 Guoqing Zhang
Guoqing Zhang Beibei Yan
Beibei Yan Yanan Guo
Yanan Guo Hang Yang
Hang Yang Jindong Li
Jindong Li- Department of Thoracic Surgery and Lung Transplantation, First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
EGFR TKIs are not curative, and targeted resistance inevitably results in therapeutic failure. Additionally, there are numerous uncommon EGFR mutations that are insensitive to EGFR TKIs, and there is a lack of clinical strategies to overcome these limitations. EGFR TKI and mAbs target EGFR at different sites, and a combination regimen for delaying/preventing resistance to targeted therapy or obtaining more intensive inhibition for uncommon mutations at cellular, animal and human levels has been explored. This review critically focuses on a combination strategy for uncommon EGFR mutation-positive NSCLC, and discuss the preclinical data, clinical implications, limitations and future prospects of the combination strategy.
Introduction
Over the past decades, systemic treatment strategies involving epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) have greatly improved outcomes and represent the backbone of treatment for advanced EGFR-mutant non-small-cell lung cancer (NSCLC) (1, 2). EGFR TKIs are now the standard treatment for classic mutations (such as exon 19del and exon 21 L858R point mutations), with median progression-free survival (PFS) ranging from 10-12 months (2–4) and 18.9 months (5) for first/second-generation EGFR TKIs and third-generation TKIs, respectively. However, EGFR TKIs are not curative, and targeted resistance inevitably results in therapeutic failure (6). Moreover, various primary and acquired uncommon mutations (such as EGFR-dependent or EGFR-independent mutations) have been reported, and such uncommon EGFR mutations may be associated with poor response (7) or even resistance (8) to EGFR-TKI monotherapy.
Comprehensive next-generation sequencing (NGS) has greatly promoted clinical research on targeted therapy. Strategies for targeted therapy to delay drug resistance or find effective targeted strategies for uncommon mutations have always been a hot topic in clinical research. Despite available information on resistance mechanisms and uncommon mutations, there is a paucity of clinical strategies for overcoming these limitations. To date, EGFR TKI combination therapy [with cytotoxic anticancer agents (9–12), angiogenesis inhibitors (13–16), or TKIs (17)] have been widely explored in this heterogeneous group of patients with regard to efficacy, safety and tolerability. Studies have also begun exploring combination therapy with EGFR TKIs and mAbs; we call this the “sandwich” strategy because EGFR is blocked by integrating EGFR TKIs intracellularly and EGFR mAbs extracellularly) (18, 19).
This review focuses specifically on the “sandwich” strategy for EGFR mutation-positive NSCLC, aiming to overcome drug resistance and discuss prospects for their use in clinical settings.
Mechanisms of Limited Responsiveness to EGFR-TKIs With Uncommon EGFR Mutations
Under normal physiological processes, EGFR forms a dimer when bound by ligands, such as EGF, EPF, TGFα, AR, BTC, HB-EGF and EPR, after which autophosphorylation of the tyrosine kinase domain (TKD) occurs, transmitting pro-proliferation signals in cells (20). Under circumstances of EGFR driver mutation, the TKD is homeostatically activated in a ligand-independent manner, leading to transmission of excessive pro-survival and pro-proliferation signals and resulting in cancer initiation and progression (21). The efficacy of EGFR mAbs involves blocking the binding of ligands to EGFR (part of the mechanism), inhibiting ligand-induced activation of TKD (22); the efficacy of EGFR TKIs is related to TKD binding, decreasing the relative affinity of TKD for ATP in a ligand-independent manner (23).
Intratumor heterogeneity (ITH) (24, 25) is defined as the a tumor containing different tumor cells (TCs) with different genomic features. Several studies (26, 27) have described the ITH and evolutionary process of NSCLC. Primary or acquired resistance is a direct consequence of preexisting ITH and continuous development of new therapy-resistant phenotypes. Broadly five mechanisms of drug resistance (primary and acquired) to EGFR TKIs have been reported, as follows (8): 1. EGFR-dependent mutations (5) (including exon 18 point mutation (L718Q, G724S), exon 19 point mutation (D761Y, L747S/P), exon 20 point mutation (S768I, T790M, L792F/H, G796S/R/D, C797S), exon 21 point mutation (V843I, T854A), exon 20ins mutation (28) and EGFR amplification (8, 29), with some complex EGFR mutations reported to be responsible for resistance acquisition (8, 30)); 2. EGFR-independent mutations (8), such as activation of alternative signaling pathways, including RET amplification, MET amplification, and HER2 amplification; 3. Mutations in downstream signaling genes (31) such as BRAF/KRAS mutation, PIK3CA mutation, and RAS/RAF/MEK/ERK mutation; 4. phenotype alteration, such as small-cell lung cancer (SCLC) transformation (29); 5. enhancement of autophagy (6) (Figure 1). Furthermore, these mechanisms may be coactivated in a single case, and their crosstalk can further complicate treatment and worsen patient outcome.
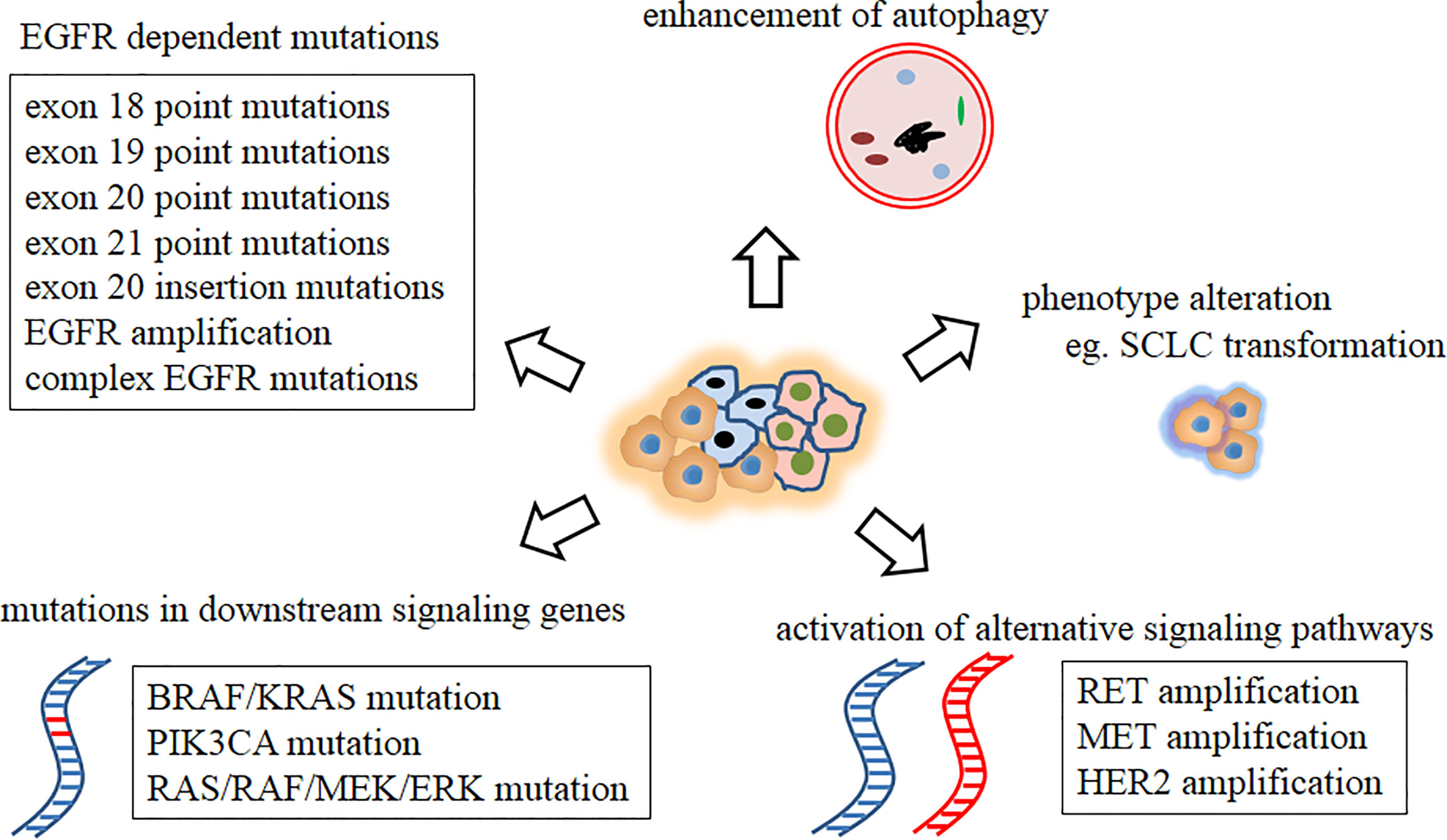
Figure 1 Mechanisms of drug resistance to EGFR TKIs. EGFR, epidermal growth factor receptor; SCLC, small cell lung cancer; TKI, tyrosine kinase inhibitor.
