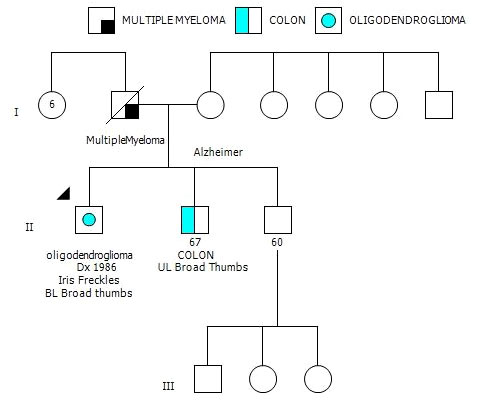
Figure 4 Three-generation family history pedigree.
The patient was counseled on the implications of the PMS2 germline mutation, and he was advised to undergo a colonoscopy as surveillance in accordance with the new germline finding. Although he does not have children, the patient believed that it was important for him to share the information with his brother who had a diagnosis of colon cancer. He also planned to share his genetic testing results with his primary care physician so that his medical care can be customized to this condition.
In this case report, we describe the case of a 65-year-old man diagnosed with a recurrent oligodendroglioma, IDH-mutant and 1p/19q co-deleted, CNS WHO grade 3. The tumor also harbored a pathogenic mutation in PMS2, a DNA MMR gene, which was determined to be a germline mutation. PMS2 is associated with LS and in turn, with a predisposition to several types of cancers, including brain tumors.
Although uncommon, several types of brain tumors are associated with hereditary cancer syndromes like LS. Specifically, LS confers a 4-fold increased risk of developing brain tumors (12, 13). While glioblastoma is the most frequent histological subtype of brain tumors that are found in LS families (56%), other diffuse astrocytomas (22%) and oligodendrogliomas (9%) are also often found (12). Previous studies that have looked at the association between brain tumors and LS have found that 68% of patients have mutations in MSH2, 15% had mutations in MLH1, 15% in MSH6, and 2% in PMS2 (12), with most showing a loss of corresponding protein expression.
Due to the reduced penetrance and genetic testing complications of monoallelic PMS2 mutations, biallelic PMS2 mutations are more commonly detected and better characterized, especially in constitutional MMR deficiency (CMMRD) syndrome (14–16). Several studies have examined the association between biallelic PMS2 mutations and brain tumors (16–18). One such study reported a case of two affected sisters, one who died of a grade 3 oligodendroglioma and colorectal tumor (the proband), and the other who died of a neuroblastoma at a very early age, with no history of tumors in their parents. The proband was compound heterozygous for PMS2 (19). Another study in a five-generation Pakistani family revealed six affected individuals who died of brain tumors at very young ages, of which three had biallelic PMS2 mutations (20).
Carriers of monoallelic PMS2 mutations are at an increased risk for cancer, however, less is known about monoallelic PMS2 mutations and their association with brain tumors (21). A report focused on characterizing monoallelic PMS2 mutations found that only 6.2% of the class 4/5 variant carriers had a family history that fulfilled the Amsterdam diagnosis criteria (10). Another study that aimed to explore the clinical phenotype of germline PMS2 mutations uncovered that if clinicians relied solely on Amsterdam criteria to identify potential PMS2 mutation carriers, 90.9% of the mutation carriers in their study would not have been identified, or if clinicians relied on revised Bethesda guidelines alone, 25% of the mutation carriers in their study would have been missed (22). Our patient also did not have a clear family history suggestive of LS. Despite the PMS2 germline mutation, there was still positive protein expression in the proband, potentially describing why the patient did not have a strong LS phenotype. Moreover, retained protein expression of PMS2 also makes it hard to exclude the possibility of a coincidence in the occurrence of two moderately rare but unrelated diseases. Senter et al. further demonstrated that PMS2-associated LS presents highly variable clinical characteristics. For example, the age of the first LS-associated tumor in monoallelic PMS2 carriers varies from 23-77 years (22). Since half of the carriers are diagnosed with cancer before the age of 45, the general population screening recommendations for colon cancer beginning at the age of 45 would not be adequate for families with a PMS2 mutation.
The specific PMS2 mutation in the patient reported here has a gnomAD allele frequency of 0.0004%, underscoring its rarity. To date, it has only been previously reported in few other cases – in an individual with suspected LS (23), in a patient with colorectal cancer and LS (10), and in a 75-year-old patient with sarcoma (24). There are multiple layers of rarity in our case – the low incidence of oligodendrogliomas diagnosed with LS, the rare association of monoallelic PMS2 mutations with brain tumors, and the extremely low gnomAD allele frequency of the c.251-2A>T mutation in PMS2.