Targeting kinases with small molecular TKI directed at their well-characterized ATP-binding pockets or allosteric pockets has been carried out (32). Structures of uncommon mutations may interfere with the binding of targeted drugs to ATP-binding pockets or allosteric pockets, which may be a reason for the limited responsiveness to EGFR TKIs. Cases of uncommon EGFR mutations are highly heterogeneous (33). Specifically, these uncommon mutations consist of gatekeeper mutation (T790M), mutation causing steric hindrance (L718Q, L844V), mutation modifying the TKI-binding site (L798I, C797S), solvent-front mutation (G796S/R/D) and mutation within the hinge region (L792F/H). The T790M mutation in EGFR is located at a special position; it is often referred to as the “gatekeeper residue”, a structural location documented to interfere with inhibitor binding (20). By in silico protein structure modeling for TKI binding, Yang and colleagues revealed that the L718Q and L792H substitutions prevent osimertinib (a third-generation EGFR TKI) binding by introducing spatial confliction and decreasing local hydrophobicity. Furthermore, the L792 and L718 mutations markedly increase the half inhibitory concentration (IC50) of osimertinib in vitro (31), consistent with the in vivo conclusion. L844V mutation is reported to reduce WZ4002 (a third-generation EGFR TKI) binding and alter hydrophobic contacts with its inhibitor (34). As the second-generation irreversible EGFR TKI afatinib/dacomitinib and the third-generation EGFR TKI osimertinib bind covalently to Cys797 in the ATP-binding pocket (35), the occurrence of secondary mutations near the binding site (C797S (36), L798I (37)) theoretically leads to drug resistance. Uchibori et al. proved that C797S mutations reduce the affinity between osimertinib and the EGFR kinase domain and increase the relative affinity for ATP (38).
Possibility of the “Sandwich” Strategy in NSCLC
Using a highly sensitive locked nucleic acid (LNA)-based method, T790M has been detected in up to 68% of cases of EGFR TKI acquired resistance (39). Regarding drug resistance mechanisms to TKIs, EGFR-dependent mutations are observed in most cases (T790M for first/second-generation and loss of T790M and other EGFR uncommon and complex mutations for third-generation) (29). These data suggest instability of the EGFR signaling pathway or insufficient inhibition of EGFR by TKIs. Thus, it may be reasonable to combine EGFR TKI and EGFR mAbs for more intensive consolidation therapy in selected patients. Currently, EGFR TKIs used for the “sandwich” strategy include gefitinib, erlotinib, afatinib, EAI045, brigatinib and lazertinib; EGFR mAbs include cetuximab, necitumumab, panitumumab and amivantamab.
EGFR TKI and EGFR mAbs both target EGFR; however, some of their mechanisms of action and their blocking effects do not completely overlap (Figure 2). Low-molecular-weight TKIs block EGFR signaling by either competing with ATP (20) or changing the structure of EGFR (known as allosteric inhibition) (40). For high-molecular-weight mAbs, EGFR mAbs have the following mechanisms in addition to direct tumor inhibition by preventing ligand binding by blocking the ligand-binding extracellular domain. Under normal biological processes, the binding of ligand to EGFR results in cell surface receptor number downregulation via internalization of the ligand–receptor complex, which is eventually degraded in lysosomes (41). EGFR mAbs have the capacity to form receptor-containing complexes, which attenuate EGFR pathway signaling through receptor internalization. Additionally, studies have shown that EGFR TKIs can induce EGFR upregulation (8, 42–44), and EGFR mAbs have been proven to be able to decrease the level of cell surface EGFR (38) and abrogate the increase in TKI-induced EGFR transcription (44). From an immunological perspective, mAbs (such as cetuximab and necitumumab) of the IgG1 subtype have the ability to preferentially enhance affinity toward binding Fc with FcγR, leading to antibody-dependent cellular cytotoxicity (ADCC) or antibody-dependent cellular phagocytosis (ADCP) of tumor cells (45, 46). Interestingly, the human IgG2 isotype EGFR antibody panitumumab has Fc-mediated effector functions (45, 47). The mechanistic explanation is that panitumumab is highly potent in recruiting myeloid effector cells (such as PMN and M1 macrophages) for tumor cell killing by ADCC and ADCP (48). Therefore, theoretically, EGFR TKIs and mAbs exert synergistic antitumor effects while lowering the dose required for efficacy.
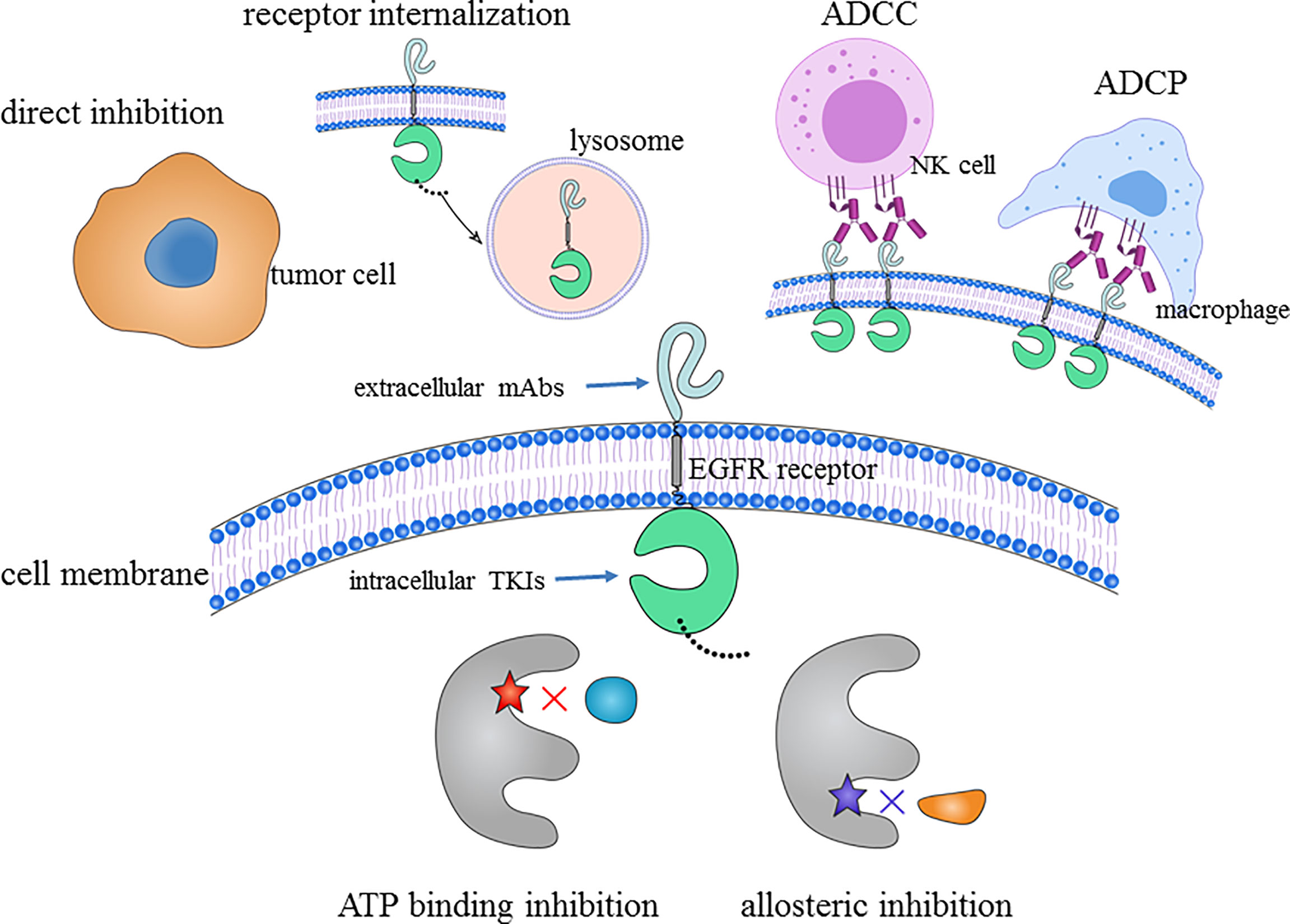
Figure 2 Antitumor mechanisms of the “sandwich” strategy: extracellular domain targeted by EGFR mAbs and intracellular domain targeted by EGFR TKIs. ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; EGFR, epidermal growth factor receptor; mAb, monoclonal antibody; NK, natural killer; TKI, tyrosine kinase inhibitor.
EGFR TKI and EGFR mAb target EGFR at different sites, and preclinical and clinical studies have revealed shared and complementary mechanisms of action, resulting in superior inhibition of EGFR, MAPK (mitogen-activated protein kinase), Akt phosphorylation, induction of apoptosis and vascularization inhibition of xenografts (49). The “sandwich” strategy has been explored in delaying/preventing resistance to targeted therapy or obtaining more intensive inhibition for uncommon mutations at the cellular, animal and human levels.`
Preclinical Data of the “Sandwich” Strategy
Early in the last century, scientists realized the unique capability of this combination regimen for EGFR inhibition (50). Numerous studies have demonstrated the benefit of EGFR TKI plus EGFR mAbs in lung cancer using in vitro and in vivo model systems (Table 1). Huang et al. found that gefitinib/erlotinib plus cetuximab shows significant cell proliferation inhibition, apoptosis promotion and tumor suppression in lung cancer cell lines and human lung cancer xenografts (51).
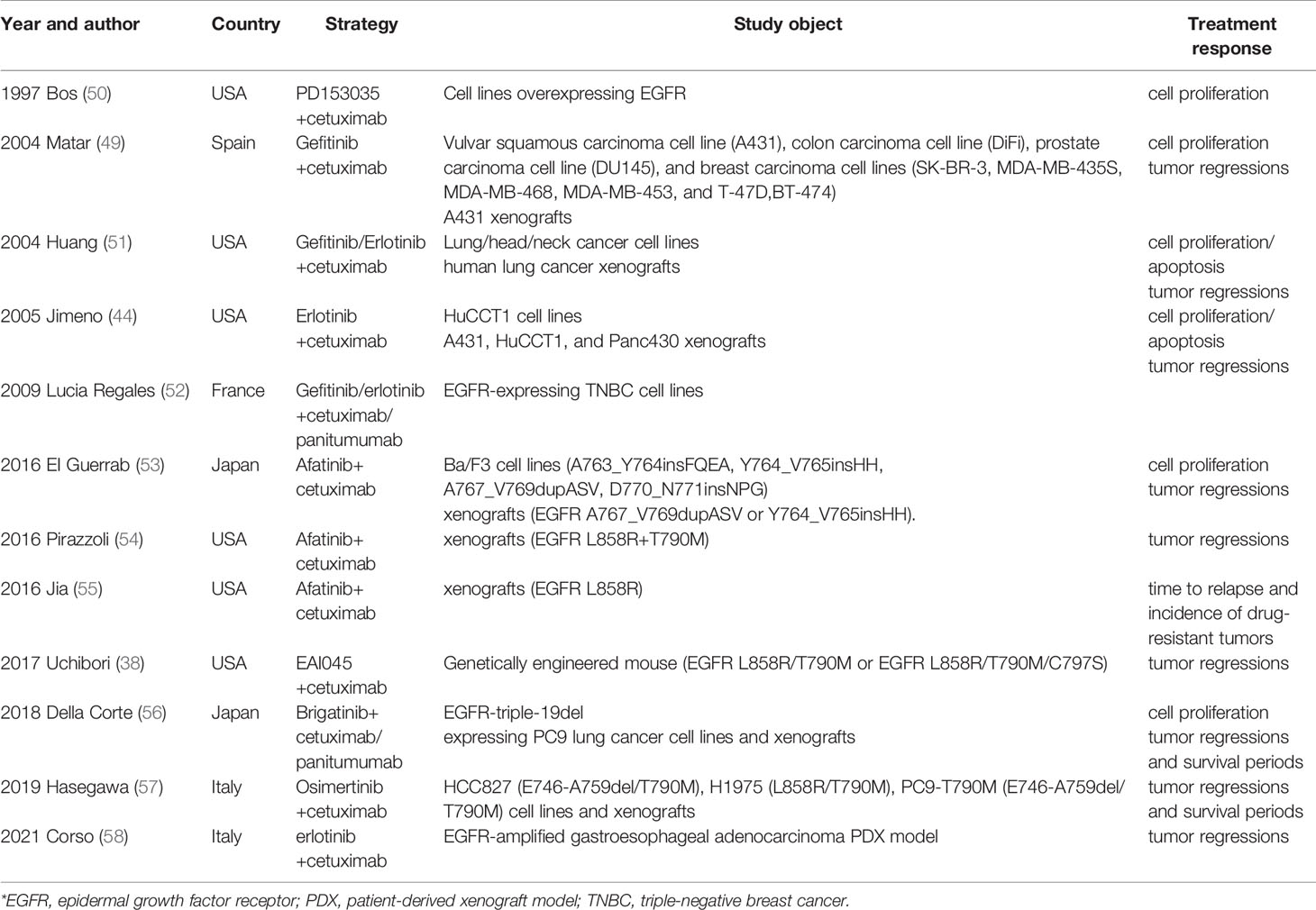
Table 1 Preclinical data evaluating the “sandwich” strategy.
Subsequent studies began to focus on specific types of mutations. EGFR exon 20ins is reportedly resistant to EGFR TKIs, and the afatinib- or osimertinib-based “sandwich strategy” has been proven to induce a more potent inhibitory effect against several EGFR exon 20ins mutations than either therapy alone in Ba/F3 cells (A763_Y764insFQEA, Y764_V765insHH, A767_V769dupASV, and D770_N771insNPG) and BALB/c-nu mice (EGFR A767_V769dupASV and EGFR Y764_V765insHH) (57). A similar study evaluated combinations of osimertinib + cetuximab in several NSCLC models with certain complex mutations (E746-A759del/T790M, L858R/T790M, and E746-A759del/T790M) as second-line treatment following development of resistance to osimertinib, and the results demonstrated that osimertinib + cetuximab may be a novel effective therapeutic option (56) Nevertheless, targeted treatment for patients with cis-C797S/T790M/classic mutations is difficult, and no effective therapeutic strategies have been reported. Uchibori et al. found brigatinib to be effective against cis-C797S/T790M/classic mutations in vitro and in vivo, and its effect was markedly enhanced by combination with cetuximab/panitumumab (38). Allosteric inhibitors also play an important role in the “sandwich” strategy. For example, EAI045 is an allosteric inhibitor targeting drug-resistant EGFR mutants with high selectivity and simultaneously spares wild-type EGFR. Jia et al. (55) tested the in vitro and in vivo efficacy of EAI045 in genetically engineered Ba/F3 cells and a mouse model, and only EAI045 in combination with cetuximab was effective for NSCLC driven by EGFR L858R/T790M and L858R/T790M/C797S.
These preclinical studies suggest a synergistic effect between EGFR TKIs and EGFR mAbs. However, it is not yet clear whether we can translate this research evidence from the preclinical stage to humans.
Clinical Implications of the “Sandwich” Strategy
A study evaluated the efficacy of gefitinib combined with cetuximab for patients with advanced/metastatic NSCLC previously treated with platinum-based chemotherapy without EGFR mutation or amplification, and the results showed that none achieved partial response (PR) (59). A similar result was obtained in a study evaluating erlotinib combined with cetuximab for patients with acquired resistance to erlotinib (ORR=0%) (60). Interestingly, the “sandwich strategy” based on afatinib exhibited significant activity, with an ORR of 29% and a median PFS of 4.7 months in advanced EGFR-mutant NSCLC and acquired resistance to erlotinib/gefitinib. Furthermore, the results showed comparable treatment effects in patients with T790M-postive or -negative tumors (60). Inspired by the afatinib-based “sandwich strategy”, the randomized phase II, multicenter trial SWOG S1403 evaluated the benefit of afatinib plus cetuximab compared with afatinib alone as first-line treatment targeting classic mutations (exon 19del and exon 21 L858R). Unfortunately, the results showed that the addition of cetuximab to afatinib did not improve outcomes, with 30% of patients in the combination regimen discontinuing cetuximab due to intolerable adverse events (AEs) (61). As an EGFR-MET bispecific antibody, amivantamab (approved by the FDA in 2021) has also been explored in combination with lazertinib in cases progressing on osimertinib (CHRYSALIS), and encouraging preliminary antitumor activity was observed, with an ORR of 36% (62, 63). CHRYSALIS-2 (64) is an ongoing phase 1/1b open-label study evaluating the effects of amivantamab + lazertinib in patients with EGFR-mutant NSCLC (first-line therapy) and a phase 3 study (MARIPOSA) in ongoing to assess activity of the combination in EGFR-mutant NSCLC (first-line therapy) (65).
Van Veggel et al. reported four stage IV NSCLC patients with EGFR exon 20ins treated with afatinib (40 mg/d) and cetuximab (250-500 mg, q2w); 3/4 patients (Ser768_Asp770dup, Asn771_His773dup and Ala767_Val769dup) showed a partial response (PR), whereas the remaining (His773dup) showed stable disease (SD); the mPFS was 5.4 months (66). Osimertinib (80 mg/d) plus cetuximab (400 mg, q2w) is also effective for the complex mutation EGFR A767_S768insSVD and EGFRex20-ins, and the authors concluded that high-dose osimertinib (160 mg/d) can be considered for such patients, with good tolerance (67).
C797S and T790M mutations may occur either in cis or trans in NSCLC (68). In general, tumors harboring the trans-C797S/T790M/classic mutation are sensitive to a combination of first- and third-generation TKIs (69–71), and cis-C797S/T790M/classic mutations are resistant to first/third- and first plus third-generation EGFR TKIs (36, 72). Preclinical studies have demonstrated brigatinib plus cetuximab to be effective for tumors with cis-C797S/T790M/classic mutations in vitro and in vivo (38). Wang et al. reported the first clinical evidence for the combination of brigatinib (90 mg/d) and cetuximab (600 mg, q4w) against the cis-C797S/T790M/19del mutation, with a remarkable PFS of 9 months (36). Interestingly, trans-C797S/T790M/classic mutations treated with a combination of first- and third-generation TKIs resulted in rapid drug resistance [PFS was 1 month in a case reported by Arulananda (71)] or evolved to cis-C797S/T790M/classic mutations in a short time (69, 70). Zhou et al. found that addition of bevacizumab to osimertinib and brigatinib may delay drug resistance (69). Overall, further research is needed to assess whether it is reasonable to apply brigatinib and cetuximab early before drug resistance occurs under such circumstances.
For some rare complex EGFR mutations, EGFR TKIs plus EGFR mAbs are still therapeutically effective. We have reported a rare case of lung adenocarcinoma with a novel EGFR–intergenic region (IGR) (SEC61G) fusion (the mutation frequency was 79.8%) and EGFR amplification (copy number: 5.6). The patients received first-line targeted combination therapy with gefitinib (250 mg/d) and cetuximab (500 mg/m2, q2w) and achieved PR according to RECIST guidelines (19).
Interestingly, several preclinical trials of combinations of various EGFR TKIs and EGFR mAb targeting EGFR were successful but failed to translate into clinically significant results (59, 61, 73, 74) (Table 2). There are several reasons for such inconsistencies when comparing preclinical and clinical studies.