These findings emphasize that despite the uncommon nature of hereditary cancer syndromes in neuro-oncology, genetic testing and counseling can help identify genetic conditions that may require specific surveillance regimens, in turn improving patient care. Since early detection could lead to early intervention and routine surveillance, brain imaging may be indicated in some patients with LS.
The data analyzed in this study is subject to the following licenses/restrictions: a. The reported PMS2 mutation in this manuscript is from the germline whole exome sequencing. Other than this alteration, the rest of the dataset cannot be shared with the public, even by request. b. Requests to access these datasets should be directed to Jing Wu, amluZy53dTNAbmloLmdvdg==.
The studies involving human participants were reviewed and approved by the Institutional Review Board of the National Institutes of Health. The participant provided the written informed consent to participate in this study.
MM, MRay, YP, OK, ZS, and JW drafted the manuscript. MQ, MRaf, LX, JK, MT, ZA, and KA performed pathologic and molecular diagnostic testing. TP, BO, KZ, JH, TA, MG, and JW were involved in patient care. All authors contributed to the article and approved the submitted version.
This research was supported by the NIH Intramural Research Program and Lasker Clinical Research Scholar Program. Clinical genetics and genetic counseling were supported by the NCI-CONNECT program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the united states in 2014-2018. Neuro Oncol (2021) 23(12 Suppl 2):iii1–iii105. doi: 10.1093/neuonc/noab200
2. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
3. Lynch HT, de la Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet (1999) 36(11):801–18.
4. de Jong AE, Hendriks YM, Kleibeuker JH, de Boer SY, Cats A, Griffioen G, et al. Decrease in mortality in lynch syndrome families because of surveillance. Gastroenterology (2006) 130(3):665–71. doi: 10.1053/j.gastro.2005.11.032
5. Haraldsdottir S, Rafnar T, Frankel WL, Einarsdottir S, Sigurdsson A, Hampel H, et al. Comprehensive population-wide analysis of lynch syndrome in Iceland reveals founder mutations in MSH6 and PMS2. Nat Commun (2017) 8:14755. doi: 10.1038/ncomms14755
6. Vasen HF, Sanders EA, Taal BG, Nagengast FM, Griffioen G, Menko FH, et al. The risk of brain tumours in hereditary non-polyposis colorectal cancer (HNPCC). Int J Cancer (1996) 65(4):422–5. doi: 10.1002/(SICI)1097-0215(19960208)65:4<422::AID-IJC4>3.0.CO;2-Z
7. Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer (1999) 81(2):214–8. doi: 10.1002/(SICI)1097-0215(19990412)81:2<214::AID-IJC8>3.0.CO;2-L
8. Watson P, Riley B. The tumor spectrum in the lynch syndrome. Fam Cancer (2005) 4(3):245–8. doi: 10.1007/s10689-004-7994-z
9. Talseth-Palmer BA, McPhillips M, Groombridge C, Spigelman A, Scott RJ. MSH6 and PMS2 mutation positive Australian lynch syndrome families: novel mutations, cancer risk and age of diagnosis of colorectal cancer. Hered Cancer Clin Pract (2010) 8(1):5. doi: 10.1186/1897-4287-8-5
10. Wang Q, Leclerc J, Bougeard G, Olschwang S, Vasseur S, Cassinari K, et al. Characterisation of heterozygous PMS2 variants in French patients with lynch syndrome. J Med Genet (2020) 57(7):487–99. doi: 10.1136/jmedgenet-2019-106256
11. ten Broeke SW, Brohet RM, Tops CM, van der Klift HM, Velthuizen ME, Bernstein I, et al. Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. J Clin Oncol (2015) 33(4):319–25. doi: 10.1200/JCO.2014.57.8088
12. Therkildsen C, Ladelund S, Rambech E, Persson A, Petersen A, Nilbert M. Glioblastomas, astrocytomas and oligodendrogliomas linked to lynch syndrome. Eur J Neurol (2015) 22(4):717–24. doi: 10.1111/ene.12647