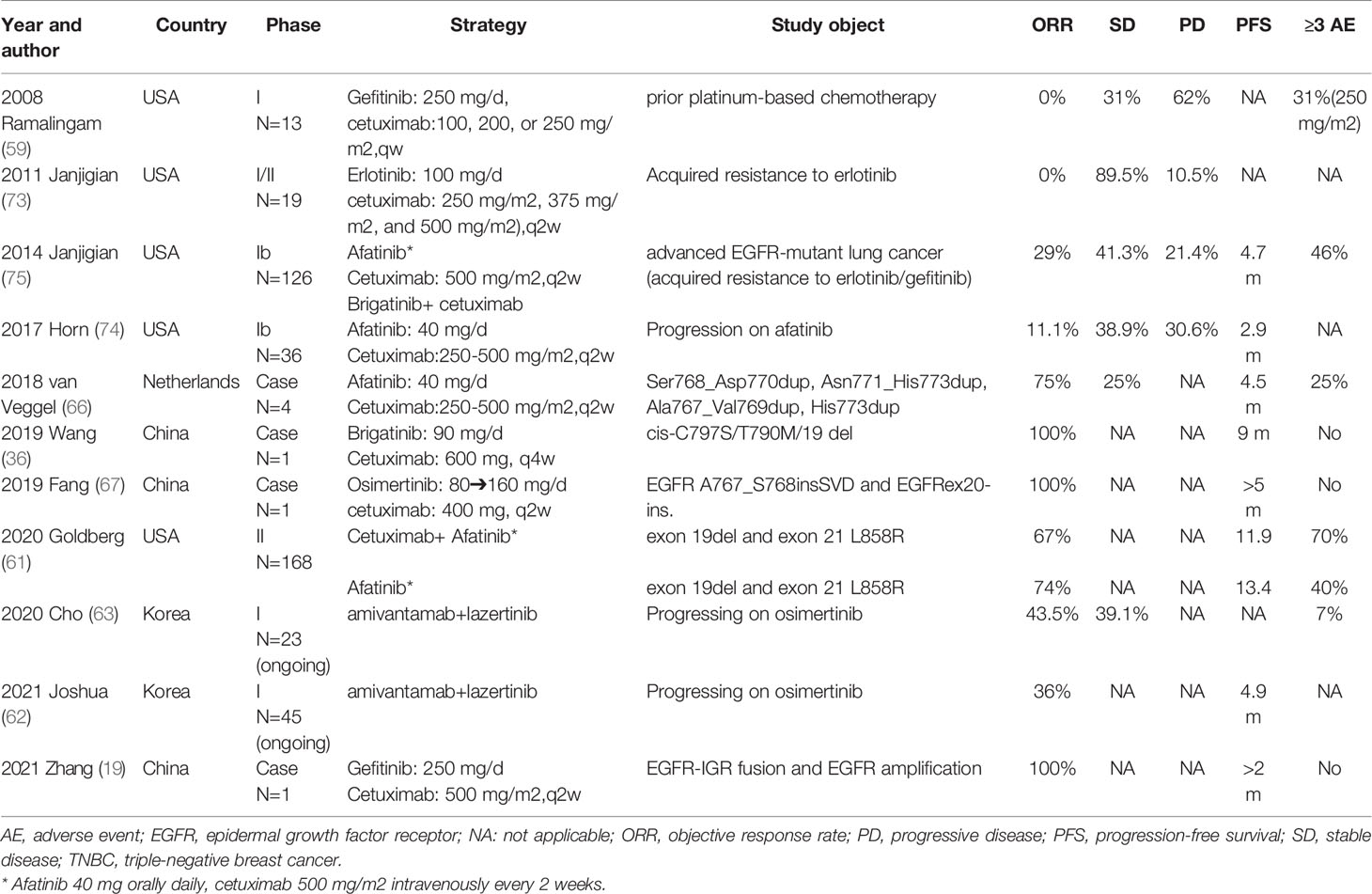
Table 2 Clinical data evaluating the “sandwich” strategy.
First, studies with significant results usually included specific mutation sites, either preclinical or clinical. That is, this combination is effective against only specific uncommon EGFR mutations. For example, although EAI045 in combination with cetuximab can dramatically inhibit tumors harboring EGFR L858R/T79M/C797S in mice, this combination does not well target 19del/T79M/C797S (55). Second, there is limited drug dosage in humans. For example, afatinib and dacomitinib are effective at inhibiting EGFR T790M in vitro and in preclinical models, but their clinical utility for EGFR T790M is hampered because the clinical doses required to effectively inhibit this mutation in vivo are high and cause severe toxicity (35). In general, efficacy is significantly related to dosage: high doses of EGFR TKIs and mAb result in optimal regression of large tumors in animals (49), but high dosages of drugs usually lead to complicated toxicity situations in humans (59). Third, a more complex tumor microenvironment exists in the human body, and no animal model is a complete replica for a process within humans (76). Indeed, one study showed that only approximately 1/3 of animal research translates into successful human research (77). Fourth, when we compare preclinical and clinical studies, mutation in the subjects included (if any) may have a critical difference in the success or failure of the experiments/clinical trials. A “sandwich” strategy that achieves more precise intervention (EGFR mutation before treatment) will result in better disease control (75). An erlotinib/afatinib-based “sandwich” strategy for patients with acquired resistance to erlotinib (73)/afatinib (74) has significantly reduced effectiveness. When we compare SWOG S1403 (first line: treatment naïve mutation) (61) with the study by Janjigian (second line: acquired resistance) (75), we conclude the existence of differences in the biology of untreated disease compared with that of acquired resistance (uncommon mutations may be the targeted beneficiary group by the “sandwich” strategy). In terms of medication, the second-generation irreversible pantarget inhibitor afatinib may confer more complete inhibition of ErbB family members. Cetuximab was used at a higher dose (500 mg/m2) in the afatinib-based “sandwich” strategy (59, 73, 75) (Table 1). Fifth, negative experimental results are less likely to be published (78), which may create the illusion of success for all published preclinical studies.
“Sandwich” Strategy for Other Tumors
In addition to NSCLC, several malignancies, such as glioblastoma multiforme, glioblastoma, breast cancer, vulvar squamous carcinoma, prostate carcinoma, head and neck cancer, and gastrointestinal cancers (stomach, colorectal, and pancreatic carcinomas), are associated with EGFR mutation or amplification. Therefore, EGFR-targeted therapy should theoretically be effective.
As early as 2004, Matar et al. explored the blocking effect of gefitinib and cetuximab on the EGFR pathway using human cancer cell lines (vulvar squamous carcinoma, prostate carcinoma cells and breast carcinoma cells) and A431 (vulvar squamous carcinoma) xenografts in nude mice (49). The results demonstrated that the “sandwich” strategy resulted in a synergistic effect with regard to the inhibition of cell proliferation and regression of tumors by targeting EGFR (49). Similar conclusions were reported with the gefitinib/erlotinib and cetuximab combination for head/neck cancer (51). Triple-negative breast cancer (TNBC) is characterized by poor prognosis, and a study suggested that the “sandwich” strategy might result in an enhanced antitumor effect in vitro (53).
With regard to the gastrointestinal digestive system, colorectal cancer escapes EGFR blockade by downstream signaling activation (RAS-MEK), and combination of EGFR blockade and MEK blockade can prevent resistance both in vitro and in vivo (79). Additionally, gefitinib and cetuximab can inhibit colon carcinoma cell proliferation and promote tumor regression in nude mice (49). EGFR amplification predicts aggressive biological behavior and poor prognosis. Preclinical trials performed on PDX models with EGFR-amplified gastroesophageal adenocarcinoma revealed that the cetuximab and lapatinib/erlotinib combination still results in a deep and durable response (58). Similarly, in a study evaluating the biliary tract cancer cell line HuCCT1 (in vitro cell proliferation inhibition and apoptosis promotion) and pancreatic cancer cell line Panc430 xenografts, tumor growth in vivo was significantly decreased with combined therapy (gefitinib/erlotinib and cetuximab) (44).
However, the data about the “sandwich” strategy are fairly limited in clinical settings. Most of the studies have focused on EGFR TKIs or EGFR mAbs, rather than their combination. A randomized phase 2 study (NCT01919879) comparing cetuximab and cetuximab plus afatinib in refractory wtKRAS metastatic colorectal cancer has been completed, but the results have not yet been reported (80).
Limitations and Prospects of the “Sandwich” Strategy
The “sandwich” strategy exerts a stronger antitumor effect than a single drug and has certain clinical prospects for NSCLC, especially in patients with certain kinds of uncommon EGFR mutations. However, this combination is far from optimal, which limits its wide clinical use.
Relatively Severe Toxicity of the “Sandwich” Strategy
The ultimate goal of targeted therapy is low toxicity and high efficiency. The most prominent problem of the “sandwich” strategy is its relatively increased inhibition of wild-type EGFR, leading to intolerable toxicity. Based on clinical data for patients with acquired resistance to first-generation TKIs, therapy-related grade ≥3 adverse events of the afatinib-based “sandwich” strategy occurred in 46%; 36% required a dose reduction, and 13% discontinued therapy due to intolerable AEs (75). Based on SWOG S1403, therapy-related grade ≥3 AEs with afatinib plus cetuximab and afatinib occurred in 70% and 40% of patients, respectively, and a dose reduction of afatinib to 30 mg occurred in 56.7% and 26.2% of patients, respectively; 30% discontinued cetuximab therapy due to intolerable AEs in the combination group (61). It is noteworthy that the first/third-generation TKI-based “sandwich” strategy appears to have good tolerance, with only 31% reporting ≥3 AEs (59). However, the sample size for this combination is relatively small, involving mostly case reports (19, 59, 67, 73). Considering the rate of grade ≥3 AEs with TKI monotherapy (gefitinib 31% (81), erlotinib 45% (2), afatinib 36-40% (4), osimertinib 34% (5)), we conclude that AEs of the first/third-generation TKI-based “sandwich” strategy may be clinically acceptable but that the afatinib-based “sandwich” strategy significantly increases the occurrence of AEs compared to monotherapy. Thus, considering the various options for patients harboring common EGFR mutations as well as T790M mutations, the “sandwich” strategy may not be a priority selection regarding effectiveness and safety. Recent studies have shown that the novel “sandwich” strategy, amivantamab+lazertinib, may significantly reduce AEs, with grade ≥3 AEs occurring in only 7% of patients. Therefore, novel, more effective and safer therapeutic strategies may provide new insight into the “sandwich” strategy.
Drug Resistance of the “Sandwich” Strategy
Resistance to targeted therapy is inevitable, and “sandwich” strategies are no exception. In the era of precision targeted therapy, gene mutations should be rechecked after drug resistance to guide subsequent treatment. Due to limited clinical application, the mechanism of drug resistance of the “sandwich” strategy is far from understood, and it seems that EGFR-independent mutations will be the majority because of sufficient inhibition of EGFR. However, Pirazzoli et al. proved that an afatinib-based “sandwich” strategy does not suppress emergence of T790M KRAS mutations (such as G12R, G12V and G12D) and that EGFR amplification is associated with resistance to an afatinib-based “sandwich” strategy (54). Additionally, mutations in downstream signaling genes (RAS/RAF/MEK/ERK, PI3K/AKT/mTOR) are induced (54). Combined targeting with three drugs (osimertinib, bevacizumab and brigatinib) in patients with cis-C797S/T790 M/L858R mutation seems to delay the time to incidence of drug resistance (72). Preclinical data suggest that the afatinib-based “sandwich” strategy markedly delays drug resistance in transgenic mice harboring EGFR L858R (54); however, clinical studies have not yielded similar results. Inspired by a study by Zhao et al. (72), introduction of vascular endothelial growth factor receptor (VEGFR) TKIs may modify the resistance mechanism, which requires further study.
Future Prospects of Targeted Therapy
Historically, reversal of drug resistance to first-generation TKIs (with T790M as the most common resistance mutation) has been explored by implementing the “sandwich” strategy (73, 75) because there are no better treatment options. However, with the development of targeted therapies, a large number of targeted drugs (such as osimertinib (82), almonertinib (83), avitinib (84), and furmonertinib (85)) have been developed with high selectivity for the mutant type, which further reduces the value of such a strategy for this kind of mutation. for the C797S mutation, brigatinib has been shown to be effective at inhibiting cis-C797S/T790M/del19 but spares wild-type cells (38). Another novel highly potent selective 4th-generation EGFR TKI BLU-945 also has promising activity for the treatment of EGFR T790M/C797S resistant NSCLC, with high selectivity, and the ongoing SYMPHONY will reveal more data (86). The novel EGFR TKI mobocertinib (87) and third-generation TKI furmonertinib (approval in China in March 2021 (88)) (89, 90) have been shown to be effective for EGFR exon 20ins disease. Studies have shown that the effect of treatment with immunotherapy in patients harboring EGFR exon 20ins mutations is poor (91) and can even lead to hyperprogressive disease (92). Interestingly, the EGFR–MET-targeted bispecific antibody amivantamab (approved by the FDA in May 2021) yielded robust and durable responses to EGFR exon 20ins-mutated tumors after progression on platinum-based chemotherapy (74, 93).
Numerous nonsensitive mutations exist, and combination treatment may drastically reduce the number of remaining tumor cells compared to a single drug. Combination drugs often indicate combined AEs. Theoretically, a single agent targeting nonsensitive mutations may be of great significance, similar to bispecific antibodies in the field of cancer immunotherapy, such as amivantamab (31, 34).
What is the potential for the “sandwich” strategy? First, as we described, EGFR TKI and EGFR mAbs may exert synergistic antitumor effects, suggesting the possibility that we can reduce the dose of a drug to control AEs without impairing the therapeutic effect. Second, with the precision of medical interventions, treatment for tumors with specific uncommon EGFR mutations will undoubtedly be tailored more individually, especially with the “sandwich” strategy (relatively severe toxicity). Third, research and development of new drugs with high efficacy and low toxicity may offer breakthroughs in the “sandwich” strategy. A novel combination amivantamab + lazertinib is being explored as a first-line or multiple-line treatment, with promising therapeutic effects.
As another cornerstone of therapy, the role of chemotherapy in NSCLC should not be ignored. Nonetheless, studies have shown that TKIs are not inferior to or even better than chemotherapy with respect to disease control in patients with uncommon EGFR mutations (such as exon 20ins, L861Q and G719X) (94, 95). Studies have also demonstrated that chemotherapy for patients with uncommon mutations may result in significantly better survival (96, 97) than with EGFR-TKIs, despite worse mPFS (97). Therefore, it is currently unknown how to best sequence these different therapies. There is also a lack of evidence with respect to the advantages and disadvantages of chemotherapy and the “sandwich” strategy. Further studies are needed to investigate appropriate drug sequences.
Conclusions
In this review, we focus on several EGFR-dependent mutations for which a “sandwich” strategy is possible. Our review highlights that dual EGFR inhibition is particularly meaningful in this patient population. We conclude that the “sandwich” strategy may have limited benefit in patients with previously untargeted classic EGFR mutations and is expected to improve the prognosis of NSCLC patients harboring certain uncommon EGFR mutations. However, their relatively severe toxicity limits their clinical application, and there is an urgent need to develop new targeted drugs with less inhibition of wild-type EGFR.
Author Contributions
GZ, BY, and YG contributed to conception and design of the study. GZ, BY, YG and HY organized the database. HY performed the statistical analysis. GZ wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This study was supported by the Key Scientific Research Projects of Institutions of Higher Learning in Henan Province (No. 21A320032).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Dr Hui Ma (Department of Respiratory Disease, People’s Hospital of Zhengzhou) for her valuable suggestion.
References
1. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall Survival With Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N Engl J Med (2020) 382:41–50. doi: 10.1056/NEJMoa1913662
2. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib Versus Standard Chemotherapy as First-Line Treatment for European Patients With Advanced EGFR Mutation-Positive non-Small-Cell Lung Cancer (EURTAC): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol (2012) 13:239–46. doi: 10.1016/s1470-2045(11)70393-x
3. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or Carboplatin-Paclitaxel in Pulmonary Adenocarcinoma. N Engl J Med (2009) 361:947–57. doi: 10.1056/NEJMoa0810699
4. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib Versus Cisplatin Plus Gemcitabine for First-Line Treatment of Asian Patients With Advanced non-Small-Cell Lung Cancer Harbouring EGFR Mutations (LUX-Lung 6): An Open-Label, Randomised Phase 3 Trial. Lancet Oncol (2014) 15:213–22. doi: 10.1016/s1470-2045(13)70604-1
5. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med (2018) 378:113–25. doi: 10.1056/NEJMoa1713137
6. Kwon Y, Kim M, Jung H, Kim Y, Jeoung D. Targeting Autophagy for Overcoming Resistance to Anti-EGFR Treatments. Cancers (2019) 11(9):1374–7. doi: 10.3390/cancers11091374
7. JC Y, Schuler M, Popat S, Miura S, Heeke S, Park K, et al. Afatinib for the Treatment of NSCLC Harboring Uncommon EGFR Mutations: A Database of 693 Cases. J Thorac Oncol (2020) 15:803–15. doi: 10.1016/j.jtho.2019.12.126
8. Zhang YC, Chen ZH, Zhang XC, Xu CR, Yan HH, Xie Z, et al. Analysis of Resistance Mechanisms to Abivertinib, a Third-Generation EGFR Tyrosine Kinase Inhibitor, in Patients With EGFR T790M-Positive non-Small Cell Lung Cancer From a Phase I Trial. EBioMedicine (2019) 43:180–7. doi: 10.1016/j.ebiom.2019.04.030