13. Koornstra JJ, Mourits MJ, Sijmons RH, Leliveld AM, Hollema H, Kleibeuker JH. Management of extracolonic tumours in patients with lynch syndrome. Lancet Oncol (2009) 10(4):400–8. doi: 10.1016/S1470-2045(09)70041-5
14. Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle N, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'care for CMMRD' (C4CMMRD). J Med Genet (2014) 51(6):355–65. doi: 10.1136/jmedgenet-2014-102284
15. Ramchander NC, Ryan NA, Crosbie EJ, Evans DG. Homozygous germ-line mutation of the PMS2 mismatch repair gene: a unique case report of constitutional mismatch repair deficiency (CMMRD). BMC Med Genet (2017) 18(1):40. doi: 10.1186/s12881-017-0391-x
16. De Vos M, Hayward BE, Charlton R, Taylor GR, Glaser AW, Picton S, et al. PMS2 mutations in childhood cancer. J Natl Cancer Inst (2006) 98(5):358–61. doi: 10.1093/jnci/djj073
17. Kruger S, Kinzel M, Walldorf C, Gottschling S, Bier A, Tinschert S, et al. Homozygous PMS2 germline mutations in two families with early-onset haematological malignancy, brain tumours, HNPCC-associated tumours, and signs of neurofibromatosis type 1. Eur J Hum Genet (2008) 16(1):62–72. doi: 10.1038/sj.ejhg.5201923
18. Agostini M, Tibiletti MG, Lucci-Cordisco E, Chiaravalli A, Morreau H, Furlan D, et al. Two PMS2 mutations in a turcot syndrome family with small bowel cancers. Am J Gastroenterol (2005) 100(8):1886–91. doi: 10.1111/j.1572-0241.2005.50441.x
19. De Rosa M, Fasano C, Panariello L, Scarano MI, Belli G, Iannelli A, et al. Evidence for a recessive inheritance of turcot's syndrome caused by compound heterozygous mutations within the PMS2 gene. Oncogene (2000) 19(13):1719–23. doi: 10.1038/sj.onc.1203447
20. Baig SM, Fatima A, Tariq M, Khan TN, Ali Z, Faheem M, et al. Hereditary brain tumor with a homozygous germline mutation in PMS2: pedigree analysis and prenatal screening in a family with constitutional mismatch repair deficiency (CMMRD) syndrome. Fam Cancer (2019) 18(2):261–5. doi: 10.1007/s10689-018-0112-4
21. Goodenberger ML, Thomas BC, Riegert-Johnson D, Boland CR, Plon SE, Clendenning M, et al. PMS2 monoallelic mutation carriers: the known unknown. Genet Med (2016) 18(1):13–9. doi: 10.1038/gim.2015.27
22. Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD, et al. The clinical phenotype of lynch syndrome due to germ-line PMS2 mutations. Gastroenterology (2008) 135(2):419–28. doi: 10.1053/j.gastro.2008.04.026
23. Yurgelun MB, Allen B, Kaldate RR, Bowles KR, Judkins T, Kaushik P, et al. Identification of a variety of mutations in cancer predisposition genes in patients with suspected lynch syndrome. Gastroenterology (2015) 149(3):604–13 e20. doi: 10.1053/j.gastro.2015.05.006
Keywords: lynch syndrome, PMS2, CNS, oligodendroglioma, IDH-mutant and 1p/19q-codeleted
Citation: Merchant M, Raygada M, Pang Y, Quezado M, Raffeld M, Xi L, Kim J, Tyagi M, Abdullaev Z, Kim O, Sergi Z, Pillai T, Ozer B, Zaghloul K, Heiss JD, Armstrong TS, Gilbert MR, Aldape K and Wu J (2022) Case report: Oligodendroglioma, IDH-mutant and 1p/19q-codeleted, associated with a germline mutation in PMS2. Front. Oncol. 12:954879. doi: 10.3389/fonc.2022.954879
Received: 27 May 2022; Accepted: 30 June 2022;
Published: 02 August 2022.
Edited by:
Laura Gatti, IRCCS Carlo Besta Neurological Institute Foundation, ItalyReviewed by:
Angela Mastronuzzi, Bambino Gesù Children’s Hospital (IRCCS), ItalyCopyright © 2022 Merchant, Raygada, Pang, Quezado, Raffeld, Xi, Kim, Tyagi, Abdullaev, Kim, Sergi, Pillai, Ozer, Zaghloul, Heiss, Armstrong, Gilbert, Aldape and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Wu, amluZy53dTNAbmloLmdvdg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.