9. Gatzemeier U, Pluzanska A, Szczesna A, Kaukel E, Roubec J, De Rosa F, et al. Phase III Study of Erlotinib in Combination With Cisplatin and Gemcitabine in Advanced non-Small-Cell Lung Cancer: The Tarceva Lung Cancer Investigation Trial. J Clin Oncol Off J Am Soc Clin Oncol (2007) 25:1545–52. doi: 10.1200/jco.2005.05.1474
10. Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, et al. Gefitinib in Combination With Gemcitabine and Cisplatin in Advanced non-Small-Cell Lung Cancer: A Phase III Trial–INTACT 1. J Clin Oncol (2004) 22:777–84. doi: 10.1200/jco.2004.08.001
11. Herbst RS, Giaccone G, Schiller JH, Natale RB, Miller V, Manegold C, et al. Gefitinib in Combination With Paclitaxel and Carboplatin in Advanced non-Small-Cell Lung Cancer: A Phase III Trial–INTACT 2. J Clin Oncol Off J Am Soc Clin Oncol (2004) 22:785–94. doi: 10.1200/jco.2004.07.215
12. Herbst RS, Prager D, Hermann R, Fehrenbacher L, Johnson BE, Sandler A, et al. TRIBUTE: A Phase III Trial of Erlotinib Hydrochloride (OSI-774) Combined With Carboplatin and Paclitaxel Chemotherapy in Advanced non-Small-Cell Lung Cancer. J Clin Oncol (2005) 23:5892–9. doi: 10.1200/jco.2005.02.840
13. Akamatsu H, Toi Y, Hayashi H, Fujimoto D, Tachihara M, Furuya N, et al. Efficacy of Osimertinib Plus Bevacizumab vs Osimertinib in Patients With EGFR T790M-Mutated Non-Small Cell Lung Cancer Previously Treated With Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor: West Japan Oncology Group 8715l Phase 2 Randomized Clinical Trial. JAMA Oncol (2021) 7:386–94. doi: 10.1001/jamaoncol.2020.6758
14. Nakagawa K, Garon EB, Seto T, Nishio M, Ponce Aix S, Paz-Ares L, et al. Ramucirumab Plus Erlotinib in Patients With Untreated, EGFR-Mutated, Advanced non-Small-Cell Lung Cancer (RELAY): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2019) 20:1655–69. doi: 10.1016/s1470-2045(19)30634-5
15. Saito H, Fukuhara T, Furuya N, Watanabe K, Sugawara S, Iwasawa S, et al. Erlotinib Plus Bevacizumab Versus Erlotinib Alone in Patients With EGFR-Positive Advanced non-Squamous non-Small-Cell Lung Cancer (NEJ026): Interim Analysis of an Open-Label, Randomised, Multicentre, Phase 3 Trial. Lancet Oncol (2019) 20:625–35. doi: 10.1016/s1470-2045(19)30035-x
16. Kawashima Y, Fukuhara T, Saito H, Furuya N, Watanabe K, Sugawara S, et al. Bevacizumab Plus Erlotinib Versus Erlotinib Alone in Japanese Patients With Advanced, Metastatic, EGFR-Mutant non-Small-Cell Lung Cancer (NEJ026): Overall Survival Analysis of an Open-Label, Randomised, Multicentre, Phase 3 Trial. Lancet Respir Med (2022) 10:72–82. doi: 10.1016/s2213-2600(21)00166-1
17. Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition With Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov (2018) 8:1529–39. doi: 10.1158/2159-8290.cd-18-1022
18. Goldberg S, Redman M, Lilenbaum R, Politi K, Stinchcombe T, Horn L, et al. EGFRRandomized Trial of Afatinib Plus Cetuximab Versus Afatinib Alone for First-Line Treatment of -Mutant Non-Small-Cell Lung Cancer: Final Results From SWOG S1403. J Clin Oncol (2020) 38:(34):4076–85. doi: 10.1200/jco.20.01149
19. Zhang G, Xia P, Zhao S, Yuan L, Wang X, Li X, et al. Gefitinib Combined With Cetuximab for the Treatment of Lung Adenocarcinoma Harboring the EGFR-Intergenic Region (SEC61G) Fusion and EGFR Amplification. Oncol (2021) 26:e1898–902. doi: 10.1002/onco.13921
20. Kumar A, Petri ET, Halmos B, Boggon TJ. Structure and Clinical Relevance of the Epidermal Growth Factor Receptor in Human Cancer. J Clin Oncol (2008) 26:1742–51. doi: 10.1200/jco.2007.12.1178
21. Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell (2006) 125:1137–49. doi: 10.1016/j.cell.2006.05.013
22. Mendelsohn J. Epidermal Growth Factor Receptor Inhibition by a Monoclonal Antibody as Anticancer Therapy. Clin Cancer Res (1997) 3:2703–7.
23. Wheeler DL, Dunn EF, Harari PM. Understanding Resistance to EGFR Inhibitors-Impact on Future Treatment Strategies. Nat Rev Clin Oncol (2010) 7:493–507. doi: 10.1038/nrclinonc.2010.97
24. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. New Engl J Med (2012) 366:883–92. doi: 10.1056/NEJMoa1113205
25. Marusyk A, Janiszewska M, Polyak K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell (2020) 37:471–84. doi: 10.1016/j.ccell.2020.03.007
26. Hu X, Fujimoto J, Ying L, Fukuoka J, Ashizawa K, Sun W, et al. Multi-Region Exome Sequencing Reveals Genomic Evolution From Preneoplasia to Lung Adenocarcinoma. Nat Commun (2019) 10:2978. doi: 10.1038/s41467-019-10877-8
27. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. New Engl J Med (2017) 376:2109–21. doi: 10.1056/NEJMoa1616288
28. Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL, et al. Structural, Biochemical, and Clinical Characterization of Epidermal Growth Factor Receptor (EGFR) Exon 20 Insertion Mutations in Lung Cancer. Sci Trans Med (2013) 5:216ra177. doi: 10.1126/scitranslmed.3007205
29. Lazzari C, Gregorc V, Karachaliou N, Rosell R, Santarpia M. Mechanisms of Resistance to Osimertinib. J Thorac Dis (2020) 12:2851–8. doi: 10.21037/jtd.2019.08.30
30. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in non-Small Cell Lung Cancer Harboring EGFR T790M. Nat Med (2015) 21:560–2. doi: 10.1038/nm.3854
31. Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin Cancer Res (2018) 24:3097–107. doi: 10.1158/1078-0432.ccr-17-2310
32. Kannan S, Fox SJ, Verma CS. Exploring Gatekeeper Mutations in EGFR Through Computer Simulations. J Chem Inf Model (2019) 59:2850–8. doi: 10.1021/acs.jcim.9b00361
33. Russo A, Franchina T, Ricciardi G, Battaglia A, Picciotto M, Adamo V. Heterogeneous Responses to Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs) in Patients With Uncommon EGFR Mutations: New Insights and Future Perspectives in This Complex Clinical Scenario. Int J Mol Sci (2019) 20(6):1431–50. doi: 10.3390/ijms20061431
34. Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, et al. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res (2015) 21:3913–23. doi: 10.1158/1078-0432.ccr-14-2789
35. Skoulidis F, Papadimitrakopoulou VA. Targeting the Gatekeeper: Osimertinib in EGFR T790M Mutation-Positive Non-Small Cell Lung Cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2017) 23:618–22. doi: 10.1158/1078-0432.ccr-15-2815
36. Wang X, Zhou L, Yin JC, Wu X, Shao YW, Gao B. Lung Adenocarcinoma Harboring EGFR 19del/C797S/T790M Triple Mutations Responds to Brigatinib and Anti-EGFR Antibody Combination Therapy. J Thorac Oncol (2019) 14:e85–8. doi: 10.1016/j.jtho.2019.01.015
37. Park HR, Kim TM, Lee Y, Kim S, Park S, Ju YS, et al. Acquired Resistance to Third-Generation EGFR Tyrosine Kinase Inhibitors in Patients With De Novo EGFR(T790M)-Mutant NSCLC. J Thorac Oncol (2021) 16:1859–71. doi: 10.1016/j.jtho.2021.06.013
38. Uchibori K, Inase N, Araki M, Kamada M, Sato S, Okuno Y, et al. Brigatinib Combined With Anti-EGFR Antibody Overcomes Osimertinib Resistance in EGFR-Mutated non-Small-Cell Lung Cancer. Nat Commun (2017) 8:14768. doi: 10.1038/ncomms14768
39. Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, et al. Rebiopsy of Lung Cancer Patients With Acquired Resistance to EGFR Inhibitors and Enhanced Detection of the T790M Mutation Using a Locked Nucleic Acid-Based Assay. Clin Cancer Res (2011) 17:1169–80. doi: 10.1158/1078-0432.ccr-10-2277
40. Laufkötter O, Hu H, Miljković F, Bajorath J. Structure- and Similarity-Based Survey of Allosteric Kinase Inhibitors, Activators, and Closely Related Compounds. J Medicinal Chem (2022) 65:922–34. doi: 10.1021/acs.jmedchem.0c02076
41. Carpenter G. Receptors for Epidermal Growth Factor and Other Polypeptide Mitogens. Annu Rev Biochem (1987) 56:881–914. doi: 10.1146/annurev.bi.56.070187.004313
42. Tan AR, Yang X, Hewitt SM, Berman A, Lepper ER, Sparreboom A, et al. Evaluation of Biologic End Points and Pharmacokinetics in Patients With Metastatic Breast Cancer After Treatment With Erlotinib, an Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor. J Clin Oncol (2004) 22:3080–90. doi: 10.1200/jco.2004.08.189
43. Gregorc V, Ceresoli GL, Floriani I, Spreafico A, Bencardino KB, Ludovini V, et al. Effects of Gefitinib on Serum Epidermal Growth Factor Receptor and HER2 in Patients With Advanced non-Small Cell Lung Cancer. Clin Cancer Res (2004) 10:6006–12. doi: 10.1158/1078-0432.ccr-03-0770
44. Jimeno A, Rubio-Viqueira B, Amador ML, Oppenheimer D, Bouraoud N, Kulesza P, et al. Epidermal Growth Factor Receptor Dynamics Influences Response to Epidermal Growth Factor Receptor Targeted Agents. Cancer Res (2005) 65:3003–10. doi: 10.1158/0008-5472.can-04-3586
45. Graziano RF, Engelhardt JJ. Role of Fcγrs in Antibody-Based Cancer Therapy. Curr Topics Microbiol Immunol (2019) 423:13–34. doi: 10.1007/82_2019_150
46. Kimura H, Sakai K, Arao T, Shimoyama T, Tamura T, Nishio K. Antibody-Dependent Cellular Cytotoxicity of Cetuximab Against Tumor Cells With Wild-Type or Mutant Epidermal Growth Factor Receptor. Cancer Sci (2007) 98:1275–80. doi: 10.1111/j.1349-7006.2007.00510.x
47. Gruijs M, Braster R, Overdijk MB, Hellingman T, Verploegen S, Korthouwer R, et al. Epidermal Growth Factor Receptor as Target for Perioperative Elimination of Circulating Colorectal Cancer Cells. J Oncol (2022) 2022:3577928. doi: 10.1155/2022/3577928
48. Rösner T, Kahle S, Montenegro F, Matlung HL, Jansen JHM, Evers M, et al. Immune Effector Functions of Human IgG2 Antibodies Against EGFR. Mol Cancer Ther (2019) 18:75–88. doi: 10.1158/1535-7163.mct-18-0341
49. Matar P, Rojo F, Cassia R, Moreno-Bueno G, Di Cosimo S, Tabernero J, et al. Combined Epidermal Growth Factor Receptor Targeting With the Tyrosine Kinase Inhibitor Gefitinib (ZD1839) and the Monoclonal Antibody Cetuximab (IMC-C225): Superiority Over Single-Agent Receptor Targeting. Clin Cancer Res (2004) 10:6487–501. doi: 10.1158/1078-0432.ccr-04-0870
50. Bos M, Mendelsohn J, Kim YM, Albanell J, Fry DW, Baselga J. PD153035, a Tyrosine Kinase Inhibitor, Prevents Epidermal Growth Factor Receptor Activation and Inhibits Growth of Cancer Cells in a Receptor Number-Dependent Manner. Clin Cancer Res (1997) 3:2099–106.
51. Huang S, Armstrong EA, Benavente S, Chinnaiyan P, Harari PM. Dual-Agent Molecular Targeting of the Epidermal Growth Factor Receptor (EGFR): Combining Anti-EGFR Antibody With Tyrosine Kinase Inhibitor. Cancer Res (2004) 64:5355–62. doi: 10.1158/0008-5472.can-04-0562
52. Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al. Dual Targeting of EGFR can Overcome a Major Drug Resistance Mutation in Mouse Models of EGFR Mutant Lung Cancer. J Clin Invest (2009) 119:3000–10. doi: 10.1172/jci38746
53. El Guerrab A, Bamdad M, Kwiatkowski F, Bignon YJ, Penault-Llorca F, Aubel C. Anti-EGFR Monoclonal Antibodies and EGFR Tyrosine Kinase Inhibitors as Combination Therapy for Triple-Negative Breast Cancer. Oncotarget (2016) 7:73618–37. doi: 10.18632/oncotarget.12037
54. Pirazzoli V, Ayeni D, Meador CB, Sanganahalli BG, Hyder F, de Stanchina E, et al. Afatinib Plus Cetuximab Delays Resistance Compared to Single-Agent Erlotinib or Afatinib in Mouse Models of TKI-Naïve EGFR L858R-Induced Lung Adenocarcinoma. Clin Cancer Res (2016) 22:426–35. doi: 10.1158/1078-0432.ccr-15-0620
55. Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, et al. Overcoming EGFR(T790M) and EGFR(C797S) Resistance With Mutant-Selective Allosteric Inhibitors. Nature (2016) 534:129–32. doi: 10.1038/nature17960
56. Della Corte CM, Ciaramella V, Cardone C, La Monica S, Alfieri R, Petronini PG, et al. Antitumor Efficacy of Dual Blockade of EGFR Signaling by Osimertinib in Combination With Selumetinib or Cetuximab in Activated EGFR Human NCLC Tumor Models. J Thorac Oncol (2018) 13:810–20. doi: 10.1016/j.jtho.2018.02.025
57. Hasegawa H, Yasuda H, Hamamoto J, Masuzawa K, Tani T, Nukaga S, et al. Efficacy of Afatinib or Osimertinib Plus Cetuximab Combination Therapy for non-Small-Cell Lung Cancer With EGFR Exon 20 Insertion Mutations. Lung Cancer (Amsterdam Netherlands) (2019) 127:146–52. doi: 10.1016/j.lungcan.2018.11.039
58. Corso S, Pietrantonio F, Apicella M, Migliore C, Conticelli D, Petrelli A, et al. Optimized EGFR Blockade Strategies in EGFR Addicted Gastroesophageal Adenocarcinomas. Clin Cancer Res (2021) 27:3126–40. doi: 10.1158/1078-0432.ccr-20-0121
59. Ramalingam S, Forster J, Naret C, Evans T, Sulecki M, Lu H, et al. Dual Inhibition of the Epidermal Growth Factor Receptor With Cetuximab, an IgG1 Monoclonal Antibody, and Gefitinib, a Tyrosine Kinase Inhibitor, in Patients With Refractory non-Small Cell Lung Cancer (NSCLC): A Phase I Study. J Thorac Oncol (2008) 3:258–64. doi: 10.1097/JTO.0b013e3181653d1b
60. Baselga J, Scho¨ffski P, Rojo F. A Phase I Pharmacokinetic (PK) and Molecular Pharmacodynamic (PD) Study of the Combination of Two Anti-EGFR Therapies, the Monoclonal Antibody (MAb) Cetuximab and the Tyrosine Kinase Inhibitor (TKI) Gefitinib (G), in Patients (Pts) With Advanced Colorectal (CRC), Head and Neck (HNC), and non-Small Cell Lung Cancer (NSCLC). Proc Am Soc Clin Oncol (2006) 24:abstr 3006.
61. Goldberg SB, Redman MW, Lilenbaum R, Politi K, Stinchcombe TE, Horn L, et al. Randomized Trial of Afatinib Plus Cetuximab Versus Afatinib Alone for First-Line Treatment of EGFR-Mutant Non-Small-Cell Lung Cancer: Final Results From SWOG S1403. J Clin Oncol (2020) 38:4076–85. doi: 10.1200/jco.20.01149
62. Bauml J, Cho BC, Park K, Lee KH, CHO EK, Kim D-W, et al. Amivantamab in Combination With Lazertinib for the Treatment of Osimertinib-Relapsed, Chemotherapy-Naïve EGFR Mutant (EGFRm) non-Small Cell Lung Cancer (NSCLC) and Potential Biomarkers for Response. J Clin Oncol (2021) 39:9006–6. doi: 10.1200/JCO.2021.39.15_suppl.9006
63. Cho BC, Lee KH, Cho EK, Kim DW, Lee JS, Han JY, et al. 1258o Amivantamab (JNJ-61186372), an EGFR-MET Bispecific Antibody, in Combination With Lazertinib, a 3rd-Generation Tyrosine Kinase Inhibitor (TKI), in Advanced EGFR NSCLC. Ann Oncol (2020) 31:S813. doi: 10.1016/j.annonc.2020.08.1572
64. Shu CA, Goto K, Cho BC, Griesinger F, Yang JC-H, Felip E, et al. CHRYSALIS-2: A Phase 1/1b Study of Lazertinib as Monotherapy and in Combination With Amivantamab in Patients With EGFR-Mutant NSCLC. J Clin Oncol (2021) 39:TPS9132–TPS9132. doi: 10.1200/JCO.2021.39.15_suppl.TPS9132
65. Cho BC, Felip E, Hayashi H, Thomas M, Lu S, Besse B, et al. MARIPOSA: Phase 3 Study of First-Line Amivantamab + Lazertinib Versus Osimertinib in EGFR-Mutant non-Small-Cell Lung Cancer. Future Oncol (London England) (2022) 18:639–47. doi: 10.2217/fon-2021-0923
66. van Veggel B, de Langen AJ, Hashemi SMS, Monkhorst K, Heideman DAM, Thunnissen E, et al. Afatinib and Cetuximab in Four Patients With EGFR Exon 20 Insertion-Positive Advanced NSCLC. J Thorac Oncol (2018) 13:1222–6. doi: 10.1016/j.jtho.2018.04.012
67. Fang W, Huang Y, Gan J, Hong S, Zhang LA. Patient With EGFR Exon 20 Insertion-Mutant Non-Small Cell Lung Cancer Responded to Osimertinib Plus Cetuximab Combination Therapy. J Thorac Oncol (2019) 14:e201–2. doi: 10.1016/j.jtho.2019.04.013
68. Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, et al. The Allelic Context of the C797S Mutation Acquired Upon Treatment With Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin Cancer Res (2015) 21:3924–33. doi: 10.1158/1078-0432.ccr-15-0560
69. Zhou Z, Zhao Y, Shen S, Gu L, Niu X, Xu Y, et al. Durable Clinical Response of Lung Adenocarcinoma Harboring EGFR 19del/T790M/in Trans-C797S to Combination Therapy of First- and Third-Generation EGFR Tyrosine Kinase Inhibitors. J Thorac Oncol (2019) 14:e157–9. doi: 10.1016/j.jtho.2019.04.020
70. Wang Z, Yang JJ, Huang J, Ye JY, Zhang XC, Tu HY, et al. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance. J Thorac Oncol (2017) 12:1723–7. doi: 10.1016/j.jtho.2017.06.017
71. Arulananda S, Do H, Musafer A, Mitchell P, Dobrovic A, John T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-Mutated Non-Small Cell Lung Cancer. J Thorac Oncol (2017) 12:1728–32. doi: 10.1016/j.jtho.2017.08.006
72. Zhao J, Zou M, Lv J, Han Y, Wang G, Wang G. Effective Treatment of Pulmonary Adenocarcinoma Harboring Triple EGFR Mutations of L858R, T790M, and Cis-C797S by Osimertinib, Bevacizumab, and Brigatinib Combination Therapy: A Case Report. OncoTargets Ther (2018) 11:5545–50. doi: 10.2147/ott.s170358
73. Janjigian YY, Azzoli CG, Krug LM, Pereira LK, Rizvi NA, Pietanza MC, et al. Phase I/II Trial of Cetuximab and Erlotinib in Patients With Lung Adenocarcinoma and Acquired Resistance to Erlotinib. Clin Cancer Res (2011) 17:2521–7. doi: 10.1158/1078-0432.ccr-10-2662
74. Friedlaender A, Subbiah V, Russo A, Banna GL, Malapelle U, Rolfo C, et al. EGFR and HER2 Exon 20 Insertions in Solid Tumours: From Biology to Treatment. Nat Rev Clin Oncol (2022) 19:51–69. doi: 10.1038/s41571-021-00558-1
75. Janjigian YY, Smit EF, Groen HJ, Horn L, Gettinger S, Camidge DR, et al. Dual Inhibition of EGFR With Afatinib and Cetuximab in Kinase Inhibitor-Resistant EGFR-Mutant Lung Cancer With and Without T790M Mutations. Cancer Discovery (2014) 4:1036–45. doi: 10.1158/2159-8290.cd-14-0326
76. Arora T, Mehta AK, Joshi V, Mehta KD, Rathor N, Mediratta PK, et al. Substitute of Animals in Drug Research: An Approach Towards Fulfillment of 4r's. Indian J Pharm Sci (2011) 73:1–6. doi: 10.4103/0250-474x.89750
77. Hackam DG, Redelmeier DA. Translation of Research Evidence From Animals to Humans. Jama (2006) 296:1731–2. doi: 10.1001/jama.296.14.1731
78. Joober R, Schmitz N, Annable L, Boksa P. Publication Bias: What are the Challenges and can They be Overcome? J Psychiatry Neurosci JPN (2012) 37:149–52. doi: 10.1503/jpn.120065
79. Misale S, Bozic I, Tong J, Peraza-Penton A, Lallo A, Baldi F, et al. Vertical Suppression of the EGFR Pathway Prevents Onset of Resistance in Colorectal Cancers. Nat Commun (2015) 6:8305. doi: 10.1038/ncomms9305
80. Senellart H, Samalin E, Adenis A, Malka D, Francois E, de la Fouchardiere C, et al. UCGI 25: A Multicentric Randomized Phase II Trial Evaluating Dual Targeting of the Epidermal Growth Factor (EGFR) Using the Combination of Cetuximab and Afatinib Versus Cetuximab Alone in Patients (Pts) Wih Chemotherapy Refractory wtRAS Metastatic Colorectal Cancer (mCRC). J Clin Oncol (2014) 32:TPS3666–TPS3666. doi: 10.1200/jco.2014.32.15_suppl.tps3666
81. Hosomi Y, Morita S, Sugawara S, Kato T, Fukuhara T, Gemma A, et al. Gefitinib Alone Versus Gefitinib Plus Chemotherapy for Non-Small-Cell Lung Cancer With Mutated Epidermal Growth Factor Receptor: NEJ009 Study. J Clin Oncol (2020) 38:115–23. doi: 10.1200/jco.19.01488
82. Sequist LV, Besse B, Lynch TJ, Miller VA, Wong KK, Gitlitz B, et al. Neratinib, an Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor: Results of a Phase II Trial in Patients With Advanced non-Small-Cell Lung Cancer. J Clin Oncol (2010) 28:3076–83. doi: 10.1200/jco.2009.27.9414
83. Nishino M, Suda K, Kobayashi Y, Ohara S, Fujino T, Koga T, et al. Effects of Secondary EGFR Mutations on Resistance Against Upfront Osimertinib in Cells With EGFR-Activating Mutations In Vitro. Lung Cancer (Amsterdam Netherlands) (2018) 126:149–55. doi: 10.1016/j.lungcan.2018.10.026
84. Yang X, Huang C, Chen R, Zhao J. Resolving Resistance to Osimertinib Therapy With Afatinib in an NSCLC Patient With EGFR L718Q Mutation. Clin Lung Cancer (2020) 21:e258–60. doi: 10.1016/j.cllc.2019.12.002
85. Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, et al. Irreversible Inhibitors of the EGF Receptor may Circumvent Acquired Resistance to Gefitinib. Proc Natl Acad Sci U.S.A. (2005) 102:7665–70. doi: 10.1073/pnas.0502860102
86. Spigel D, Goto K, Camidge DR, Elamin Y, de Langen AJ, Leighl NB, et al. Abstract P230: A Phase 1/2 Study of BLU-945, a Highly Potent and Selective Inhibitor of Epidermal Growth Factor Receptor (EGFR) Resistance Mutations, in Patients With EGFR-Mutant non-Small Cell Lung Cancer (NSCLC). (2021), P230–0. doi: 10.1158/1535-7163.targ-21-p230
87. Riely GJ, Neal JW, Camidge DR, Spira AI, Piotrowska Z, Costa DB, et al. Activity and Safety of Mobocertinib (TAK-788) in Previously Treated Non-Small Cell Lung Cancer With EGFR Exon 20 Insertion Mutations From a Phase I/II Trial. Cancer Discovery (2021) 11:1688–99. doi: 10.1158/2159-8290.cd-20-1598
88. Deeks ED. Furmonertinib: First Approval. Drugs (2021) 81:1775–80. doi: 10.1007/s40265-021-01588-w
89. Jiang W, Sha M, Chen C. Successful Salvage Therapy With a High Dose of Furmonertinib in a Case of Lung Adenocarcinoma Harboring EGFR Exon 20 Insertion. Am J Ther (2022). doi: 10.1097/mjt.0000000000001504
90. Jia K, Yang S, Chen B, Yu J, Wu Y, Li W, et al. Advanced Lung Adenocarcinoma Patient With EGFR Exon 20 Insertion Benefits From High-Dose Furmonertinib for Nine Months After Progression From Mobocertinib: A Case Report. Ann Trans Med (2022) 10:386. doi: 10.21037/atm-22-1167
91. Metro G, Baglivo S, Bellezza G, Mandarano M, Gili A, Marchetti G, et al. Sensitivity to Immune Checkpoint Blockade in Advanced Non-Small Cell Lung Cancer Patients With EGFR Exon 20 Insertion Mutations. Genes (2021) 12(5):679–89. doi: 10.3390/genes12050679
92. Huang X, Xia L, Lan F, Shao YW, Li W, Xia Y. Treatment of Nivolumab Results in Hyperprogressive Disease in a Patient Harboring EGFR Exon 20 Insertion and MYC Amplification. J Thorac Oncol (2019) 14:e189–91. doi: 10.1016/j.jtho.2019.04.009
93. Park K, Haura EB, Leighl NB, Mitchell P, Shu CA, Girard N, et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J Clin Oncol (2021) 39:3391–402. doi: 10.1200/jco.21.00662
94. Shi J, Yang H, Jiang T, Li X, Zhao C, Zhang L, et al. Uncommon EGFR Mutations in a Cohort of Chinese NSCLC Patients and Outcomes of First-Line EGFR-TKIs and Platinum-Based Chemotherapy. Chin J Cancer Res = Chung-kuo yen cheng yen chiu (2017) 29:543–52. doi: 10.21147/j.issn.1000-9604.2017.06.09
95. Chantharasamee J, Poungvarin N, Danchaivijitr P, Techawatanawanna S. Clinical Outcome of Treatment of Metastatic non-Small Cell Lung Cancer in Patients Harboring Uncommon EGFR Mutation. BMC Cancer (2019) 19:701. doi: 10.1186/s12885-019-5913-9
96. Watanabe S, Minegishi Y, Yoshizawa H, Maemondo M, Inoue A, Sugawara S, et al. Effectiveness of Gefitinib Against non-Small-Cell Lung Cancer With the Uncommon EGFR Mutations G719X and L861Q. J Thorac Oncol (2014) 9:189–94. doi: 10.1097/jto.0000000000000048
97. Li H, Wang C, Wang Z, Hu Y, Zhang G, Zhang M, et al. Efficacy and Long-Term Survival of Advanced Lung Adenocarcinoma Patients With Uncommon EGFR Mutations Treated With 1st Generation EGFR-TKIs Compared With Chemotherapy as First-Line Therapy. Lung Cancer (Amsterdam Netherlands) (2019) 130:42–9. doi: 10.1016/j.lungcan.2019.02.001
Keywords: epidermal growth factor receptor, tyrosine kinase inhibitor, uncommon mutation, NSCLC, drug resistance
Citation: Zhang G, Yan B, Guo Y, Yang H and Li J (2022) “Sandwich” Strategy to Intensify EGFR Blockade by Concurrent Tyrosine Kinase Inhibitor and Monoclonal Antibody Treatment in Highly Selected Patients. Front. Oncol. 12:952939. doi: 10.3389/fonc.2022.952939
Received: 25 May 2022; Accepted: 16 June 2022;
Published: 12 July 2022.
Edited by:
Ashleigh Poh, Olivia Newton-John Cancer Research Institute, AustraliaReviewed by:
Ryan O’Keefe, Olivia Newton-John Cancer Research Institute, AustraliaSai Vara Prasad Chitti, La Trobe University, Australia
Copyright © 2022 Zhang, Yan, Guo, Yang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jindong Li, MTM1OTg4MjA1ODlAMTYzLmNvbQ==
†These authors have contributed equally to this work